 Cristina Ruiz-Ruiz
Cristina Ruiz-Ruiz Francesco Calzaferri
Francesco Calzaferri Antonio G. García
Antonio G. García- 1Instituto Teófilo Hernando and Departamento de Farmacología, Facultad de Medicina, Universidad Autónoma de Madrid, Madrid, Spain
- 2Instituto de Investigación Sanitaria, Hospital Universitario de La Princesa, Universidad Autónoma de Madrid, Madrid, Spain
This review focuses on the purinergic ionotropic receptor P2X7 (P2X7R) as a potential target for developing drugs that delay the onset and/or disease progression in patients with amyotrophic lateral sclerosis (ALS). Description of clinical and genetic ALS features is followed by an analysis of advantages and drawbacks of transgenic mouse models of disease based on mutations in a bunch of proteins, particularly Cu/Zn superoxide dismutase (SOD1), TAR-DNA binding protein-43 (TDP-43), Fused in Sarcoma/Translocated in Sarcoma (FUS), and Chromosome 9 open reading frame 72 (C9orf72). Though of limited value, these models are however critical to study the proof of concept of new compounds, before reaching clinical trials. The authors also provide a description of ALS pathogenesis including protein aggregation, calcium-dependent excitotoxicity, dysfunction of calcium-binding proteins, ultrastructural mitochondrial alterations, disruption of mitochondrial calcium handling, and overproduction of reactive oxygen species (ROS). Understanding disease pathogenic pathways may ease the identification of new drug targets. Subsequently, neuroinflammation linked with P2X7Rs in ALS pathogenesis is described in order to understand the rationale of placing the use of P2X7R antagonists as a new therapeutic pharmacological approach to ALS. This is the basis for the hypothesis that a P2X7R blocker could mitigate the neuroinflammatory state, indirectly leading to neuroprotection and higher motoneuron survival in ALS patients.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease with upper and lower motor neuron (MN) loss in the cerebral cortex, brainstem, and spinal cord. Clinical symptoms start at 55 ± 15 years, with gradual loss of daily motor activities, ambulation, speech and swallowing impairment, compromised respiration, progressive paralysis, and death within 3–5 years after diagnosis. Due to this prompt death, disease prevalence is low; in the UK, the incidence is 1 in 472 women and 1 in 350 men (Alonso et al., 2009), while in the USA, there are about 16,000 affected patients and 5,000 new diagnosed cases every year (The ALS association, 2020).
For the last 25 years, the only pharmacological treatment available has been riluzole, a drug that blocks glutamate release from presynaptic terminals and accelerates glutamate clearance from the synapse. Riluzole also elicits partial blockade of presynaptic sodium channels and inhibits postsynaptic N-methyl-D-aspartate (NMDA) receptors (Doble, 1996). Unfortunately, riluzole is only able to prolong patients’ life for approximately 2 months (Miller et al., 2003).
A second drug, edaravone, was approved by the U.S. Food and Drug Administration (FDA) in 2017, with antioxidant effects that presumably preserve MN viability (Cruz, 2018). This approval generated great controversy inasmuch as it was based in a single clinical trial (CT) performed in Japanese patients, with genetic background distinct from that of Caucasian patients. Furthermore, ALS incidence is lower in Asian population, compared with European/American population (Chio et al., 2013). In any case, it is worth mentioning that edaravone has already been approved in Japan (2015), South Korea (2015), USA (2017), Canada (2018), Switzerland (2019), and China (2019).
In light of the scarce therapeutic tools currently available to delay the appearance of symptoms or disease progression, we will analyze here the following aspects of ALS to be able to extract meaningful conclusions for new drug therapy approaches: (i) genetic features; (ii) mouse models of ALS, based on gene mutations found in patients; (iii) pathogenic signaling pathways involved in MN death, and potential drug targets; (iv) neuroinflammation and the role of the purinergic P2X7 receptor (P2X7R) in disease pathogenesis; (v) available ligands for P2X7Rs; (vi) proof of concept of efficacy of some P2X7R antagonists in mouse models of ALS; (vii) past and ongoing CTs; and (viii) conclusions and perspectives.
Genetics of ALS
A family history is present in 5–10% of patients of ALS (familial ALS or fALS). The first mutations reported were those in the gene coding for Cu/Zn superoxide dismutase (SOD1) protein, which account for around 15% of all fALS and 1–2% of the sporadic cases (sporadic ALS or sALS) in European population (Rosen et al., 1993; Stephenson and Amor, 2017; Zou et al., 2017). Interestingly, these mutations (nearly 200 different mutations have been reported) do not impair the enzymatic activity of SOD1. Rather, they cause its pathological aggregation in the cytosol or within mitochondria, leading to MN toxicity (Monk and Shaw, 2006). The observation that all cell types express SOD1, yet only MNs degenerate, is puzzling (Pasinelli and Brown, 2006). This could be due to the low capacity of Ca2+ buffering exhibited by these neurons (Bezprozvanny, 2009; De Diego et al., 2012).
Around 40 different mutations in the TARDBP gene, which encodes for the transactive response (TAR)-DNA binding protein-43 (TDP-43), have been described. Mutations account for the 4–5% of fALS and <1% of sALS in European population (Arai et al., 2006; Neumann et al., 2006; Zou et al., 2017). These mutations cause the aggregation of TDP-43 protein in the cell cytoplasm, which has important functions in RNA processing and regulation. Interestingly, 97% of ALS patients present TDP-43 inclusions that are only absent in patients carrying SOD1 mutations, revealing the high relevance of this protein in ALS pathogenesis.
Mutations in the Fused in Sarcoma/Translocated in Sarcoma (FUS/TLS) gene have also been linked to the pathogenesis of ALS (Lagier-Tourenne and Cleveland, 2009). Mutations in FUS are present in 3% of fALS and <1% of sALS European patients (Zou et al., 2017), and they are responsible for the early-onset forms of the disease. This protein has relevant functions in RNA splicing and transport, and it also forms aggregates in the cell cytoplasm of ALS patients with this mutation.
Hexanucleotide repeats (GGGGCC) in intron 1 of chromosome 9 open reading frame 72 (C9orf72) gene were identified as the major cause of ALS, being present in 30–40% of fALS and 5% of sALS cases in European population (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Majounie et al., 2012; Zou et al., 2017). Hexanucleotide repeats seem to cause RNA toxicity due to the expansion of sense and antisense RNAs, the aggregation of RNA binding proteins, and the translation of repeat-association non-ATG (RAN) proteins. Patients harboring these mutations also present TDP-43 protein inclusions in the cytoplasm (Liu Y. et al., 2016).
Modeling ALS Genetics in Mice
The ALS mutations mentioned above have been identified as leading causes of disease, and based on them, several mouse models have been developed. Ideally, mouse models should present all the hallmarks of the human disease, namely, MN loss, muscle weakness and atrophy, metabolic deficits, TDP-43 inclusions and proteinopathy, gliosis, and changes in innate immunity. Even though available models are far from being perfect, they have been useful to understand ALS pathogenesis, to identify new drug targets with therapeutic potential, as well as to establish the proof of concept of the efficacy and safety of lead compounds (Stephenson and Amor, 2017). This is particularly true for the SOD1G93A mice that is by far the model that has been, and it is being used more frequently.
The discovery of new genes linked to ALS (Chia et al., 2018) and the fast development of gene editing techniques such as CRISPR/Cas9 will surely increase the number of ALS mouse models, giving rise to a drug testing platform that includes the high variability of the human disease. We describe below the models that are more frequently used, emphasizing their advantages and caveats for drug discovery, previously described in two detailed recent reviews (Stephenson and Amor, 2017; Lutz, 2018).
SOD1 Models
SOD1 overexpressing mouse models were developed in the first place, and they are still the most used when it comes to ALS drug discovery (Rosen et al., 1993; Gurney et al., 1994). In particular, the SOD1G93A mouse model has been the most used and studied of all of them, as it closely resembles the disease progression in humans. These mice develop a progressive MN disease with adult onset and reduced lifespan. Pathogenically, SOD1G93A mice show early onset astrogliosis and microgliosis, glutamate-induced excitotoxicity, deficits in axonal transport, axonal denervation, protein aggregation, aberrant neurofilament processing, and mitochondrial vacuolization. All these processes result in the selective loss of spinal cord MN and the marked wasting, paralysis and atrophy of the forelimbs and hindlimbs. Thus, this model exhibits almost all the hallmarks of ALS (Philips and Rothstein, 2015; Lutz, 2018).
However, some disadvantages are found in this model. As mutations in SOD1 are found in a minority of ALS cases, the model is not representative of the whole patient population. This is of great importance, as SOD1 mutations do not result in TDP-43 inclusions in the SOD1G93A model, with this hallmark of disease pathogenesis being not commonly present in either SOD1 patients or mice (Stephenson and Amor, 2017). However, other SOD1 models such as SOD1G86S do exhibit TDP-43 inclusions and could be used to overcome this pitfall (Jeon et al., 2019). Furthermore, while patients develop the disease with basal quantities of the mutated protein, mice show very high overexpression of the mutated SOD1 protein (Lutz, 2018). In addition, in mice, the disease always commences in lumbar spinal cord MNs, while in humans, ALS onset may be initiated in cortical, bulbar, or spinal cord MNs (Philips and Rothstein, 2015).
TDP-43 Models
Mouse ALS models based in TARDBP mutations, encoding for TDP-43 protein, have failed in most cases to mimic the human disease, the overexpressing model TDP-43A315T being the best one in resembling it. These mice show an adult onset, with progressive and fatal neurodegeneration. Mice present TDP-43 aggregates, activation of astroglia and microglia, as well as MN loss. However, survival is impaired by a severe gastrointestinal pathology that can cause their sudden death previous to neurodegeneration (Wegorzewska et al., 2009; Herdewyn et al., 2014). Further limitations of the model are the overexpression of mutated TDP-43 and a poor loss of spinal cord MNs.
FUS Models
The majority of mouse models based in human FUS mutations do not present a complete ALS-like phenotype, neither progression to paralysis or disease end stage. The model that more closely resembles fALS is the wild type (WT) overexpressing mouse or hFUSWT. Mice develop paralysis, weight loss, muscle atrophy, and tremor. Moreover, MN loss, protein aggregation, and gliosis are also present. The major caveat of the model is that it does not carry a disease-linked mutation and, hence, the underlying pathology may not be the same as in the human disease (Mitchell et al., 2013).
C9orf72 Models
Again, most C9orf72-based models failed to develop an ALS-like pathology. The most suitable model is the h(G4C2)37-500, which shows muscle weakness, paralysis, weight loss, cognitive deficits, and premature death. Moreover, mice show histopathological hallmarks of the disease, such as MN loss, TDP-43 proteinopathy, and gliosis. However, this pathology is only developed by a 30–35% of female subset of mice. Another 40% of females and around 45% of males develop a milder pathology that in some cases progresses to the disease end point (Liu Y. et al., 2016). This heterogeneity seriously limits the use of this model to test the effect of compounds within a drug development program.
ALS Pathogenesis
ALS pathogenesis is far from being completely understood. Even though the disease is known to be multifactorial and multisystem, it is still unknown why the selective death of MNs occurs. Our limited knowledge on disease pathogenesis could explain why over 50 CTs with a large variety of compounds targeting different receptors and signaling pathways have provided negative outcomes in ALS patients. We will review here some features of disease pathogenesis such as cell Ca2+ dyshomeostasis, mitochondrial Ca2+ handling, and production of reactive oxygen species (ROS). Neuroinflammation and the role of P2X7 receptor (P2X7R) will be commented in “Neuroinflammation and P2X7 Receptors in ALS Pathogenesis” section.
Calcium Dyshomeostasis: The Excitotoxic Hypothesis
The excitotoxic hypothesis implies that alterations of some of the multiple receptors, ion channels, and calcium-binding proteins (CBPs), could be the result of some of the ALS mutations above described and the aggregation of pathological proteins (Appel et al., 2001; Van Den Bosch et al., 2006; Bezprozvanny, 2009; Grosskreutz et al., 2010; De Diego et al., 2012). Several factors support the assumption that augmented glutamatergic neurotransmission and Ca2+-dependent excitotoxicity play central relevant roles in MN degeneration (Patai et al., 2017). They are as follows: (i) high levels of excitatory amino acids have been found in the cerebrospinal fluid (CSF) of ALS patients (Plaitakis and Caroscio, 1987; Rothstein et al., 1991; Fiszman et al., 2010); (ii) CSF from ALS patients exerts MN death in vitro (Terro et al., 1996; Sen et al., 2005; Anneser et al., 2006; Gunasekaran et al., 2009; Yáñez et al., 2011); (iii) selective loss of astroglial excitatory amino acid transporter 2 (EAAT2) in motor cortex and spinal cord is found in both sporadic and fALS (Rothstein et al., 1995; Fray et al., 1998; Lin et al., 1998; Sasaki et al., 2000) with concomitant impairment of astrocytes’ ability to remove glutamate from the synaptic cleft; (iv) a defective glutamate clearance was detected in synaptosomes from brain areas and spinal cord of sALS patients (Rothstein et al., 1992), but prolonged in vivo blockade of astroglial EAAT2 with concomitant extracellular glutamate increase was innocuous for spinal MNs in rats (Tovar et al., 2009); and (v) α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) rather than NMDARs seem to contribute more to MN cytotoxicity, firstly because they are expressed at higher density in cultured MNs (Carriedo et al., 1996; Van Den Bosch and Robberecht, 2000) and, secondly, because of their higher Ca2+ permeability due to the lack of a GluR2 subunit in the tetrameric receptor complex (Van Damme et al., 2002). Consistent with this are the observations of lower GluR2 mRNA levels in MNs compared with other neurons (Van Damme et al., 2002), and the lower AMPAR mRNA in human spinal MNs (Heath and Shaw, 2002). Furthermore, the lack of GluR2 in SOD1G93A mice accelerated MN loss (Van Damme et al., 2005), and conversely, GluR2 overexpression augmented MN lifespan (Tateno et al., 2004). Of physiological and pharmacological interest are the observations that the GluR2 subunit is regulated by the vascular endothelial growth factor (VEGF; Bogaert et al., 2010) as well as by the brain-derived neurotrophic factor (BDNF) and glia cell-derived neurotrophic factor (GDNF; Brené et al., 2000). Of note is the fact that these growth factors also contribute to maintain MNs in good health. This is the case for VEGF (Wang et al., 2016), BDNF (Lanuza et al., 2019), and GDNF (Thomsen et al., 2018).
Strong or weak capacity to bind free Ca2+ ions in the cytosol ([Ca2+]c) by CBPs has immediate consequences for neuron vulnerability during Ca2+ loads in ALS-linked MN degeneration. For instance, cortical, lower cranial, and spinal MNs die soon after ALS onset, which correlates with their lack of expression of the CBPs parvalbumin and calbindin-D28k. In contrast, MNs from Onuf’s nucleus, oculomotor, trochlear, and abducens nerves that express high levels of parvalbumin and/or calbindin-D28k are resistant to damage or are injured at advanced disease stages (Alexianu et al., 1994). Moreover, in brain slices, the expression level of CBPs by different types of neurons correlate with their specific vulnerability to Ca2+ loads (Ho et al., 1996; Beers et al., 2001; Van Den Bosch et al., 2002). In line with these results, another experiment showed that cross breeding SOD1 mutant mice with mice overexpressing parvalbumin in spinal MNs resulted in a delay of disease onset and a longer MN survival (Beers et al., 2001).
Altered Mitochondrial Calcium Handling
Mitochondria are involved in nearly all types of cell death, including necrosis, apoptosis, and necroptosis (Kroemer et al., 1998; Galluzzi and Kroemer, 2008). But notably, the redistribution of mutated TDP-43 from the nucleus to the cytoplasm is being considered a pathological hallmark for most forms of ALS (Kabashi et al., 2008; Sreedharan et al., 2008). A recent study demonstrates that TDP-43 toxicity resides in its mitochondrial location. The aberrant protein specifically impairs the Complex I of the mitochondrial oxidative phosphorylation system (OXPHOS) by preferentially binding to mitochondrial transcribed ND3/6 mRNAs as well as by inhibiting their translation, leading to mitochondrial dysfunction and neurodegeneration. The interest of this study resides in the additional observation that the suppression of WT or mutant TDP-43 in mitochondria restores neuronal viability. This provides a rationale for reducing the aggregated TDP-43 protein inside the mitochondria as a therapeutic approach for ALS (Wang et al., 2016).
Moreover, mitochondria in MNs from SOD1G93A and TDP-43 mice show a swelling shape, have internal vacuoles and disorganized cristae, and aggregate in abnormal clusters. This causes a defect in mitochondrial axonal transport towards the synaptic nerve terminals, leading to neuronal metabolic alterations (Magrane et al., 2014). Mitochondria also show aberrant fission and fusion dynamics that are necessary for damaged mitochondria confinement and repair (Smith et al., 2019).
The Link Between ROS and Mitochondrial Calcium Cycling
ROS are natural metabolic products of cell functions and particularly of oxidative phosphorylation itself. However, impairment of mitochondrial electron transport chain generates an excess of ROS that can damage cell proteins, membrane lipids, and nucleic acids. In fact, several markers of ROS damage, i.e., 3-nitrotyrosine, 8-hydroxy-2′-deoxyguanosine, and 4-hydroxynonenal, have been found in blood, CSF, and urine of ALS patients, as well as in their spinal MNs (Smith et al., 1998; Ihara et al., 2005; Mitsumoto et al., 2008).
Oxidative stress is also caused by a defect in the cellular defense machinery against ROS. Such is the case of SOD1 that catalyzes the conversion of superoxide radicals (O2−) into hydrogen peroxide (H2O2). Physiological reduction of H2O2 into oxygen and water through the action of glutathione and catalase offers cell protection from ROS. Interestingly, the mutation of SOD1 in ALS does not result in a loss of its enzymatic function, but rather, it enhances its ability to oxidize cellular antioxidants, reducing molecular oxygen to O2− (Liochev and Fridovich, 2003). Nevertheless, the MN toxicity linked to mutated SOD1 is mainly due to the cytoplasmic aggregates created by the aberrant protein (Hardiman et al., 2017).
Large Ca2+ loads occurring upon cell stimulation are cleared up by mitochondria through the high-capacity mitochondrial Ca2+ uniporter (MCU; Herrington et al., 1996; Xu et al., 1997; Montero et al., 2000). The Ca2+ accumulated in the mitochondrial matrix stimulates respiration and ATP synthesis, to couple cell activity and bioenergetics needs (Gunter et al., 1994; Rizzuto et al., 2000). During the excitotoxic process, this accumulation could lead to excessive mitochondrial Ca2+ load, oxidative stress, and cell death (Cano-Abad et al., 2001; Orrenius et al., 2003).
Correlation between Ca2+ influx with massive mitochondrial Ca2+ uptake also occurs in hypoglossal MNs (Ladewig et al., 2003). Such mitochondrial Ca2+ uptake is comparatively small in highly Ca2+-buffered ALS-resistant spinal MNs (Carriedo et al., 2000), suggesting that mitochondria could partially compensate for the weak cytosolic Ca2+ buffering in vulnerable neurons. In fact, following Ca2+ entry through voltage-activated calcium channels (VACCs), substantial mitochondrial Ca2+ uptake occurs in weakly buffered neurons (Bergmann and Keller, 2004). Mitochondria may exert an additional control of Ca2+ homeostasis through ROS-dependent regulation of excitability. Thus, enhanced ROS production occurs upon inhibition of respiration by sodium cyanide. ROS induces the opening of sodium channels, augmentation of action potential firing and enhanced voltage-dependent Ca2+ influx (Bergmann and Keller, 2004). Some studies support the idea that the production of ROS is increased in spinal MN mitochondria as a result of Ca2+ overload following excitotoxic stimulation of AMPA/kainate receptors (Carriedo et al., 2000).
Mitochondrial damage elicited by mutant SOD1 aggregates found in the mitochondrial matrix of transgenic mice (Jaarsma et al., 2001; Pasinelli et al., 2004) could decrease the enzymatic activity of the electron transport chain at complexes I, II, and IV (Jung et al., 2002; Mattiazzi et al., 2002). In fact, mutant SOD1 could disrupt the association of complex IV (cytochrome c) with the inner mitochondrial membrane, thereby interfering with respiration (Kirkinezos et al., 2005). This leads to increased ROS production as shown in mutant SOD1-expressing MNs cultures (Kruman et al., 1999) as well as in MNs of brain slices where complex IV was inhibited by sodium cyanide (Bergmann and Keller, 2004).
It seems that ROS produced in MNs can diffuse to neighboring astrocytes to cause oxidative disruption of glutamate transporters (Rao et al., 2003). In turn, this will increase the local extracellular concentration of glutamate, thereby enhancing local excitotoxicity in a vicious circle of MN damage. This model integrates the hypothesis of Ca2+-dependent excitotoxicity and oxidative damage. In this frame, distorted mitochondrial respiration sensitizes MNs to glutamate stimulation and to environmental toxins, thus increasing their vulnerability (Kruman et al., 1999; Andreassen et al., 2001). In this direction, it was observed that in cultured neurons, the chronic mitochondrial inhibition induced by malonate or sodium azide led to selective MN death; free-radical scavengers and AMPAR blockers protected such neurons from death (Kaal et al., 2000).
Neuroinflammation and P2X7 Receptors in ALS Pathogenesis
The purinergic P2X7 ionotropic receptor (P2X7R) is considered as one of the main players of inflammation (Di Virgilio, 2015). In fact, this receptor is expressed in immune and inflammatory cells such as dendritic cells, osteoclasts, microglia, mast cells, natural killer cells, or T and B lymphocytes. Additionally, P2X7Rs are upregulated in inflammatory processes (Ferrari et al., 2006).
As a result of cell stress or tissue damage, large amounts of ATP are released into the extracellular space that stimulates the low-affinity target P2X7R, the main sensor for ATP during inflammation. This receptor is also the main trigger of the protective/regenerative immune response, consisting in the maturation and release of several interleukins, mainly IL-1β (Adinolfi et al., 2018).
The maturation of IL-1β requires the activation of the NLRP3 inflammasome, an event that requires Na+ influx accompanied by water, Ca2+ influx, and K+ efflux. The intracellular K+ drop is, in fact, the best established mechanism underlying the P2X7R-mediated formation of the NLRP3 inflammasome (Compan et al., 2012; Katsnelson et al., 2015; Jo et al., 2016; Karmakar et al., 2016). This is supported by experiments showing that not only ATP, but also other agents that induce K+ efflux, such as the ionophore nigericin or crystalline molecules, elicit inflammasome activation too (Compan et al., 2012; Yaron et al., 2015). Noteworthy, ROS, also, induce inflammasome aggregation and IL-1β release due to P2X7R activation (Hung et al., 2013; Minkiewicz et al., 2013).
The role of P2X7Rs in neuroinflammation is supported by the fact that they are expressed in various cells of the central nervous system (CNS), namely, astrocytes, microglia, and oligodendrocytes (Sperlagh et al., 2006; Kaczmarek-Hajek et al., 2018), where they mediate inflammasome signaling (Franceschini et al., 2015). The presence of reactive astrocytes and microglia define the neuroinflammatory process in the CNS. As peripheral immune and endothelial cells, activated microglia and astrocytes produce pro-inflammatory cytokines (IL-1β, IL-16, TNF-α), chemokines (CCL2, CCL9, CCL1), ROS, and secondary messengers (nitric oxide, prostaglandins; DiSabato et al., 2016). However, the activation of P2X7Rs in these cells is known to produce dual and divergent effects depending on the duration of the inflammatory stimulus and the disease stage, presymptomatic or symptomatic.
How neuroinflammation contributes to neurodegeneration has recently been analyzed by Ransohoff (2016). Thus, a chronic mild activation of glial cells via P2X7Rs leads, in the long term, to synaptic dysfunction, synapse loss, and neuronal death. The fact that glial cells exhibit a low overall turnover rate make them more susceptible to the neuroinflammatory effects of age, injury, or stress in neurodegenerative diseases (Ajami et al., 2007).
A dual effect of P2X7R-mediated neuroinflammation seems to operate during the time course of ALS. This could be explained in the context of two different phenotypes that microglia develop upon P2X7R activation. The M2 phenotype or anti-inflammatory predominates at earlier disease stages, while the pro-inflammatory M1 phenotype is prevalent at later disease stages in SOD1G93A mice (Liao et al., 2012; Parisi et al., 2016). Consistent with this is an experiment done in SOD1G93A mice with genetic ablation of P2X7Rs; exacerbation of gliosis and enhanced MN death were unexpectedly found in these mice (Apolloni et al., 2013a). Another experiment supporting duality indicated that short P2X7R stimulation augmented autophagy and M2 microglial markers. However, continued P2X7R stimulation impaired autophagy, suggesting a microglial shift to the M1 phenotype. A dual report that implies duality of action at central and peripheral tissues indicated a positive role of P2X7Rs on SOD1G93A muscles (Fabbrizio et al., 2020).
A few preclinical and clinical experiments support the implication of P2X7Rs in ALS pathogenesis. So, the pro-inflammatory action of microglial P2X7Rs was augmented in SOD1G93A mice (D’Ambrosi et al., 2009). Furthermore, spinal cord pathology was ameliorated by P2X7R antagonism, also in these mice (Apolloni et al., 2014). Consistent with this was the observation that P2X7R activation by high ATP concentrations activated NOX2 and the kinase ERK1/2 in the microglia of SOD1G93A mice, thus provoking an increase of ROS (Apolloni et al., 2013b). Additionally, in co-cultures of astrocytes and MNs from SOD1G93A mice, cell stimulation with P2X7R agonists ATP and 3′-O-(4-Benzoyl)benzoyl ATP (BzATP) elicited a neurotoxic phenotype that was prevented by the P2X7R blocker Brilliant Blue G (BBG; Gandelman et al., 2010). However, unexpected P2X7R down-regulation and Ca2+ dyshomeostasis was found in peripheral monocytes of ALS patients (Liu J. et al., 2016). This contrasts with another study showing up-regulation in spinal cord tissue of post-mortem ALS patients (Yiangou et al., 2006).
This cumulative set of data mostly from preclinical in vitro and in vivo models of ALS, plus a scarce number of clinical studies in ALS patients, support the view in the sense that P2X7Rs mediate the activation of astrocytes and microglia, giving rise to a chronic neuroinflammatory state that is critical in the progression of ALS pathology. However, it should be kept in mind that dependent on disease stage, microglial activation may be anti-inflammatory (early stage, M2 phenotype) or pro-inflammatory (late stage, M1 phenotype).
Some P2X7R Blockers With Favorable Pharmacokinetics
Due to its involvement in neuroinflammation, the P2X7R is considered an adequate target for the treatment of several neurodegenerative and neurological diseases, as well as mood disorders (Díaz-Hernández et al., 2009; Hempel et al., 2013; Bhattacharya and Biber, 2016; Jimenez-Pacheco et al., 2016; Volonte et al., 2016; Cieslak and Wojtczak, 2018; Cieslak et al., 2019). Yet, preclinical data in mouse models of ALS leave some uncertainty about it, as previously described. Moreover, P2X7R antagonists have not succeeded in any CT for CNS disorders so far (ClinicalTrials.gov - NIH U.S. National Library of Medicine, 2020). Despite the notable number of P2X7R blockers already existing as pharmacological tools, not all meet the pharmacokinetic (PK) characteristics that are necessary to become a potential drug for the treatment of neurodegenerative diseases, including ALS. This is the case of BBG that has been amply used as a P2X7R blocker only because it is cheap and readily available. However, this compound has a poor selectivity towards P2X7Rs, blocking also P2X4, P2X5, and rat P2Y1 and P2Y2, while potentiating the human P2Y1 at 1–3 μM concentrations (Jacobson et al., 2002, 2006; Jacobson, 2010). Research has led nowadays to very potent antagonists, but unfortunately, this is not enough. Between the main problems of these compounds, there is a lack of favorable PK properties, such as low blood–brain barrier (BBB) permeability and scarce stability in blood and brain tissue, which determine a drop in drug half-life and CNS residence time. Additionally, pronounced differences have been found in potencies and affinities among P2X7R in mouse, rat, and human. Such is the case of AZ11645373 (Figure 1) that blocks the human P2X7R at nanomolar concentrations and the mouse P2X7R at micromolar concentrations and has no effect on the rat orthologous (Michel et al., 2009). Another example is compound GW791343, which is a potent allosteric inhibitor of the human receptor, but also a positive allosteric modulator of rat P2X7R (Jacobson, 2010). This complicates the scenario for the development of a drug for clinical use and diminishes the possibility for a new compound of being selected for preclinical and clinical studies.
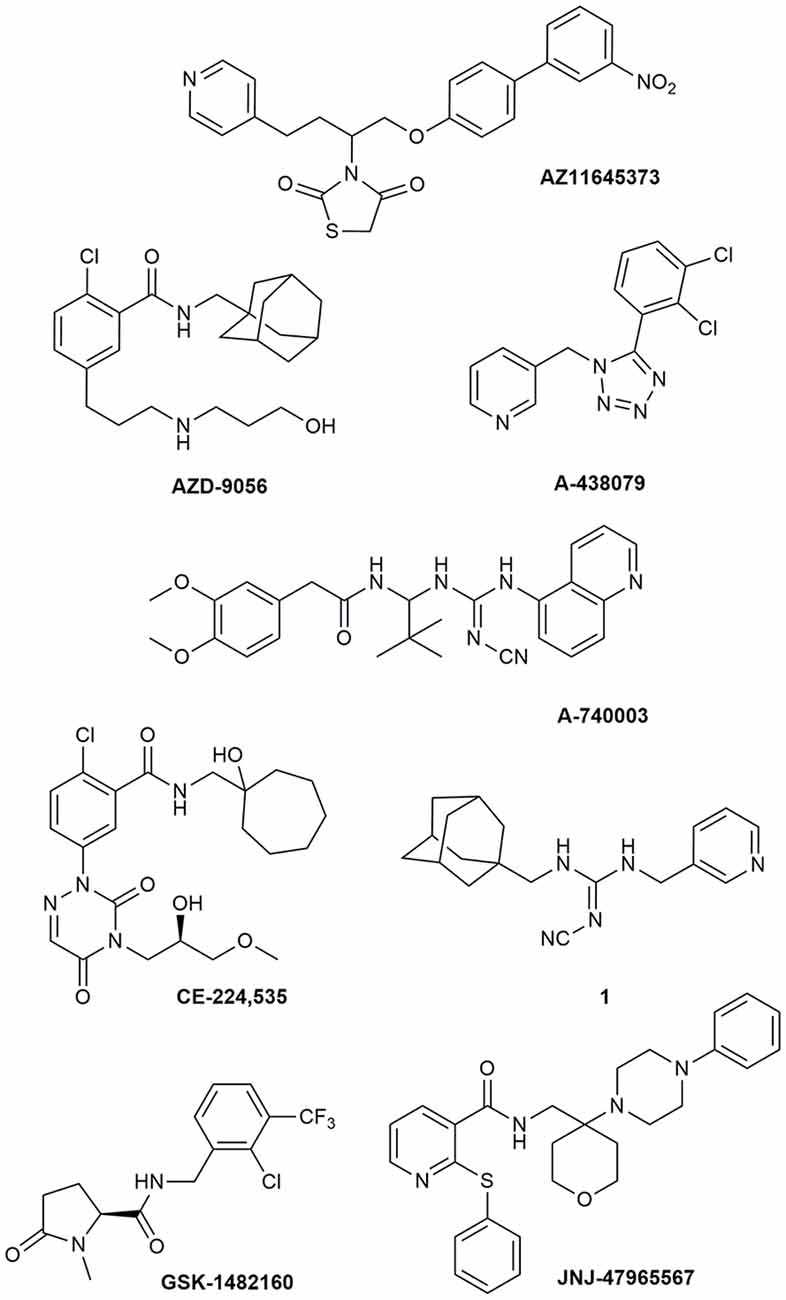
Figure 1. P2X7R antagonists lacking adequate pharmacokinetic properties for the treatment of CNS disorders.
During around the last 10–15 years, the interest in finding new P2X7R clinical candidates has shifted from no-BBB-permeable antagonists for the treatment of peripheral inflammatory diseases (e.g., AZD-9056 from AstraZeneca, tried in CTs—Figure 1) to BBB-permeable blockers, mainly due to the increasing awareness of P2X7R role in CNS disorders (Bhattacharya and Biber, 2016; Bhattacharya, 2018).
However, just increasing lipophilicity is not always highly recommended for the development of BBB-permeable drugs. In fact, this can penalize other important PK properties like solubility, enzyme metabolism, blood protein binding, and half-life. After the development of the high lipophilic o-chlorobenzamide P2X7R antagonists by Pfizer, their attempts were focused on the increase of molecular polarity, giving a good clinical candidate that, however, was no longer CNS penetrant (CE-244,535—Figure 1; Duplantier et al., 2011).
Abbott Laboratories developed several potent P2X7R antagonists, mainly tetrazoles and cyanoguanidines. Thus, tetrazole derivative A-438079 (Figure 1) was very appealing for its small molecular weight, good potency, and efficacy in reverting allodynia in the Chung model of neuropathic pain. Nevertheless, its half-life of 1 h and low bioavailability after intraperitoneal (ip) administration was discouraging (Guile et al., 2009). Cyanoguanidine A-740003 (Figure 1) was found to have a better half-life after iv administration (t½ = 4 h) but not a good BBB permeability, as confirmed by its radiotracer [11C]A-740003 (Janssen et al., 2014). Cyanoguanidine A-804598 (Figure 2) has the best balance of pharmacodynamics and PK properties. In fact, it is potent (IC50 of around 10 nM for mouse, rat, and human species, vs. the potency of A-740003 ranging from 44 to 150 nM in the different orthologs), selective for the P2X7R, and brain-permeable, persisting in brain tissues at least for 1 h after administration, as shown in ex vivo studies of receptor occupancy (Able et al., 2011). More recently, Abbott has developed new potent P2X7R antagonists by adding the adamantine core to cyanoguanidines (compound 1—Figure 1), but without great PK improvements (e.g., t½ = 0.22 h; O’Brien-Brown et al., 2017).
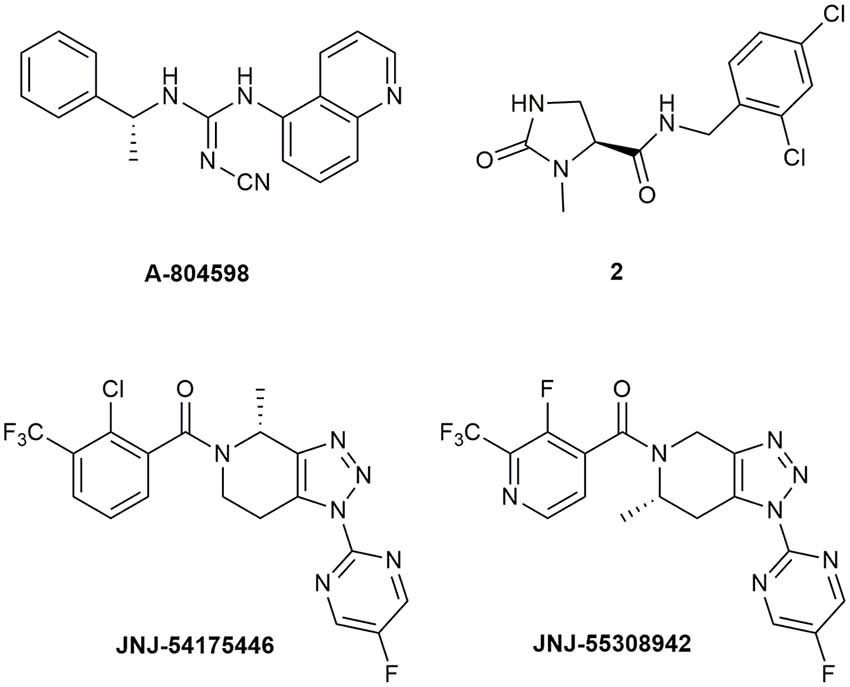
Figure 2. P2X7R antagonists with optimal pharmacokinetic properties for the treatment of CNS disorders.
GlaxoSmithKline developed a series of compounds with excellent physicochemical properties, such as the BBB-permeable GSK-1482160 in rats (Figure 1; Abdi et al., 2010). Unfortunately, deeper studies on its metabolism pathway explained why some parameters, like its half-life, worsened in dog and monkey models, blocking its clinical development. Anyway, new chemical modifications succeeded in overcoming this problem. Among the compounds of this new series, the oxoimidazolidine 2 (Figure 2) stands out (Abberley et al., 2010).
It is a fact that Janssen Pharmaceuticals has probably developed the most interesting P2X7R antagonists as clinical candidates for the treatment of CNS diseases. One of the most used ligands in basic and preclinical research is JNJ-47965567 (Figure 1). Unfortunately, its poor oral bioavailability, as well as the lack of deeper details on its residence time in brain, stopped its clinical progress (Bhattacharya et al., 2013). However, new series of dihydro- and tetrahydro-triazolopyridines were discovered and extensively optimized. Among these, JNJ-54175446 and JNJ-55308942 (Figure 2) have optimal PK properties like solubility, metabolism profile, bioavailability, and half-life, as well as good BBB permeability and tolerability (Letavic et al., 2017; Chrovian et al., 2018). Proof of this is the fact that they were chosen as clinical candidates and that JNJ-55308942 has completed three phase I CTs, while JNJ-54175446 is currently in a phase II CT for major depression (ClinicalTrials.gov - NIH U.S. National Library of Medicine, 2020; Recourt et al., 2020).
Proof of Concept of P2X7Rs as a Druggable Target in ALS
The proof of concept that a P2X7R antagonist could have promising therapeutic potential in ALS has been tested in the SOD1G93A mouse model of ALS (Table 1). In a first study, BBG was administered intraperitoneally at a dose of 45.5 mg/kg every 48 h, starting at postnatal day 90 (P90), a late pre-onset disease stage. The treatment improved deficits in motor performance to a greater extent in males than in females, but no effect on survival was observed (Cervetto et al., 2013).
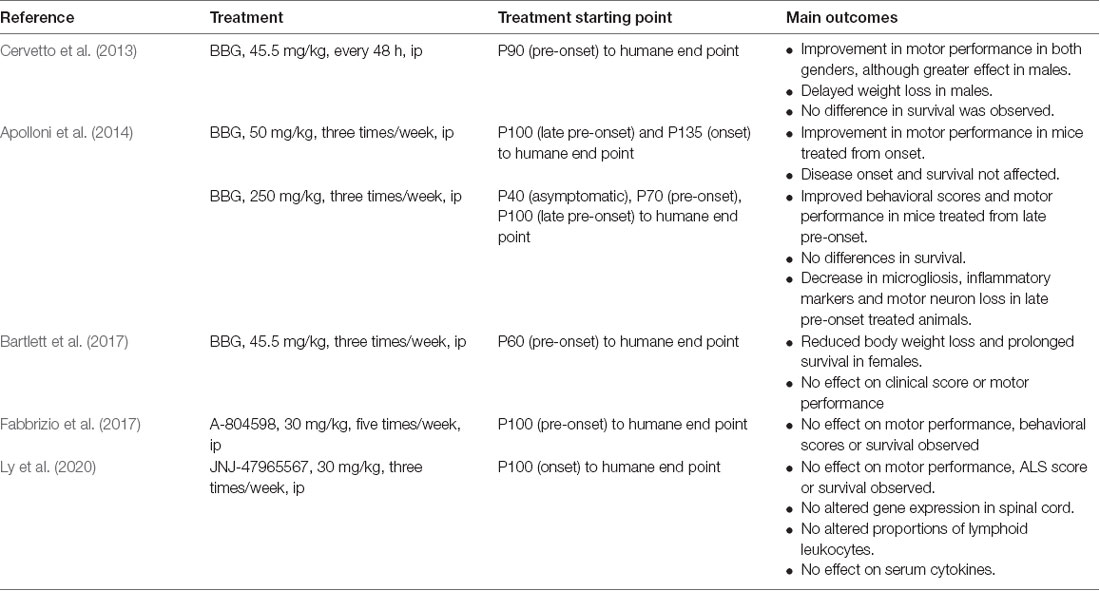
Table 1. Proof of concept of P2X7R antagonism on clinical outcomes in SOD1G93A mice.
Taking into account the dual role of P2X7Rs (protection at early stages and promotion of cell death at advanced disease stages), an experiment was designed changing the dose and defining different start points of BBG administration (250 mg/kg, 3 times/week). Starting at late pre-onset stage (P100), higher MN survival and reduced microgliosis were observed in the spinal cord of SODG93A mice. Moreover, disease onset was delayed and motor performance was improved in both males and females, although survival was not affected. This was not the case when BBG was administered at pre-symptomatic stages, supporting the hypothesis of the dual role of P2X7R in ALS (Apolloni et al., 2014).
In a third experiment, BBG administration at pre-onset stage or P62–P64 (45.5 mg/kg, three times/week) reduced body weight loss in females but not in males, a sign of delayed muscle loss. Survival was also augmented in females but not in males, and motor performance was unaffected in either sex (Bartlett et al., 2017).
Finally, two experiments have used more potent and specific P2X7R antagonists. When the antagonist A-804598 was administered to female mice five times/week at 30 mg/kg, no effects on motor performance, disease onset, or survival were observed (Fabbrizio et al., 2017). Same negative results were obtained with the administration of the antagonist JNJ-47965567 from P100, three times/week at 30 mg/kg (Ly et al., 2020).
These erratic, yet mild positive outcomes suggest that the experimental conditions of future experiments should be considered as follows: (i) to adjust drug administration starting point; (ii) to define better the dosing and frequency of administration, according to (iii) the PK drug properties, including the BBB permeability and CNS residence time, longer half-life and solubility; and (iv) to include both females and males and, if possible, different ALS mouse models.
From Bench to Clinical Trials
Recent reviews (Petrov et al., 2017; Andrews et al., 2019; Chipika et al., 2019) analyze the outcomes of over 50 CTs performed in ALS patients, since riluzole was available. Various druggable potential targets have been addressed: (1) antiglutamatergic compounds (riluzole, memantine, talampanel, ceftriaxone); (2) antioxidant agents (edaravone, coenzyme Q, creatine); (3) anti-inflammatory drugs (valproic acid, NP001, glatiramer acetate, minocycline, pioglitazone, erythropoietin, celecoxib); (4) neurotrophic factors (IGF-1, CNTF, BDNF); (5) lithium; (6) inhibitors of kinase (masitinib, fasudil); and (7) neuroprotective compounds (xaliproden, dexpramipexole, olesoxime, omigapil).
Despite riluzole and edaravone being approved by the FDA after having been tested in CTs, they gave poor clinical outcomes. In the case of riluzole, two out of three late-stage CTs reported negative outcomes. Similarly, in the case of edaravone, two out of three phase III CTs also reported negative results. The rest of the compounds and drugs tested provided negative results in CTs.
Masitinib is emerging as a singular agent to mitigate microgliosis and neuroinflammation. The compound also prolongs survival when administered to SOD1G93A ALS mice at the onset of paralysis (Trias et al., 2016). An add-on riluzole therapy phase III CT reported significant outcomes in ALS patients treated with masitinib [Masitinib in Combination With Riluzole for the Treatment of Patients Suffering From Amyotrophic Lateral Sclerosis (ALS), 2018].
Conclusions and Perspectives
Much is known about clinical features and pathogenic pathways of ALS. Cumulating knowledge, derived from in vitro and in vivo disease models, as well as from patients and postmortem tissues, outlines the sequence of the pathophysiological events involved in ALS. These could be hypothetically ordered as follows: (1) cytosolic and mitochondrial protein aggregation, affecting mitochondrial ultrastructure and function; (2) distorted mitochondrial calcium handling and circulation; (3) deficits in ATP generation and excess of ROS production; (4) reactive microglia and production of inflammatory mediators; and (5) enhanced MN vulnerability and death.
Regarding disease pathogenesis, a crucial open question remains: are the pathogenic features common for both sporadic and familial ALS? And also, why do only MNs die, if mutated proteins are expressed in all cell types? Given the complexity of ALS pathogenesis, why have CTs been performed using single-target compounds in the last 20 years? In light of the chronic inflammatory background, should we target with neuroprotective agents only the MNs, or the activated glia, or both? Much further research is required before we can answer these questions.
Author Contributions
CR-R and FC contributed equally to this manuscript. They both searched for scientific literature concerning ALS pathogenesis and P2X7R involvement. CR-R reviewed the most considerable ALS mouse models used in research, while FC made a summary of the molecular and pharmacokinetic profiles of the main P2X7 receptor blockers. AG re-elaborated and drafted the final form of the manuscript, reviewing also the subsequent versions.
Funding
This project has received funding from: (1) the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 766124; (2) the Spanish Ministry of Science, Technology and Innovation SAF2016-78892R; and (3) Fundación Teófilo Hernando.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
[Ca2+]c, Ca2+ concentration in the cytosol; ALS, amyotrophic lateral sclerosis; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; BBB, blood–brain barrier; BBG, Brilliant Blue G; BDNF, brain-derived neurotrophic factor; BzATP, 3’-O-(4-benzoyl)benzoyl-ATP; C9orf72, Chromosome 9 open reading frame 72; CBPs, calcium-binding proteins; CNS, central nervous system; COX-2, cyclooxygenase-2; CSF, cerebrospinal fluid; CT, clinical trial; EAAT2, excitatory amino acid transporter 2; fALS, familial ALS; FDA, U.S. Food and Drug Administration; FUS/TLS, Fused in Sarcoma/Translocated in Sarcoma; GDNF, glia cell-derived neurotrophic factor; H2O2, hydrogen peroxide; HIV, human immunodeficiency virus; ip, intraperitoneal; iv, intravenous; MCU, mitochondrial Ca2+ uniporter; mNCX, mitochondrial Na+/Ca2+ exchanger; MN, motor neuron; NMDA, N-methyl-D-aspartate; NOX, NADPH oxidase; O2−, superoxide radical; OXPHOS, mitochondrial oxidative phosphorylation system; P2X7R, purinergic P2X7 receptor; P90, postnatal day 90; PET, positron emission tomography; PK, pharmacokinetics; R, receptor; RAN, repeat-association non-ATG; ROS, reactive oxygen species; sALS, sporadic ALS; SOD1, Cu/Zn superoxide dismutase; TAR, transactive response; TDP-43, TAR-DNA binding protein-43; VACCs, voltage-activated calcium channels; VEGF, vascular endothelial growth factor; WT, wild type.
References
Abberley, L., Bebius, A., Beswick, P. J., Billinton, A., Collis, K. L., Dean, D. K., et al. (2010). Identification of 2-oxo-N-(phenylmethyl)-4-imidazolidinecarboxamide antagonists of the P2X(7) receptor. Bioorg. Med. Chem. Lett. 22, 6370–6374. doi: 10.1016/j.bmcl.2010.09.101
Abdi, M. H., Beswick, P. J., Billinton, A., Chambers, L. J., Charlton, A., Collins, S. D., et al. (2010). Discovery and structure-activity relationships of a series of pyroglutamic acid amide antagonists of the P2X7 receptor. Bioorg. Med. Chem. Lett. 20, 5080–5084. doi: 10.1016/j.bmcl.2010.07.033
Able, S. L., Fish, R. L., Bye, H., Booth, L., Logan, Y. R., Nathaniel, C., et al. (2011). Receptor localization, native tissue binding and ex vivo occupancy for centrally penetrant P2X7 antagonists in the rat. Br. J. Pharmacol. 2, 405–414. doi: 10.1111/j.1476-5381.2010.01025.x
Adinolfi, E., Giuliani, A. L., De Marchi, E., Pegoraro, A., Orioli, E., and Di Virgilio, F. (2018). The P2X7 receptor: a main player in inflammation. Biochem. Pharmacol. 151, 234–244. doi: 10.1016/j.bcp.2017.12.021
Ajami, B., Bennett, J. L., Krieger, C., Tetzlaff, W., and Rossi, F. M. (2007). Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 10, 1538–1543. doi: 10.3410/f.1097339.553379
Alexianu, M. E., Ho, B. K., Mohamed, A. H., La Bella, V., Smith, R. G., and Appel, S. H. (1994). The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann. Neurol. 36, 846–858. doi: 10.1002/ana.410360608
Alonso, A., Logroscino, G., Jick, S. S., and Hernan, M. A. (2009). Incidence and lifetime risk of motor neuron disease in the United Kingdom: a population-based study. Eur. J. Neurol. 16, 745–751. doi: 10.1111/j.1468-1331.2009.02586.x
Andreassen, O. A., Ferrante, R. J., Klivenyi, P., Klein, A. M., Dedeoglu, A., Albers, D. S., et al. (2001). Transgenic ALS mice show increased vulnerability to the mitochondrial toxins MPTP and 3-nitropropionic acid. Exp. Neurol. 2, 356–363. doi: 10.1006/exnr.2001.7627
Andrews, J. A., Bruijn, L. I., and Shefner, J. M. (2019). ALS drug development guidances and trial guidelines: consensus and opportunities for alignment. Neurology 93, 66–71. doi: 10.1212/WNL.0000000000007695
Anneser, J. M., Chahli, C., and Borasio, G. D. (2006). Protective effect of metabotropic glutamate receptor inhibition on amyotrophic lateral sclerosis-cerebrospinal fluid toxicity in vitro. Neuroscience 141, 1879–1886. doi: 10.1016/j.neuroscience.2006.05.044
Apolloni, S., Amadio, S., Montilli, C., Volonte, C., and D’Ambrosi, N. (2013a). Ablation of P2X7 receptor exacerbates gliosis and motoneuron death in the SOD1–G93A mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 22, 4102–4116. doi: 10.1093/hmg/ddt259
Apolloni, S., Parisi, C., Pesaresi, M. G., Rossi, S., Carri, M. T., Cozzolino, M., et al. (2013b). The NADPH oxidase pathway is dysregulated by the P2X7 receptor in the SOD1#x02013;G93A microglia model of amyotrophic lateral sclerosis. J. Immunol. 10, 5187–5195. doi: 10.4049/jimmunol.1203262
Apolloni, S., Amadio, S., Parisi, C., Matteucci, A., Potenza, R. L., Armida, M., et al. (2014). Spinal cord pathology is ameliorated by P2X7 antagonism in a SOD1-mutant mouse model of amyotrophic lateral sclerosis. Dis. Model Mech. 9, 1101–1109. doi: 10.1242/dmm.017038.
Appel, S. H., Beers, D., Siklos, L., Engelhardt, J. I., and Mosier, D. R. (2001). Calcium: the darth vader of ALS. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2, S47–54. doi: 10.1080/14660820152415744
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., et al. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 3, 602–611. doi: 10.1016/j.bbrc.2006.10.093
Bartlett, R., Sluyter, V., Watson, D., Sluyter, R., and Yerbury, J. J. (2017). P2X7 antagonism using brilliant blue G reduces body weight loss and prolongs survival in female SOD1(G93A) amyotrophic lateral sclerosis mice. PeerJ. 5:e3064. doi: 10.7717/peerj.3064
Beers, D. R., Ho, B. K., Siklos, L., Alexianu, M. E., Mosier, D. R., Mohamed, A. H., et al. (2001). Parvalbumin overexpression alters immune-mediated increases in intracellular calcium and delays disease onset in a transgenic model of familial amyotrophic lateral sclerosis. J. Neurochem. 3, 499–509. doi: 10.1046/j.1471-4159.2001.00582.x
Bergmann, F., and Keller, B. U. (2004). Impact of mitochondrial inhibition on excitability and cytosolic Ca2+ levels in brainstem motoneurones from mouse. J. Physiol. 555, 45–59. doi: 10.1113/jphysiol.2003.053900
Bezprozvanny, I. (2009). Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 15, 89–100. doi: 10.1016/j.molmed.2009.01.001
Bhattacharya, A. (2018). Recent advances in CNS P2X7 physiology and pharmacology: focus on neuropsychiatric disorders. Front. Pharmacol. 9:30. doi: 10.3389/fphar.2018.00030
Bhattacharya, A., and Biber, K. (2016). The microglial ATP-gated ion channel P2X7 as a CNS drug target. Glia 64, 1772–1787. doi: 10.1002/glia.23001
Bhattacharya, A., Wang, Q., Ao, H., Shoblock, J. R., Lord, B., Aluisio, L., et al. (2013). Pharmacological characterization of a novel centrally permeable P2X7 receptor antagonist: JNJ-47965567. Br. J. Pharmacol. 3, 624–640. doi: 10.1111/bph.12314
Bogaert, E., Van Damme, P., Poesen, K., Dhondt, J., Hersmus, N., Kiraly, D., et al. (2010). VEGF protects motor neurons against excitotoxicity by upregulation of GluR2. Neurobiol. Aging 12, 2185–2191. doi: 10.1016/j.neurobiolaging.2008.12.007
Brené, S., Messer, C., Okado, H., Hartley, M., Heinemann, S. F., and Nestler, E. J. (2000). Regulation of GluR2 promoter activity by neurotrophic factors via a neuron-restrictive silencer element. Eur. J. Neurosci. 12, 1525–1533. doi: 10.1046/j.1460-9568.2000.00040.x
Cano-Abad, M. F., Villarroya, M., Garcia, A. G., Gabilan, N. H., and Lopez, M. G. (2001). Calcium entry through L-type calcium channels causes mitochondrial disruption and chromaffin cell death. J. Biol. Chem. 276, 39695–39704. doi: 10.1074/jbc.m102334200
Carriedo, S. G., Sensi, S. L., Yin, H. Z., and Weiss, J. H. (2000). AMPA exposures induce mitochondrial Ca2+ overload and ROS generation in spinal motor neurons in vitro. J. Neurosci. 20, 240–250. doi: 10.1523/jneurosci.20-01-00240.2000
Carriedo, S. G., Yin, H. Z., and Weiss, J. H. (1996). Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J. Neurosci. 16, 4069–4079. doi: 10.1523/jneurosci.16-13-04069.1996
Cervetto, C., Frattaroli, D., Maura, G., and Marcoli, M. (2013). Motor neuron dysfunction in a mouse model of ALS: gender-dependent effect of P2X7 antagonism. Toxicology 311, 69–77. doi: 10.1016/j.tox.2013.04.004
Chia, R., Chio, A., and Traynor, B. J. (2018). Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 17, 94–102. doi: 10.1016/s1474-4422(17)30401-5
Chio, A., Logroscino, G., Traynor, B. J., Collins, J., Simeone, J. C., Goldstein, L. A., et al. (2013). Global epidemiology of amyotrophic lateral sclerosis: a systematic review of the published literature. Neuroepidemiology 2, 118–130. doi: 10.1159/000351153
Chipika, R. H., Finegan, E., Li Hi Shing, S., Hardiman, O., and Bede, P. (2019). Tracking a fast-moving disease: longitudinal markers, monitoring and clinical trial endpoints in ALS. Front. Neurol. 10:229. doi: 10.3389/fneur.2019.00229
Chrovian, C. C., Soyode-Johnson, A., Peterson, A. A., Gelin, C. F., Deng, X., Dvorak, C. A., et al. (2018). A dipolar cycloaddition reaction to access 6-methyl-4,5,6,7-tetrahydro-1H-[1,2,3]triazolo[4,5-c]pyridines enables the discovery synthesis and preclinical profiling of a P2X7 antagonist clinical candidate. J. Med. Chem. 1, 207–223. doi: 10.1021/acs.jmedchem.7b01279
Cieslak, M., Roszek, K., and Wujak, M. (2019). Purinergic implication in amyotrophic lateral sclerosis-from pathological mechanisms to therapeutic perspectives. Purinergic Signal. 15, 1–15. doi: 10.1007/s11302-018-9633-4
Cieslak, M., and Wojtczak, A. (2018). Role of purinergic receptors in the Alzheimer’s disease. Purinergic Signal. 14, 331–344. doi: 10.1007/s11302-018-9629-0
ClinicalTrials.gov - NIH U.S. National Library of Medicine (2020). Available online at: https://clinicaltrials.gov/. Accessed February 5, 2020
Compan, V., Baroja-Mazo, A., Lopez-Castejon, G., Gomez, A. I., Martinez, C. M., Angosto, D., et al. (2012). Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 3, 487–500. doi: 10.1016/j.immuni.2012.06.013
Cruz, M. P. (2018). Edaravone (Radicava): a novel neuroprotective agent for the treatment of amyotrophic lateral sclerosis. P. T. 43, 25–28. Available online at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5737249/
D’Ambrosi, N., Finocchi, P., Apolloni, S., Cozzolino, M., Ferri, A., Padovano, V., et al. (2009). The proinflammatory action of microglial P2 receptors is enhanced in SOD1 models for amyotrophic lateral sclerosis. J. Immunol. 7, 4648–4656. doi: 10.4049/jimmunol.0901212
De Diego, A. M. G., Lorrio, S., Hernández-Guijo, J. M., Gandía, L., and García, A. G. (2012). “Multitarget drugs for stabilization of calcium cycling and neuroprotection in neurodegenerative diseases and stroke,” in Therapeutic Targets: Modulation, Inhibition and Activation eds L. Botana, M. Loza (Hoboken, NJ: Wiley and Sons Inc.), 123–200.
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2, 245–256. doi: 10.1016/j.neuron.2011.09.011
Díaz-Hernández, M., Díez-Zaera, M., Sánchez-Nogueiro, J., Gómez-Villafuertes, R., Canals, J. M., Alberch, J., et al. (2009). Altered P2X7-receptor level and function in mouse models of Huntington’s disease and therapeutic efficacy of antagonist administration. FASEB J. 6, 1893–1906. doi: 10.1096/fj.08-122275
DiSabato, D. J., Quan, N., and Godbout, J. P. (2016). Neuroinflammation: the devil is in the details. J. Neurochem. 139, 136–153. doi: 10.1111/jnc.13607
Di Virgilio, F. (2015). P2X receptors and inflammation. Curr. Med. Chem. 22, 866–877. doi: 10.2174/0929867322666141210155311
Doble, A. (1996). The pharmacology and mechanism of action of riluzole. Neurology 47, S233–S241. doi: 10.1212/wnl.47.6_suppl_4.233s
Duplantier, A. J., Dombroski, M. A., Subramanyam, C., Beaulieu, A. M., Chang, S. P., Gabel, C. A., et al. (2011). Optimization of the physicochemical and pharmacokinetic attributes in a 6-azauracil series of P2X7 receptor antagonists leading to the discovery of the clinical candidate CE-224,535. Bioorg. Med. Chem. Lett. 12, 3708–3711. doi: 10.1016/j.bmcl.2011.04.077
Fabbrizio, P., Amadio, S., Apolloni, S., and Volonte, C. (2017). P2X7 receptor activation modulates autophagy in SOD1–G93A mouse microglia. Front. Cell. Neurosci. 11:249. doi: 10.3389/fncel.2017.00249
Fabbrizio, P., Apolloni, S., Bianchi, A., Salvatori, I., Valle, C., Lanzuolo, C., et al. (2020). P2X7 activation enhances skeletal muscle metabolism and regeneration in SOD1G93A mouse model of amyotrophic lateral sclerosis. Brain Pathol. 2, 272–282. doi: 10.1111/bpa.12774
Ferrari, D., Pizzirani, C., Adinolfi, E., Lemoli, R. M., Curti, A., Idzko, M., et al. (2006). The P2X7 receptor: a key player in IL-1 processing and release. J. Immunol. 7, 3877–3883. doi: 10.4049/jimmunol.176.7.3877
Fiszman, M. L., Ricart, K. C., Latini, A., Rodriguez, G., and Sica, R. E. (2010). in vitro neurotoxic properties and excitatory aminoacids concentration in the cerebrospinal fluid of amyotrophic lateral sclerosis patients. Relationship with the degree of certainty of disease diagnoses. Acta Neurol. Scand. 121, 120–126. doi: 10.1111/j.1600-0404.2009.01200.x
Franceschini, A., Capece, M., Chiozzi, P., Falzoni, S., Sanz, J. M., Sarti, A. C., et al. (2015). The P2X7 receptor directly interacts with the NLRP3 inflammasome scaffold protein. FASEB J. 6, 2450–2461. doi: 10.1096/fj.14-268714
Fray, A. E., Ince, P. G., Banner, S. J., Milton, I. D., Usher, P. A., Cookson, M. R., et al. (1998). The expression of the glial glutamate transporter protein EAAT2 in motor neuron disease: an immunohistochemical study. Eur. J. Neurosci. 8, 2481–2489. doi: 10.1046/j.1460-9568.1998.00273.x
Galluzzi, L., and Kroemer, G. (2008). Necroptosis: a specialized pathway of programmed necrosis. Cell 135, 1161–1163. doi: 10.1016/j.cell.2008.12.004
Gandelman, M., Peluffo, H., Beckman, J. S., Cassina, P., and Barbeito, L. (2010). Extracellular ATP and the P2X7 receptor in astrocyte-mediated motor neuron death: implications for amyotrophic lateral sclerosis. J. Neuroinflammation 7:33. doi: 10.1186/1742-2094-7-33
Grosskreutz, J., Van Den Bosch, L., and Keller, B. U. (2010). Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 47, 165–174. doi: 10.1016/j.ceca.2009.12.002
Guile, S. D., Alcaraz, L., Birkinshaw, T. N., Bowers, K. C., Ebden, M. R., Furber, M., et al. (2009). Antagonists of the P2X(7) receptor. From lead identification to drug development. J. Med. Chem. 10, 3123–3141. 10.1021/jm801528x
Gunasekaran, R., Narayani, R. S., Vijayalakshmi, K., Alladi, P. A., Shobha, K., Nalini, A., et al. (2009). Exposure to cerebrospinal fluid of sporadic amyotrophic lateral sclerosis patients alters Nav1.6 and Kv1.6 channel expression in rat spinal motor neurons. Brain Res. 1255, 170–179. doi: 10.1016/j.brainres.2008.11.099
Gunter, T. E., Gunter, K. K., Sheu, S. S., and Gavin, C. E. (1994). Mitochondrial calcium transport: physiological and pathological relevance. Am. J. Physiol. 267, C313–C339. doi: 10.1152/ajpcell.1994.267.2.C313
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 5166, 1772–1775. doi: 10.1126/science.266.5190.1586-a
Hardiman, O., Al-Chalabi, A., Chio, A., Corr, E. M., Logroscino, G., Robberecht, W., et al. (2017). Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 3:17071. doi: 10.1038/nrdp.2017.71
Heath, P. R., and Shaw, P. J. (2002). Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis. Muscle Nerve 26, 438–458. doi: 10.1002/mus.10186
Hempel, C., Norenberg, W., Sobottka, H., Urban, N., Nicke, A., Fischer, W., et al. (2013). The phenothiazine-class antipsychotic drugs prochlorperazine and trifluoperazine are potent allosteric modulators of the human P2X7 receptor. Neuropharmacology 75, 365–379. doi: 10.1016/j.neuropharm.2013.07.027
Herdewyn, S., Cirillo, C., Van Den Bosch, L., Robberecht, W., Vanden Berghe, P., and Van Damme, P. (2014). Prevention of intestinal obstruction reveals progressive neurodegeneration in mutant TDP-43 (A315T) mice. Mol. Neurodegener. 9:24. doi: 10.1186/1750-1326-9-24
Herrington, J., Park, Y. B., Babcock, D. F., and Hille, B. (1996). Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron 16, 219–228. doi: 10.1016/s0896-6273(00)80038-0
Ho, B. K., Alexianu, M. E., Colom, L. V., Mohamed, A. H., Serrano, F., and Appel, S. H. (1996). Expression of calbindin-D28K in motoneuron hybrid cells after retroviral infection with calbindin-D28K cDNA prevents amyotrophic lateral sclerosis IgG-mediated cytotoxicity. Proc. Natl. Acad. Sci. U S A 93, 6796–6801. doi: 10.1073/pnas.93.13.6796
Hung, S. C., Choi, C. H., Said-Sadier, N., Johnson, L., Atanasova, K. R., Sellami, H., et al. (2013). P2X4 assembles with P2X7 and pannexin-1 in gingival epithelial cells and modulates ATP-induced reactive oxygen species production and inflammasome activation. PLoS One 8:e70210. doi: 10.1371/journal.pone.0070210
Ihara, Y., Nobukuni, K., Takata, H., and Hayabara, T. (2005). Oxidative stress and metal content in blood and cerebrospinal fluid of amyotrophic lateral sclerosis patients with and without a Cu, Zn-superoxide dismutase mutation. Neurol. Res. 27, 105–108. doi: 10.1179/016164105x18430
Jaarsma, D., Rognoni, F., van Duijn, W., Verspaget, H. W., Haasdijk, E. D., and Holstege, J. C. (2001). CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol. 102, 293–305. doi: 10.1007/s004010100399
Jacobson, K. A. (2010). P2X and P2Y receptors. Available online at: https://www.tocris.com/literature/scientific-reviews/p2x-and-p2y-receptors. Accessed April 28, 2020
Jacobson, K. A., Costanzi, S., Joshi, B. V., Besada, P., Shin, D. H., Ko, H., et al. (2006). Agonists and antagonists for P2 receptors. Novartis Found. Symp. 276, 58–68; discussion-72, 107–112, 275–281. doi: 10.1002/9780470032244.ch6
Jacobson, K. A., Jarvis, M. F., and Williams, M. (2002). Purine and pyrimidine (P2) receptors as drug targets. J. Med. Chem. 45, 4057–4093. doi: 10.1021/jm020046y
Janssen, B., Vugts, D. J., Funke, U., Spaans, A., Schuit, R. C., Kooijman, E., et al. (2014). Synthesis and initial preclinical evaluation of the P2X7 receptor antagonist [(1)(1)C]A-740003 as a novel tracer of neuroinflammation. J. Labelled Comp. Radiopharm. 8, 509–516. doi: 10.1002/jlcr.3206
Jeon, G. S., Shim, Y. M., Lee, D. Y., Kim, J. S., Kang, M., Ahn, S. H., et al. (2019). Pathological modification of TDP-43 in amyotrophic lateral sclerosis with SOD1 mutations. Mol. Neurobiol. 56, 2007–2021. doi: 10.1007/s12035-018-1218-2
Jimenez-Pacheco, A., Diaz-Hernandez, M., Arribas-Blazquez, M., Sanz-Rodriguez, A., Olivos-Ore, L. A., Artalejo, A. R., et al. (2016). Transient P2X7 receptor antagonism produces lasting reductions in spontaneous seizures and gliosis in experimental temporal lobe epilepsy. J. Neurosci. 22, 5920–5932. doi: 10.1523/JNEUROSCI.4009-15.2016
Jo, E. K., Kim, J. K., Shin, D. M., and Sasakawa, C. (2016). Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 13, 148–159. doi: 10.1038/cmi.2015.95
Jung, C., Higgins, C. M., and Xu, Z. (2002). Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. J. Neurochem. 83, 535–545. doi: 10.1046/j.1471-4159.2002.01112.x
Kaal, E. C., Vlug, A. S., Versleijen, M. W., Kuilman, M., Joosten, E. A., and Bar, P. R. (2000). Chronic mitochondrial inhibition induces selective motoneuron death in vitro: a new model for amyotrophic lateral sclerosis. J. Neurochem. 74, 1158–1165. doi: 10.1046/j.1471-4159.2000.741158.x
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., McConkey, B. J., Vande Velde, C., et al. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 5, 572–574. doi: 10.1038/ng.132
Kaczmarek-Hajek, K., Zhang, J., Kopp, R., Grosche, A., Rissiek, B., Saul, A., et al. (2018). Re-evaluation of neuronal P2X7 expression using novel mouse models and a P2X7-specific nanobody. Elife 7:e36217. doi: 10.7554/eLife.36217
Karmakar, M., Katsnelson, M. A., Dubyak, G. R., and Pearlman, E. (2016). Neutrophil P2X7 receptors mediate NLRP3 inflammasome-dependent IL-1beta secretion in response to ATP. Nat. Commun. 7:10555. doi: 10.1038/ncomms10555
Katsnelson, M. A., Rucker, L. G., Russo, H. M., and Dubyak, G. R. (2015). K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 194, 3937–3952. doi: 10.4049/jimmunol.1402658
Kirkinezos, I. G., Bacman, S. R., Hernandez, D., Oca-Cossio, J., Arias, L. J., Perez-Pinzon, M. A., et al. (2005). Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J. Neurosci. 1, 164–172. doi: 10.1523/JNEUROSCI.3829-04.2005
Kroemer, G., Dallaporta, B., and Resche-Rigon, M. (1998). The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 60, 619–642. doi: 10.1146/annurev.physiol.60.1.619
Kruman, I. I., Pedersen, W. A., Springer, J. E., and Mattson, M. P. (1999). ALS-linked Cu/Zn-SOD mutation increases vulnerability of motor neurons to excitotoxicity by a mechanism involving increased oxidative stress and perturbed calcium homeostasis. Exp. Neurol. 160, 28–39. doi: 10.1006/exnr.1999.7190
Ladewig, T., Kloppenburg, P., Lalley, P. M., Zipfel, W. R., Webb, W. W., and Keller, B. U. (2003). Spatial profiles of store-dependent calcium release in motoneurones of the nucleus hypoglossus from newborn mouse. J. Physiol. 547, 775–787. doi: 10.1113/jphysiol.2002.033605
Lagier-Tourenne, C., and Cleveland, D. W. (2009). Rethinking ALS: the FUS about TDP-43. Cell 136, 1001–1004. doi: 10.1016/j.cell.2009.03.006
Lanuza, M. A., Just-Borras, L., Hurtado, E., Cilleros-Mane, V., Tomas, M., Garcia, N., et al. (2019). The impact of kinases in amyotrophic lateral sclerosis at the neuromuscular synapse: insights into BDNF/TrkB and PKC signaling. Cells 8:1578. doi: 10.3390/cells8121578
Letavic, M. A., Savall, B. M., Allison, B. D., Aluisio, L., Andres, J. I., De Angelis, M., et al. (2017). 4-Methyl-6,7-dihydro-4H-triazolo[4,5-c]pyridine-Based P2X7 Receptor antagonists: optimization of pharmacokinetic properties leading to the identification of a clinical candidate. J. Med. Chem. 11, 4559–4572. doi: 10.1021/acs.jmedchem.7b00408
Liao, B., Zhao, W., Beers, D. R., Henkel, J. S., and Appel, S. H. (2012). Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp. Neurol. 237, 147–152. doi: 10.1016/j.expneurol.2012.06.011
Lin, C. L., Bristol, L. A., Jin, L., Dykes-Hoberg, M., Crawford, T., Clawson, L., et al. (1998). Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 3, 589–602. doi: 10.1016/s0896-6273(00)80997-6
Liochev, S. I., and Fridovich, I. (2003). Mutant Cu,Zn superoxide dismutases and familial amyotrophic lateral sclerosis: evaluation of oxidative hypotheses. Free Radic. Biol. Med. 34, 1383–1389. doi: 10.1016/s0891-5849(03)00153-9
Liu, Y., Pattamatta, A., Zu, T., Reid, T., Bardhi, O., Borchelt, D. R., et al. (2016). C9orf72 BAC mouse model with motor deficits and neurodegenerative features of ALS/FTD. Neuron 3, 521–534. doi: 10.1016/j.neuron.2016.04.005
Liu, J., Prell, T., Stubendorff, B., Keiner, S., Ringer, T., Gunkel, A., et al. (2016b). Down-regulation of purinergic P2X7 receptor expression and intracellular calcium dysregulation in peripheral blood mononuclear cells of patients with amyotrophic lateral sclerosis. Neurosci. Lett. 630, 77–83. doi: 10.1016/j.neulet.2016.07.039
Lutz, C. (2018). Mouse models of ALS: Past, present and future. Brain Res. 1693, 1–10. doi: 10.1016/j.brainres.2018.03.024
Ly, D., Dongol, A., Cuthbertson, P., Guy, T. V., Geraghty, N. J., Sophocleous, R. A., et al. (2020). The P2X7 receptor antagonist JNJ-47965567 administered thrice weekly from disease onset does not alter progression of amyotrophic lateral sclerosis in SOD1(G93A) mice. Purinergic Signal. 1, 109–122. doi: 10.1007/s11302-020-09692-4
Magrane, J., Cortez, C., Gan, W. B., and Manfredi, G. (2014). Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 23, 1413–1424. doi: 10.1093/hmg/ddt528
Majounie, E., Renton, A. E., Mok, K., Dopper, E. G., Waite, A., Rollinson, S., et al. (2012). Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 4, 323–330. doi: 10.1016/j.yneu.2012.05.040
Masitinib in Combination With Riluzole for the Treatment of Patients Suffering From Amyotrophic Lateral Sclerosis (ALS) (2018). [Internet]. Available online at: https://clinicaltrials.gov/ct2/show/NCT02588677?term=masitinibandcond=ALSanddraw=2andrank=1. Accessed April 28, 2020
Mattiazzi, M., D’Aurelio, M., Gajewski, C. D., Martushova, K., Kiaei, M., Beal, M. F., et al. (2002). Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 33, 29626–29633. doi: 10.1074/jbc.m203065200
Michel, A. D., Ng, S. W., Roman, S., Clay, W. C., Dean, D. K., and Walter, D. S. (2009). Mechanism of action of species-selective P2X(7) receptor antagonists. Br. J. Pharmacol. 156, 1312–1325. doi: 10.1111/j.1476-5381.2009.00135.x
Miller, R. G., Mitchell, J. D., Lyon, M., and Moore, D. H. (2003). Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Amyotroph. Lateral Scler. Other Motor Neuron Disord. 4, 191–206. Available online at: https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD001447.pub3/full
Minkiewicz, J., de Rivero Vaccari, J. P., and Keane, R. W. (2013). Human astrocytes express a novel NLRP2 inflammasome. Glia 61, 1113–1121. doi: 10.1002/glia.22499
Mitchell, J. C., McGoldrick, P., Vance, C., Hortobagyi, T., Sreedharan, J., Rogelj, B., et al. (2013). Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol. 2, 273–288. doi: 10.1007/s00401-012-1043-z
Mitsumoto, H., Santella, R. M., Liu, X., Bogdanov, M., Zipprich, J., Wu, H. C., et al. (2008). Oxidative stress biomarkers in sporadic ALS. Amyotroph. Lateral Scler. 3, 177–183. doi: 10.1080/17482960801933942
Monk, P. N., and Shaw, P. J. (2006). ALS: life and death in a bad neighborhood. Nat. Med. 12, 885–887. doi: 10.1038/nm0806-885
Montero, M., Alonso, M. T., Carnicero, E., Cuchillo-Ibanez, I., Albillos, A., Garcia, A. G., et al. (2000). Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat. Cell Biol. 2, 57–61. doi: 10.1038/35000001
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 5796, 130–133. doi: 10.1126/science.1134108
O’Brien-Brown, J., Jackson, A., Reekie, T. A., Barron, M. L., Werry, E. L., Schiavini, P., et al. (2017). Discovery and pharmacological evaluation of a novel series of adamantyl cyanoguanidines as P2X7 receptor antagonists. Eur. J. Med. Chem. 130, 433–439. doi: 10.1016/j.ejmech.2017.02.060.
Orrenius, S., Zhivotovsky, B., and Nicotera, P. (2003). Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 4, 552–565. doi: 10.1038/nrm1150
Parisi, C., Napoli, G., Amadio, S., Spalloni, A., Apolloni, S., Longone, P., et al. (2016). MicroRNA-125b regulates microglia activation and motor neuron death in ALS. Cell Death Differ. 3, 531–541. doi: 10.1038/cdd.2015.153
Pasinelli, P., Belford, M. E., Lennon, N., Bacskai, B. J., Hyman, B. T., Trotti, D., et al. (2004). Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 1, 19–30. doi: 10.1016/j.neuron.2004.06.021
Pasinelli, P., and Brown, R. H. (2006). Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 7, 710–723. doi: 10.1038/nrn1971
Patai, R., Nogradi, B., Engelhardt, J. I., and Siklos, L. (2017). Calcium in the pathomechanism of amyotrophic lateral sclerosis - Taking center stage? Biochem. Biophys. Res. Commun. 483, 1031–1039. doi: 10.1016/j.bbrc.2016.08.089
Petrov, D., Mansfield, C., Moussy, A., and Hermine, O. (2017). ALS clinical trials review: 20 years of failure. are we any closer to registering a new treatment? Front. Aging Neurosci. 9:68. doi: 10.3389/fnagi.2017.00068
Philips, T., and Rothstein, J. D. (2015). Rodent models of amyotrophic lateral sclerosis. Curr. Protoc. Pharmacol. 69, 5.67.1–5.67.21. doi: 10.1002/0471141755.ph0567s69
Plaitakis, A., and Caroscio, J. T. (1987). Abnormal glutamate metabolism in amyotrophic lateral sclerosis. Ann. Neurol. 22, 575–579. doi: 10.1002/ana.410220503
Ransohoff, R. M. (2016). How neuroinflammation contributes to neurodegeneration. Science 353, 777–783. doi: 10.1126/science.aag2590
Rao, S. D., Yin, H. Z., and Weiss, J. H. (2003). Disruption of glial glutamate transport by reactive oxygen species produced in motor neurons. J. Neurosci. 23, 2627–2633. doi: 10.1523/JNEUROSCI.23-07-02627.2003
Recourt, K., van der Aart, J., Jacobs, G., de Kam, M., Drevets, W., van Nueten, L., et al. (2020). Characterisation of the pharmacodynamic effects of the P2X7 receptor antagonist JNJ-54175446 using an oral dexamphetamine challenge model in healthy males in a randomised, double-blind, placebo-controlled, multiple ascending dose trial. J. Psychopharmacol. 2020:269881120914206. doi: 10.1177/0269881120914206
Renton, A. E., Majounie, E., Waite, A., Simon-Sanchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2, 257–268. doi: 10.1016/j.neuron.2011.09.010
Rizzuto, R., Bernardi, P., and Pozzan, T. (2000). Mitochondria as all-round players of the calcium game. J. Physiol. 529, 37–47. doi: 10.1111/j.1469-7793.2000.00037.x
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., et al. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 6415, 59–62. doi: 10.1038/362059a0
Rothstein, J. D., Kuncl, R., Chaudhry, V., Clawson, L., Cornblath, D. R., Coyle, J. T., et al. (1991). Excitatory amino acids in amyotrophic lateral sclerosis: an update. Ann. Neurol. 2, 224–225. doi: 10.1002/ana.410300223
Rothstein, J. D., Martin, L. J., and Kuncl, R. W. (1992). Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 326, 1464–1468. doi: 10.1056/NEJM199205283262204
Rothstein, J. D., Van Kammen, M., Levey, A. I., Martin, L. J., and Kuncl, R. W. (1995). Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 38, 73–84. doi: 10.1002/ana.410380114
Sasaki, S., Komori, T., and Iwata, M. (2000). Excitatory amino acid transporter 1 and 2 immunoreactivity in the spinal cord in amyotrophic lateral sclerosis. Acta Neuropathol. 100, 138–144. doi: 10.1007/s004019900159
Sen, I., Nalini, A., Joshi, N. B., and Joshi, P. G. (2005). Cerebrospinal fluid from amyotrophic lateral sclerosis patients preferentially elevates intracellular calcium and toxicity in motor neurons via AMPA/kainate receptor. J. Neurol. Sci. 235, 45–54. doi: 10.1016/j.jns.2005.03.049
Smith, R. G., Henry, Y. K., Mattson, M. P., and Appel, S. H. (1998). Presence of 4-hydroxynonenal in cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Ann Neurol. 44, 696–699. doi: 10.1002/ana.410440419
Smith, E. F., Shaw, P. J., and De Vos, K. J. (2019). The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 710:132933. doi: 10.1016/j.neulet.2017.06.052
Sperlagh, B., Vizi, E. S., Wirkner, K., and Illes, P. (2006). P2X7 receptors in the nervous system. Prog. Neurobiol. 78, 327–346. doi: 10.1016/j.pneurobio.2006.03.007
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., et al. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 5870, 1668–1672. doi: 10.1126/science.1154584
Stephenson, J., and Amor, S. (2017). Modelling amyotrophic lateral sclerosis in mice. Drug Discov. Today Dis. Model. 25–26, 35–45. doi: 10.1016/j.ddmod.2018.10.001
Tateno, M., Sadakata, H., Tanaka, M., Itohara, S., Shin, R. M., Miura, M., et al. (2004). Calcium-permeable AMPA receptors promote misfolding of mutant SOD1 protein and development of amyotrophic lateral sclerosis in a transgenic mouse model. Hum. Mol. Genet. 19, 2183–2196. doi: 10.1093/hmg/ddh246
Terro, F., Lesort, M., Viader, F., Ludolph, A., and Hugon, J. (1996). Antioxidant drugs block in vitro the neurotoxicity of CSF from patients with amyotrophic lateral sclerosis. Neuroreport 7, 1970–1972.
The ALS association. (2020). Available online at: http://www.alsa.org/about-als/. Accessed April 28, 2020
Thomsen, G. M., Avalos, P., Ma, A. A., Alkaslasi, M., Cho, N., Wyss, L., et al. (2018). Transplantation of neural progenitor cells expressing glial cell line-derived neurotrophic factor into the motor cortex as a strategy to treat amyotrophic lateral sclerosis. Stem Cells 7, 1122–1131. doi: 10.1002/stem.2825
Tovar, Y. R. L. B., Santa-Cruz, L. D., Zepeda, A., and Tapia, R. (2009). Chronic elevation of extracellular glutamate due to transport blockade is innocuous for spinal motoneurons in vivo. Neurochem. Int. 54, 186–191. doi: 10.1016/j.neuint.2008.09.015
Trias, E., Ibarburu, S., Barreto-Nunez, R., Babdor, J., Maciel, T. T., Guillo, M., et al. (2016). Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J. Neuroinflammation 1:177. doi: 10.1186/s12974-016-0620-9
Van Damme, P., Braeken, D., Callewaert, G., Robberecht, W., and Van Den Bosch, L. (2005). GluR2 deficiency accelerates motor neuron degeneration in a mouse model of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 64, 605–612. doi: 10.1097/01.jnen.0000171647.09589.07
Van Damme, P., Van Den Bosch, L., Van Houtte, E., Callewaert, G., and Robberecht, W. (2002). GluR2-dependent properties of AMPA receptors determine the selective vulnerability of motor neurons to excitotoxicity. J. Neurophysiol. 88, 1279–1287. doi: 10.1152/jn.2002.88.3.1279
Van Den Bosch, L., and Robberecht, W. (2000). Different receptors mediate motor neuron death induced by short and long exposures to excitotoxicity. Brain Res. Bull. 53, 383–388. doi: 10.1016/s0361-9230(00)00371-3
Van Den Bosch, L., Schwaller, B., Vleminckx, V., Meijers, B., Stork, S., Ruehlicke, T., et al. (2002). Protective effect of parvalbumin on excitotoxic motor neuron death. Exp. Neurol. 2, 150–161. doi: 10.1006/exnr.2001.7858
Van Den Bosch, L., Van Damme, P., Bogaert, E., and Robberecht, W. (2006). The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1762, 1068–1082. doi: 10.1016/j.bbadis.2006.05.002
Volonte, C., Apolloni, S., Parisi, C., and Amadio, S. (2016). Purinergic contribution to amyotrophic lateral sclerosis. Neuropharmacology 104, 180–193. doi: 10.1016/j.neuropharm.2015.10.026
Wang, W., Wang, L., Lu, J., Siedlak, S. L., Fujioka, H., Liang, J., et al. (2016). The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 8, 869–878. doi: 10.1038/nm.4130
Wegorzewska, I., Bell, S., Cairns, N. J., Miller, T. M., and Baloh, R. H. (2009). TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. U S A 106, 18809–18814. doi: 10.1073/pnas.0908767106
Xu, T., Naraghi, M., Kang, H., and Neher, E. (1997). Kinetic studies of Ca2+ binding and Ca2+ clearance in the cytosol of adrenal chromaffin cells. Biophys. J. 73, 532–545. doi: 10.1016/s0006-3495(97)78091-3
Yáñez, M., Galan, L., Matias-Guiu, J., Vela, A., Guerrero, A., and Garcia, A. G. (2011). CSF from amyotrophic lateral sclerosis patients produces glutamate independent death of rat motor brain cortical neurons: protection by resveratrol but not riluzole. Brain Res. 1423, 77–86. doi: 10.1016/j.brainres.2011.09.025
Yaron, J. R., Gangaraju, S., Rao, M. Y., Kong, X., Zhang, L., Su, F., et al. (2015). K(+) regulates Ca2+ to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis. 6:e1954. doi: 10.3410/f.725889052.793519729
Yiangou, Y., Facer, P., Durrenberger, P., Chessell, I. P., Naylor, A., Bountra, C., et al. (2006). COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 6:12. doi: 10.1186/1471-2377-6-12
Keywords: amyotrophic lateral sclerosis, ALS, neuroinflammation, P2X7, P2X7 receptor antagonists, calcium dyshomeostasis, mitochondrial disruption, motoneuron death
Citation: Ruiz-Ruiz C, Calzaferri F and García AG (2020) P2X7 Receptor Antagonism as a Potential Therapy in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 13:93. doi: 10.3389/fnmol.2020.00093
Received: 25 March 2020; Accepted: 05 May 2020;
Published: 12 June 2020.
Edited by:
Miguel Diaz-Hernandez, Complutense University of Madrid, SpainReviewed by:
Ronald Sluyter, University of Wollongong, AustraliaCinzia Volonté, National Research Council (CNR), Italy
Copyright © 2020 Ruiz-Ruiz, Calzaferri and García. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio G. García, YWdnQHVhbS5lcw==
† These authors have contributed equally to this work