 Tariq Ahmed1,2
Tariq Ahmed1,2 Ann Van der Jeugd2,3
Ann Van der Jeugd2,3 Raphaëlle Caillierez4,5
Raphaëlle Caillierez4,5 Luc Buée4,5
Luc Buée4,5 David Blum4,5Rudi D’Hooge2,3
David Blum4,5Rudi D’Hooge2,3 Detlef Balschun1,2*
Detlef Balschun1,2*- 1Brain and Cognition, Faculty of Psychology and Educational Sciences, KU Leuven, Leuven, Belgium
- 2Leuven Brain Institute, Leuven, Belgium
- 3Laboratory of Biological Psychology, Brain and Cognition, Faculty of Psychology and Educational Sciences, KU Leuven, Leuven, Belgium
- 4Univ. Lille, Inserm, CHU Lille, U1172—LilNCog—Lille Neuroscience and Cognition, Lille, France
- 5Alzheimer and Tauopathies, LabEx DISTALZ, Lille, France
A major goal in diseases is identifying a potential therapeutic agent that is cost-effective and can remedy some, if not all, disease symptoms. In Alzheimer’s disease (AD), aggregation of hyperphosphorylated tau protein is one of the neuropathological hallmarks, and Tau pathology correlates better with cognitive impairments in AD patients than amyloid-β load, supporting a key role of tau-related mechanisms. Selenium is a non-metallic trace element that is incorporated in the brain into selenoproteins. Chronic treatment with sodium selenate, a non-toxic selenium compound, was recently reported to rescue behavioral phenotypes in tau mouse models. Here, we focused on the effects of chronic selenate application on synaptic transmission and synaptic plasticity in THY-Tau22 mice, a transgenic animal model of tauopathies. Three months with a supplement of sodium selenate in the drinking water (12 μg/ml) restored not only impaired neurocognitive functions but also rescued long-term depression (LTD), a major form of synaptic plasticity. Furthermore, selenate reduced the inactive demethylated catalytic subunit of protein phosphatase 2A (PP2A) in THY-Tau22 without affecting total PP2A.Our study provides evidence that chronic dietary selenate rescues functional synaptic deficits of tauopathy and identifies activation of PP2A as the putative mechanism.
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder affecting predominantly brain regions that are required for information processing and memory such as hippocampus and cortex (Squire et al., 2007; Hyman et al., 2012). The phenotypes of AD include deficits in declarative (short- and long-term) memory and disruption in synaptic plasticity, a cellular correlate of memory and undoubtedly one of the first functions to be affected in AD (Selkoe, 2002; Hoover et al., 2010; Takeuchi et al., 2013; Scheltens et al., 2016; Forner et al., 2017). At the neuropathological level, AD is defined by extracellular Aβ plaques, formed as a result of the misprocessing of amyloid precursor protein (APP) as well as aggregation of hyperphosphorylated tau proteins into neurofibrillary tangles (NFTs; Buée et al., 2010; Querfurth and LaFerla, 2010; Masters et al., 2015). The spatiotemporal progression of NFTs from the entorhinal cortex and the hippocampus to the isocortical areas has been reported to correlate well with cognitive deficits and disease progression. Thus, it is Tau pathology and not Aβ-mediated functional deterioration that turned out to correlate better with the severity of dementia (Gomez-Isla et al., 1997; Braak et al., 1998; Nelson et al., 2012; Brier et al., 2016). In accordance with the lower correlation between Aβ-pathology and AD progression, attempts to delay or reverse AD pathology by either removing Aβ or reducing its production had so far not the expected outcome in clinical studies (Lovestone and Manji, 2020; Sabbagh, 2020). It was therefore logical that tau has become the next main target for AD modifying strategies. Apart from the many ongoing Tau-targeting passive and active immunization studies, the diversity of mechanisms that contribute to Tau pathology (e.g., hyperphosphorylation, acetylation, N-glycosylation, and truncation) offers multiple targets for therapy development (Congdon and Sigurdsson, 2018). Strategies that target early pathological Tau mechanisms such as Tau hyperphosphorylation and aggregation meet ideally the requirement to shift the time of therapeutic interventions to very early stages of AD. Not only the degree of hyperphosphorylation is decisive for Tau pathology but also the specific pattern of Tau hyperphosphorylation, i.e., which of the many phosphorylations sites of Tau (Sergeant et al., 2008) are (hyper)phosphorylated under the particular pathological condition. These pathological Tau phosphorylation patterns are the result of an intricate balance between the activity of Tau kinases, such as glycogen synthase kinase-3β (GSK3β) as the major Tau kinase, and phosphatases with protein phosphatase 2A (PP2A) as the main representative (Liu et al., 2005; Kremer et al., 2011; Martin et al., 2013). Thus, therapeutic reduction of Tau hyperphosphorylation and subsequent aggregation can be either achieved by inhibition of Tau kinases or activation of phosphatases. Suitable candidate therapeutic compounds should be easy-to-administer, free of toxic side-effects in a broad dose range and show excellent bioavailability and CNS penetrating properties.
All these criteria are met by sodium selenate, an oxidized form of selenium, which was reported to be a selective activator of PP2A (Corcoran et al., 2010b; van Eersel et al., 2010; Cardoso et al., 2019). This is confirmed by a Phase IIa control trial that assessed the safety and tolerability of sodium selenate in forty patients with mild to moderate AD, aged ≥55 years, and found the compound to be safe and well-tolerated (Malpas et al., 2016).
PP2A accounts for ~71% of total tau phosphatase activity in the human brain (Liu et al., 2005); and its expression and/or activity have been found significantly decreased under the conditions of AD pathology (see Lambrecht et al., 2013; Taleski and Sontag, 2018 for further references), which should promote hyperphosphorylation of Tau by disturbing the balance between Tau kinase and phosphatase activities mentioned above. PP2A phosphatases are a diverse family of holoenzymes, comprised of at least two subunits, a scaffolding A subunit and a catalytic C subunit. This dimer can further associate with one of a diverse range (<20) of regulatory B-subunits to form the typical mammalian heterotrimeric holoenzyme. Within these trimeric complexes, the B-type subunits determine important properties like the catalytic activity, substrate specificity, and subcellular localization, and hence, the physiological functions of the holoenzyme (Lambrecht et al., 2013; Taleski and Sontag, 2018). Important to mention, the predominant neuronal PP2A holoenzyme that shows the highest tau phosphatase activity and the strongest affinity for tau is the one that contains the Ba (or PPP2R2A or PR55) regulatory subunit (Sontag et al., 1996, 1999; Xu et al., 2008). Important for a candidate therapeutic compound, sodium selenate was found to be non-toxic in primary rat hippocampal slice cultures up to a concentration of 100 μM (Corcoran et al., 2010b; van Eersel et al., 2010), while other selenium compounds such as sodium selenite and selenomethionine showed significant toxicity under the same conditions (Corcoran et al., 2010b; van Eersel et al., 2010).
Here, we examined for the first time the effects of a chronic 3-month dietary application of this compound on the disease phenotype of a tauopathy AD mouse model, THY-Tau22 mice (Schindowski et al., 2006), with the focus on synaptic transmission and plasticity. The selenate treatment started at 9 months, an age at which THY-Tau22 mice progressively develop AD-like hippocampal Tau pathology associated with severe deficits in memory underlined by synaptic impairments (Schindowski et al., 2006; Van der Jeugd et al., 2011, 2013; Burnouf et al., 2013). Our data demonstrate that a chronic dietary supplement with sodium selenate rescues deficits in hippocampal synaptic plasticity and neurocognition in THY-Tau22 mice and disclose an activation of PP2A as the putative underlying mechanism.
Materials and Methods
Animals
Twenty male heterozygous transgenic THY-Tau22 mice and a corresponding number of wildtype (WT) sibling controls were used in the present study. THY-Tau22 and WT littermate mice were generated as described previously (Schindowski et al., 2006). THY-Tau22 mice (C57Bl6/J background) overexpress a 1N4R human mutated Tau isoform (G272V and P301S) under a Thy1.2-promoter. All animals were kept in standard animal cages under conventional laboratory conditions (12 h/12 h light-dark cycle, lights on 08:00–20:00, 22°C), with ad libitum access to food and water, unless stated otherwise. Behavioral tests were conducted during the light phase of their activity cycle. All behavioral and electrophysiological experiments have been reviewed and approved by the animal experiments committee of the University of Leuven, Belgium, and were carried out following the European Directive 2010/63/EU.
Chronic Treatment With Sodium Selenate (Na2SO4)
At the age of 9 months, 12 weeks before the start of experiments, the mice were subdivided into four groups of 10 mice. One group from each genotype was provided with 12 μg/ml sodium selenate in the drinking water (Corcoran et al., 2010b; van Eersel et al., 2010). The other half received normal drinking water. We chose sodium selenate because the closely related inorganic form sodium selenite and the main organic form selenomethionine are toxic in primary hippocampal slice cultures at comparable and even lower concentrations (Corcoran et al., 2010b).
Time-Line
Behavioral testing [Cage Activity, Rotating Rod, Open Field, Morris Water Maze (MWM) and Passive Avoidance] was performed successively as depicted in Figure 1A. Mice were handled for 3 days before the start of the experiments (including weighing the mice and color-coding their tails for further identification). Before each test, animals were habituated to the experimental room for at least 30 min.
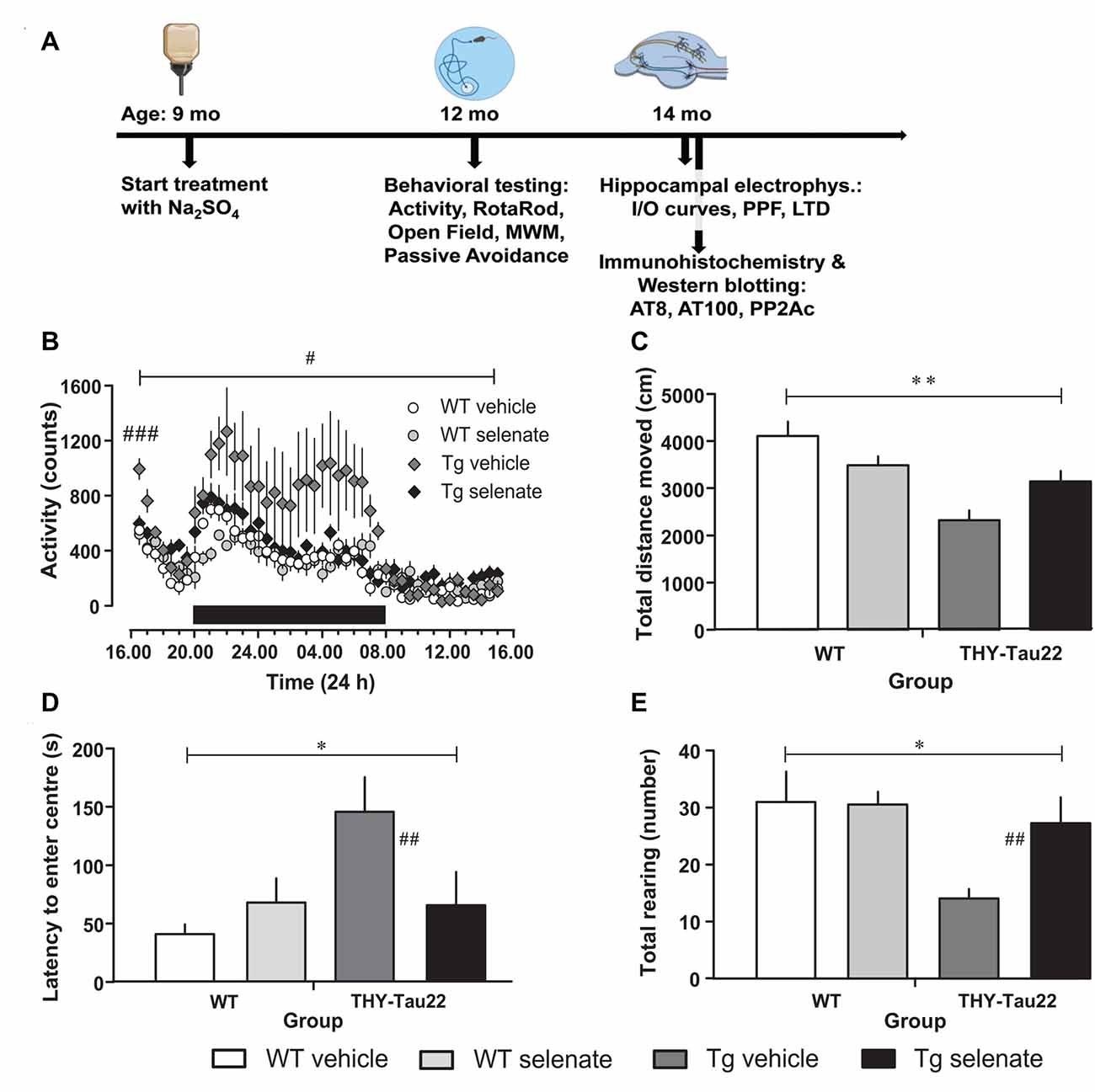
Figure 1. Selenate treatment restores behavioral and neurocognitive deficits in THY-Tau22 (henceforth referred to as Tg) mice to levels comparable with wildtype (WT) controls. (A) Timeline of the experiments. The behavioral experiments started at the age of 12 months, the electrophysiological examination at 14 months. At the same time, tissue samples were collected for immunohistochemistry and Western blotting. (B) Analysis of cage activity revealed higher activity of Tg mice at the beginning of the recording period and increased nocturnal activity as compared with WT controls. These differences were abolished by chronic selenate treatment. Note that selenate treatment did not affect WT animals. (C–E) Open-field behavior. Tg animals traveled less in the arena (C) showed higher latencies to enter the center of the arena (D) and displayed less exploratory rearings in the center (E) as compared with WT mice. Selenate treatment remediated these behavior changes. Mean ± SEM is given; group sizes: cage activity n = 10 per group; open field test Tg-veh n = 9, Tg-sel n = 10, WT-veh n = 8, WT-sel n = 10. Bar with # indicates the significance level p ≤ 0.05 in RM-ANOVA with a Dunnett post hoc test using Tg vehicle as comparison; bars with a * and ** indicate p ≤ 0.01 and p ≤ 0.001, respectively, in the Two-way ANOVA; # and ## represent p ≤ 0.01 and p ≤ 0.001, respectively, in the Dunnett post hoc test.
Behavioral Studies
Neuromotor Tests
Cage Activity
To measure diurnal cage activity, mice were placed individually in 26.7 cm × 20.7 cm transparent cages (floor area 370 cm2) that were positioned between three infrared photo beams. Beam crossings that represented locomotor cage activity were registered for each 30-min interval during a 23-h recording period, using an interfaced PC (Goddyn et al., 2006).
Rotating Rod
Previous studies on the Rotating Rod (Rotarod, MED Associates) documented no motor deficits for THY-Tau22 (Van der Jeugd et al., 2011) but to exclude any effect of the selenate treatment on motor coordination, the rotarod study was repeated.
Open Field
Open field exploration was monitored using a brightly lit 50 cm × 50 cm arena. Before the experiment, mice underwent 30 min of dark adaptation inside a dark cupboard. Each mouse was placed in the arena for 10 min. Numbers of entries in the corners and the center (defined as a circle with a 30 cm diameter), the latency of the first corner and center approach, as well as time spent, and velocity in corners and center were measured (Noldus Ethovision, The Netherlands).
Neurocognitive Tests
Morris Water Maze
Spatial and reference memory abilities were examined in the standard hidden-platform acquisition and retention version of the MWM. Training consisted of five consecutive days of four trials (120 s each) from different starting positions. The time required to locate the hidden escape platform (escape latency in seconds), distance traveled (path length in centimeter), and swimming speed (velocity in cm/s) were recorded (Noldus Ethovision, The Netherlands). After 2 days of rest, a probe trial with the platform removed was performed for 100 s. The time (s) the animals spent in each of the four quadrants, and the frequency the mice swam over the former platform position (n) were measured (Van der Jeugd et al., 2011).
Passive Avoidance
Single-trial passive avoidance learning was examined in a step-through box with a small illuminated compartment and a larger dark compartment with a grid-floor. The grid-floor was connected with a constant current shocker (MED Associates Inc., St. Albans, USA). Animals were dark-adapted for 30 min and then placed in the small illuminated compartment. After 5 s, the sliding door to the dark compartment was opened and the entry latency was recorded. When mice entered the dark compartment, a mild foot shock (0.3 mA, 1 s) was delivered. Retention was tested 24 h later according to the same procedure, excepting that animals did not receive a shock. The entry latencies were recorded with a cut-off of 300 s.
In vitro Hippocampal Synaptic Plasticity
After completion of behavioral tests, animals were killed by cervical dislocation and the brain rapidly isolated out into ice-cold (4°C) artificial cerebrospinal fluid (ACSF), oxygen saturated with carbogen (95% O2/5% CO2). ACSF consisted of (in mM): 124 NaCl, 4.9 KCl, 24.6 NaHCO3, 1.20, KH2PO4, 2.0 CaCl2, 2.0 MgCl2, 10.0 glucose, pH 7.4. Transverse slices (400 μm thick) were prepared from the dorsal area of the right hippocampus with a tissue chopper and placed into a submerged-type chamber, where they were kept at 32°C and continuously perfused with ACSF at a flow-rate of 2.2 ml/min. After about 90 min incubation, one slice was arbitrarily selected and a lacquer-coated monopolar stainless steel or tungsten electrode was placed in CA1 stratum radiatum for stimulation in constant current mode. For the recording of field excitatory postsynaptic potentials (fEPSPs), a glass electrode (filled with ACSF, 3–7 MΩ) was placed in the stratum radiatum. The time course of the field EPSP was measured as the descending slope function for all sets of experiments. After a further hour of incubation, input/output curves were established and the stimulation strength was adjusted to elicit an fEPSP-slope of 35% of the maximum and was kept constant throughout the experiment. Paired pulse facilitation was investigated by applying two pulses in rapid succession (interpulse intervals of 10, 20, 50, 100, 200, and 500 ms, respectively) at 120 s intervals. During baseline recording, three single stimuli (0.1 ms pulse width; 10 s interval) were measured every 5 min and averaged. LTD was induced by low-frequency stimulation (LFS) consisting of 1,200 pulses at 2 Hz (0.2 ms pulse-width) and repeated three times with a 10-min interval between completion of one LFS-train and the start of the successive one (Balschun et al., 2003; Ahmed et al., 2011, 2015). Immediately after each conditioning stimulus, evoked responses were monitored at 2, 5, and 8 min and then subsequently every 5 min recording up to 4 h after the first conditioning stimulus.
Immunohistochemistry and Western Blotting
For immunohistological experiments, the tissue was prepared as described in Leboucher et al. (2013). Animals were killed by cervical dislocation, brains were removed, one hemisphere was postfixed for 7 days in 4% paraformaldehyde and then incubated in 20% sucrose for 24 h before being frozen at −40°C in isopentane (methyl butane) and kept frozen at −80°C until use. Free-floating coronal sections (40 μm) were obtained using a cryostat (Leica). The sections of interest were used for free-floating immunohistochemistry using AT8 (Pierce MN-1020, recognizes pS202/T205, 1/200) and AT100 antibodies (Thermo Fisher Scientific, recognizes pT212/pS214; 1/1,000) and finally mounted on Superfrost slides respectively. Of note, although these are mouse IgG1 antibodies, they were proven to work fine in THY-Tau22 mice without causing any disturbing background staining (Leboucher et al., 2013). Staining pixel count was performed blindly by setting the threshold to the same value for each section as previously described using ImageJ software (Scion Software).
The other hemisphere was used for Western blotting as previously described (Laurent et al., 2014). Briefly, tissue was homogenized in 200 μl Tris buffer (pH 7.4) containing 10% sucrose and protease inhibitors (Complete; Roche Diagnostics, Meylan, France), sonicated and kept at −80°C until use. Protein amounts were evaluated using the BCA assay (Pierce, Rockford, IL, USA). Proteins were diluted with lithium dodecyl sulfate buffer (LDS) 2× supplemented with reducing agents (Invitrogen) and then separated on NuPage Novex gels (Invitrogen) for Western blot analysis. Proteins were transferred to nitrocellulose membranes, which were then blocked (5% non-fat dry milk in TNT:Tris-HCl 15 mM, pH 8, NaCl 140 mM, 0.05% Tween) and incubated with primary and secondary antibodies. Signals were visualized using chemiluminescence kits (ECLTM, Amersham Velizy-Villacoublay, Villacoublay, France) and a LAS3000 imaging system (Fujifilm, Tokyo, Japan). We evaluated the expression of the catalytic subunit of PP2A (PP2Ac; Millipore, 1/1,000) as well as PP2Ac demethylation (demethyl PP2Ac; Santa Cruz, 1/1,000) in THY-Tau22 selenate-treated and vehicle-treated mice. We performed two separate Western blots for each antibody using three samples of each group, normalized to GAPDH. The PP2Ac/GAPDH and Demethy-PP2A/GAPDH ratios were calculated as well as the double ratio. PP2A demethylation (Leu 309) was taken as an index of its activity. Indeed, PP2A activity is enhanced by the methylation of its catalytic subunit (PP2Ac) and is conversely decreased by its demethylation (Papon et al., 2013; Sontag and Sontag, 2014).
Statistical Analysis
Statistical analyses were performed by using IBM SPSS 19 (IBM SPSS, Armonk, NY, USA) and GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA). Unless otherwise stated, all statistics were done with Two-Way ANOVA or repeated measures ANOVA (RM-ANOVA). For single between-group comparisons, t-tests with Welch‘s correction were used.
Results
Neuromotor Tests
When cage activity was monitored in the current study, we found a difference between groups (RM-ANOVA over the whole recording time F(3,30) = 3.258, p = 0.035). Post hoc Dunnett tests confirmed that only vehicle-treated THY-Tau22 mice (henceforth referred to as Tg mice for the remainder of the manuscript) demonstrated a significantly different level of exploratory activity compared with age-matched WT mice (p = 0.032; Figure 1B). This higher activity level of vehicle-treated Tg mice is not simply due to an increase in the mean activity level but primarily due to a higher initial and nocturnal activity. Noticeably, upon placement, all mice showed increased activity values (when compared to the activity level in the afternoon of the next day), which gradually disappeared. The high initial values and the decay of activity levels during this phase, which we consider as habituation to the novel environment, are apparently different in Tg vehicle-treated mice from the other genotypes as confirmed by a multivariate ANOVA of the first hour of recording (F(3,33) = 6.545, p = 0.002; post hoc Dunnett test vs. vehicle-treated Tg mice, p = 0.001). Chronic treatment with sodium selenate corrected for the increased initial and nocturnal activity of THY-Tau22 mice.
Next, we tested general motor abilities and coordination in the rotarod test and found no differences. Thus, all four groups were equally capable of remaining on the rotating rod (F(3,33) = 0.747, p > 0.05; data not shown). Previous investigations of the ambulatory activity of 10-month-old THY-Tau22 mice had demonstrated an overall increased activity in Tg mice (Van Der Jeugd et al., 2013).
In the Open field, the total path length was significantly different between groups (F(3,32) = 7.623, p = 0.001; Figure 1C). Vehicle-treated Tg animals tended to travel less distance in the arena compared with treated transgenic animals (post hoc Dunnett test p = 0.06). Time to approach the center of the arena and number of exploratory rearings in the center were different between groups (overall ANOVA F(3,32) = 5.700, p = 0.03 for latency to the center and F(3,32) = 3.723, p = 0.021 for rearing in the center). Non-treated Tg mice stayed longer away from and displayed less exploratory rearings in the center as compared to the vehicle-treated Tg animals (post hoc Dunnett tests p = 0.007 for center approach latency; Figure 1D and p = 0.01 for rearing; Figure 1E). Interestingly, after chronic selenate treatment, these behavioral changes were normalized to the level of WT values.
Neurocognitive Tests
We assessed spatial learning and memory in the established hippocampus-dependent water maze paradigm. Looking at both the time to locate the hidden platform and the path length traveled to reach it, we found significant differences between the groups (overall RM-ANOVA F(3,33) = 44.542, p = 0.0001 for escape latency and F(3,33) = 43.466, p = 0.0001 for distance traveled). Post hoc Dunnett tests revealed that spatial acquisition in vehicle-treated Tg mice at 12–13 months of age was severely impaired compared with the other groups [Figure 2A, group effect: escape latency (s): p = 0.001; path length (cm): p = 0.001, latter data not shown]. This impairment was not due to motor deficits because swim speed did not differ across groups (p > 0.05 data not shown). Noteworthy, there was a pronounced and significant improvement in spatial learning of selenate-treated Tg mice which was comparable with the WT-groups. The comparison of vehicle- with selenate-treated Tg mice confirmed the rescue effect of chronic selenate treatment because the latter was statistically indistinguishable from the two WT groups in terms of escape latency and path length (Figure 2A). In agreement with acquisition performance, spatial retention during probe trials was significantly different between the genotypes for the target quadrant (two-way ANOVA F(3,32) = 9.775, p = 0.004). However, there was also a significant effect of group detected for the opposite quadrant (F(3,32) = 15.778, p < 0.001), and interaction effect (F(3,32) = 27.388, p < 0.001). Dunnett tests indicated that vehicle-treated THY-Tau22 failed to develop a preference for the target quadrant in contrast to the other three groups (p = 0.001 for comparisons of vehicle-treated THY-Tau22 with the other three groups, Figures 2B–F). Vehicle-treated THY-Tau22 mice displayed instead a preference for the opposite quadrant (Tg_veh vs. Tg_sel p = 0.001, vs. wt_veh p = 0.002, vs. wt_sel p = 0.005). Analysis of the latency to cross the former platform position revealed a trend towards poorer spatial reference memory in the vehicle-treated Tg mice compared to the other groups (Two-way ANOVA F(3,32) = 3.893, p = 0.057).
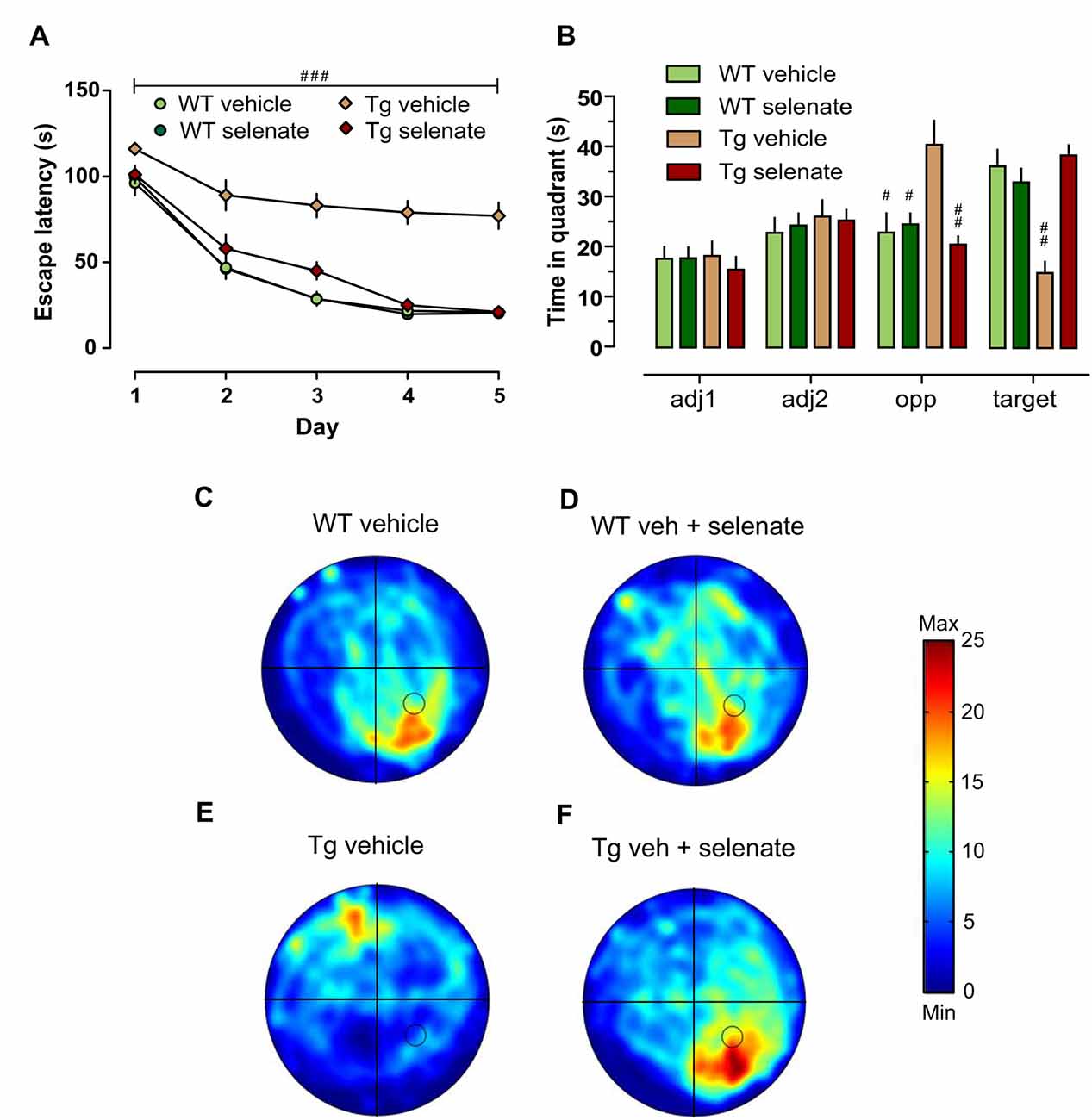
Figure 2. Impaired spatial learning and memory of Tg mice in the Morris water maze (MWM) was restored by treatment with selenate. (A) Tg mice provided with dietary selenate displayed escape latencies for locating the hidden platform comparable with the selenate-treated WT and non-treated WT groups. In contrast, the vehicle-treated Tg group were poor learners. (B) In the probe test, both WT groups and selenate-treated Tg groups confirmed memory for the platform location by spending significantly more time in the platform quadrant compared to vehicle-treated Tg mice. The vehicle-treated Tg group, in contrast, spent significantly less time in the target quadrant and showed a preference for the opposite quadrant instead (adj = adjacent 1/2, opp = opposite and target; hashes denote statistically significant differences between vehicle-treated Tg mice and the other three groups). (C–F) Examples of heat maps of the swim patterns during probe tests. The location of the platform is marked in the heat map as a black circle in the bottom right quadrant. (C) WT vehicle, (D) WT selenate-treated, (E) Tg vehicle, (F) Tg selenate-treated. Note the clear target preference of Tg after selenate treatment in panel (F) as compared to vehicle-treated Tg in (E). The heat map scale bar indicates the time in seconds. Mean ± SEM is given; group sizes: Tg-veh n = 9, Tg-sel n = 10, WT-veh n = 8, WT-sel n = 9. Bar with ### indicates significance level p ≤ 0.001 of RM-ANOVA with a Dunnett post hoc test using Tg vehicle as comparison; # and ## represent p ≤ 0.01 and p ≤ 0.001, respectively, in the Dunnett post hoc test.
Finally, in passive avoidance learning, a one-trial learning test, we found differences between the groups (overall ANOVA F(3,33) = 32.348, p = 0.0001). Thus, the two WT-groups exhibited clear memory recall of the shock experienced 24 h earlier, whereas vehicle-treated Tg mice failed to remember as indicated by the short-latency to enter the dark compartment. Notably, in selenate-treated Tg animals, retention memory was restored to WT levels, resulting in a highly significant effect of treatment (Figure 3, post hoc Dunnett tests for vehicle-treated Tg compared to the other three groups p = 0.0001).
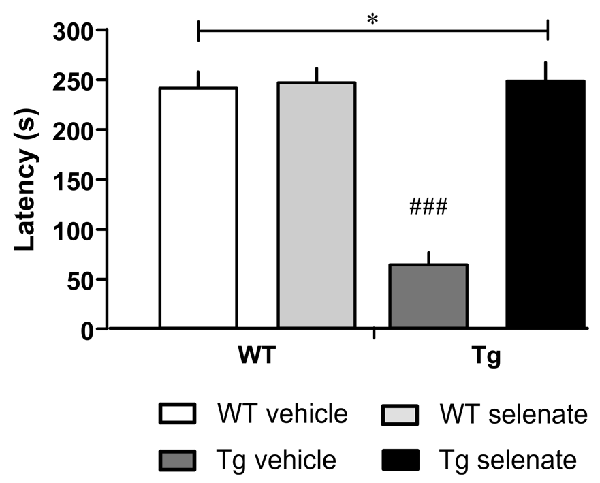
Figure 3. Selenate restored impaired retention memory of Tg mice in the Passive Avoidance test. Mean ± SEM is given. Group sizes: Tg-veh n = 9, Tg-sel n = 10, WT-veh n = 8, WT-sel n = 10. A bar with a * indicates p ≤ 0.01 in the Two-way ANOVA; ### represents p ≤ 0.0001 in the Dunnett post hoc test.
Electrophysiological Studies
Several reports have established that chronic sodium selenate treatment rescues certain behavioral phenotypes in tau mice models, including reducing NFT load in neurons (Corcoran et al., 2010b; van Eersel et al., 2010). However, the effects of chronic selenate application on synaptic transmission and synaptic plasticity have not yet been investigated. In a recent study, we reported that transient bath-application of selenate rescued an impaired long-term depression (LTD), one of the major types of synaptic plasticity, in Tg mice in vitro (Ahmed et al., 2015). Here, we tested the effects of oral chronic selenate application on synaptic transmission and plasticity in the CA1-region of hippocampal slices ex vivo after in vivo treatment. At 9 months, the age at which the selenate treatment was started, Tg mice show already prominent signs of AD-like hippocampal Tau pathology as hyperphosphorylation, abnormal conformational changes, and aggregation of tau, associated with severe memory deficits and synaptic impairments. However, there are no indications of neuronal loss or neurodegeneration (Schindowski et al., 2006; Belarbi et al., 2009; Van der Jeugd et al., 2011; Burnouf et al., 2013).
When basal synaptic transmission was inspected (Figure 4A), selenate treatment caused in Tg mice higher EPSP slopes in response to increasing stimulation strengths compared with the other three groups. Two-way ANOVA confirmed a significant effect of group (F(3,35) = 3.389, p < 0.029) and post hoc Dunnett test revealed significant differences between selenate-treated WT and Tg mice (p ≤ 0.020). We then studied paired-pulse responses, a form of presynaptically mediated short-term plasticity. Tg mice displayed rather high paired-pulse ratios at short inter-pulse intervals of 10 ms and 20 ms independent of whether or not the animals had received selenate (Figure 4B). The increased paired-pulse values of selenate-treated Tg mice were statistically different from selenate-treated WT animals at 10 and 20 ms (10 ms: p = 0.0248, 20 ms: p = 0.0339, Tg sel n = 12, WT sel n = 12, Two-Way ANOVA plus Dunnett post hoc test compared with WT-selenate). Differences of similar size occurred also when the vehicle groups were compared, but the higher variability prevented statistical significance. While there were no significant differences at interpulse intervals of 50 ms and 100 ms, treatment with selenate led to significantly increased values in WT mice at 200 ms (Figure 4B, p = 0.0242, WT sel n = 12, WT veh n = 8, Two-Way ANOVA plus Dunnett post hoc comparison. A similar trend in Tg mice at 200 ms was not significant.
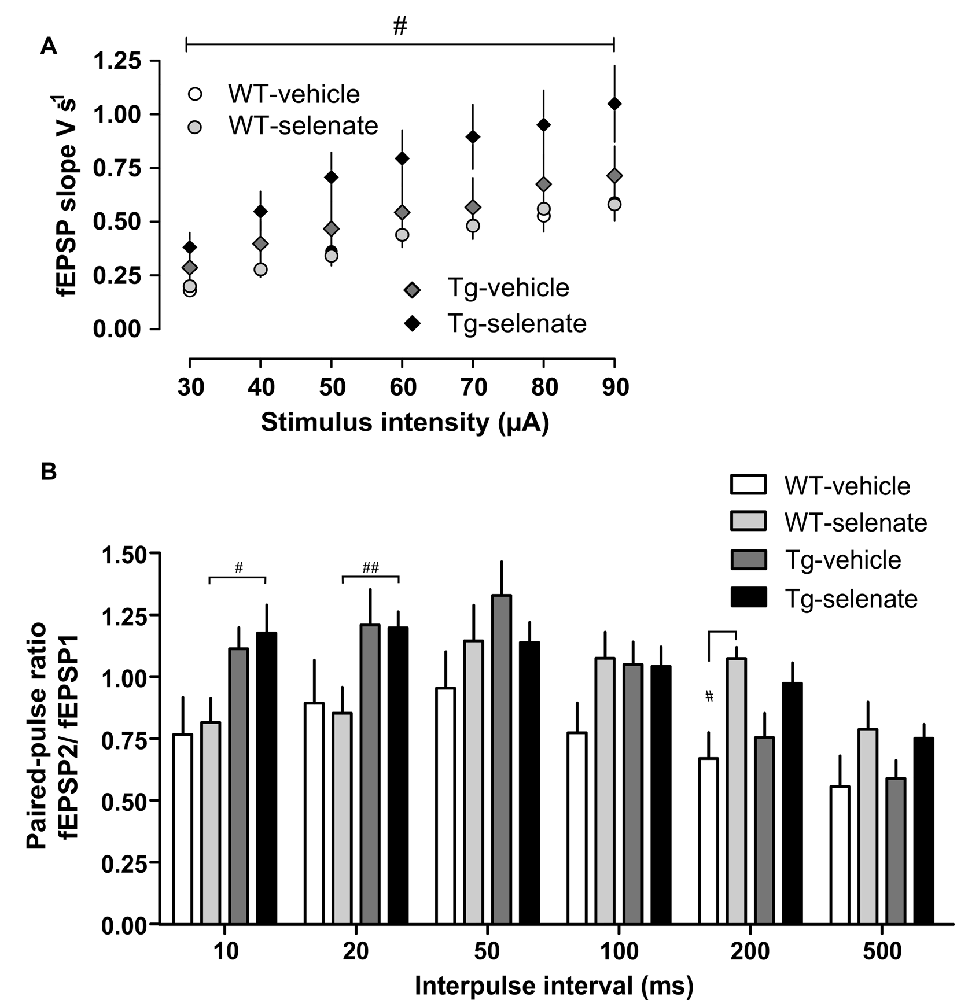
Figure 4. Enhanced basal synaptic transmission by selenate treatment in Tg mice. (A) Chronic selenate application increased basal synaptic transmission in Tg mice. Two-way ANOVA confirmed a significant between-group difference (F(3,35) = 3.389, p < 0.029) and post hoc comparisons revealed a significant enhancement of synaptic transmission in selenate-treated Tg mice (n = 10) compared to selenate-treated WT mice (n = 8; p ≤ 0.020, Dunnett test with WT-sel as the control group). (B) Selenate-treated Tg mice (n = 12) had higher paired-pulse values than selenate-treated WT mice (n = 12) at short interpulse intervals of 10 and 20 ms. Interestingly, selenate caused a significant enhancement of paired-pulse values in WT mice at 200 ms. Two-Way ANOVA plus Dunnett post hoc test compared with WT-selenate. Mean ± SEM is given. Bar with #indicates significance level ≤0.05 of Dunnett post hoc test, likewise #p ≤ 0.05; ##p ≤ 0.01.
Next, we examined long-term synaptic plasticity. We reported earlier that the triple repetition of low-frequency stimulation (LFS) at 2 Hz was successful in inducing late-phase LTD (L-LTD) in young and middle-aged mice (Balschun et al., 2003; Van der Jeugd et al., 2011; Ahmed et al., 2015). In the current study, this robust L-LTD was expressed in WT mice but was severely impaired in Tg animals (Figure 5A). Although Tg vehicle mice attained 2 min after completion of the first LFS-train very similar values as WT vehicle animals (Tg vehicle 61 ± 3%, n = 10, WT vehicle 66 ± 8%, n = 8), the depression could not be maintained (last 30 min: Tg vehicle 117 ± 8, n = 10; WT-vehicle 70 ± 7%). Statistical analysis of LTD of all groups after the induction (60–240 min) with RM-ANOVA plus Tukey’s post hoc comparison, revealed a significant effect of group (F(3,28) = 6.843), p = 0.001 and significant differences between vehicle-treated Tg animals and all other groups (Tg-sel p = 0.027; WT-vehicle p = 0.002; WT-sel 0.008). While the chronic oral application of sodium selenate had virtually no effect on WT mice, it completely rescued the L-LTD in Tg animals (Figure 5B) as convincingly demonstrated by the exact match with the LTD of the selenate-treated WT counterparts (Figure 5C).
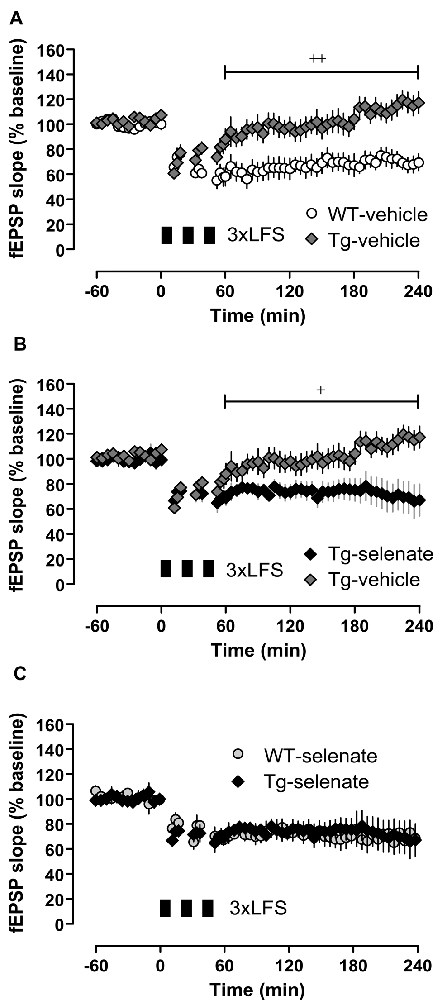
Figure 5. Chronic selenate treatment restored synaptic plasticity [long-term depression (LTD)] in Tg mice. RM-ANOVA of all four groups confirmed a significant main effect of group F(2,28) = 6.843, p = 0.001. (A) Vehicle-treated Tg mice (n = 10) failed to express robust LTD which was intact in vehicle-treated WT siblings (n = 8). (B) Selenate rescued LTD in Tg (n = 6) compared with vehicle-treated siblings (n = 10). (C) The rescued LTD in selenate-treated Tg mice matched exactly the time-course of LTD in selenate-treated WT animals (n = 8). Mean ± SEM is given. Bar with cross(es) indicates a significant difference in Tukey’s post hoc multiple comparisons, +p ≤ 0.05, ++p ≤ 0.01.
Immunohistochemistry and Western Blotting
We selected AT8 as the antibody of choice (Su et al., 1994; Augustinack et al., 2002; Deters et al., 2009) for the histological examination because several studies suggest that PP2Ac dephosphorylates primarily the AT8 epitopes S202/T205 while other phosphorylation sites that become hyperphosphorylated during AD, detected by antibodies such as AT100, AT180, 12E8, and PHF1, are not affected (Kins et al., 2001).
Histological examination of sections from vehicle-treated (n = 3) and selenate-treated Tg animals (n = 3) with AT8, a human tau pathological marker of S202/T205 (Figure 6A), revealed a significant decrease in selenate-treated mice (p = 0.027, t-test; Figure 6B). In contrast, no significant effect was found with AT100, which recognizes Tau phosphorylation epitopes pT212/pS214 (p = 0.245, t-test, data not shown).
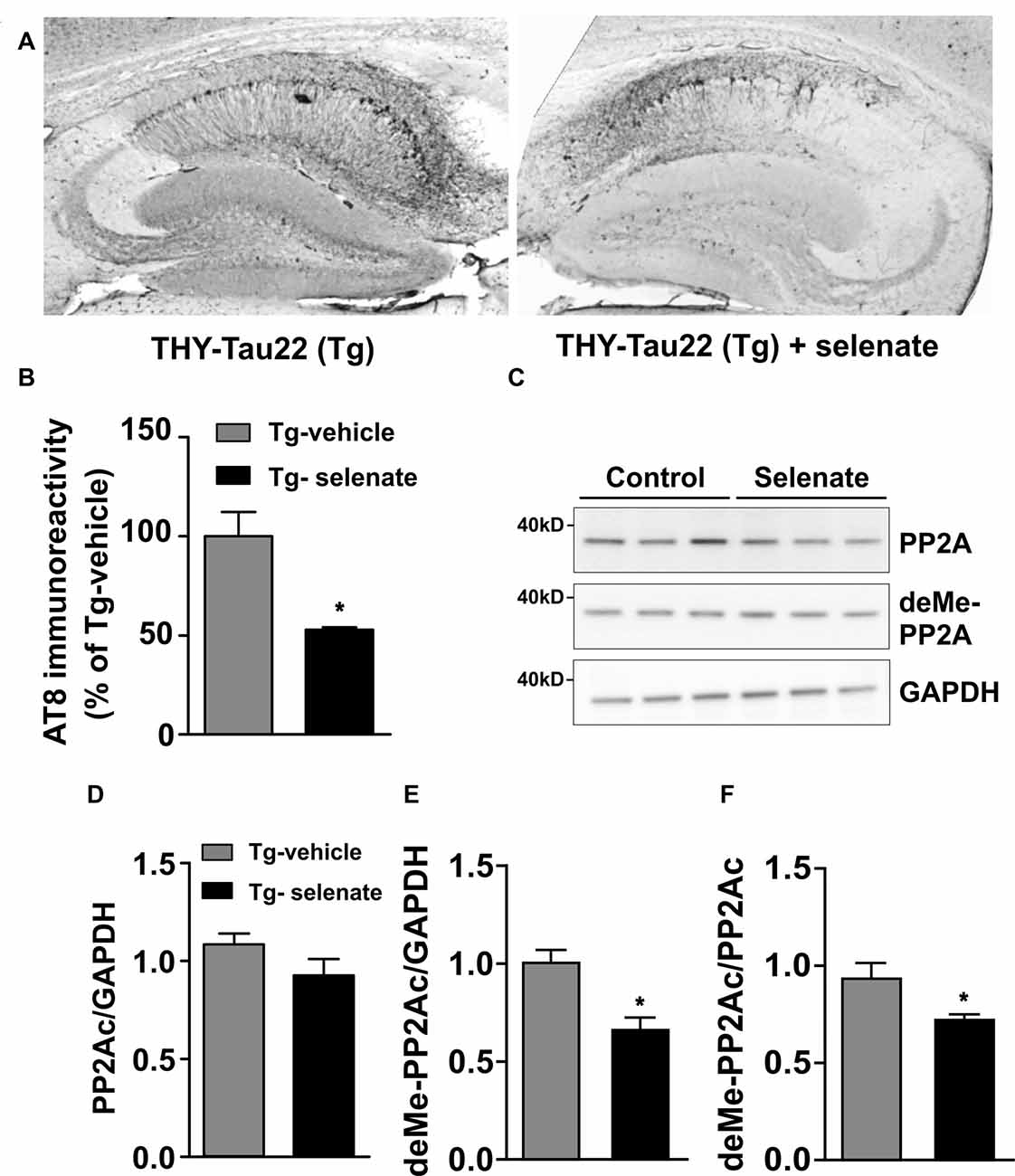
Figure 6. Selenate reduces AT8 immunoreactivity and antagonizes PP2A-demethylation in Tg mice. (A) Representative immunohistochemical sections from a vehicle-treated Tg mouse (left) and an animal that received selenate (right). Note the reduced AT8 immunoreactivity in the mouse treated with selenate. (B) Quantification of immunohistochemical AT8-staining from both groups. Selenate treatment reduced AT8-immunoreactivity to about 50% (p = 0.0027). (C) Immunoblots of PP2Ac and demethylated PP2Ac in vehicle-treated and selenate-treated Tg. (D,E) Quantification of the levels of PP2Ac and demethylated PP2Ac, respectively. The amount of demethylated PP2Ac is significantly lower after treatment with selenate, supporting higher PP2A activity under treatment. (F) The amount of demethylated PP2Ac (deMe-PP2Ac) normalized to the amount of PP2AC. Mean ± SEM is given. *p < 0.05, n = 3 per group.
Since several reports have documented a downregulation of PP2Ac in murine models of tauopathies (Kins et al., 2001; Liu et al., 2005; Sontag and Sontag, 2014), we evaluated total levels of PP2Ac (normalized to GAPDH) in vehicle-treated and selenate-treated Tg mice by Western blotting (Figure 6C) and found them not being statistically different (Tg vehicle-treated: 1.08 ± 0.05, n = 3, Tg selenate-treated: 0.92 ± 0.08, n = 3, p = 0.174, t-tests; Figure 6D). When demethylation of PP2Ac (inactive PP2Ac) was quantified, a statistically significant reduction by selenate treatment was confirmed (Tg vehicle-treated: 0.93 ± 0.08, n = 3, Tg selenate-treated: 0.67 ± 0.05, n = 3, p = 0.049, t-test; Figure 6E). This difference persisted when the values of deMe-PP2Ac were normalized to the respective values of PP2Ac (Tg vehicle-treated: 0.94 ± 0.07, Tg selenate-treated: 0.732 ± 0.02, p = 0.042, t-test; Figure 6F. Interestingly, when all individual pairs of AT8 and deMePP2Ac values were subjected to correlation analysis, a significant Pearson correlation coefficient of r = 0.919 (p = 0.0096) was obtained.
Discussion
One of the major goals of this study was to establish whether sodium selenate is a suitable candidate for an effective, mechanism-based, and inexpensive treatment to correct not only motor and behavioral phenotypes, but also synaptic deficits in a murine model of tauopathy and AD (Loef et al., 2011). The data presented here clearly demonstrate this outcome. In this study, we used male mice. It must be noted in this context, that there is an intense debate about gender effects on AD. A recent review by Nebel et al. (2018) mentions that about two-thirds of the more than 5.5 Americans afflicted with AD are women. However, a worldwide meta-analysis by Fiest et al. (2016) did not find a significantly higher prevalence of AD in women than in men after controlling for sex differences in longevity. Likewise, in two meta-analyses of worldwide studies of frontotemporal dementia, a major tauopathy, no significant gender difference in prevalence was found (Onyike and Diehl-Schmid, 2013; Hogan et al., 2016).
We found that vehicle-treated Tg mice displayed a marked increase in locomotor and exploratory activity during the initial habituation to the test environment and the dark phase of their activity cycle. This higher activity level, which was corrected by selenate treatment, is reminiscent of reports of hyperactivity in other Tau mouse models such as P301S (Przybyla et al., 2016), rTg4510 (Joly-Amado et al., 2016; Jul et al., 2016; Blackmore et al., 2017; Wang et al., 2018), MAPT-N296H (Wobst et al., 2017), N279K (Taniguchi et al., 2005) and a triple repeat mutant tau transgenic mouse line (Rockenstein et al., 2015). Of note, a Mapt−/− model on a B6129PF3/J genetic background also displayed hyperactive behavior (Biundo et al., 2018). Hyperactivity was also observed in mouse models that combine Aβ- and Tau pathology such as the triple transgenic mouse line that overexpresses mutant tau via the taup301L transgene (Pietropaolo et al., 2008; Baeta-Corral and Gimenez-Llort, 2014; Cañete et al., 2015). As discussed in a recent review by Kosel et al. (2020), hyperactivity is a common feature of AD mouse models, including early sleep-wake disturbances and nocturnal hyperactivity. Interestingly, in a mouse model that expresses both the APP/PS1 transgene and wild-type human tau under the control of an inducible promoter, Aβ and tau were found to work cooperatively to generate hyperactive behavior (Pickett et al., 2019).
In line with other reports (Corcoran et al., 2010b; van Eersel et al., 2010; Shultz et al., 2015), chronic selenate treatment had no effects on motor control. However, we found that this compound normalized the reduced exploration of Tg in the open field as evidenced by similar values as WT controls for the number of rearings, total distance moved, and latency to enter the center of the arena. The latter could be considered as an anxiolytic effect of selenate. The difference in activity in the cage activity and the open field test can be explained by the set-up of both tests. The activity cage looks alike their home cage in the animalium: both are transparent cages composed out of the same plastic material with an identical mesh grid top and provided with the same bedding and dim-lit. In contrast, the open field arena indeed is a more anxious environment because it is composed of a non-transparent large box without bedding and brightly lit. Also, before the Open Field test, mice undergo 30 min of dark adaptation inside a dark cupboard. Taken all of this together, we, therefore, think that the mice are more anxious in the open field which is reflected in the decreased distance traveled and increased latency to enter the center of the open field the two main parameters for an anxiogenic phenotype.
We further observed that selenate treatment facilitated/rescued the contextual awareness of Tg mice in the passive avoidance test resulting in a similar performance as WT mice. Deficits in contextual hippocampus-dependent functions have been reported for several tauopathy mouse models (Fujio et al., 2007; Sydow et al., 2011; Van der Jeugd et al., 2011; Van Der Jeugd et al., 2012). The clear improvement of spatial reference memory of Tg mice in the MWM by chronic selenate treatment is corroborated by similar findings in another tauopathy mouse model (TMTH) in response to selenate (Corcoran et al., 2010b), and by marked improvements in spatial learning after treating 3xTg mice with selenomethionine (Song et al., 2014; Zhang et al., 2017), sodium selenate (Van Der Jeugd et al., 2018) and Ebselen, a lipid-soluble low molecular weight selenoorganic compound (Xie et al., 2017), respectively. However, in these studies, mitigated or rescued pathology in 3xTg mice could also be caused by dietary selenate or selenomethionine repressing amyloid-β formation and deposition via down-regulation of β-secretase (BACE1) expression and decreased APP cleavage (Zhang et al., 2016; Jin et al., 2017). In our study with Tg mice, rescuing effects mediated by changed amyloid-β production can be excluded.
Experimental evidence supports the activation of protein phosphatase A (PP2A) by selenate (Corcoran et al., 2010b; van Eersel et al., 2010; Brozmanova, 2011; Shultz et al., 2015; Jin et al., 2017) and selenomethionine (Zhang et al., 2017), but the precise mechanism remains unclear. Interestingly in this context, Nicholls et al. (2008) postulated a role for PP2A in cognitive tasks such as the spatial version of the MWM. In our experiments, Tg mice presented severe deficits in acquisition and probe trial performance in the MWM, i.e., under the conditions of a progressing tauopathy when PP2A activity has been described to be downregulated (Sontag and Sontag, 2014). Upon selenate treatment, we observed a restoration of spatial memory of Tg to WT-levels in the probe trials during acquisition. Thus, it appears reasonable to assume that increased PP2Ac activity can rescue or reinstate molecular processes that are important for the formation and consolidation of spatial memory.
In our electrophysiological measurements of synaptic readouts, we observed that selenate significantly increased basal synaptic transmission in Tg, as becoming overt in the input-output curves. Furthermore, we recorded increased paired-pulse ratios in Tg mice at short interpulse intervals pointing to decreased recurrent inhibition. This could be due to the loss of interneurons due to their high vulnerability during progressing AD/tau pathology as reported for several AD mouse models (Loreth et al., 2012). Such a decline in interneuron numbers is considered a major reason for the abundant occurrence of epileptic seizures in AD patients, which has been replicated in certain murine AD models (Palop and Mucke, 2016). However, in the hippocampus of Tg mice no signs of neuronal loss have been detected, but an increase in astroglial cells (Van der Jeugd et al., 2011).
Recently, Shultz et al. (2015) have reported that sodium selenate treatment increases expression of the PP2A/PR55B regulatory subunit after traumatic brain injury, arguably another tauopathy. Of note, the PP2A isoform that contains the PR55 (or PPP2R2A or Ba) regulatory subunit is the predominant neuronal PP2A holoenzyme showing the highest tau phosphatase activity and the strongest affinity for tau (Sontag et al., 1996, 1999; Xu et al., 2008). Whereas we did not measure the expression of this subunit, we compared overall changes of the catalytic subunit, PP2Ac, and found no differences between genotypes. However, when we assayed for demethylated PP2Ac (Leu309; Sontag et al., 2013), we recorded a significant decrease upon selenate treatment compared with vehicle-treated Tg, which is in agreement with published reports that methylation of the catalytic subunit PP2Ac at Leu309 by leucine carboxyl methyltransferase 1 (LCMT1) is required for enhanced PP2Ac activity and the assembly and stabilization of PP2A/Ba holoenzymes (Sontag, 2001; Sontag et al., 2004a; Sontag and Sontag, 2014). Interestingly, it has been documented that Tg mice have increased GSK3β activity (Ahmed et al., 2015) and enhanced GSK3β activity has been linked to reduced PP2Ac activity as found here (Qian et al., 2010; Yao et al., 2012).
Presynaptic mechanisms are unlikely to be included in the selenate effects as no differences in the paired-pulse ratios (PPR) at the 50 ms inter-pulse interval were found between treated and vehicle-treated Tg groups. Intriguingly, in WT mice selenate enhanced PPR at an interpulse interval of 200 ms, pointing to selenate-mediated effects on GABAB-mediated inhibition.
In Tg mice treated with selenate, we observed a restoration of LTD comparable with WT levels while selenate did not affect WT synaptic plasticity. This LTD rescue “phenomenon” was similar to the one that we have previously documented in response to a transient bath-application of selenate, which notably overcame okadaic acid inhibition (Ahmed et al., 2015). Further, histological evidence using the AT8 antibody, one established pathological marker for the tau phosphorylation site Ser202/Thr205 which is closely associated with PP2Ac activity (Kins et al., 2003; Deters et al., 2009), clearly identified reduced phosphorylation of this epitope after chronic selenate treatment. Recent studies have shown that the AT8 binding motif does not only include the well-known pS202/pT205 epitope but also pS208 which is important for the affinity and kinetics of AT8 binding (Malia et al., 2016). Since phosphorylation of S208 was suggested to enhance the formation of tau filaments that lead to NFTs (Xia et al., 2020), PP2Ac likely controls early hyperphosphorylation steps in tau pathogenesis that promote aggregation but are still reversible. There is also evidence from a mouse model expressing a repressible human tau variant, that the pathological processes responsible for cognitive decline and the processes underlying NFT-formation may dissociate at a certain stage of Tau pathology (SantaCruz et al., 2005).
Given the current state of knowledge, it is rather difficult to identify a certain mechanism that is causally responsible for the selenate-mediated rescue effect on synaptic plasticity and cognition. Selenate, has been used as an anti-cancer agent for many years (Corcoran et al., 2010a; Brozmanova, 2011; Rayman, 2012) and was found to be incorporated into proteins as selenocysteine. The latter is a major component of the anti-oxidant agent’s glutathione peroxidases (GP) and thioredoxin reductases (TR) which are well represented in the brain (Hoppe et al., 2008; Bellinger et al., 2009; Pitts et al., 2014). Both enzymes are greatly reduced in AD and linked to a reduction in PP2A methylation (Tolstykh et al., 2000; Vafai and Stock, 2002; Morris, 2003). Sontag et al. (2010) described a link between high levels of S-adenosylhomocysteine and a reduction in PP2A methylation levels. Moreover, PP2A methylation is under the tight control of protein phosphatase methylesterase-1 (PME1) and leucine carboxyl methyltransferase-1 (LCMT1; Sontag et al., 2010). Overexpression of PME1 in murine models had detrimental consequences on neurocognition and synaptic plasticity while overexpression of LCMT1 had beneficial effects (Nicholls et al., 2016). In AD, decreased levels of LCMT1 and methylated PP2A were described to be correlated with reduced amounts and the severity of phospho-tau pathology (Sontag et al., 2004a,b; Taleski and Sontag, 2018). Thus, changes in the amount of PP2A/Ba may lead to changes in PP2A methylation which in turn may compromise the intricate balance between the activity of tau kinases and phosphatases (Taleski and Sontag, 2018). Thus, these reports indicate that PP2A methylation is a defining factor in tauopathies.
In conclusion, we demonstrate for the first time that a 3-month treatment with dietary selenate rescues deficits in synaptic plasticity as well as learning and memory in Tg mice at an advanced stage of tauopathy, which led already to severe functional deficits at the synaptic, behavioral, and cognitive level. The completely recovered LTD demonstrates a rescue of the deleterious effects of progressive tauopathy at the level of the synapse, the main site of functional deterioration that leads to cognitive decline. This is most likely due to a reduction in the demethylated, catalytically inactive catalytic subunit PP2Ac, which shifts the equilibrium in favor of the methylated PP2Ac and thus to a higher PP2A activity. Consequently, this leads to a decrease in hyperphosphorylated, but not misconformed Tau as supported by the significant reduction in AT-8 but not in AT-100 immunostaining. Together with the clear rescuing effects on synaptic plasticity and cognitive measures in our study, this reinforces the idea that the detrimental impact of pathological Tau towards plasticity and memory is likely related to non-aggregated soluble Tau species and recommends selenate as a candidate compound for human Tau-targeting clinical trials.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors upon reasonable request.
Ethics Statement
The animal study was reviewed and approved by the Animal experiments committee of the University of Leuven, Belgium and were carried out in accordance with the European Directive 2010/63/EU.
Author Contributions
TA developed the idea for these studies. AVDJ and RD’H designed the behavioral experiments which were conducted by AVDJ. The electrophysiological study was designed by TA and DBa, the biochemical and histological assays by DBl and LB. TA performed the electrophysiological measurements, DBl the biochemical and histological assays. AVDJ and DBa analyzed the behavioral data, TA, and DBa the electrophysiological experiments and DBl and RC the biochemical and histological assays with input from LB. DBa, TA, and AVDJ wrote the article with contributions from DBl and LB.
Funding
This work was supported by the Research Foundation-Flanders (FWO grants G.0327.08 to RD’H and DBa, G.0D76.14 to DBa, and FWO fellowship to AVDJ 1157812N). DBl and LB received support from: I-Site ULNE, France Alzheimer/Fondation de France, FHU VasCog research network (Lille, France), programs d’investissements d’avenir LabEx (excellence laboratory) DISTALZ (Development of Innovative Strategies for a Transdisciplinary approach to ALZheimer’s disease), Agence Nationale de la Recherche (ANR; GRAND, SPREADTAU to LB, ADORATAU and ADORASTrAU to DBl), Fondation pour la Recherche Médicale, Vaincre Alzheimer, Fondation Plan Alzheimer as well as Inserm, CNRS, Université Lille, Lille Métropole Communauté Urbaine, Région Hauts-de-France, DN2M.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the animal care facility (animal facilities of Université de Lille–Inserm) of “Plateformes en Biologie Santé de Lille” and M. Besegher-Dumoulin, J. Devassine, Y. Lepage, C. Meunier, C. Degraeve, and D. Taillieu for transgenic mouse production and care. Sebastien Carrier, Nele De Ruyck, and Leen Van Aerschot are acknowledged for mouse care and genotyping.
References
Ahmed, T., Blum, D., Burnouf, S., Demeyer, D., Buée-Scherrer, V., D’Hooge, R., et al. (2015). Rescue of impaired late-phase long-term depression in a tau transgenic mouse model. Neurobiol. Aging 36, 730–739. doi: 10.1016/j.neurobiolaging.2014.09.015
Ahmed, T., Sabanov, V., D’Hooge, R., and Balschun, D. (2011). An N-methyl-D-aspartate-receptor dependent, late-phase long-term depression in middle-aged mice identifies no GluN2-subunit bias. Neuroscience 185, 27–38. doi: 10.1016/j.neuroscience.2011.04.006
Augustinack, J. C., Schneider, A., Mandelkow, E. M., and Hyman, B. T. (2002). Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 103, 26–35. doi: 10.1007/s004010100423
Baeta-Corral, R., and Gimenez-Llort, L. (2014). Bizarre behaviors and risk assessment in 3xTg-AD mice at early stages of the disease. Behav. Brain Res. 258, 97–105. doi: 10.1016/j.bbr.2013.10.017
Balschun, D., Wolfer, D. P., Gass, P., Mantamadiotis, T., Welzl, H., Schutz, G., et al. (2003). Does cAMP response element-binding protein have a pivotal role in hippocampal synaptic plasticity and hippocampus-dependent memory? J. Neurosci 23, 6304–6314. doi: 10.1523/JNEUROSCI.23-15-06304.2003
Belarbi, K., Schindowski, K., Burnouf, S., Caillierez, R., Grosjean, M. E., Demeyer, D., et al. (2009). Early tau pathology involving the septo-hippocampal pathway in a tau transgenic model: relevance to Alzheimer’s disease. Curr. Alzheimer Res. 6, 152–157. doi: 10.2174/156720509787602843
Bellinger, F. P., Raman, A. V., Reeves, M. A., and Berry, M. J. (2009). Regulation and function of selenoproteins in human disease. Biochem. J. 422, 11–22. doi: 10.1042/bj20090219
Biundo, F., Del, P. D., Zhang, H., Arancio, O., and D’Adamio, L. (2018). A role for tau in learning, memory and synaptic plasticity. Sci. Rep. 8:3184. doi: 10.1038/s41598-018-21596-3
Blackmore, T., Meftah, S., Murray, T. K., Craig, P. J., Blockeel, A., Phillips, K., et al. (2017). Tracking progressive pathological and functional decline in the rTg4510 mouse model of tauopathy. Alzheimers Res. Ther. 20:77. doi: 10.1186/s13195-017-0306-2
Braak, H., de Vos, R. A., Jansen, E. N., Bratzke, H., and Braak, E. (1998). Neuropathological hallmarks of Alzheimer’s and Parkinson’s diseases. Prog. Brain Res. 117, 267–285. doi: 10.1016/s0079-6123(08)64021-2
Brier, M. R., Gordon, B., Friedrichsen, K., McCarthy, J., Stern, A., Christensen, J., et al. (2016). Tau and Aβ imaging, CSF measures and cognition in Alzheimer’s disease. Sci. Transl. Med. 8:338ra66. doi: 10.1126/scitranslmed.aaf2362
Buée, L., Troquier, L., Burnouf, S., Belarbi, K., Van der, J. A., Ahmed, T., et al. (2010). From tau phosphorylation to tau aggregation: what about neuronal death? Biochem. Soc. Trans. 38, 967–972. doi: 10.1042/bst0380967
Burnouf, S., Martire, A., Derisbourg, M., Laurent, C., Belarbi, K., Leboucher, A., et al. (2013). NMDA receptor dysfunction contributes to impaired brain-derived neurotrophic factor-induced facilitation of hippocampal synaptic transmission in a tau transgenic model. Aging Cell 12, 11–23. doi: 10.1111/acel.12018
Cañete, T., Blázquez, G., Tobeña, A., Giménez-Llort, L., and Fernández-Teruel, A. (2015). Cognitive and emotional alterations in young Alzheimer’s disease (3xTgAD) mice: effects of neonatal handling stimulation and sexual dimorphism. Behav. Brain Res. 281, 156–171. doi: 10.1016/j.bbr.2014.11.004
Cardoso, B. R., Roberts, B. R., Malpas, C. B., Vivash, L., Genc, S., Saling, M. M., et al. (2019). Supranutritional sodium selenate supplementation delivers selenium to the central nervous system: results from a randomized controlled pilot trial in Alzheimer’s disease. Neurotherapeutics 16, 192–202. doi: 10.1007/s13311-018-0662-z
Congdon, E. E., and Sigurdsson, E. M. (2018). Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 14, 399–415. doi: 10.1038/s41582-018-0013-z
Corcoran, N. M., Hovens, C. M., Michael, M., Rosenthal, M. A., and Costello, A. J. (2010a). Open-label, phase I dose-escalation study of sodium selenate, a novel activator of PP2A, in patients with castration-resistant prostate cancer. Br. J. Cancer 103, 462–468. doi: 10.1038/sj.bjc.6605798
Corcoran, N. M., Martin, D., Hutter-Paier, B., Windisch, M., Nguyen, T., Nheu, L., et al. (2010b). Sodium selenate specifically activates PP2A phosphatase, dephosphorylates tau and reverses memory deficits in an Alzheimer’s disease model. J. Clin. Neurosci. 17, 1025–1033. doi: 10.1016/j.jocn.2010.04.020
Deters, N., Ittner, L. M., and Götz, J. (2009). Substrate-specific reduction of PP2A activity exaggerates tau pathology. Biochem. Biophys. Res. Commun. 379, 400–405. doi: 10.1016/j.bbrc.2008.12.140
Fiest, K. M., Roberts, J. I., Maxwell, C. J., Hogan, D. B., Smith, E. E., Frolkis, A., et al. (2016). The prevalence and incidence of dementia due to Alzheimer’s disease: a systematic review and meta-analysis. Can. J. Neurol. Sci. 43, S51–S82. doi: 10.1017/cjn.2016.36
Forner, S., Baglietto-Vargas, D., Martini, A. C., Trujillo-Estrada, L., and LaFerla, F. M. (2017). Synaptic impairment in Alzheimer’s disease: a dysregulated symphony. Trends Neurosci. 40, 347–357. doi: 10.1016/j.tins.2017.04.002
Fujio, J., Hosono, H., Ishiguro, K., Ikegami, S., and Fujita, S. C. (2007). Tau phosphorylation in the mouse brain during aversive conditioning. Neurochem. Int. 51, 200–208. doi: 10.1016/j.neuint.2007.04.024
Goddyn, H., Leo, S., Meert, T., and D’Hooge, R. (2006). Differences in behavioural test battery performance between mice with hippocampal and cerebellar lesions. Behav. Brain Res. 173, 138–147. doi: 10.1016/j.bbr.2006.06.016
Gomez-Isla, T., Hollister, R., West, H., Mui, S., Growdon, J. H., Petersen, R. C., et al. (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann. Neurol. 41, 17–24. doi: 10.1002/ana.410410106
Hogan, D. B., Jette, N., Fiest, K. M., Roberts, J. I., Pearson, D., Smith, E. E., et al. (2016). The prevalence and incidence of frontotemporal dementia: a systematic review. Can. J. Neurol. Sci. 43, S96–S109. doi: 10.1017/cjn.2016.25
Hoover, B. R., Reed, M. N., Su, J., Penrod, R. D., Kotilinek, L. A., Grant, M. K., et al. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081. doi: 10.1016/j.neuron.2010.11.030
Hoppe, B., Bräuer, A. U., Kühbacher, M., Savaskan, N. E., Behne, D., and Kyriakopoulos, A. (2008). Biochemical analysis of selenoprotein expression in brain cell lines and in distinct brain regions. Cell Tissue Res. 332, 403–414. doi: 10.1007/s00441-008-0575-y
Hyman, B. T., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Carrillo, M. C., et al. (2012). National Institute on aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 8, 1–13. doi: 10.1016/j.jalz.2011.10.007
Jin, N., Zhu, H., Liang, X., Huang, W., Xie, Q., Xiao, P., et al. (2017). Sodium selenate activated Wnt/β-catenin signaling and repressed amyloid-β formation in a triple transgenic mouse model of Alzheimer’s disease. Exp. Neurol. 297, 36–49. doi: 10.1016/j.expneurol.2017.07.006
Joly-Amado, A., Serraneau, K. S., Brownlow, M., de Evsikova, C. M., Speakman, J. R., Gordon, M. N., et al. (2016). Metabolic changes over the course of aging in a mouse model of tau deposition. Neurobiol. Aging 44, 62–73. doi: 10.1016/j.neurobiolaging.2016.04.013
Jul, P., Volbracht, C., de Jong, I. E. M., Helboe, L., Elvang, A. B., and Pedersen, J. T. (2016). Hyperactivity with agitative-like behavior in a mouse tauopathy model. J. Alzheimers Dis. 49, 783–795. doi: 10.3233/jad-150292
Kins, S., Crameri, A., Evans, D. R., Hemmings, B. A., Nitsch, R. M., and Gotz, J. (2001). Reduced protein phosphatase 2A activity induces hyperphosphorylation and altered compartmentalization of tau in transgenic mice. J. Biol. Chem. 276, 38193–38200. doi: 10.1074/jbc.M102621200
Kins, S., Kurosinski, P., Nitsch, R. M., and Gotz, J. (2003). Activation of the ERK and JNK signaling pathways caused by neuron-specific inhibition of PP2A in transgenic mice. Am. J. Pathol. 163, 833–843. doi: 10.1016/s0002-9440(10)63444-x
Kosel, F., Pelley, J. M. S., and Franklin, T. B. (2020). Behavioural and psychological symptoms of dementia in mouse models of Alzheimer’s disease-related pathology. Neurosci. Biobehav. Rev. 112, 634–647. doi: 10.1016/j.neubiorev.2020.02.012
Kremer, A., Louis, J. V., Jaworski, T., and Van, L. F. (2011). GSK3 and Alzheimer’s disease: facts and fiction. Front. Mol. Neurosci. 4:17. doi: 10.3389/fnmol.2011.00017
Lambrecht, C., Haesen, D., Sents, W., Ivanova, E., and Janssens, V. (2013). Structure, regulation and pharmacological modulation of PP2A phosphatases. Methods Mol. Biol. 1053, 283–305. doi: 10.1007/978-1-62703-562-0_17
Laurent, C., Eddarkaoui, S., Derisbourg, M., Leboucher, A., Demeyer, D., Carrier, S., et al. (2014). Beneficial effects of caffeine in a transgenic model of Alzheimer’s disease-like tau pathology. Neurobiol. Aging 35, 2079–2090. doi: 10.1016/j.neurobiolaging.2014.03.027
Leboucher, A., Laurent, C., Fernandez-Gomez, F.-J., Burnouf, S., Troquier, L., Eddarkaoui, S., et al. (2013). Detrimental effects of diet-induced obesity on tau pathology are independent of insulin resistance in tau transgenic mice. Diabetes 62, 1681–1688. doi: 10.2337/db12-0866
Liu, F., Grundke-Iqbal, I., Iqbal, K., and Gong, C. X. (2005). Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 22, 1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x
Loef, M., Schrauzer, G. N., and Walach, H. (2011). Selenium and Alzheimer’s disease: a systematic review. J. Alzheimers Dis. 26, 81–104. doi: 10.3233/JAD-2011-110414
Loreth, D., Ozmen, L., Revel, F. G., Knoflach, F., Wetzel, P., Frotscher, M., et al. (2012). Selective degeneration of septal and hippocampal GABAergic neurons in a mouse model of amyloidosis and tauopathy. Neurobiol. Dis. 47, 1–12. doi: 10.1016/j.nbd.2012.03.011
Lovestone, S., and Manji, H. K. (2020). Will we have a drug for Alzheimer’s disease by 2030? the view from pharma. Clin. Pharmacol. Ther. 107, 79–81. doi: 10.1002/cpt.1685
Malia, T. J., Teplyakov, A., Ernst, R., Wu, S. J., Lacy, E. R., Liu, X., et al. (2016). Epitope mapping and structural basis for the recognition of phosphorylated tau by the anti-tau antibody AT8. Proteins 84, 427–434. doi: 10.1002/prot.24988
Malpas, C. B., Vivash, L., Genc, S., Saling, M. M., Desmond, P., Steward, C., et al. (2016). A phase IIa randomized control trial of VEL015 (sodium selenate) in mild-moderate Alzheimer’s disease. J. Alzheimers Dis. 54, 223–232. doi: 10.3233/jad-160544
Martin, L., Latypova, X., Wilson, C. M., Magnaudeix, A., Perrin, M.-L., Yardin, C., et al. (2013). Tau protein kinases: involvement in Alzheimer’s disease. Ageing Res. Rev. 12, 289–309. doi: 10.1016/j.arr.2012.06.003
Masters, C. L., Bateman, R., Blennow, K., Rowe, C. C., Sperling, R. A., and Cummings, J. L. (2015). Alzheimer’s disease. Nat. Rev. Dis. Primers 1:15056. doi: 10.1038/nrdp.2015.56
Morris, M. S. (2003). Homocysteine and Alzheimer’s disease. Lancet Neurol. 2, 425–428. doi: 10.1016/s1474-4422(03)00438-1
Nebel, R. A., Aggarwal, N. T., Barnes, L. L., Gallagher, A., Goldstein, J. M., Kantarci, K., et al. (2018). Understanding the impact of sex and gender in Alzheimer’s disease: a call to action. Alzheimers Dement. 14, 1171–1183. doi: 10.1016/j.jalz.2018.04.008
Nelson, P. T., Alafuzoff, I., Bigio, E. H., Bouras, C., Braak, H., Cairns, N. J., et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381. doi: 10.1097/NEN.0b013e31825018f7
Nicholls, R. E., Alarcon, J. M., Malleret, G., Carroll, R. C., Grody, M., Vronskaya, S., et al. (2008). Transgenic mice lacking NMDAR-dependent LTD exhibit deficits in behavioral flexibility. Neuron 58, 104–117. doi: 10.1016/j.neuron.2008.01.039
Nicholls, R. E., Sontag, J.-M., Zhang, H., Staniszewski, A., Yan, S., Kim, C. Y., et al. (2016). PP2A methylation controls sensitivity and resistance to β-amyloid-induced cognitive and electrophysiological impairments. Proc. Natl. Acad. Sci. U S A 113, 3347–3352. doi: 10.1073/pnas.1521018113
Onyike, C. U., and Diehl-Schmid, J. (2013). The epidemiology of frontotemporal dementia. Int. Rev. Psychiatry. 25, 130–137. doi: 10.3109/09540261.2013.776523
Palop, J. J., and Mucke, L. (2016). Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 17, 777–792. doi: 10.1038/nrn.2016.141
Papon, M. A., El Khoury, N. B., Marcouiller, F., Julien, C., Morin, F., Bretteville, A., et al. (2013). Deregulation of protein phosphatase 2A and hyperphosphorylation of tau protein following onset of diabetes in NOD mice. Diabetes 62, 609–617. doi: 10.2337/db12-0187
Pickett, E. K., Herrmann, A. G., McQueen, J., Abt, K., Dando, O., Tulloch, J., et al. (2019). Amyloid beta and tau cooperate to cause reversible behavioral and transcriptional deficits in a model of Alzheimer’s disease. Cell Rep. 29, 3592.e5–3604.e5. doi: 10.1016/j.celrep.2019.11.044
Pietropaolo, S., Feldon, J., and Yee, B. K. (2008). Age-dependent phenotypic characteristics of a triple transgenic mouse model of Alzheimer disease. Behav. Neurosci. 122, 733–747. doi: 10.1037/a0012520
Pitts, M. W., Byrns, C. N., Ogawa-Wong, A. N., Kremer, P., and Berry, M. J. (2014). Selenoproteins in nervous system development and function. Biol. Trace Elem. Res. 161, 231–245. doi: 10.1007/s12011-014-0060-2
Przybyla, M., Stevens, C. H., van der Hoven, J., Harasta, A., Bi, M., Ittner, A., et al. (2016). Disinhibition-like behavior in a P301S mutant tau transgenic mouse model of frontotemporal dementia. Neurosci. Lett. 19, 24–29. doi: 10.1016/j.neulet.2016.08.007
Qian, W., Shi, J., Yin, X., Iqbal, K., Grundke-Iqbal, I., Gong, C. X., et al. (2010). PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3β. J. Alzheimers Dis. 19, 1221–1229. doi: 10.3233/jad-2010-1317
Querfurth, H. W., and LaFerla, F. M. (2010). Alzheimer’s disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Rayman, M. P. (2012). Selenium and human health. Lancet 379, 1256–1268. doi: 10.1016/S0140-6736(11)61452-9
Rockenstein, E., Overk, C. R., Ubhi, K., Mante, M., Patrick, C., Adame, A., et al. (2015). A novel triple repeat mutant tau transgenic model that mimics aspects of pick’s disease and fronto-temporal tauopathies. PLoS One 10:e0121570. doi: 10.1371/journal.pone.0121570
Sabbagh, M. N. (2020). Editorial: Alzheimer’s disease drug development pipeline 2020. J. Prev. Alzheimers Dis. 7, 66–67. doi: 10.14283/jpad.2020.12
SantaCruz, K., Lewis, J., Spires, T., Paulson, J., Kotilinek, L., Ingelsson, M., et al. (2005). Tau suppression in a neurodegenerative mouse model improves memory function. Science 309, 476–481. doi: 10.1126/science.1113694
Scheltens, P., Blennow, K., Breteler, M. M. B., de Strooper, B., Frisoni, G. B., Salloway, S., et al. (2016). Alzheimer’s disease. Lancet 388, 505–517. doi: 10.1016/S0140-6736(15)01124-1
Schindowski, K., Bretteville, A., Leroy, K., Begard, S., Brion, J. P., Hamdane, M., et al. (2006). Alzheimer’s disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am. J. Pathol. 169, 599–616. doi: 10.2353/ajpath.2006.060002
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Sergeant, N., Bretteville, A., Hamdane, M., Caillet-Boudin, M.-L., Grognet, P., Bombois, S., et al. (2008). Biochemistry of tau in Alzheimer’s disease and related neurological disorders. Expert. Rev. Proteomics 5, 207–224. doi: 10.1586/14789450.5.2.207
Shultz, S. R., Wright, D. K., Zheng, P., Stuchbery, R., Liu, S. J., Sashindranath, M., et al. (2015). Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain 138, 1297–1313. doi: 10.1093/brain/awv053
Song, G., Zhang, Z., Wen, L., Chen, C., Shi, Q., Zhang, Y., et al. (2014). Selenomethionine ameliorates cognitive decline, reduces tau hyperphosphorylation and reverses synaptic deficit in the triple transgenic mouse model of Alzheimer’s disease. J. Alzheimers Dis. 41, 85–99. doi: 10.3233/jad-131805
Sontag, E. (2001). Protein phosphatase 2A: the trojan horse of cellular signaling. Cell. Signal. 13, 7–16. doi: 10.1016/s0898-6568(00)00123-6
Sontag, E., Hladik, C., Montgomery, L., Luangpirom, A., Mudrak, I., Ogris, E., et al. (2004a). Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J. Neuropathol. Exp. Neurol. 63, 1080–1091. doi: 10.1093/jnen/63.10.1080
Sontag, E., Luangpirom, A., Hladik, C., Mudrak, I., Ogris, E., Speciale, S., et al. (2004b). Altered expression levels of the protein phosphatase 2A ABαC enzyme are associated with Alzheimer disease pathology. J. Neuropathol. Exp. Neurol. 63, 287–301. doi: 10.1093/jnen/63.4.287
Sontag, E., Nunbhakdi-Craig, V., Lee, G., Bloom, G. S., and Mumby, M. C. (1996). Regulation of the phosphorylation state and microtubule-binding activity of tau by protein phosphatase 2A. Neuron 17, 1201–1207. doi: 10.1016/s0896-6273(00)80250-0
Sontag, E., Nunbhakdi-Craig, V., Lee, G., Brandt, R., Kamibayashi, C., Kuret, J., et al. (1999). Molecular interactions among protein phosphatase 2A, tau and microtubules. Implications for the regulation of tau phosphorylation and the development of tauopathies. J. Biol. Chem. 274, 25490–25498. doi: 10.1074/jbc.274.36.25490
Sontag, J.-M., and Sontag, E. (2014). Protein phosphatase 2A dysfunction in Alzheimer’s disease. Front. Mol. Neurosci. 7:16. doi: 10.3389/fnmol.2014.00016
Sontag, J.-M., Nunbhakdi-Craig, V., Mitterhuber, M., Ogris, E., and Sontag, E. (2010). Regulation of protein phosphatase 2A methylation by LCMT1 and PME-1 plays a critical role in differentiation of neuroblastoma cells. J. Neurochem. 115, 1455–1465. doi: 10.1111/j.1471-4159.2010.07049.x
Sontag, J.-M., Nunbhakdi-Craig, V., and Sontag, E. (2013). Leucine carboxyl methyltransferase 1 (LCMT1)-dependent methylation regulates the association of protein phosphatase 2A and tau protein with plasma membrane microdomains in neuroblastoma cells. J. Biol. Chem. 288, 27396–27405. doi: 10.1074/jbc.m113.490102
Squire, L. R., Wixted, J. T., and Clark, R. E. (2007). Recognition memory and the medial temporal lobe: a new perspective. Nat. Rev. Neurosci. 8, 872–883. doi: 10.1038/nrn2154
Su, J. H., Cummings, B. J., and Cotman, C. W. (1994). Early phosphorylation of tau in Alzheimer’s disease occurs at ser-202 and is preferentially located within neurites. Neuroreport 5, 2358–2362. doi: 10.1097/00001756-199411000-00037
Sydow, A., Van Der Jeugd, A., Zheng, F., Ahmed, T., Balschun, D., Petrova, O., et al. (2011). Tau-induced defects in synaptic plasticity, learning and memory are reversible in transgenic mice after switching off the toxic tau mutant. J. Neurosci. 31, 2511–2525. doi: 10.1523/JNEUROSCI.5245-10.2011
Takeuchi, T., Duszkiewicz, A. J., and Morris, R. G. (2013). The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130288. doi: 10.1098/rstb.2013.0288
Taleski, G., and Sontag, E. (2018). Protein phosphatase 2A and tau: an orchestrated “pas de deux”. FEBS Lett. 592, 1079–1095. doi: 10.1002/1873-3468.12907
Taniguchi, T., Doe, N., Matsuyama, S., Kitamura, Y., Mori, H., Saito, N., et al. (2005). Transgenic mice expressing mutant (N279K) human tau show mutation dependent cognitive deficits without neurofibrillary tangle formation. FEBS Lett. 579, 5704–5712. doi: 10.1016/j.febslet.2005.09.047
Tolstykh, T., Lee, J., Vafai, S., and Stock, J. B. (2000). Carboxyl methylation regulates phosphoprotein phosphatase 2A by controlling the association of regulatory B subunits. EMBO J. 19, 5682–5691. doi: 10.1093/emboj/19.21.5682
Vafai, S. B., and Stock, J. B. (2002). Protein phosphatase 2A methylation: a link between elevated plasma homocysteine and Alzheimer’s disease. FEBS Lett. 518, 1–4. doi: 10.1016/s0014-5793(02)02702-3
Van der Jeugd, A., Ahmed, T., Burnouf, S., Belarbi, K., Hamdame, M., Grosjean, M. E., et al. (2011). Hippocampal tauopathy in tau transgenic mice coincides with impaired hippocampus-dependent learning and memory and attenuated late-phase long-term depression of synaptic transmission. Neurobiol. Learn. Mem. 95, 296–304. doi: 10.1016/j.nlm.2010.12.005
Van Der Jeugd, A., Blum, D., Raison, S., Eddarkaoui, S., Buée, L., and D’Hooge, R. (2013). Observations in THY-Tau22 mice that resemble behavioral and psychological signs and symptoms of dementia. Behav. Brain Res. 242, 34–39. doi: 10.1016/j.bbr.2012.12.008
Van Der Jeugd, A., Hochgrafe, K., Ahmed, T., Decker, J. M., Sydow, A., Hofmann, A., et al. (2012). Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human tau. Acta Neuropathol. 123, 787–805. doi: 10.1007/s00401-012-0987-3
Van Der Jeugd, A., Parra-Damas, A., Baeta-Corral, R., Soto-Faguas, C. M., Ahmed, T., LaFerla, F. M., et al. (2018). Reversal of memory and neuropsychiatric symptoms and reduced tau pathology by selenium in 3xTg-AD mice. Sci. Rep. 8:6431. doi: 10.1038/s41598-018-24741-0
van Eersel, J., Ke, Y. D., Liu, X., Delerue, F., Kril, J. J., Gotz, J., et al. (2010). Sodium selenate mitigates tau pathology, neurodegeneration and functional deficits in Alzheimer’s disease models. Proc. Natl. Acad. Sci. U S A 107, 13888–13893. doi: 10.1073/pnas.1009038107
Wang, X., Smith, K., Pearson, M., Hughes, A., Cosden, M. L., Marcus, J., et al. (2018). Early intervention of tau pathology prevents behavioral changes in the rTg4510 mouse model of tauopathy. PLoS One 13:e0195486. doi: 10.1371/journal.pone.0195486
Wobst, H. J., Denk, F., Oliver, P. L., Livieratos, A., Taylor, T. N., Knudsen, M. H., et al. (2017). Increased 4R tau expression and behavioural changes in a novel MAPT-N296H genomic mouse model of tauopathy. Sci. Rep. 7:43198. doi: 10.1038/srep43198
Xia, Y., Prokop, S., Gorion, K. M., Kim, J. D., Sorrentino, Z. A., Bell, B. M., et al. (2020). Tau ser208 phosphorylation promotes aggregation and reveals neuropathologic diversity in Alzheimer’s disease and other tauopathies. Acta Neuropathol. Commun. 8:88. doi: 10.1186/s40478-020-00967-w
Xie, Y., Tan, Y., Zheng, Y., Du, X., and Liu, Q. (2017). Ebselen ameliorates β-amyloid pathology, tau pathology and cognitive impairment in triple-transgenic Alzheimer’s disease mice. J. Biol. Inorg. Chem. 22, 851–865. doi: 10.1007/s00775-017-1463-2
Xu, Y., Chen, Y., Zhang, P., Jeffrey, P. D., and Shi, Y. (2008). Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated tau dephosphorylation. Mol. Cell. 31, 873–885. doi: 10.1016/j.molcel.2008.08.006
Yao, X. Q., Li, X. C., Zhang, X. X., Yin, Y. Y., Liu, B., Luo, D. J., et al. (2012). Glycogen synthase kinase-3β regulates leucine-309 demethylation of protein phosphatase-2A via PPMT1 and PME-1. FEBS Lett. 586, 2522–2528. doi: 10.1016/j.febslet.2012.06.018
Zhang, Z. H., Chen, C., Wu, Q. Y., Zheng, R., Liu, Q., Ni, J. Z., et al. (2016). Selenomethionine reduces the deposition of beta-amyloid plaques by modulating beta-secretase and enhancing selenoenzymatic activity in a mouse model of Alzheimer’s disease. Metallomics 8, 782–789. doi: 10.1039/c6mt00117c
Keywords: Alzheimer’s disease, chronic oral treatment, synaptic transmission, synaptic plasticity, long-term depression, neurocognitive functions, tau hyperphosphorylation, protein phosphatase 2A (PP2A)
Citation: Ahmed T, Van der Jeugd A, Caillierez R, Buée L, Blum D, D’Hooge R and Balschun D (2020) Chronic Sodium Selenate Treatment Restores Deficits in Cognition and Synaptic Plasticity in a Murine Model of Tauopathy. Front. Mol. Neurosci. 13:570223. doi: 10.3389/fnmol.2020.570223
Received: 06 June 2020; Accepted: 26 August 2020;
Published: 08 October 2020.
Edited by:
Isabel Lastres-Becker, Autonomous University of Madrid, SpainReviewed by:
Mickael Audrain, Icahn School of Medicine at Mount Sinai, United StatesOlga Kokiko-Cochran, The Ohio State University, United States
Copyright © 2020 Ahmed, Van der Jeugd, Caillierez, Buée, Blum, D’Hooge and Balschun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Detlef Balschun, ZGV0bGVmLmJhbHNjaHVuQGt1bGV1dmVuLmJl