 Parnayan Syed
Parnayan Syed Nela Durisic
Nela Durisic Robert J. Harvey
Robert J. Harvey Pankaj Sah1,4
Pankaj Sah1,4 Joseph W. Lynch
Joseph W. Lynch- 1Queensland Brain Institute, The University of Queensland, Brisbane, QLD, Australia
- 2School of Health and Behavioural Sciences, University of the Sunshine Coast, Maroochydore, QLD, Australia
- 3Sunshine Coast Health Institute, Birtinya, QLD, Australia
- 4 Department of Biology, Joint Center for Neuroscience and Neural Engineering, Southern University of Science and Technology, Shenzhen, China
Missense mutations T166M, Q242L, T336M, and Y474C in the GABAA receptor (GABAAR) α3 subunit gene are associated with epileptic seizures, dysmorphic features, intellectual disability, and developmental delay. When incorporated into GABAARs expressed in oocytes, all mutations are known to reduce GABA-evoked whole-cell currents. However, their impact on the properties of inhibitory synaptic currents (IPSCs) is unknown, largely because it is difficult to establish, much less control, the stoichiometry of GABAAR expressed in native neuronal synapses. To circumvent this problem, we employed a HEK293 cell-neuron co-culture expression system that permits the recording of IPSCs mediated by a pure population of GABAARs with a defined stoichiometry. We first demonstrated that IPSCs mediated by α3-containing GABAARs (α3β3γ2) decay significantly slower than those mediated by α1-containing isoforms (α1β2γ2 or α1β3γ2). GABAAR α3 mutations did not affect IPSC peak amplitudes or 10–90% rise times, but three of the mutations affected IPSC decay. T336M significantly accelerated the IPSC decay rate whereas T166M and Y474C had the opposite effect. The acceleration of IPSC decay kinetics caused by the T366M mutation was returned to wild-type-like values by the anti-epileptic medication, midazolam. Quantification experiments in HEK293 cells revealed a significant reduction in cell-surface expression for all mutants, in agreement with previous oocyte data. Taken together, our results show that impaired surface expression and altered IPSC decay rates could both be significant factors underlying the pathologies associated with these mutations.
Introduction
Type A γ-aminobutyric acid receptors (GABAARs) are pentameric ligand-gated ion channels found at a majority of inhibitory synapses in the central nervous system. They are anion-selective channels that mediate fast synaptic inhibitory neurotransmission on a millisecond timescale and are crucial for maintaining the excitatory/inhibitory balance of activity in the brain. GABAARs exhibit a vast heterogeneity, due to the large number of subunits which can combine in a myriad of combinations giving rise to unique subtypes. In humans, there are six α subunits, three β and three γ subunits, three ρ and one each of ρ, θ, δ and ε (McKernan and Whiting, 1996). Each of these subunits consists of a hydrophilic extracellular N-terminal domain containing a Cys-loop, followed by four α-helical transmembrane domains (TM1–4) and an extracellular C-terminus. TM2 lines the integral ion channel and the intracellular loop between TM3 and TM4 interacts with various proteins involved in receptor trafficking, phosphorylation, and clustering (Kasaragod and Schindelin, 2019).
The subunit composition of a receptor dictates its kinetic, pharmacological, and membrane surface localization properties. The α subunit, for example, determines isoform-selective pharmacology (Sieghart and Sperk, 2002), and synaptic localization of the receptor via direct interaction with gephyrin (Saiepour et al., 2010; Brady and Jacob, 2015; Gao and Heldt, 2016). Although the α1, α2 and α5 subunits have been extensively studied (Browne et al., 2001; Sieghart and Sperk, 2002; Jacob, 2019), relatively little is known about the α3 subunits. The α3 subunit is selectively distributed being primarily found at inhibitory synapses of the reticular thalamic nucleus (Pirker et al., 2000), the suprachiasmatic nucleus (Ono et al., 2018), and reelin-positive cells in the medial entorhinal cortex (Berggaard et al., 2019). It has also been shown to be expressed in the basolateral amygdala where it contributes to tonic inhibition (Marowsky et al., 2012). The α3 subunit is thought to be critical during brain development, as it is one of the most widely expressed α subunits in the brain during embryonic and early postnatal ages in the rat (Laurie et al., 1992; Wisden et al., 1992). It also undergoes A-to-I RNA-editing in the transmembrane domain, which may contribute towards synapse formation and maintenance of excitatory/inhibitory balance during development (Rula et al., 2008).
Alterations in GABAergic synaptic clustering, diffusion, or receptor kinetics can lead to several neurological diseases resulting from a loss of inhibitory control exerted by these receptors (Rudolph and Möhler, 2014). The study of disease-associated mutations in GABAAR subunit genes is important for understanding the role of individual GABAAR isoforms in the underlying pathogenesis of neurological disorders (Maljevic et al., 2019). This in turn can help us to understand the roles of particular isoforms under normal physiological conditions.
A recent study identified four missense mutations (T166M, Q242L, T336M, and Y474C) in the α3 subunit gene (GABRA3) that were associated with epileptic seizures, dysmorphic features, intellectual disability, and developmental delay (Niturad et al., 2017). All mutations were shown to substantially reduce total whole-cell currents for α3β2γ2S GABAARs expressed in Xenopus oocytes, while paradoxically increasing GABA sensitivity for the Q242L, T336M, and Y474C mutants. The genetic, clinical, and functional characteristics of the mutations are summarized in Table 1.
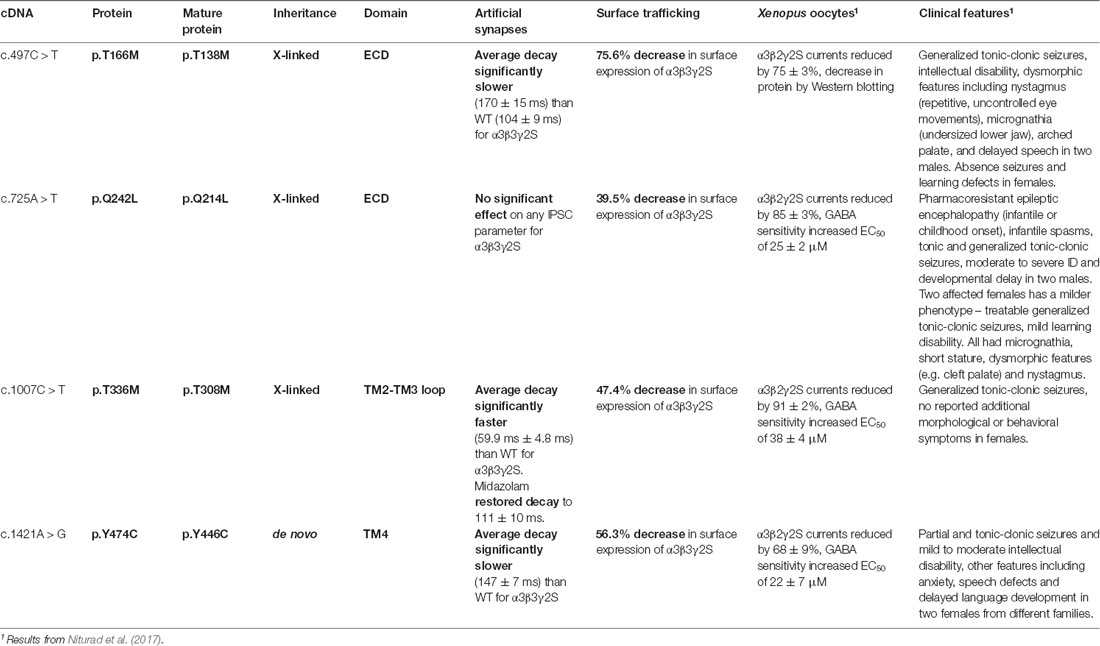
Table 1. γ-aminobutyric acid receptor (GABAAR) alpha3 subunit mutations.
Importantly, the effects of the mutations on the properties of inhibitory synaptic currents (IPSCs) mediated by synaptic GABAARs is not known. Unfortunately, it is difficult to study individual, defined GABAAR isoforms in native neurons due to the multitude of other isoforms present, and the difficulty in pharmacologically or genetically isolating the isoform of interest at particular synapses. Also, GABRA3 is subject to X-inactivation in all tissues except peripheral blood (Cotton et al., 2015), which may explain the variability in disease phenotype in males and females with a given GABAAR α3 mutations. X-inactivation is the process by which one of the copies of the X chromosome is randomly silenced in female cells during development. Males are expected to be hemizygous for GABRA3 mutations (i.e., both α3 subunits in an α3β2γ2 heteropentamer will be mutant as they derive from a single X chromosome). However, female mutation carriers are likely to express either wild-type or mutant GABAARs (again with two mutant α3 subunits) in a mosaic pattern in different neurons in the brain, because X inactivation creates two populations of cells that differ in terms of the “active” X chromosome. To avoid these uncertainties and reproduce the mutant GABAAR subtypes found in vivo, we employed a HEK293 cell—neuron co-culture expression system that permits the recording of IPSCs from a pure population of GABAARs with a defined stoichiometry (Dixon et al., 2014, 2015).
Materials and Methods
Cell Culture and Transfection
HEK293AD cells were used for all electrophysiological experiments. The cells were cultured in monolayers in T75 flasks with Dulbecco’s Modified Eagle Medium (DMEM), supplemented with 10% fetal bovine serum. The cells were kept in a 5% CO2 incubator at 37°C and passaged at least once a week. Trypsinized cells from the flask were plated onto 35 mm dishes and transfected at 50–70% confluency using a calcium phosphate precipitation method. The transfected cells were incubated overnight in a 3% CO2 incubator with the transfection mix for 16–20 h and then washed with divalent cation-free phosphate-buffered saline (PBS) to terminate the transfection. The plasmids used were: human α1 (pcDNA3.1), human GABAAR β3 (pCMV6) and γ2S (pcDNA3.1), Neuroligin 2A (pNICE) and empty pEGFP. The wild-type (WT) human GABAAR α3 construct was generously provided by Dr. Philip Ahring (University of Sydney). Mutations were introduced using site-directed mutagenesis and confirmed via sequencing of the entire plasmid. The α, β and γ subunits were transfected with Neuroligin 2A and GFP at a ratio of 1:1:4:0.5:0.5 to optimize the incorporation of the γ subunit into triheteromers. Cells for immunocytochemistry were transfected in the same plasmid ratio but using Lipofectamine 2000 (Invitrogen) according to manufacturer instructions (total DNA:lipofectamine 1:1 ng/μl). In these experiments, transfected cells were incubated overnight in a 5% CO2 incubator with the transfection mix for 4–6 h and then washed with divalent cation-free PBS to terminate the transfection.
Artificial Synapse Formation
Cortical neurons were harvested from Wistar rat embryos of both sexes at embryonic day 18 (University of Queensland, Institutional Breeding Colony). Euthanasia of timed-pregnant rats was performed via CO2 inhalation. All experiments were performed following relevant guidelines and regulations as approved by the University of Queensland Animal Ethics Committee (approval number: QBI/142/16/NHMRC/ARC). Cerebral tissue was extracted from the embryos as per protocol (Fuchs et al., 2013), and trypsinized using 0.25% trypsin-EDTA (Thermo Fisher, Australia). The tissue was triturated gently in DMEM and the neuronal suspension was centrifuged three times to maximize live-cell sedimentation. The cells were then counted and 70,000-80,000 neurons were plated onto 12 mm coverslips coated with poly-D-lysine in 4-well dishes. The medium was replaced with Neurobasal medium supplemented with 1% GlutaMAX and 2% B-27 24 h later, and half of it was again removed a week later and topped up with the freshly prepared medium. All components of the neuronal media were purchased from Thermo Fisher, Australia. Neurons were grown for 3–5 weeks at 37°C in a 5% CO2 incubator before being used for experiments. Transfected HEK293 cells were resuspended in the neuronal medium and plated onto neurons. The co-cultures were incubated overnight to allow for synapse formation and used over the next 1–3 days for electrophysiological recordings.
Electrophysiology
Whole-cell patch-clamp electrophysiology experiments were performed at room temperature (20–23°C) on the transfected HEK293AD cells using an Axopatch 1D amplifier and pClamp10 software (Molecular Devices, San Jose, CA, USA). Cells were placed in a bath and continuously perfused with extracellular solution containing (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 D-glucose, adjusted to pH 7.4 with NaOH. Patch pipettes, fabricated from borosilicate glass capillaries (Harvard Apparatus, Holliston, MA, USA), were pulled using a horizontal puller (Sutter Instruments, Novato, CA, USA), with the resistance of 3–6 MΩ, and fire-polished. The pipettes were filled with an intercellular solution containing (in mM): 145 CsCl, 2 CaCl2, 2 MgCl2, 10 EGTA, 2 MgATP, adjusted to pH 7.4 with CsOH.
Spontaneous GABAergic IPSCs were recorded at a holding potential of −70 mV; signals were filtered at 5 kHz and sampled at 20 kHz. Recordings with series resistance above 20 MΩ were discarded. The capacitance of the HEK293 cells was typically 20 pF, resulting in a typical corner frequency of 398 Hz. Because this was satisfactory for our experiments, series resistance compensation was not applied.
Midazolam (Sigma) was prepared in stock solutions of 10 mM in dimethylsulfoxide and stored at −20°C. Stock solutions were diluted to the desired concentration in extracellular solution on the day of recording. Typically, at least 3 min of spontaneous activity was recorded before and during drug application. To preserve network activity for spontaneous recordings, a drug solution was targeted to the recorded cell while the extracellular solution was washed over the surrounding area.
IPSC decay time constants, 10–90% rise times, and peak amplitudes were calculated using Axograph X (Axograph Scientific, Australia), as has been described previously (Dixon et al., 2015). Peak amplitudes were detected with a 2:1 signal to noise ratio as the threshold, and all peaks manually examined to select well-separated events. Parameters calculated by Axograph X for each event were averaged to determine the final values.
Statistical analysis and graph plots were performed using SigmaPlot 13 (Systat Software, San Jose, CA, USA). One-way ANOVA was used for group comparisons, and p < 0.05 taken to be statistically significant. Data are presented as mean ± SEM unless otherwise stated.
Surface Labeling of Receptors
Unpermeabilized HEK293 cells were fixed with 4% paraformaldehyde 2 days post-transfection for 10 min and then washed with PBS and labeled with rabbit anti-GABA α3 antibody (Synaptic Systems), diluted 1:500 in blocking solution (1% bovine serum albumin) at 37°C overnight. Cells were then washed with PBS, incubated with goat anti-rabbit antibody conjugated with Alexa555 (Thermo Fisher Scientific) for 3 h, and mounted onto glass slides using DABCO (220 mM in 90% glycerol). Each isoform was tested across at least two separate transfections and immunocytochemistry sample preparation.
Imaging and Analysis
All imaging was performed using the LSM 510 Meta inverted point-scanning laser confocal microscope (Carl Zeiss), fitted with a 63× 1.4 NA oil-immersion objective. The 488 nm laser was used to capture images of cells expressing GFP, and the 514 nm laser to visualize GABAARs containing the α3 subunit expressed at the cell surface. All exposure parameters were kept the same across all experiments to enable comparative levels of expression via fluorescence intensity measurement.
A custom code in ImageJ Macro language (IJM) was written in ImageJ (Fiji) to analyze and quantify the fluorescence from GABAAR α3 labelings for each cell (available on request). Global background subtraction was done for each image, and cells selected using regions of interest (ROI). The mean gray value of these ROIs was calculated and averaged. Mean, medians, and interquartile ranges were calculated across each dataset and one-way ANOVA on ranks used to determine statistical significance.
Results
Electrophysiological Characterization of Wild-Type α3β3γ2 GABAARs
We recorded spontaneous IPSCs in HEK293 cells expressing α3β3γ2 GABAARs, a physiologically-relevant receptor isoform found at central synapses (Fritschy and Mohler, 1995). We compared the kinetics of IPSCs mediated by this combination with those mediated by α1β2γ2, the most widely-expressed synaptic GABAAR subtype in the brain (Sieghart and Sperk, 2002). These results are summarized in Figure 1. Sample recordings shown on two different time-bases of IPSCs from HEK293 cells expressing wild-type (WT) receptors show markedly different decay kinetics for α1β2γ2 and α3β3γ2 GABAARs (Figure 1A). All isolated events from each of the recordings were averaged to produce a single digitally-averaged synaptic current waveform from each cell. These waveforms are shown normalized and superimposed in Figure 1B. The average peak IPSC amplitude, 10–90% rise time, and decay time constants from each cell are displayed in Figures 1C–E, where each data point represents the average from all well-isolated IPSCs recorded from a single cell. The comparison of α3β3γ2 with α1β2γ2 showed a significantly faster mean rise time (3.96 ± 0.29 vs. 1.76 ± 0.14 ms) and decay time constant (104.0 ± 8.7 vs. 23.7 ± 1.5 ms) for α1 containing receptors, whereas their respective IPSC amplitudes varied widely and were not significantly different to each other. To determine the contribution of β3 to these differences, we also compared these properties against those of α1β3γ2. These results (Figure 1), show that although the rise time of α1β3γ2 was also slow (3.68 ± 0.22 ms) compared to α1β2γ2, the decay time of α3-containing receptors was much slower than α1β3γ2 receptors (30.5 ± 3.6 ms).
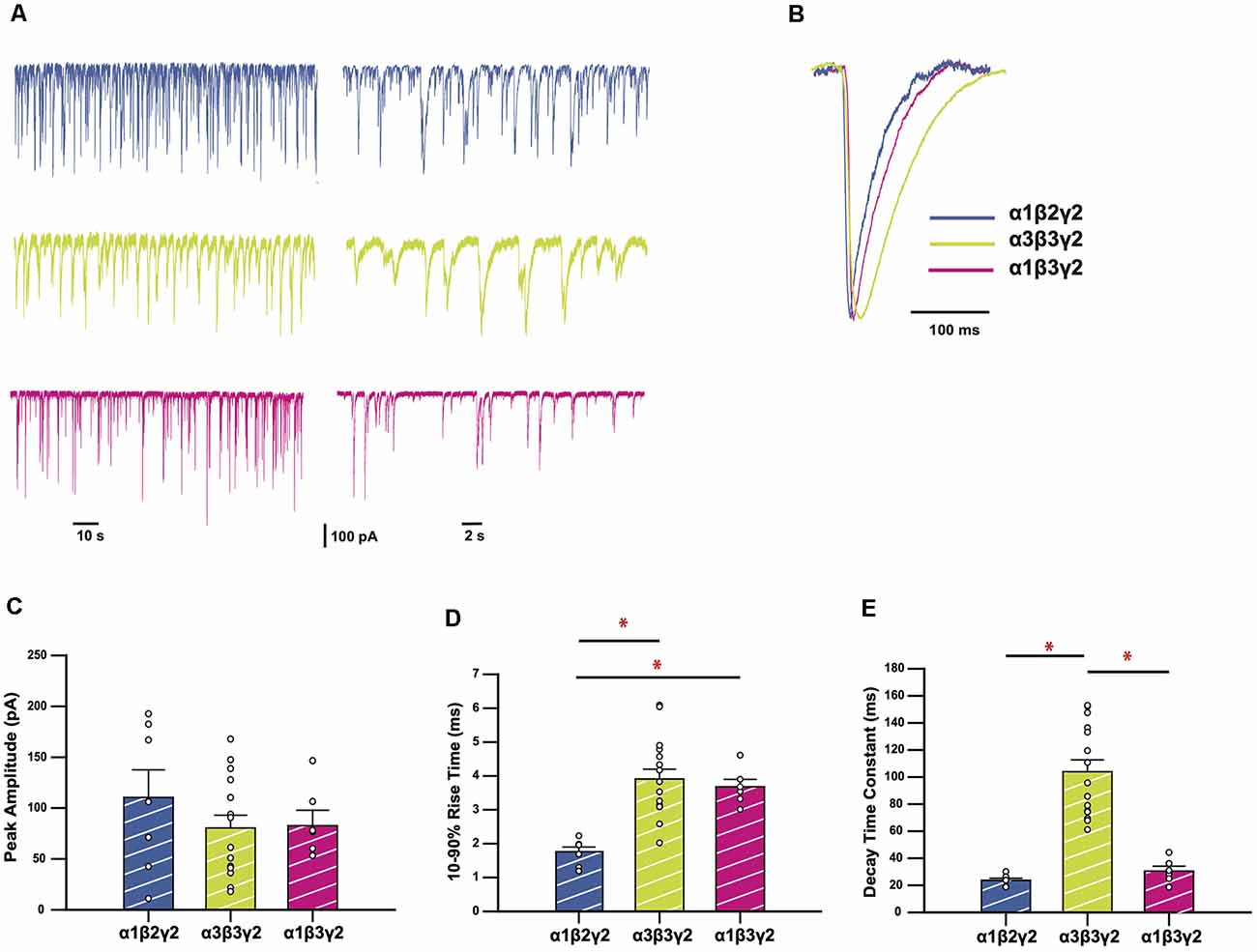
Figure 1. GABAA receptors (GABAARs) containing the α3 subunit generate inhibitory synaptic currents (IPSCs) with slower rise times and decay time constants. (A) Representative traces recorded from HEK293 cells expressing the GABAAR isoforms as indicated. (B) Averaged and normalized IPSCs from individual cells, overlayed for comparison. (C–E) Bar plots comparing mean peak amplitudes (C), 10–90% rise times (D), and decay time constants (E) of wild-type (WT) GABAAR isoforms. Error bars represent SEM and n values were, respectively 7, 15, and 6. Statistically significant results (p < 0.05) are indicated with an asterisk.
Effects of GABAAR α3 Subunit T166M, Q242L, T336M, and Y474C Mutations on IPSC Kinetics
We analyzed four GABAAR α3 subunit missense mutations (T166M, Q242L, T336M, and Y474C) that have previously been identified in patients with epileptic seizures, dysmorphic features, intellectual disability, and developmental delay (Niturad et al., 2017; Table 1). Their locations on the GABAAR α3 subunit polypeptide are shown in Figure 2. To understand the effect of these mutations we recorded IPSCs from co-culture synapses in which HEK293 cells had been transfected with the mutant GABAAR α3 subunits in combination with β3 and γ2 subunits.
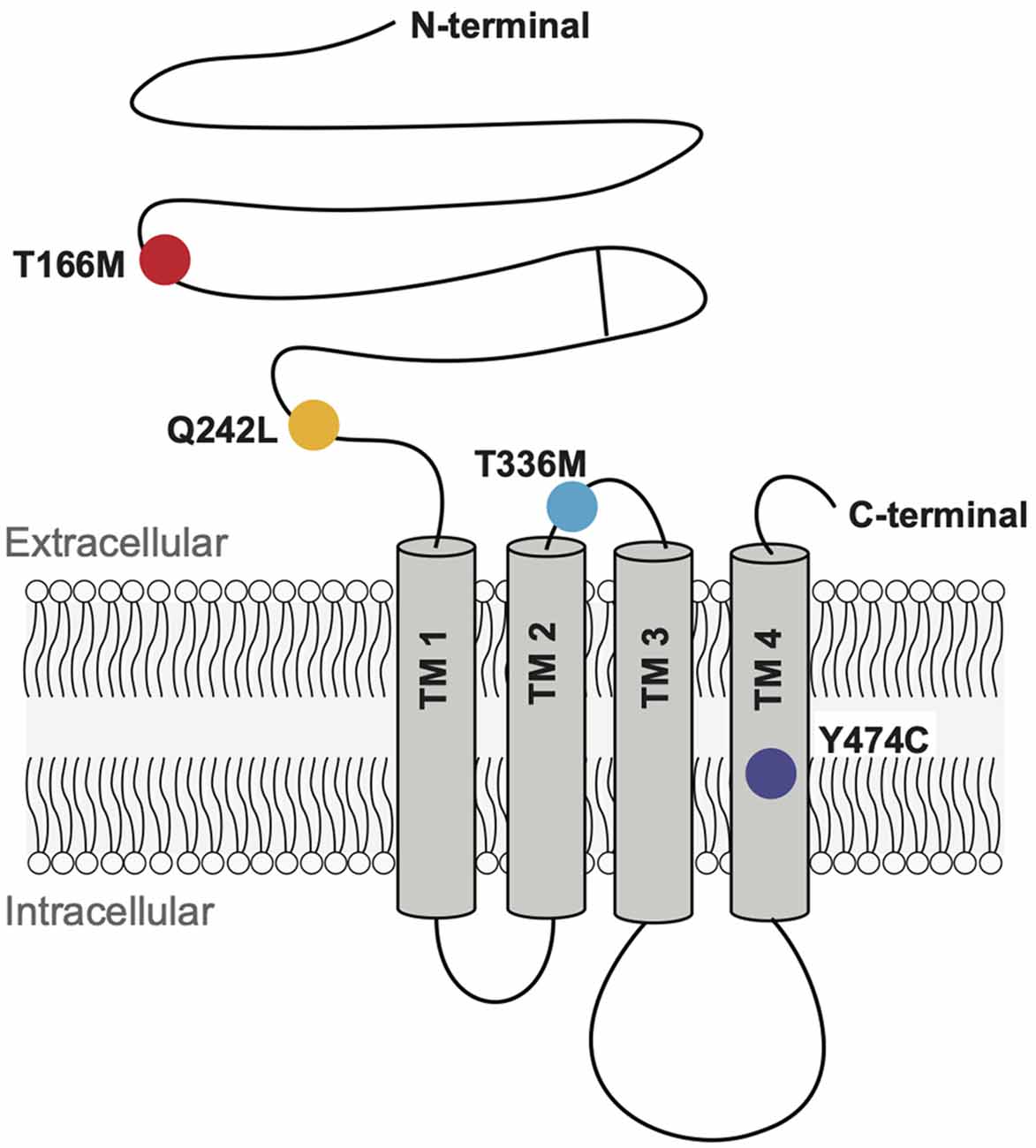
Figure 2. Schematic representation of a GABAAR subunit showing the positions of the four mutant residues investigated here.
Sample recordings of IPSCs shown on two different time-bases are shown in Figure 3A. Average peak IPSC amplitude, 10–90% rise time, and decay time constants from all events averaged from each cell are displayed in Figures 3C–E, where each data point represents the average of all IPSCs recorded from a single cell. The results indicate that while the average peak amplitudes and 10–90% rise times of all GABAAR α3 mutants showed no significant differences concerning WT receptors, the decay times of T166M and Y474C were significantly slower (170.6 ± 15.0 and 146.8 ± 7.1 ms respectively) than WT (104.0 ± 8.7 ms) whereas T336M was significantly faster (59.9 ± 4.8 ms). Q242L, on the other hand, had no significant effect on any measured IPSC parameter in these experiments. All these results are summarized in Table 1.
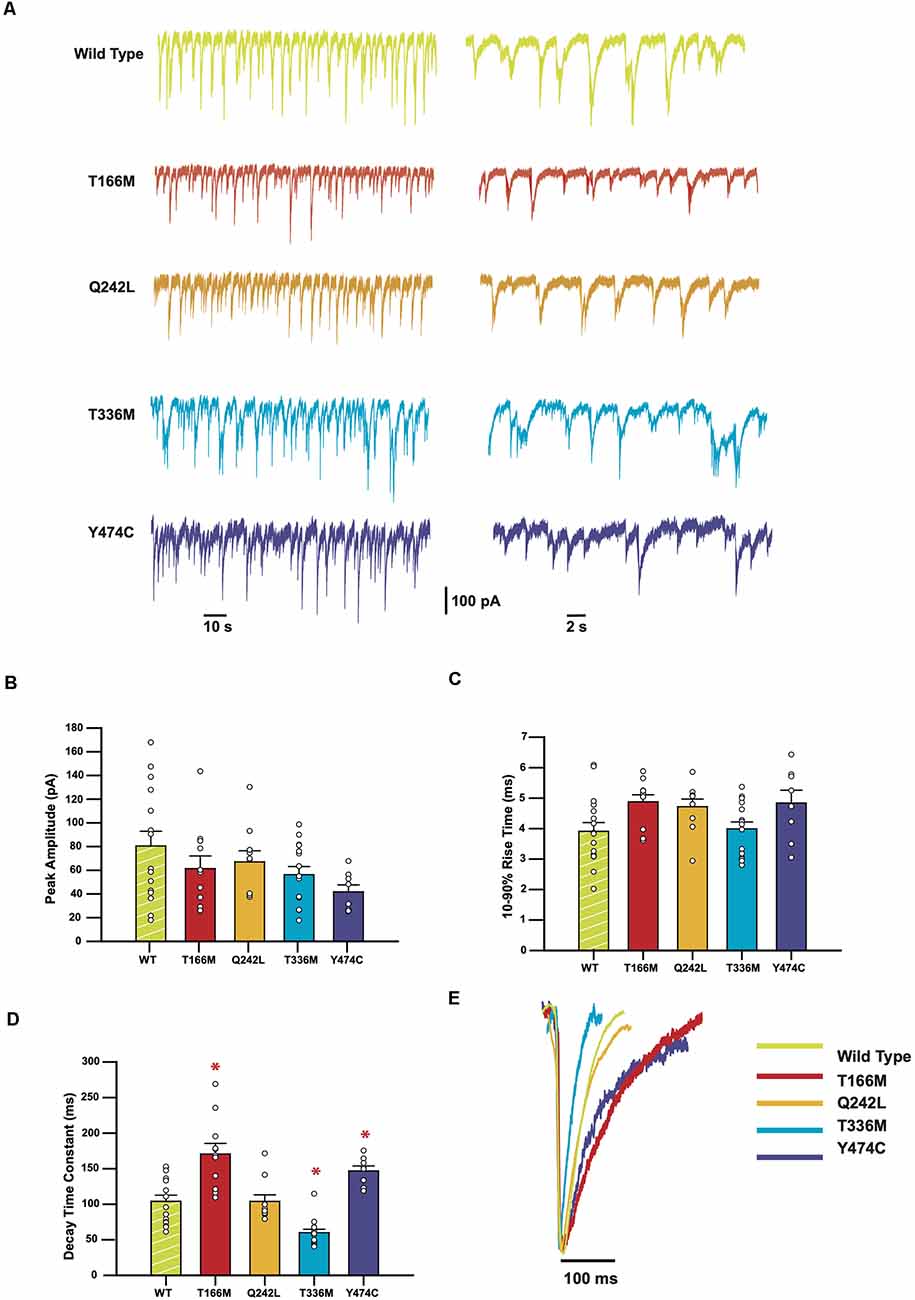
Figure 3. IPSCs mediated by GABAAR epilepsy mutants show a variety of changes in kinetic parameters. (A) Representative traces from HEK293 cells expressing WT or mutant α3 subunits, along with β3 and γ2. (B–D) Bar plots comparing the kinetic properties of the mutants compared to WT α3-containing GABAARs. Error bars represent SEM and n values were, respectively 15, 11, 9, 15, and 8. Statistically significant results (p < 0.05) are indicated with an asterisk. (E) Averaged normalized currents from single cells overlayed to compare the shape of the IPSCs recorded from each mutant compared to the WT.
Midazolam Treatment of T336M
Midazolam, a benzodiazepine, is a widely used anti-epileptic in infants and children that is thought to act via its action on GABAARs. Since the T336M mutation significantly increased the decay rate of IPSCs mediated by α3β3γ2 GABAARs, we tested whether the decay rate could be normalized using 1 μM midazolam. To account for variability between HEK293 cell batches, activity levels of neuronal cultures, and transfection efficiencies, the WT dataset was repeated under the same conditions for this set of experiments. A sample recording showing the effect of midazolam is shown in Figure 4A. Figure 4B shows that in the presence of midazolam, the average amplitude of IPSCs mediated by T336M-containing receptors increased from 19.3 ± 3.3 − 24.9 ± 5.7 pA, though this difference is not statistically significant and remains less than the WT average amplitude of 33.3 ± 5.0 pA. Similarly, the average IPSC rise time was not affected by midazolam (Figure 4C). However, the average decay time constant of IPSCs increased from 60.2 ± 4.2 − 110.5 ± 9.9 ms, which is comparable to the average decay time of WT α3β3γ2 GABAARs (108.1 ± 8.9 ms; Figure 4D). These results show that midazolam can restore the decay times of the T336M mutant GABAARs to values comparable with WT receptors.
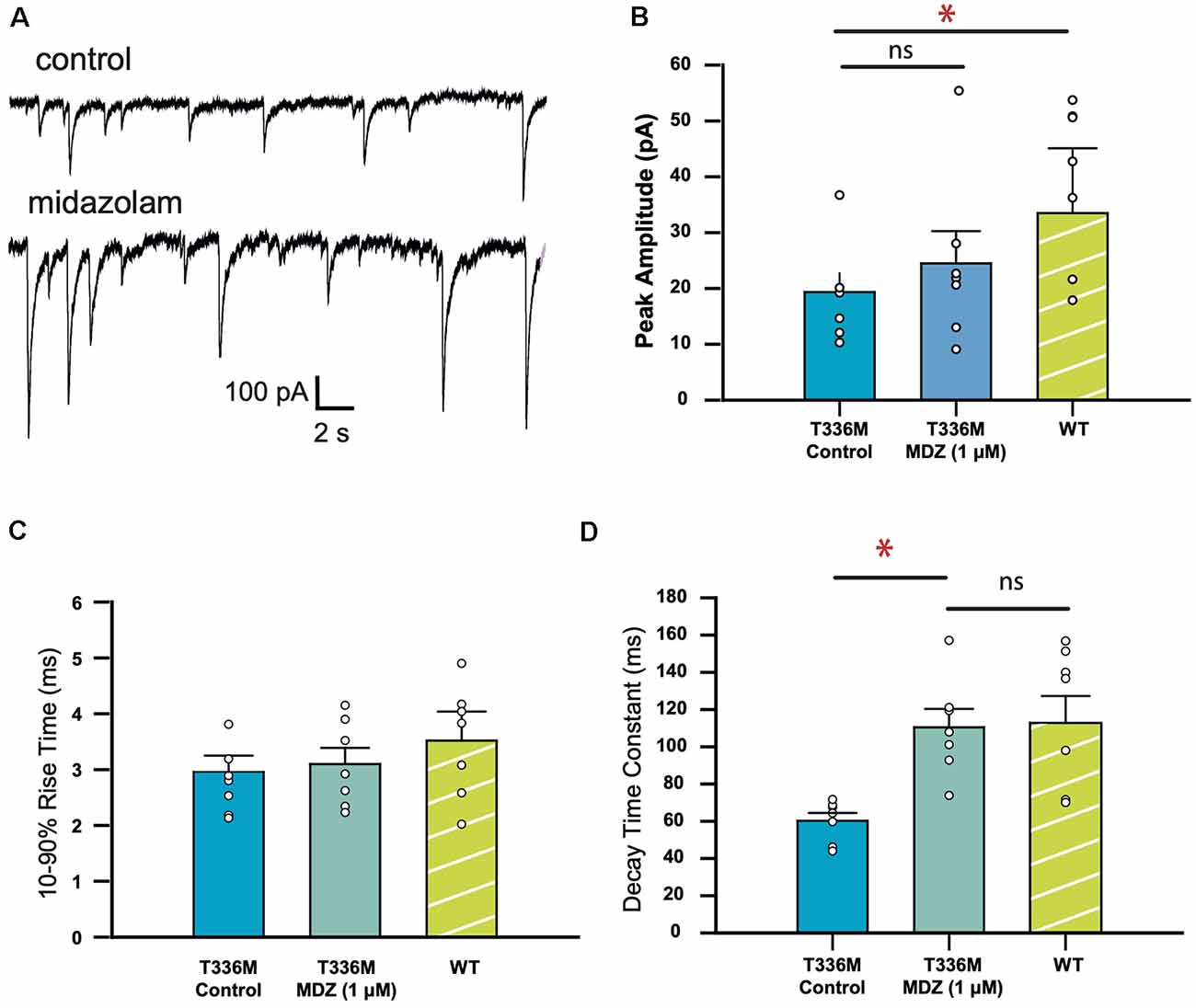
Figure 4. Midazolam restores the kinetic properties of GABAARs incorporating the GABAAR α3 subunit T336M mutation. (A) Representative traces recorded from a HEK293 cell expressing the T336M mutant, before and after midazolam treatment. (B–D) Mean kinetic properties of the T336M mutant before and after treatment with 1 μM midazolam. Control WT data are included for comparison (*p < 0.05; ns = no significant difference; n = 6 for all constructs).
Surface Expression of the GABAAR α3 Subunit T166M, Q242L, T336M, and Y474C Mutants
Because of the varying effects of GABAAR α3 mutations on the kinetic properties of IPSCs and the complete lack of any observable effect for the Q242L mutation, we examined other mechanisms by which these mutations might be affecting receptor function. To this end, we used immunocytochemistry to quantify the surface expression for each mutant receptor and compared it to WT α3β3γ2 GABAARs. Transient transfection of transmembrane proteins in HEK293 cells often results in a large variation in surface expression even when the transfection is carefully optimized (Ooi et al., 2016). We, therefore, imaged large numbers of immunostained cells to obtain an accurate representation of the population. Exposure settings of the camera were kept constant throughout all experiments, and our inclusion criterion stipulated that any cell that showed an immunofluorescent signal discernible from the background would be included. In electrophysiology measurements, only cells with the highest levels of expression (judged by their fluorescence intensity) were selected for patching because those cells have the highest probability of forming synapses with the neurons and produce the most robust IPSCs.
Representative images of cells expressing high and low levels of α3β3γ2 receptors are shown in Figure 5A. For comparison with electrophysiology measurements, we pooled twenty cells with the highest GABAAR surface expression from immunostaining experiments for the WT and each mutant (Figure 5B). The fluorescence intensity median (interquartile range), for WT α3β3γ2 was 28.2 (21.8–32.9) × 102 a.u. All mutants had significantly lower fluorescence intensity, with median (interquartile range) values as follows: Q242L: 9.4 (8.2–13.4) × 102 a.u., T336M: 12.5 (9.7–16.4) × 102 a.u., and Y474C: 13.6 (9.9–16.8) × 102 a.u. An exception was T166M: 23.3 (19.6–28.0) × 102 a.u., which also trended towards lower expression than WT, but not to a statistically significant degree. However, when we included all imaged cells, we observed a significant decrease in the surface expression of all mutants with an overall reduction in median values to 75.6% for T166M, 39.5% for Q242L, 47.4% for T336M, and 56.3% for Y474C compared to WT α3β3γ2 (Figure 5C).
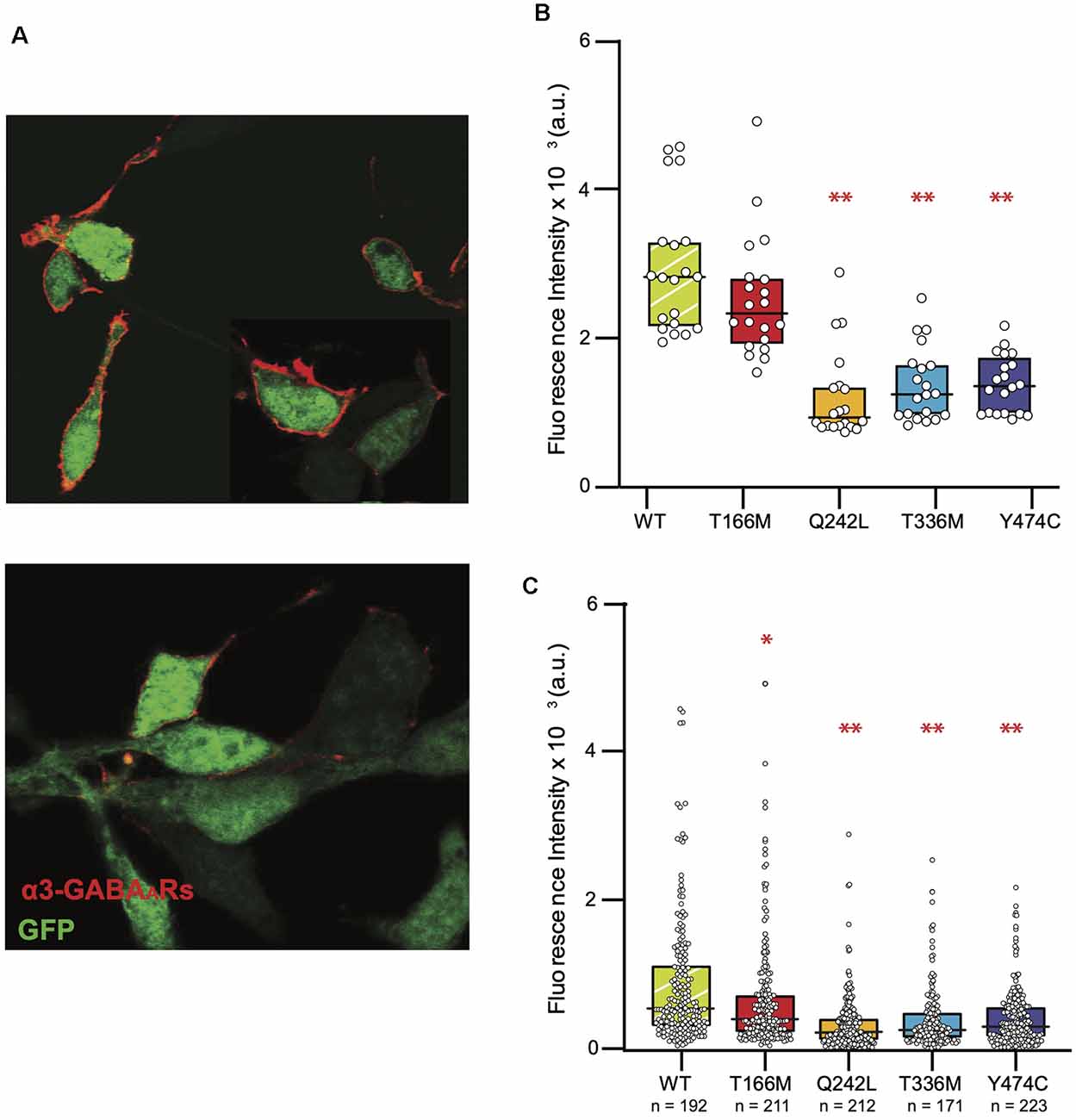
Figure 5. Reduced surface expression of mutant GABAARs in HEK293 cells. (A) Representative images of cells expressing α3β3γ2 receptors (red). Intracellular GFP (green) was used as an indicator of transfection efficiency. The top panel shows a sample image of cells showing a high level of expression and the bottom panel shows cells that had very low levels to no discernible expression levels. (B) Fluorescence intensity of the brightest cells for each isoform (n = 20 for all constructs) is shown as these cells are typically used to measure IPSCs in the “artificial synapse” system. The boxplots indicate the median and interquartile range, one-way ANOVA on ranks, **p < 0.001. (C) Full distribution of GABAAR surface expression in HEK293 cells shows reduced surface expression of all mutants compared to the WT, one-way ANOVA on ranks, *p < 0.05, **P < 0.001. Median (interquartile range) values are as follows: WT: 5.29 (3.01–11.4 × 102) a.u., T166M: 4.0 (2.14–7.31) × 102 a.u., Q2442L: 2.06 (1.05–4.19) × 102 a.u., T336M: 2.51 (1.97–5.0) × 102 a.u., Y474C: 2.98 (1.5–5.75) × 102 a.u.
Discussion
GABAARs Containing the α3 Subunit Have Slow Decay Kinetics
This study aimed to elucidate the functional characteristics of α3β3γ2 GABAAR-mediated IPSCs and the effects of selected pathogenic mutations. α3-containing GABAARs are the main inhibitory synaptic receptors in the reticular thalamic nucleus, and are important for controlling the excitability of thalamocortical networks (Browne et al., 2001; Crabtree, 2018). However, IPSCs mediated by these receptors have not previously been studied in isolation. Inhibitory synapses within the thalamocortical network, like most GABAergic synapses in the brain, present heterogeneity in terms of subunit composition (Fritschy and Mohler, 1995). In the ventrobasal nucleus of the thalamus, α1-containing receptors mediate phasic inhibition and α4-containing receptors are involved in tonic inhibition (Jia et al., 2005). Studies of thalamocortical activity in α3-knockout mice have not been able to conclusively resolve the functional impact of these receptors, due to other compensatory mechanisms (Winsky-Sommerer et al., 2008; Schofield et al., 2009). In the present study, we used the HEK293-neuronal co-culture technique, because it allowed IPSCs mediated by a defined GABAAR isoform to be studied in isolation.
We demonstrated that IPSCs mediated by α3β3γ2 GABAARs decay slower than the other synaptically-abundant GABAARs, α1β2γ2 and α1β3γ2 (McKernan and Whiting, 1996; Hutcheon et al., 2004; Daniel et al., 2010). These results are in agreement with previously published studies that show that GABAergic IPSCs in the reticular thalamic neurons, which express mostly α3-containing receptors at synapses (Çavdar et al., 2014), are slower than those of neurons in the ventrobasal nucleus, where the α1-containing isoforms predominate (Schofield and Huguenard, 2007). It has also been shown in a single-channel study that α3-containing GABAARs have longer active periods, and higher intraburst open probabilities compared to α1-containing GABAARs, which results in slower deactivation of the channel (Keramidas and Harrison, 2010). The β3 subunit has also been shown to contribute to slow activation and decay kinetics of GABAARs in a co-culture system (Chen et al., 2017b). To rule out the contribution of the β3 subunit in the slow deactivation time of α3β3γ2 receptors, we also compared their IPSC kinetics with those of α1β3γ2 receptors. This analysis revealed that while the rise times are similar for the two β3-containing isoforms, α1β3γ2 receptors have decay times that are more comparable with α1- than α3-containing GABAARs. A comparison between all three receptor types shows that the α3 contributes to the slow IPSC decay time in a statistically significant manner.
Effects of Pathogenic Mutations on IPSCs Mediated by α3-Containing GABAARs
Using the same methods, we tested how the mutations T166M, Q242L, T336M, and Y474C, identified in patients with epileptic seizures, dysmorphic features, intellectual disability, and developmental delay (Niturad et al., 2017) affected GABAAR mediated synaptic currents. These mutations did not affect average peak amplitudes or 10–90% rise time of synaptic currents, but all mutants, except Q242L, affected the decay time constants of the IPSCs (Table 1). The mutation Q242L, in the extracellular loop, associated with tonic-clonic seizures, dysmorphic features, intellectual disability, and developmental delay did not significantly affect any of the tested IPSC kinetic parameters. The mutant T336M, located in the TM2-TM3 loop, also associated with generalized tonic-clonic seizures, resulted in a significantly reduced IPSC decay time constant. This is similar to what has been observed for several epilepsy-related mutations in other GABAAR subunits (Fisher, 2004; Chen et al., 2017a; Dixon et al., 2017). The faster decay of the IPSC results in an overall loss of inhibitory control, which is likely to be the main cause of epilepsies associated with GABAAR mutations (Hernandez and Macdonald, 2019). A common pharmacological intervention for epilepsy is the use of benzodiazepines, which act on GABAARs and potentiate the influx of chloride ions (Ochoa and Kilgo, 2016). Midazolam is one such compound that has been used for the treatment of status epilepticus for decades (Smith and Brown, 2017). We tested whether this drug could restore the decay time of the mutant T336M GABAARs towards WT values, and found that indeed it does potentiate the mutant receptor to a level that could increase inhibitory control in the brain.
The mutations T166M in the extracellular domain, and Y474C in the TM4 domain, are associated with absence and complex partial seizures respectively, but also comorbidities such as autism, anxiety, dysmorphic features, and mild to moderate intellectual disability. Paradoxically, these mutations resulted in a slower decay time constant for the IPSCs. This has been previously observed for other epilepsy-causing mutants in GABAAR subunits, notably γ2 R43Q (Bowser et al., 2002), which is associated with febrile seizures and childhood absence epilepsy (Wallace et al., 2001). This apparent gain-of-function does not explain the associated pathology. However, further studies have shown that γ2 R43Q is retained in the endoplasmic reticulum (Durisic et al., 2018), resulting in reduced surface expression and synaptic targeting of the assembled receptor complexes (Frugier et al., 2007), which may explain the resulting loss of inhibitory control underlying the epileptic phenotypes.
These studies, as well as previous work on α3 mutations that demonstrated reduced whole-cell currents, and increased GABA sensitivity for Q242L, T336M, and Y474C (Niturad et al., 2017), led us to explore the effect of these mutants on cell-surface expression.
Analysis of Cell-Surface Expression Efficiency
Our quantification experiments in HEK293 cells showed a significant reduction in surface expression for all mutants. This effect was most profound for mutation Q242L. This is corroborated by previous work in oocytes which showed reduced whole-cell currents for these mutants (Niturad et al., 2017). Decreased cell surface expression reduces overall inhibition, resulting in the manifestation of epilepsy phenotypes (Fisher, 2004; Durisic et al., 2018; Shi et al., 2019). However, it may also result in the preferential expression of receptors lacking the α3 subunit which could in turn alter IPSC kinetics. This is certainly a possibility in neurons where compensatory upregulation of other α subunits may occur. However, such a mechanism is unlikely to explain our results as HEK293 cells express no endogenous α subunits and functional synaptic receptors do not form without these.
Paradoxically, the peak amplitude of IPSCs recorded from co-cultures was not reduced in magnitude (Figure 3B). Thus, they do not reflect the observed differences in cell surface expression between the WT and mutant α3-GABAARs for two reasons. First, in co-culture preparations, we typically focused on cells with the highest expression level as those cells are most likely to form synapses with neuronal presynaptic terminals. While the highest expressing HEK293 cells allow for the recording of robust synaptic currents, they also attract multiple synaptic contacts (Leacock et al., 2018). As a result, electrophysiological recordings from these cells are typically characterized by overlapping events as seen in Figure 2A. These events, often with the largest amplitudes, are discarded in the analysis because they do not allow for accurate calculation of the rise and decay times. Thus, while cell-surface expression and IPSC amplitudes are expected to correlate linearly, the different analysis of the two datasets shows different trends and the expression levels may be underestimated when judged from the peak amplitude of IPSCs measured in highly-expressing cells.
Second, it is not known if the extracellular N-terminal domain of the α3 subunit is involved in synapse formation in HEK293 cells. While it has been shown that N-terminal domains of GABAAR α1, β2, and γ2 subunits are directly involved in synaptic contact formation (Oh et al., 2016; Lu et al., 2017), very little is known about the role of α3 subunit in synaptogenesis. Nevertheless, it is noteworthy that the GABAAR isoforms studied in these experiments did contain the γ2 subunit, which could potentially increase GABAAR clustering at postsynaptic densities (Schweizer et al., 2003), as has been shown for α1β3γ2 receptors (Brown et al., 2016). This would increase the IPSC peak amplitude even when the overall expression level in HEK293 cells is very small. Therefore, the direct correlation between the peak amplitude of IPSCs recorded from co-cultures and GABAAR surface expression levels may be lost.
Immunostaining experiments are unaffected by these factors and enable the detection of very low cell-surface expression levels, which results in a more accurate measure of GABAAR levels. Taken together, our results suggest that receptor trafficking to the cell surface could be a significant factor underlying the pathologies associated with these mutations. However, a detailed study examining the subcellular localization of these mutants and quantification of cell-surface expression levels using biotinylation or differential labeling is still warranted. Defects in receptor trafficking could be corrected using drugs such as SAHA, which can rescue misfolded proteins from the ER and restore expression levels of low-expressing GABAAR mutants (Chen et al., 2017a; Durisic et al., 2018). Patients carrying mutations like Q242L, which causes low expression levels without affecting channel kinetics, could potentially benefit from these types of therapies.
Conclusion
This study reveals unique kinetic properties of IPSCs mediated by α3-containing GABAARs in a co-culture system, and shows that they have slower decay kinetics than other synaptic GABAAR isoforms. We also show that disease-causing mutations affecting the GABAAR α3 subunit have significant but varied effects on the functional properties of IPSCs mediated by α3-containing GABAARs. Of particular note, the acceleration of IPSC decay kinetics caused by the T366M mutation was returned to WT-like levels by the antiepileptic drug, midazolam. Finally, we showed that all mutations studied induced a significant reduction in cell-surface expression of GABAA receptors, which indicates that effective pharmacotherapies should target deficient channel kinetics, whilst restoring the cell-surface expression of mutant subunits.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by University of Queensland Animal Ethics Committee (approval number: QBI/142/16/NHMRC/ARC).
Author Contributions
All authors contributed to experimental design. RH and P. Syed performed the molecular biology. P. Syed performed all experiments. P. Syed and ND analyzed the data. P. Syed and JL wrote the first draft of the manuscript. All authors were involved in revising the manuscript for important intellectual content, and all authors approved the final version to be published. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Health and Medical Research Council (1058542 to JL, 1147600 to ND, 1156673 to RH). P. Sah was supported by grants from the Australian National Health and Medical Research Council and the Australian Research Council (CE140100007). P. Syed was supported by an Australian Government Research Training Program Scholarship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Berggaard, N., Witter, M. P., and van der Want, J. J. L. (2019). GABAA receptor subunit α3 in network dynamics in the medial entorhinal cortex. Front. Syst. Neurosci. 13:10. doi: 10.3389/fnsys.2019.00010
Bowser, D. N., Wagner, D. A., Czajkowski, C., Cromer, B. A., Parker, M. W., Wallace, R. H., et al. (2002). Altered kinetics and benzodiazepine sensitivity of a GABAA receptor subunit mutation γ2R43Q found in human epilepsy. Proc. Natl. Acad. Sci. U S A 99, 15170–15175. doi: 10.1073/pnas.212320199
Brady, M. L., and Jacob, T. C. (2015). Synaptic localization of α5 GABAA receptors via gephyrin interaction regulates dendritic outgrowth and spine maturation. Dev. Neurobiol. 75, 1241–1251. doi: 10.1002/dneu.22280
Brown, L. E., Nicholson, M. W., Arama, J. E., Mercer, A., Thomson, A. M., and Jovanovic, J. N. (2016). γ-Aminobutyric acid type A (GABAA) receptor subunits play a direct structural role in synaptic contact formation via their N-terminal extracellular domains. J. Biol. Chem. 291, 13926–13942. doi: 10.1074/jbc.M116.714790
Browne, S. H., Kang, J., Akk, G., Chiang, L. W., Schulman, H., Huguenard, J. R., et al. (2001). Kinetic and pharmacological properties of GABAA receptors in single thalamic neurons and GABAA subunit expression. J. Neurophysiol. 86, 2312–2322. doi: 10.1152/jn.2001.86.5.2312
Çavdar, S., Hacioğlu Bay, H., Yildiz, S. D., Akakin, D., Sirvanci, S., and Onat, F. (2014). Comparison of numbers of interneurons in three thalamic nuclei of normal and epileptic rats. Neurosci. Bull. 30, 451–460. doi: 10.1007/s12264-013-1402-3
Chen, X., Durisic, N., Lynch, J. W., and Keramidas, A. (2017a). Inhibitory synapse deficits caused by familial α1 GABAA receptor mutations in epilepsy. Neurobiol. Dis. 108, 213–224. doi: 10.1016/j.nbd.2017.08.020
Chen, X., Keramidas, A., and Lynch, J. W. (2017b). Physiological and pharmacological properties of inhibitory postsynaptic currents mediated by α5β1γ2, α5β2γ2 and α5β3γ2 GABAA receptors. Neuropharmacology 125, 243–253. doi: 10.1016/j.neuropharm.2017.07.027
Cotton, A. M., Price, E. M., Jones, J. M., Balaton, B. P., Kobor, M. S., and Brown, C. J. (2015). Landscape of DNA methylation on the X chromosome reflects CpG density, functional chromatin state and X-chromosome inactivation. Hum. Mol. Genet. 24, 1528–1539. doi: 10.1093/hmg/ddu564
Crabtree, J. W. (2018). Functional diversity of thalamic reticular subnetworks. Front. Syst. Neurosci. 12:41. doi: 10.3389/fnsys.2018.00041
Daniel, C., Wahlstedt, H., Ohlson, J., Björk, P., and Ohman, M. (2010). Adenosine-to-inosine RNA editing affects trafficking of the γ-aminobutyric acid type A (GABAA) receptor. J. Biol. Chem. 286, 2031–2040. doi: 10.1074/jbc.M110.130096
Dixon, C. L., Sah, P., Keramidas, A., Lynch, J. W., and Durisic, N. (2017). γ1-containing GABAA receptors cluster at synapses where they mediate slower synaptic currents than γ2-containing GABAA receptors. Front. Mol. Neurosci. 10:178. doi: 10.3389/fnmol.2017.00178
Dixon, C. L., Sah, P., Lynch, J. W., and Keramidas, A. (2014). GABAA receptor α and γ subunits shape synaptic currents via different mechanisms. J. Biol. Chem. 289, 5399–5411. doi: 10.1074/jbc.M113.514695
Dixon, C. L., Zhang, Y., and Lynch, J. W. (2015). Generation of functional inhibitory synapses incorporating defined combinations of GABAA or glycine receptor subunits. Front. Mol. Neurosci. 8:80. doi: 10.3389/fnmol.2015.00080
Durisic, N., Keramidas, A., Dixon, C. L., and Lynch, J. W. (2018). SAHA (Vorinostat) corrects inhibitory synaptic deficits caused by missense epilepsy mutations to the GABAA receptor γ2 subunit. Front. Mol. Neurosci. 11:89. doi: 10.3389/fnmol.2018.00089
Fisher, J. L. (2004). A mutation in the GABAA receptor α1 subunit linked to human epilepsy affects channel gating properties. Neuropharmacology 46, 629–637. doi: 10.1016/j.neuropharm.2003.11.015
Fritschy, J. M., and Mohler, H. (1995). GABAA receptor heterogeneity in the adult rat brain: differential regional and cellular distribution of seven major subunits. J. Comp. Neurol. 359, 154–194. doi: 10.1002/cne.903590111
Frugier, G., Coussen, F., Giraud, M.-F., Odessa, M.-F., Emerit, M. B., Boué-Grabot, E., et al. (2007). A γ2R43Q mutation, linked to epilepsy in humans, alters GABAA receptor assembly and modifies subunit composition on the cell surface. J. Biol. Chem. 282, 3819–3828. doi: 10.1074/jbc.M608910200
Fuchs, C., Coussen, F., Giraud, M.-F., Odessa, M.-F., Emerit, M. B., Boué-Grabot, E., et al. (2013). GABAA receptors can initiate the formation of functional inhibitory GABAergic synapses. Eur. J. Neurosci. 38, 3146–3158. doi: 10.1111/ejn.12331
Gao, Y., and Heldt, S. A. (2016). Enrichment of GABAA receptor α-subunits on the axonal initial segment shows regional differences. Front. Cell. Neurosci. 10:39. doi: 10.3389/fncel.2016.00039
Hernandez, C. C., and Macdonald, R. L. (2019). A structural look at GABAA receptor mutations linked to epilepsy syndromes. Brain Res. 1714, 234–247. doi: 10.1016/j.brainres.2019.03.004
Hutcheon, B., Fritschy, J. M., and Poulter, M. O. (2004). Organization of GABAA receptor α-subunit clustering in the developing rat neocortex and hippocampus. Eur. J. Neurosci. 19, 2475–2487. doi: 10.1111/j.0953-816X.2004.03349.x
Jacob, T. C. (2019). Neurobiology and therapeutic potential of α5-GABA type A receptors. Front. Mol. Neurosci. 12:179. doi: 10.3389/fnmol.2019.00179
Jia, F., Pignataro, L., Schofield, C. M., Yue, M., Harrison, N. L., and Goldstein, P. A. (2005). An extrasynaptic GABAA receptor mediates tonic inhibition in thalamic VB neurons. J. Neurophysiol. 94, 4491–4501. doi: 10.1152/jn.00421.2005
Kasaragod, V. B., and Schindelin, H. (2019). Structure of heteropentameric GABAA receptors and receptor-anchoring properties of gephyrin. Front. Mol. Neurosci. 12:191. doi: 10.3389/fnmol.2019.00191
Keramidas, A., and Harrison, N. L. (2010). The activation mechanism of α1β2γ2S and α3β3γ2S GABAA receptors. J. Gen. Physiol. 135, 59–75. doi: 10.1085/jgp.200910317
Laurie, D. J., Wisden, W., and Seeburg, P. H. (1992). The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain: III. Embryonic and postnatal development. J. Neurosci. 12, 4151–4172. doi: 10.1523/JNEUROSCI.12-11-04151.1992
Leacock, S., Syed, P., James, V. M., Bode, A., Kawakami, K., Keramidas, A., et al. (2018). Structure/function studies of the α4 subunit reveal evolutionary loss of a GlyR subtype involved in startle and escape responses. Front. Mol. Neurosci. 11:23. doi: 10.3389/fnmol.2018.00023
Lu, W., Bromley-Coolidge, S., and Li, J. (2017). Regulation of GABAergic synapse development by postsynaptic membrane proteins. Brain Res. Bull. 129, 30–42. doi: 10.1016/j.brainresbull.2016.07.004
Maljevic, S., Møller, R. S., Reid, C. A., Pérez-Palma, E., Lal, D., May, P., et al. (2019). Spectrum of GABAA receptor variants in epilepsy. Curr. Opin. Neurol. 32, 183–190. doi: 10.1097/WCO.0000000000000657
Marowsky, A., Rudolph, U., Fritschy, J.-M., and Arand, M. (2012). Tonic inhibition in principal cells of the amygdala: a central role for α3 subunit-containing GABAA receptors. J. Neurosci. 32, 8611–8619. doi: 10.1523/JNEUROSCI.4404-11.2012
McKernan, R. M., and Whiting, P. J. (1996). Which GABAA receptor subtypes really occur in the brain? Trends Neurosci. 19, 139–143. doi: 10.1016/s0166-2236(96)80023-3
Niturad, C. E., Lev, D., Kalscheuer, V. M., Charzewska, A., Schubert, J., Lerman-Sagie, T., et al. (2017). Rare GABRA3 variants are associated with epileptic seizures, encephalopathy and dysmorphic features. Brain 140, 2879–2894. doi: 10.1093/brain/awx236
Ochoa, J. G., and Kilgo, W. A. (2016). The role of benzodiazepines in the treatment of epilepsy. Curr. Treat. Options Neurol. 18:18. doi: 10.1007/s11940-016-0401-x
Oh, W. C., Lutzu, S., Castillo, P. E., and Kwon, H.-B. (2016). De novo synaptogenesis induced by GABA in the developing mouse cortex. Science 353, 1037–1040. doi: 10.1126/science.aaf5206
Ono, D., Honma, K.-I., Yanagawa, Y., Yamanaka, A., and Honma, S. (2018). Role of GABA in the regulation of the central circadian clock of the suprachiasmatic nucleus. J. Physiol. Sci. 68, 333–343. doi: 10.1007/s12576-018-0604-x
Ooi, A., Wong, A., Esau, L., Lemtiri-Chlieh, F., and Gehring, C. (2016). A guide to transient expression of membrane proteins in HEK-293 cells for functional characterization. Front. Physiol. 7:300. doi: 10.3389/fphys.2016.00300
Pirker, S., Schwarzer, C., Wieselthaler, A., Sieghart, W., and Sperk, G. (2000). GABAA receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience 101, 815–850. doi: 10.1016/s0306-4522(00)00442-5
Rudolph, U., and Möhler, H. (2014). GABAA receptor subtypes: therapeutic potential in down syndrome, affective disorders, schizophrenia, and autism. Annu. Rev. Pharmacol. Toxicol. 54, 483–507. doi: 10.1146/annurev-pharmtox-011613-135947
Rula, E. Y., Lagrange, A. H., Jacobs, M. M., Hu, N., Macdonald, R. L., and Emeson, R. B. (2008). Developmental modulation of GABAA receptor function by RNA editing. J. Neurosci. 28, 6196–6201. doi: 10.1523/JNEUROSCI.0443-08.2008
Saiepour, L., Fuchs, C., Patrizi, A., Sassoè-Pognetto, M., Harvey, R. J., and Harvey, K. (2010). Complex role of collybistin and gephyrin in GABAA receptor clustering. J. Biol. Chem. 285, 29623–29631. doi: 10.1074/jbc.M110.121368
Schofield, C. M., and Huguenard, J. R. (2007). GABA affinity shapes IPSCs in thalamic nuclei. J. Neurosci. 27, 7954–7962. doi: 10.1523/JNEUROSCI.0377-07.2007
Schofield, C. M., Kleiman-Weiner, M., Rudolph, U., and Huguenard, J. R. (2009). A gain in GABAA receptor synaptic strength in thalamus reduces oscillatory activity and absence seizures. Proc. Natl. Acad. Sci. U S A 106, 7630–7635. doi: 10.1073/pnas.0811326106
Schweizer, C., Balsiger, S., Bluethmann, H., Mansuy, I. M., Fritschy, J.-M., Mohler, H., et al. (2003). The γ2 subunit of GABAA receptors is required for maintenance of receptors at mature synapses. Mol. Cell. Neurosci. 24, 442–450. doi: 10.1016/s1044-7431(03)00202-1
Shi, Y.-W., Zhang, Q., Cai, K., Poliquin, S., Shen, W., Winters, N., et al. (2019). Synaptic clustering differences due to different GABRB3 mutations cause variable epilepsy syndromes. Brain 142, 3028–3044. doi: 10.1093/brain/awz250
Sieghart, W., and Sperk, G. (2002). Subunit composition, distribution and function of GABAA receptor subtypes. Curr. Top. Med. Chem. 2, 795–816. doi: 10.2174/1568026023393507
Smith, R., and Brown, J. (2017). Midazolam for status epilepticus. Aust. Prescr. 40, 23–25. doi: 10.18773/austprescr.2017.005
Wallace, R. H., Marini, C., Petrou, S., Harkin, L. A., Bowser, D. N., Panchal, R. G., et al. (2001). Mutant GABAA receptor γ2-subunit in childhood absence epilepsy and febrile seizures. Nat. Genet. 28, 49–52. doi: 10.1038/ng0501-49
Winsky-Sommerer, R., Knapman, A., Fedele, D. E., Schofield, C. M., Vyazovskiy, V. V., Rudolph, U., et al. (2008). Normal sleep homeostasis and lack of epilepsy phenotype in GABAA receptor α3 subunit-knockout mice. Neuroscience 154, 595–605. doi: 10.1016/j.neuroscience.2008.03.081
Keywords: α3 subunit, GABAA receptor, GABRA3, IPSC, missense mutation, epilepsy
Citation: Syed P, Durisic N, Harvey RJ, Sah P and Lynch JW (2020) Effects of GABAA Receptor α3 Subunit Epilepsy Mutations on Inhibitory Synaptic Signaling. Front. Mol. Neurosci. 13:602559. doi: 10.3389/fnmol.2020.602559
Received: 03 September 2020; Accepted: 02 October 2020;
Published: 20 November 2020.
Edited by:
Daniel F. Gilbert, Bruker Daltonik GmbH, GermanyReviewed by:
Stephan Alexander Pless, University of Copenhagen, DenmarkVolker Eulenburg, University Hospital Leipzig, Germany
Copyright © 2020 Syed, Durisic, Harvey, Sah and Lynch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph W. Lynch, ai5seW5jaEB1cS5lZHUuYXU=