 Bonnie Alberry
Bonnie Alberry Benjamin I. Laufer
Benjamin I. Laufer Eric Chater-Diehl
Eric Chater-Diehl Shiva M. Singh
Shiva M. Singh- 1Department of Biology, Faculty of Science, The University of Western Ontario, London, ON, Canada
- 2Department of Medical Microbiology and Immunology, School of Medicine, University of California, Davis, Davis, CA, United States
- 3Genome Center, University of California, Davis, Davis, CA, United States
- 4MIND Institute, University of California, Davis, Davis, CA, United States
- 5Genetics and Genome Biology, Research Institute, The Hospital for Sick Children, Toronto, ON, Canada
Neurodevelopment in humans is a long, elaborate, and highly coordinated process involving three trimesters of prenatal development followed by decades of postnatal development and maturation. Throughout this period, the brain is highly sensitive and responsive to the external environment, which may provide a range of inputs leading to positive or negative outcomes. Fetal alcohol spectrum disorders (FASD) result from prenatal alcohol exposure (PAE). Although the molecular mechanisms of FASD are not fully characterized, they involve alterations to the regulation of gene expression via epigenetic marks. As in the prenatal stages, the postnatal period of neurodevelopment is also sensitive to environmental inputs. Often this sensitivity is reflected in children facing adverse conditions, such as maternal separation. This exposure to early life stress (ELS) is implicated in the manifestation of various behavioral abnormalities. Most FASD research has focused exclusively on the effect of prenatal ethanol exposure in isolation. Here, we review the research into the effect of prenatal ethanol exposure and ELS, with a focus on the continuum of epigenomic and transcriptomic alterations. Interestingly, a select few experiments have assessed the cumulative effect of prenatal alcohol and postnatal maternal separation stress. Regulatory regions of different sets of genes are affected by both treatments independently, and a unique set of genes are affected by the combination of treatments. Notably, epigenetic and gene expression changes converge at the clustered protocadherin locus and oxidative stress pathway. Functional studies using epigenetic editing may elucidate individual contributions of regulatory regions for hub genes and further profiling efforts may lead to the development of non-invasive methods to identify children at risk. Taken together, the results favor the potential to improve neurodevelopmental outcomes by epigenetic management of children born with FASD using favorable postnatal conditions with or without therapeutic interventions.
Introduction
Human neurodevelopment involves highly orchestrated molecular processes directed by coordinated changes in gene expression. It begins early in ontogeny with the organization of the neural tube and lasts decades. Mammalian neurodevelopment involves a series of events beginning with neurogenesis, followed by cell migration and differentiation, finishing with synaptogenesis and synaptic rearrangement. Neurogenesis is the emergence of neurons from neural progenitor cells in the ventricular zone of what will become the ventricles. Neurogenesis peaks in humans during gestation and continues for years after birth in specific brain regions (Semple et al., 2013). This process is characterized by the accumulation of DNA methylation and histone post-translational modification (PTMs; Lim et al., 2009; Pereira et al., 2010; Wu et al., 2010). Next, the cells must migrate out of this ventricular zone to form the cortex, a process that peaks during early postnatal development (Sanai et al., 2011). Cell migration is mediated by gene expression events, noncoding RNA, and the removal of histone post-translational modifications (Li et al., 2020). Depending on location, the cells will differentiate into neurons, growing axons and dendrites, ultimately forming synapses with other cells. Synapse formation and proper function are also mediated by the removal of histone post-translational modifications and associated regulation of gene expression (Tang et al., 2017). Following competition for neurotrophic factors, apoptotic cell death occurs in neurons that have failed to establish effective communications. Finally, synaptic rearrangement occurs with the formation of new synapses and pruning of inefficient or unused synapses. Synapse formation and pruning begin after 20 gestational weeks and peaks between 8 months and 4 years of age depending on brain region (Lenroot and Giedd, 2006). These processes continue throughout adolescence and into adulthood, albeit at reduced rates. Most notably, maturation of the prefrontal cortex peaks in adolescence and continues for approximately 25 years in humans (Arain et al., 2013). The orchestration of neurodevelopmental cellular processes is sensitive to the prenatal and postnatal environment over time (Tau and Peterson, 2010). The effect of the environment during development has been recognized in many neurodevelopmental disorders. Unfortunately, nearly 18% of children in the United States have developmental disabilities that may be a result of environmental factors (Zablotsky et al., 2019). For example, exposure to early life stress (ELS) via neglect or abuse increases the risk of psychiatric disorders later in life (Kisely et al., 2018). Child maltreatment, including physical abuse, emotional abuse, and neglect is associated with later depression, anxiety, post-traumatic stress disorder (PTSD), drug use, suicide attempts, as well as sexually transmitted infections, and risky sexual behavior (Norman et al., 2012; Kisely et al., 2018).
Many neurodevelopmental disorders have multiple and complex contributors. They result from a combination of biological, psychological, social, and environmental risk factors. A broad range of environmental risks can impact neurodevelopment at different stages. One major environmental contributor is maternal alcohol consumption during pregnancy, which is associated with numerous detrimental outcomes, including stillbirth (Cornman-Homonoff et al., 2012), spontaneous abortion (Kesmodel et al., 2002), premature birth (Sokol et al., 2007), birth defects (O’Leary et al., 2010), and growth delays (Sabra et al., 2018). Specifically, prenatal alcohol exposure (PAE) causes fetal alcohol spectrum disorders (FASD), a common, heterogeneous set of neurodevelopmental disorders that begin in utero, manifest during childhood, and last a lifetime. As with many neurodevelopmental disorders, the stages of neurodevelopment are influenced by epigenetic control and disrupted in FASD. In this review article, we discuss the context of a developmental epigenetic continuum that is vulnerable to environmental impacts contributing to the manifestation of FASD.
Neurodevelopment and Fetal Alcohol Spectrum Disorders (FASD)
FASD is an umbrella term for a collection of impairments, including developmental delays, growth restrictions, physical abnormalities, and behavioral deficits that include intellectual disability (Streissguth and O’Malley, 2000; Sokol et al., 2003; Chudley et al., 2005). FASD manifestation is highly heterogeneous, which may be a result of variable dose and timing, as well as other risk factors. Notably, many individuals with FASD only display behavioral and intellectual disabilities that are not accompanied by any obvious physical abnormalities.
FASD is a serious societal concern. It is the leading preventable cause of intellectual disability in the western world. In Canada, an estimated 10% of pregnant women consume alcohol (Popova et al., 2017), and the prevalence of an FASD diagnosis in Canadian 7 to 9-year-olds is estimated between 2 and 3% (Popova et al., 2018). Similarly, in a cross-sectional study of four communities in the United States, the estimated prevalence of FASD is 1.1–5% (May et al., 2018). Despite societal efforts to raise awareness of the risks, gestational alcohol use persists in North America. Unfortunately, this incidence may have increased in recent years. Of the medical harms identified by a recent increase in alcohol-related emergency room visits, suspected fetal damage including fetal alcohol syndrome rose 2,133.3% between 2003 and 2016 in Ontario, Canada (Myran et al., 2019). Given that this concerning trend may extend to populations beyond Ontario, it does not bode well for a focus on FASD prevention via alcohol avoidance (Alberry and Singh, 2019).
FASD remains a costly societal burden throughout an affected individual’s lifetime. Individuals with FASD often suffer from poor judgment, are easily distracted, and have difficulty perceiving social cues (Streissguth et al., 1991). Additionally, they often have poor academic performance, social deficiencies, intellectual disability, as well as early and repeated delinquency (Fast et al., 1999; Fast and Conry, 2004). Beyond personal consequences, the economic burden of FASD in Canada in 2013, including costs due to productivity losses, the correctional system, and health care, was approximately $1.8 billion (Popova et al., 2016). In a comparison of economic costs between the United States, Canada, Sweden, and New Zealand, the annual mean cost per person is from $12,000 to $68,000 US dollars per individual per year (Greenmyer et al., 2018). Individuals with FASD have high rates of physical and mental health comorbidities, and high rates of substance use (Popova et al., 2021). FASD diagnosis is challenging as there are no reliable biomarkers nor effective treatments. Additionally, many disorders are comorbid with FASD, with attention-deficit/hyperactivity disorder occurring in 50% of people with FASD (Weyrauch et al., 2017). It will require a concerted effort to prevent or attenuate the detrimental effect of PAE on the developing brain.
Children born with FASD are often born in an unstable home environment that may include various early life stressors. Children with FASD often enter childcare systems such as foster care or orphanages (Lange et al., 2013). Children with FASD have high rates of early life adversity and are less likely to live with both biological parents compared to children without FASD (Flannigan et al., 2021). Specifically, in a recent cross-sectional study, 75% of individuals with FASD were involved with the child welfare system, and these individuals are more than four times more lately to also have an anxiety disorder (Popova et al., 2021). Unfortunately, children with PAE often have comorbid prenatal and postnatal risk factors, including drug exposures and nutritional deficiencies. In a Canadian report of children with confirmed PAE, 95% had other prenatal substance exposure, with 61% experiencing deprivation as a failure to meet basic needs before 2 years old (Lebel et al., 2019). In a study from Washington State, 74% of children with confirmed PAE were not living with their birth parents, with over four out-of-home placements on average per child (Hemingway et al., 2020). In a recent study from one national medical adoption unit in Israel, 20.2% of children had a known history of PAE, and of these just 22.2% had no discernable abnormalities (Tenenbaum et al., 2020). Furthermore, many children without a known PAE history otherwise fit the FASD diagnostic criteria, supporting the notion that FASD is underdiagnosed.
Cumulative Impacts: Prenatal Alcohol Exposure (PAE) Followed by Early Life Stress (ELS)
Little is known about how ELS negatively impacts children born with FASD (Price et al., 2017). Following PAE and an ELS, such as abuse or neglect during early development, children are more likely to have impaired speech (Coggins et al., 2007), as well as behavioral deficits including impaired memory and attention (Henry et al., 2007; Koponen et al., 2009, 2013). More recently, research has highlighted how common mental health problems are in youth with PAE. While an FASD diagnosis is associated with increased risk for the development of comorbid neurodevelopmental disorders, additional adverse childhood events further increased the rates of neurodevelopmental disorders in people born with FASD (Kambeitz et al., 2019). PAE alone does not sufficiently predict the rate of later disorders and youth with adverse childhood experiences, specifically out-of-home care, have the highest risk of mental health problems (Koponen et al., 2020). Additionally, children with PAE and subsequent postnatal adversity have different brain structures and symptom profiles than children without added postnatal adversity (Andre et al., 2020). Together, these reports highlight the importance of the postnatal environment in the manifestation of FASD. The postnatal environment may improve or worsen the outcome of a child born with FASD but remains to be established. Research involving humans is challenging and often prohibitive. As a result, animal models have become essential tools for characterizing these environmental impacts on neurodevelopment.
Modeling FASD Including ELS in Rodents
While animal models are a useful tool to explore molecular mechanisms involved in the development of FASD, few have considered the postnatal environment as a modulator of outcome severity. Given the rate of ELS in individuals with FASD, research is now focused on this interaction. The interaction between PAE and postnatal stress has been explored at different age equivalents, including adolescence (Comeau et al., 2014), and adulthood (Hellemans et al., 2008; Uban et al., 2013; Gangisetty et al., 2014; Lan et al., 2015). Our model of PAE and maternal separation stress found sex-dependent changes in behavior following the combination of treatments, with both treatments resulting in changes in activity and learning deficits (Alberry and Singh, 2016). In a novel open field environment, females are more active than males, but not following PAE. There was also increased exploratory behavior in the open field following PAE. Further, PAE and maternal separation stress both result in overnight hypoactivity in a home cage environment, as well as sex-dependent learning deficits (Alberry and Singh, 2016). In a similar rat model from another group, only the combination of PAE and postnatal stress was sufficient to alter anxiety-like behavior (Biggio et al., 2018). In a separate rat model including PAE and ELS, there is altered social recognition memory and hypothalamic neuropeptide expression (Holman et al., 2021).
ELS reduces serum cytokine levels in control rats, but not following PAE (Raineki et al., 2017). Also, serum levels of C-reactive protein, a marker of inflammation, are higher following PAE and higher still with the combination of PAE and ELS. In the amygdala, PAE reduced CXCL1 and IL-10, further emphasizing the potential role of an immune response in FASD (Raineki et al., 2017). Similarly, in our FASD mouse model that includes ELS, changes in hippocampal gene expression are associated with anxiety-like behavior and treatment, specifically genes important for transcriptional regulation and neurodevelopment (Alberry et al., 2020). In a rat model, only the combination of PAE and ELS results in decreased allopregnanolone, a neuroactive steroid (Biggio et al., 2018). While foot shock stress increases plasma allopregnanolone and corticosterone, this response is exaggerated in animals following PAE and ELS (Biggio et al., 2018). The results discussed here are summarized in Table 1, where models of FASD that include ELS differ in rodent species and strains, exposure paradigms, postnatal stressors, sex, timing of assessment, behaviors studied, and molecular features evaluated. The experimental differences may account for the discrepancies in outcomes described in the literature. The gene expression changes following PAE and ELS accompany comprehensive demonstration that different regimes of PAE result in genome-wide changes in brain gene expression in rodents at different stages of neurodevelopment (Kleiber et al., 2012, 2013; Mantha et al., 2014; Gangisetty et al., 2015; Chastain et al., 2019; Lucia et al., 2019; Ieraci and Herrera, 2020; Shivakumar et al., 2020). As gene expression changes likely underlie changes in behavior, the focus becomes on mechanisms of gene expression regulation throughout neurodevelopment.
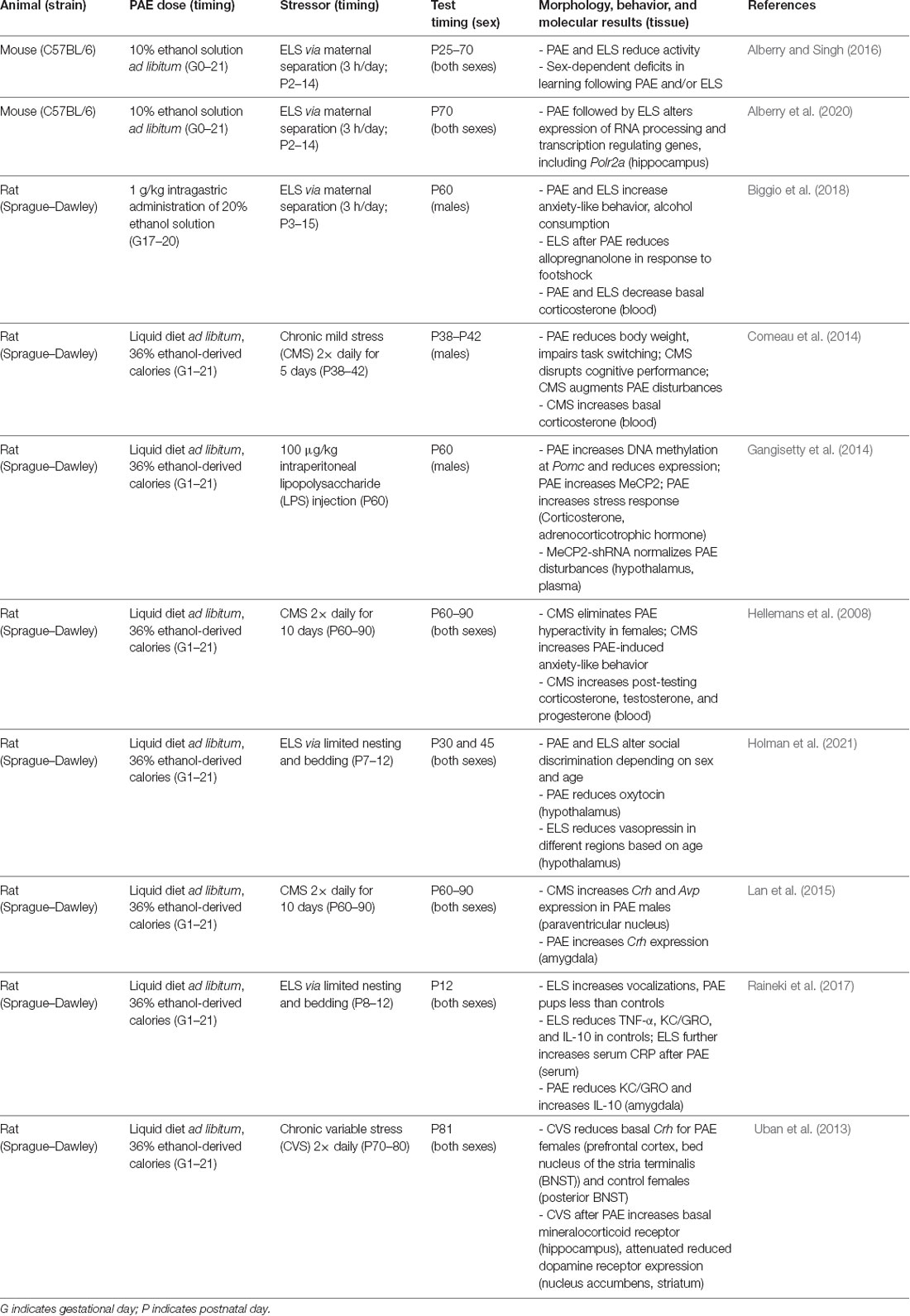
Table 1. A summary of rodent models of FASD that include PAE as well as a postnatal stressor in the literature.
Epigenetics: The Interface Between Genome and Environment
The epigenome plays an essential role in mammalian neurodevelopment by regulating the expression of genes required for development and tissue specificity. Epigenetic marks act in concert to regulate chromatin structure, which consists mainly of DNA and associated proteins. Epigenetic marks that regulate transcription include the covalent modification of DNA in the form of DNA cytosine methylation, the PTM of histone protein tails, and non-coding RNAs (ncRNA) that can serve as scaffolds between DNA and proteins. Also included in the scope of the epigenome are ncRNAs that act at the post-transcriptional level to interfere with the translation of mRNA into proteins, such as microRNAs (miRNAs).
The epigenome is dynamic and functions as a responsive interface between the environment (internal or external) and the genome. It enables prenatal and postnatal events to shape neurodevelopment and related behavioral endophenotypes. At each stage of neurodevelopment, epigenetic players are at work. Neurogenesis relies on intergenic de novo DNA methylation by DNMT3A in neural stem cells to maintain active chromatin states for essential genes (Wu et al., 2010). TET1 deficiency impairs neurogenesis and leads to the downregulation of neural progenitor proliferation genes (Zhang et al., 2013), indicating the importance of the TET family of enzymes responsible for DNA demethylation. Histone modifications also contribute to neurogenesis, given that a knockout of the H3K4me3 methyltransferase, Mll1, fails to activate the neurogenesis regulator Dlx2 (Lim et al., 2009). Similarly, the histone methyltransferase of polycomb repressive complex 2, EZH2, is critical for maintaining the timing and number of cells produced from progenitor cells (Pereira et al., 2010). Cell migration is regulated by long ncRNA (lncRNA) via cis-activation of the lysine demethylase Kdm2b, a critical factor in the migration of cortical projection neurons (Li et al., 2020). The histone H3K27 demethylase, lysine demethylase 6a (Kdm6a), is critical for appropriate synapse formation and transmission through the regulation of genes such as the neurotransmitter receptor Htr5b (Tang et al., 2017).
Importantly, many changes in expression following PAE occur for genes with key epigenetic roles, which include proteins important for reading, writing, and erasing epigenetic modifications as well as related ncRNAs. Here, we discuss epigenetic results involving ncRNA, DNA methylation, and histone PTMs in the context of modeling FASD. The results from models discussed in this section are summarized in Table 2, where animal models of FASD differ in species and strains, exposure paradigms, sex, timing of assessment, and molecular features evaluated. These variations may account for differences reported in the literature.
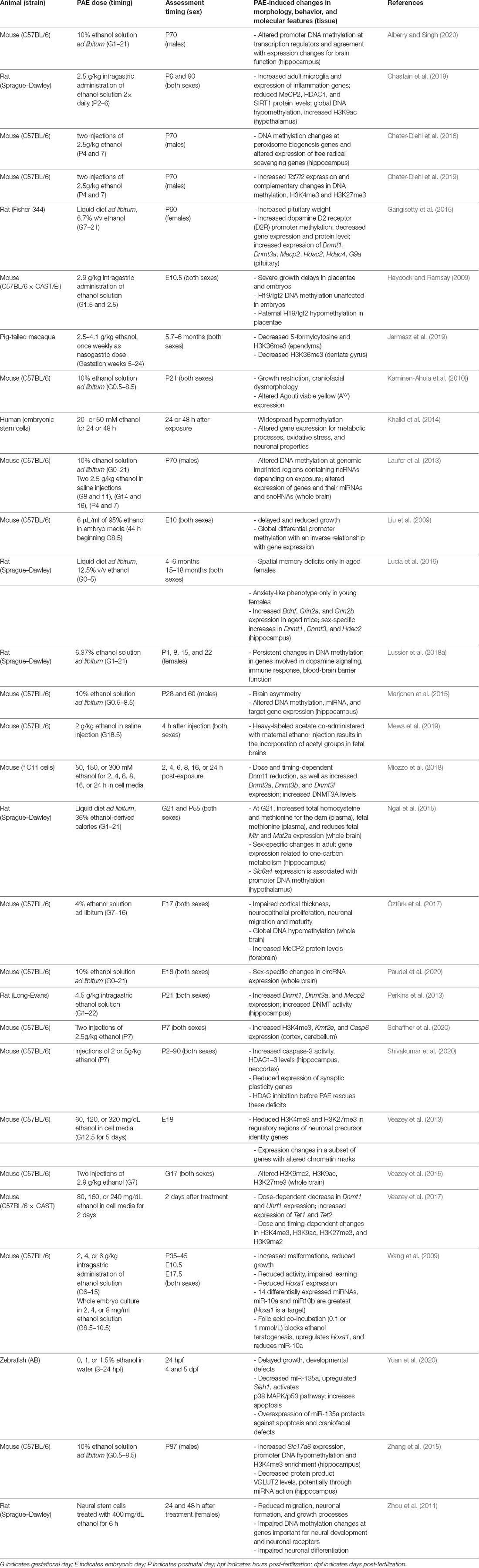
Table 2. Summary of animal models of FASD that investigate miRNAs, DNA methylation, and histone post-translational modifications in the literature.
Noncoding RNA
Cellular phenotypic outcomes are often the result of inter-related epigenetic mechanisms. For example, noncoding RNAs are important for gene expression regulation at the transcriptional and post-transcriptional levels. They include long non-coding RNAs (lncRNAs; Lee, 2012), microRNAs (miRNAs; Chuang and Jones, 2007), short interfering RNAs (siRNAs), and piwi-interacting RNAs (piRNAs; Holoch and Moazed, 2015). Individual miRNAs also have critical roles in neurodevelopment, particularly for neurogenesis and cell fate specification (Coolen and Bally-Cuif, 2009). More specifically, MeCP2-regulated miRNAs influence neurogenesis and neuronal migration (Mellios et al., 2018). Specifically, in the context of this review, many ncRNAs are environmentally responsive. Interestingly. Circular RNAs (circRNAs) have also been implicated as differentially expressed in the brain in a PAE rat model (Paudel et al., 2020).
The noncoding RNA research in FASD has been focused on the relationship between microRNAs and their mRNA gene targets. Following PAE, genomically imprinted clusters of miRNAs and small nucleolar RNAs (snoRNAs) are deregulated in the adult mouse brain (Laufer et al., 2013). The miRNAs and their predicted mRNA targets are reciprocally differentially expressed and associated with neurodevelopmental events. In a trimester one-equivalent exposure model, microRNAs important for cell state and dendritic spines in the hippocampus are differentially expressed following PAE (Marjonen et al., 2015). In a rat model, third trimester-equivalent ethanol exposure results in increased variance in miRNA expression and increased expression of miR-200c, a miRNA important for neurogenesis (Balaraman et al., 2017). In zebrafish, miR-135a is downregulated following ethanol treatment, and its overexpression reduces the associated apoptosis in neural crest cells, growth restrictions, and craniofacial defects (Yuan et al., 2020).
Profiling of miRNA following PAE also holds translational potential. In a recent rat model of PAE, the composition of exosomal RNA in the amniotic fluid was altered following maternal ethanol exposure, with some putative functional roles for differentially expressed miRNAs on stem cell regulation (Tavanasefat et al., 2020). Also, infant circulating extracellular miRNA are predictive of PAE-induced growth restriction and cognitive impairment (Mahnke et al., 2021). Finally, differential miRNA and target gene expression resulting from PAE can be reversed by folic acid supplementation, which is notable given that folic acid is involved in establishing DNA methylation (Wang et al., 2009).
DNA Methylation
DNA cytosine methylation is a strong candidate for a biological mechanism of action for PAE because ethanol impairs folate transport to the developing fetus (Hutson et al., 2012), a key methyl donor important for DNA methylation. PAE in a mouse model results in changes in expression of the DNA methylation-dependent metastable epiallele, Agouti viable yellow (Avy), indicating that DNA methylation changes occur after PAE (Kaminen-Ahola et al., 2010).
In several studies, PAE alters the expression of DNA methyltransferases, the writers of DNA methylation. In a rat model, PAE results in increased gene expression of DNA methyltransferases, Dnmt1 and Dnmt3a, alongside increased DNMT protein activity (Perkins et al., 2013). In a murine cell culture model, there is a dose-dependent increase in Dnmt3a, Dnmt3b, and Dnmt3l expression, with increased DNMT3A protein abundance (Miozzo et al., 2018). While others have found dose-dependent decreased expression of Dnmt1 (Veazey et al., 2017). Moreover, increased gene expression of Dnmt1 and Dnmt3a in male rat hippocampus following early PAE (Lucia et al., 2019) or continuous PAE (Gangisetty et al., 2015) has been observed.
Other DNA methylation genes have been implicated in FASD. In rodent models, PAE results in increased expression of the DNA methylation reader methyl CpG binding protein 2 (Mecp2) at both the gene (Perkins et al., 2013; Gangisetty et al., 2015) and protein level (Öztürk et al., 2017; Chastain et al., 2019). In a mouse embryonic stem cell model, increased expression of DNA methylation erasers (demethylation genes) Tet1 and Tet2 occurs following ethanol treatment (Veazey et al., 2017).
PAE is also known to alter methylation at genes that undergo genomic imprinting, which typically have distinct DNA methylation profiles at regulatory elements for each allele that allow for a parent-of-origin specific gene expression. Several studies regarding DNA methylation have focused attention on a specific imprinted locus in the genome, insulin-like growth factor 2, IGF2/H19. H19 is a long noncoding RNA only transcribed from the maternal allele, while the paternal H19 is normally methylated at the promoter and not expressed. Paternal H19 allele methylation allows for the expression of paternal IGF2, which is not typically maternally expressed (DeChiara et al., 1991). An examination of genome-wide promoter DNA methylation identified hypermethylation at this locus in adult mouse brains following PAE via continuous preference drinking (Laufer et al., 2013). Also, targeted bisulfite sequencing of this locus in placentae revealed hypomethylation of the paternal allele in mid-gestation mouse hybrid embryos following pre-implantation alcohol exposure (Haycock and Ramsay, 2009). Furthermore, a genome-wide assay of CpG methylation identified hypomethylation at this locus in the saliva of young children born with FASD (Portales-Casamar et al., 2016). In addition, a polymorphism at this locus is associated with placental DNA methylation and head circumference following PAE (Marjonen et al., 2017). The differences between epigenetic alterations at this locus may be attributed to variations in study design across experiments.
Gene promoter DNA methylation changes following ethanol exposure have been reported in several models. Following ethanol treatment of rat neural stem cells, there is promoter hypermethylation of the chromatin remodeling complex member SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily A, member 2 (Smarca2), together with adhesion and polarity genes DiGeorge syndrome critical region gene 2 (Dgcr2), and Par-6 family cell polarity regulator alpha (Pard6a, Zhou et al., 2011). Conversely, transcription factors cut like homeobox 2 (Cux2), POU class 4 homeobox 3 (Pou4f3), and SRY-box transcription factor 7 (Sox7), are each hypomethylated following ethanol treatment (Zhou et al., 2011). Each of these genes is also critical for neurodevelopment. In a trimester one-equivalent model, there are corresponding changes in DNA methylation status at candidate genes with altered gene expression in the hippocampus, but also in peripheral tissue (Marjonen et al., 2015). Similarly, decreased hypothalamic serotonin transporter (Slc6a4) mRNA following PAE was accompanied by increased promoter methylation (Ngai et al., 2015), and increased vesicular glutamate transporter (Slc17a6) alongside decreased promoter methylation (Zhang et al., 2015). Binge-like trimester three-equivalent ethanol exposure results in altered promoter DNA methylation, particularly for oxidative stress and peroxisome biogenesis genes (Chater-Diehl et al., 2016).
In our mouse model of continuous PAE, we found altered promoter methylation at genes important for transcriptional regulation. Specifically, PAE led to decreased promoter methylation of polycomb repressive complex 1 members, including chromobox 4 and 8 (Cbx4, Cbx8) polycomb group ring finger 6 (Pcgf6), and ring finger protein 1 (Ring1), as well as transcription factor II D complex members general transcription factor II A, 2 (Gtf2a2), and TATA-box binding protein associated factors 8, 10, and 11 (Taf8, Taf10, Taf11; Alberry and Singh, 2020). Additionally, there is decreased promoter methylation of nine members of the RNA polymerase II transcription factor complex, including the aforementioned Gtf2a2, Taf8, Taf10, Taf11, as well as pygopus 2 (Pygo2), nuclear transcription factor-Y gamma (Nfyc), peroxisome proliferator activator receptor delta (Ppard), and nuclear receptor subfamily 1, group H, member 4 and group D, member 2 (Nr1h4, Nr1d2; Alberry and Singh, 2020).
Global changes and regional genome-wide differences in DNA methylation following PAE have repeatedly been reported in rodents (Liu et al., 2009; Laufer et al., 2013; Öztürk et al., 2017; Lussier et al., 2018a), human embryonic stem cells (Khalid et al., 2014), and nonhuman primate brain (Jarmasz et al., 2019). In human post-mortem brain samples, reduced DNA methylation in the CA1 region of the hippocampus is associated with PAE (Jarmasz et al., 2019).
In terms of translation potential, research aimed at finding a specific DNA methylation signature in peripheral tissue for FASD has had limited success identifying sites that discriminate children with an FASD diagnosis from those without (Laufer et al., 2015; Portales-Casamar et al., 2016; Lussier et al., 2018b). Also, DNA methylation profiling of blood from children with FASD was able to identify sites related to FASD sub-phenotypes (Cobben et al., 2019), although a strong profile was not observed in cord blood (Sharp et al., 2018). While not yet specific or accurate enough for diagnosis likely due to a combination of effects from genotype, the tissue of origin, and other exposures, these studies suggest the potential for future biomarker discovery. As detection technology and knowledge of DNA methylation states and cofactors improve, there will be improvements in the precision of biomarker detection. Interestingly, while research has established genome-wide DNA methylation profiles as epigenetic clocks for biological aging, there appears to be accelerated epigenetic aging in individuals with FASD (Okazaki et al., 2020).
Using a mouse model combining PAE and ELS, we found different sets of genes with altered promoter methylation following prenatal ethanol exposure, postnatal maternal separation stress, and the combination of treatments (Alberry and Singh, 2020). The combination of PAE and ELS results in decreased promoter methylation of genes critical for neuronal migration, including sema domain, transmembrane domain (TM), and cytoplasmic domain, (semaphorin) 6A (Sema6a), unc-51-like kinase 4 (Ulk4), disabled 2 interacting protein (Dab2ip), and rho/rac guanine nucleotide exchange factor (GEF) 2 (Arhgef2). Conversely, the combination of PAE and ELS led to increased promoter methylation of genes important for immune response, including 21 members of the cytokine-cytokine receptor interaction KEGG pathway (Alberry and Singh, 2020). While there is a minimal correspondence between promoter DNA methylation and gene expression, genes involved are critical for brain development and function. These results argue that while promoter methylation and reciprocal alterations to gene expression persist following PAE and ELS, the mechanisms are complex. Gene expression is representative of transient states occurring at that time, while DNA methylation is also reflective of past events and the priming of future ones. Furthermore, some gene expression profiles require activation by specific events. Finally, this discrepancy also suggests that other epigenetic modifications contribute to the lasting changes in gene expression and ultimately the behavioral deficits that occur in FASD, and these include histone PTMs.
Histone Post-translational Modifications (PTMs)
Histone PTMs are another strong candidate for the mechanism of action following PAE (Chater-Diehl et al., 2017). Modifications to histone tails at specific genomic loci are the foundation of epigenetic mechanisms, with a wide variety of known potential modifications (Allis and Jenuwein, 2016). While modifications can include methylation that is dependent on the same methyl source as DNA methylation, histone acetylation is another common modification. In pregnant mice, exposure to isotope-labeled ethanol results in labeled acetyl groups being incorporated as histone acetylation in the gestating fetal brains (Mews et al., 2019).
PAE is often associated with altered expression of histone modifiers, including deacetylases and methyltransferases, and genome-wide changes in the abundance of modifications. Early embryonic PAE leads to increased Hdac2 expression in rat hippocampus (Lucia et al., 2019), and continuous PAE results in its increase in female pituitary along with increased Hdac4 and G9a (Gangisetty et al., 2015). Binge-like trimester three-equivalent PAE in rats leads to increased H3 lysine 9 acetylation (H3K9ac) and decreased protein levels of HDAC1 and SIRT1 in the hypothalamus (Chastain et al., 2019), alongside changes in other modifications that may correlate with the phenotype (Veazey et al., 2015). In mice, PAE is associated with increased H3 lysine 4 trimethylation (H3K4me3) levels and increased expression of the chromatin modification gene lysine (K)-specific methyltransferase 2E, Kmt2e (Schaffner et al., 2020). It also leads to increased levels of chromatin modifiers, including histone deacetylases (HDAC1-3), and reduced histone 3 (H3) and histone 4 (H4) acetylation in the hippocampus and neocortex (Shivakumar et al., 2020). Furthermore, decreased hippocampal and temporal lobe ependyma H3K36me3 has been observed in a nonhuman primate model of FASD (Jarmasz et al., 2019). In human post-mortem brain samples, PAE is associated with region-specific decreases in H3K4me3, H3K9ac, H3K27ac, H4K12ac, and H4K16ac (Jarmasz et al., 2019).
Altered histone PTMs at specific genes are regularly reported in the literature. In a neurosphere cell culture model, Homeobox genes important for neurogenesis have altered histone H3K4 or H3K27 trimethylation following ethanol treatment (Veazey et al., 2013). While there are often changes in histone PTMs at specific genes, these do not regularly translate into altered expression (Veazey et al., 2017). Following binge-like trimester three-equivalent ethanol exposure, many genes have been implicated by regional differences in H3K4 and H3K27 methylation, particularly increased H3K4 methylation and decreased H3K27 methylation (Chater-Diehl et al., 2016). Interestingly, the Wnt transcription factor Tcf7l2 is differentially expressed and also has complementary changes in DNA methylation and histone modifications in this model (Chater-Diehl et al., 2016, 2019). These results suggest changes in gene-specific expression in response to PAE may be the result of a variety of epigenetic mechanisms. Although DNA methylation, non-coding RNAs, and histone PTMs have all been implicated, their inter-relationship is not known. The results summarized however emphasize the effect of alcohol on neurodevelopment is likely realized by a combination of epigenetic alterations. Some of these effects are genome-wide, while others represent relatively focused effects. As examples, we will discuss two specific observations involving clustered protocadherins and genes involved in oxidative stress.
Clustered Protocadherins in FASD and ELS
Protocadherins are a group of cell-cell adhesion molecules that are highly expressed in the brain and are involved in establishing single-cell neuronal identity. Many of these protocadherins are encoded in a large genomic locus that consists of three multi-gene clusters (Chen and Maniatis, 2013). Epigenetic deregulation of this locus in humans has been implicated in numerous diseases and disorders, including following PAE and ELS (El Hajj et al., 2017). Our group was the first to identify a PAE-associated profile of DNA promoter hypermethylation at the clustered protocadherin locus, which was observed in both the brains of adult mice following PAE via continuous preference drinking, as well as the buccal swabs of children born with FASD (Laufer et al., 2015). This result has been replicated in the saliva of children with FASD (Portales-Casamar et al., 2016; Lussier et al., 2018b). However, these studies have noted variation in the specific differentially methylated CpG sites within the locus. This observed variation in single CpG sites across experiments is in line with the stochastic nature of methylation establishment at this locus (Toyoda et al., 2014; Canzio et al., 2019) and highlights that this profile is apparent the level of larger differentially methylated regions rather than single CpG sites. Other factors, such as genetic background, sex, and age, may also contribute to this profile. Additionally, the differences between studies may be related to the nature of cellular content differences, as either buccal epithelial swabs (Laufer et al., 2015) or saliva were assayed (Portales-Casamar et al., 2016; Lussier et al., 2018b). Both sample sources are heterogeneous in that they contain epithelial cells and blood leukocytes, however, buccal swabs have a higher proportion of epithelial cells than saliva samples (Theda et al., 2018). This difference in the cellular composition of oral sample sources may explain some of the differences between studies, particularly when considering the mechanism that results in clustered protocadherin hypermethylation. We hypothesize that the clustered protocadherin hypermethylation profile observed in oral sample sources is from buccal epithelial cells given that both buccal epithelial and brain cells are derived from the ectoderm during development, which creates the potential for buccal epithelial cells to contain epigenetic “footprints” of early neurodevelopmental disruptions (Smith et al., 2015). This hypothesis is supported by our observations in the hippocampus of adult mice following binge ethanol exposure in the trimester three equivalent period. Following PAE, promoters in the clustered protocadherin locus are hypermethylated and have decreased levels of H3K4me3 and H3K27me3 (Chater-Diehl et al., 2016). This finding is notable given that these two histone PTMs make up bivalent chromatin, which plays a key role in development (Bernstein et al., 2006). Together, these results suggest a putative mechanism, where bivalent chromatin is not properly established during neurodevelopment. As a result, DNA methyltransferase 3L (DNMT3L) can recognize the unmodified histone H3 (H3K4me0) and stimulate DNA methyltransferase 3A (DNMT3A) to methylate the locus (Ooi et al., 2007; Otani et al., 2009; Zhang et al., 2010). This action is expected to decrease gene expression (Figure 1), which is a general trend observed across diverse exposure paradigms. This includes from adult mouse whole brain following a trimester 3 equivalent binge-like exposure (Kleiber et al., 2013), and more recently in adult mouse hippocampus following continuous PAE (Alberry et al., 2020). This is exemplified in adult mouse brains from our continuous preference drinking paradigm, where PAE resulted in increased methylation (p < 0.01) at the Pcdhb2 promoter and a corresponding decrease in gene expression (fold change = −1.23, p < 0.05; Laufer et al., 2013).
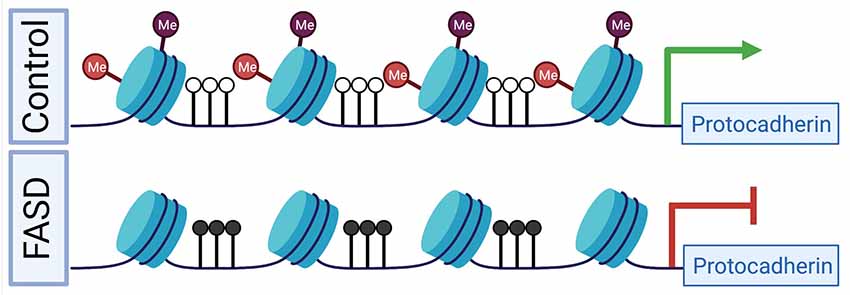
Figure 1. A putative mechanism for differential epigenetic and transcriptomic profiles at the clustered protocadherin locus in fetal alcohol spectrum disorders (FASD). During development in healthy controls, the locus is marked by bivalent histone PTMs, which prevent DNA hypermethylation and poises genes for high expression levels. In FASD, prenatal alcohol exposure (PAE) results in the depletion of bivalent histone PTMs, which leads to DNA hypermethylation and reduced gene expression. Red lollipops represent H3K27me3, purple lollipops represent H3K4me3, black lollipops represent DNA hypermethylation, and white lollipops represent DNA hypomethylation. The green arrow represents normal gene expression, while the red inhibitor shape indicated reduced gene expression. Figure 1 was created with BioRender.com.
The clustered protocadherins have also been implicated in ELS, where a comparison of the hippocampal tissue from individuals that have experienced abuse as children and rats that received low levels of maternal care revealed DNA promoter hypermethylation and gene body DNA hypomethylation at the locus, corresponding with a repressive effect (Suderman et al., 2012). An integrative analysis of this broad signature of differential DNA methylation in adult hippocampal tissue from rats revealed that low levels of maternal care were also associated with H3K9ac (a histone PTM associated with transcriptional activation) and gene expression at the clustered protocadherins (McGowan et al., 2011).
In an FASD model that includes PAE and ELS in adult mouse hippocampus both hyper- and hypo-methylation in the protocadherin region as well as decreased gene expression resulted from PAE (Alberry and Singh, 2020; Alberry et al., 2020). ELS alone and the combination of PAE and ELS resulted in hypermethylation of the locus and decreased expression of a transcript (Alberry and Singh, 2020; Alberry et al., 2020). Interestingly, transcripts from this locus were within a gene co-expression module that correlates with treatment and anxiety-like behavior (Alberry et al., 2020). Taken together, these results suggest that the clustered protocadherins are sensitive to PAE, ELS, and the cumulative impact of the two treatments. Ultimately, the alterations to epigenomic and transcriptomic profiles of the clustered protocadherin locus likely lead to changes in neuronal identity and synaptic connections, as well as disrupt neural circuits during neurodevelopment.
Oxidative Stress Pathways in FASD and ELS
Oxidative stress is a well-characterized part of FASD etiology. At the cellular level, ethanol acts directly on mitochondria to produce superoxide, hydroxide, and nitric oxide radicals (Wu and Cederbaum, 2003). Metabolism of ethanol produces oxidized products (Mansouri et al., 2001) and acetaldehyde in the brain, further increasing the formation of reactive oxygen species (Shaw, 1989). Oxidative damage can lead to blood-brain barrier impairment, inflammation, and increased apoptosis (Haorah et al., 2008), key features of FASD etiology (Barak et al., 1996). Importantly, increased oxidative stress is also a key feature of ELS. Studies have found altered oxidative stress in adult male mice following early maternal separation (Malcon et al., 2020) and in plasma of human adolescents reporting childhood maltreatment (do Prado et al., 2016). Lasting oxidative damage to the brain occurs in mice exposed to maternal separation (Réus et al., 2017). Interestingly, treatment with voluntary exercise in adolescence leads to some recovery of mitochondrial function after maternal separation in rats (Sahafi et al., 2018). At the gene expression and epigenetic levels, oxidative stress genes are often implicated after PAE. We found altered expression, DNA methylation, and histone PTMs at oxidative stress and free radical scavenging genes in adult mouse hippocampus following trimester three equivalent ethanol exposure (Chater-Diehl et al., 2016). Other groups have also identified similar results. An integrative analysis of DNA methylation, miRNA expression, and gene expression from rats that received augmented maternal care demonstrated that key oxidative stress genes were affected (Vogel Ciernia et al., 2018). Taken together, these studies show that oxidative stress is a key component of both PAE and ELS, which is also reflected in transcriptomic and epigenomic studies. Targeting such mechanisms may be a promising avenue for therapeutic strategies in the future.
Light at The End of The Tunnel
FASD is a common neurodevelopmental disorder that represents a societal burden with no effective treatments. Yet, it is entirely preventable. It requires the avoidance of alcohol before and during pregnancy. This is a challenging proposition given that the use of alcohol in reproductive-age women is increasing and many pregnancies are unintended, which is expected to lead to a corresponding increase in FASD (Alberry and Singh, 2019). Under the circumstances, there is a need to better understand the molecular mechanisms involved and develop novel treatment modalities. The research discussed in this review argues that multiple epigenetic mechanisms are involved in the initiation, progression, and maintenance of aberrant phenotypes associated with FASD. Epigenetic mechanisms are dynamic and operational during the neurodevelopmental continuum spanning prenatal and postnatal periods (Figure 2). It offers the potential to reverse the outcome via epigenetic manipulation.
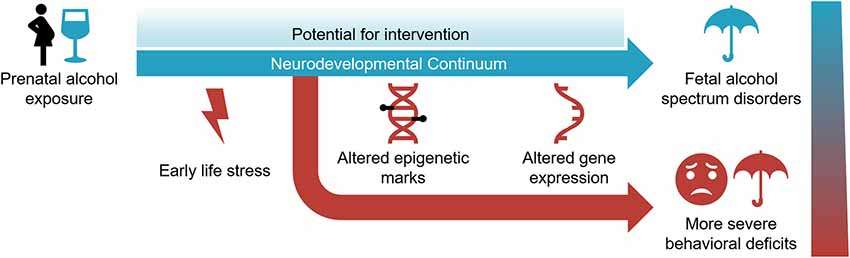
Figure 2. The neurodevelopmental continuum is susceptible to PAE and early life stress (ELS) in the development and severity of fetal alcohol spectrum disorder. Epigenetic modifications and gene expression are affected by these environmental exposures. While it represents a sensitive period for environmental assaults, the dynamic nature of the neurodevelopmental continuum also offers potential for intervention.
To this end, research regarding the prevention of FASD has centered on increasing the availability of methyl donors during development through supplementation of known methyl donors such as folate and choline. In an embryonic cell culture model, co-administration of folic acid prevents the morphological delays and deficits, together with the downregulation of Hoxa1 and its miRNA, miR-10a (Wang et al., 2009). Where PAE results in malformations, offspring of pregnant mice on a diet supplemented with choline, betaine, folic acid, vitamin B12, L-methionine, and zinc, followed by PAE have fewer malformations than those with PAE alone (Downing et al., 2011). Gestational choline supplementation in a rat model reduces ethanol-associated alterations in gene expression and protein levels of DNA methylation genes Dnmt1 and Mecp2 in the hypothalamus (Bekdash et al., 2013). Similarly, choline supplementation prevents the ethanol-associated changes in gene expression of histone methylation genes Set7/9, G9a, and Setdb1, as well as the detected changes in histone PTMs, H3K4me2, 3 and H3K9me2 (Bekdash et al., 2013). Also, choline supplementation blocks the ethanol-associated increase in POMC promoter methylation and corresponding decreased gene expression in the hypothalamus (Bekdash et al., 2013). Betaine supplementation during embryonic ethanol exposure in an avian embryo model reduces malformation defects observed following ethanol alone (Karunamuni et al., 2017). Following PAE, choline supplementation and working memory training in adolescent rats sufficiently improve the cognitive flexibility and connectivity deficits measured in adulthood (Waddell et al., 2020). Choline supplementation as an intervention for women drinking during pregnancy has been minimally investigated. However, choline-supplemented infants have improved weight and head circumference at 6.5 and 12 months, in addition to better visual recognition memory compared to placebo infants (Jacobson et al., 2018). In a trial of choline administration in 2.5- to 5-year-olds with FASD, children who receive 500 mg of choline daily for 9 months have higher non-verbal intelligence, visual-spatial skills, working memory, verbal memory, and fewer symptoms associated with ADHD than the placebo group when assessed 4 years later (Wozniak et al., 2020). While the methyl-donor supplementation hypothesis has gained support, the precise mechanism of action is still unknown. Still, it remains an exciting avenue for potential prevention of FASD-related deficits following PAE. However, it remains an exciting avenue for potential prevention of FASD-related deficits following PAE. Further understanding of how the postnatal environment may modulate FASD-related deficits will come from animal models. In one study, decreased dendritic complexity in the hippocampus associated with binge-like trimester three-equivalent exposure was avoided by exercise and environmental complexity (Boschen et al., 2017). Such results on epigenetic interventions are encouraging for most neurodevelopmental disorders that result from environmental exposures. This optimism must be tempered with the observation that for most of these studies, a few metrics show improvement but many outcomes of PAE remain. This is likely due to structural changes occurring in the brain following neuronal apoptosis that are not possible to rectify by any manner of available postnatal treatments. This is not to negate the positive outcomes of these studies but should focus our attention on the prenatal fetal environment for treatments during this time.
Further consideration should also be paid to the types of experiments used in FASD research. To date, most studies have used bulk tissue or whole embryo inputs for molecular experiments. These have provided many valuable insights to this point, however, given the heterogeneity of the effects of PAE and preferential apoptosis of specific cell types, more focused studies are needed. Specifically, single-cell epigenetic and transcriptomic techniques are now viable for most FASD models. Single-cell or single-cell-type resolution may address many outstanding questions about the outcomes of ethanol exposure at the molecular level. Furthermore, single-cell studies and improved bioinformatic cellular deconvolution methods will aid future translational studies examining the DNA methylation signature of FASD in saliva (Turinsky et al., 2019) and other non-invasively obtained samples.
Conclusion
Neurodevelopment in mammals is a long-lasting, highly orchestrated, and coordinated continuum. It is highly sensitive and responsive to environmental exposures that may determine the outcome and resulting brain function, including neurodevelopmental disorders such as FASD. FASD is highly heterogeneous and a direct result of maternal alcohol consumption during pregnancy. Further, a stressful postnatal environment may worsen the outcomes. This review emphasizes the role of dynamic epigenetic changes during the neurodevelopmental continuum, from prenatal to postnatal stages in the development of FASD, including ncRNAs, DNA methylation, and histone PTMs. We argue that FASD is an epigenetic disorder. Additionally, the early postnatal environment may determine the severity of the effect of PAE. It may be possible to moderate this effect by postnatal epigenetic modifications, which may or may not be driven by pharmaceutical interventions. This perspective offers new hope for the development of effective treatment for FASD. Finally, the strategy of epigenetic intervention discussed here may apply to a variety of neurodevelopmental disorders involving epigenetic marks caused by environmental exposures.
Author Contributions
All authors contributed to the design and organization of this review. All authors performed a literature review, as well as interpretation and synthesis of findings. BA created the first draft, with all authors substantially contributing to revisions. All authors have approved the final revisions prior to submission. All authors contributed to the article and approved the submitted version.
Funding
We wish to acknowledge support from the Natural Sciences and Engineering Research Council (NSERC) of Canada, the Canadian Institutes of Health Research (CHIR), and the Ontario Mental Health Foundation (OMHF). BL was supported by a CIHR Banting postdoctoral fellowship (BPF-162684).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alberry, B. L. J., Castellani, C. A., and Singh, S. M. (2020). Hippocampal transcriptome analysis following maternal separation implicates altered RNA processing in a mouse model of fetal alcohol spectrum disorder. J. Neurodev. Disord. 12:15. doi: 10.1186/s11689-020-09316-3
Alberry, B., and Singh, S. M. (2016). Developmental and behavioral consequences of early life maternal separation stress in a mouse model of fetal alcohol spectrum disorder. Behav. Brain Res. 308, 94–103. doi: 10.1016/j.bbr.2016.04.031
Alberry, B., and Singh, S. M. (2019). Alarming ontario drinking statistics obligate reappraisal of the impact of alcohol on newborns. CMAJ 191:E1283. doi: 10.1503/cmaj.73305
Alberry, B. L. J., and Singh, S. M. (2020). Hippocampal DNA methylation in a mouse model of fetal alcohol spectrum disorder that includes maternal separation stress only partially explains changes in gene expression. Front. Genet. 11:70. doi: 10.3389/fgene.2020.00070
Allis, C. D., and Jenuwein, T. (2016). The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487–500. doi: 10.1038/nrg.2016.59
Andre, Q. R., McMorris, C. A., Kar, P., Ritter, C., Gibbard, W. B., Tortorelli, C., et al. (2020). Different brain profiles in children with prenatal alcohol exposure with or without early adverse exposures. Hum. Brain Mapp. 41, 4375–4385. doi: 10.1001/jamapsychiatry.2021.0480
Arain, M., Haque, M., Johal, L., Mathur, P., Nel, W., Rais, A., et al. (2013). Maturation of the adolescent brain. Neuropsychiatr. Dis. Treat. 9, 449–461. doi: 10.2147/NDT.S39776
Balaraman, S., Idrus, N. M., Miranda, R. C., and Thomas, J. D. (2017). Postnatal choline supplementation selectively attenuates hippocampal microRNA alterations associated with developmental alcohol exposure. Alcohol 60, 159–167. doi: 10.1016/j.alcohol.2016.12.006
Barak, A. J., Beckenhauer, H. C., and Tuma, D. J. (1996). Betaine effects on hepatic methionine metabolism elicited by short-term ethanol feeding. Alcohol 13, 483–486. doi: 10.1016/0741-8329(96)00040-7
Bekdash, R. A., Zhang, C., and Sarkar, D. K. (2013). Gestational choline supplementation normalized fetal alcohol-induced alterations in histone modifications, DNA methylation and proopiomelanocortin (POMC) gene expression in beta-endorphin-producing POMC neurons of the hypothalamus. Alcohol. Clin. Exp. Res. 37, 1133–1142. doi: 10.1111/acer.12082
Bernstein, B. E., Mikkelsen, T. S., Xie, X., Kamal, M., Huebert, D. J., Cuff, J., et al. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326. doi: 10.1016/j.cell.2006.02.041
Biggio, F., Talani, G., Locci, V., Pisu, M. G., Boero, G., Ciarlo, B., et al. (2018). Low doses of prenatal ethanol exposure and maternal separation alter HPA axis function and ethanol consumption in adult male rats. Neuropharmacology 131, 271–281. doi: 10.1016/j.neuropharm.2017.12.005
Boschen, K. E., McKeown, S. E., Roth, T. L., and Klintsova, A. Y. (2017). Impact of exercise and a complex environment on hippocampal dendritic morphology, Bdnf gene expression and DNA methylation in male rat pups neonatally exposed to alcohol. Dev. Neurobiol. 77, 708–725. doi: 10.1002/dneu.22448
Canzio, D., Nwakeze, C. L., Horta, A., Rajkumar, S. M., Coffey, E. L., Duffy, E. E., et al. (2019). Antisense lncRNA transcription mediates dna demethylation to drive stochastic protocadherin α promoter choice. Cell 177, 639.e15–653.e15. doi: 10.1016/j.cell.2019.03.008
Chastain, L. G., Franklin, T., Gangisetty, O., Cabrera, M. A., Mukherjee, S., Shrivastava, P., et al. (2019). Early life alcohol exposure primes hypothalamic microglia to later-life hypersensitivity to immune stress: possible epigenetic mechanism. Neuropsychopharmacology 44, 1579–1588. doi: 10.1038/s41386-019-0326-7
Chater-Diehl, E. J., Laufer, B. I., Castellani, C. A., Alberry, B. L., and Singh, S. M. (2016). Alteration of gene expression, DNA methylation and histone methylation in free radical scavenging networks in adult mouse hippocampus following fetal alcohol exposure. PLoS One 11:e0154836. doi: 10.1371/journal.pone.0154836
Chater-Diehl, E. J., Laufer, B. I., and Singh, S. M. (2017). Changes to histone modifications following prenatal alcohol exposure: An emerging picture. Alcohol 60, 41–52. doi: 10.1016/j.alcohol.2017.01.005
Chater-Diehl, E., Sokolowski, D., Alberry, B., and Singh, S. M. (2019). Coordinated Tcf7l2 regulation in a mouse model implicates Wnt signaling in fetal alcohol spectrum disorders. Biochem. Cell Biol. 97, 375–379. doi: 10.1139/bcb-2018-0215
Chen, W. V., and Maniatis, T. (2013). Clustered protocadherins. Development 140, 3297–3302. doi: 10.1242/dev.090621
Chuang, J. C., and Jones, P. A. (2007). Epigenetics and microRNAs. Pediatr. Res. 61, 24R–29R. doi: 10.1203/pdr.0b013e3180457684
Chudley, A. E., Conry, J., Cook, J. L., Loock, C., Rosales, T., LeBlanc, N., et al. (2005). Fetal alcohol spectrum disorder: canadian guidelines for diagnosis. Can. Med. Assoc. J. 172, S1–S21. doi: 10.1503/cmaj.1040302
Cobben, J. M., Krzyzewska, I. M., Venema, A., Mul, A. N., Polstra, A., Postma, A. V., et al. (2019). DNA methylation abundantly associates with fetal alcohol spectrum disorder and its subphenotypes. Epigenomics 11, 767–785. doi: 10.2217/epi-2018-0221
Coggins, T. E., Timler, G. R., and Olswang, L. B. (2007). A state of double jeopardy: impact of prenatal alcohol exposure and adverse environments on the social communicative abilities of school-age children with fetal alcohol spectrum disorder. Lang. Speech Hear. Serv. Sch. 38, 117–127. doi: 10.1044/0161-1461(2007/012)
Comeau, W. L., Winstanley, C. A., and Weinberg, J. (2014). Prenatal alcohol exposure and adolescent stress—unmasking persistent attentional deficits in rats. Eur. J. Neurosci. 40, 3078–3095. doi: 10.1111/ejn.12671
Coolen, M., and Bally-Cuif, L. (2009). MicroRNAs in brain development and physiology. Curr. Opin. Neurobiol. 19, 461–470. doi: 10.1016/j.conb.2009.09.006
Cornman-Homonoff, J., Kuehn, D., Aros, S., Carter, T. C., Conley, M. R., Troendle, J., et al. (2012). Heavy prenatal alcohol exposure and risk of stillbirth and preterm delivery. J. Matern. Neonatal Med. 25, 860–863. doi: 10.3109/14767058.2011.587559
DeChiara, T. M., Robertson, E. J., and Efstratiadis, A. (1991). Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64, 849–859. doi: 10.1016/0092-8674(91)90513-x
do Prado, C. H., Grassi-Oliveira, R., Wieck, A., Zaparte, A., Filho, L. D., da Silva Morrone, M., et al. (2016). The impact of childhood maltreatment on redox state: Relationship with oxidative damage and antioxidant defenses in adolescents with no psychiatric disorder. Neurosci. Lett. 617, 173–177. doi: 10.1016/j.neulet.2016.01.062
Downing, C., Johnson, T. E., Larson, C., Leakey, T. I., Siegfried, R. N., Rafferty, T. M., et al. (2011). Subtle decreases in DNA methylation and gene expression at the mouse Igf2 locus following prenatal alcohol exposure: effects of a methyl-supplemented diet. Alcohol 45, 65–71. doi: 10.1016/j.alcohol.2010.07.006
El Hajj, N., Dittrich, M., and Haaf, T. (2017). Epigenetic dysregulation of protocadherins in human disease. Semin. Cell Dev. Biol. 69, 172–182. doi: 10.1016/j.semcdb.2017.07.007
Fast, D. K., and Conry, J. (2004). The challenge of fetal alcohol syndrome in the criminal legal system. Addict. Biol. 9, 161–168. doi: 10.1080/13556210410001717042
Fast, D. K., Conry, J., and Loock, C. A. (1999). Identifying fetal alcohol syndrome among youth in the criminal justice system. J. Dev. Behav. Pediatr. 20, 370–372. doi: 10.1097/00004703-199910000-00012
Flannigan, K., Kapasi, A., Pei, J., Murdoch, I., Andrew, G., and Rasmussen, C. (2021). Characterizing adverse childhood experiences among children and adolescents with prenatal alcohol exposure and fetal alcohol spectrum disorder. Child Abuse Negl. 112:104888. doi: 10.1016/j.chiabu.2020.104888
Gangisetty, O., Bekdash, R., Maglakelidze, G., and Sarkar, D. K. (2014). Fetal alcohol exposure alters proopiomelanocortin gene expression and hypothalamic-pituitary-adrenal axis function via increasing MeCP2 expression in the hypothalamus. PLoS One 9:e113228. doi: 10.1371/journal.pone.0113228
Gangisetty, O., Wynne, O., Jabbar, S., Nasello, C., and Sarkar, D. K. (2015). Fetal alcohol exposure reduces dopamine receptor d2 and increases pituitary weight and prolactin production via epigenetic mechanisms. PLoS One 10:e0140699. doi: 10.1371/journal.pone.0140699
Greenmyer, J. R., Klug, M. G., Kambeitz, C., Popova, S., and Burd, L. (2018). A multicountry updated assessment of the economic impact of fetal alcohol spectrum disorder: costs for children and adults. J. Addict. Med. 12, 466–473. doi: 10.1097/ADM.0000000000000438
Haorah, J., Schall, K., Ramirez, S. H., and Persidsky, Y. (2008). Activation of protein tyrosine kinases and matrix metalloproteinases causes blood-brain barrier injury: novel mechanism for neurodegeneration associated with alcohol abuse. Glia 56, 78–88. doi: 10.1002/glia.20596
Haycock, P. C., and Ramsay, M. (2009). Exposure of mouse embryos to ethanol during preimplantation development: effect on DNA methylation in the H19 imprinting control region. Biol. Reprod. 81, 618–627. doi: 10.1095/biolreprod.108.074682
Hellemans, K. G., Verma, P., Yoon, E., Yu, W., and Weinberg, J. (2008). Prenatal alcohol exposure increases vulnerability to stress and anxiety-like disorders in adulthood. Ann. N Y Acad. Sci. 1144, 154–175. doi: 10.1196/annals.1418.016
Hemingway, S. J. A., Davies, J. K., Jirikowic, T., and Olson, E. M. (2020). What proportion of the brain structural and functional abnormalities observed among children with fetal alcohol spectrum disorder is explained by their prenatal alcohol exposure and their other prenatal and postnatal risks? Adv. Pediatr. Res. 7:41. Available online at: https://pubmed.ncbi.nlm.nih.gov/33335991/.
Henry, J., Sloane, M., and Black-Pond, C. (2007). Neurobiology and neurodevelopmental impact of childhood traumatic stress and prenatal alcohol exposure. Lang. Speech. Hear. Serv. Sch. 38, 99–108. doi: 10.1044/0161-1461(2007/010)
Holman, P. J., Raineki, C., Chao, A., Grewal, R., Haghighat, S., Fung, C., et al. (2021). Altered social recognition memory and hypothalamic neuropeptide expression in adolescent male and female rats following prenatal alcohol exposure and/or early-life adversity. Psychoneuroendocrinology 126:105146. doi: 10.1016/j.psyneuen.2021.105146
Holoch, D., and Moazed, D. (2015). RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 16, 71–84. doi: 10.1038/nrg3863
Hutson, J. R., Stade, B., Lehotay, D. C., Collier, C. P., and Kapur, B. M. (2012). Folic acid transport to the human fetus is decreased in pregnancies with chronic alcohol exposure. PLoS One 7:e38057. doi: 10.1371/journal.pone.0038057
Ieraci, A., and Herrera, D. G. (2020). Early postnatal ethanol exposure in mice induces sex-dependent memory impairment and reduction of hippocampal NMDA-R2B expression in adulthood. Neuroscience 427, 105–115. doi: 10.1016/j.neuroscience.2019.11.045
Jacobson, S. W., Carter, R. C., Molteno, C. D., Stanton, M. E., Herbert, J. S., Lindinger, N. M., et al. (2018). Efficacy of maternal choline supplementation during pregnancy in mitigating adverse effects of prenatal alcohol exposure on growth and cognitive function: a randomized, double-blind, placebo-controlled clinical trial. Alcohol. Clin. Exp. Res. 42, 1327–1341. doi: 10.1111/acer.13769
Jarmasz, J. S., Stirton, H., Basalah, D., Davie, J. R., Clarren, S. K., Astley, S. J., et al. (2019). Global DNA methylation and histone posttranslational modifications in human and nonhuman primate brain in association with prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 43, 1145–1162. doi: 10.1111/acer.14052
Kambeitz, C., Klug, M. G., Greenmyer, J., Popova, S., and Burd, L. (2019). Association of adverse childhood experiences and neurodevelopmental disorders in people with fetal alcohol spectrum disorders (FASD) and non-FASD controls. BMC Pediatr. 19:498. doi: 10.1186/s12887-019-1878-8
Kaminen-Ahola, N., Ahola, A., Maga, M., Mallitt, K.-A. A., Fahey, P., Cox, T. C., et al. (2010). Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet. 6:e1000811. doi: 10.1371/journal.pgen.1000811
Karunamuni, G., Sheehan, M. M., Doughman, Y. Q., Gu, S., Sun, J., Li, Y., et al. (2017). Supplementation with the methyl donor betaine prevents congenital defects induced by prenatal alcohol exposure. Alcohol. Clin. Exp. Res. 41, 1917–1927. doi: 10.1111/acer.13495
Kesmodel, U., Wisborg, K., Olsen, S. F., Henriksen, T. B., and Secher, N. J. (2002). Moderate alcohol intake in pregnancy and the risk of spontaneous abortion. Alcohol Alcohol. 37, 87–92. doi: 10.1093/alcalc/37.1.87
Khalid, O., Kim, J. J., Kim, H.-S. S., Hoang, M., Tu, T. G., Elie, O., et al. (2014). Gene expression signatures affected by alcohol-induced DNA methylomic deregulation in human embryonic stem cells. Stem Cell Res. 12, 791–806. doi: 10.1016/j.scr.2014.03.009
Kisely, S., Abajobir, A. A., Mills, R., Strathearn, L., Clavarino, A., and Najman, J. M. (2018). Child maltreatment and mental health problems in adulthood: birth cohort study. Br. J. Psychiatry 213, 698–703. doi: 10.1192/bjp.2018.207
Kleiber, M. L., Laufer, B. I., Wright, E., Diehl, E. J., and Singh, S. M. (2012). Long-term alterations to the brain transcriptome in a maternal voluntary consumption model of fetal alcohol spectrum disorders. Brain Res. 1458, 18–33. doi: 10.1016/j.brainres.2012.04.016
Kleiber, M. L., Mantha, K., Stringer, R. L., and Singh, S. M. (2013). Neurodevelopmental alcohol exposure elicits long-term changes to gene expression that alter distinct molecular pathways dependent on timing of exposure. J. Neurodev. Disord. 5:6. doi: 10.1186/1866-1955-5-6
Koponen, A. M., Kalland, M., and Autti-Rämö, I. (2009). Caregiving environment and socio-emotional development of foster-placed FASD-children. Child. Youth Serv. Rev. 31, 1049–1056. doi: 10.1016/j.childyouth.2009.05.006
Koponen, A. M., Kalland, M., Autti-Rämö, I., Laamanen, R., and Suominen, S. (2013). Socio-emotional development of children with foetal alcohol spectrum disorders in long-term foster family care: a qualitative study. Nord. Soc. Work Res. 3, 38–58. doi: 10.1080/2156857X.2013.766234
Koponen, A. M., Nissinen, N.-M. M., Gissler, M., Autti-Rämö, I., Sarkola, T., and Kahila, H. (2020). Prenatal substance exposure, adverse childhood experiences and diagnosed mental and behavioral disorders—a longitudinal register-based matched cohort study in Finland. SSM—Popul. Heal. 11, 100625. doi: 10.1016/j.ssmph.2020.100625
Lan, N., Hellemans, K. G. C., Ellis, L., and Weinberg, J. (2015). Exposure to chronic mild stress differentially alters corticotropin-releasing hormone and arginine vasopressin mRNA expression in the stress-responsive neurocircuitry of male and female rats prenatally exposed to alcohol. Alcohol. Clin. Exp. Res. 39, 2414–2421. doi: 10.1111/acer.12916
Lange, S., Shield, K., Rehm, J., and Popova, S. (2013). Prevalence of fetal alcohol spectrum disorders in child care settings: a meta-analysis. Pediatrics 132, e950–e980. doi: 10.1542/peds.2013-0066
Laufer, B. I., Kapalanga, J., Castellani, C. A., Diehl, E. J., Yan, L., and Singh, S. M. (2015). Associative DNA methylation changes in children with prenatal alcohol exposure. Epigenomics 7, 1259–1274. doi: 10.2217/epi.15.60
Laufer, B. I., Mantha, K., Kleiber, M. L., Diehl, E. J., Addison, S. M., and Singh, S. M. (2013). Long-lasting alterations to DNA methylation and ncRNAs could underlie the effects of fetal alcohol exposure in mice. Dis. Model. Mech. 6, 977–992. doi: 10.1242/dmm.010975
Lebel, C. A., McMorris, C. A., Kar, P., Ritter, C., Andre, Q., Tortorelli, C., et al. (2019). Characterizing adverse prenatal and postnatal experiences in children. Birth Defects Res. 111, 848–858. doi: 10.1002/bdr2.1464
Lee, J. T. (2012). Epigenetic regulation by long noncoding RNAs. Science 338, 1435–1439. doi: 10.1126/science.1231776
Lenroot, R. K., and Giedd, J. N. (2006). Brain development in children and adolescents: insights from anatomical magnetic resonance imaging. Neurosci. Biobehav. Rev. 30, 718–729. doi: 10.1016/j.neubiorev.2006.06.001
Li, W., Shen, W., Zhang, B., Tian, K., Li, Y., Mu, L., et al. (2020). Long non-coding RNA LncKdm2b regulates cortical neuronal differentiation by cis-activating Kdm2b. Protein Cell 11, 161–186. doi: 10.1007/s13238-019-0650-z
Lim, D. A., Huang, Y. C., Swigut, T., Mirick, A. L., Garcia-Verdugo, J. M., Wysocka, J., et al. (2009). Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 458, 529–533. doi: 10.1038/nature07726
Liu, Y., Balaraman, Y., Wang, G., Nephew, K. P., and Zhou, F. C. (2009). Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation. Epigenetics 4, 500–511. doi: 10.4161/epi.4.7.9925
Lucia, D., Burgess, D., Cullen, C. L., Dorey, E. S., Rawashdeh, O., and Moritz, K. M. (2019). Periconceptional maternal alcohol consumption leads to behavioural changes in adult and aged offspring and alters the expression of hippocampal genes associated with learning and memory and regulators of the epigenome. Behav. Brain Res. 362, 249–257. doi: 10.1016/j.bbr.2019.01.009
Lussier, A. A., Bodnar, T. S., Mingay, M., Morin, A. M., Hirst, M., Kobor, M. S., et al. (2018a). Prenatal alcohol exposure: profiling developmental DNA methylation patterns in central and peripheral tissues. Front. Genet. 9:610. doi: 10.3389/fgene.2018.00610
Lussier, A. A., Morin, A. M., MacIsaac, J. L., Salmon, J., Weinberg, J., Reynolds, J. N., et al. (2018b). DNA methylation as a predictor of fetal alcohol spectrum disorder. Clin. Epigenetics 10:5. doi: 10.1186/s13148-018-0439-6
Mahnke, A. H., Sideridis, G. D., Salem, N. A., Tseng, A. M., Carter, R. C., Dodge, N. C., et al. (2021). Infant circulating MicroRNAs as biomarkers of effect in fetal alcohol spectrum disorders. Sci. Rep. 11:1429. doi: 10.1038/s41598-020-80734-y
Malcon, L. M. C., Wearick-Silva, L. E., Zaparte, A., Orso, R., Luft, C., Tractenberg, S. G., et al. (2020). Maternal separation induces long-term oxidative stress alterations and increases anxiety-like behavior of male Balb/cJ mice. Exp. Brain Res. 238, 2097–2107. doi: 10.1007/s00221-020-05859-y
Mansouri, A., Demeilliers, C., Amsellem, S., Pessayre, D., and Fromenty, B. (2001). Acute ethanol administration oxidatively damages and depletes mitochondrial DNA in mouse liver, brain, heart and skeletal muscles: protective effects of antioxidants. J. Pharmacol. Exp. Ther. 298, 737–743. Available online at: https://jpet.aspetjournals.org/content/298/2/737.long.
Mantha, K., Laufer, B. I., and Singh, S. M. (2014). Molecular changes during neurodevelopment following second-trimester binge ethanol exposure in a mouse model of fetal alcohol spectrum disorder: from immediate effects to long-term adaptation. Dev. Neurosci. 36, 29–43. doi: 10.1159/000357496
Marjonen, H., Kahila, H., and Kaminen-Ahola, N. (2017). rs10732516 polymorphism at the IGF2/H19 locus associates with a genotype-specific trend in placental DNA methylation and head circumference of prenatally alcohol-exposed newborns. Hum. Reprod. Open 2017:hox014. doi: 10.1093/hropen/hox014
Marjonen, H., Sierra, A., Nyman, A., Rogojin, V., Gröhn, O., Linden, A.-M. M., et al. (2015). Early maternal alcohol consumption alters hippocampal DNA methylation, gene expression and volume in a mouse model. PLoS One 10:e0124931. doi: 10.1371/journal.pone.0124931
May, P. A., Chambers, C. D., Kalberg, W. O., Zellner, J., Feldman, H., Buckley, D., et al. (2018). Prevalence of fetal alcohol spectrum disorders in 4 US communities. JAMA 319, 474–482. doi: 10.1001/jama.2017.21896
McGowan, P. O., Suderman, M., Sasaki, A., Huang, T. C. T., Hallett, M., Meaney, M. J., et al. (2011). Broad epigenetic signature of maternal care in the brain of adult rats. PLoS One 6:e14739. doi: 10.1371/journal.pone.0014739
Mellios, N., Feldman, D. A., Sheridan, S. D., Ip, J. P. K., Kwok, S., Amoah, S. K., et al. (2018). MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol. Psychiatry 23, 1051–1065. doi: 10.1038/mp.2017.86
Mews, P., Egervari, G., Nativio, R., Sidoli, S., Donahue, G., Lombroso, S. I., et al. (2019). Alcohol metabolism contributes to brain histone acetylation. Nature 574, 717–721. doi: 10.1038/s41586-019-1700-7
Miozzo, F., Arnould, H., de Thonel, A., Schang, A.-L. L., Sabéran-Djoneidi, D., Baudry, A., et al. (2018). Alcohol exposure promotes DNA methyltransferase DNMT3A upregulation through reactive oxygen species-dependent mechanisms. Cell Stress Chaperones 23, 115–126. doi: 10.1007/s12192-017-0829-2
Myran, D. T., Hsu, A. T., Smith, G., and Tanuseputro, P. (2019). Rates of emergency department visits attributable to alcohol use in Ontario from 2003 to 2016: a retrospective population-level study. CMAJ 191, E804–E810. doi: 10.1503/cmaj.181575
Ngai, Y. F., Sulistyoningrum, D. C., O’Neill, R., Innis, S. M., Weinberg, J., and Devlin, A. M. (2015). Prenatal alcohol exposure alters methyl metabolism and programs serotonin transporter and glucocorticoid receptor expression in brain. Am. J. Physiol. Integr. Comp. Physiol. 309, R613–R622. doi: 10.1152/ajpregu.00075.2015
Norman, R. E., Byambaa, M., De, R., Butchart, A., Scott, J., and Vos, T. (2012). The long-term health consequences of child physical abuse, emotional abuse and neglect: a systematic review and meta-analysis. PLoS Med. 9:e1001349. doi: 10.1371/journal.pmed.1001349
O’Leary, C. M., Nassar, N., Kurinczuk, J. J., De Klerk, N., Geelhoed, E., Elliott, E. J., et al. (2010). Prenatal alcohol exposure and risk of birth defects. Pediatrics 126, e843–e850. doi: 10.1542/peds.2010-0256
Okazaki, S., Otsuka, I., Shinko, Y., Horai, T., Hirata, T., Yamaki, N., et al. (2020). Epigenetic clock analysis in children with fetal alcohol spectrum disorder. Alcohol. Clin. Exp. Res. 45, 329–337. doi: 10.1111/acer.14532
Ooi, S. K. T., Qiu, C., Bernstein, E., Li, K., Jia, D., Yang, Z., et al. (2007). DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448, 714–717. doi: 10.1038/nature05987
Otani, J., Nankumo, T., Arita, K., Inamoto, S., Ariyoshi, M., and Shirakawa, M. (2009). Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep. 10, 1235–1241. doi: 10.1038/embor.2009.218
Öztürk, N. C., Resendiz, M., Öztürk, H., and Zhou, F. C. (2017). DNA Methylation program in normal and alcohol-induced thinning cortex. Alcohol 60, 135–147. doi: 10.1016/j.alcohol.2017.01.006
Paudel, P., Pierotti, C., Lozano, E., Amoah, S. K., Gardiner, A. S., Caldwell, K. K., et al. (2020). Prenatal alcohol exposure results in sex-specific alterations in circular RNA expression in the developing mouse brain. Front. Neurosci. 14:581895. doi: 10.3389/fnins.2020.581895
Pereira, J. D., Sansom, S. N., Smith, J., Dobenecker, M. W., Tarakhovsky, A., and Livesey, F. J. (2010). Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc. Natl. Acad. Sci. U S A 107, 15957–15962. doi: 10.1073/pnas.1002530107
Perkins, A., Lehmann, C., Lawrence, R. C., and Kelly, S. J. (2013). Alcohol exposure during development: impact on the epigenome. Int. J. Dev. Neurosci. 31, 391–397. doi: 10.1016/j.ijdevneu.2013.03.010
Popova, S., Lange, S., Burd, L., and Rehm, J. (2016). The economic burden of fetal alcohol spectrum disorder in Canada in 2013. Alcohol Alcohol. 51, 367–375. doi: 10.1093/alcalc/agv117
Popova, S., Lange, S., Chudley, A. E., Reynolds, J. N., Rehm, J., May, P. A., et al. (2018). World health organization international study on the prevalence of fetal alcohol spectrum disorder (FASD). Centre Addict. Ment. Health Available online at: www.camh.ca. Accessed April 25, 2019.
Popova, S., Lange, S., Probst, C., Gmel, G., and Rehm, J. (2017). Global prevalence of alcohol use and binge drinking during pregnancy and fetal alcohol spectrum disorder. Biochem. Cell Biol. 96, 237–240. doi: 10.1139/bcb-2017-0077
Popova, S., Temple, V., Dozet, D., O’Hanlon, G., Toews, C., and Rehm, J. (2021). Health, social and legal outcomes of individuals with diagnosed or at risk for fetal alcohol spectrum disorder: canadian example. Drug Alcohol Depend. 219:108487. doi: 10.1016/j.drugalcdep.2020.108487
Portales-Casamar, E., Lussier, A. A., Jones, M. J., MacIsaac, J. L., Edgar, R. D., Mah, S. M., et al. (2016). DNA methylation signature of human fetal alcohol spectrum disorder. Epigenetics Chromatin 9:25. doi: 10.1186/s13072-016-0074-4
Price, A., Cook, P. A., Norgate, S., and Mukherjee, R. (2017). Prenatal alcohol exposure and traumatic childhood experiences: a systematic review. Neurosci. Biobehav. Rev. 80, 89–98. doi: 10.1016/j.neubiorev.2017.05.018
Raineki, C., Bodnar, T. S., Holman, P. J., Baglot, S. L., Lan, N., and Weinberg, J. (2017). Effects of early-life adversity on immune function are mediated by prenatal environment: role of prenatal alcohol exposure. Brain. Behav. Immun. 66, 210–220. doi: 10.1016/j.bbi.2017.07.001
Réus, G. Z., Fernandes, G. C., de Moura, A. B., Silva, R. H., Darabas, A. C., de Souza, T. G., et al. (2017). Early life experience contributes to the developmental programming of depressive-like behaviour, neuroinflammation and oxidative stress. J. Psychiatr. Res. 95, 196–207. doi: 10.1515/jcim-2020-0448
Sabra, S., Malmqvist, E., Almeida, L., Gratacos, E., and Gomez Roig, M. D. (2018). Differential correlations between maternal hair levels of tobacco and alcohol with fetal growth restriction clinical subtypes. Alcohol 70, 43–49. doi: 10.1016/j.alcohol.2018.01.001
Sahafi, E., Peeri, M., Hosseini, M. J., and Azarbyjani, M. A. (2018). Cardiac oxidative stress following maternal separation stress was mitigated following adolescent voluntary exercise in adult male rat. Physiol. Behav. 183, 39–45. doi: 10.1016/j.physbeh.2017.10.022
Sanai, N., Nguyen, T., Ihrie, R. A., Mirzadeh, Z., Tsai, H. H., Wong, M., et al. (2011). Corridors of migrating neurons in the human brain and their decline during infancy. Nature 478, 382–386. doi: 10.1038/nature10487
Schaffner, S. L., Lussier, A. A., Baker, J. A., Goldowitz, D., Hamre, K. M., and Kobor, M. S. (2020). Neonatal alcohol exposure in mice induces select differentiation- and apoptosis-related chromatin changes both independent of and dependent on sex. Front. Genet. 11:35. doi: 10.3389/fgene.2020.00035
Semple, B. D., Blomgren, K., Gimlin, K., Ferriero, D. M., and Noble-Haeusslein, L. J. (2013). Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 106–107, 1–16. doi: 10.1016/j.pneurobio.2013.04.001
Sharp, G. C., Arathimos, R., Reese, S. E., Page, C. M., Felix, J., Küpers, L. K., et al. (2018). Maternal alcohol consumption and offspring DNA methylation: findings from six general population-based birth cohorts. Epigenomics 10, 27–42. doi: 10.2217/epi-2017-0095
Shaw, S. (1989). Lipid peroxidation, iron mobilization and radical generation induced by alcohol. Free Radic. Biol. Med. 7, 541–547. doi: 10.1016/0891-5849(89)90030-0
Shivakumar, M., Subbanna, S., Joshi, V., and Basavarajappa, B. S. (2020). Postnatal ethanol exposure activates HDAC-mediated histone deacetylation, impairs synaptic plasticity gene expression and behavior in mice. Int. J. Neuropsychopharmacol. 23, 324–338. doi: 10.1093/ijnp/pyaa017
Smith, A. K., Kilaru, V., Klengel, T., Mercer, K. B., Bradley, B., Conneely, K. N., et al. (2015). DNA extracted from saliva for methylation studies of psychiatric traits: evidence tissue specificity and relatedness to brain. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 168, 36–44. doi: 10.1002/ajmg.b.32278
Sokol, R. J., Delaney-Black, V., and Nordstrom, B. (2003). Fetal alcohol spectrum disorder. JAMA 290, 2996–2999. doi: 10.1001/jama.290.22.2996
Sokol, R. J., Janisse, J. J., Louis, J. M., Bailey, B. N., Ager, J., Jacobson, S. W., et al. (2007). Extreme prematurity: an alcohol-related birth effect. Alcohol. Clin. Exp. Res. 31, 1031–1037. doi: 10.1111/j.1530-0277.2007.00384.x
Streissguth, A. P., Aase, J. M., Clarren, S. K., Randels, S. P., Ladue, R. A., and Smith, D. F. (1991). Fetal alcohol syndrome in adolescents and adults. J. Am. Med. Assoc. 265, 1961–1967.
Streissguth, A. P., and O’Malley, K. (2000). Neuropsychiatric implications and long-term consequences of fetal alcohol spectrum disorders. Semin. Clin. Neuropsychiatry 5, 177–190. doi: 10.1053/scnp.2000.6729
Suderman, M., McGowan, P. O., Sasaki, A., Huang, T. C. T., Hallett, M. T., Meaney, M. J., et al. (2012). Conserved epigenetic sensitivity to early life experience in the rat and human hippocampus. Proc. Natl. Acad. Sci. U S A 109, 17266–17272. doi: 10.1073/pnas.1121260109
Tang, G. B., Zeng, Y. Q., Liu, P. P., Mi, T. W., Zhang, S. F., Dai, S. K., et al. (2017). The histone H3K27 demethylase UTX regulates synaptic plasticity and cognitive behaviors in mice. Front. Mol. Neurosci. 10:267. doi: 10.3389/fnmol.2017.00267
Tau, G. Z., and Peterson, B. S. (2010). Normal development of brain circuits. Neuropsychopharmacology 35, 147–168. doi: 10.1038/npp.2009.115
Tavanasefat, H., Li, F., Koyano, K., Gourtani, B. K., Marty, V., Mulpuri, Y., et al. (2020). Molecular consequences of fetal alcohol exposure on amniotic exosomal miRNAs with functional implications for stem cell potency and differentiation. PLoS One 15:e0242276. doi: 10.1371/journal.pone.0242276
Tenenbaum, A., Mandel, A., Dor, T., Sapir, A., Sapir-Bodnaro, O., Hertz, P., et al. (2020). Fetal alcohol spectrum disorder among pre-adopted and foster children. BMC Pediatr. 20:275. doi: 10.1186/s12887-020-02164-z
Theda, C., Hwang, S. H., Czajko, A., Loke, Y. J., Leong, P., and Craig, J. M. (2018). Quantitation of the cellular content of saliva and buccal swab samples. Sci. Rep. 8:6944. doi: 10.1038/s41598-018-25311-0
Toyoda, S., Kawaguchi, M., Kobayashi, T., Tarusawa, E., Toyama, T., Okano, M., et al. (2014). Developmental epigenetic modification regulates stochastic expression of clustered Protocadherin genes, generating single neuron diversity. Neuron 82, 94–108. doi: 10.1016/j.neuron.2014.02.005
Turinsky, A. L., Butcher, D. T., Choufani, S., Weksberg, R., and Brudno, M. (2019). Don’t brush off buccal data heterogeneity. Epigenetics 14, 109–117. doi: 10.1080/15592294.2019.1581592
Uban, K. A., Comeau, W. L., Ellis, L. A., Galea, L. A., and Weinberg, J. (2013). Basal regulation of HPA and dopamine systems is altered differentially in males and females by prenatal alcohol exposure and chronic variable stress. Psychoneuroendocrinology 38, 1953–1966. doi: 10.1016/j.psyneuen.2013.02.017
Veazey, K. J., Carnahan, M. N., Muller, D., Miranda, R. C., and Golding, M. C. (2013). Alcohol-induced epigenetic alterations to developmentally crucial genes regulating neural stemness and differentiation. Alcohol. Clin. Exp. Res. 37, 1111–1122. doi: 10.1111/acer.12080
Veazey, K. J., Parnell, S. E., Miranda, R. C., and Golding, M. C. (2015). Dose-dependent alcohol-induced alterations in chromatin structure persist beyond the window of exposure and correlate with fetal alcohol syndrome birth defects. Epigenetics Chromatin 8:39. doi: 10.1186/s13072-015-0031-7
Veazey, K. J., Wang, H., Bedi, Y. S., Skiles, W. M., Chang, R. C.-A., and Golding, M. C. (2017). Disconnect between alcohol-induced alterations in chromatin structure and gene transcription in a mouse embryonic stem cell model of exposure. Alcohol 60, 121–133. doi: 10.1016/j.alcohol.2017.01.007
Vogel Ciernia, A., Laufer, B. I., Dunaway, K. W., Mordaunt, C. E., Coulson, R. L., Totah, T. S., et al. (2018). Experience-dependent neuroplasticity of the developing hypothalamus: integrative epigenomic approaches. Epigenetics 13, 318–330. doi: 10.1080/15592294.2018.1451720
Waddell, J., Hill, E., Tang, S., Jiang, L., Xu, S., and Mooney, S. M. (2020). Choline plus working memory training improves prenatal alcohol-induced deficits in cognitive flexibility and functional connectivity in adulthood in rats. Nutrients 12, 1–18. doi: 10.3390/nu12113513
Wang, L. L., Zhang, Z., Li, Q., Yang, R., Pei, X., Xu, Y., et al. (2009). Ethanol exposure induces differential microRNA and target gene expression and teratogenic effects which can be suppressed by folic acid supplementation. Hum. Reprod. 24, 562–579. doi: 10.1093/humrep/den439
Weyrauch, D., Schwartz, M., Hart, B., Klug, M. G., and Burd, L. (2017). Comorbid mental disorders in fetal alcohol spectrum disorders: a systematic review. J. Dev. Behav. Pediatr. 38, 283–291. doi: 10.1097/DBP.0000000000000440
Wozniak, J. R., Fink, B. A., Fuglestad, A. J., Eckerle, J. K., Boys, C. J., Sandness, K. E., et al. (2020). Four-year follow-up of a randomized controlled trial of choline for neurodevelopment in fetal alcohol spectrum disorder. J. Neurodev. Disord. 12:9. doi: 10.1186/s11689-020-09312-7
Wu, D., and Cederbaum, A. I. (2003). Alcohol, oxidative stress and free radical damage. Alcohol Res. Health 27, 277–284. Available online at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6668865/.
Wu, H., Coskun, V., Tao, J., Xie, W., Ge, W., Yoshikawa, K., et al. (2010). Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science 329, 444–447. doi: 10.1126/science.1190485
Yuan, F., Yun, Y., Fan, H., Li, Y., Lu, L., Liu, J., et al. (2020). MicroRNA-135a protects against ethanol-induced apoptosis in neural crest cells and craniofacial defects in zebrafish by modulating the Siah1/p38/p53 pathway. Front. Cell Dev. Biol. 8:583959. doi: 10.3389/fcell.2020.583959
Zablotsky, B., Black, L. I., Maenner, M. J., Schieve, L. A., Danielson, M. L., Bitsko, R. H., et al. (2019). Prevalence and trends of developmental disabilities among children in the United States: 2009–2017. Pediatrics 144:e20190811. doi: 10.1542/peds.2019-0811
Zhang, R. R., Cui, Q. Y., Murai, K., Lim, Y. C., Smith, Z. D., Jin, S., et al. (2013). Tet1 regulates adult hippocampal neurogenesis and cognition. Cell Stem Cell 13, 237–245. doi: 10.1016/j.stem.2013.05.006
Zhang, C. R., Ho, M. F., Vega, M. C. S., Burne, T. H. J., and Chong, S. (2015). Prenatal ethanol exposure alters adult hippocampal VGLUT2 expression with concomitant changes in promoter DNA methylation, H3K4 trimethylation and miR-467b-5p levels. Epigenetics Chromatin 8:40. doi: 10.1186/s13072-015-0032-6
Zhang, Y., Jurkowska, R., Soeroes, S., Rajavelu, A., Dhayalan, A., Bock, I., et al. (2010). Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 38, 4246–4253. doi: 10.1093/nar/gkq147
Keywords: fetal alcohol spectrum disorders (FASD), epigenetics, neurodevelopment, prenatal alcohol exposure (PAE), early life stress (ELS), oxidative stress, DNA methylation, clustered protocadherins (Pcdhs)
Citation: Alberry B, Laufer BI, Chater-Diehl E and Singh SM (2021) Epigenetic Impacts of Early Life Stress in Fetal Alcohol Spectrum Disorders Shape the Neurodevelopmental Continuum. Front. Mol. Neurosci. 14:671891. doi: 10.3389/fnmol.2021.671891
Received: 24 February 2021; Accepted: 30 April 2021;
Published: 03 June 2021.
Edited by:
Fabio Coppedè, University of Pisa, ItalyReviewed by:
David F. Werner, Binghamton University, United StatesSerge McGraw, Université de Montréal, Canada
Copyright 2021 Alberry, Laufer, Chater-Diehl and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shiva M. Singh,c3NpbmdoQHV3by5jYQ==