Abstract
Cell types rich in mitochondria, including neurons, display a high energy demand and a need for calcium buffering. The importance of mitochondria for proper neuronal function is stressed by the occurrence of neurological defects in patients suffering from a great variety of diseases caused by mutations in mitochondrial genes. Genetic and pharmacological evidence also reveal a role of these organelles in various aspects of neuronal physiology and in the pathogenesis of neurodegenerative disorders. Yet the mechanisms by which mitochondria can affect neurotransmission largely remain to be elucidated. In this review we focus on experimental data that suggest a critical function of synaptic mitochondria in the function and organization of synaptic vesicle pools, and in neurotransmitter release during intense neuronal activity. We discuss how calcium handling, ATP production and other mitochondrial mechanisms may influence synaptic vesicle pool organization and synaptic function. Given the link between synaptic mitochondrial function and neuronal communication, efforts toward better understanding mitochondrial biology may lead to novel therapeutic approaches of neurological disorders including Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis and psychiatric disorders that are at least in part caused by mitochondrial deficits.
Introduction
Mitochondria are energy-generating organelles found in virtually all eukaryotic cell types. They are strikingly abundant in neurons and in particular at chemical synapses (Palay, 1956) where neuronal communication takes place. It has long been recognized that mitochondria are important for the maintenance of neuronal integrity and function, as diseases caused by a mitochondrial failure often display a neurodegenerative component affecting the central or peripheral nervous system (DiMauro and Schon, 2008). Conversely, mitochondrial defects are frequently observed in tissue samples collected from patients with a neurodegenerative disorder (Chen and Chan, 2009) and the involvement of mitochondrial dysfunction in a large number of diseases likely remains gravely underestimated (Table 1). These observations suggest an intimate involvement of mitochondria in maintaining neuronal integrity, possibly in part by controlling neurotransmission, yet the exact function of these organelles in synaptic activity has long remained elusive.
Table 1
| Disorder | Characteristics | Genes | Mitochondrial link | |||
|---|---|---|---|---|---|---|
| NEURODEGENERATION | ||||||
| Charcot-Marie-Tooth type 2A2 (CMT2A2) | Affecting sensory and motorneurons of the distal extremetities | Mitofusin 2 | Fusion (Chen and Chan, 2005, 2009) | |||
| Autosomal dominant optic atrophy (DOA) | Degeneration of retinal ganglia cells which leads to optic nerve atrophy | OPA1 | Fusion (Chen and Chan, 2005, 2009) | |||
| Parkinson's disease | Degeneration of dopaminergic neurons in the substantia nigra | Pinkl, DJ-1, Parkin | Electron transport chain, oxidative stress, mitophagy (Kahle et al., 2009; Morais et al., 2009; Tanaka, 2010) | |||
| Huntington's disease | Progressive loss of striatal and cortical neurons | Huntingtin | Electron transport chain, Ca2+ storage (Petrozzi et al., 2007; Bossy-Wetzel et al., 2008) | |||
| Alzheimer's disease | Neuronal death in cerebral cortex | APP, Presenilin | Fusion/fission (Wang et al., 2009) | |||
| Amyotrophic lateral sclerosis | Degeneration of anterior horn cells of the spinal chord and cortical motorneurons | SOD1 | Oxidative phosphorylation (Petrozzi et al., 2007) | |||
| Leigh's Syndrome | Necrotizing encephalopathy | A broad variety of genes | Multiple complexes of the electron transport chain (Finsterer, 2008) | |||
| Multiple sclerosis | Demyelination, neuroaxonal depletion | A broad variety of genes | Multiple complexes of the electron transport chain (Kalman et al., 2007) | |||
| PSYCHIATRIC DISORDER | ||||||
| Autism | Behavioural syndrome | A broad variety of genes | Electron transport chain (Correia et al., 2006) | |||
| Schizophrenia | Disorder of thought | DISC1 | Oxidative phosphorylation, splicing isoforms targeted to mitochondria (Shao et al., 2008; Ben-Shachar, 2009) | |||
| CHRONICAL NEUROLOGICAL DISORDER | ||||||
| Epilepsy | Recurrent seizures | A broad variety of genes | Oxidative phosphorylation (Kudin et al., 2009) | |||
Neurological and psychiatric disorders associated with mitochondrial dysfunction.
Disorders are listed in this table along with their most characteristic phenotype, when known, their causative gene(s) and the mitochondrial features reported to be defective. The mitochondrial defect might be the direct cause of the disease or the result of an upstream dysfunction in the pathology.
Mitochondria are dynamic organelles that divide, fuse and are redistributed within the cell in response to various physiological cues. Besides their predominant role in energy metabolism (Figure 1), they are involved in many other cellular processes that are also relevant for neuronal function, including the regulation of calcium homeostasis and a form of programmed cell death (or apoptosis). Defects in all these functions as well as in mitochondrial fusion or fission are predicted to affect various aspects of neuronal physiology (DiMauro and Schon, 2008; Chen and Chan, 2009). However, unraveling the link between a mitochondrial defect and its consequences on neuronal function or survival has proven a complex task, mostly because different mitochondrial functions are interdependent (see for example Niemann et al., 2005; Graier et al., 2007; Gunter and Sheu, 2009 see also below). Experimental results obtained using drugs or mutations that interfere with one aspect of mitochondrial physiology thus need to be interpreted with care, as they may have numerous side effects.
Figure 1
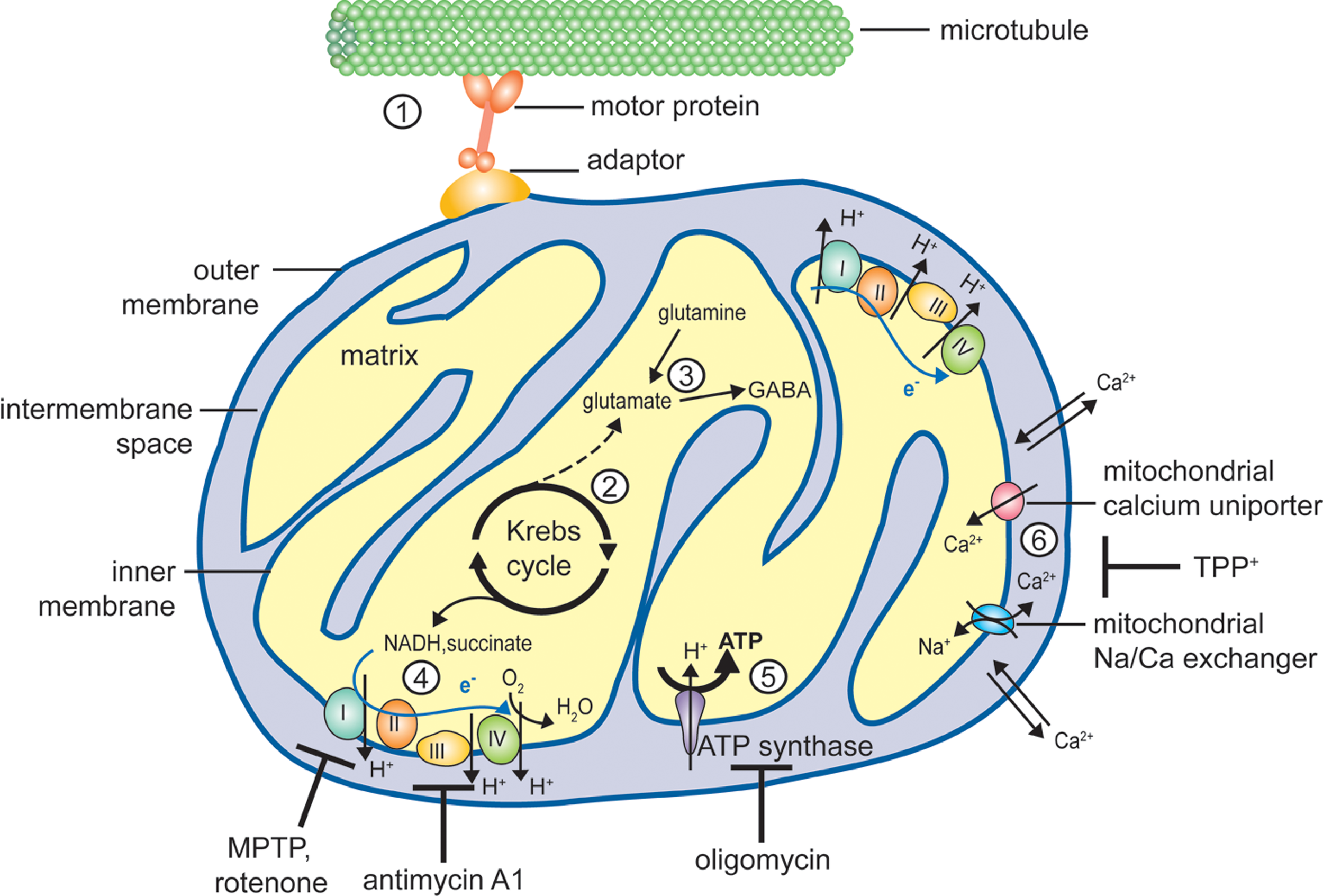
Mitochondrial functions susceptible to affect neurotransmission. Schematic representation of a mitochondrion illustrating the different mitochondrial functions discussed in the text. (1) Mitochondria are dynamic organelles, able to move bidirectionally along microtubules tracts with the help of motor proteins (kinesin for anterograde and dynein for retrograde axonal transport, respectively) and adaptor proteins (e.g., Milton and the associated Rho-GTPase Miro or syntabulin). Short-distance transport along actin filaments is mediated by myosin motors (not shown). (2) The Krebs cycle takes place in the mitochondrial matrix, generating α-ketoglutarate and electron donors (NADH and succinate). (3) Transamination of α-ketoglutarate yields the neurotransmitter glutamate, which can also be formed by glutamine hydrolysis. Glutamate can be further decarboxylated to yield GABA, another neurotransmitter. (4) The electron transport chain (ETC), consisting of 4 multiprotein complexes, mediates electron transfer (blue arrow) from NADH and succinate to the electron acceptor O2. Numerous drugs target components of the ETC, for example MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) and rotenone affect complex I while antimycin A1 inhibits complex III. Electron transfer is coupled with proton extrusion out of the matrix toward the intermembrane space, producing an electrochemical gradient across the inner membrane (about – 200 mV). (5) Protons ultimately return to the matrix through the ATP synthase, using the free energy produced by the ETC to drive ATP synthesis. Oligomycin is an inhibitor of the ATP synthase. (6) Mitochondria are also involved in calcium homeostasis, and Ca2+ ions can be sequestered in the matrix under the form of a reversible phosphate complex. While the outer mitochondrial membrane is rather permeable to calcium, Ca2+ entry across the inner mitochondrial membrane is mediated by a uniporter, and its extrusion largely relies on a sodium/calcium exchanger. Mitochondrial calcium transport can be inhibited by TPP+ (tetraphenylphosphonium).
This review focuses on the role of presynaptic mitochondria in the control of synaptic vesicle (SV) function. In general, SVs can be attributed to three functionally different pools, termed the readily releasable pool, the recycling pool and the reserve pool (RP) (Rizzoli and Betz, 2005). Together, the readily releasable pool and the recycling pool, sometimes referred to as the exo-endo cycling pool (ECP), typically represent about 10–20% of the SVs and are released under moderate or intense neuronal activity, whereas vesicles from the RP are recruited only upon high frequency stimulation (Figure 2) (Kuromi and Kidokoro, 2005; Rizzoli and Betz, 2005). Based on these characteristics, different stimulation protocols have been developed that allow the specific mobilization of SVs from different pools as well as their labeling with fluorescent dyes (Kuromi and Kidokoro, 2002; Verstreken et al., 2008). Because the synaptic vesicle cycle involves numerous ATP-consuming steps and is tightly controlled by the cytosolic calcium concentration ([Ca2+]cyto), vesicle cycling appears to be highly dependent on mitochondrial function. In the following sections we will review recent reports indicating how mitochondria can affect the distribution in pools and release properties of SVs. In particular, we reexamine experimental evidence which together indicate that these organelles are essential for synaptic vesicle release under conditions of intense neuronal activity through vesicle mobilization from the RP. Current studies dedicated to this link between mitochondria and the SV cycle will hopefully lead to new therapeutic strategies for the handling of neurological disorders characterized by a mitochondrial defect and which have a tremendous social impact, like Alzheimer's, Huntington's and Parkinson's diseases.
Figure 2
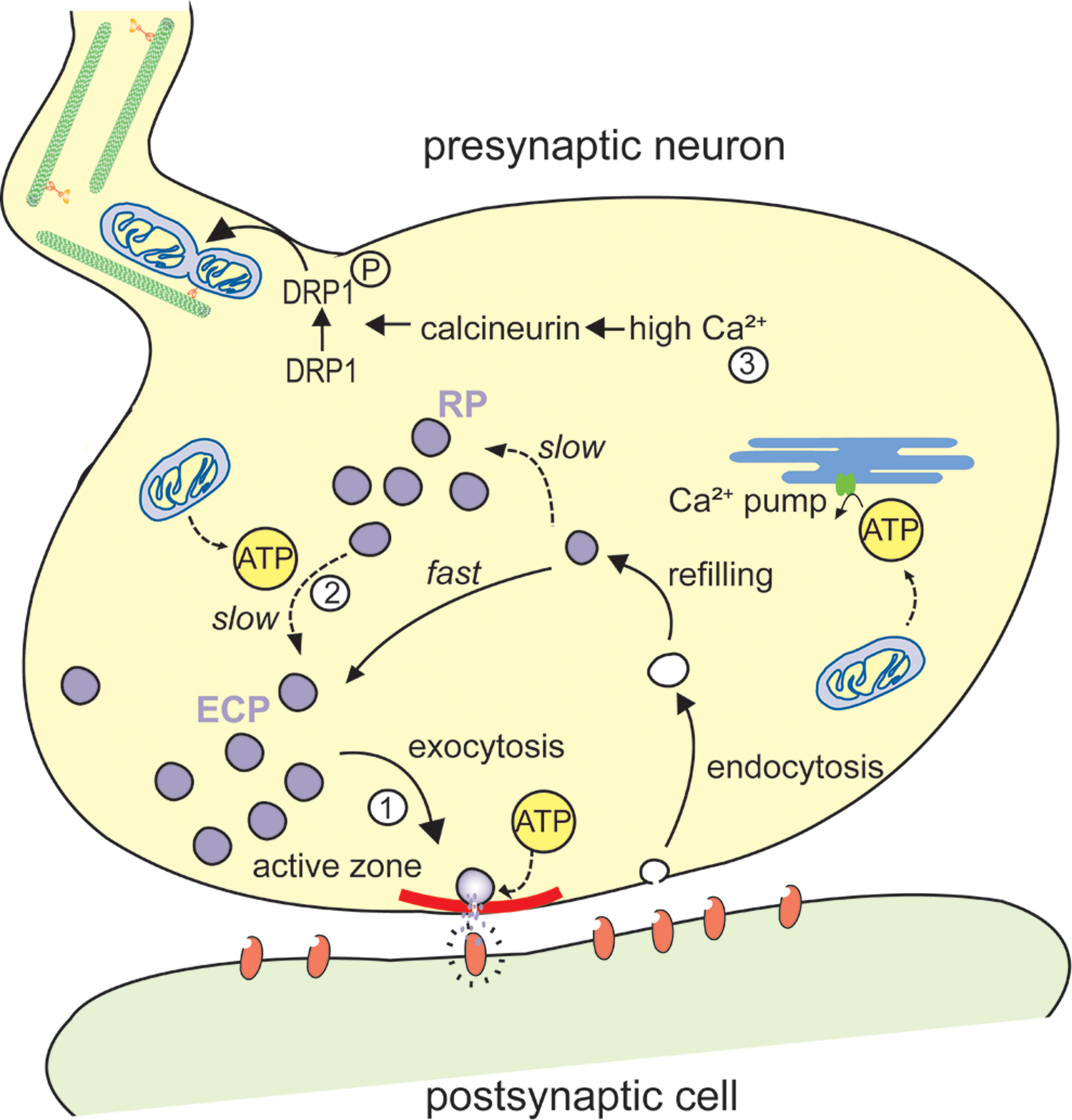
Schematic representation of a mitochondrial involvement in the organization of synaptic vesicle pools. (1) Synaptic vesicles (SVs) from the exo-endo cycling pool (ECP) participate in neurotransmitter release at active zones (in red). Upon arrival of an action potential at the nerve terminal, these vesicles fuse with the presynaptic membrane and discharge neurotransmitters in the synaptic cleft. (2) In contrast, SVs from the reserve pool (RP), which contains the majority of vesicles in most synapses, are tethered to cytoskeletal elements and are only recruited to the active zone during intense stimulation. This recruitment of the RP depends on ATP produced by the mitochondria. (3) The ATP produced by the mitochondria also plays an important role for preynaptic Ca2+ buffering in the endoplasmic reticulum (ER). High Ca2+, induced by exo-endocytosis, activate calcineurin that in turn activates and dephosphorylates DRP1, resulting in mitochondrial fission and transport.
Calcium Buffering by Presynaptic Mitochondria Influences Exo- and Endocytosis of SVs
In excitable cells, depolarization of the plasma membrane triggers the opening of voltage-gated calcium channels, leading to Ca2+ entry and a subsequent rise of the [Ca2+]cyto that initiates various downstream cellular responses. In neurons, calcium signaling is involved in neurotransmitter release from the presynaptic terminals, in vesicle recycling via endocytosis and in long-term neuronal responses like neurite growth and degeneration, or modulation of synaptic strength (Ghosh and Greenberg, 1995; Zucker, 1999; Jonas, 2006; Yao et al., 2009). Similarly to the endoplasmic reticulum (ER), mitochondria are able to sequester and release Ca2+ ions and thereby modulate calcium-dependent signaling events. Finally, mitochondria may also contribute to calcium homeostasis by providing ATP to fuel the plasma membrane Ca2+-ATPase (Zenisek and Matthews, 2000). Considerable effort has thus been invested in studying how presynaptic mitochondria control the time-course and the amplitude of calcium signaling.
Mitochondrial calcium uptake is driven by the negative potential established across the inner mitochondrial membrane (Figure 1) and can be inhibited by drugs that dissipate the proton gradient responsible for this potential (Gunter et al., 2000; Graier et al., 2007). The outer mitochondrial membrane is relatively permeable to calcium ions, so that Ca2+ accumulation in the mitochondrial matrix mainly depends on a mitochondrial calcium uniporter (MCU) that resides in the inner membrane (Figure 1). The MCU shows a very low affinity for calcium ions (Km estimated between 1 and 189 μM in different mitochondrial preparations) (Gunter and Sheu, 2009), that can be overcome by the proximity between mitochondria and the Ca2+ source, i.e., the cytosolic orifice of calcium channels at the mitochondria-ER (see below) or mitochondria-plasma membrane contact sites, where local subdomains of high Ca2+ concentration can be generated (Rizzuto et al., 1999; Montero et al., 2000). In heart and liver cells, an additional calcium uptake mechanism that does not rely on the MCU, termed rapid uptake mode, has also been described (Buntinas et al., 2001). Once accumulated in the matrix, calcium ions can be buffered by forming reversible Ca2+-phosphate complexes, or be exported back to the cytosol by a Na+/Ca2+ exchanger (Figure 1) (Gunter and Sheu, 2009). Interestingly, mitochondrial transport along microtubule tracks is negatively regulated by calcium (Yi et al., 2004) (see also below), and in Drosophila Ca2+ ions were found to disrupt the interaction between the motor protein kinesin and the Miro GTPase, which is required for the transport of these organelles along microtubules (Figure 1) (MacAskill et al., 2009). Local subcellular regions of elevated [Ca2+]cyto building up during the opening of voltage-gated plasma membrane Ca2+ channel – as observed upon arrival of an action potential at the nerve terminal – might thus cause mitochondria to pause and transiently accumulate close to active zones where SVs will fuse.
In neurons, [Ca2+]cyto peaks stimulate SV exocytosis and hence neurotransmitter release. This effect is mediated by synaptotagmin, a Ca2+ sensor that resides in the vesicle membrane and promotes SNARE-mediated fusion with the presynaptic membrane (Tucker et al., 2004; Chicka et al., 2008).
By sequestering Ca2+, presynaptic mitochondria can rapidly buffer the [Ca2+]cyto and thus diminish the SV release probability, thereby accelerating recovery of neurotransmission after nerve stimulation. This role of mitochondria in Ca2+ buffering is prominent in chromaffin cells (Montero et al., 2000) and in certain types of synapses, for example at the rat calyx of Held (Billups and Forsythe, 2002) and at the mouse neuromuscular junction (NMJ) (David and Barrett, 2003). In these synapses, pharmacological inhibition of mitochondrial Ca2+ uptake slows down the decay of the [Ca2+]cyto peak observed after nerve stimulation. This in turn affects neurotransmission, as synaptic depression is more rapid (David and Barrett, 2003) and under these conditions recovery from synaptic depression after a stimulus train is slowed down (Billups and Forsythe, 2002), suggesting that the prolonged presence of free calcium ions in the cytosol stimulates the depletion of the pool of available SVs. A first effect of mitochondrial calcium uptake is thus to restrict the period of neurotransmitter release after the arrival of an action potential at the nerve terminal.
Stimulation of SV exocytosis by cytosolic calcium ions is also at the heart of the so-called “residual calcium hypothesis”, according to which residual calcium ions present in the cytoplasm during the decay of the [Ca2+]cyto peak after each action potential increase the probability of neurotransmitter release (Katz and Miledi, 1968). Consistently, slow Ca2+ release from intracellular stores – i.e., the ER and/or mitochondria – after the stimulus end causes a persistent elevation of [Ca2+]cyto that in turn stimulates SV exocytosis (Tang and Zucker, 1997; Yang et al., 2003). This has been well described at the crayfish NMJ, where inhibition of mitochondrial Ca2+ uptake leads to a higher [Ca2+]cyto during tetanic stimulation, as in the work from the groups of Forsythe and Barrett described above, and increases short-term augmentation (Tang and Zucker, 1997). In contrast, decay of the [Ca2+]cyto peak after tetanus is faster than in untreated neurons, thereby reducing long-term potentiation (Tang and Zucker, 1997). Recently, the importance of mitochondria in controlling presynaptic [Ca2+]cyto and hence in synaptic plasticity has been illustrated in mammals as well. Defective calcium buffering at mouse hippocampal presynaptic terminals, resulting from a lack of the microtubule-binding protein syntaphilin required for axonal docking of mitochondria (Figure 1), was shown to cause an elevation of [Ca2+]cyto during intense stimulation, prolonging the facilitation phenomenon observed in control neurons (Kang et al., 2008). In short, a second consequence of calcium buffering by mitochondria is that neurotransmitter release can be prolonged after the stimulus end as Ca2+ ions are progressively unloaded from the mitochondrial matrix into the cytosol.
There are thus both a negative and a positive effect of mitochondrial calcium sequestration on synaptic activity. According to the residual calcium hypothesis, a defect in calcium buffering is expected to favor recovery from depression, as indeed observed by several groups (Dittman and Regehr, 1998; Wang and Kaczmarek, 1998). This however contrasts with the results from the groups of Barrett and Forsythe discussed above, according to which mitochondrial calcium buffering shortens synaptic depression (Billups and Forsythe, 2002; David and Barrett, 2003). This apparent discrepancy is most likely due to the use of different stimulation protocols in various model systems, and to the different approaches used to observe calcium buffering, either via mobile calcium buffers (Dittman and Regehr, 1998; Wang and Kaczmarek, 1998) or specifically by the mitochondria (Billups and Forsythe, 2002; David and Barrett, 2003).
More importantly, this controversy also stresses the versatile role of calcium in the SV cycle. Indeed, Ca2+ ions not only influence exocytosis of SVs but also their recycling via endocytosis (Yao et al., 2009). The [Ca2+]cyto required for proper endocytosis at synapses is much lower than the concentration needed to cause SV exocytosis and is actually close to the intra-axonal [Ca2+]cyto (Shupliakov, 2009), but there is a positive correlation between the intracellular calcium concentration and the endocytic rate (Wu et al., 2009). Experimental data collected e.g., from the frog NMJ (Ceccarelli and Hurlbut, 1980), giant lamprey synapse (Gad et al., 1998) and rat hippocampal neurons (Balaji et al., 2008) suggest that extracellular Ca2+ might actually be required only to increase the endocytic capacity of the presynaptic membrane under conditions of intense stimulation, in order to compensate the large influx of exocytic material (Dittman and Ryan, 2009). The molecular mechanism of this regulation is only starting to be elucidated, but it appears that the calcium-binding protein calmodulin initiates endocytosis at the synapse and facilitates both plasma membrane invagination and fission (Wu et al., 2009). Moreover, other Ca2+-binding endocytic proteins including synaptotagmin and endophilin have been identified (Brose et al., 1992; Chen et al., 2003). In conclusion, the overall effect of Ca2+ ions on neurotransmitter release should thus be regarded as a balance between the stimulation of SV exocytosis, which can promote either recovery from depression or SV depletion depending on the experimental conditions, and a positive effect on SV recycling.
Notably, deregulated mitochondrial Ca2+ uptake can have deleterious consequences for neurons. When mitochondria are overloaded with Ca2+, they undergo a dramatic event termed mitochondrial permeability transition (MPT), i.e., the formation of non-selective pores in the inner mitochondrial membrane that allows the entry of any molecule smaller than 1,500 Dalton into the mitochondrial matrix, causing an increase in osmotic pressure leading to mitochondrial swelling and rupture of the outer mitochondrial membrane (Norenberg and Rao, 2007). Strikingly, synaptic mitochondria are able to buffer less Ca2+ ions and are more prone to display MPT than non-synaptic mitochondria isolated from the same sample of rat cortical brain tissue (Brown et al., 2006) or mouse sympathetic neurons (Núñez et al., 2007). The reason for this difference in Ca2+-handling capacity remains unclear, but it might favor Ca2+-dependent exocytosis at the presynaptic sites by maintaining a higher [Ca2+]cyto. Yet a reduced Ca2+-buffering capacity of synaptic mitochondria, caused under pathogenic conditions or by aging, may presumably increase their susceptibility to MPT and subsequent release of cell-death molecules (see below), leading to synapse degeneration as observed e.g., in Alzheimer's disease (Selkoe, 2002), schizophrenia (Glantz et al., 2006) or Huntington's disease (Damiano et al., 2010).
In some synapses like e.g., the Drosophila NMJ mitochondrial contribution to Ca2+ buffering appears to be only minor and/or easily compensated by alternative mechanisms, at least during mild stimulation (Guo et al., 2005; Verstreken et al., 2005; Lnenicka et al., 2006). Inhibiting mitochondrial Ca2+ uptake in fly motor nerve terminals by adding TPP+ (tetraphenylphosphonium) (Verstreken et al., 2005) or antimycin A1 (Lnenicka et al., 2006) (Figure 1) does indeed not significantly influence calcium clearance or SV mobilization and recycling after a 10 Hz train stimulus. In contrast, decay of the [Ca2+]cyto peak is significantly slowed down when the Ca2+ ATPase mediating calcium uptake into the ER is pharmacologically blocked or, to an even greater extent, when the plasma membrane Ca2+ ATPase (PMCA) is inhibited (Lnenicka et al., 2006). Although Ca2+ clearance is thus mainly ensured by the PMCA (Lnenicka et al., 2006), calcium storage in the ER lumen contributes to evoked neurotransmitter release at the Drosophila NMJ, supposedly by allowing the [Ca2+]cyto to remain elevated after the stimulus end (Sanyal et al., 2005). Similarly, in frog motor nerve terminals intracellular Ca2+ release from the ER after cessation of stimulation leads to post-tetanic potentiation (PTP) (Narita et al., 2000). In this context, it is worth noting that a very close physical connection between the ER and mitochondria can be observed in many cell types, including neurons (Mironov and Symonchuk, 2006). Such contacts, termed mitochondria-associated ER membrane (MAM), allow the bidirectional exchange of phospholipids and Ca2+ ions between these organelles (Hayashi et al., 2009b; Kornmann et al., 2009; Rizzutto et al., 2009). In mouse respiratory neurons, where both the ER and mitochondria contribute to calcium-dependent modulation of synaptic activity, some of the Ca2+ ions transiently accumulated within ER vesicles in response to a stimulus are subsequently transferred to the mitochondrial matrix (Mironov and Symonchuk, 2006). If this transfer is prevented, the peak of [Ca2+]mito observed after stimulation is smaller compared to controls and neurotransmitter release is reduced (Mironov and Symonchuk, 2006). Given the low affinity of the mitochondrial calcium uptake system, MAMs may thus play a critical role in the capacity of mitochondria to promptly respond to a change in [Ca2+]cyto and thereby affect many aspects of mitochondrial biology (Hayashi et al., 2009b), including the control of SV exocytosis and/or endocytosis.
Finally, in addition to their direct effects on exocytosis and endocytosis, Ca2+ ions can also regulate the SV cycle indirectly by modulating mitochondrial bioenergetics. Three enzymes of the Krebs cycle residing in the mitochondrial matrix are indeed activated in a calcium-dependent manner (Duchen, 1992), so that mitochondrial calcium uptake increases the flux of electrons through the electron transport chain (ETC) and hence ATP production (Figure 1) (Graier et al., 2007; Gunter and Sheu, 2009). As a consequence, drugs preventing mitochondrial calcium uptake also negatively affect ATP production, thereby complicating the interpretation of experimental data. Thus it is advisable to look at the effects of these drugs under conditions where the cytosolic ATP level is artificially preserved.
In conclusion, Ca2+ ions play a versatile role in several steps of the SV cycle through different Ca2+ sensors. Mitochondria thus affect exocytosis and endocytosis of SVs through their ability to regulate Ca2+ homeostasis. However, the relative contribution of the ER and mitochondria to synaptic Ca2+-buffering appears to vary between different neuronal systems and might be influenced by the integration of different signaling pathways (Rizzuto, 2003).
Mitochondrial ATP Production is Required for SV Mobilization and Recycling Under Intense Stimulation
The most well-known function of mitochondria is to produce energy, and in neurons they generate the majority of ATP (about 90 %) via aerobic metabolism (Figure 1), the rest being provided by glycolysis in the cytosol (Mathews and Van Holde, 1995). This energy supply is critical since proper neuronal function requires ATP in order to fuel ion pumps, to organize cytoskeletal components, to support numerous phosphorylation reactions (Attwell and Laughlin, 2001) and for many steps in the SV cycle including myosin-dependent axonal transport of SVs, their SNARE-mediated fusion with the plasma membrane, membrane scission, vesicle uncoating and refilling of vesicles with neurotransmitter (Murthy and Camilli, 2003).
Because of the highly specialized architecture of neurons, with their long processes contacting cells located far away from the cell body, ATP diffusion from a central location to the distal synapses is thought to be too slow to meet these energy requirements (Kuiper et al., 2008). Accordingly, in neurons mitochondrial biogenesis occurs mostly in the soma (Davis and Clayton, 1996) but these organelles can subsequently be transported along cytoskeleton tracks in the neuronal processes (Figure 1) (Hollenbeck and Saxton, 2005), where they show saltatory bidirectional movement, with frequent pauses and changes of direction (Cai and Sheng, 2009). Mitochondria are then specifically retained in regions of the axon where the energy demand is high (Hollenbeck, 2005), including synapses (Palay, 1956; Nguyen et al., 1997) and in particular near the active zones where SVs are released (Rowland et al., 2000; Perkins et al., 2010). Moreover, there is a positive correlation between anterograde axonal transport of mitochondria and the electrical potential across their inner membrane (Miller and Sheetz, 2004) which drives ATP production (Alfonzo and Racker, 1979) (Figure 1), and mitochondria with a high potential tend to accumulate at synapses (Lee and Peng, 2006). Finally, there appears to be a direct relationship between the frequency and/or the intensity of activity in a given synapse and the number and metabolic activity of presynaptic mitochondria, both under physiological and pathological conditions (Atwood et al., 1986; Nguyen et al., 1997; Brodin et al., 1999; Dubinsky, 2009). All these observations point to a central role of mitochondrial ATP production in neuronal transmission, but the exact molecular mechanisms involved in this regulation are only beginning to be deciphered.
The number of presynaptic mitochondria is partially regulated by DRP1 (dynamin related protein) (Verstreken et al., 2005). This protein mediates mitochondrial fission (Praefcke and McMahon, 2004; Rube and van der Bliek, 2004) and its activity is controlled by calcineurin, a Ca2+ responsive phosphatase (Cereghetti et al., 2008). High Ca2+ leads to calcineurin activation (Guerini, 1997) and in turn to dephosphorylation and activation of DRP1 and thus mitochondrial fission, which has been associated with the transport of mitochondria into the synapse (Verstreken et al., 2005; Cereghetti et al., 2008) (Figure 2). Taken together, these data support a model where dynamic mitochondria distribution in neurons is adjusted to meet the energy requirement for proper neuronal communication. Several studies now start to shed light on the exact nature of the presynaptic events that most critically depend on mitochondrial ATP production.
At Drosophila NMJ synapses, SVs can be assigned to different, but spatially mixed functional vesicle pools, the ECP and RP (Kuromi and Kidokoro, 2005). Several mutants have been used to examine how these two vesicles pools are affected by mitochondrial dysfunction. The first observation that was made is that synaptic communication is rapidly inhibited when the activity of the mitochondrial ATP translocase is switched off by means of the thermosensitive mutation sesB (stress sensitive B), suggesting that the cytosolic pool of ATP at synaptic terminals is turned over rapidly under basal conditions (Rikhy et al., 2003; Trotta et al., 2004), at least at high temperature. Further studies showed that these mutants have problems maintaining SV cycling during periods of high demand. Vesicle trafficking defects are also observed when mitochondrial morphology is altered due to impaired fission in thermosensitive drp1 mutant flies (Rikhy et al., 2007). Together, these observations suggest that mitochondria extensively control neuronal function, maybe by influencing SV pools. Because both the sesB and drp1 mutations enhance the phenotype of the paralytic mutant shibire, where endocytosis is known to be blocked at the restrictive temperature (Hinshaw and Schmid, 1995), one possibility proposed by the authors is that reduced ATP levels could affect endocytosis, thereby preventing efficient SV recycling (Rikhy et al., 2003, 2007).
Remarkably, in the absence of presynaptic mitochondria in drp1 or miro (mitochondrial Rho-GTPase) loss-of-function mutant flies basal synaptic activity at the Drosophila neuromuscular junction (NMJ) appears not to be significantly altered. In these flies mitochondrial fission or axonal mitochondrial transport are inhibited, respectively, resulting in synapses largely devoid of mitochondria (Guo et al., 2005; Verstreken et al., 2005). In contrast, these mutants show impaired neurotransmission during tetanic stimulation (Guo et al., 2005; Verstreken et al., 2005) and this phenotype can be largely restored by supplementing synapses with exogenous ATP (Verstreken et al., 2005), suggesting that energy production by presynaptic mitochondria is essential only during intense neuronal activity. Failure of the drp1 mutant to sustain neuronal activity during intense stimulation is due to a defect in RP mobilization, while basal exocytosis and endocytosis of ECP vesicles is unaffected (Verstreken et al., 2005). Similar effects on RP vesicle mobilization are observed at wild-type NMJs treated with oligomycin, an inhibitor of the mitochondrial ATP synthase (Figure 1), and forward-filling drp1 motor neurons with ATP restores the vesicle trafficking defects in the RP (Verstreken et al., 2005). Thus, mitochondrial ATP production is specifically required for mobilization of the RP, a process believed to underlie PTP (Kuromi and Kidokoro, 2005). In particular, ATP appears to be needed for RP vesicle propulsion along actin filaments mediated by the myosin ATPase complex (Verstreken et al., 2005). An additional role of ATP in RP mobilization is exerted after its conversion into cAMP by phosphodiesterase and adenylate cyclase. cAMP indeed activates the PKA kinase, which mediates unthetering of RP vesicles and allows their subsequent recruitment at the active zone (Kuromi and Kidokoro, 2005). A very similar dependence of neurotransmission on mitochondrial ATP production has recently been observed in mammalian neurons, where anterograde transport of mitochondria along the axon relies on the adaptor protein syntabulin, linking these organelles to the kinesin heavy chain (Figure 1) (Cai et al., 2005). Knock-down of syntabulin or disruption of its interaction with kinesin impair neurotransmitter release under intense stimulation conditions and this defect is partially rescued by exogenous ATP addition (Ma et al., 2009), paralleling the effects of the fly drp1 mutation (Verstreken et al., 2005).
Interestingly, in rat hippocampal neurons the GTPase activity of Drp1 appears to be positively regulated by the antiapoptotic protein Bcl-xL (Li et al., 2008). By activating Drp1, Bcl-xL promotes mitochondrial fission and recruitment at developing synaptic sites, and is believed to thereby increase ATP availability to fuel synaptogenesis and further synaptic activity (Jonas et al., 2003; Li et al., 2008). Overexpression of either Bcl-xL or Drp1 in rat hippocampal neurons leads to the formation of larger SV clusters compared to controls. Conceivably, the presence of an increased number of mitochondria at the synapses may stimulate the formation of a larger RP and/or favor SV recycling (Li et al., 2008). This is likely to be attributed to increased ATP release from mitochondria since, at least at the squid giant synapse, the positive effects of Bcl-xL overexpression on neurotransmission can be mimicked by ATP injection (Jonas et al., 2003).
That mitochondrial energy production is required for the mobilization of SVs is also supported by numerous studies investigating the spatial organization of presynaptic mitochondria and SVs. In the cat calyx of Held presynaptic mitochondria are positioned about 200 nm away from the plasma membrane, next to the active zones, within a characteristic structure named mitochondrion-associated adherens complex (MAC) (Rowland et al., 2000). Compared to other mitochondria in the same neuron, MAC mitochondria display a higher density of crista-membranes, suggesting increased metabolic activity (Perkins et al., 2010). Strikingly, dense populations of SVs are connected to cytoskeletal elements emanating from MACs, which appear to form a barrier between vesicle pools associated with neighboring active zones (Perkins et al., 2010). As suggested by the authors, this physical separation might contribute to the different vesicle release probability observed between different active zones. At the calyx of Held from rat, presynaptic mitochondria are located within so-called “swellings”, where they are surrounded by active zones and a large population of SVs thought to represent the RP (Wimmer et al., 2006). In both types of calyces, this particular distribution of mitochondria relative to synaptic vesicles might contribute to trap ATP and glutamate – which is generated in mitochondria from precursors emanating from the Krebs cycle or from glutamine produced by glial cells (Figure 1) – in restricted areas, thereby facilitating vesicle refilling and mobilization, ensuring sustained synaptic transmission during intense neuronal activity.
In conclusion, although most processes involved in the SV cycle are known to be ATP-dependent, experimental evidence point to a critical role of mitochondrial ATP production mostly in the establishment of large SV pools and in sustained neurotransmitter release during intense neuronal activity, i.e., for mobilization of RP vesicles and for vesicle recycling via endocytosis. In contrast, low ATP levels produced by glycolysis in the cytosol appear to be sufficient to fuel other steps of the SV cycle (Calupca et al., 2001), and this is likely accomplished by optimal positioning of glycolytic enzymes close to the ATP consumption sites (Ishida et al., 2009).
Mitochondria and Disease
As described above, energy generation by mitochondria is of major importance for the SV cycle in neurons, and one can expect that disturbed ATP production may have deleterious consequences for neurons in health and disease. Defects in most of the complexes of the ETC (Figure 1) are indeed associated with different neurodegenerative diseases and numerous mutations in mitochondrial-associated proteins have been linked to a variety of neurological diseases (Ly and Verstreken, 2006).
One of the best known examples of this is Parkinsonism. The first evidence that mitochondria are involved in Parkinson's disease (PD) came from the observation that mitochondrial drugs like MPTP and rotenone that specifically affect complex I of the ETC (Figure 1) cause PD-like symptoms, including degeneration of dopaminergic neurons (German et al., 1989; Betarbet et al., 2000; Panov et al., 2005). It was later shown that mutations in the Parkinson related gene pink1 similarly lead to a reduced complex I activity (Gautier et al., 2008; Morais et al., 2009). The product of two other Parkinson related genes, parkin and DJ-1, also appear to be involved in the regulation of mitochondrial functions (Bonifati et al., 2003; Hayashi et al., 2009a; Wild and Dikic, 2010). Furthermore, amyotrophic lateral sclerosis (ALS), Leigh's syndrome and multiple sclerosis can also be caused by defects in several complexes of the ETC (Kalman et al., 2007; Petrozzi et al., 2007; Finsterer, 2008).
Aside from direct ETC dysfunction that leads to reduced ATP production, disturbed mitochondrial transport to sites of high energy demand, including synapses, may also lead to local ATP depletion and thus synaptic transmission deficits. As discussed before, DRP1, a regulator of mitochondrial fission appears to be a key regulatory switch controlling mitochondrial transport and thus synaptic ATP production, possibly in response to calcium influx and neuronal activity (Lee et al., 2004; Verstreken et al., 2005; Cereghetti et al., 2008). While to date no inherited disease resulting directly from mutations in genes regulating fission are known; increased levels of DRP1, and thus presumably also increased mitochondrial fission and synaptic transport has been implicated in alleviating Parkinson disease-like phenotypes in flies, yet it is still unclear whether these effects are direct or not (Morais et al., 2009; Poole et al., 2010). Indeed, increased DRP1 activity is predicted to augment synaptic ATP production by concentrating mitochondria at active synapses, in turn facilitating SV mobilization.
The converse process, mitochondrial fusion, results in lower mitochondrial membrane potential and thus reduced ATP production (Chen and Chan, 2005) and is associated with neurodegeneration. Three proteins are required for mitochondrial fusion: OPA1, mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) (Chen and Chan, 2005). The latter two proteins are also required for axonal transport of mitochondria (Misko et al., 2010). MFN2 has been identified as one of the genes causing Charcot-Marie-Tooth type 2A (CMT2A) syndrome. CMT2A is a neuropathy that affects sensory and motor neurons of the distal extremities and is characterized by progressive weakness of the muscles followed by muscular atrophy (Zuchner et al., 2004). Mutations in OPA1 cause autosomal dominant optic atrophy (DOA), characterized by a degeneration of retinal ganglia cells leading to optic nerve atrophy (Alexander et al., 2000; Delettre et al., 2000). At first sight it might seem that mutations in two genes encoding proteins involved in mitochondrial fusion cause two diseases with differential phenotypes. Yet CMT2A patients have been described to suffer from optic atrophy (Banchs et al., 2008). Likewise, some DOA patients express additional symptoms including peripheral motor-sensory neuropathy (Amati-Bonneau et al., 2008). Linking these mitochondrial fusion related diseases to defects in neuronal function, the reduced ATP levels as a consequence of less mitochondrial fusion may be responsible for defects in SV release and/or recycling under intense stimulation. Taken together, these data support the hypothesis that ATP production may play a critical role in neuronal communication during intense stimulation and that mitochondria play a specific role in regulating synaptic strength, suggesting that neurological disorders with defects in presynaptic mitochondrial function or mitochondrial transport also display abnormalities in the SV cycle.
One puzzling feature common to all the diseases discussed here is the selective tissue vulnerability associated with mitochondrial defects. First of all, the particular mode of mitochondrial inheritance might contribute to the appearance of clinical manifestations only in a specific cell population, since stochastic distribution of mitochondria between daughter cells can lead to very different ratio of mutant to wild-type organelles after cytokinesis (Chan, 2006). Such a genetic drift has indeed been observed for some mitochondrial encephalomyopathies where mitochondrial DNA carrying the causative mutation accumulates in a specific region of the brain (Kurenai et al., 2000; Betts et al., 2006), although the underlying mechanisms remains unknown.
Moreover, compared to other cell types neurons appear particularly susceptible to mitochondrial dysfunction. This is most likely related to their very high energy demands and their specific architecture, as mitochondria must travel long distances from the cell body to reach regions of intense ATP consumption like the presynaptic sites. Defective mitochondrial fusion and/or fission – and hence altered mitochondria recruitment at the nerve terminals – is thus particularly detrimental for these cells (Guo et al., 2005; Verstreken et al., 2005). This is expected to be even more critical for neurons possessing a very long axon, like those innervating distal muscles that are affected in patients with CMT disease (Misko et al., 2010).
Finally, in addition to this cellular context the tissue context might also influence the sensitivity of some neurons to mitochondrial dysfunction. For example, dopaminergic neurons which are progressively lost in PD are innervated by glutamatergic neurons, likely making them more susceptible to glutamate excitotoxicity and hence to calcium overload (Caudle and Zhang, 2009). A defect in mitochondrial calcium handling capacity and/or bioenergetics would in this model predispose dopaminergic neurons to excitotoxic damage and be particularly detrimental for them compared to other cell types (Greene and Greenamyre, 1996; Nicholls et al., 2007). We thus surmise that dysfunction or degeneration of specific neuronal populations in specific neurological diseases may be a consequence of both cell autonomous effects that inherently regulate mitochondrial function and cell non-autonomous effects, where the particular cellular context a neuron finds itself in may (over-) stress mitochondrial function.
In conclusion, there is now accumulating evidence that the capacity of presynaptic mitochondria to produce ATP and to buffer Ca2+ is of major importance to maintain neuronal communication, at least in part by controlling SVs mobilization and recycling. Mitochondrial dysfunctions linked to neurodegenerative diseases are thus likely to exert their effect at least in part through an alteration of the SV cycle. In the light of the literature detailed above, it seems reasonable to speculate that disease-related mutations that eventually cause a reduction of mitochondrial ATP production and calcium buffering capacity in the nerve terminals would negatively affect neurotransmission by preventing replenishment of the ECP and vesicle mobilization from the RP, as indeed observed in pink1 mutant Drosophila where expression of a clinical mutant fails to rescue RP mobilization (Morais et al., 2009). This model does not exclude that other mechanisms such as reactive oxygen species production or enhanced neuronal death are involved. These aspects lie outside the scope of this review and have been excellently reviewed by others (see for example Yuan and Yankner, 2000; Benn and Woolf, 2004; Naoi et al., 2009). Further work is now needed to shed light on the exact implications of altered mitochondrial metabolism and dynamics in the pathogenesis of neuronal disorders, and hopefully to generate more efficient treatments for these diseases.
Statements
Acknowledgments
We like to thank the members of the Verstreken lab for stimulating discussions. Work in the Verstreken lab is supported by a Marie Curie Excellence grant (MEXT-CT-2006-042267); an FWO grant (G.0747.09); the Research Fund KU Leuven; a Methusalem grant of the Flemish Government; the Francqui Foundation an ERC starting grant and VIB. Melissa Vos is a fellow of the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen) and Elsa Lauwers is a Research Fellow of the National Fund for Scientific Research, Belgium.
Conflict of interest
The authors declare the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Alexander C. Votruba M. Pesch U. E. A. Thiselton D. L. Mayer S. Moore A. Rodriguez M. Kellner U. Leo-Kottler B. Auburger G. Bhattacharya S. S. Wissinger B. (2000). OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet.26, 211–215.10.1038/79944
2
Alfonzo M. Racker E. (1979). Components and mechanism of action of ATP-driven proton pumps. Can. J. Biochem.57, 1351–1358.10.1139/o79-180
3
Amati-Bonneau P. Valentino M. L. Reynier P. Gallardo M. E. Bornstein B. Boissiere A. Campos Y. Rivera H. de la Aleja J. G. Carroccia R. Iommarini L. Labauge P. Figarella-Branger D. Marcorelles P. Furby A. Beauvais K. Letournel F. Liguori R. La Morgia C. Montagna P. Liguori M. Zanna C. Rugolo M. Cossarizza A. Wissinger B. Verny C. Schwarzenbacher R. Martin M. A. Arenas J. Ayuso C. Garesse R. Lenaers G. Bonneau D. Carelli V. (2008). OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain131, 338–351.10.1093/brain/awm298
4
Attwell D. Laughlin S. B. (2001). An Energy Budget for Signaling in the Grey Matter of the Brain. J. Cereb. Blood Flow Metab.21, 1133–1145.10.1097/00004647-200110000-00001
5
Atwood H. L. Wojtowicz J. M. John R. S. Ronald J. B. (1986). “Short-term and long-term plasticity and physiological differentiation of crustacean motor synapses,” in International Review of Neurobiology, ed. BradleyJ. R. S. a. R. J. (Orlando: Academic Press), 275–362.
6
Balaji J. Armbruster M. Ryan T. A. (2008). Calcium Control of Endocytic Capacity at a CNS Synapse. J. Neurosci.28, 6742–6749.10.1523/JNEUROSCI.1082-08.2008
7
Banchs I. Casasnovas C. Montero J. Martínez-Matos J. A. Volpini V. (2008). Two Spanish families with Charcot-Marie-Tooth type 2A: clinical, electrophysiological and molecular findings. Neuromuscul. Disord.18, 974–978.10.1016/j.nmd.2008.09.006
8
Benn S. C. Woolf C. J. (2004). Adult neuron survival strategies [mdash] slamming on the brakes. Nat. Rev. Neurosci.5, 686–700.10.1038/nrn1477
9
Ben-Shachar D. (2009). The interplay between mitochondrial complex I, dopamine and Sp1 in schizophrenia. J. Neural Transm.116, 1383–1396.10.1007/s00702-009-0319-5
10
Betarbet R. Sherer T. B. MacKenzie G. Garcia-Osuna M. Panov A. V. Greenamyre J. T. (2000). Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci.3, 1301–1306.10.1038/81834
11
Betts J. Jaros E. Perry R. H. Schaefer A. M. Taylor R. W. Abdel-All Z. Lightowlers R. N. Turnbull D. M. (2006). Molecular neuropathology of MELAS: level of heteroplasmy in individual neurons and evidence of extensive vascular involvement. Neuropathol. Appl. Neurobiol.32, 359–373.10.1111/j.1365-2990.2006.00731.x
12
Billups B. Forsythe I. D. (2002). Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J. Neurosci.22, 5840–5847.
13
Bonifati V. Rizzu P. Squitieri F. Krieger E. Vanacore N. Swieten J. C. Brice A. Duijn C. M. Oostra B. Meco G. Heutink P. (2003). DJ-1(PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol. Sci.24, 159–160.10.1007/s10072-003-0108-0
14
Bossy-Wetzel E. Petrilli A. Knott A. B. (2008). Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci.31, 609–616.10.1016/j.tins.2008.09.004
15
Brodin L. Bakeeva L. Shupliakov O. (1999). Presynaptic mitochondria and the temporal pattern of neurotransmitter release. Philos. Trans. R. Soc. Lond., B, Biol. Sci.354, 365–372.10.1098/rstb.1999.0388
16
Brose N. Petrenko A. Sudhof T. Jahn R. (1992). Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science256, 1021–1025.10.1126/science.1589771
17
Brown M. R. Sullivan P. G. Geddes J. W. (2006). Synaptic mitochondria are more susceptible to Ca2+ overload than nonsynaptic mitochondria. J. Biol. Chem.281, 11658–11668.10.1074/jbc.M510303200
18
Buntinas L. Gunter K. K. Sparagna G. C. Gunter T. E. (2001). The rapid mode of calcium uptake into heart mitochondria (RaM): comparison to RaM in liver mitochondria. Biochim. Biophys.1504, 248–261.10.1016/S0005-2728(00)00254-1
19
Cai Q. Gerwin C. Sheng Z. (2005). Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J. Cell Biol.170, 959–969.10.1083/jcb.200506042
20
Cai Q. Sheng Z.-H. (2009). Mitochondrial transport and docking in axons. Exp. Neurol.218, 257–267.10.1016/j.expneurol.2009.03.024
21
Calupca M. A. Prior C. Merriam L. A. Hendricks G. M. Parsons R. L. (2001). Presynaptic function is altered in snake K+-depolarized motor nerve terminals containing compromised mitochondria. J. Physiol.532, 217–227.10.1111/j.1469-7793.2001.0217g.x
22
Caudle W. M. Zhang J. (2009). Glutamate, excitotoxicity, and programmed cell death in parkinson disease. Exp. Neurol.220, 230–233.10.1016/j.expneurol.2009.09.027
23
Ceccarelli B. Hurlbut W. (1980). Ca2+-dependent recycling of synaptic vesicles at the frog neuromuscular junction. J. Cell Biol.87, 297–303.10.1083/jcb.87.1.297
24
Cereghetti G. M. Stangherlin A. de Brito O. M. Chang C. R. Blackstone C. Bernardi P. Scorrano L. (2008). Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. U.S.A.105, 15803–15808.10.1073/pnas.0808249105
25
Chan D. C. (2006). Mitochondria: dynamic organelles in disease, aging, and development. Cell125, 1241–1252.
26
Chen H. Chan D. C. (2005). Emerging functions of mammalian mitochondrial fusion and fission. Hum. Mol. Genet.14, 7.10.1093/hmg/ddi270
27
Chen H. Chan D. C. (2009). Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum. Mol. Genet.18, 8.10.1093/hmg/ddp326
28
Chen Y. Deng L. Maeno-Hikichi Y. Lai M. Chang S. Chen G. Zhang J. -f. (2003). Formation of an endophlin-Ca2+ channel complex is critical for clathrin-mediated synaptic vesicle endocytosis. Cell115, 37–48.
29
Chicka M. C. Hui E. Liu H. Chapman E. R. (2008). Synaptotagmin arrests the SNARE complex before triggering fast, efficient membrane fusion in response to Ca2+. Nat. Struct. Mol. Biol.15, 827–835.10.1038/nsmb.1463
30
Correia C. Coutinho A. Diogo L. Grazina M. Marques C. Miguel T. Ataíde A. Almeida J. Borges L. Oliveira C. Oliveira G. Vicente A. (2006). Brief report: high frequency of biochemical markers for mitochondrial dysfunction in autism: no association with the mitochondrial aspartate/glutamate carrier SLC25A12 gene. J. Autism Dev. Disord.36, 1137–1140.10.1007/s10803-006-0138-6
31
Damiano M. Galvan L. Déglon N. Brouillet E. (2010). Mitochondria in Huntington's disease. Biochim. Biophys. Acta1802, 52–61.
32
David G. Barrett E. F. (2003). Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J. Physiol.548, 425–438.10.1113/jphysiol.2002.035196
33
Davis A. Clayton D. (1996). In situ localization of mitochondrial DNA replication in intact mammalian cells. J. Cell Biol.135, 883–893.10.1083/jcb.135.4.883
34
Delettre C. Lenaers G. Griffoin J.-M. Gigarel N. Lorenzo C. Belenguer P. Pelloquin L. Grosgeorge J. Turc-Carel C. Perret E. Astarie-Dequeker C. Lasquellec L. Arnaud B. Ducommun B. Kaplan J. Hamel C. P. (2000). Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat. Genet.26, 207–210.10.1038/79936
35
DiMauro S. Schon E. A. (2008). Mitochondrial Disorders in the Nervous System. Annu. Rev. Neurosci.31, 91–123.10.1146/annurev.neuro.30.051606.094302
36
Dittman J. Ryan T. A. (2009). Molecular Circuitry of Endocytosis at Nerve Terminals. Annu. Rev. Cell Dev. Biol.25, 133–160.10.1146/annurev.cellbio.042308.113302
37
Dittman J. S. Regehr W. G. (1998). Calcium dependence and recovery kinetics of presynaptic depression at the climbing fiber to purkinje cell synapse. J. Neurosci.18, 6147–6162.
38
Dubinsky J. M. (2009). Heterogeneity of nervous system mitochondria: location, location, location!Exp. Neurol.218, 293–307.10.1016/j.expneurol.2009.05.020
39
Duchen M. (1992). Ca(2+)-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem. J.283, 41–50.
40
Finsterer J. (2008). Leigh and leigh-like syndrome in children and adults. Pediatr. Neurol.39, 223–235.10.1016/j.pediatrneurol.2008.07.013
41
Gad H. Löw P. Zotova E. Brodin L. Shupliakov O. (1998). Dissociation between Ca2+-triggered synaptic vesicle exocytosis and clathrin-mediated endocytosis at a central synapse. Neuron21, 607–616.10.1016/S0896-6273(00)80570-X
42
Gautier C. A. Kitada T. Shen J. (2008). Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. U.S.A.105, 11364–11369.10.1073/pnas.0802076105
43
German D. C. Manaye K. Smith W. K. Woodward D. J. Saper C. B. (1989). Midbrain dopaminergic cell loss in parkinson's disease: computer visualization. Ann. Neurol.26, 507–514.10.1002/ana.410260403
44
Ghosh A. Greenberg M. (1995). Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science268, 239–247.10.1126/science.7716515
45
Glantz L. A. Gilmore J. H. Lieberman J. A. Jarskog L. F. (2006). Apoptotic mechanisms and the synaptic pathology of schizophrenia. Schizophr. Res.81, 47–63.10.1016/j.schres.2005.08.014
46
Graier W. Frieden M. Malli R. (2007). Mitochondria and Ca2+ signaling: old guests, new functions. Pflugers Arch.455, 375–396.10.1007/s00424-007-0296-1
47
Greene J. G. Greenamyre J. T. (1996). Bioenergetics and glutamate excitotoxicity. Prog. Neurobiol.48, 613–634.10.1016/0301-0082(96)00006-8
48
Guerini D. (1997). Calcineurin: Not Just a Simple Protein Phosphatase. Biochem. Biophys. Res. Commun.235, 271–275.10.1006/bbrc.1997.6802
49
Gunter T. E. Buntinas L. Sparagna G. Eliseev R. Gunter K. (2000). Mitochondrial calcium transport: mechanisms and functions. Cell Calcium28, 285–296.10.1054/ceca.2000.0168
50
Gunter T. E. Sheu S.-S. (2009). Characteristics and possible functions of mitochondrial Ca2+ transport mechanisms. Biochim. Biophys. Acta.1787, 1291–1308.10.1016/j.bbabio.2008.12.011
51
Guo X. Macleod G. Wellington A. Hu F. Panchumarthi S. Schoenfield M. Marin L. Charlton M. Atwood H. Zinsmaier K. (2005). The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron47, 379–393.10.1016/j.neuron.2005.06.027
52
Hayashi T. Ishimori C. Takahashi-Niki K. Taira T. Kim Y.-c. Maita H. Maita C. Ariga H. Iguchi-Ariga S. M. M. (2009a). DJ-1 binds to mitochondrial complex I and maintains its activity. Biochem. Biophys. Res. Commun.390, 667–672.10.1016/j.bbrc.2009.10.025
53
Hayashi T. Rizzuto R. Hajnoczky G. Su T.-P. (2009b). MAM: more than just a housekeeper. Trends Cell Biol.19, 81–88.10.1016/j.tcb.2008.12.002
54
Hinshaw J. E. Schmid S. L. (1995). Dynamin self-assembles into rings suggesting a mechanism for coated vesicle budding. Nature374, 190–192.10.1038/374190a0
55
Hollenbeck P. J. (2005). Mitochondria and neurotransmission: evacuating the synapse. Neuron47, 3.10.1016/j.neuron.2005.07.017
56
Hollenbeck P. J. Saxton W. M. (2005). The axonal transport of mitochondria. J. Cell. Sci.118, 5411–5419.10.1242/jcs.02745
57
Ishida A. Noda Y. Ueda T. (2009). Synaptic vesicle-bound pyruvate kinase can support vesicular glutamate uptake. Neurochem. Res.34, 807–818.10.1007/s11064-008-9833-3
58
Jonas E. (2006). BCL-xL regulates synaptic plasticity. Mol. Interv.6, 208–222.10.1124/mi.6.4.7
59
Jonas E. A. Hoit D. Hickman J. A. Brandt T. A. Polster B. M. Fannjiang Y. McCarthy E. Montanez M. K. Hardwick J. M. Kaczmarek L. K. (2003). Modulation of synaptic transmission by the BCL-2 family protein BCL-xL. J. Neurosci.23, 8423–8431.
60
Kahle P. J. Waak J. Gasser T. (2009). DJ-1 and prevention of oxidative stress in Parkinson's disease and other age-related disorders. Free Radic. Biol. Med.47, 1354–1361.10.1016/j.freeradbiomed.2009.08.003
61
Kalman B. Laitinen K. Komoly S. (2007). The involvement of mitochondria in the pathogenesis of multiple sclerosis. J. Neuroimmunol.188, 1–12.10.1016/j.jneuroim.2007.03.020
62
Kang J.-S. Tian J.-H. Pan P.-Y. Zald P. Li C. Deng C. Sheng Z.-H. (2008). Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell132, 137–148.10.1016/j.cell.2007.11.024
63
Katz B. Miledi R. (1968). The role of calcium in neuromuscular facilitation. J. Physiol.195, 481–492.
64
Kornmann B. Currie E. Collins S. R. Schuldiner M. Nunnari J. Weissman J. S. Walter P. (2009). An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science325, 477–481.10.1126/science.1175088
65
Kudin A. P. Zsurka G. Elger C. E. Kunz W. S. (2009). Mitochondrial involvement in temporal lobe epilepsy. Exp. Neurol.218, 326–332.10.1016/j.expneurol.2009.02.014
66
Kuiper J. Oerlemans F. Fransen J. Wieringa B. (2008). Creatine kinase B deficient neurons exhibit an increased fraction of motile mitochondria. BMC Neurosci.9, 73.10.1186/1471-2202-9-73
67
Kurenai T. Eric A. S. Salvatore D. Eduardo B. (2000). Kearns-Sayre syndrome: oncocytic transformation of choroid plexus epithelium. J. Neurol. Sci.178, 29–36.10.1016/S0022-510X(00)00354-3
68
Kuromi H. Kidokoro Y. (2002). Selective replenishment of two vesicle pools depends on the source of Ca2+ at the Drosophila synapse. Neuron35, 333–343.10.1016/S0896-6273(02)00777-8
69
Kuromi H. Kidokoro Y. (2005). Exocytosis and endocytosis of synaptic vesicles and functional roles of vesicle pools: lessons from the drosophila neuromuscular junction. Neuroscientist.11, 138–147.10.1177/1073858404271679
70
Lee C. W. Peng H. B. (2006). Mitochondrial clustering at the vertebrate neuromuscular junction during presynaptic differentiation. J. Neurobiol.66, 522–536.10.1002/neu.20245
71
Lee Y.-j. Jeong S.-Y. Karbowski M. Smith C. L. Youle R. J. (2004). roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell15, 5001–5011.10.1091/mbc.E04-04-0294
72
Li H. Chen Y. Jones A. F. Sanger R. H. Collis L. P. Flannery R. McNay E. C. Yu T. Schwarzenbacher R. Bossy B. Bossy-Wetzel E. Bennett M. V. L. Pypaert M. Hickman J. A. Smith P. J. S. Hardwick J. M. Jonas E. A. (2008). Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A.105, 2169–2174.10.1073/pnas.0711647105
73
Lnenicka G. A. Grizzaffi J. Lee B. Rumpal N. (2006). Ca2⩲ dynamics along identified synaptic terminals in Drosophila larvae. J. Neurosci.26, 12283–12293.10.1523/JNEUROSCI.2665-06.2006
74
Ly C. V. Verstreken P. (2006). Mitochondria at the Synapse. Neuroscientist12, 291–299.10.1177/1073858406287661
75
Ma H. Cai Q. Lu W. Sheng Z.-H. Mochida S. (2009). KIF5B Motor Adaptor Syntabulin Maintains Synaptic Transmission in Sympathetic Neurons. J. Neurosci.29, 13019–13029.10.1523/JNEUROSCI.2517-09.2009
76
MacAskill A. F. Rinholm J. E. Twelvetrees A. E. Arancibia-Carcamo I. L. Muir J. Fransson A. Aspenstrom P. Attwell D. Kittler J. T. (2009). Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron61, 541–555.10.1016/j.neuron.2009.01.030
77
Mathews C. Van Holde K. (1995). Biochemistry, 2nd Edn. New York: Benjamin/Cummings.
78
Miller K. E. Sheetz M. P. (2004). Axonal mitochondrial transport and potential are correlated. J. Cell. Sci.117, 2791–2804.10.1242/jcs.01130
79
Mironov S. L. Symonchuk N. (2006). ER vesicles and mitochondria move and communicate at synapses. J. Cell. Sci.119, 4926–4934.10.1242/jcs.03254
80
Misko A. Jiang S. Wegorzewska I. Milbrandt J. Baloh R. H. (2010). Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the miro/milton complex. J. Neurosci.30, 4232–4240.10.1523/JNEUROSCI.6248-09.2010
81
Montero M. Alonso M. T. Carnicero E. Cuchillo-Ibanez I. Albillos A. Garcia A. G. Garcia-Sancho J. Alvarez J. (2000). Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat. Cell Biol.2, 57–61.10.1038/35000001
82
Morais V. A. Verstreken P. Roethig A. Smet J. Snellinx A. Vanbrabant M. Haddad D. Frezza C. Mandemakers W. Vogt-Weisenhorn D. Van Coster R. Wurst W. Scorrano L. De Strooper B. (2009). Parkinson's disease mutations in PINK1 result in decreased complex I activity and deficient synaptic function. EMBO Mol. Med.1, 99–111.10.1002/emmm.200900006
83
Murthy V. N. Camilli P. D. (2003). Cell biology of the presynaptic terminal. Annu. Rev. Neurosci.26, 701–728.10.1146/annurev.neuro.26.041002.131445
84
Naoi M. Maruyama W. Yi H. Inaba K. Akao Y. Shamoto-Nagai M. (2009). Mitochondria in neurodegenerative disorders: regulation of the redox state and death signaling leading to neuronal death and survival. J. Neural Transm.116, 1371–1381.10.1007/s00702-009-0309-7
85
Narita K. Akita T. Hachisuka J. Huang S.-M. Ochi K. Kuba K. (2000). Functional coupling of Ca2+ channels to ryanodine receptors at presynaptic terminals: amplification of exocytosis and plasticity. J. Gen. Physiol.115, 519–532.10.1085/jgp.115.4.519
86
Nguyen P. V. Marin L. Atwood H. L. (1997). Synaptic physiology and mitochondrial function in crayfish tonic and phasic motor neurons. J. Neurophysiol.78, 281–294.
87
Nicholls D. G. Johnson-Cadwell L. Vesce S. Jekabsons M. Yadava N. (2007). Bioenergetics of mitochondria in cultured neurons and their role in glutamate excitotoxicity. J. Neurosci. Res.85, 3206–3212.10.1002/jnr.21290
88
Niemann A. Ruegg M. La Padula V. Schenone A. Suter U. (2005). Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network. J. Cell Biol.170, 1067–1078.10.1083/jcb.200507087
89
Norenberg M. D. Rao K. V. R. (2007). The mitochondrial permeability transition in neurologic disease. Neurochem. Int.50, 983–997.10.1016/j.neuint.2007.02.008
90
Núñez L. Senovilla L. Sanz-Blasco S. Chamero P. Alonso M. Villalobos C. García-Sancho J. (2007). Bioluminescence imaging of mitochondrial Ca2+ dynamics in soma and neurites of individual adult mouse sympathetic neurons. J. Physiol.580, 385–395.10.1113/jphysiol.2006.126524
91
Palay S. L. (1956). Synapses in the central nervous system. J. Cell Biol.2, 193–202.10.1083/jcb.2.4.193
92
Panov A. Dikalov S. Shalbuyeva N. Taylor G. Sherer T. Greenamyre J. T. (2005). Rotenone model of parkinson disease. J. Biol. Chem.280, 42026–42035.10.1074/jbc.M508628200
93
Perkins G. A. Tjong J. Brown J. M. Poquiz P. H. Scott R. T. Kolson D. R. Ellisman M. H. Spirou G. A. (2010). The micro-architecture of mitochondria at active zones: electron tomography reveals novel anchoring scaffolds and cristae structured for high-rate metabolism. J. Neurosci.30, 1015–1026.10.1523/JNEUROSCI.1517-09.2010
94
Petrozzi L. Ricci G. Giglioli N. Siciliano G. Mancuso M. (2007). Mitochondria and neurodegeneration. Biosci. Rep.27, 87–104.10.1007/s10540-007-9038-z
95
Poole A. C. Thomas R. E. Yu S. Vincow E. S. Pallanck L. (2010). The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/Parkin pathway. PLoS ONE5, e10054.10.1371/journal.pone.0010054
96
Praefcke G. J. K. McMahon H. T. (2004). The dynamin superfamily: universal membrane tubulation and fission molecules?Nat. Rev. Mol. Cell Biol.5, 133–147.10.1038/nrm1313
97
Rikhy R. Kamat S. Ramagiri S. Sriram V. Krishnan K. S. (2007). Mutations in dynamin-related protein result in gross changes in mitochondrial morphology and affect synaptic vesicle recycling at the Drosophila neuromuscular junction. Genes Brain Behav.6, 42–53.10.1111/j.1601-183X.2006.00218.x
98
Rikhy R. Ramaswami M. Krishnan K. S. (2003). A temperature-sensitive allele of drosophila sesb reveals acute functions for the mitochondrial adenine nucleotide translocase in synaptic transmission and dynamin regulation. Genetics165, 1243–1253.
99
Rizzoli S. O. Betz W. J. (2005). Synaptic vesicle pools. Nat. Rev. Neurosci.6, 57–69.10.1038/nrn1583
100
Rizzuto R. (2003). Calcium mobilization from mitochondria in synaptic transmitter release. J. Cell Biol.163, 441–443.10.1083/jcb.200309111
101
Rizzuto R. Pinton P. Brini M. Chiesa A. Filippin L. Pozzan T. (1999). Mitochondria as biosensors of calcium microdomains. Cell Calcium26, 193–200.10.1054/ceca.1999.0076
102
Rowland K. C. Irby N. K. Spirou G. A. (2000). Specialized synapse-associated structures within the calyx of held. J. Neurosci.20, 9135–9144.
103
Rube D. van der Bliek A. (2004). Mitochondrial morphology is dynamic and varied. Mol. Cell. Biochem.256–257, 331–339.10.1023/B:MCBI.0000009879.01256.f6
104
Sanyal S. Consoulas C. Kuromi H. Basole A. Mukai L. Kidokoro Y. Krishnan K. S. Ramaswami M. (2005). Analysis of conditional paralytic mutants in Drosophila sarco-endoplasmic reticulum calcium ATPase reveals novel mechanisms for regulating membrane excitability. Genetics169, 737–750.10.1534/genetics.104.031930
105
Selkoe D. J. (2002). Alzheimer's disease is a synaptic failure. Science298, 789–791.10.1126/science.1074069
106
Shao L. Martin M. V. Watson S. J. Schatzberg A. Akil H. Myers R. M. Jones E. G. Bunney W. E. Vawter M. P. (2008). Mitochondrial involvement in psychiatric disorders. Ann. Med.40, 281–295.10.1080/07853890801923753
107
Shupliakov O. (2009). The synaptic vesicle cluster: a source of endocytic proteins during neurotransmitter release. Neuroscience158, 204–210.10.1016/j.neuroscience.2008.03.035
108
Tanaka A. (2010). Parkin-mediated selective mitochondrial autophagy, mitophagy: Parkin purges damaged organelles from the vital mitochondrial network. FEBS Letters584, 1386–1392.10.1016/j.febslet.2010.02.060
109
Tang Y.-g. Zucker R. S. (1997). Mitochondrial Involvement in Post-Tetanic Potentiation of Synaptic Transmission. Neuron18, 483–491.10.1016/S0896-6273(00)81248-9
110
Trotta N. Rodesch C. K. Fergestad T. Broadie K. (2004). Cellular bases of activity-dependent paralysis in Drosophila stress-sensitive mutants. J. Neurobiol.60, 328–347.10.1002/neu.20017
111
Tucker W. C. Weber T. Chapman E. R. (2004). Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science304, 435–438.10.1126/science.1097196
112
Verstreken P. Ly C. V. Venken K. J. T. Koh T.-W. Zhou Y. Bellen H. J. (2005). Synaptic mitochondria are critical for mobilization of reserve pool vesicles at drosophila neuromuscular junctions. Neuron47, 365–378.10.1016/j.neuron.2005.06.018
113
Verstreken P. Ohyama T. Bellen H. J. (2008). “FM 1-43 labeling of synaptic vesicle pools at the drosophila neuromuscular junction,” in Exocytosis and Endocytosis, ed. IvanovA. I. (Totowa: Humana Press), 349–369.
114
Wang L.-Y. Kaczmarek L. K. (1998). High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature394, 384–388.10.1038/28645
115
Wang X. Su B. Lee H.-g. Li X. Perry G. Smith M. A. Zhu X. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J. Neurosci.29, 9090–9103.10.1523/JNEUROSCI.1357-09.2009
116
Wild P. Dikic I. (2010). Mitochondria get a Parkin’ ticket. Nat. Cell Biol.12, 104–106.10.1038/ncb0210-104
117
Wimmer V. C. Horstmann H. Groh A. Kuner T. (2006). Donut-like topology of synaptic vesicles with a central cluster of mitochondria wrapped into membrane protrusions: a novel structure-function module of the adult calyx of held. J. Neurosci.26, 109–116.10.1523/JNEUROSCI.3268-05.2006
118
Wu X.-S. McNeil B. D. Xu J. Fan J. Xue L. Melicoff E. Adachi R. Bai L. Wu L.-G. (2009). Ca2+ and calmodulin initiate all forms of endocytosis during depolarization at a nerve terminal. Nat. Neurosci.12, 1003–1010.10.1038/nn.2355
119
Yang F. He X.-p. Russell J. Lu B. (2003). Ca2+ influx-independent synaptic potentiation mediated by mitochondrial Na+-Ca2+ exchanger and protein kinase C. J. Cell Biol.163, 511–523.10.1083/jcb.200307027
120
Yao C.-K. Lin Y. Q. Ly C. V. Ohyama T. Haueter C. M. Moiseenkova-Bell V. Y. Wensel T. G. Bellen H. J. (2009). A synaptic vesicle-associated Ca2+ channel promotes endocytosis and couples exocytosis to endocytosis. Cell138, 947–960.
121
Yi M. Weaver D. Hajnoczky G. (2004). Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J. Cell Biol.167, 661–672.10.1083/jcb.200406038
122
Yuan J. Yankner B. A. (2000). Apoptosis in the nervous system. Nature407, 802–809.10.1038/35037739
123
Zenisek D. Matthews G. (2000). The role of mitochondria in presynaptic calcium handling at a ribbon synapse. Neuron25, 229–237.10.1016/S0896-6273(00)80885-5
124
Zuchner S. Mersiyanova I. V. Muglia M. Bissar-Tadmouri N. Rochelle J. Dadali E. L. Zappia M. Nelis E. Patitucci A. Senderek J. Parman Y. Evgrafov O. Jonghe P. D. Takahashi Y. Tsuji S. Pericak-Vance M. A. Quattrone A. Battologlu E. Polyakov A. V. Timmerman V. Schroder J. M. Vance J. M. (2004). Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet.36, 449–451.10.1038/ng1341
125
Zucker R. S. (1999). Calcium- and activity-dependent synaptic plasticity. Curr. Opin. Neurobiol.9, 305–313.10.1016/S0959-4388(99)80045-2
Summary
Keywords
mitochondria, neurotransmission, reserve pool, synapse
Citation
Vos M, Lauwers E and Verstreken P (2010) Synaptic Mitochondria in Synaptic Transmission and Organization of Vesicle Pools in Health and Disease. Front. Syn. Neurosci. 2:139. doi: 10.3389/fnsyn.2010.00139
Received
12 May 2010
Accepted
09 August 2010
Published
22 September 2010
Volume
2 - 2010
Edited by
Robert Renden, Heidelberg University, Germany; UCB Pharma SA, Belgium
Reviewed by
George A. Spirou, West Virginia University, USA; Josef Kittler, University College London, UK; Gregory T. Macleod, The University of Texas Health Science Center at San Antonio, USA
Copyright
© 2010 Vos, Lauwers and Verstreken.
This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Patrik Verstreken, Department of Molecular and Developmental Genetics, VIB, Laboratory of Neuronal Communication, Herestraat 49, bus 602, 3000 Leuven, Belgium. e-mail: patrik.verstreken@cme.vib-kuleuven.be
†Melissa Vos and Lauwers Elsa have contributed equally to this work.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.