 Kalpana Rajanala
Kalpana Rajanala Farokh Dotiwala
Farokh Dotiwala Arun Upadhyay
Arun Upadhyay- Research and Development, Ocugen Inc., Malvern, PA, United States
Geographic atrophy (GA) is an advanced stage of age-related macular degeneration (AMD) that leads to gradual and permanent vision loss. GA is characterized by the loss of photoreceptor cells and retinal pigment epithelium (RPE), leading to distinct atrophic patches in the macula, which tends to increase with time. Patients with geographic atrophy often experience a gradual and painless loss of central vision, resulting in difficulty reading, recognizing faces, or performing activities that require detailed vision. The primary risk factor for the development of geographic atrophy is advanced age; however, other risk factors, such as family history, smoking, and certain genetic variations, are also associated with AMD. Diagnosis is usually based on a comprehensive eye examination, including imaging tests such as fundus photography, optical coherence tomography (OCT), and fluorescein angiography. Numerous clinical trials are underway, targeting identified molecular pathways associated with GA that are promising. Recent approvals of Syfovre and Izervay by the FDA for the treatment of GA provide hope to affected patients. Administration of these drugs resulted in slowing the rate of progression of the disease. Though these products provide treatment benefits to the patients, they do not offer a cure for geographic atrophy and are limited in efficacy. Considering these safety concerns and limited treatment benefits, there is still a significant need for therapeutics with improved efficacy, safety profiles, and better patient compliance. This comprehensive review discusses pathophysiology, currently approved products, their limitations, and potential future treatment strategies for GA.
Introduction
Age-related macular degeneration (AMD) is a progressive retinal neurodegenerative disease that involves the loss of photoreceptor cells and supportive retinal pigmented epithelial cells (RPE). The RPE has multiple functions, it acts as a blood-retina barrier, nourishes the photoreceptors, and is responsible for the phagocytosis of debris and wound healing (1). Several external and intrinsic risk factors (summarized in Figure 1) contribute to the pathophysiology of AMD, resulting in localized inflammation and neurodegeneration of the macula (the central part of the retina). The onset of the disease is characterized by the formation of lipid-rich extracellular deposits called drusen particles (2). AMD affects 28 million people worldwide and causes loss of vision in the population aged over 50 (3). The Clinical AMD staging system (4), based on the size of drusen particles, as identified by the color fundus photography, classifies the disease into the following grades: Grade 1, which has no drusen or few drusen deposits with sizes less than 63 µm. The presence of drusen between 63 to 124 µm is staged as Grade 2 or early AMD. Grade 3 or intermediate AMD is identified by the size of drusen ≥ 124 µm. The presence of drusen >125 µm is classified as Grade 4 or Geographic Atrophy (GA). Grade 5 is AMD with choroidal neovascularization (CNV), which is also known as wet or exudative AMD (Figure 1) (1, 4).
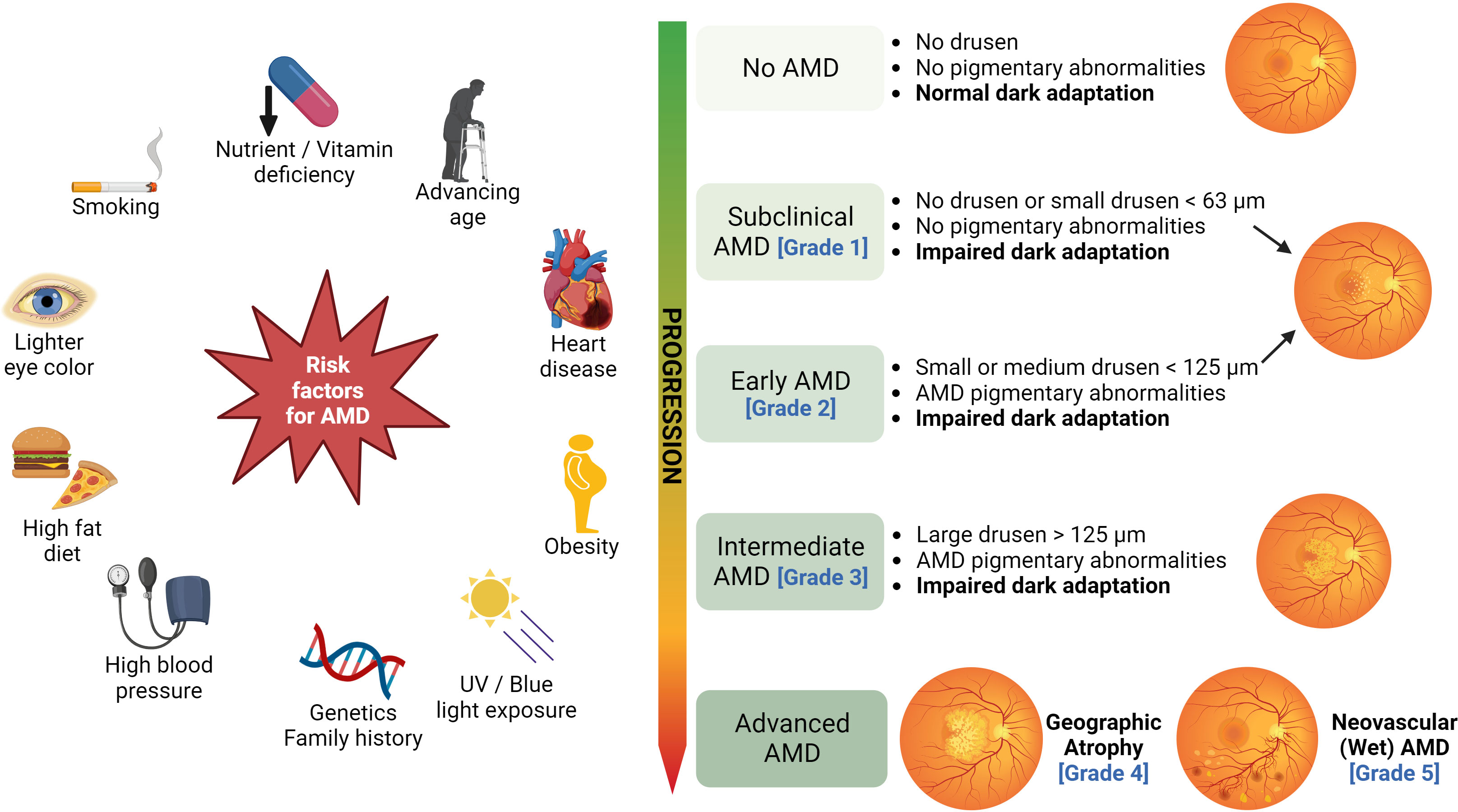
Figure 1 Risk factors and clinical stages of AMD: Several risk factors, such as cigarette smoking, advancing age, nutrient deficiency, high-fat diet, excessive UV/blue light exposure, obesity, and genetics contribute to the development of AMD. The clinical AMD staging system based on the size of drusen particles is illustrated on the right (figure generated using bioRender.com).
GA or dry/non-exudative AMD with or without foveal involvement is an advanced form of AMD, which affects over 8 million people globally, and it is known to cause permanent visual impairment (3, 5). The term “geographic” is used because the atrophic areas typically appear as well-defined, irregularly shaped lesions that may resemble the outlines of a map. These lesions can vary in size and may progress over time. The incidence of GA is more prevalent in the European population compared to the Asian, African, and Hispanic people (6, 7). Especially, GA incidence was shown to increase four-fold every ten years from the ages 50 to 80 in the European population (8, 9). GA affects the outermost layers of the retina, leading to a progressive decline in retinal pigment epithelial cells (RPE), photoreceptor cells, and reduced vessel density in the choriocapillaris (3, 10). The foveal region is located at the center of the macula, it comprises a high density of cone photoreceptors and is responsible for central and color vision. The rate of progression of GA varies according to the size and the location of the atrophy and the extent of foveal involvement (11, 12). GA lesions initially appear in the perifoveal macula, excluding the foveal center, and expand over time, usually within 1.5 to 2.4 years, to include the fovea (9, 12). Perifoveal atrophy causes impairment in reading, driving, and low-light vision, and atrophy extending to the foveal region can cause severe impairment in central vision (13).
Globally, the aging population is estimated to increase over the coming decades, and the incidence of GA is projected to rise (6). To date, intravitreal pegcetacoplan (Syfovre; Apellis Pharmaceuticals, Inc., approved in 2023) and Iveric Bio’s Izervay are commercially available in the United States to slow down the progression of GA. Therefore, there is an emerging need to discover new approaches to treat and manage this disease. This review discusses the risk factors, pathophysiology, and potential treatment strategies for GA.
Diagnosis of geographic atrophy
Visual acuity
GA is diagnosed by visual function tests to assess the extent of visual impairment. The best corrected visual acuity (BCVA) test utilizes the Snellen chart or the early treatment diabetic retinopathy study chart to measure how many letters a patient can read from a distance. Depending on the extent of foveal involvement, the patients may be able to read the letters and maintain visual acuity (VA). Reading speed can also be used to assess the progression of GA, where the number of correctly read words are quantified over a specific time. In a study, it was found that in patients with GA, the reading rate decreased significantly from 110 words per minute (wpm) at baseline to 51 wpm at two years (13). Low-luminance visual acuity (LLVA) measures visual function in low light by using a neutral density filter, which reduces the luminance by 2 log units and is conducted like BCVA (14). The difference between the patient’s BCVA and LLVA scores is termed low-luminance deficit (LLD), which is predictive of lesion enlargement in GA and subsequent vision loss (14, 15).
A patient’s central visual field can be examined by the Amsler grid, in which the graph lines can be distinguished accurately by the healthy eye. However, in patients with AMD, the lines may appear wavy, distorted, or blurred, and the grid may appear to have “holes,” dark areas, or scotomas, which are indicative of visual impairment (9).
Another visual function that is impaired in GA is dark adaptation. In the test to determine this function, the eye is exposed to a bright light to bleach most of the rhodopsin from rod and cone photoreceptors, and subsequently, the eye is stimulated with a bright light of ~420 nm wavelength against a pitch-black background. Delays in dark adaptation are observed in AMD patients with increasing severity of the disease determined by the presence of subretinal drusen deposits, drusen grade, and RPE pigment changes (16).
Microperimetry
Microperimetry utilizes varying light intensities to stimulate different regions of the macula and is used to measure retinal sensitivity. It combines perimetry and retinal imaging to measure the sensitivity of the macula. It involves the projection of light stimuli on different points of the retina to generate a detailed “map” of the macular sensitivity to light and performs a fixation analysis at each moment of the examination. Microperimetry uses different technologies to assess residual visual function and functional vision. It has been used for the study of retinal macular disorders such as AMD, diabetic macular edema (DME), and macular dystrophies. It is specifically indicated for the early monitoring and diagnosis of pathologies affecting the macula and evaluating detailed vision and reading ability, along with other precision activities. A decrease in retinal sensitivity correlates with lesion enlargement and is associated with GA progression over time (17). Recently, automated perimetry was combined with scanning ophthalmoscopy to perform the structure-function analysis of GA (18). The correlation between the functional data and the anatomic morphology that can be generated by this technique aids in assessing the changes in GA lesion progression. In patients with GA, assessment of mean retinal sensitivity, pointwise or regional sensitivity, perilesional sensitivity, and fixation stability can characterize the severity of the disease (19).
The clinical features of macular degeneration or geographic atrophy can be characterized by the imaging tests described below.
Color fundus photography
Retinal fundus photography provides two-dimensional (2D) color images of the retina as taken by a fundus camera. The images can be used to visualize abnormalities such as the presence of drusen, Lipofuscin granules, lipids, blood, scar tissue, and regions of atrophy (20). In Geographic atrophy, the drusen appear as round yellow lesions in the images, and the atrophic RPE can be distinguished by hypopigmentation (21). This method can be used to categorize drusen according to their size. However, the images obtained by FP lack depth, and they do not provide detailed quantitative information (21). Due to these limitations, this technique is often combined with other diagnostic methods for accuracy.
Fundus autofluorescence
Another non-invasive imaging technique that is extensively popular in both clinical and research settings due to its ability to aid in the diagnosis and management of a variety of retinal disorders is fundus autofluorescence (FAF). It imparts better detection of exudative retinal diseases such as choroidal neovascularization, and a density map of lipofuscin, the predominant ocular fluorophore, in the retinal pigment epithelium. This provides information on the metabolic state and overall health of the RPE and, indirectly, the photoreceptor layer. Abnormal autofluorescence (AF) patterns on FAF imaging can act as markers for retinal disease. Since many retinal pathologies lead to RPE dysfunction and an accumulation of lipofuscin, abnormal AF patterns can help in the diagnosis and monitoring of retinal disease progression. FAF imaging is ideal for detecting orange pigment, which is sometimes subtle or near-invisible funduscopically, especially in deep or amelanotic lesions. FAF patterns may be linked to disease progression in patients with AMD and may thereby help clinicians determine appropriate therapeutic courses in the future. Fundus autofluorescence occurs due to drusen (lipid) deposits in the retinal pigment epithelium (22) comprising of lipids and fluorophores such as Lipofuscin (LF) (23), and A2E (N-retinylidene-N-retinylethanolamine) (24). Areas of atrophy in dry AMD appear as regions with reduced fluorescent signal due to the loss of RPE cells and photoreceptors and thus can be detected by FAF (25). The patterns of FAF associated with GA lesions can be correlated with the enlargement rate, with more diffuse hyper-autofluorescence changes generally representing a faster rate of progression (25). FAF can also be used to determine the size of GA lesions and the area of GA lesion progression. A recent meta-analysis implicated that determining the mean GA growth rate by FAF can be an important primary outcome measure for GA treatment trials (26). A novel quantitative FAF method (qAF) combines autofluorescence intensities measured by scanning ophthalmoscope with spatial information, which can be helpful in establishing structural correlations and allows for distinguishing changes related to disease progression (27). The limitations of FAF imaging include low autofluorescence signal compared to CFP, high image noise, low contrast, and interference from the anterior segment (28).
In summary, FAF is a valuable tool for clinicians in diagnosing and managing retinal diseases. It provides a density map of lipofuscin, detects abnormal patterns of autofluorescence, and helps visualize orange pigment. FAF imaging aids in the evaluation of prognosis in patients with age-related macular degeneration. However, there are some limitations to this imaging technique, including approximately two orders of magnitude lesser signal strength than the peak signal of fluorescein angiography. FAF imaging uses short-wavelength excitation light in the blue range, which limits the clear visualization of the central retina due to the absorption of blue light by macular pigment. This leads to limited visualization of the fovea and parafoveal regions in the retina.
Optical coherence tomography
Optical Coherence Tomography (OCT), a non-invasive imaging procedure, utilizes infrared light to create a cross-sectional image of the retina, and the accuracy of the retinal view generated by OCT is at least 10-15 microns (29). The images obtained by OCT can characterize the retina layers in the form of hypo- or hyper-reflective bands. OCT is a sensitive examining tool for GA evaluation as it provides a detailed characterization of retinal layers (3). This method can be used to visualize drusen deposits, choroidal neovascularization, detachment of RPE, and subretinal fluid (21, 30). The choroidal vascularity index, as determined by structural OCT analysis, can be used to predict GA progression (31). Reticular pseudo drusen (RPD) are located above the RPE and hence are distinguished from conventional drusen (32, 33). The presence of RPD is usually associated with the progression of atrophy to the foveal region. RPD cannot be assessed by clinical examination; however, it can be visualized by FAF and OCT (34). Due to the faster acquisition and the fine resolution of retinal images, OCT has emerged as a comfortable and preferred modality to assess GA lesions in patients (35). Technological advances in SD-OCT have improved the imaging performance and clinical usability of the technique, and improved visualization of photoreceptors, retinal pigment epithelium, and Bruch’s membrane may facilitate earlier detection and treatment of retinal diseases.
Based on the OCT findings and the anatomical layers affected, GA is classified into four categories: incomplete RPE and outer retinal atrophy (iRORA), complete RPE and outer retinal atrophy (cRORA), incomplete retinal atrophy, complete retinal atrophy (3, 36, 37). A region of choroidal signal hyper-transmission along with a corresponding zone of disruption or attenuation of the RPE layer and the presence of drusen and degenerated photoreceptors is defined as iRORA (38). The presence of these typical findings is a risk factor for the progression to cRORA, which is characterized by a region of hyper-transmission and the zone of disrupted or attenuated RPE of over 250 µm, with the presence of degenerated photoreceptors and an intact RPE (36–38).
Molecular mechanisms underlying the pathophysiology of geographic atrophy
Several internal metabolic and oxidative stressors and external stressors such as advanced age and smoking are risk factors and trigger the pathogenesis of GA (11) (Figure 1). In early AMD, the macula has an abnormal RPE pigment distribution and drusen deposition in the space between the basal lamina of RPE and the inner collagenous layer of Bruch’s membrane (2). With age, these stressors induce damage to the RPE and cause the accumulation of extracellular deposits in the form of drusen (39, 40). Drusen particles are made up of lipids, proteins, cellular debris, β-amyloid deposits, apolipoproteins, iron, and zinc ions (41). Large or soft drusen with indistinct borders are a major risk factor for AMD progression. A2E was shown to increase the lysosomal pH of the RPE cells and thereby impair the ability of lysosomal degradation (42, 43). In addition, LF generates reactive oxygen species (ROS) upon photoinduction (44) that induces damage to the RPE cells (43). LF accumulation occurs over time resulting from the phagocytosis of membranous discs that are shed from retinal photoreceptors, and due to the inability of the RPE cells to degrade these granules. Excessive exposure to blue light was shown to damage RPE, while overexposure to white light induced photoreceptor cell damage. Activation of the inflammatory pathways in the presence of extrinsic and genetic risk factors contributes to GA pathogenesis.
Vitamin A (retinol), a cofactor in the phototransduction cycle, is a crucial component of the photopigment (11-cis-retinal). Absorption of a photon in the neural retinal outer segment releases retinaldehyde from the opsin protein. This results in the photoisomerization of 11-cis-retinal to all-trans-retinal, which subsequently results in the conversion of the visual pigment to the signaling form, meta-rhodopsin II, and transformation of light photons to electrical signals (45). Vitamin A is shuttled across the retina (between the photoreceptors and the RPE) as various isoforms: all-trans-retinol, all-trans-retinyl esters, all-trans-retinaldehyde, 11-cis-retinol, 11-cis-retinyl esters, 11-cis-retinaldehyde, and retinoic acid as a part of the visual cycle (46). Several biochemical pathways are involved in vitamin A transfer and storage in the RPE and transfer to the photoreceptors (47). During the visual cycle, a portion of this retinaldehyde condenses with phosphatidylethanolamine (PE) to form retinaldehyde-PE, leading to the formation of vitamin A dimer by-products, N-retinylidene-N-retinylethanolamine (A2E) (Figure 2), and all-trans-retinaldehyde dimer (ATR-dimer) (48). At the end of the visual cycle, specific enzymes carry out the regeneration of the active form of vitamin A (11-cis-retinal), the recycling of inactive forms, and the removal of toxic by-products (47). Accelerated dimerization of vitamin A is responsible for the formation of granular lipofuscin deposits in the eye, causing RPE atrophy (49). Abnormal accumulation of toxic dimers, such as A2E, causes protracted degeneration of the RPE and is associated with the etiology of AMD and Stargardt disease (49, 50).
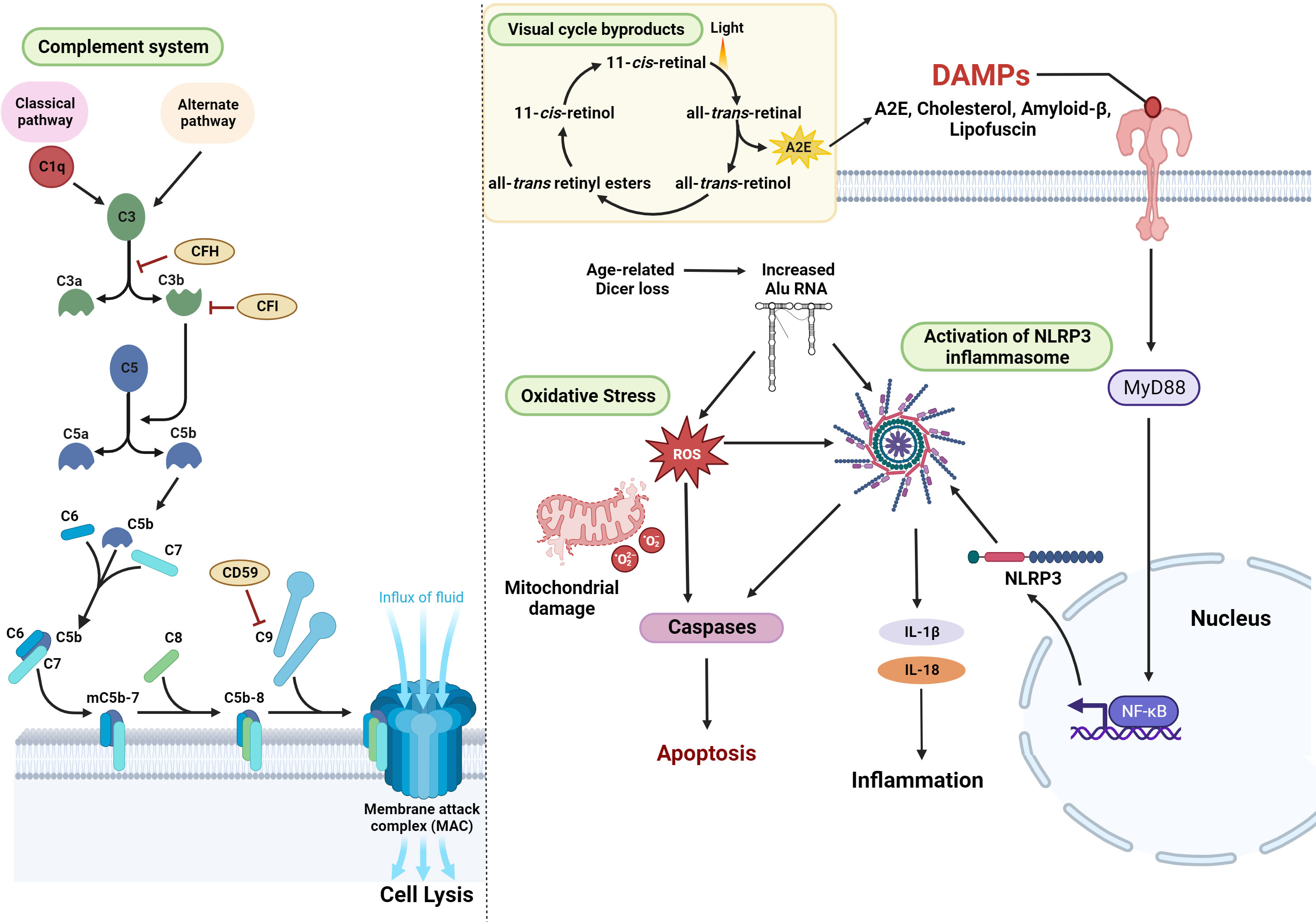
Figure 2 Cellular pathways associated with the development and progression of GA – Activation of the complement pathway, resulting in lytic cell death through MAC, is one of the major mechanisms associated with the pathology of AMD. Mitochondrial oxidative stress and the generation of ROS are also key contributing factors in the etiology of GA. In addition, damage-associated molecular patterns (DAMPs) such as cholesterol, amyloid-β Lipofuscin, or A2E (a byproduct of the visual cycle) activate Myd88 signaling and NFkB-induced transcription leading to the enhanced expression of NLRP3. Alternatively, age-related Dicer deficiency results in an abnormal increase in Alu RNA transcripts. Alu RNA-induced Myd88 signaling or the direct activation of NLRP3 results in inflammation, contributing to the disease pathogenesis (figure generated using bioRender.com).
The complement pathway
Activation of complement cascade, a noncellular component of the innate immune system, is integral to the pathogenesis of GA (51). The complement system consists of over 30 circulating systemic proteins, which are associated with inflammation, opsonization, phagocytosis, and cell death and are responsible for the detection and clearance of pathogens (52). The activation of the complement system occurs via three pathways – the classical pathway, which is activated by antigen-antibody complexes, the lectin pathway, which is activated by the polysaccharides on microbes or the alternative pathway, which is activated by the cell surfaces of foreign pathogens (53). C1q is an initiating factor of the classical complement pathway, which is activated by components of drusen such as lysophospholipids (generated by lipase degradation of lipoproteins) and β-amyloid deposits (54, 55). Accumulation of C1q with age and inflammation induces retinal damage and is a key factor linked to the initiation and progression of GA (56). Initiation of the pathway ultimately results in the formation of multi-subunit serine protease complexes known as the C3 and C5 convertases, that act on the central complement proteins C3 and C5 (52). Cleavage of complement factor C3 into pro-inflammatory anaphylatoxin C3a and opsonin C3b induces inflammation and opsonizes the cells for phagocytosis. In AMD patients, components of the complement system C5, C3, and its fragments were detected in the subretinal space and the drusen particles (56, 57). C3 and its activation products were shown to induce multiple pathways, including inflammasome activation, recruitment of macrophages and microglia to the subretinal space, aberrant turnover of lysosomes and activation of intracellular C3 mechanisms within the RPE, such as the mTOR pathway (58–60). Complement factor B is a 93-kDa single-chain glycoprotein required for the activation of the alternative pathway. Association of factor B (FB) with protein C3b results in its cleavage by complement factor D to release an N-terminal FBa and the bound carboxyl-terminal serine proteinase FBb fragments to form the C3bBb C3 convertase (61). This convertase can cause further amplification of the complement pathway and the formation of MAC. Elevated plasma levels of activation fragments FBa and FBb, predominantly located in the vitreous, Bruch’s membrane, and choroidal vasculature, have been reported in patients with AMD (61, 62). The cleavage product C3b leads to the activation of C5 convertase, which acts on C5 to generate the pro-inflammatory anaphylatoxin C5a (which binds C5aR on immune cells) and the cleavage product C5b. Subsequently, C5b recruits C6, C7, and C8, and polymerization of 12-18 molecules of C9 forms C5b-9 to form a pore called the membrane attack complex (MAC). Accumulation of MAC beyond a certain threshold leads to disruption of the membrane and results in lytic cell death. CD59 is a glycoprotein that inhibits the polymerization of C9, preventing the formation of C5b-9 and MAC (63). The complement cascade is inactivated by regulatory factors like complement factors H and I (CFH and CFI). Negative regulation of the pathway by CFH results in the inactivation of C3b (iC3b), preventing the generation of the C5 convertase and thereby inhibiting the generation of MAC (Figure 2). RPE dysfunction results in a compromise in the blood-retinal barrier and allows leakage of serum proteins and components of the classical and alternative complement pathways into the retina from the underlying choriocapillaris, making it susceptible to the MAC (64). Studies in mice deficient in complement component receptors such as C3aR and C5aR demonstrated that these mice developed early and progressive retinal degeneration, suggesting the importance of this pathway in preserving retinal structure and function (65). In another independent study, C3 knockout mice exhibited severe photoreceptor loss, thickening of Bruch’s membrane, and high levels of retinal inflammation compared to control groups (66). In addition, reduced retinal function, thinning of retinal layers with age, and impaired signaling in the retinal INL and ONL were observed in complement knockout mice (67). These findings indicate that complement pathway components are critical for maintaining retinal homeostasis and integrity during the aging process. Therefore, it is imperative that the critical role of early complement factors in retinal biology should be carefully considered while developing therapies for AMD. Several GA-linked genetic risk factors pertaining to the complement cascade can cause dysregulation of this pathway, resulting in hyperactivation and inflammation, leading to retinal cell death, which is a characteristic feature of GA.
Activation of the NLRP3 inflammasome
A multiprotein scaffold of the cellular stress sensor NLRP3 (nucleotide-binding domain, leucine-rich–containing family, pyrin domain–containing-3), adaptor ASC (apoptosis-associated speck-like protein), and the effector caspase 1 constitute the NLRP3 inflammasome (68). Activation of the inflammasome by damage-associated molecular patterns (DAMPs) like cholesterol crystals, lipofuscin, or the presence of drusen components such as Aβ-peptide 1–40 or C1q (68, 69) results in elevated interleukin (IL)-1b and IL-18 cytokine levels which mediate innate and adaptive immune pathways (68, 70–72).
Risk factors, such as aging, cause a buildup of cellular oxidative stress and lead to downregulation of the post-transcriptional expression of the miRNA processing enzyme, DICER1 (73). In GA, repetitive transposable elements of non-coding RNA, termed Alu RNA, were shown to accumulate in RPE due to a loss of DICER1 (74, 75). Alu RNA hinders the expression of ROS scavengers like superoxide dismutase 2 (SOD2) and endothelial nitric oxide synthase (eNOS) by directly intervening with their transcription and translation initiation (76). Alu RNA induces the priming of NLRP3 inflammasome via the pro-inflammatory transcription factor nuclear factor kappa B (NF-κB) pathway, promoting the transcription of inflammasome components. In GA, the RPE cell death is facilitated by apoptosis mediated by Caspase-3, which is triggered by MyD88 signaling via the IL-18 receptor (69, 74, 75) (Figure 2).
Oxidative stress
Cigarette smoking was shown to be a major contributor leading to the development of AMD in at least 27% of the cases (77). Components in cigarette smoke such as hydroquinone (HQ) cause cellular oxidative damage, resulting in the generation of reactive oxygen species (ROS) (78). In addition, photoreceptor shedding, excessive light exposure, and aging also contribute to oxidative stress in the retina (79). Increased intracellular oxidative stress causes mitochondrial DNA (mtDNA) damage, oxidation of mitochondrial proteins and lipids, and structural damage to the mitochondria, subsequently resulting in mitochondrial dysfunction (Figure 2). In addition, mitochondrial ROS generation can also be mediated by Alu RNA via voltage-dependent anion channels (VDAC)-1 and -2, which are major channels at the outer mitochondrial membrane for the exchange of metabolites that can subsequently impair mitochondrial potential (80). Photoreceptors utilize glucose for glycolysis and produce large amounts of lactate, which is then transported to the RPE cells. The RPE cells subsequently use this lactate for energy production through oxidative phosphorylation by mitochondria (81). Mitochondrial oxidative stress was shown to increase the glycolytic metabolism in the RPE due to metabolic reprogramming, thereby leading to its dysfunction and ultimately resulting in the disruption of the photoreceptors.(82) The metabolic shift in RPE correlated with severe disruption of photoreceptor mitochondria involving downregulation of translocase of the outer mitochondrial membrane 20 (TOMM20) expression and reduced COXIII/β-actin levels, which are critical for maintaining mitochondrial morphology (82). Cytosolic mtDNA released due to mitochondrial damage causes activation of cyclic GMP-AMP synthase (cGAS)-driven type I interferon signaling (IFN), thereby resulting in NLRP3 inflammasome activation (83).
Retinoic acid receptor-related orphan receptor alpha (RORα) is a nuclear receptor that regulates inflammatory and cholesterol metabolism pathways (84). RORα also regulates lipid metabolism and lipoproteins, such as high-density lipoprotein, serum amyloid A, and apolipoprotein A1 (85). RORα-mediated transcriptional inhibition of the NFκB pathway inhibits TNF-α induced expression of inflammatory cytokines such as IL-6, IL-8, and COX-2 (86). Gene expression data and linkage analysis revealed RORα SNPs rs4335725 and rs12900948 are associated with AMD (87). In addition, RORα was shown to negatively regulate pathological neovascularization by modulating inflammatory response in the choroid/RPE complex (88, 89).
Role of microglia and monocytes in AMD pathobiology
Retinal microglia maintain homeostasis by eliminating toxic waste, surveying the environment for damage, and mediating innate immune response (90). When exposed to genetic and environmental risk factors, microglia express pathogenic cytokines, recruit monocytes to sub-retinal space, and affect photoreceptor and RPE integrity (91). Under stress conditions, the hyperactive immune response triggered by infiltrated mononuclear phagocytes, such as microglia, macrophages, and monocytes, activate NF-κB signaling and result in the secretion of inflammatory cytokines, and contribute to retinal degeneration (92). Deficiency in microglial checkpoint genes such as CX3CR1 (Chemokine Receptor-1) was implicated in lowering the threshold of microglial activation and inducing age-related infiltration of mononuclear phagocytes (92). CX3CR1 SNPs were found to be associated with an increased incidence of AMD (93).
Genetic factors associated with GA
Genome-wide association studies indicate different genes associated with the prevalence of late AMD and progression of GA (60, 94, 95). The risk of AMD is amplified in the presence of genetic variants that regulate various facets of the complement system, including factors that negatively regulate complement activation, such as CFH and CFI. These factors are involved in convertase formation like complement factor B (CFB), C2 and C3, and C5b-9, which is a part of the MAC (96–99). A study comparing 3235 cases of GA and 10,749 cases with choroidal neovascularization identified 52 independently associated single nucleotide polymorphisms (SNPs) across 34 genetic loci (94). These SNPs were found in CFH, C2/CFB, CFI, C3, and C9 genes involved in the complement pathway, APOE, LIPC, CETP, and BAIAP2L2 genes involved in the lipid metabolism and transport pathways, COL8A1, COL10A1, TIMP3, ADAMTS9, TGFBR1, HTRA1, and B3GALTL genes involved in the remodeling and maintenance of extracellular matrix, RAD51B and TNFRSF10A genes involved DNA repair and cell survival, and VEGFA, TGFBR1, and ADAMTS9 genes involved in angiogenesis (2) (Figure 3).
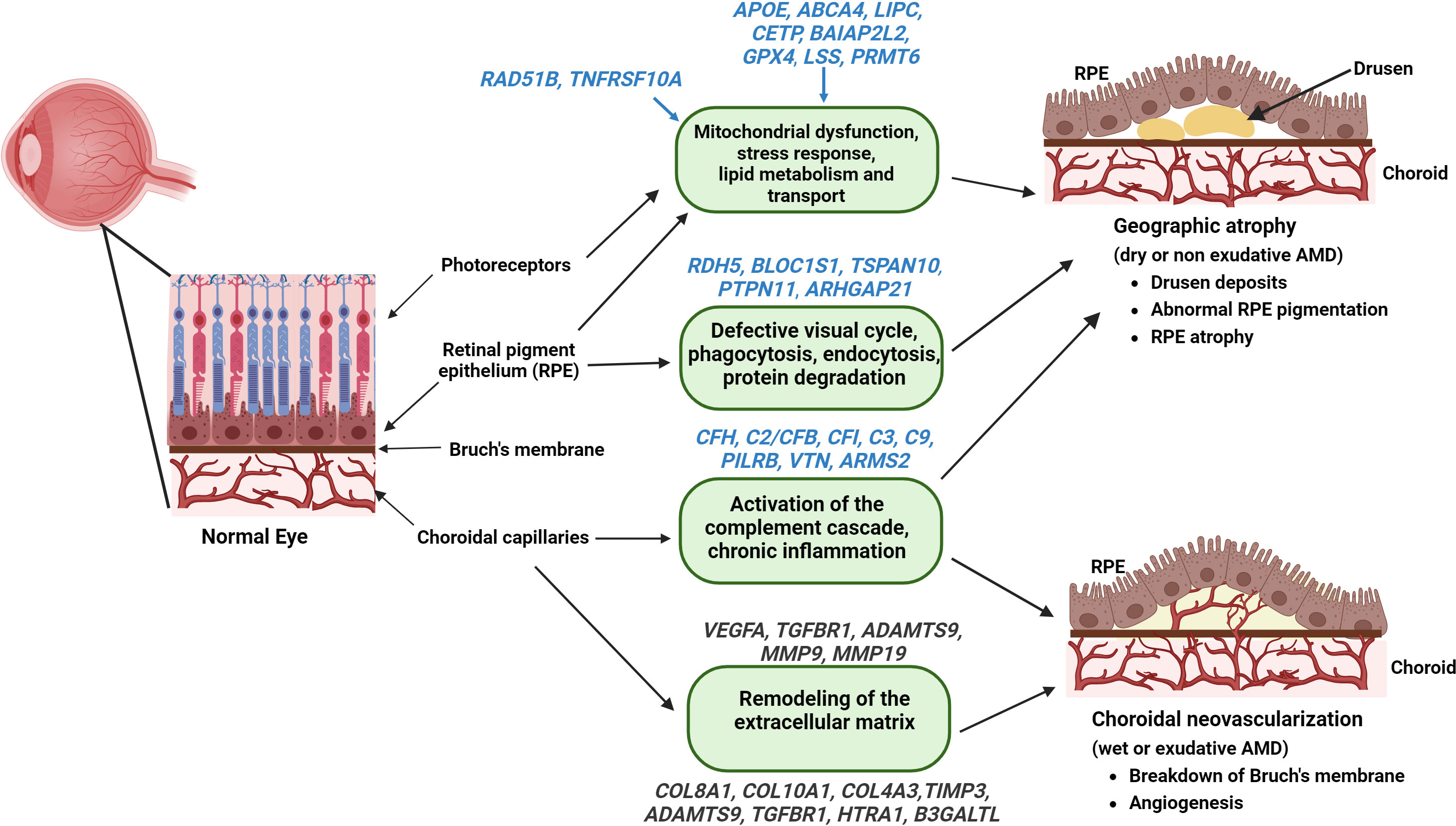
Figure 3 Genes associated with the pathophysiology of AMD – Identified genes (with established and putative roles) correlated with their functions that contribute to the pathogenesis of GA (highlighted in blue) and wet AMD (highlighted in Grey) are illustrated (figure generated using bioRender.com).
A common variant in the complement cascade gene, CFH (complement factor H), on chromosome 1 at the 1q31.3 locus, variant rs1061170 (p.Tyr402His), was strongly associated with an increased risk for developing AMD (96, 100–102). One CFH missense variant, rs121913059 (p.Arg1210Cys), was also linked to AMD (94). The p.Arg1210Cys and p.Tyr402His variants reduce the binding of CFH, resulting in reduced function, thereby preventing complement system inhibition in local chronic inflammation (103).
Among the identified lead variants associated with GA, the high-temperature requirement factor A1 (HTRA1) and the age-related maculopathy susceptibility 2 (ARMS2) are two tightly linked genes located on chromosome 10q26 (Chr10 locus). ARMS2 protein was shown to regulate complement activation by recruiting complement activator properdin, increasing C3b surface opsonization and phagocytosis. An SNP rs11200638 in the 10q26 genetic locus located at the promoter region of the HTRA1 gene increased the expression of HTRA1 in the RPE (104, 105). Two other polymorphisms in the 10q26 genetic locus, an ARMS2 SNP variant Ala69Ser, (rs10490924), and an insertion/deletion polymorphism (del443ins54) in 3′UTR of ARMS2 were identified to associate with AMD (94, 106). HTRAs are a conserved group of serine proteases containing a protease and a PDZ domain in the C-terminal region that function in substrate binding, oligomerization, and protein translocation (107). Substrates of HTRA1 include fibronectin, transforming growth factor Beta (TGF-β), clusterin, an inhibitor of angiogenesis thrombospondin (TSP1), and an extracellular matrix protein EFEMP1 (108) HTRA1 inhibits the canonical Wnt signaling pathway and modulates the TGF-β pathway. HTRA1 regulates angiogenesis by interacting with various members of the TGF-β family, such as TGF β1, TGF β2, activin, BMP4, and growth differentiation factor 5 (GDF5) and is associated with neovascular AMD (109). Aberrant accumulation of HTRA1 and its substrates, such as EFEMP1, form drusen-like deposits and activate the complement pathway (110). Overexpression of HTRA1 results in polypoidal lesions, the formation of subretinal deposits, and vascular abnormalities that are associated with AMD (111, 112). Oxidative stress, one of the major causative factors for AMD, induces HTRA1 expression in the RPE cells and increases cell senescence via the p38/MAPK pathway (113).
Other identified lead variants are CETP, MMP9, and SYN3-TIMP3 loci, which demonstrated a significant difference between the AMD disease subtypes (94). In addition, the protein arginine methyltransferase 6 gene (PRMT6; chromosome 1p13.3) and the lanosterol synthase gene (LSS; chromosome 21q22.3) were proposed as the functionally relevant genes associated with the progression of GA (95). The variant rs42450006 upstream of MMP9 was shown to be specifically associated with choroidal neovascularization but not with GA (94). Retinal transcriptome and expression quantitative trait loci (eQTL) analysis identified PILRB/PILRA (paired immunoglobin like type 2 receptor beta/alpha), B3GLCT (beta 3-glucosyltransferase), BLOC1S1 (biogenesis of lysosome related organelles complex 1 subunit 1), TMEM199 (transmembrane protein 199), and TSPAN10 (tetraspanin 10) as additional putative causal genes for AMD (114, 115).
Potential biomarkers for the detection of AMD
Retinal biomarkers can immensely contribute to the early detection of AMD and are evolving research areas research. Imaging biomarkers such as drusen characteristics, size, and area of drusen and pigmentary changes identified by color fundus photography can be used to classify stages of AMD (116). The most widely used imaging technique is OCT, which can be used to detect biomarkers such as drusen volume, hyper-reflective foci, hyper-transmission defects, reticular pseudo drusen, and iRORA (116). In the context of GA, FAF patterns are extremely helpful markers for the evaluation of atrophic areas and disease progression (117). Multifocal electroretinography (mfERG) measures photoreceptor signaling in response to a light stimulus using contact electrodes in the cornea (118). The functional areas of the retina and their sensitivity can be mapped by this technique by varying the intensity of the light stimulus and thereby can be used for early detection of AMD (119). Assessment of visual function by testing for visual acuity, dark adaptation, contrast sensitivity, and evaluation of retinal sensitivity by microperimetry can be specific and significant markers that indicate different stages of disease progression (19, 120).
Studies indicate a strong association between the family history of AMD and the incidence of the disease. The risk of AMD increases 4-fold for those with a family history and nearly 27 times for those with an affected parent (121). Therefore, the incorporation of comprehensive natural history studies and identification of associated genetic biomarkers will aid in early detection. SNPs in genes with specific functions in the retina, such as ABCA4 and TIMP-3, genes pertaining to complement pathway, such as CFI, C3, and CFH, genes related to lipid metabolism, such as ApoE, and immune function related genes, such as ARMS2/HTRA1 can be potential genetic biomarkers (122).
Identifying serum biomarkers for AMD can aid in monitoring disease progression and treatment response. Evaluation of levels of C-reactive protein (CRP), cholesterol, Interferon-γ, Homocysteine, and proinflammatory cytokines such as IL-8 can be indicative of AMD risk and can be developed as screening tools (122). The various biomarkers that can be used for early diagnosis are summarized in Figure 4.
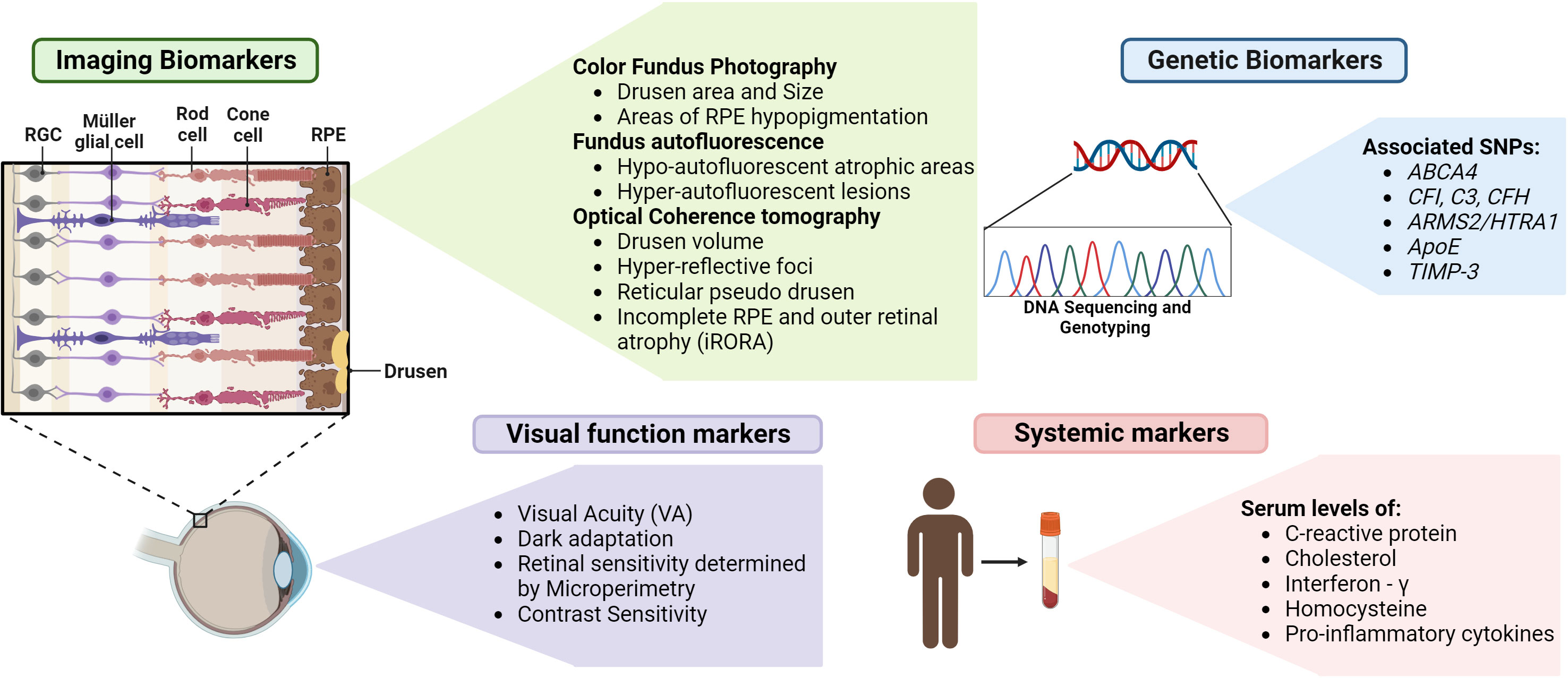
Figure 4 Biomarkers for detection of GA – Various biomarkers indicated above can assist in the early detection, evaluation of GA progression and treatment outcomes (figure generated using bioRender.com).
Treatment of GA and ongoing clinical trials
Despite an increased understanding of the underlying risk factors and the inflammatory pathways contributing to the pathophysiology of GA, limited treatment options are available to combat the disease. Several therapeutic strategies that target the complement pathway and reduce inflammation that are under clinical trials for the treatment of GA are listed in Figure 5.
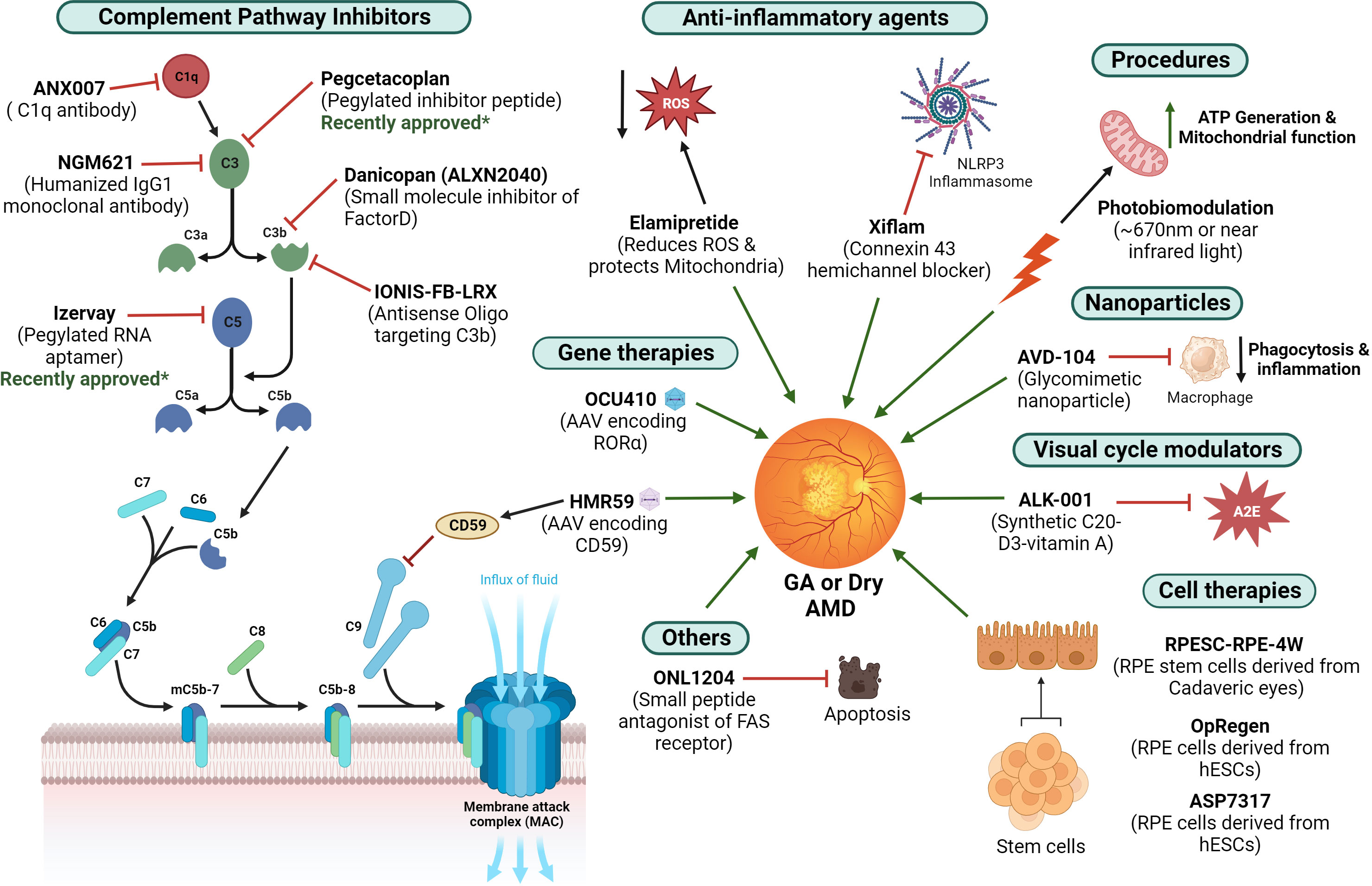
Figure 5 Current treatment strategies for GA – Different therapeutic agents under ongoing clinical trials that are being investigated for the treatment of GA are illustrated (figure generated using bioRender.com). The therapies approved recently are also indicated.
Inhibition of the complement pathway
C3 inhibition
Pegcetacoplan is a synthetic pegylated complement C3 inhibitor peptide that was developed by Apellis Pharmaceuticals. The phase III OAKS and DERBY studies (NCT03525600, NCT03525613) in which the patients with GA were given both monthly and bimonthly doses of pegcetacoplan demonstrated a clinically significant reduction in GA lesion growth. Recently, intravitreal pegcetacoplan (Syfovre; Apellis Pharmaceuticals, Inc.) was approved by the FDA for patients with GA. However, in clinical trials, by month 24, administration of Syfovre was associated with a higher rate of progression to neovascular AMD or choroidal neovascularization (12% when administered monthly, 7% when administered every other month).
NGM621 is a humanized immunoglobulin G1 (IgG1) monoclonal antibody that aims to inhibit C3 activity that is developed by NGM Biopharmaceuticals. CATALINA Phase II trial (NTC04465955) evaluated NGM621 for treatment of GA and failed to meet its primary endpoint - the change in GA lesion area as measured by FAF over the 52 weeks of treatment. Further update on the trial results is awaited as the evaluation of specified secondary endpoints and post-hoc analyses are underway.
C5 inhibition
Another component of the complement system being targeted is C5. An Avacincaptad Pegol (ACP, a pegylated RNA aptamer developed by Iveric Bio, Inc.), which binds and inhibits C5, was under clinical trials. (123) Phase II/III GATHER1 (n=286, NCT02686658), which included 286 patients, and Phase III GATHER2, including a larger cohort of 448 patients (NCT04435366), which administered 2 mg of the avacincaptad pegol or sham monthly were also conducted. Data from GATHER2 showed that treated patients had a 14.3% reduction in the average rate of GA area growth over 12 months. FDA approved this treatment recently for patients with GA and is now marketed as Izervay. However, by month 12, the use of Izervay was linked to increased rates of wet AMD or choroidal neovascularization (7% when administered monthly and 4% in the sham group) in clinical trials.
C1q inhibition
Inhibiting C1q activation prevents the classical complement activation cascade (which includes C4, C3, and C5), implicated in retinal tissue damage. (124) Pharmacological inhibition of C1q was shown to preserve the function of photoreceptor cells and prevent their progressive loss, even if the photooxidative damage was induced before the administration of the inhibitor. (125) A high-affinity antibody Fab fragment binding to C1q, ANX007, was tested by intravitreal (IVT) administration in cynomolgus monkeys in a study, which was reported to be well tolerated and resulted in near complete C1q inhibition (124). A Phase 2 clinical trial of ANX007, ARCHER study (NCT04656561), comparing the safety and efficacy of ANX007 administered either monthly or every other month, was conducted in 270 patients with GA. Results from the 12-month treatment period demonstrated a statistically significant, dose-dependent preservation of visual function in the patients treated monthly. However, the rate of lesion growth was not reduced by this treatment (Annexonbio.com). Based on the results of this trial, ANX007 is still being developed as a potential therapy for the preservation of visual acuity in patients with GA.
Factor D inhibition
Factor D is an essential enzyme that cleaves factor B bound to C3b and mediates the activation of the alternate complement pathway. Danicopan (ALXN2040) is a small molecule inhibitor that has high affinity binding to factor D and can thereby inhibit alternate pathway activation, ultimately blocking the inflammatory response and cell lysis (126). Alexion Pharmaceuticals is conducting a dose-finding phase II clinical trial for oral Danicopan for GA patients without foveal involvement (NCT05019521).
Gene therapy to inhibit the complement pathway
CD59 is a glycosylphosphatidylinositol (GPI)-anchored membrane protein that prevents the incorporation of C9 into the C5b-8 complex, thereby inhibiting the action of the MAC on cell membranes (127). Hemera Biosciences developed HMR59 (AAVCAGsCD59), which is an intravitreally administered adeno-associated virus 2 (AAV2) vector that delivers the soluble form of CD59 (sCD59). HMR-1001 (NCT03144999), a phase I safety and efficacy study, reported that patients with GA showed a 23% reduction in GA progression when administered HMR59. However, 27% of the patients given HMR59 reported mild vitritis and uveitis, requiring 6–8 weeks of anti-inflammatory therapy (128).
GT005, developed by Gyroscope Therapeutics, is a recombinant non-replicating AAV vector encoding human CFI, an inhibitor of the complement system. Preliminary results of phase I/II clinical trial, FOCUS (NCT03846193) delivered GT005 via a subretinal injection in 28 patients with GA reported a significant (122%) increase in CFI levels and up to a 46% reduction in the levels of key C3 breakdown proteins associated with complement activation compared to baseline levels (Gyroscopetx.com) (128). Other ongoing phase II clinical trials for GT005 include EXPLORE (NCT04437368), to evaluate patients with GA who have low levels or rare variants of CFI, and HORIZON (NCT04566445), which evaluated a broader group of patients with GA secondary to AMD. The Explore and Horizon trials were terminated recently as they did not meet the efficacy outcome.
Antisense oligonucleotides to inhibit the alternate complement pathway
Complement factor B is required for the activation of the alternative complement pathway. In a study, second-generation ASOs targeting factor B were administered subcutaneously in healthy mice or monkeys, resulting in a significant decrease in both ocular and plasma factor B protein levels (129). IONIS-FB-LRX, developed by Ionis Pharmaceuticals and Roche, is a 2’-O-methoxyethyl (2’MOE) second-generation ASO (20 bp) conjugated to an N- acetyl galactosamine (GalNAc) ligand (for efficient liver targeting), is an antisense inhibitor of complement factor B (128). The GOLDEN STUDY is a phase 2 clinical trial assessing the safety and efficacy of IONIS-FB-LRX for patients with GA (NCT03815825).
Anti- oxidative agents
Elamipretide, developed by Stealth Biotherapeutics, is a small tetrapeptide (D-Arg-dimethylTyr-Lys-Phe-NH2) that reduces the production of ROS and protects mitochondrial function. The phase II clinical trial, ReCLAIM-2 study (NCT03891875) in 180 patients who received a daily 40mg subcutaneous injection of Elamipretide for 48 weeks failed to meet its primary endpoints—change in low-luminance visual acuity (LLVA) and GA lesion size. However, the latest data from study participants was promising and demonstrated >2-line improvement in LLVA, so the program is being continued by Stealth Biotherapeutics.
Anti- inflammatory agents
NLRP3 inflammasome complex is activated and assembled by Connexin43 hemichannel-mediated ATP release, and inhibition of connexin 43 hemichannels reduces inflammation in cells (130). Oral connexin 43 hemichannel blocker, tonabersat (Xiflam) by InflammX, is under Phase IIb clinical trials for treating intermediate dry AMD and GA; however, no trials are registered at ClinicalTrials.gov.
Retinoic acid-related orphan receptor α (RORα) is a nuclear hormone receptor involved in the suppression of inflammatory cytokine expression and inhibition of complement factor activation (86, 88, 131). OCU410, which delivers RORα, is being developed as a modifier gene therapy for GA by Ocugen. A recent study demonstrated that OCU410 treatment decreased drusen-like deposition in Abca4-/- mouse retinas and improved retinal function (131).
Cell therapy for GA
Luxa biotechnology is investigating the efficacy of transplanting allogeneic retinal pigment epithelium stem cell (RPESC)-derived RPE cells isolated from the RPE layer of human cadaveric eyes (RPESC-RPE-4W) in the macular region of the eye. Phase I/II clinical trial evaluating the safety and tolerability of RPESC-RPE-4W as therapy for dry AMD or GA is ongoing (NCT04627428).
OpRegen, developed by Lineage Cell Therapeutics in partnership with Genentech/Roche, uses human embryonic stem cell (hESC) derived RPE cells. The Phase I/II trial (NCT02286089) administered RPE cells sub-retinally as a cell suspension in 24 patients with dry AMD. The trial reported no cases of rejection, acute or delayed intraocular inflammation, or sustained increases in intraocular pressure after OpRegen transplantation. Currently, a Phase IIa clinical trial (NCT05626114) to further assess the safety of subretinal surgical delivery and to test the preliminary activity of OpRegen in participants with GA is underway.
ASP7317 (RPE cell program, MA09-hRPE) is a cell therapy developed by Astellas Institute for Regenerative Medicine. A phase Ib clinical trial for subretinal administration of RPE cells derived from hESCs to evaluate their efficacy in slowing or reversing the atrophic lesion growth in dry AMD is under progress (NCT03178149).
Modulators of the visual cycle
Fenretinide or 4-HPR (N-(4-hydroxyphenyl) retinamide) is a synthetic derivative of vitamin A which competes with retinol and binds to retinol-binding protein (RBP) (132). The formation of the RBP-Fenretinide complex results in the reduction in retinol delivery and decreased accumulation of toxic visual cycle byproducts. A phase II study which administered different doses of oral Fenretinide in GA patients established that there was a reduction in GA lesion growth rate and neovascularization in treated subjects (133).
A synthetic vitamin A with deuterium at the C20 position (C20-D3-vitamin A or ALK-001) was shown to slow down the formation of toxic vitamin A dimers by 4-5 fold (46, 48). The oral formulation of ALK-001 is currently under phase 3 clinical trial (NCT03845582) for the treatment of GA (134).
Other treatments
Belite Bio developed Tinlarebant, an oral, small-molecule antagonist of retinol-binding protein 4 (RBP4). A phase I trial (NCT03735810) in 71 patients confirmed the safety, tolerability, and achievement of a potential therapeutic level of the drug. A phase III trial is being planned for this drug for patients with GA.
ONL1204, a small peptide antagonist of fragment apoptosis stimulator (FAS) receptor, which can inhibit Fas receptor-mediated apoptosis, is under development by ONL therapeutics. In preclinical AMD models, ONL1204 was shown to protect the RPE cells (Cera.org.au). Intravitreal administration of ONL1204 is under phase I clinical trial for patients with GA (NCT04744662).
A glycomimetic sialic-acid coated nanoparticle, AVD-104, can specifically bind to receptors on activated macrophages and repolarize them to a resting state, thereby hindering their phagocytic function (avicedarx.com). AVD-104, which was also shown to inhibit the complement cascade and reduce the inflammatory damage in preclinical retinal models, is being investigated by Aviceda therapeutics for the treatment of GA. A phase II clinical trial was recently initiated to assess the efficacy and pharmacokinetics of AVD-104 in GA patients via intravitreal administration (NCT05839041).
Cholesterol-lowering statins can be efficacious for clearing up lipid debris in patients with AMD (135). In a study with high-risk AMD patients, high-dose atorvastatin (80mg) was shown to reduce the accumulation of lipid deposits, improve visual acuity, and slow the progression to advanced disease (136). Druslov Therapeutics is planning a Phase II clinical trial for Ocustatin.
Toxic oxidation of polyunsaturated fatty acids in photoreceptor membranes, such as docosahexaenoic acid (DHA), is one of the contributing factors for retinal degeneration. Deuterated DHA is resistant to lipid peroxidation and can have protective effects against retinal degeneration (137). Biojiva is exploring this as a possible therapy for dry AMD.
During aging, reduced expression of the ELOVL2 (Elongation of very long chain fatty acids-Like 2) gene results in a decline in long and very long-chain polyunsaturated fatty acids (PUFA’s), which is associated with AMD pathogenesis (138). Visgenx Inc. is planning subretinal delivery of VGX-0111 (carrying an ELOVL2 transgene) as a treatment for dry AMD.
A large Age-Related Eye Disease Study (AREDS) reported that daily intake of certain nutritional vitamins and minerals lowered the rate of drusen formation and decreased the risk of wet AMD and vision loss. These are marketed as AREDS 2 supplements and are available for use in patients with a high risk of developing AMD.
Procedures
Exposing the retina to low-intensity light (~670 nm or near-infrared light) was shown to improve retinal mitochondrial function due to the absorption of light by cytochrome C oxidase, which results in its increased expression and enhances cellular respiration and ATP generation (139, 140). This process, also known as photobiomodulation, is being investigated as a potential therapy for GA. The Lightsite I clinical trial utilizing the LumiThera LT 300 Light Delivery System, which exposes patients with dry AMD to wavelengths of light ranging from 500-1000 nm, was shown to significantly reduce drusen thickness and volume and improve clinical outcomes (141) (NCT02725762). The Lightsite III clinical trial (NCT04065490) utilizing the Valeda Light Delivery System, which delivers 590, 660, and 850 nm of light to the study eye of patients with dry AMD, met its primary efficacy endpoint and demonstrated improved anatomical and clinical outcomes (142).
Future directions
Currently, there are two FDA-approved treatments for GA that target the complement pathway: Pegcetacoplan and Izervay. While these complement inhibitors have been approved and show promise to slow down lesion growth, their efficacy is limited, and they do not work for all patients. Also, these treatments are not the cure and only slow the progression of the disease. These treatments require frequent visits to the clinic and can be uncomfortable for patients because of multiple frequent administration during treatments. No therapy is currently able to repair the impaired retinal pigment epithelium (RPE) and outer retinal layers that are affected by GA.
In summary, the current treatment options for GA have limitations, including the lack of a cure, limited efficacy, the requirement for frequent administration, unknown factors underlying the disease, and the inability to repair the damage caused by GA. Extensive research has contributed to new GA classification based on advanced imaging techniques and phenotypic characteristics. Understanding and identifying genetic predispositions for diagnosis and designing specific treatment approaches is imperative. Promising therapies targeting multiple signaling pathways are underway to establish different strategies to treat the disease through gene therapy, stem cell therapy, and neuroprotective agents.
Author contributions
KR: Writing – original draft, Writing – review & editing. FD: Writing – review & editing. AU: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
KR, FD, and AU were employed by Ocugen Inc., which is currently developing gene therapy for AMD.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Arya M, Sabrosa AS, Duker JS, Waheed NK. Choriocapillaris changes in dry age-related macular degeneration and geographic atrophy: a review. Eye Vis (Lond) (2018) 5:22. doi: 10.1186/s40662-018-0118-x
2. Fritsche LG, Fariss RN, Stambolian D, Abecasis GR, Curcio CA, Swaroop A. Age-related macular degeneration: genetics and biology coming together. Annu Rev Genomics Hum Genet (2014) 15:151–71. doi: 10.1146/annurev-genom-090413-025610
3. Clevenger L, Rachitskaya A. Identifying geographic atrophy. Curr Opin Ophthalmol (2023) 34(3):195–202. doi: 10.1097/ICU.0000000000000952
4. Seddon JM, Sharma S, Adelman RA. Evaluation of the clinical age-related maculopathy staging system. Ophthalmology (2006) 113(2):260–6. doi: 10.1016/j.ophtha.2005.11.001
5. Rein DB, Wittenborn JS, Burke-Conte Z, Gulia R, Robalik T, Ehrlich JR, et al. Prevalence of age-related macular degeneration in the US in 2019. JAMA Ophthalmol (2022) 140(12):1202–8. doi: 10.1001/jamaophthalmol.2022.4401
6. Wong WL, Su X, Li X, Cheung CM, Klein R, Cheng CY, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health (2014) 2(2):e106–116. doi: 10.1016/S2214-109X(13)70145-1
7. Rim TH, Kawasaki R, Tham YC, Kang SW, Ruamviboonsuk P, Bikbov MM, et al. Prevalence and pattern of geographic atrophy in asia: the asian eye epidemiology consortium. Ophthalmology (2020) 127(10):1371–81. doi: 10.1016/j.ophtha.2020.04.019
8. Rudnicka AR, Jarrar Z, Wormald R, Cook DG, Fletcher A, Owen CG. Age and gender variations in age-related macular degeneration prevalence in populations of European ancestry: a meta-analysis. Ophthalmology (2012) 119(3):571–80. doi: 10.1016/j.ophtha.2011.09.027
9. Bakri SJ, Bektas M, Sharp D, Luo R, Sarda SP, Khan S. Geographic atrophy: Mechanism of disease, pathophysiology, and role of the complement system. J Manag Care Spec Pharm (2023) 29(5-a Suppl):S2–S11. doi: 10.18553/jmcp.2023.29.5-a.s2
10. Bagheri S, Lains I, Silverman RF, Kim I, Eliott D, Silva R, et al. Percentage of foveal vs total macular geographic atrophy as a predictor of visual acuity in age-related macular degeneration. J Vitreoretin Dis (2019) 3(5):278–82. doi: 10.1177/2474126419859454
11. Boyer DS, Schmidt-Erfurth U, van Lookeren Campagne M, Henry EC, Brittain C. The pathophysiology of geographic atrophy secondary to age-related macular degeneration and the complement pathway as a therapeutic target. Retina (2017) 37(5):819–35. doi: 10.1097/IAE.0000000000001392
12. Fleckenstein M, Mitchell P, Freund KB, Sadda S, Holz FG, Brittain C, et al. The progression of geographic atrophy secondary to age-related macular degeneration. Ophthalmology (2018) 125(3):369–90. doi: 10.1016/j.ophtha.2017.08.038
13. Sunness JS, Rubin GS, Applegate CA, Bressler NM, Marsh MJ, Hawkins BS, et al. Visual function abnormalities and prognosis in eyes with age-related geographic atrophy of the macula and good visual acuity. Ophthalmology (1997) 104(10):1677–91. doi: 10.1016/S0161-6420(97)30079-7
14. Sunness JS, Rubin GS, Broman A, Applegate CA, Bressler NM, Hawkins BS. Low luminance visual dysfunction as a predictor of subsequent visual acuity loss from geographic atrophy in age-related macular degeneration. Ophthalmology (2008) 115(9):1480–1488, 1488 e1481-1482. doi: 10.1016/j.ophtha.2008.03.009
15. Wu Z, Ayton LN, Luu CD, Guymer RH. Longitudinal changes in microperimetry and low luminance visual acuity in age-related macular degeneration. JAMA Ophthalmol (2015) 133(4):442–8. doi: 10.1001/jamaophthalmol.2014.5963
16. Nigalye AK, Hess K, Pundlik SJ, Jeffrey BG, Cukras CA, Husain D. Dark adaptation and its role in age-related macular degeneration. J Clin Med (2022) 11(5). doi: 10.3390/jcm11051358
17. Meleth AD, Mettu P, Agron E, Chew EY, Sadda SR, Ferris FL, et al. Changes in retinal sensitivity in geographic atrophy progression as measured by microperimetry. Invest Ophthalmol Vis Sci (2011) 52(2):1119–26. doi: 10.1167/iovs.10-6075
18. Farci R, Carta A, Fogagnolo P, Rossetti LM, Fossarello M. Compass fundus-guided perimetry in geographic atrophy. J Ophthalmol (2022) 2022:1315588. doi: 10.1155/2022/1315588
19. Csaky KG, Patel PJ, Sepah YJ, Birch DG, Do DV, Ip MS, et al. Microperimetry for geographic atrophy secondary to age-related macular degeneration. Surv Ophthalmol (2019) 64(3):353–64. doi: 10.1016/j.survophthal.2019.01.014
20. Gobel AP, Fleckenstein M, Schmitz-Valckenberg S, Brinkmann CK, Holz FG. Imaging geographic atrophy in age-related macular degeneration. Ophthalmologica (2011) 226(4):182–90. doi: 10.1159/000330420
21. Fernandes AR, Zielinska A, Sanchez-Lopez E, Dos Santos T, Garcia ML, Silva AM, et al. Exudative versus nonexudative age-related macular degeneration: physiopathology and treatment options. Int J Mol Sci (2022) 23(5). doi: 10.3390/ijms23052592
22. Jeong YJ, Hong IH, Chung JK, Kim KL, Kim HK, Park SP. Predictors for the progression of geographic atrophy in patients with age-related macular degeneration: fundus autofluorescence study with modified fundus camera. Eye (Lond) (2014) 28(2):209–18. doi: 10.1038/eye.2013.275
23. Feeney-Burns L, Berman ER, Rothman H. Lipofuscin of human retinal pigment epithelium. Am J Ophthalmol (1980) 90(6):783–91. doi: 10.1016/S0002-9394(14)75193-1
24. Eldred GE, Lasky MR. Retinal age pigments generated by self-assembling lysosomotropic detergents. Nature (1993) 361(6414):724–6. doi: 10.1038/361724a0
25. Holz FG, Bellmann C, Margaritidis M, Schutt F, Otto TP, Volcker HE. Patterns of increased in vivo fundus autofluorescence in the junctional zone of geographic atrophy of the retinal pigment epithelium associated with age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol (1999) 237(2):145–52. doi: 10.1007/s004170050209
26. Wang J, Ying GS. Growth rate of geographic atrophy secondary to age-related macular degeneration: A meta-analysis of natural history studies and implications for designing future trials. Ophthalmic Res (2021) 64(2):205–15. doi: 10.1159/000510507
27. Sparrow JR, Duncker T, Schuerch K, Paavo M, de Carvalho JRL. Lessons learned from quantitative fundus autofluorescence. Prog Retin Eye Res (2020) 74:100774. doi: 10.1016/j.preteyeres.2019.100774
28. Pole C, Ameri H. Fundus autofluorescence and clinical applications. J Ophthalmic Vis Res (2021) 16(3):432–61. doi: 10.18502/jovr.v16i3.9439
29. Huang D, Swanson EA, Lin CP, Schuman JS, Stinson WG, Chang W, et al. Optical coherence tomography. Science (1991) 254(5035):1178–81. doi: 10.1126/science.1957169
30. Talks SJ, Aftab AM, Ashfaq I, Soomro T. The role of new imaging methods in managing age-related macular degeneration. Asia Pac J Ophthalmol (Phila) (2017) 6(6):498–507.
31. Sacconi R, Battista M, Borrelli E, Senni C, Tombolini B, Grosso D, et al. Choroidal vascularity index is associated with geographic atrophy progression. Retina (2022) 42(2):381–7. doi: 10.1097/IAE.0000000000003305
32. Goh KL, Chen FK, Balaratnasingam C, Abbott CJ, Hodgson LAB, Guymer RH, et al. Cuticular drusen in age-related macular degeneration: association with progression and impact on visual sensitivity. Ophthalmology (2022) 129(6):653–60. doi: 10.1016/j.ophtha.2022.01.028
33. Liu J, Laiginhas R, Shen M, Shi Y, Li J, Trivizki O, et al. Multimodal imaging and en face OCT detection of calcified drusen in eyes with age-related macular degeneration. Ophthalmol Sci (2022) 2(2). doi: 10.1016/j.xops.2022.100162
34. Agron E, Domalpally A, Cukras CA, Clemons TE, Chen Q, Lu Z, et al. Reticular pseudodrusen: the third macular risk feature for progression to late age-related macular degeneration: age-related eye disease study 2 report 30. Ophthalmology (2022) 129(10):1107–19. doi: 10.1016/j.ophtha.2022.05.021
35. Holz FG, Sadda SR, Staurenghi G, Lindner M, Bird AC, Blodi BA, et al. Imaging protocols in clinical studies in advanced age-related macular degeneration: recommendations from classification of atrophy consensus meetings. Ophthalmology (2017) 124(4):464–78. doi: 10.1016/j.ophtha.2016.12.002
36. Sadda SR, Guymer R, Holz FG, Schmitz-Valckenberg S, Curcio CA, Bird AC, et al. Consensus definition for atrophy associated with age-related macular degeneration on OCT: classification of atrophy report 3. Ophthalmology (2018) 125(4):537–48. doi: 10.1016/j.ophtha.2017.09.028
37. Jaffe GJ, Chakravarthy U, Freund KB, Guymer RH, Holz FG, Liakopoulos S, et al. Imaging features associated with progression to geographic atrophy in age-related macular degeneration: classification of atrophy meeting report 5. Ophthalmol Retina (2021) 5(9):855–67. doi: 10.1016/j.oret.2020.12.009
38. Guymer RH, Rosenfeld PJ, Curcio CA, Holz FG, Staurenghi G, Freund KB, et al. Incomplete retinal pigment epithelial and outer retinal atrophy in age-related macular degeneration: classification of atrophy meeting report 4. Ophthalmology (2020) 127(3):394–409. doi: 10.1016/j.ophtha.2019.09.035
39. Moreno-Garcia A, Kun A, Calero O, Medina M, Calero M. An overview of the role of lipofuscin in age-related neurodegeneration. Front Neurosci (2018) 12:464. doi: 10.3389/fnins.2018.00464
40. Zhang X, Sivaprasad S. Drusen and pachydrusen: the definition, pathogenesis, and clinical significance. Eye (Lond) (2021) 35(1):121–33. doi: 10.1038/s41433-020-01265-4
41. Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U.S.A. (2002) 99(23):14682–7. doi: 10.1073/pnas.222551899
42. Holz FG, Schutt F, Kopitz J, Eldred GE, Kruse FE, Volcker HE, et al. Inhibition of lysosomal degradative functions in RPE cells by a retinoid component of lipofuscin. Invest Ophthalmol Vis Sci (1999) 40(3):737–43.
43. Schutt F, Davies S, Kopitz J, Holz FG, Boulton ME. Photodamage to human RPE cells by A2-E, a retinoid component of lipofuscin. Invest Ophthalmol Vis Sci (2000) 41(8):2303–8.
44. Rozanowska M, Jarvis-Evans J, Korytowski W, Boulton ME, Burke JM, Sarna T. Blue light-induced reactivity of retinal age pigment. In vitro generation of oxygen-reactive species. J Biol Chem (1995) 270(32):18825–30. doi: 10.1074/jbc.270.32.18825
45. Saari JC. Vitamin A metabolism in rod and cone visual cycles. Annu Rev Nutr (2012) 32:125–45. doi: 10.1146/annurev-nutr-071811-150748
46. Zhang D, Robinson K, Washington I. C20D3-vitamin A prevents retinal pigment epithelium atrophic changes in a mouse model. Transl Vis Sci Technol (2021) 10(14):8. doi: 10.1167/tvst.10.14.8
47. Sajovic J, Meglic A, Glavac D, Markelj S, Hawlina M, Fakin A. The role of vitamin A in retinal diseases. Int J Mol Sci (2022) 23(3). doi: 10.3390/ijms23031014
48. Kaufman Y, Ma L, Washington I. Deuterium enrichment of vitamin A at the C20 position slows the formation of detrimental vitamin A dimers in wild-type rodents. J Biol Chem (2011) 286(10):7958–65. doi: 10.1074/jbc.M110.178640
49. Mihai DM, Washington I. Vitamin A dimers trigger the protracted death of retinal pigment epithelium cells. Cell Death Dis (2014) 5(7):e1348. doi: 10.1038/cddis.2014.314
50. Penn J, Mihai DM, Washington I. Morphological and physiological retinal degeneration induced by intravenous delivery of vitamin A dimers in rabbits. Dis Model Mech (2015) 8(2):131–8. doi: 10.1242/dmm.017194
51. Lynch AM, Mandava N, Patnaik JL, Frazer-Abel AA, Wagner BD, Palestine AG, et al. Systemic activation of the complement system in patients with advanced age-related macular degeneration. Eur J Ophthalmol (2020) 30(5):1061–8. doi: 10.1177/1120672119857896
52. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol (2010) 11(9):785–97. doi: 10.1038/ni.1923
53. Park YG, Park YS, Kim IB. Complement system and potential therapeutics in age-related macular degeneration. Int J Mol Sci (2021) 22(13). doi: 10.3390/ijms22136851
54. Tacnet-Delorme P, Chevallier S, Arlaud GJ. Beta-amyloid fibrils activate the C1 complex of complement under physiological conditions: evidence for a binding site for A beta on the C1q globular regions. J Immunol (2001) 167(11):6374–81. doi: 10.4049/jimmunol.167.11.6374
55. Ma W, Paik DC, Barile GR. Bioactive lysophospholipids generated by hepatic lipase degradation of lipoproteins lead to complement activation via the classical pathway. Invest Ophthalmol Vis Sci (2014) 55(10):6187–93. doi: 10.1167/iovs.14-14352
56. Yednock T, Fong DS, Lad EM. C1q and the classical complement cascade in geographic atrophy secondary to age-related macular degeneration. Int J Retina Vitreous (2022) 8(1):79. doi: 10.1186/s40942-022-00431-y
57. Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol (2002) 134(3):411–31. doi: 10.1016/S0002-9394(02)01624-0
58. Kim BJ, Liu T, Mastellos DC, Lambris JD. Emerging opportunities for C3 inhibition in the eye. Semin Immunol (2022) 59:101633. doi: 10.1016/j.smim.2022.101633
59. de Jong S, Tang J, Clark SJ. Age-related macular degeneration: A disease of extracellular complement amplification. Immunol Rev (2023) 313(1):279–97. doi: 10.1111/imr.13145
60. Keenan TDL. Geographic atrophy in age-related macular degeneration: A tale of two stages. Ophthalmol Sci (2023) 3(3):100306. doi: 10.1016/j.xops.2023.100306
61. Loyet KM, Deforge LE, Katschke KJ Jr., Diehl L, Graham RR, Pao L, et al. Activation of the alternative complement pathway in vitreous is controlled by genetics in age-related macular degeneration. Invest Ophthalmol Vis Sci (2012) 53(10):6628–37. doi: 10.1167/iovs.12-9587
62. Reynolds R, Hartnett ME, Atkinson JP, Giclas PC, Rosner B, Seddon JM. Plasma complement components and activation fragments: associations with age-related macular degeneration genotypes and phenotypes. Invest Ophthalmol Vis Sci (2009) 50(12):5818–27. doi: 10.1167/iovs.09-3928
63. Rollins SA, Sims PJ. The complement-inhibitory activity of CD59 resides in its capacity to block incorporation of C9 into membrane C5b-9. J Immunol (1990) 144(9):3478–83. doi: 10.4049/jimmunol.144.9.3478
64. Katschke KJ Jr., Xi H, Cox C, Truong T, Malato Y, Lee WP, et al. Classical and alternative complement activation on photoreceptor outer segments drives monocyte-dependent retinal atrophy. Sci Rep (2018) 8(1):7348. doi: 10.1038/s41598-018-25557-8
65. Yu M, Zou W, Peachey NS, McIntyre TM, Liu J. A novel role of complement in retinal degeneration. Invest Ophthalmol Vis Sci (2012) 53(12):7684–92. doi: 10.1167/iovs.12-10069
66. Hoh Kam J, Lenassi E, Malik TH, Pickering MC, Jeffery G. Complement component C3 plays a critical role in protecting the aging retina in a murine model of age-related macular degeneration. Am J Pathol (2013) 183(2):480–92. doi: 10.1016/j.ajpath.2013.04.008
67. Mukai R, Okunuki Y, Husain D, Kim CB, Lambris JD, Connor KM. The complement system is critical in maintaining retinal integrity during aging. Front Aging Neurosci (2018) 10:15. doi: 10.3389/fnagi.2018.00015
68. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
69. Celkova L, Doyle SL, Campbell M. NLRP3 inflammasome and pathobiology in AMD. J Clin Med (2015) 4(1):172–92. doi: 10.3390/jcm4010172
70. Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med (2012) 18(5):791–8. doi: 10.1038/nm.2717
71. Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Kohl J, et al. C3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood (2013) 122(20):3473–81. doi: 10.1182/blood-2013-05-502229
72. Liu RT, Gao J, Cao S, Sandhu N, Cui JZ, Chou CL, et al. Inflammatory mediators induced by amyloid-beta in the retina and RPE in vivo: implications for inflammasome activation in age-related macular degeneration. Invest Ophthalmol Vis Sci (2013) 54(3):2225–37. doi: 10.1167/iovs.12-10849
73. Wiesen JL, Tomasi TB. Dicer is regulated by cellular stresses and interferons. Mol Immunol (2009) 46(6):1222–8. doi: 10.1016/j.molimm.2008.11.012
74. Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature (2011) 471(7338):325–30. doi: 10.1038/nature09830
75. Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell (2012) 149(4):847–59. doi: 10.1016/j.cell.2012.03.036
76. Wang W, Wang WH, Azadzoi KM, Dai P, Wang Q, Sun JB, et al. Alu RNA accumulation in hyperglycemia augments oxidative stress and impairs eNOS and SOD2 expression in endothelial cells. Mol Cell Endocrinol (2016) 426:91–100. doi: 10.1016/j.mce.2016.02.008
77. Chakravarthy U, Augood C, Bentham GC, de Jong PT, Rahu M, Seland J, et al. Cigarette smoking and age-related macular degeneration in the EUREYE Study. Ophthalmology (2007) 114(6):1157–63. doi: 10.1016/j.ophtha.2006.09.022
78. Pryor WA, Hales BJ, Premovic PI, Church DF. The radicals in cigarette tar: their nature and suggested physiological implications. Science (1983) 220(4595):425–7. doi: 10.1126/science.6301009
79. Cai J, Nelson KC, Wu M, Sternberg P Jr., Jones DP. Oxidative damage and protection of the RPE. Prog Retin Eye Res (2000) 19(2):205–21. doi: 10.1016/S1350-9462(99)00009-9
80. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469(7329):221–5. doi: 10.1038/nature09663
81. Kanow MA, Giarmarco MM, Jankowski CS, Tsantilas K, Engel AL, Du J, et al. Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. Elife (2017) 6. doi: 10.7554/eLife.28899
82. Brown EE, DeWeerd AJ, Ildefonso CJ, Lewin AS, Ash JD. Mitochondrial oxidative stress in the retinal pigment epithelium (RPE) led to metabolic dysfunction in both the RPE and retinal photoreceptors. Redox Biol (2019) 24:101201. doi: 10.1016/j.redox.2019.101201
83. Kerur N, Fukuda S, Banerjee D, Kim Y, Fu D, Apicella I, et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat Med (2018) 24(1):50–61. doi: 10.1038/nm.4450
84. Boukhtouche F, Mariani J, Tedgui A. The “CholesteROR” protective pathway in the vascular system. Arterioscler Thromb Vasc Biol (2004) 24(4):637–43. doi: 10.1161/01.ATV.0000119355.56036.de
85. Lau P, Fitzsimmons RL, Raichur S, Wang SC, Lechtken A, Muscat GE. The orphan nuclear receptor, RORalpha, regulates gene expression that controls lipid metabolism: staggerer (SG/SG) mice are resistant to diet-induced obesity. J Biol Chem (2008) 283(26):18411–21. doi: 10.1074/jbc.M710526200
86. Delerive P, Monte D, Dubois G, Trottein F, Fruchart-Najib J, Mariani J, et al. The orphan nuclear receptor ROR alpha is a negative regulator of the inflammatory response. EMBO Rep (2001) 2(1):42–8. doi: 10.1093/embo-reports/kve007
87. Silveira AC, Morrison MA, Ji F, Xu H, Reinecke JB, Adams SM, et al. Convergence of linkage, gene expression and association data demonstrates the influence of the RAR-related orphan receptor alpha (RORA) gene on neovascular AMD: a systems biology based approach. Vision Res (2010) 50(7):698–715. doi: 10.1016/j.visres.2009.09.016
88. Sun Y, Liu CH, SanGiovanni JP, Evans LP, Tian KT, Zhang B, et al. Nuclear receptor RORalpha regulates pathologic retinal angiogenesis by modulating SOCS3-dependent inflammation. Proc Natl Acad Sci U.S.A. (2015) 112(33):10401–6. doi: 10.1073/pnas.1504387112
89. Liu CH, Yemanyi F, Bora K, Kushwah N, Blomfield AK, Kamenecka TM, et al. Genetic deficiency and pharmacological modulation of RORalpha regulate laser-induced choroidal neovascularization. Aging (Albany NY) (2023) 15(1):37–52. doi: 10.18632/aging.204480
90. Karlstetter M, Scholz R, Rutar M, Wong WT, Provis JM, Langmann T. Retinal microglia: just bystander or target for therapy. Prog Retin Eye Res (2015) 45:30–57. doi: 10.1016/j.preteyeres.2014.11.004
91. Fletcher EL. Contribution of microglia and monocytes to the development and progression of age related macular degeneration. Ophthalmic Physiol Opt (2020) 40(2):128–39. doi: 10.1111/opo.12671
92. Yu C, Roubeix C, Sennlaub F, Saban DR. Microglia versus monocytes: distinct roles in degenerative diseases of the retina. Trends Neurosci (2020) 43(6):433–49. doi: 10.1016/j.tins.2020.03.012
93. Zhang R, Wang LY, Wang YF, Wu CR, Lei CL, Wang MX, et al. Associations between the T280M and V249I SNPs in CX3CR1 and the risk of age-related macular degeneration. Invest Ophthalmol Vis Sci (2015) 56(9):5590–8. doi: 10.1167/iovs.15-16830
94. Fritsche LG, Igl W, Bailey JN, Grassmann F, Sengupta S, Bragg-Gresham JL, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet (2016) 48(2):134–43. doi: 10.1038/ng.3448
95. Grassmann F, Harsch S, Brandl C, Kiel C, Nurnberg P, Toliat MR, et al. Assessment of novel genome-wide significant gene loci and lesion growth in geographic atrophy secondary to age-related macular degeneration. JAMA Ophthalmol (2019) 137(8):867–76. doi: 10.1001/jamaophthalmol.2019.1318
96. Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U.S.A. (2005) 102(20):7227–32. doi: 10.1073/pnas.0501536102
97. Seddon JM, Yu Y, Miller EC, Reynolds R, Tan PL, Gowrisankar S, et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat Genet (2013) 45(11):1366–70. doi: 10.1038/ng.2741
98. Hoffman JD, Cooke Bailey JN, D’Aoust L, Cade W, Ayala-Haedo J, Fuzzell D, et al. Rare complement factor H variant associated with age-related macular degeneration in the Amish. Invest Ophthalmol Vis Sci (2014) 55(7):4455–60. doi: 10.1167/iovs.13-13684
99. Kavanagh D, Yu Y, Schramm EC, Triebwasser M, Wagner EK, Raychaudhuri S, et al. Rare genetic variants in the CFI gene are associated with advanced age-related macular degeneration and commonly result in reduced serum factor I levels. Hum Mol Genet (2015) 24(13):3861–70. doi: 10.1093/hmg/ddv091
100. Edwards AO, Ritter R, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science (2005) 308(5720):421–4. doi: 10.1126/science.1110189
101. Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science (2005) 308(5720):419–21. doi: 10.1126/science.1110359
102. Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, et al. Complement factor H polymorphism in age-related macular degeneration. Science (2005) 308(5720):385–9. doi: 10.1126/science.1109557
103. Lores-Motta L, van Beek AE, Willems E, Zandstra J, van Mierlo G, Einhaus A, et al. Common haplotypes at the CFH locus and low-frequency variants in CFHR2 and CFHR5 associate with systemic FHR concentrations and age-related macular degeneration. Am J Hum Genet (2021) 108(8):1367–84. doi: 10.1016/j.ajhg.2021.06.002
104. Yang Z, Camp NJ, Sun H, Tong Z, Gibbs D, Cameron DJ, et al. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science (2006) 314(5801):992–3. doi: 10.1126/science.1133811
105. Chen W, Xu W, Tao Q, Liu J, Li X, Gan X, et al. Meta-analysis of the association of the HTRA1 polymorphisms with the risk of age-related macular degeneration. Exp Eye Res (2009) 89(3):292–300. doi: 10.1016/j.exer.2008.10.017
106. Rivera A, Fisher SA, Fritsche LG, Keilhauer CN, Lichtner P, Meitinger T, et al. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet (2005) 14(21):3227–36. doi: 10.1093/hmg/ddi353
107. Oka C, Tsujimoto R, Kajikawa M, Koshiba-Takeuchi K, Ina J, Yano M, et al. HtrA1 serine protease inhibits signaling mediated by Tgfbeta family proteins. Development (2004) 131(5):1041–53. doi: 10.1242/dev.00999
108. Lin MK, Yang J, Hsu CW, Gore A, Bassuk AG, Brown LM, et al. HTRA1, an age-related macular degeneration protease, processes extracellular matrix proteins EFEMP1 and TSP1. Aging Cell (2018) 17(4):e12710. doi: 10.1111/acel.12710
109. Zhang L, Lim SL, Du H, Zhang M, Kozak I, Hannum G, et al. High temperature requirement factor A1 (HTRA1) gene regulates angiogenesis through transforming growth factor-beta family member growth differentiation factor 6. J Biol Chem (2012) 287(2):1520–6. doi: 10.1074/jbc.M111.275990
110. Marmorstein LY, Munier FL, Arsenijevic Y, Schorderet DF, McLaughlin PJ, Chung D, et al. Aberrant accumulation of EFEMP1 underlies drusen formation in Malattia Leventinese and age-related macular degeneration. Proc Natl Acad Sci U.S.A. (2002) 99(20):13067–72. doi: 10.1073/pnas.202491599
111. Jones A, Kumar S, Zhang N, Tong Z, Yang JH, Watt C, et al. Increased expression of multifunctional serine protease, HTRA1, in retinal pigment epithelium induces polypoidal choroidal vasculopathy in mice. Proc Natl Acad Sci U.S.A. (2011) 108(35):14578–83. doi: 10.1073/pnas.1102853108
112. Nakayama M, Iejima D, Akahori M, Kamei J, Goto A, Iwata T. Overexpression of HtrA1 and exposure to mainstream cigarette smoke leads to choroidal neovascularization and subretinal deposits in aged mice. Invest Ophthalmol Vis Sci (2014) 55(10):6514–23. doi: 10.1167/iovs.14-14453
113. Supanji, Shimomachi M, Hasan MZ, Kawaichi M, Oka C. HtrA1 is induced by oxidative stress and enhances cell senescence through p38 MAPK pathway. Exp Eye Res (2013) 112:79–92. doi: 10.1016/j.exer.2013.04.013
114. Ratnapriya R, Sosina OA, Starostik MR, Kwicklis M, Kapphahn RJ, Fritsche LG, et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat Genet (2019) 51(4):606–10. doi: 10.1038/s41588-019-0351-9
115. Orozco LD, Chen HH, Cox C, Katschke KJ Jr., Arceo R, Espiritu C, et al. Integration of eQTL and a single-cell atlas in the human eye identifies causal genes for age-related macular degeneration. Cell Rep (2020) 30(4):1246–1259 e1246. doi: 10.1016/j.celrep.2019.12.082
116. Flores R, Carneiro A, Tenreiro S, Seabra MC. Retinal progression biomarkers of early and intermediate age-related macular degeneration. Life (Basel) (2021) 12(1). doi: 10.3390/life12010036
117. Cachulo L, Silva R, Fonseca P, Pires I, Carvajal-Gonzalez S, Bernardes R, et al. Early markers of choroidal neovascularization in the fellow eye of patients with unilateral exudative age-related macular degeneration. Ophthalmologica (2011) 225(3):144–9. doi: 10.1159/000321064
118. Birch DG, Jost BF, Fish GE. The focal electroretinogram in fellow eyes of patients with idiopathic macular holes. Arch Ophthalmol (1988) 106(11):1558–63. doi: 10.1001/archopht.1988.01060140726043
119. Sadda SR, Chakravarthy U, Birch DG, Staurenghi G, Henry EC, Brittain C. Clinical endpoints for the study of geographic atrophy secondary to age-related macular degeneration. Retina (2016) 36(10):1806–22. doi: 10.1097/IAE.0000000000001283
120. Dimitrov PN, Robman LD, Varsamidis M, Aung KZ, Makeyeva GA, Guymer RH, et al. Visual function tests as potential biomarkers in age-related macular degeneration. Invest Ophthalmol Vis Sci (2011) 52(13):9457–69. doi: 10.1167/iovs.10-7043
121. Shahid H, Khan JC, Cipriani V, Sepp T, Matharu BK, Bunce C, et al. Age-related macular degeneration: the importance of family history as a risk factor. Br J Ophthalmol (2012) 96(3):427–31. doi: 10.1136/bjophthalmol-2011-300193
122. Lambert NG, ElShelmani H, Singh MK, Mansergh FC, Wride MA, Padilla M, et al. Risk factors and biomarkers of age-related macular degeneration. Prog Retin Eye Res (2016) 54:64–102. doi: 10.1016/j.preteyeres.2016.04.003
123. Halawa OA, Lin JB, Miller JW, Vavvas DG. A review of completed and ongoing complement inhibitor trials for geographic atrophy secondary to age-related macular degeneration. J Clin Med (2021) 10(12). doi: 10.3390/jcm10122580
124. Grover A, Sankaranarayanan S, Mathur V, Suri P, Qiu H, Andrews-Zwilling Y, et al. Pharmacokinetic and target engagement measures of ANX007, an anti-C1q antibody fragment, following intravitreal administration in nonhuman primates. Invest Ophthalmol Vis Sci (2023) 64(2):3. doi: 10.1167/iovs.64.2.3
125. Jiao H, Rutar M, Fernando N, Yednock T, Sankaranarayanan S, Aggio-Bruce R, et al. Subretinal macrophages produce classical complement activator C1q leading to the progression of focal retinal degeneration. Mol Neurodegener (2018) 13(1):45. doi: 10.1186/s13024-018-0278-0
126. Boyer DD, Ko YP, Podos SD, Cartwright ME, Gao X, Wiles JA, et al. Danicopan, an oral complement factor D inhibitor, exhibits high and sustained exposure in ocular tissues in preclinical studies. Transl Vis Sci Technol (2022) 11(10):37. doi: 10.1167/tvst.11.10.37
127. Farkas I, Baranyi L, Ishikawa Y, Okada N, Bohata C, Budai D, et al. CD59 blocks not only the insertion of C9 into MAC but inhibits ion channel formation by homologous C5b-8 as well as C5b-9. J Physiol (2002) 539(Pt 2):537–45. doi: 10.1113/jphysiol.2001.013381
128. Dreismann AK, Hallam TM, Tam LC, Nguyen CV, Hughes JP, Ellis S, et al. Gene targeting as a therapeutic avenue in diseases mediated by the complement alternative pathway. Immunol Rev (2023) 313(1):402–19. doi: 10.1111/imr.13149
129. Grossman TR, Carrer M, Shen L, Johnson RB, Hettrick LA, Henry SP, et al. Reduction in ocular complement factor B protein in mice and monkeys by systemic administration of factor B antisense oligonucleotide. Mol Vis (2017) 23:561–71.
130. Mugisho OO, Rupenthal ID, Paquet-Durand F, Acosta ML, Green CR. Targeting connexin hemichannels to control the inflammasome: the correlation between connexin43 and NLRP3 expression in chronic eye disease. Expert Opin Ther Targets (2019) 23(10):855–63. doi: 10.1080/14728222.2019.1673368
131. Akula M, McNamee S, Chan NPM, DeAngelis MM, Haider NB. RORA modifier gene therapy rescues retinal degeneration in a juvenile AMD mouse model of stargardt disease. Invest Ophthalmol Visual Sci (2023) 64(8):3842–2.
132. Berni R, Formelli F. In vitro interaction of fenretinide with plasma retinol-binding protein and its functional consequences. FEBS Lett (1992) 308(1):43–5. doi: 10.1016/0014-5793(92)81046-O
133. Mata NL, Lichter JB, Vogel R, Han Y, Bui TV, Singerman LJ. Investigation of oral fenretinide for treatment of geographic atrophy in age-related macular degeneration. Retina (2013) 33(3):498–507. doi: 10.1097/IAE.0b013e318265801d
134. Richard AJ, Duker JS, Reichel E. Geographic atrophy: where we are now and where we are going. Curr Opin Ophthalmol (2021) 32(3):247–52. doi: 10.1097/ICU.0000000000000763
135. Guymer RH, Baird PN, Varsamidis M, Busija L, Dimitrov PN, Aung KZ, et al. Proof of concept, randomized, placebo-controlled study of the effect of simvastatin on the course of age-related macular degeneration. PloS One (2013) 8(12):e83759. doi: 10.1371/journal.pone.0083759
136. Vavvas DG, Daniels AB, Kapsala ZG, Goldfarb JW, Ganotakis E, Loewenstein JI, et al. Regression of some high-risk features of age-related macular degeneration (AMD) in patients receiving intensive statin treatment. EBioMedicine (2016) 5:198–203. doi: 10.1016/j.ebiom.2016.01.033
137. Liu Y, Bell BA, Song Y, Zhang K, Anderson B, Axelsen PH, et al. Deuterated docosahexaenoic acid protects against oxidative stress and geographic atrophy-like retinal degeneration in a mouse model with iron overload. Aging Cell (2022) 21(4):e13579. doi: 10.1111/acel.13579
138. Skowronska-Krawczyk D, Chao DL. Long-chain polyunsaturated fatty acids and age-related macular degeneration. Adv Exp Med Biol (2019) 1185:39–43. doi: 10.1007/978-3-030-27378-1_7
139. Begum R, Powner MB, Hudson N, Hogg C, Jeffery G. Treatment with 670 nm light up regulates cytochrome C oxidase expression and reduces inflammation in an age-related macular degeneration model. PloS One (2013) 8(2):e57828. doi: 10.1371/journal.pone.0057828
140. Sivapathasuntharam C, Sivaprasad S, Hogg C, Jeffery G. Aging retinal function is improved by near infrared light (670 nm) that is associated with corrected mitochondrial decline. Neurobiol Aging (2017) 52:66–70. doi: 10.1016/j.neurobiolaging.2017.01.001
141. Munk MR, Markowitz SN, Devenyi R, Croissant C, Tedford S, Ruckert R, et al. Final analysis of LIGHTSITE I: A double-masked, randomized, sham-controlled study with photobiomodulation in dry age-related macular degeneration subjects. Invest Ophthalmol Visual Sci (2018) 59(9):2415–5.
Keywords: geographic atrophy, macular degeneration, drusen, inflammation, retinal biomarkers, gene therapy
Citation: Rajanala K, Dotiwala F and Upadhyay A (2023) Geographic atrophy: pathophysiology and current therapeutic strategies. Front. Ophthalmol. 3:1327883. doi: 10.3389/fopht.2023.1327883
Received: 25 October 2023; Accepted: 22 November 2023;
Published: 05 December 2023.
Edited by:
Alfredo Martínez, Centro de Investigación Biomédica de La Rioja, SpainReviewed by:
Ryo Mukai, Fukushima Medical University, JapanGlenn Prazere Lobo, University of Minnesota Twin Cities, United States
Copyright © 2023 Rajanala, Dotiwala and Upadhyay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arun Upadhyay, QXJ1bkBvY3VnZW4uY29t