 Ingrid M. Bonilla1,2
Ingrid M. Bonilla1,2


- 1 College of Pharmacy, The Ohio State University, Columbus, OH, USA
- 2 Dorothy M. Davis Heart and Lung Research Institute, The Ohio State University, Columbus, OH, USA
- 3 Glaxo Smith Kline, Ware, UK
- 4 College of Medicine, The Ohio State University, Columbus, OH, USA
Oxidative stress has been implicated in the pathogenesis of atrial fibrillation. There are multiple systems in the myocardium which contribute to redox homeostasis, and loss of homeostasis can result in oxidative stress. Potential sources of oxidants include nitric oxide synthases (NOS), which normally produce nitric oxide in the heart. Two NOS isoforms (1 and 3) are normally expressed in the heart. During pathologies such as heart failure, there is induction of NOS 2 in multiple cell types in the myocardium. In certain conditions, the NOS enzymes may become uncoupled, shifting from production of nitric oxide to superoxide anion, a potent free radical and oxidant. Multiple lines of evidence suggest a role for NOS in the pathogenesis of atrial fibrillation. Therapeutic approaches to reduce atrial fibrillation by modulation of NOS activity may be beneficial, although further investigation of this strategy is needed.
Introduction
Atrial fibrillation is the manifestation of multiple forms of pathologic processes. There are multiple known antecedents to atrial fibrillation including valve disease, increasing age, diabetes, and heart failure. It is notable that these antecedents are all associated with the development of oxidative stress (Van Wagoner, 2008). While many antecedents of atrial fibrillation are associated with oxidative stress, atrial fibrillation itself may also result in oxidative stress. Collectively, these data suggest a role for oxidative stress in the initiation and possibly sustenance of atrial fibrillation.
Current pharmacologic therapies for atrial fibrillation are drugs which modulate ion channel function. Unfortunately, current therapeutic approaches are sub-optimal due to lack of efficacy as well as adverse events, including proarrhythmia (Dobrev and Nattel, 2011; Schotten et al., 2011). An alternative approach, “upstream therapy” has been suggested to mitigate the signaling events resulting in pathologic alterations favoring the development of AF (Komatsu et al., 2009, 2011; Savelieva et al., 2011). Upstream therapy has been proposed as a means to provide a safer, more effective approach to prevent and/or treat atrial fibrillation. An example of an upstream approach is targeting oxidative stress and the consequent effects of reactive species on ion channels, ion currents, structural remodeling, and/or contractile function.
Redox Balance and Oxidative Stress
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are normal products of cellular metabolism, and there are multiple sources of ROS and RNS in the myocardium (Figure 1). When the oxidizing species exceed the reducing capacity of the cell, there is a loss of redox homeostasis. Excess ROS results in oxidative stress, while excess RNS results in nitrosative stress. There are multiple potential sources of reactive species in the myocardium including the mitochondrial electron transport chain, xanthine oxidase, NADPH oxidases, and uncoupled nitric oxide synthases (NOS), while the antioxidant (reducing) defenses include glutathione, superoxide dismutase, and thioredoxin. In addition to oxidative stress, it has recently been suggested that excess reducing equivalents may result in “reductive stress” (Rajasekaran et al., 2008; Zhang et al., 2010, 2012). A role for reductive stress in cardiomyopathic alterations has been suggested by these reports, but the contribution to arrhythmogenesis is undefined at the present time, and is beyond the scope of this review. This review is focused on the role of NOS as a source of ROS and RNS, and the evidence linking NOS to the pathogenesis of atrial fibrillation.
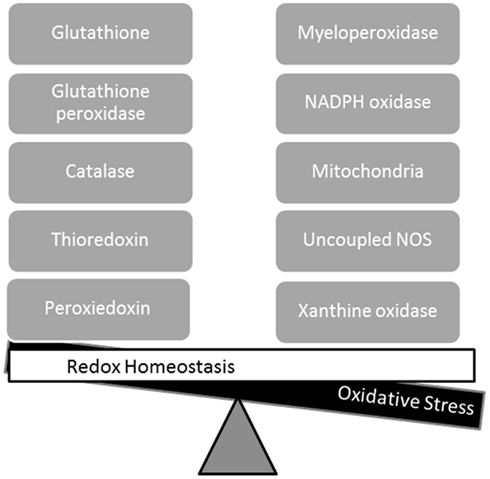
Figure 1. Redox homeostasis is maintained by balanced interactions of reducing (left side) and oxidizing pathways (right side). Oxidative stress results from an imbalance in which oxidizing species exceed the reducing capacity of the cell.
Nitric Oxide Synthase
There are three isoforms of nitric oxide (NO) synthase: NOS1, neuronal NOS (nNOS); NOS2, inducible NOS (iNOS); and NOS3, endothelial NOS (eNOS); the isoform names are based on the tissues in which they were first described. The heart normally expresses the constitutive enzymes NOS1 and NOS3. NOS2 is inducible and only expressed only during inflammatory and/or pathologic states such as hypertrophy or heart failure (Haywood et al., 1996; Vejlstrup et al., 1998; Nishijima et al., 2011). There are postulated intracellular compartments in cardiac myocytes for NOS1 (sarcoplasmic reticulum) and NOS3 (caveolae), while NOS2 is ubiquitously distributed in the cytosol of cardiac myocytes (Barouch et al., 2002; Ziolo and Bers, 2003). Under normal conditions, NOS can mediate effects through stimulatory effects of NO on guanalyte cyclase, or through nitration of tyrosine or nitrosylation of cysteine residues.
NO is a diffusible and highly reactive radical, and the half-life is therefore very short (∼5 s). The substrate for NOS is L-arginine, which is converted to citrulline with the concomitant production of nitric oxide. Substrate level regulation of NOS isoforms can occur via competitive inhibition by the endogenous methylarginines. Dimethylarginines are endogenous analogs of arginine. Asymmetric dimethylarginine (ADMA) is an inhibitor of NO synthase enzymatic activity and under conditions of NOS substrate (arginine) depletion ADMA can result in “uncoupling” of NOS. NOS uncoupling results in a shift of enzymatic activity such that NOS electron transfer is shifted from L-arginine to molecular oxygen yielding superoxide anion a potent oxidant. Tetrahydrobiopterin (BH4) is a cofactor for NOS activity, and when depleted, or oxidized to BH2 may also initiate NOS oxidase activity (see Figure 2). Recently an additional pathway for shifting NOS activity (specifically eNOS) from NO to production has been described, S-glutathionylation (Chen et al., 2010; Zweier et al., 2011). S-glutathionylation of eNOS is reversible and occurs when glutathione is bound to a protein thiol. This provides an alternative means for uncoupling of NOS activity, and the implications for modulation of other NOS isoforms are not clear at the present time.
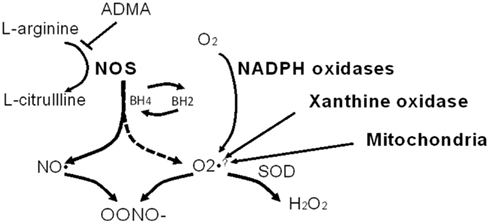
Figure 2. Nitric oxide synthase (NOS) as a source of oxidants. L-arginine is the substrate for NOS and in the presence of the NOS cofactor, tetrahydrobiopterin (BH4), NOS produces nitric oxide (NO). When L-arginine and/or BH4 is/are depleted, NOS becomes uncoupled (dashed arrow) and NOS produces superoxide anion at the expense of NO. Note that asymmetric dimethyl arginine (ADMA) can inhibit NOS activity. NO and combine to form peroxynitrite (OONO−) which is highly reactive, and nitrates tyrosine residues (to form 3-nitrotyrosine). Other abbreviations: SOD, superoxide dismutase; H2O2, hydrogen peroxide. Other non-NOS sources of are noted as well.
Electrophysiologic Effects of NO
The electrophysiologic effects of nitric oxide have been assessed using various NO donors, and there are multiple NO donors and scavengers which can be used to study the effects of NO signaling. The use of donors is necessary due to the high reactivity and short half-life of NO. An important consideration in the use of NO donors is the rate of release and the production of by-products, which may exert their own biological effects. A recent review (Tamargo et al., 2010) describes the NO modulation of cardiac ion channels and currents. The effects of NO on cardiac ion currents are species-dependent and can be highly variable, e.g., ICa-L. See Table 1 for a summary of effects on cardiac ion currents.
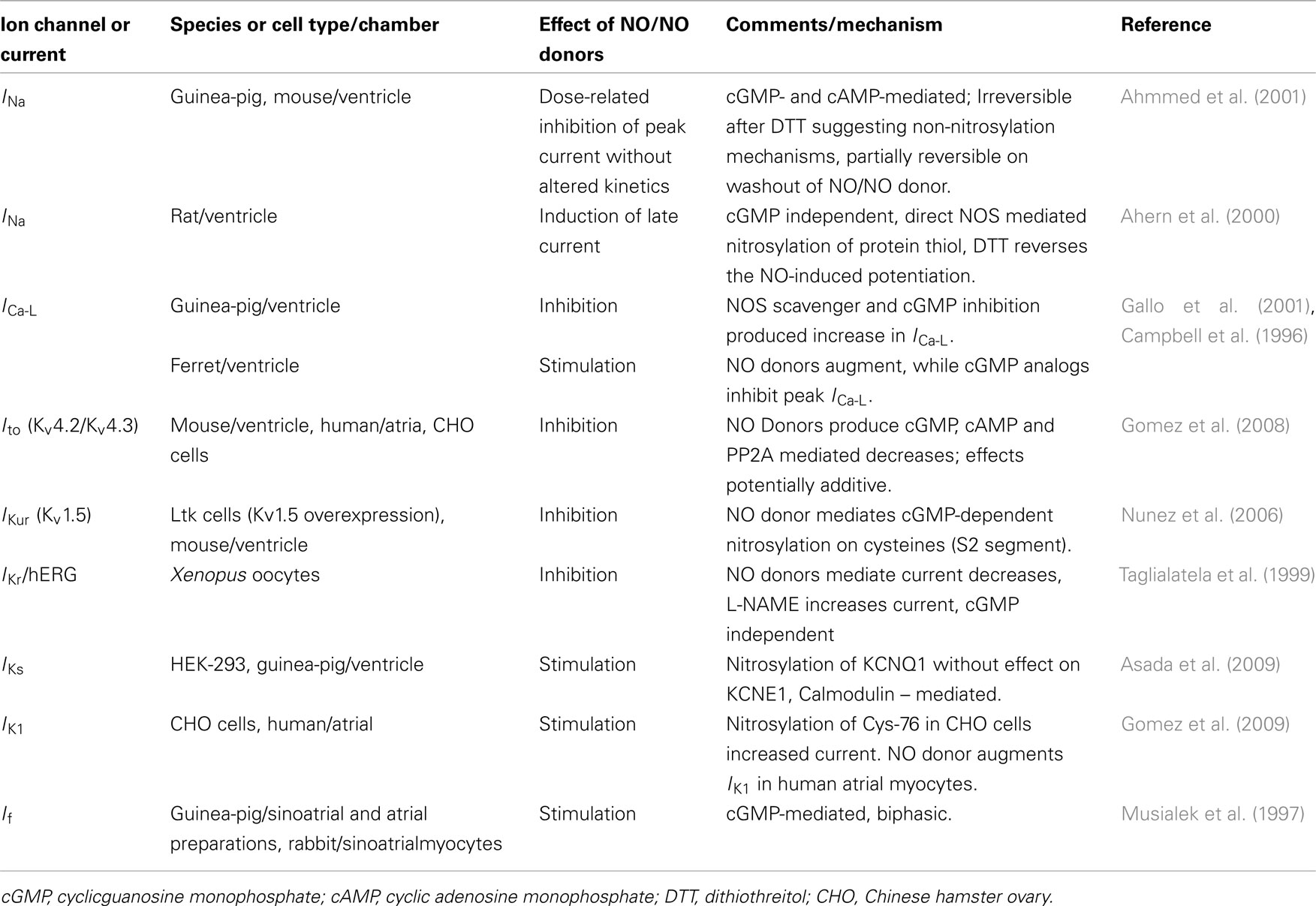
Table 1. Electrophysiologic effects of NO.
Electrophysiologic Effects of Peroxynitrite
SIN-1 (3-morpholinosydnonimine) is often described as a peroxynitrite donor, as it releases both NO and superoxide anion, which reacts at a diffusion limited rate to form the highly reactive peroxynitrite (OONO−). Peroxynitrite is highly diffusible and rapidly modifies tyrosine residues via nitration (resulting in 3-nitrotyrosine, 3-NT), and forms the highly stable modified amino acid, 3-NT. SIN-1 may also act as an NO donor, rather than a peroxynitrite donor in certain conditions. Similar to the effects of NO on cardiac ion currents, the effects of SIN-1 may be chamber and species-dependent. Table 2 provides an overview of electrophysiologic effects of SIN-1.
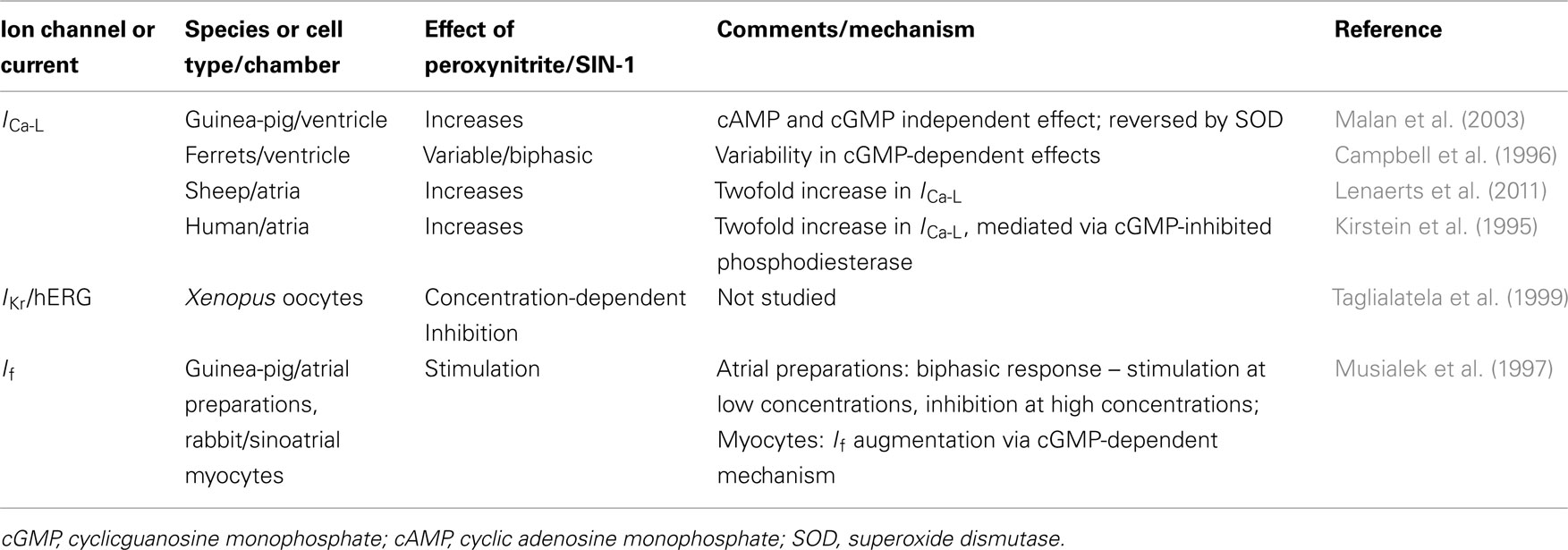
Table 2. Electrophysiologic effects of SIN-1.
Evidence for SIN-1 as a mediator of atrial myocyte calcium handling is provided by a recent paper in a sheep model of atrial tachycardia (4 h of rapid atrial pacing; Lenaerts et al., 2011). Infusion of molsidomine (SIN-1 prodrug) prevented the tachypacing-induced reduction of atrial ERP. This was attributable to direct electrophysiologic effects of the drug as it resulted in monophasic action potential prolongation in the right atrium of control sheep in sinus rhythm. Complementary experiments in isolated sheep atrial myocytes with SIN-1 found increases in ICa-L and the amplitude of the calcium transient.
The relevance of peroxynitrite to human atrial fibrillation has been examined, and 3-nitrotyrosine, a biomarker for OONO− formation, is indeed increased in atrial tissues from patients with persistent AF (Mihm et al., 2001). Furthermore, there was evidence that nitration contributed to the impaired contractile function associated with AF (Mihm et al., 2001). Collectively, these data support a role for acute and chronic modulation of atrial electrophysiology and function by OONO−.
NOS2 in Atrial Pathology
NOS2 can be inducibly expressed in macrophages, neutrophils, endothelial cells, vascular smooth muscle cells, and cardiomyocytes. NOS2 uncoupling in macrophages has been shown to catalyze the production of and OONO−, which are important in immune response and cytotoxicity of macrophages (Xia and Zweier, 1997). This is of interest as human atrial samples from patients with AF were recently shown to have increased immune cell infiltration compared to those from patients without AF (Yamashita et al., 2010). In this report, recruitment of immune cells to the atrial myocardium was shown to be mediated by intracellular adhesion molecule (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and monocyte chemotactic protein-1 (MCP-1) expression. The immunologically active monocytes and macrophages were found in a gradient with the highest concentration in the endocardium with evidence of migration to midmyocardium (Yamashita et al., 2010). This suggests a potential role for immune cells as contributors to oxidative stress-mediated atrial pathogenesis and AF.
While in normal hearts, NOS2 gene and protein expression is minimal or undetectable, it can be highly expressed during heart failure, including human heart failure (Winlaw et al., 1994; Haywood et al., 1996; Vejlstrup et al., 1998). Increased cytokines in heart failure (e.g., interleukin-6 and tumor necrosis factor-alpha) induce NOS2 expression (Balligand et al., 1994; Umar and van der Laarse, 2010). In end-stage human heart failure, NOS2 is increased in all four chambers (Haywood et al., 1996). The cellular location of NOS2 is not consistently described, however, diffuse distribution in myocytes has been reported, suggesting a role for NOS2-dependent modulation of atrial myocyte pathology (Barouch et al., 2002; Ziolo and Bers, 2003).
In human samples from patients with permanent AF, there was induction of NOS2 and increased 3-nitrotyrosine (OONO− biomarker) expression in the right atrium, compared to those with normal sinus rhythm (Han et al., 2008). In contrast, eNOS expression did not differ as a function of AF (Han et al., 2008). The LV size and ejection fraction was comparable in the permanent AF and control group, suggesting that the induction of iNOS was not a function of heart failure, but rather of AF itself.
We recently reported induction of NOS2 expression in the failing canine atria (4 months tachypacing-induced heart failure; Nishijima et al., 2011). This was associated with shortening of the atrial effective refractory period and the development of a substrate for inducible atrial fibrillation. We found that chronic heart failure led to depletion of BH4 in the left atrium, while L-arginine was unchanged. There was attendant evidence of NOS uncoupling as there was reduced NO production and increased production in atrial tissue, with evidence of increased atrial oxidative stress measured by electron paramagenetic resonance (EPR) spectroscopy. Repletion of BH4 with chronic oral treatment normalized NOS enzyme activity and EPR spectra, while reducing the inducibility of atrial fibrillation. The mechanisms for the in vivo electrophysiologic responses were assessed in left atrial myocytes, where we observed attenuation of the heart failure induced changes in electrophysiology. Thus, this supports a role for induction of NOS2 as a contributor to the substrate for atrial fibrillation in chronic heart failure.
eNOS in Atrial Pathology
Cai et al. (2002) studied a porcine model of 6 weeks of atrial fibrillation. The left atrium was found to produce a significant amount of NO, relative to the right atrium or aorta. Atrial fibrillation resulted in reduced left atrial endocardial eNOS and a significant reduction in NO production (Cai et al., 2002). In contrast, a paper in a 7-day canine atrial tachypacing model of AF reported increases in left atrial iNOS and eNOS (Shiroshita-Takeshita et al., 2006). Therapy to reduce eNOS expression (prednisone) was associated with attenuation of atrial electrophysiologic remodeling (less inducibility of AF, less shortening of the atrial ERP, and less reduction in rate adaptation of the atrial ERP) compared to control.
The association between eNOS polymorphisms and AF has been examined in clinical cohorts with non-valvular AF (Gensini et al., 2003; Fatini et al., 2006; Giusti et al., 2007). The first report examined the association between T-786C, G894T, and 4a/4b polymorphisms in Caucasians with AF, polymorphisms associated with altered NO production by eNOS (Gensini et al., 2003). This study compared patients with AF to normal controls, and did not find any association between eNOS genotype polymorphisms and AF. A second study of 331 patients with non-valvular AF examined T-786C, 4a/4b, and G894T eNOS genotype polymorphisms relative to controls, as well as polymorphisms in minK, the beta subunit of the IKs channel(Fatini et al., 2006). An interaction between minK S38G polymorphism and eNOS -786C gene variants was observed, resulting in a significant increase in the risk of AF, while the eNOS variant alone had a modest association with an increased risk of AF (OR = 1.50). The other two eNOS polymorphisms were not associated with the development of AF. A subsequent study found an association between the eNOS -786C genotype and homocysteine concentrations (Giusti et al., 2007). In this cohort, AF patients had elevated concentrations of homocysteine. While there was an association between the -786C genotype and homocysteine concentration, no association was found between the eNOS polymorphism and AF in this study. A study of patients with heart failure and atrial fibrillation also examined the role of T-786C, G894T, and intron 4b/a eNOS polymorphisms (Bedi et al., 2006). In this study, there was an association between the eNOS 894 T/T genotype and AF (OR = 3.2). Collectively, these data suggest a possible modulatory effect of eNOS gene polymorphisms on AF risk, which are likely to depend on interactions with other polymorphisms or variables to mediate an altered risk of AF.
Endogenous Methylarginines and Atrial Pathology
Symmetric dimethylarginine (SDMA) does not directly impact NOS enzymatic activity but can modulate NO formation via inhibiting cellular uptake of the NOS substrate, L-arginine. A study of ischemic stroke survivors found that those with atrial fibrillation had increased circulating SDMA compared to those in normal rhythm, while ADMA did not differ between the two groups.(Schulze et al., 2010) Elevated SDMA was also a predictor of all-cause mortality following ischemic stroke in this cohort. It is difficult to predict the impact of elevated SDMA on atrial function, as there is ample reserve of arginine in cardiac tissue.
Systemic concentrations of ADMA, the endogenous L-arginine analog, have been shown to be elevated in patients with heart failure (Usui et al., 1998), and in patients with persistent AF, with ADMA concentrations reduced following cardioversion (Goette et al., 2012). In parallel studies in a porcine right atrial tachypacing model (7 h), systemic ADMA concentrations were elevated. Further investigation is needed to understand the relationship between dimethylarginines and NOS modulation in atrial fibrillation.
NADPH Oxidase and NOS in Atrial Pathology
In addition to NOS, other sources of superoxide anion have been shown to participate in the pathology of AF. Time-dependent alterations in ROS sources during AF were examined in a tachypacing-induced AF chronic goat model (Reilly et al., 2011). Superoxide anion was increased after 2 weeks of AF, and did not increase further when assessed at 6 months of AF. The increase in superoxide was NADPH oxidase dependent at 2 weeks of AF (but not at 6 months of AF), and NOS- and mitochondria-dependent after 6 months of AF. Thus, the duration of AF may itself be an important mediator of ROS source. Complementary studies in atrial tissues from humans undergoing cardiac surgery found higher basal superoxide production in patients who developed post-operative AF, which was NADPH oxidase dependent; while atrial samples from patients with permanent AF had NOS- and mitochondrial-dependent increases in superoxide production. This suggests that the efficacy of ROS-specific therapeutic approaches may be highly dependent on the duration of AF, and that initiation, maintenance, and permanent sustenance of the arrhythmia may result from variable forms of ROS signaling. The authors also suggest that structural remodeling may contribute to the observed shift in ROS source.
A study in human right atrial samples examined sources of superoxide anion in atrial fibrillation by comparing samples from those with AF to those in sinus rhythm, with no history of atrial arrhythmias (Kim et al., 2005). In this report, NADPH oxidase was found to be a major source of superoxide, and NO synthases were also found to be a source of superoxide, consistent with NOS uncoupling in human AF.
Therapeutics
There are preclinical (e.g., prednisone or BH4 as reviewed above) as well as clinical studies describing modulation of NOS pathways as a therapeutic strategy in AF. In this section we will discuss other therapeutic approaches to ameliorate AF through manipulation of NOS pathways. An important caveat is that many of these approaches are non-specific and may affect multiple pathways which participate in AF pathogenesis.
Antioxidant Vitamins
There have been multiple trials of antioxidant therapies to prevent atrial fibrillation, many focusing on post-operative AF. Many of the trials have been limited by small cohorts, heterogeneous populations, variable therapeutic regimens, and variable adherence to the therapeutic regimen. A recent meta-analysis suggests that prophylaxis with the antioxidant vitamins C and E may be antiarrhythmic in the post-operative period (Harling et al., 2011). This meta-analysis of 567 patients found that the odds-ratio for the development of post-operative AF was 0.43 (95% CI 0.29–0.99). A sufficiently powered study with a standardized dosing regimen and appropriately controlled study design is required to definitively determine the role of peri-operative antioxidants in the prophylaxis of post-operative AF.
HMG–CoA Reductase Inhibitors (Statins)
In observational studies, statins [3-hydroxyl-3-methyl coenzyme A (HMG–CoA) reductase inhibitors have been shown to reduce the incidence of AF; Young-Xu et al., 2003; Hanna et al., 2006]. In contrast a recent interventional trial to assess the efficacy of atorvastatin did not find a reduction in the recurrence of AF after cardioversion (Negi et al., 2011), and was associated with reductions in inflammatory, but not oxidative stress biomarkers. The discrepancy in clinical trial results may reflect temporal differences in the pathologic processes contributing to AF.
Acute application of fluvastatin has been shown to reduce automaticity in rabbit pulmonary veins, an important source for initiation of atrial fibrillation; this effect was attributed to modulation of eNOS activity via phosphorylation of the enzyme through an IP3 Kinase/AKT pathway (Hu et al., 2009). In a 2-week ventricular tachypacing model of heart failure, treatment with simvastatin was found to reduce the inducibility of AF (Shiroshita-Takeshita et al., 2007). Interestingly, in this 2 weeks model, simvastatin also reduced eNOS, iNOS, and 3-nitrotyrosine in the left ventricle, but not the left atrium. The beneficial effects of simvastatin treatment on the atrial fibrillation substrate were attributed to attenuation of atrial fibrosis, and conduction abnormalities, as well as improved LV function. An investigation of angiotensin II-induced atrial remodeling in an eNOS knockout mouse model found that statin treatment reduced atrial remodeling and AF (Yagi et al., 2010). This eNOS-independent beneficial effect of pitavastatin was attributed to reductions in TGF-β1 and Smad 2/3 signaling as well as reduced rac1 activation. Notably, the potential role of angiotensin II-induction of iNOS (Vaziri et al., 2002) was not evaluated in this model system.
A recent report suggests that treatment with the HMG–CoA reductase inhibitor, rosuvastatin, alters ADMA concentrations (Li et al., 2012). In this study a canine rapid atrial pacing model of AF was used (400 BPM for 6 weeks), with treatment with rosuvastatin started 3 days prior to the initiation of pacing. The serum concentration of ADMA was reduced and the concentration of NO was increased in the rosuvastatin-treated group compared to the control group. This was accompanied by a reduction in pacing-induced increases in atrial interstitial fibrosis, apoptosis, atrial dilatation, and was also associated with improved atrial contractile function.
Collectively, these studies suggest that statins may exert beneficial effects on the atrial substrate for AF through multiple mechanisms, including modulation of NOS activity. Based on current literature, further investigation of statins as an upstream therapy to reduce AF is warranted.
Fish Oil and Omega-3 Polyunsaturated Fatty Acids
The essential polyunsaturated fatty acids, eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA), are not synthesized by mammals. Therefore, dietary intake or supplements are required for these fatty acids. The most common sources are consumption of marine vertebrates or fish, or fish oil or omega-3 polyunsaturated fatty acid supplements.
Prophylactic dietary treatment with fish oil in a canine model of post-cardiac surgery canine model was reported to reduce post-operative AF (Mayyas et al., 2011). The reduction in AF was attributable in part to reduced inflammatory changes accompanied by less induction of atrial iNOS. Eicosapentaneoic acid (EPA), a component of the omega-3 polyunsaturated fatty acids found in fish oil, has recently been found to reduce the spontaneous activity in pulmonary veins (Suenari et al., 2011). Unlike the canine study of fish oil, this study used acute application of EPA to rabbit pulmonary vein preparations. Acute application of EPA was found to reduce delayed afterdepolarizations; the effects of EPA on PV electrophysiology were somewhat abrogated by the NOS inhibitor, L-NAME, suggesting that the effects of EPA were NOS mediated. The results of this study must be interpreted with caution since the effects of acute application of EPA are likely to differ from chronic treatment where the EPA is incorporated into the tissues (Billman et al., 2010).
Conclusion
NO signaling is an important component of the homeostatic regulation of the cardiovascular system. Uncoupled NO synthase is an important contributor to oxidative stress in the atrium, and several lines of evidence (preclinical and clinical) implicate a role for NOS signaling in the pathogenesis of atrial fibrillation. The role of specific NOS isoforms is difficult to definitively define at the present time, but may be highly significant based on the intracellular distribution of various isoforms.
Therapeutic interventions to modulate NOS function have had variable efficacy. Considering the complex system for cardiac redox balance, and the interactions between the redox pathways, modulation of a single pathway is a complex challenge. The variability in response to therapeutic interventions likely reflects the complex nature of the problem, the requisite role of NO signaling in maintenance of normal function, and confounding effects by off-target effects.
Our current understanding of NOS pathway signaling in normal atrial function as well as in AF is incomplete. It appears that there may be chamber-specific effects which may further complicate therapeutic manipulation of NOS signaling. These factors currently limit the ability to design optimal therapeutic interventions. Thus, much remains to be learned and further investigation is warranted.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH/NHLBI (R01 HL089836).
References
Ahern, G. P., Hsu, S. F., Klyachko, V. A., and Jackson, M. B. (2000). Induction of persistent sodium current by exogenous and endogenous nitric oxide. J. Biol. Chem. 275, 28810–28815.
Ahmmed, G. U., Xu, Y., Hong, D. P., Zhang, Z., Eiserich, J., and Chiamvimonvat, N. (2001). Nitric oxide modulates cardiac Na(+) channel via protein kinase A and protein kinase G. Circ. Res. 89, 1005–1013.
Asada, K., Kurokawa, J., and Furukawa, T. (2009). Redox- and calmodulin-dependent S-nitrosylation of the KCNQ1 channel. J. Biol. Chem. 284, 6014–6020.
Balligand, J. L., Ungureanu-Longrois, D., Simmons, W. W., Pimental, D., Malinski, T. A., Kapturczak, M., Taha, Z., Lowenstein, C. J., Davidoff, A. J., and Kelly, R. A. (1994). Cytokine-inducible nitric oxide synthase (iNOS) expression in cardiac myocytes. Characterization and regulation of iNOS expression and detection of iNOS activity in single cardiac myocytes in vitro. J. Biol. Chem. 269, 27580–27588.
Barouch, L. A., Harrison, R. W., Skaf, M. W., Rosas, G. O., Cappola, T. P., Kobeissi, Z. A., Hobai, I. A., Lemmon, C. A., Burnett, A. L., O’Rourke, B., Rodriguez, E. R., Huang, P. L., Lima, J. A., Berkowitz, D. E., and Hare, J. M. (2002). Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature 416, 337–339.
Bedi, M., McNamara, D., London, B., and Schwartzman, D. (2006). Genetic susceptibility to atrial fibrillation in patients with congestive heart failure. Heart Rhythm 3, 808–812.
Billman, G. E., Nishijima, Y., Belevych, A. E., Terentyev, D., Xu, Y., Haizlip, K. M., Monasky, M. M., Hiranandani, N., Harris, W. S., Gyorke, S., Carnes, C. A., and Janssen, P. M. (2010). Effects of dietary omega-3 fatty acids on ventricular function in dogs with healed myocardial infarctions: in vivo and in vitro studies. Am. J. Physiol. Heart Circ. Physiol. 298, H1219–H1228.
Cai, H., Li, Z., Goette, A., Mera, F., Honeycutt, C., Feterik, K., Wilcox, J. N., Dudley, S. C. Jr., Harrison, D. G., and Langberg, J. J. (2002). Downregulation of endocardial nitric oxide synthase expression and nitric oxide production in atrial fibrillation: potential mechanisms for atrial thrombosis and stroke. Circulation 106, 2854–2858.
Campbell, D. L., Stamler, J. S., and Strauss, H. C. (1996). Redox modulation of L-type calcium channels in ferret ventricular myocytes. Dual mechanism regulation by nitric oxide and S-nitrosothiols. J. Gen. Physiol. 108, 277–293.
Chen, C. A., Wang, T. Y., Varadharaj, S., Reyes, L. A., Hemann, C., Talukder, M. A., Chen, Y. R., Druhan, L. J., and Zweier, J. L. (2010). S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468, 1115–1118.
Dobrev, D., and Nattel, S. (2011). New insights into the molecular basis of atrial fibrillation: mechanistic and therapeutic implications. Cardiovasc. Res. 89, 689–691.
Fatini, C., Sticchi, E., Genuardi, M., Sofi, F., Gensini, F., Gori, A. M., Lenti, M., Michelucci, A., Abbate, R., and Gensini, G. F. (2006). Analysis of minK and eNOS genes as candidate loci for predisposition to non-valvular atrial fibrillation. Eur. Heart J. 27, 1712–1718.
Gallo, M. P., Malan, D., Bedendi, I., Biasin, C., Alloatti, G., and Levi, R. C. (2001). Regulation of cardiac calcium current by NO and cGMP-modulating agents. Pflugers Arch. 441, 621–628.
Gensini, F., Padeletti, L., Fatini, C., Sticchi, E., Gensini, G. F., and Michelucci, A. (2003). Angiotensin-converting enzyme and endothelial nitric oxide synthase polymorphisms in patients with atrial fibrillation. Pacing Clin. Electrophysiol. 26, 295–298.
Giusti, B., Gori, A. M., Marcucci, R., Sestini, I., Saracini, C., Sticchi, E., Gensini, F., Fatini, C., Abbate, R., and Gensini, G. F. (2007). Role of C677T and A1298C MTHFR, A2756G MTR and -786 C/T eNOS gene polymorphisms in atrial fibrillation susceptibility. PLoS ONE 2, e495.
Goette, A., Hammwohner, M., Bukowska, A., Scalera, F., Martens-Lobenhoffer, J., Dobrev, D., Ravens, U., Weinert, S., Medunjanin, S., Lendeckel, U., and Bode-Boger, S. M. (2012). The impact of rapid atrial pacing on ADMA and endothelial NOS. Int. J. Cardiol. 154, 141–146.
Gomez, R., Caballero, R., Barana, A., Amoros, I., Calvo, E., Lopez, J. A., Klein, H., Vaquero, M., Osuna, L., Atienza, F., Almendral, J., Pinto, A., Tamargo, J., and Delpon, E. (2009). Nitric oxide increases cardiac IK1 by nitrosylation of cysteine 76 of Kir2.1 channels. Circ. Res. 105, 383–392.
Gomez, R., Nunez, L., Vaquero, M., Amoros, I., Barana, A., de, P. T., Macaya, C., Maroto, L., Rodriguez, E., Caballero, R., Lopez-Farre, A., Tamargo, J., and Delpon, E. (2008). Nitric oxide inhibits Kv4.3 and human cardiac transient outward potassium current (Ito1). Cardiovasc. Res. 80, 375–384.
Han, W., Fu, S., Wei, N., Xie, B., Li, W., Yang, S., Li, Y., Liang, Z., and Huo, H. (2008). Nitric oxide overproduction derived from inducible nitric oxide synthase increases cardiomyocyte apoptosis in human atrial fibrillation. Int. J. Cardiol. 130, 165–173.
Hanna, I. R., Heeke, B., Bush, H., Brosius, L., King-Hageman, D., Dudley, S. C. Jr., Beshai, J. F., and Langberg, J. J. (2006). Lipid-lowering drug use is associated with reduced prevalence of atrial fibrillation in patients with left ventricular systolic dysfunction. Heart Rhythm 3, 881–886.
Harling, L., Rasoli, S., Vecht, J. A., Ashrafian, H., Kourliouros, A., and Athanasiou, T. (2011). Do antioxidant vitamins have an anti-arrhythmic effect following cardiac surgery? A meta-analysis of randomised controlled trials. Heart 97, 1636–1642.
Haywood, G. A., Tsao, P. S., von der Leyen, H. E., Mann, M. J., Keeling, P. J., Trindade, P. T., Lewis, N. P., Byrne, C. D., Rickenbacher, P. R., Bishopric, N. H., Cooke, J. P., McKenna, W. J., and Fowler, M. B. (1996). Expression of inducible nitric oxide synthase in human heart failure. Circulation 93, 1087–1094.
Hu, Y. F., Chen, Y. C., Cheng, C. C., Higa, S., Chen, Y. J., and Chen, S. A. (2009). Fluvastatin reduces pulmonary vein spontaneous activity through nitric oxide pathway. J. Cardiovasc. Electrophysiol. 20, 200–206.
Kim, Y. M., Guzik, T. J., Zhang, Y. H., Zhang, M. H., Kattach, H., Ratnatunga, C., Pillai, R., Channon, K. M., and Casadei, B. (2005). A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ. Res. 97, 629–636.
Kirstein, M., Rivet-Bastide, M., Hatem, S., Benardeau, A., Mercadier, J. J., and Fischmeister, R. (1995). Nitric oxide regulates the calcium current in isolated human atrial myocytes. J. Clin. Invest. 95, 794–802.
Komatsu, T., Tachibana, H., Sato, Y., Ozawa, M., Kunugida, F., Orii, M., and Nakamura, M. (2009). Long-term efficacy of upstream therapy using angiotensin-converting enzyme inhibitors and statins in combination with antiarrhythmic agents for the treatment of paroxysmal atrial fibrillation. Int. Heart J. 50, 465–476.
Komatsu, T., Tachibana, H., Sato, Y., Ozawa, M., Kunugita, F., and Nakamura, M. (2011). Long-term efficacy of upstream therapy with lipophilic or hydrophilic statins on antiarrhythmic drugs in patients with paroxysmal atrial fibrillation: comparison between atorvastatin and pravastatin. Int. Heart J. 52, 359–365.
Lenaerts, I., Holemans, P., Pokreisz, P., Sipido, K. R., Janssens, S., Heidbuchel, H., and Willems, R. (2011). Nitric oxide delays atrial tachycardia-induced electrical remodelling in a sheep model. Europace 13, 747–754.
Li, J., Xia, W., Feng, W., and Qu, X. (2012). Effects of rosuvastatin on serum asymmetric dimethylarginine levels and atrial structural remodeling in atrial fibrillation dogs. Pacing Clin. Electrophysiol.
Malan, D., Levi, R. C., Alloatti, G., Marcantoni, A., Bedendi, I., and Gallo, M. P. (2003). Cyclic AMP and cyclic GMP independent stimulation of ventricular calcium current by peroxynitrite donors in guinea pig myocytes. J. Cell Physiol. 197, 284–296.
Mayyas, F., Sakurai, S., Ram, R., Rennison, J. H., Hwang, E. S., Castel, L., Lovano, B., Brennan, M. L., Bibus, D., Lands, B., Barnard, J., Chung, M. K., and Van Wagoner, D. R. (2011). Dietary omega3 fatty acids modulate the substrate for post-operative atrial fibrillation in a canine cardiac surgery model. Cardiovasc. Res. 89, 852–861.
Mihm, M. J., Yu, F., Carnes, C. A., Reiser, P. J., McCarthy, P. M., Van Wagoner, D. R., and Bauer, J. A. (2001). Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation 104, 174–180.
Musialek, P., Lei, M., Brown, H. F., Paterson, D. J., and Casadei, B. (1997). Nitric oxide can increase heart rate by stimulating the hyperpolarization-activated inward current, I(f). Circ. Res. 81, 60–68.
Negi, S., Shukrullah, I., Veledar, E., Bloom, H. L., Jones, D. P., and Dudley, S. C. (2011). Statin therapy for the prevention of atrial fibrillation trial (SToP AF trial). J. Cardiovasc. Electrophysiol. 22, 414–419.
Nishijima, Y., Sridhar, A., Bonilla, I., Velayutham, M., Khan, M., Terentyeva, R., Li, C., Kuppusamy, P., Elton, T. S., Terentyev, D., Gyorke, S., Zweier, J. L., Cardounel, A. J., and Carnes, C. A. (2011). Tetrahydrobiopterin depletion and NOS2 uncoupling contribute to heart failure-induced alterations in atrial electrophysiology. Cardiovasc. Res. 91, 71–79.
Nunez, L., Vaquero, M., Gomez, R., Caballero, R., Mateos-Caceres, P., Macaya, C., Iriepa, I., Galvez, E., Lopez-Farre, A., Tamargo, J., and Delpon, E. (2006). Nitric oxide blocks hKv1.5 channels by S-nitrosylation and by a cyclic GMP-dependent mechanism. Cardiovasc. Res. 72, 80–89.
Rajasekaran, N. S., Firpo, M. A., Milash, B. A., Weiss, R. B., and Benjamin, I. J. (2008). Global expression profiling identifies a novel biosignature for protein aggregation R120GCryAB cardiomyopathy in mice. Physiol. Genomics 35, 165–172.
Reilly, S. N., Jayaram, R., Nahar, K., Antoniades, C., Verheule, S., Channon, K. M., Alp, N. J., Schotten, U., and Casadei, B. (2011). Atrial sources of reactive oxygen species vary with the duration and substrate of atrial fibrillation: implications for the antiarrhythmic effect of statins. Circulation 124, 1107–1117.
Savelieva, I., Kakouros, N., Kourliouros, A., and Camm, A. J. (2011). Upstream therapies for management of atrial fibrillation: review of clinical evidence and implications for European Society of Cardiology guidelines. Part I: primary prevention. Europace 13, 308–328.
Schotten, U., Verheule, S., Kirchhof, P., and Goette, A. (2011). Pathophysiological mechanisms of atrial fibrillation: a translational appraisal. Physiol. Rev. 91, 265–325.
Schulze, F., Carter, A. M., Schwedhelm, E., Ajjan, R., Maas, R., von Holten, R. A., Atzler, D., Grant, P. J., and Boger, R. H. (2010). Symmetric dimethylarginine predicts all-cause mortality following ischemic stroke. Atherosclerosis 208, 518–523.
Shiroshita-Takeshita, A., Brundel, B. J., Burstein, B., Leung, T. K., Mitamura, H., Ogawa, S., and Nattel, S. (2007). Effects of simvastatin on the development of the atrial fibrillation substrate in dogs with congestive heart failure. Cardiovasc. Res. 74, 75–84.
Shiroshita-Takeshita, A., Brundel, B. J., Lavoie, J., and Nattel, S. (2006). Prednisone prevents atrial fibrillation promotion by atrial tachycardia remodeling in dogs. Cardiovasc. Res. 69, 865–875.
Suenari, K., Chen, Y. C., Kao, Y. H., Cheng, C. C., Lin, Y. K., Kihara, Y., Chen, Y. J., and Chen, S. A. (2011). Eicosapentaenoic acid reduces the pulmonary vein arrhythmias through nitric oxide. Life Sci. 89, 129–136.
Taglialatela, M., Pannaccione, A., Iossa, S., Castaldo, P., and Annunziato, L. (1999). Modulation of the K(+) channels encoded by the human ether-a-gogo-related gene-1 (hERG1) by nitric oxide. Mol. Pharmacol. 56, 1298–1308.
Tamargo, J., Caballero, R., Gomez, R., and Delpon, E. (2010). Cardiac electrophysiological effects of nitric oxide. Cardiovasc. Res. 87, 593–600.
Umar, S., and van der Laarse, A. (2010). Nitric oxide and nitric oxide synthase isoforms in the normal, hypertrophic, and failing heart. Mol. Cell. Biochem. 333, 191–201.
Usui, M., Matsuoka, H., Miyazaki, H., Ueda, S., Okuda, S., and Imaizumi, T. (1998). Increased endogenous nitric oxide synthase inhibitor in patients with congestive heart failure. Life Sci. 62, 2425–2430.
Van Wagoner, D. R. (2008). Oxidative stress and inflammation in atrial fibrillation: role in pathogenesis and potential as a therapeutic target. J. Cardiovasc. Pharmacol. 52, 306–313.
Vaziri, N. D., Wang, X. Q., Ni, Z. N., Kivlighn, S., and Shahinfar, S. (2002). Effects of aging and AT-1 receptor blockade on NO synthase expression and renal function in SHR. Biochim. Biophys. Acta 153–161.
Vejlstrup, N. G., Bouloumie, A., Boesgaard, S., Andersen, C. B., Nielsen-Kudsk, J. E., Mortensen, S. A., Kent, J. D., Harrison, D. G., Busse, R., and Aldershvile, J. (1998). Inducible nitric oxide synthase (iNOS) in the human heart: expression and localization in congestive heart failure. J. Mol. Cell. Cardiol. 30, 1215–1223.
Winlaw, D. S., Smythe, G. A., Keogh, A. M., Schyvens, C. G., Spratt, P. M., and Macdonald, P. S. (1994). Increased nitric oxide production in heart failure. Lancet 344, 373–374.
Xia, Y., and Zweier, J. L. (1997). Superoxide and peroxynitrite generation from inducible nitric oxide synthase in macrophages. Proc. Natl. Acad. Sci. U.S.A. 94, 6954–6958.
Yagi, S., Akaike, M., Aihara, K., Ishikawa, K., Iwase, T., Ikeda, Y., Soeki, T., Yoshida, S., Sumitomo-Ueda, Y., Matsumoto, T., and Sata, M. (2010). Endothelial nitric oxide synthase-independent protective action of statin against angiotensin II-induced atrial remodeling via reduced oxidant injury. Hypertension 55, 918–923.
Yamashita, T., Sekiguchi, A., Iwasaki, Y. K., Date, T., Sagara, K., Tanabe, H., Suma, H., Sawada, H., and Aizawa, T. (2010). Recruitment of immune cells across atrial endocardium in human atrial fibrillation. Circ. J. 74, 262–270.
Young-Xu, Y., Jabbour, S., Goldberg, R., Blatt, C. M., Graboys, T., Bilchik, B., and Ravid, S. (2003). Usefulness of statin drugs in protecting against atrial fibrillation in patients with coronary artery disease. Am. J. Cardiol. 92, 1379–1383.
Zhang, H., Limphong, P., Pieper, J., Liu, Q., Rodesch, C. K., Christians, E., and Benjamin, I. J. (2012). Glutathione-dependent reductive stress triggers mitochondrial oxidation and cytotoxicity. FASEB J. 26, 1442–1451.
Zhang, X., Min, X., Li, C., Benjamin, I. J., Qian, B., Zhang, X., Ding, Z., Gao, X., Yao, Y., Ma, Y., Cheng, Y., and Liu, L. (2010). Involvement of reductive stress in the cardiomyopathy in transgenic mice with cardiac-specific overexpression of heat shock protein 27. Hypertension 55, 1412–1417.
Ziolo, M. T., and Bers, D. M. (2003). The real estate of NOS signaling: location, location, location. Circ. Res. 92, 1279–1281.
Keywords: atrial fibrillation, nitric oxide synthase, oxidative stress, electrophysiology, therapeutics
Citation: Bonilla IM, Sridhar A, Györke S, Cardounel AJ and Carnes CA (2012) Nitric oxide synthases and atrial fibrillation. Front. Physio. 3:105. doi: 10.3389/fphys.2012.00105
Received: 10 March 2012; Paper pending published: 02 April 2012;
Accepted: 02 April 2012; Published online: 23 April 2012.
Edited by:
David R. Van Wagoner, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, USAReviewed by:
Rob Gourdie, Medical University of South Carolina, USADavid R. Van Wagoner, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, USA
Copyright: © 2012 Bonilla, Sridhar, Györke, Cardounel and Carnes. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Cynthia A. Carnes, College of Pharmacy, The Ohio State University, 500 West 12th Avenue, Columbus, OH 43210, USA. e-mail:Y2FybmVzLjRAb3N1LmVkdQ==