 Mariam El Assar1
Mariam El Assar1 Javier Angulo1,2
Javier Angulo1,2 Susana Vallejo3
Susana Vallejo3 Concepción Peiró3
Concepción Peiró3 Carlos F. Sánchez-Ferrer3
Carlos F. Sánchez-Ferrer3 Leocadio Rodríguez-Mañas1,4*
Leocadio Rodríguez-Mañas1,4*- 1 Fundación para la Investigación Biomédica, Hospital Universitario de Getafe, Madrid, Spain
- 2 Instituto Ramón y Cajal de Investigación Sanitaria, Madrid, Spain
- 3 Departamento de Farmacología, Facultad de Medicina, Universidad Autónoma de Madrid, Madrid, Spain
- 4 Servicio de Geriatría, Hospital Universitario de Getafe, Madrid, Spain
Vascular aging is a key process determining health status of aged population. Aging is an independent cardiovascular risk factor associated to an impairment of endothelial function, which is a very early and important event leading to cardiovascular disease. Vascular aging, formerly being considered an immutable and inexorable risk factor, is now viewed as a target process for intervention in order to achieve a healthier old age. A further knowledge of the mechanisms underlying the age-related vascular dysfunction is required to design an adequate therapeutic strategy to prevent or restore this impairment of vascular functionality. Among the proposed mechanisms that contribute to age-dependent endothelial dysfunction, this review is focused on the following aspects occurring into the vascular wall: (1) the reduction of nitric oxide (NO) bioavailability, caused by diminished NO synthesis and/or by augmented NO scavenging due to oxidative stress, leading to peroxynitrite formation (ONOO−); (2) the possible sources involved in the enhancement of oxidative stress; (3) the increased activity of vasoconstrictor factors; and (4) the development of a low-grade pro-inflammatory environment. Synergisms and interactions between all these pathways are also analyzed. Finally, a brief summary of some cellular mechanisms related to endothelial cell senescence (including telomere and telomerase, stress-induced senescence, as well as sirtuins) are implemented, as they are likely involved in the age-dependent endothelial dysfunction, as well as in the lower vascular repairing capacity observed in the elderly. Prevention or reversion of those mechanisms leading to endothelial dysfunction through life style modifications or pharmacological interventions could markedly improve cardiovascular health in older people.
The concept that a man (or a woman) is as old as his arteries are, coined by Georges Canabis and reformulated by Sir William Osler more than 100 years ago, can be considered nowadays quite a valid approximation. Vascular aging represents the process that more importantly impacts on the health status of elderly people. The aging process is the main risk factor for the development of cardiovascular diseases (CVD), explaining 50% of clinical CVD present in the elderly. In fact, aging is associated with complex structural and functional changes in the vasculature independently of other risk factors, such as hypertension, diabetes, or hypercholesterolemia (Barodka et al., 2011). The old vision of vascular aging considers it as an inevitable process generated through a series of inexorable mechanisms. These thoughts have evolved to the current position that assumes that the knowledge of the molecular mechanisms involved in the age-related vascular dysfunction will contribute to understand the extent and nature of these alterations. Therefore, strategies to attenuate the effect of aging in the vasculature could be potentially developed, preserving the quality of life, and alleviating CVD in the elderly population (Najjar et al., 2005). Furthermore, preservation of vascular function in aging should not only reduce deaths and disabilities secondary to cardiovascular events but it also should influence other aspects of the aging process that leads to loss of function and/or disability. In fact, loss of cardiovascular health is associated to increased risk of defective motor capacity and cognitive frailty (Panza et al., 2006; Kim et al., 2011; Watson et al., 2011). Thus, it seems to be now quite clear that robustness in advanced age cannot be achieved without preservation of vascular function. Dominant aspects of vascular aging include increased arterial stiffness, dilation of central elastic arteries, and endothelial dysfunction (Kotsis et al., 2011), although there is increasing evidence suggesting that all these processes are closely related (van Bussel et al., 2011). The present work will briefly review the most important mechanisms that have been related to the development of the age-dependent endothelial dysfunction.
Vascular Dysfunction Associated to the Aging Process
Vascular aging is characterized by functional and structural changes of the endothelium and smooth muscle cells that form the vascular wall, as well as by alterations of the communication routes between these two cell layers. Functional disturbances, together with other factors associated with aging, will contribute to the development of structural alterations, which in turn contribute to vascular stiffness and to an additional impairment of the endothelial function. Moreover, since endothelial dysfunction is associated with the major causes of morbidity and mortality, the maintenance of a correct function of this vascular layer is thought to be an essential determinant of healthy aging (Virdis et al., 2010; Toda, 2012). The evidence showing the presence of endothelial dysfunction in the aged vasculature is very important since the late 1980s in animal models and since the middle 1990s in humans. In particular, the reduction in the endothelium-dependent vasodilatations has been consistently described both in vitro and in vivo in different vascular beds from old animals and elderly humans (Matz and Andriantsitohaina, 2003; Brandes et al., 2005; Rodriguez-Mañas et al., 2009; Toda, 2012). These evidences demonstrated that aging is an independent factor associated with endothelial dysfunction even in the absence of other cardiovascular risk factors (Rodriguez-Mañas et al., 2009). The impairment of endothelial function is a progressive phenomenon starting in the middle age and, at present, it is considered as one of the main mechanisms by which aging increases the risk of CVD and the development of atherosclerosis in humans. Therefore, those approaches aimed to preserve or improve the endothelial function would be fundamental for the prevention of vascular diseases in the elderly. The reported scientific evidences indicate that the pathogenesis of the age-dependent endothelial dysfunction is clearly multifactorial, with several pathophysiological mechanisms contributing to the functional deterioration of vascular endothelial cells (Figure 1). These pathways are briefly summarized as follows.
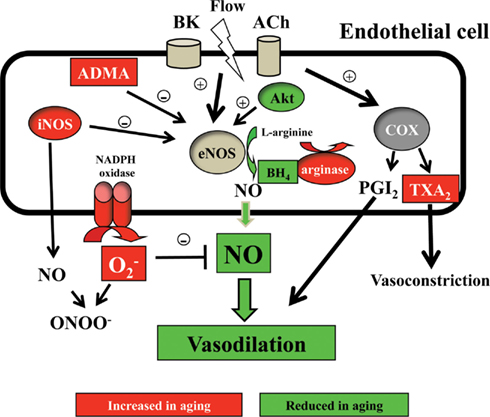
Figure 1. Mechanisms involved in the aging-induced impairment of endothelial vasodilation.
Alteration of the Nitric Oxide (NO) Pathway
NO is the main vasodilator produced by the endothelium and exerts a protective role on the vessel wall. The reduction of NO availability deeply disturbs the vascular homeostasis, being involved in the development of hypertension, atherosclerosis, or diabetic vasculopathy (Sagach et al., 2006). NO is synthesized from L-arginine by the enzyme NO synthase (NOS). There are three known NOS isoforms: the constitutive endothelial (eNOS) and neuronal (nNOS) isoforms, producing regulated NO involved in regulatory or signaling pathways, and the inducible (iNOS) isoform, leading to massive NO synthesis and related with inflammatory responses. Aging is also associated with a reduction in the NO bioavailability, which is the result of the dynamic balance between its synthesis and degradation. Reduced NO production may be due to: (1) a deficiency in NOS substrates and cofactors; (2) the presence of endogenous eNOS inhibitors; and (3) a lower expression and/or activity of eNOS. On the other hand, enhanced NO degradation may be mostly due to excessive amounts of reactive oxygen species (ROS), such as superoxide anions that quench NO hampering its functional activities.
Decrease in L-Arginine Availability
The reduction of available concentration of L-arginine to be used as eNOS substrate in aging was based on one study suggesting an improvement in endothelial function in older subjects after oral administration of L-arginine (Bode-Boger et al., 2003). However, this has not been further confirmed, as no significant improvement of the impaired flow-mediated dilation in the old subjects group has been observed after the intra-braquial infusion of L-arginine, despite a 23-fold increase of its plasmatic concentrations (Gates et al., 2007). From a biochemical point of view, a reduction in the availability of the substrate is difficult to sustain because the plasmatic concentration of L-arginine is more than one order of magnitude higher than the substrate concentration required for the optimal function of the enzyme. Although a lower availability could be explained by a reduction in the transport of L-arginine to endothelial cell, there is evidence showing that age-related deterioration of endothelial function is not associated to a change in the transport of L-arginine (Ahlers et al., 2004). Other possible explanation for the lower availability of L-arginine with aging could be related to an increased expression and/or activity of arginase, the enzyme that degrades L-arginine, leading to a decrease in substrate availability for eNOS and the consequent reduction of NO synthesis (Santhanam et al., 2008). In rats, an increase in the arginase-1 activity related to age-related endothelial dysfunction has also been proved by the use of arginase inhibitors, which restore NO-mediated vasodilation (Santhanam et al., 2007). Recovering of vasodilation after arginase inhibition in elderly subjects also supports a role for this enzyme in the endothelial dysfunction associated to human vascular aging (Holowatz et al., 2006).
The Endogenous Inhibitor of NOS (ADMA)
The synthesis of NO is blocked by the inhibition of the NOS active site with guanidine-substituted analogs of L-arginine such as asymmetric dimethylarginine (ADMA). ADMA is a naturally occurring amino acid found in plasma and various tissues (Yamagishi and Matsui, 2011). An enhanced production of ADMA has been proposed to be a cardiovascular risk factor linked to different pathologies involving vascular disease and hypertension (Cooke, 2005). A possible role for this compound has been proposed in the physiological process of aging, as a positive correlation has been reported in healthy subjects between the plasmatic levels of ADMA and age (Schulze et al., 2005). Moreover, it has been described an ADMA accelerating effect of endothelial cells senescence (Bode-Boger et al., 2005). However, other authors could not find any relationship between the endothelial expression of ADMA and the development of the age-related impairment of endothelium-dependent vasodilations (Gates et al., 2007).
Reduction of Tetrahydrobiopterin (BH4)
Tetrahydrobiopterin (BH4) is a cofactor essential for NOS activity. Substantial evidence linked BH4 deficiency as a condition leading eNOS to produce less NO with the consequent deterioration of endothelial function (Vasquez-Vivar et al., 2003). In humans, the participation of BH4 deficiency in age-related endothelial dysfunction has been demonstrated, since the infusion of BH4 restores the impaired endothelium-dependent vasodilation (Higashi et al., 2006).
Endothelial NOS
There is no doubt about the existence of a deficit in the vascular NO bioavailability associated with aging, but a high number of studies evaluating the expression of eNOS in different vascular beds of aged animals have not displayed definitive results. While some authors have not observed changes of eNOS expression in mesenteric arteries (Sun et al., 2004), others have reported significant increases in the aorta (Cernadas et al., 1998) or the mesenteric arteries from old rats (Briones et al., 2005). The analysis of the eNOS mRNA levels has shown either an increase (Barton et al., 1997) or a decrease (Tang and Vanhoutte, 2008) in the aorta of aged rats. In the mesenteric microvessels from healthy young and old humans, no age-dependent changes have been detected concerning eNOS mRNA levels (Rodriguez-Mañas et al., 2009). Moreover, human endothelial cells from brachial artery and peripheral veins do not differ significantly with age in the eNOS expression (Donato et al., 2009).
In addition to transcriptional and translational regulations, eNOS presents a significant regulation at post-translational level involving PI3 kinase/Akt-dependent phosphorylation at Ser 1177, resulting in increased activity of eNOS. Although reduced Akt-dependent phosphorylation of eNOS in micro- and macro-vasculature of aged animals has been demonstrated (Soucy et al., 2006; LeBlanc et al., 2008), the involvement of reduced eNOS phosphorylation in human vascular aging needs to be confirmed.
Endothelium-Derived Hyperpolarizing Factor
Further to the release of NO and prostacyclin, the endothelium controls vascular tone by causing hyperpolarization of underlying smooth muscle cells. This process is attributed to the endothelium-derived hyperpolarizing factor (EDHF). However, this generic term can refer to different mechanisms (with vascular territory- and species-dependent variability) including arachidonate metabolites derived from COX, lipoxygenase, or cytochrome P450 oxygenase activities, H2O2, CO, H2S, and several peptides that can be released by endothelial cells. Furthermore, vasodilation can be generated by Ca2+-induced hyperpolarization of endothelial cell causing the opening of Ca2+-activated K+-channels (KCa) that produce hyperpolarization and relaxation of smooth muscle through myo-endothelial gap junction-mediated electrical coupling or by K+-efflux from the endothelial KCa and subsequent activation of inwardly rectifier K+-channels or Na+/K+-ATPase in smooth muscle (Dora et al., 2008).
Contribution of EDHF to endothelial vasodilation of human arteries has been demonstrated in different vascular regions such as coronary arteries obtained from failing hearts at transplant surgery (Nakashima et al., 1993), omental and subcutaneous microvessels (Pascoal and Umans, 1996; MacKenzie et al., 2008), and penile resistance arteries (Angulo et al., 2003). Impaired EDHF-mediated vasodilation has been observed in mesenteric (Goto et al., 2000) and renal arteries (Long et al., 2005) from aged rats. In contrast, it has been reported that EDHF partially compensates for the loss of NO- and prostacyclin-mediated cutaneous vasodilation in old mice (Gaubert et al., 2007). These conflicting results could reflect either species differences or regional heterogeneity of vascular regulation with aging. In human gastroepiploic arteries, EDHF-induced relaxation inversely correlated with the age of the patients from whom the arteries were obtained (Urakami-Harasawa et al., 1997).
Age-induced decline in EDHF-mediated vasodilation seems to be related to an up-regulated renin-angiotensin system (RAS) since chronic angiotensin converting enzyme (ACE) inhibition, as well as angiotensin-II type 1 (AT1) receptor blockade, recovered EDHF-mediated responses in mesenteric arteries from old rats (Goto et al., 2004). In this sense, the impairment of EDHF-induced vasodilation in mesenteric arteries from middle aged rats (46 weeks old) is associated with increased AT1 receptor expression and reduced angiotensin-II type 2 (AT2) receptor and ACE expression, which were prevented by antioxidant treatment with red wine polyphenols (Idris Khodja et al., 2012). As occurs in angiotensin II (Ang II)-induced hypertension (Hilgers and Webb, 2007), age-related impairment of the EDHF component of endothelial vasodilation is likely due to reduced expression of small and intermediate conductance KCa (SKCa and IKCa; Idris Khodja et al., 2012). A decline on the responses mediated by large conductance KCa (BKCa) has been reported in coronary arteries from aged rats which can be recovered by exercise training (Albarwani et al., 2010).
Contractile Factors
COX Pathway Alteration
Cyclooxygenase (COX) plays an important role in the regulation of vascular tone under normal conditions by the synthesis of different vasoactive factors, which are particularly relevant since both vasodilators (prostacyclin, PGI2) or vasoconstrictors (thromboxane A2, TXA2) could be produced, those factors being in tight balance. During aging, a shift in the balance in favor of increased contractile factors occurs and, therefore, endothelium-dependent contractions increase. In humans, the lack of prostaglandin (PGI2)-mediated vasodilatation has been reported in vivo (Schrage et al., 2007) and in mesenteric microvessels in vitro (Rodriguez-Mañas et al., 2009), while the existence of COX-derived vasoconstrictor factors associated with aging has been described in vivo by using plethysmographic studies (Taddei et al., 1997; Vanhoutte et al., 2005) and in vitro studying isolated mesenteric arteries (Rodriguez-Mañas et al., 2009). The implication of the COX pathway in the endothelial dysfunction associated with aging is reinforced by the improvement of the impaired endothelium-dependent relaxations produced by TP-receptor antagonists (Rodriguez-Mañas et al., 2009). In humans, the nature of the endothelium-dependent vasoconstrictor factor is not entirely determined but thromboxane A2 and prostaglandin H2 are clear candidates. In aged hamsters, endothelial-dependent contractions are mediated by PGF2α produced by COX-2 (Wong et al., 2009). A role for is also possible, as these ROSs have been described as COX activity-derived endothelium-dependent vasoconstrictors (Vanhoutte et al., 2005).
No consensus has so far been established regarding the COX isoform responsible for the age-related vasoconstrictions (Matz and Andriantsitohaina, 2003). The pre-incubation with the selective COX-1 inhibitor valeryl salicylate reduced the contractile response observed in the femoral artery form old rats (Shi et al., 2008), while the COX-2 inhibitor NS-398 improved endothelial dysfunction in the aged rat aorta and mesenteric arteries (de Sotomayor et al., 2005). However, other authors did not find an improvement of endothelial dysfunction by this same compound in mesenteric arteries from aged rats (Matz et al., 2000).
Controversial results have been also reported concerning the COX protein expression in the vasculature of old animals (Heymes et al., 2000; Stewart et al., 2000; Briones et al., 2005; Shi et al., 2008). No differences related to age have been detected in the expression of mRNA for COX-1 and COX-2 isoforms in human mesenteric microvessels (Rodriguez-Mañas et al., 2009). However, protein expression may be not the only factor accounting for COX-mediated effects, as the existence of post-translational changes in the activity of these enzymes cannot be ruled out. Thus, there is an important regulation of COX activity by NO and ONOO− (Upmacis et al., 2006). Furthermore, a physiological binding interaction between COX-2 and iNOS has recently been reported, bringing NO or ONOO−to activate COX-2 in a synergistic molecular interactions between these two inflammatory pathways (Kim et al., 2005).
The Renin-Angiotensin System
The RAS is critical for cardiovascular control, impacting normal physiology and disease pathogenesis. Its major actions are mediated by Ang II, acting through its AT1 and AT2 receptors (Stegbauer and Coffman, 2011). At present, it is clear that both the increased generation of cellular ROS and activation of redox-sensitive signaling cascades are critical events involved in Ang II actions (Touyz, 2003). After binding to its AT1 receptors, Ang II triggers intracellular superoxide production by activating NAD(P)H (Kimura et al., 2005) and uncoupling endothelial NOS (eNOS; Mollnau et al., 2002). Under normal physiological conditions, Ang II-mediated signaling pathways are closely regulated. However, increased renin-angiotensin system activity is implicated in several vascular disorders and there is evidence for increased vascular expression of Ang II and ACE with aging (Rajagopalan et al., 2002; Wang et al., 2003). Furthermore a role for the angiotensin system in the vascular aging-related endothelial dysfunction has been observed. Indeed Ang II is a potent inducer of endothelial dysfunction and vascular oxidative stress (Idris Khodja et al., 2012). Moreover, treatment of rats with either an ACE inhibitor or an AT1 receptor antagonist improved endothelial dysfunction (mediated by the NO and the EDHF component) in aged blood vessels, in part, by decreasing oxidative stress (Goto et al., 2000; Kansui et al., 2002; Mukai et al., 2002). A recent study done by Benigni and associates showed that mice lacking AT1A receptors had prolonged life span compared to genetically matched wild-type controls; this enhanced longevity was associated with improved cardiovascular morphology, reduced ROS production, attenuated mitochondrial loss, and enhanced levels of nicotinamide phosphoribosyltransferase (Nampt) and sirtuin-3 (Sirt3; Benigni et al., 2009). However, losartan, an AT1 receptor antagonist, had no effect on brachial flow-mediated dilation in older adults, despite reducing blood pressure and circulating inflammatory markers (Rajagopalan et al., 2002) while other AT1 receptor antagonist, valsartan, improved vascular compliance in healthy normotensive elderly individuals without affecting flow-mediated dilation (Rajagopalan et al., 2006).
Endothelin-1
Endothelin-1 (ET-1) is the most potent vasoconstrictor protein synthesized and released by endothelial cells (Yanagisawa et al., 1988). ET-1 exerts vascular actions through activation of two distinct ET-1 receptor subtypes: ETA, localized exclusively in vascular smooth muscle, and ETB which are expressed in smooth muscle as well as endothelium (Masaki et al., 1991). Both ETA and ETB receptors are coupled to phospholipase C activation leading to increased cytosolic calcium and myosin kinase phosphorylation that results in long-lasting contraction of smooth muscle (Lüscher and Barton, 2000) and vasoconstriction (Seo et al., 1994; Haynes et al., 1995). Activation of ETB receptors by ET-1 would increase intracellular calcium in endothelial cell promoting eNOS activation and vasodilation (Tsukahara et al., 1994).
Increased contractile responsiveness to ET-1 has been demonstrated in arteries from aged rats (Donato et al., 2005; Korzick et al., 2005) while ETA blockade has been shown to reverse the impairment of endothelium-dependent relaxation in carotid arteries from old mice (Donato et al., 2009). Aging is also associated with elevated plasma concentrations of ET-1 in humans (Maeda et al., 2003; Donato et al., 2009). In humans, ETA/ETB antagonists produced larger leg blood flow increases in healthy sedentary old subjects than in young subjects, suggesting elevated ET-1-mediated vasoconstrictor tone in peripheral arteries from aged humans (Thijssen et al., 2007). Similarly, forearm vasoconstriction induced by ET-1 was blunted in older sedentary subjects while ETA blockade only increased resting forearm blood flow in old sedentary subjects but not in young (Van Guilder et al., 2007). Furthermore, endothelium-dependent forearm vasodilation in older subjects negatively correlates with the expression of ET-1 in endothelial cells (Donato et al., 2009). Interestingly, exercise training decreases plasma ET-1 in old women (Maeda et al., 2003) and reduces ET-1-mediated increase in vascular tone in elderly subjects (Thijssen et al., 2007; Van Guilder et al., 2007).
Oxidative Stress
It is now clear that ROS are physiologically produced into the vascular wall in a controlled regulated manner. Under normal conditions, the endogenous antioxidant defense mechanisms, both enzymatic, such as the manganese and copper/zinc superoxide dismutase (SOD), glutathione peroxidase (GP), catalase, and non-enzymatic, vitamin C, vitamin E, uric acid, inactivate ROS and repair the possible developed tissular damage. Indeed, together with NO, physiological ROS function as cell signaling initiators by their ability to introduce reversible post-translational protein modifications (Valko et al., 2007). Nevertheless, under pathological conditions, increased ROS levels lead to accumulation of damaged/misfolded proteins, increased mutagenesis rate, inflammation, and endothelial dysfunction (Malinin et al., 2011). This enhanced oxidative stress can consequence of either an increased ROS production and/or a reduction in the antioxidant defenses.
ROS and Vascular Damage
The implication of oxidative stress in the genesis of vascular damage has been described in several pathologies, including diabetes or hypertension, and also in aging process (Ungvari et al., 2008). A large body of evidence indicates that oxidative stress and ROS production are increased during the aging process (Ungvari et al., 2010b). The results obtained from studies in which antioxidant defense levels were determined were contradictory. While some authors observed an age-related decrease in the SOD and GP activities and a decline of plasma antioxidant capacity (Goraca, 2004), others found an increase in the plasma activity of SOD and GP (Mecocci et al., 2000). The first consequence of the increase in levels is a decrease of the NO availability, leading to a decrease in the endothelium-dependent relaxations (Pacher et al., 2007). In human mesenteric microvessels, this age–related endothelial dysfunction is improved by scavenging with SOD or TEMPOL, which correlates with an increase in levels in these same vessels (Rodriguez-Mañas et al., 2009).
NO Inactivation
Accelerated degradation of NO by oxidative stress and especially by is one of the most widely accepted mechanisms involved in the alteration of the NO pathway. The diffusion-controlled reaction between NO and leads to the formation of the peroxynitrite anion (ONOO−; Yang et al., 2009a). Thus, the NO produced in vascular cells from eNOS or iNOS reacts rapidly with to form ONOO−, which is termed as a reactive nitrogen specie (RNS) because of its high reactivity with proteins, DNA, and lipids. Unlike ONOO− can easily penetrate into the cell causing oxidative modifications of macromolecules, especially lipids, DNA, and proteins via direct oxidative reactions through the nitrosylation of tyrosine and cysteine residues or via indirect radical-mediated mechanisms. These reactions trigger cellular responses ranging from subtle modulations of cell signaling to overwhelming oxidative injury, committing cells to necrosis or apoptosis. Indeed, nitrotyrosine levels reflect the impact of ONOO− on proteins and constitute a cellular marker of oxidative stress. There are convincing data showing a substantially enhanced cardiovascular ONOO− formation during aging process (Francia et al., 2004), whereas increased nitrosative stress has been demonstrated in arteries from aged animals (van der Loo et al., 2000). Higher levels of nitrotyrosine have been found in endothelial cells obtained from brachial artery from aged subjects, while nitrotyrosine levels were inversely related to flow-mediated dilation (Donato et al., 2007). In human mesenteric microvessels, an age-dependent formation of nitrotyrosine has been reported, while the endothelial dysfunction associated to elderly is partially restored by scavenging ONOO− with uric acid (Rodriguez-Mañas et al., 2009). All these evidences suggest that the relevant changes related to age that are observed in the vascular wall are driven, at least partially, by these highly reactive molecules.
Sources of Oxidative Stress
There are several main systems proposed to be the sources for the ROS increase production in the human vasculature, namely NADPH oxidase, xanthine oxidase, uncoupled NO synthase, and the mitochondrial respiratory chain (Cai and Harrison, 2000; Loscalzo, 2000; Guzik et al., 2002; Brandes et al., 2005; Lassegue and Griendling, 2010). Increased expression of the Nox-4 subunit of NADPH oxidase has been demonstrated in mesenteric microvessels from aged humans without other known cardiovascular risk factors; furthermore, the NADPH oxidase-derived ROS produced in these arteries impact vascular function, since the NADPH oxidase inhibitor apocynin improves the age-related endothelial dysfunction (Rodriguez-Mañas et al., 2009). In the aorta from aged rats, other authors noted that oxidative stress associated with aging might be related to the increased activity of the enzyme xanthine oxidase (Newaz et al., 2006). However, the role of this enzyme as a source of oxidative stress in vascular aging has not been confirmed in humans; thus, its expression in vascular endothelial cells from older subjects is not altered while in vivo inhibition of this enzyme with allopurinol does not improve aged-dependent endothelial dysfunction (Eskurza et al., 2006).
NOS uncoupling is a possible source for in vascular aging (Cai and Harrison, 2000; Brandes et al., 2005), which has been described after a decline in BH4 bioavailability that is consequence of its oxidation to BH2 by or peroxynitrite (ONOO−; Milstien and Katusic, 1999; Laursen et al., 2001; Schiffrin, 2008). As BH4 efficiently inhibits superoxide generation from the heme group at the oxygenase domain of eNOS, it can act as a redox switch in the catalytic mechanism of the enzyme, which may have important consequences in the physiology of the endothelium (Vasquez-Vivar et al., 2003). High not only reduces NO bioavailability, but the ROS and RNS also oxidize proteins and lead to eNOS uncoupling (Munzel et al., 2005). Moreover, ONOO− further uncouples eNOS by oxidizing the zinc-thiolate complex (Zou et al., 2002). Mammalian target of rapamycin (mTOR)/SK61 signaling activation has been demonstrated to cause eNOS uncoupling in HUVEC and endothelial dysfunction in aged rats (Rajapakse et al., 2011). Interestingly, although most reports point to eNOS as the isoform that can be uncoupled, producing (Brandes et al., 2005), the inducible iNOS isoform can also serve as a source of in the absence of sufficient substrate or BH4 (Loscalzo, 2000). In addition, treatment of isolated mesenteric arteries from healthy aged subjects with BH4 results in significant recovery of dysfunctional endothelium-dependent relaxations, suggesting that the enhancement of the intracellular levels of BH4 would prevent NOS uncoupling and production, in this case likely involving the inflammatory inducible iNOS isoform (Rodriguez-Mañas et al., 2009).
Another source for oxidative stress in the aging process is mitochondria. Under physiological conditions, the mitochondria continually produce large amounts of and H2O2, so that the mitochondrial DNA (mtDNA) is particularly exposed to oxidative damage. In aging there is a reduction in the number of mitochondria and an increase in the generation of dysfunctional proteins, which leads to a depletion in the energy supply and even to an increase in the production (Pang et al., 2008). Whatever the mechanism involved in the mitochondrial dysfunction, as it is the case for insulin-resistance (Abbatecola et al., 2011), the decline in mitochondrial dysfunction is able to enhance ROS production and further damage the mtDNA (Cui et al., 2012). An additional important link between mitochondrial oxidative stress and cardiovascular aging is the induction of programmed cell death. The available evidence suggests that age-associated increase in oxidative stress causes an increased rate of endothelial apoptosis (Ungvari et al., 2010b). Furthermore, recent studies show that mitochondria-derived ROS, in addition to causing oxidative damage, play an important role in activating numerous redox-sensitive transcription factors, including NF-κB and AP-1 (Ungvari et al., 2010b).
Antioxidant Therapy in Elderly Humans
Although some conflicting results showing ineffectiveness of vitamin E ingestion to improve endothelial function in older adults exist (Simons et al., 1999), more recent clinical evidences point to beneficial effects of antioxidant therapy on endothelial dysfunction related to aging. A recent clinical trial has shown the acute reversal of endothelial dysfunction in the elderly after oral administration of an antioxidant cocktail (vitamin C + vitamin E + α-lipoic acid; Wray et al., 2012), confirming previous results showing the recovery of exercise induced vasodilation of brachial artery in elderly men after administration of the same antioxidant cocktail (Donato et al., 2010). Administration of ascorbic acid has been reported to reverse vasoconstriction and improve resting leg blood in healthy older men (Jablonski et al., 2007) as well as to augment reflex cutaneous vasodilation in aged subjects (Holowatz et al., 2006). These clinical evidences demonstrate that oxidative stress may be an important target for recovering endothelial function in aging.
Inflammation
In addition to oxidative stress, the development of the so-called low-grade chronic inflammatory state is another phenomenon linked to the aging process. This has been referred as “inflamm-aging” that could be caused by a continuous antigenic load and stress (Franceschi et al., 2000). Molecular inflammation is described as an underlying mechanism of aging and age-related diseases, which may constitute the link between normal aging and age-related pathological processes. Normal aging (“usual aging”) is associated with higher levels of cytokines, especially TNF-α, IL-1β, and members of the super family of IL-6, as well as elevations of C-reactive protein (CRP) and fibrinogen (Ferrucci et al., 2005) that contribute to a pro-inflammatory microenvironment and facilitate the development of vascular dysfunction. Interactions between inflammation and oxidative stress occurs, as different redox-sensitive transcriptional factors such as AP-1 and NF-κB are activated by ROS, increasing the gene expression of cytokines (TNF-α, IL-1, and IL-6), adhesion molecules (ICAM, VCAM), and pro-inflammatory enzymes (iNOS, COX-2; Yu and Chung, 2006). Supporting the clinical relevance of these findings in humans, epidemiological data suggest an association between elevated inflammatory cytokines and mortality in the elderly people (Harris et al., 1999; Volpato et al., 2001). Inflammatory cells could also play a role in vascular dysfunction associated to aging. In this sense, in healthy subjects older than 55 years, neutrophil, eosinophil and monocyte count (within the normal ranges) as well as myeloperoxidase activity inversely correlated with forearm blood flow responses, which were improved after BH4 administration (Walker et al., 2010). This suggests that moderate increment of inflammatory cells through myeloperoxidase activity would reduce BH4 availability compromising NO-mediated vasodilation.
In experimental models, there is clear evidence concerning the relationship between age and vascular inflammation (Chung et al., 2002; Csiszar et al., 2003; d’Alessio, 2004; Ungvari et al., 2004). An age-related up-regulation in TNF-α has been described in coronary arteries, increasing endothelial cells apoptosis and leading to endothelial dysfunction (Csiszar et al., 2004). Many of the characteristics of vascular aging, including endothelial dysfunction, oxidative stress, increased apoptosis, and pro-inflammatory gene expression profile, can be mimicked by recombinant TNF-α (Csiszar et al., 2007). Moreover, chronic TNF-α inhibition improves flow-mediated arterial dilation in resistance arteries of aged animals, while reduced ICAM-1 and iNOS expression (Arenas et al., 2006).
The induction of the inflammatory iNOS isoform can be also related to the endothelial dysfunction associated with aging. Previous reports in rats suggest an age-dependent enhanced vascular expression of iNOS (Cernadas et al., 1998; Csiszar et al., 2002). Studies in human peritoneal mesothelial cells indicated an age-dependent enhancement of iNOS expression (Nevado et al., 2006), while in mesenteric microvessels from aged subjects there is an increased expression of mRNA for iNOS (Rodriguez-Mañas et al., 2009). Furthermore, the selective inhibition of this isoform partially restored the age-dependent endothelial dysfunction (Rodriguez-Mañas et al., 2009). The iNOS isoform generates high amounts of NO; therefore, its rapid reaction with generates high levels of ONOO−. As the inducible iNOS isoform has much higher catalytic activity than eNOS and consumes more substrate and cofactors, this isoenzyme is also an important source of when uncoupled (Loscalzo, 2000).
During the last years, there is increasing evidence in humans confirming the relation between age and low-grade inflammation. The expression and activity of the inflammatory transcription nuclear factor-κB (NF-κB) is augmented in human peritoneal mesothelial cells accordingly to age (Nevado et al., 2006). Moreover, a pro-inflammatory profile has been reported in the grossly normal, aged, human aortic wall (Wang et al., 2007). Aging is also associated with greater nuclear NF-κB, lower IκBα, and increased expression of pro-inflammatory cytokines in vascular endothelial cells from healthy humans, which correlates with age-related endothelial dysfunction (Donato et al., 2007, 2008, 2009). Furthermore, an in situ age-dependent NF-κB activation has been demonstrated in the vascular wall of human mesenteric microvessels from aged subjects by Southwestern histochemistry techniques, which clearly correlates with the development of endothelial dysfunction (Rodriguez-Mañas et al., 2009).
Senescence and Endothelial Dysfunction
Replicative Senescence
In the early 1960s, Hayflick and Moorhead defined cellular senescence as a condition where the cells lose its ability to proliferate, although it can be metabolically active. The underlying mechanism for this phenomenon is the telomere length shortening that occurs during each cell division until a critical length is exceeded. Without telomeres, genetic material would be lost every time a cell divides. DNA polymerase requires a RNA primer with a 3′ hydroxyl donor group to initiate DNA replication, during which the “end-replication problem” arises. Telomeres and telomerase ameliorate this problem by providing a repetitive template for enzymatic repair of the ends of chromosomes, thereby limiting the loss of genetic information during mitosis. When the telomeres are too short, cell signaling is triggered for the arrest of cell proliferation, senescence, and apoptosis. In addition to the lack of cell replication, senescent cells acquire distinct phenotypic characteristics associated to aging and age-related diseases (Ungvari et al., 2010a). Some of these phenotypic changes are related to the regenerative and angiogenic capacity of the vascular endothelium and involved in the development of atherosclerosis during aging (Erusalimsky, 2009). Indeed, more elastase and fibronectin are observed in senescent vascular smooth muscle (Minamino and Komuro, 2007).
Although senescence has been comprehensively characterized in cell culture, there is increasing evidence of the senescent phenotype in vivo, and its relevant pathophysiological implications, particularly in the cardiovascular system (Erusalimsky and Kurz, 2005). Thus, it has been described that telomere length is shortened in the endothelial cells from cadaveric samples of thoracic and iliac arteries, as well as from abdominal aorta, accordingly to the age of the patients (Aviv et al., 2001). On the other hand, telomere length is an independent predictor of heart disease related events, as its shortening is more pronounced in arteries derived from elderly patients with coronary disease and associated to endothelial dysfunction (Ogami et al., 2004; De Meyer et al., 2011). Telomere dysfunction and vascular senescence are related to enhanced ROS, decreased NO, and increased pro-inflammatory molecules, such as adhesion molecules (Minamino et al., 2004; Minamino and Komuro, 2007). In fact, ROS generation accelerates telomere shortening and senescence in HUVEC (Kurz et al., 2004).
In a search for biomarkers of CVD, the leukocyte telomere length has been related to several physiological or pathological conditions. Thus, cross sectional studies have shown that subjects with increased arterial stiffness have shorter leukocyte telomeres, indicating this may be an indicator of biological vascular aging in men (Benetos et al., 2001). It has been also reported that the leukocyte telomere length is shorter in a population of hypertensive patients than in the normotensive peers and the patients with shorter telomeres were more likely to develop atherosclerosis 5 years after (Yang et al., 2009b). Moreover, the rate of leukocyte telomere shortening predicts mortality from CVD in elderly men (Epel et al., 2009). Some criticism still exists, however, concerning the relations between telomere length and CVDs, as not always adequate correlations are found, neither an accepted pathophysiological link (Calado and Young, 2009).
Stress-Induced Senescence
Stress-induced senescence of endothelial cells can be produced as a result of various insults, including those causing intracellular oxidative stress and inflammation (Calado and Young, 2009; Erusalimsky and Skene, 2009). ROS are potential candidates responsible for senescence through varied actions but also by compromising NO availability since NO inhibits senescence in endothelial cells (Hayashi et al., 2006). In fact, ROS-induced senescence of HUVEC is antagonized by interventions resulting in increased eNOS activity (Ota et al., 2007). It has been suggested that inflammation may be a telomere-independent mechanism producing vascular cell senescence in human atherosclerotic plaques (Minamino et al., 2004). Exposure to AGE-modified proteins induces senescence of HUVEC without affecting telomeres length or telomerase activity (Patschan and Goligorsky, 2008). This type of stress-induced senescence would be secondary to ROS generation, lysosomal permeability and dysregulation of autophagy (Patschan et al., 2008). Oxidative stress seems to be involved in the genesis of both replicative and high glucose-induced senescence leading to dimethylaminohydrolase inactivation and ADMA accumulation which inhibits NO production (Scalera et al., 2008; Yuan et al., 2010). In addition, activation of Akt/mTOR pathway has been reported to contribute to vascular senescence associated to obesity (Wang et al., 2009).
Senescence of EPCs
Special relevance deserves the senescence occurring in the endothelial progenitor cells (EPCs), which are considered to contribute to endothelial repair (Griese et al., 2003; Takamiya et al., 2006; Kirton and Xu, 2010; Briasoulis et al., 2011), a phenomenon that is less efficient in aging (Williamson et al., 2012). However, although it is widely accepted the capacity of EPCs to promote neovascularization, the role of EPCs in regeneration of endothelial cells remains controversial (Pasquier and Dias, 2010; Sanchez-Guijo et al., 2010; Resch et al., 2011). Thus, advanced age is known to impair neovascularization, a process known to depend on the function of highly proliferative EPCs. There are evidences clearly demonstrating that aging impairs the function of ex vivo expanded EPCs (Keymel et al., 2008). The loss of EPCs associated to aging may be mediated by an imbalance between factors promoting growth, migration, and survival against those enhancing oxidative stress and promoting senescence (Chang et al., 2007). In this sense, hypoxia inducible factor-1α (HIF-1α) induces the expression of stromal cell-derived factor-1 (SDF-1) that enhances the recruitment of EPCs in injured or ischemic tissues in mice (Karshovska et al., 2007). There are evidences demonstrating that the levels or activity of HIF-1α decreases with aging and thereby leads to reduced levels of SDF-1 (Hoenig et al., 2008). Telomere length declines with age in EPCs from healthy men (Minamino et al., 2004; Kushner et al., 2009). Moreover, EPCs senescence is accelerated in both experimental hypertensive rats and patients with essential hypertension (Imanishi et al., 2005b). There is also a premature senescence in EPCs from diabetic patients (Rosso et al., 2006).
Telomerase
To avoid the attrition of telomeres, germinal cells and some somatic cells produces telomerase, an enzyme that catalyze DNA synthesis to maintain telomere length. Human endothelial cells and vascular smooth muscle express telomerase activity, which is activated by mitogen stimuli via PKC, this activity being reduced with age (Minamino and Komuro, 2007). Introduction of telomerase into EPCs extends life span and improves vasculogenesis of these cells (Minamino et al., 2004). It has been also described that NO activates telomerase in endothelial cells, delaying senescence (Vasa et al., 2000; Farsetti et al., 2009). Then, strategies aimed to increase endothelial NO bioavailability could be considered as possible therapies to prevent endothelial cell senescence associated with aging (Hayashi et al., 2008). By the contrary, angiotensin II-mediated oxidative stress (Imanishi et al., 2005a; Morris, 2005) and pro-atherogenic factors, like TNFα (Breitschopf et al., 2001), are major inducers for endothelial senescence, by inducing telomerase inactivation through an Akt-dependent mechanism. Antioxidants may prevent this telomerase inactivation by inhibiting the nuclear export of the catalytic subunit of the telomerase (Haendeler et al., 2004). The inactivation of telomerase induced by angiotensin II-mediated oxidative stress is related to the presence of nitrotyrosine, suggesting the involvement of peroxynitrite (Imanishi et al., 2005a; Morris, 2005). Interestingly, the endothelial cells senescence evoked by glycated collagen and the premature vascular senescence observed in type 2 diabetic rats can be prevented with peroxynitrite scavengers (Chen et al., 2002, 2007).
Sirtuins
During the past decade, research on aging, based initially on simple laboratory organisms, has identified important genes and pathways that contribute to longevity. Among these is a family of nicotinamide adenine dinucleotide (NAD)-dependent proteins termed sirtuins, which can extend the life span in model organisms and are important in mediating the beneficial effects of low-calorie diets (Guarente, 2011). There are seven identified sirtuins in mammals (Sirt1 to Sirt7), which main function might be to promote survival and stress resistance, resulting in longevity (Sedding and Haendeler, 2007; Guarente, 2011). At present, endothelial dysfunction in arteries from aged mice and humans is associated with a reduction of vascular expression of Sirt1 (Donato et al., 2011) and there is increasing evidence that the beneficial effects produced by caloric restriction on endothelial function in aged animals are related, at least in part, by up-regulation of Sirt1 (Rippe et al., 2010; Weiss and Fontana, 2011). In fact, increased Sirt1 expression in cultured endothelial cells was driven by serum from aged rats under restricted calorie intake (Csiszar et al., 2009). In human endothelial cells, over-expression of Sirt1 prevented oxidative stress-induced senescence, while its inhibition leads to premature senescence-like phenotype. Interestingly, immunosuppressant drugs, like sirolimus and everolimus, induce endothelial cellular senescence via Sirt1 down-regulation (Ota et al., 2009). By the contrary, moderate over-expression of Sirt1 in mice hearts confers stress resistance to the heart in vivo, retarding aging phenotype (Alcendor et al., 2007). It has been suggested that NO upregulate Sirt1 in human endothelial cells, therefore inhibiting oxidative stress-induced premature cell senescence (Ota et al., 2008; Potente and Dimmeler, 2008). By the other hand, Sirt1 inhibition in HUVEC results in reduced eNOS activity and senescence-like phenotype while Sirt1 induction increases eNOS activity and prevents ROS-induced senescence (Ota et al., 2007). Sirt1 plays an important role in endothelial homeostasis by regulating eNOS activity; therefore, the Sirt1/eNOS axis may be quite relevant as a potential target against vascular senescence, endothelial dysfunction, and atherosclerosis (Ota et al., 2010). Moreover, Sirt1 enhancement inhibits the expression of AT1 receptors in vascular smooth muscle cells (Miyazaki et al., 2008), reducing cell hypertrophy and neointima formation following vascular injury (Li et al., 2011a,b). All the available data suggest that sirtuins can be a unique class of proteins exerting important effects on age-related CVDs and a promising target for drug development (Guarente, 2011).
Conclusion
The better understanding of the molecular and cellular mechanisms involved in vascular aging, as well as their potential interactions, provides a growing list of factors that can be targets for specific interventions aimed at preventing and delaying the vascular aging. The NO pathway and endothelial dysfunction, oxidative stress, inflammation, telomerase, and sirtuins are among the principal mechanisms likely involved in the vascular aging process, both in healthy and pathological conditions. A better comprehension of the complex interactions between them is an important objective for future research. Changes in lifestyle, dietary changes, loss of weight (if overweight), and particularly aerobic exercise may prevent or delay the onset of endothelial dysfunction. The pharmacological approaches could also be important tools for intervention throughout the aging process, either preventing endothelial dysfunction, or treating advanced stages of endothelial dysfunction, or even acting on structural alterations already evident in the vascular wall. Although we need more data in human beings about the role of telomerase and sirtuins in healthy aging and in disease, both may be new promising targets, in addition to mechanisms already investigated, including the activation of the NO pathway or the COX inhibition, or the interference with pro-oxidant and pro-inflammatory pathways.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study is supported by grants from Ministerio de Ciencia e Innovación (Instituto de Salud Carlos III, RETICEF RD06/0013; PI08/1649; SAF2011-28011; SAF2011-24684, Spanish Government, and Sociedad Española de Farmacología/Almirall Prodesfarma.
References
Abbatecola, A. M., Paolisso, G., Fattoretti, P., Evans, W. J., Fiore, V., Dicioccio, L., and Lattanzio, F. (2011). Discovering pathways of sarcopenia in older adults: a role for insulin resistance on mitochondria dysfunction. J. Nutr. Health Aging 15, 890–895.
Ahlers, B. A., Parnell, M. M., Chin-Dusting, J. P., and Kaye, D. M. (2004). An age-related decline in endothelial function is not associated with alterations in L-arginine transport in humans. J. Hypertens. 22, 321–327.
Albarwani, S., Al-Siyabi, S., Baomar, H., and Hassan, M. O. (2010). Exercise training attenuates ageing-induced BKCa channel downregulation in rat coronary arteries. Exp. Physiol. 95, 746–755.
Alcendor, R. R., Gao, S., Zhai, P., Zablocki, D., Holle, E., Yu, X., Tian, B., Wagner, T., Vatner, S. F., and Sadoshima, J. (2007). Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 100, 1512–1521.
Angulo, J., Cuevas, P., Fernandez, A., Gabancho, S., Videla, S., and Saenz De Tejada, I. (2003). Calcium dobesilate potentiates endothelium-derived hyperpolarizing factor-mediated relaxation of human penile resistance arteries. Br. J. Pharmacol. 139, 854–862.
Arenas, I. A., Xu, Y., and Davidge, S. T. (2006). Age-associated impairment in vasorelaxation to fluid shear stress in the female vasculature is improved by TNF-alpha antagonism. Am. J. Physiol. Heart Circ. Physiol. 290, H1259–H1263.
Aviv, H., Khan, M. Y., Skurnick, J., Okuda, K., Kimura, M., Gardner, J., Priolo, L., and Aviv, A. (2001). Age dependent aneuploidy and telomere length of the human vascular endothelium. Atherosclerosis 159, 281–287.
Barodka, V. M., Joshi, B. L., Berkowitz, D. E., Hogue, C. W. Jr., and Nyhan, D. (2011). Review article: implications of vascular aging. Anesth. Analg. 112, 1048–1060.
Barton, M., Cosentino, F., Brandes, R. P., Moreau, P., Shaw, S., and Luscher, T. F. (1997). Anatomic heterogeneity of vascular aging: role of nitric oxide and endothelin. Hypertension 30, 817–824.
Benetos, A., Okuda, K., Lajemi, M., Kimura, M., Thomas, F., Skurnick, J., Labat, C., Bean, K., and Aviv, A. (2001). Telomere length as an indicator of biological aging: the gender effect and relation with pulse pressure and pulse wave velocity. Hypertension 37, 381–385.
Benigni, A., Corna, D., Zoja, C., Sonzogni, A., Latini, R., Salio, M., Conti, S., Rottoli, D., Longaretti, L., Cassis, P., Morigi, M., Coffman, T. M., and Remuzzi, G. (2009). Disruption of the Ang II type 1 receptor promotes longevity in mice. J. Clin. Invest. 119, 524–530.
Bode-Boger, S. M., Muke, J., Surdacki, A., Brabant, G., Boger, R. H., and Frolich, J. C. (2003). Oral L-arginine improves endothelial function in healthy individuals older than 70 years. Vasc. Med. 8, 77–81.
Bode-Boger, S. M., Scalera, F., and Martens-Lobenhoffer, J. (2005). Asymmetric dimethylarginine (ADMA) accelerates cell senescence. Vasc. Med. 10(Suppl. 1), S65–S71.
Breitschopf, K., Zeiher, A. M., and Dimmeler, S. (2001). Pro-atherogenic factors induce telomerase inactivation in endothelial cells through an Akt-dependent mechanism. FEBS Lett. 493, 21–25.
Briasoulis, A., Tousoulis, D., Antoniades, C., Stefanadis, C., and Papageorgiou, N. (2011). The role of endothelial progenitor cells in vascular repair after arterial injury and atherosclerotic plaque development. Cardiovasc. Ther. 29, 125–139.
Briones, A. M., Montoya, N., Giraldo, J., and Vila, E. (2005). Ageing affects nitric oxide synthase, cyclooxygenase and oxidative stress enzymes expression differently in mesenteric resistance arteries. Auton. Autacoid Pharmacol. 25, 155–162.
Cai, H., and Harrison, D. G. (2000). Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ. Res. 87, 840–844.
Cernadas, M. R., Sanchez De Miguel, L., Garcia-Duran, M., Gonzalez-Fernandez, F., Millas, I., Monton, M., Rodrigo, J., Rico, L., Fernandez, P., De Frutos, T., Rodriguez-Feo, J. A., Guerra, J., Caramelo, C., Casado, S., and Lopez, F. (1998). Expression of constitutive and inducible nitric oxide synthases in the vascular wall of young and aging rats. Circ. Res. 83, 279–286.
Chang, E. I., Loh, S. A., Ceradini, D. J., Lin, S. E., Bastidas, N., Aarabi, S., Chan, D. A., Freedman, M. L., Giaccia, A. J., and Gurtner, G. C. (2007). Age decreases endothelial progenitor cell recruitment through decreases in hypoxia-inducible factor 1alpha stabilization during ischemia. Circulation 116, 2818–2829.
Chen, J., Brodsky, S. V., Goligorsky, D. M., Hampel, D. J., Li, H., Gross, S. S., and Goligorsky, M. S. (2002). Glycated collagen I induces premature senescence-like phenotypic changes in endothelial cells. Circ. Res. 90, 1290–1298.
Chen, J., Park, H. C., Patschan, S., Brodsky, S. V., Gealikman, O., Kuo, M. C., Li, H., Addabbo, F., Zhang, F., Nasjletti, A., Gross, S. S., and Goligorsky, M. S. (2007). Premature vascular senescence in metabolic syndrome: could it be prevented and reversed by a selenorganic antioxidant and peroxynitrite scavenger ebselen? Drug Discov. Today Ther. Strateg. 4, 93–99.
Chung, H. Y., Kim, H. J., Kim, K. W., Choi, J. S., and Yu, B. P. (2002). Molecular inflammation hypothesis of aging based on the anti-aging mechanism of calorie restriction. Microsc. Res. Tech. 59, 264–272.
Csiszar, A., Labinskyy, N., Jimenez, R., Pinto, J. T., Ballabh, P., Losonczy, G., Pearson, K. J., De Cabo, R., and Ungvari, Z. (2009). Anti-oxidative and anti-inflammatory vasoprotective effects of caloric restriction in aging: role of circulating factors and SIRT1. Mech. Ageing Dev. 130, 518–527.
Csiszar, A., Labinskyy, N., Smith, K., Rivera, A., Orosz, Z., and Ungvari, Z. (2007). Vasculoprotective effects of anti-tumor necrosis factor-alpha treatment in aging. Am. J. Pathol. 170, 388–398.
Csiszar, A., Ungvari, Z., Edwards, J. G., Kaminski, P., Wolin, M. S., Koller, A., and Kaley, G. (2002). Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ. Res. 90, 1159–1166.
Csiszar, A., Ungvari, Z., Koller, A., Edwards, J. G., and Kaley, G. (2003). Aging-induced proinflammatory shift in cytokine expression profile in coronary arteries. FASEB J. 17, 1183–1185.
Csiszar, A., Ungvari, Z., Koller, A., Edwards, J. G., and Kaley, G. (2004). Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol. Genomics 17, 21–30.
Cui, H., Kong, Y., and Zhang, H. (2012). Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct. 2012, 646354.
De Meyer, T., Rietzschel, E. R., De Buyzere, M. L., Van Criekinge, W., and Bekaert, S. (2011). Telomere length and cardiovascular aging: the means to the ends? Ageing Res. Rev. 10, 297–303.
de Sotomayor, M. A., Perez-Guerrero, C., Herrrera, M. D., Jimenez, L., Marin, R., Marhuenda, E., and Andriantsitohaina, R. (2005). Improvement of age-related endothelial dysfunction by simvastatin: effect on NO and COX pathways. Br. J. Pharmacol. 146, 1130–1138.
Donato, A. J., Black, A. D., Jablonski, K. L., Gano, L. B., and Seals, D. R. (2008). Aging is associated with greater nuclear NF kappa B, reduced I kappa B alpha, and increased expression of proinflammatory cytokines in vascular endothelial cells of healthy humans. Aging Cell 7, 805–812.
Donato, A. J., Eskurza, I., Silver, A. E., Levy, A. S., Pierce, G. L., Gates, P. E., and Seals, D. R. (2007). Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB. Circ. Res. 100, 1659–1666.
Donato, A. J., Gano, L. B., Eskurza, I., Silver, A. E., Gates, P. E., Jablonski, K., and Seals, D. R. (2009). Vascular endothelial dysfunction with aging: endothelin-1 and endothelial nitric oxide synthase. Am. J. Physiol. Heart Circ. Physiol. 297, H425–H432.
Donato, A. J., Lesniewski, L. A., and Delp, M. D. (2005). The effects of aging and exercise training on endothelin-1 vasoconstrictor responses in rat skeletal muscle arterioles. Cardiovasc. Res. 66, 393–401.
Donato, A. J., Magerko, K. A., Lawson, B. R., Durrant, J. R., Lesniewski, L. A., and Seals, D. R. (2011). SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J. Physiol. (Lond.) 589, 4545–4554.
Donato, A. J., Uberoi, A., Bailey, D. M., Wray, D. W., and Richardson, R. S. (2010). Exercise-induced brachial artery vasodilation: effects of antioxidants and exercise training in elderly men. Am. J. Physiol. Heart Circ. Physiol. 298, H671–H678.
Dora, K. A., Gallagher, N. T., Mcneish, A., and Garland, C. J. (2008). Modulation of endothelial cell KCa3.1 channels during endothelium-derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ. Res. 102, 1247–1255.
Epel, E. S., Merkin, S. S., Cawthon, R., Blackburn, E. H., Adler, N. E., Pletcher, M. J., and Seeman, T. E. (2009). The rate of leukocyte telomere shortening predicts mortality from cardiovascular disease in elderly men. Aging (Albany NY) 1, 81–88.
Erusalimsky, J. D. (2009). Vascular endothelial senescence: from mechanisms to pathophysiology. J. Appl. Physiol. 106, 326–332.
Erusalimsky, J. D., and Kurz, D. J. (2005). Cellular senescence in vivo: its relevance in ageing and cardiovascular disease. Exp. Gerontol. 40, 634–642.
Erusalimsky, J. D., and Skene, C. (2009). Mechanisms of endothelial senescence. Exp. Physiol. 94, 299–304.
Eskurza, I., Kahn, Z. D., and Seals, D. R. (2006). Xanthine oxidase does not contribute to impaired peripheral conduit artery endothelium-dependent dilatation with ageing. J. Physiol. (Lond.) 571, 661–668.
Farsetti, A., Grasselli, A., Bacchetti, S., Gaetano, C., and Capogrossi, M. C. (2009). The telomerase tale in vascular aging: regulation by estrogens and nitric oxide signaling. J. Appl. Physiol. 106, 333–337.
Ferrucci, L., Corsi, A., Lauretani, F., Bandinelli, S., Bartali, B., Taub, D. D., Guralnik, J. M., and Longo, D. L. (2005). The origins of age-related proinflammatory state. Blood 105, 2294–2299.
Franceschi, C., Bonafe, M., Valensin, S., Olivieri, F., De Luca, M., Ottaviani, E., and De Benedictis, G. (2000). Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 908, 244–254.
Francia, P., Delli Gatti, C., Bachschmid, M., Martin-Padura, I., Savoia, C., Migliaccio, E., Pelicci, P. G., Schiavoni, M., Luscher, T. F., Volpe, M., and Cosentino, F. (2004). Deletion of p66shc gene protects against age-related endothelial dysfunction. Circulation 110, 2889–2895.
Gates, P. E., Boucher, M. L., Silver, A. E., Monahan, K. D., and Seals, D. R. (2007). Impaired flow-mediated dilation with age is not explained by L-arginine bioavailability or endothelial asymmetric dimethylarginine protein expression. J. Appl. Physiol. 102, 63–71.
Gaubert, M. L., Sigaudo-Roussel, D., Tartas, M., Berrut, G., Saumet, J. L., and Fromy, B. (2007). Endothelium-derived hyperpolarizing factor as an in vivo back-up mechanism in the cutaneous microcirculation in old mice. J. Physiol. 585, 617–626.
Goraca, A. (2004). Assessment of total antioxidant capacity in human plasma. Folia Med. (Plovdiv) 46, 16–21.
Goto, K., Fujii, K., Kansui, Y., and Iida, M. (2004). Changes in endothelium-derived hyperpolarizing factor in hypertension and ageing: response to chronic treatment with renin-angiotensin system inhibitors. Clin. Exp. Pharmacol. Physiol. 31, 650–655.
Goto, K., Fujii, K., Onaka, U., Abe, I., and Fujishima, M. (2000). Angiotensin-converting enzyme inhibitor prevents age-related endothelial dysfunction. Hypertension 36, 581–587.
Griese, D. P., Ehsan, A., Melo, L. G., Kong, D., Zhang, L., Mann, M. J., Pratt, R. E., Mulligan, R. C., and Dzau, V. J. (2003). Isolation and transplantation of autologous circulating endothelial cells into denuded vessels and prosthetic grafts: implications for cell-based vascular therapy. Circulation 108, 2710–2715.
Guarente, L. (2011). Franklin H. Epstein lecture: sirtuins, aging, and medicine. N. Engl. J. Med. 364, 2235–2244.
Guzik, T. J., Mussa, S., Gastaldi, D., Sadowski, J., Ratnatunga, C., Pillai, R., and Channon, K. M. (2002). Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 105, 1656–1662.
Haendeler, J., Hoffmann, J., Diehl, J. F., Vasa, M., Spyridopoulos, I., Zeiher, A. M., and Dimmeler, S. (2004). Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ. Res. 94, 768–775.
Harris, T. B., Ferrucci, L., Tracy, R. P., Corti, M. C., Wacholder, S., Ettinger, W. H. Jr., Heimovitz, H., Cohen, H. J., and Wallace, R. (1999). Associations of elevated interleukin-6 and C-reactive protein levels with mortality in the elderly. Am. J. Med. 106, 506–512.
Hayashi, T., Matsui-Hirai, H., Miyazaki-Akita, A., Fukatsu, A., Funami, J., Ding, Q. F., Kamalanathan, S., Hattori, Y., Ignarro, L. J., and Iguchi, A. (2006). Endothelial cellular senescence is inhibited by nitric oxide: implications in atherosclerosis associated with menopause and diabetes. Proc. Natl. Acad. Sci. U.S.A. 103, 17018–17023.
Hayashi, T., Yano, K., Matsui-Hirai, H., Yokoo, H., Hattori, Y., and Iguchi, A. (2008). Nitric oxide and endothelial cellular senescence. Pharmacol. Ther. 120, 333–339.
Haynes, W. G., Strachan, F. E., and Webb, D. J. (1995). Endothelin ETA and ETB receptors cause vasoconstriction of human resistance and capacitance vessels in vivo. Circulation 92, 357–363.
Heymes, C., Habib, A., Yang, D., Mathieu, E., Marotte, F., Samuel, J., and Boulanger, C. M. (2000). Cyclo-oxygenase-1 and -2 contribution to endothelial dysfunction in ageing. Br. J. Pharmacol. 131, 804–810.
Higashi, Y., Sasaki, S., Nakagawa, K., Kimura, M., Noma, K., Hara, K., Jitsuiki, D., Goto, C., Oshima, T., Chayama, K., and Yoshizumi, M. (2006). Tetrahydrobiopterin improves aging-related impairment of endothelium-dependent vasodilation through increase in nitric oxide production. Atherosclerosis 186, 390–395.
Hilgers, R. H., and Webb, R. C. (2007). Reduced expression of SKCa and IKCa channel proteins in rat small mesenteric arteries during angiotensin II-induced hypertension. Am. J. Physiol. Heart. Circ. Physiol. 292, H2275–H2284.
Hoenig, M. R., Bianchi, C., Rosenzweig, A., and Sellke, F. W. (2008). Decreased vascular repair and neovascularization with ageing: mechanisms and clinical relevance with an emphasis on hypoxia-inducible factor-1. Curr. Mol. Med. 8, 754–767.
Holowatz, L. A., Thompson, C. S., and Kenney, W. L. (2006). L-Arginine supplementation or arginase inhibition augments reflex cutaneous vasodilatation in aged human skin. J. Physiol. (Lond.) 574, 573–581.
Idris Khodja, N., Chataigneau, T., Auger, C., and Schini-Kerth, V. B. (2012). Grape-derived polyphenols improve aging-related endothelial dysfunction in rat mesenteric artery: role of oxidative stress and the angiotensin system. PLoS ONE 7, e32039.
Imanishi, T., Hano, T., and Nishio, I. (2005a). Angiotensin II accelerates endothelial progenitor cell senescence through induction of oxidative stress. J. Hypertens. 23, 97–104.
Imanishi, T., Moriwaki, C., Hano, T., and Nishio, I. (2005b). Endothelial progenitor cell senescence is accelerated in both experimental hypertensive rats and patients with essential hypertension. J. Hypertens. 23, 1831–1837.
Jablonski, K. L., Seals, D. R., Eskurza, I., Monahan, K. D., and Donato, A. J. (2007). High-dose ascorbic acid infusion abolishes chronic vasoconstriction and restores resting leg blood flow in healthy older men. J. Appl. Physiol. 103, 1715–1721.
Kansui, Y., Fujii, K., Goto, K., Abe, I., and Iida, M. (2002). Angiotensin II receptor antagonist improves age-related endothelial dysfunction. J. Hypertens. 20, 439–446.
Karshovska, E., Zernecke, A., Sevilmis, G., Millet, A., Hristov, M., Cohen, C. D., Schmid, H., Krotz, F., Sohn, H. Y., Klauss, V., Weber, C., and Schober, A. (2007). Expression of HIF-1alpha in injured arteries controls SDF-1alpha mediated neointima formation in apolipoprotein E deficient mice. Arterioscler. Thromb. Vasc. Biol. 27, 2540–2547.
Keymel, S., Kalka, C., Rassaf, T., Yeghiazarians, Y., Kelm, M., and Heiss, C. (2008). Impaired endothelial progenitor cell function predicts age-dependent carotid intimal thickening. Basic Res. Cardiol. 103, 582–586.
Kim, D. H., Newman, A. B., Hajjar, I., Strotmeyer, E. S., Klein, R., Newton, E., Sarnak, M. J., Burke, G. L., and Lipsitz, L. A. (2011). Retinal microvascular signs and functional loss in older persons: the cardiovascular health study. Stroke 42, 1589–1595.
Kim, S. F., Huri, D. A., and Snyder, S. H. (2005). Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science 310, 1966–1970.
Kimura, S., Zhang, G. X., Nishiyama, A., Shokoji, T., Yao, L., Fan, Y. Y., Rahman, M., Suzuki, T., Maeta, H., and Abe, Y. (2005). Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension 45, 860–866.
Kirton, J. P., and Xu, Q. (2010). Endothelial precursors in vascular repair. Microvasc. Res. 79, 193–199.
Kotsis, V., Stabouli, S., Karafillis, I., and Nilsson, P. (2011). Early vascular aging and the role of central blood pressure. J. Hypertens. 29, 1847–1853.
Korzick, D. H., Muller-Delp, J. M., Dougherty, P., Heaps, C. L., Bowles, D. K., and Krick, K. K. (2005). Exaggerated coronary vasoreactivity to endothelin-1 in aged rats: role of protein kinase C. Cardiovasc Res. 66, 384–392.
Kurz, D. J., Decary, S., Hong, Y., Trivier, E., Akhmedov, A., and Erusalimsky, J. D. (2004). Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J. Cell Sci. 117, 2417–2426.
Kushner, E. J., Van Guilder, G. P., Maceneaney, O. J., Cech, J. N., Stauffer, B. L., and Desouza, C. A. (2009). Aging and endothelial progenitor cell telomere length in healthy men. Clin. Chem. Lab. Med. 47, 47–50.
Lassegue, B., and Griendling, K. K. (2010). NADPH oxidases: functions and pathologies in the vasculature. Arterioscler. Thromb. Vasc. Biol. 30, 653–661.
Laursen, J. B., Somers, M., Kurz, S., Mccann, L., Warnholtz, A., Freeman, B. A., Tarpey, M., Fukai, T., and Harrison, D. G. (2001). Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation 103, 1282–1288.
LeBlanc, A. J., Shipley, R. D., Kang, L. S., and Muller-Delp, J. M. (2008). Age impairs Flk-1 signaling and NO-mediated vasodilation in coronary arterioles. Am. J. Physiol. Heart Circ. Physiol. 295, H2280–H2288.
Li, L., Gao, P., Zhang, H., Chen, H., Zheng, W., Lv, X., Xu, T., Wei, Y., Liu, D., and Liang, C. (2011a). SIRT1 inhibits angiotensin II-induced vascular smooth muscle cell hypertrophy. Acta Biochim. Biophys. Sin. (Shanghai) 43, 103–109.
Li, L., Zhang, H. N., Chen, H. Z., Gao, P., Zhu, L. H., Li, H. L., Lv, X., Zhang, Q. J., Zhang, R., Wang, Z., She, Z. G., Wei, Y. S., Du, G. H., Liu, D. P., and Liang, C. C. (2011b). SIRT1 acts as a modulator of neointima formation following vascular injury in mice. Circ. Res. 108, 1180–1189.
Long, D. A., Newaz, M. A., Prabhakar, S. S., Price, K. L., Truong, L. D., Feng, L., Mu, W., Oyekan, A. O., and Johnson, R. J. (2005). Loss of nitric oxide and endothelial-derived hyperpolarizing factor-mediated responses in aging. Kidney Int. 68, 2154–2163.
Loscalzo, J. (2000). What we know and don’t know about L-arginine and NO. Circulation 101, 2126–2129.
Lüscher, T. F., and Barton, M. (2000). Endothelins and endothelin receptor antagonists: therapeutic considerations for a novel class of cardiovascular drugs. Circulation 102, 2434–2440.
Mackenzie, A., Cooper, E. J., and Dowell, F. J. (2008). Differential effects of glucose on agonist-induced relaxations in human mesenteric and subcutaneous arteries. Br. J. Pharmacol. 153, 480–487.
Maeda, S., Tanabe, T., Miyauchi, T., Otsuki, T., Sugawara, J., Iemitsu, M., Kuno, S., Ajisaka, R., Yamaguchi, I., and Matsuda, M. (2003). Aerobic exercise training reduces plasma endothelin-1 concentration in older women. J. Appl. Physiol. 95, 336–341.
Malinin, N. L., West, X. Z., and Byzova, T. V. (2011). Oxidation as “the stress of life.” Aging (Albany NY) 3, 906–910.
Masaki, T., Kimura, S., Yanagisawa, M., and Goto, K. (1991). Molecular and cellular mechanism of endothelin regulation. Implications for vascular function. Circulation 84, 1457–1468.
Matz, R. L., and Andriantsitohaina, R. (2003). Age-related endothelial dysfunction: potential implications for pharmacotherapy. Drugs Aging 20, 527–550.
Matz, R. L., De Sotomayor, M. A., Schott, C., Stoclet, J. C., and Andriantsitohaina, R. (2000). Vascular bed heterogeneity in age-related endothelial dysfunction with respect to NO and eicosanoids. Br. J. Pharmacol. 131, 303–311.
Mecocci, P., Polidori, M. C., Troiano, L., Cherubini, A., Cecchetti, R., Pini, G., Straatman, M., Monti, D., Stahl, W., Sies, H., Franceschi, C., and Senin, U. (2000). Plasma antioxidants and longevity: a study on healthy centenarians. Free Radic. Biol. Med. 28, 1243–1248.
Milstien, S., and Katusic, Z. (1999). Oxidation of tetrahydrobiopterin by peroxynitrite: implications for vascular endothelial function. Biochem. Biophys. Res. Commun. 263, 681–684.
Minamino, T., and Komuro, I. (2007). Vascular cell senescence: contribution to atherosclerosis. Circ. Res. 100, 15–26.
Minamino, T., Miyauchi, H., Yoshida, T., Tateno, K., and Komuro, I. (2004). The role of vascular cell senescence in atherosclerosis: antisenescence as a novel therapeutic strategy for vascular aging. Curr. Vasc. Pharmacol. 2, 141–148.
Miyazaki, R., Ichiki, T., Hashimoto, T., Inanaga, K., Imayama, I., Sadoshima, J., and Sunagawa, K. (2008). SIRT1, a longevity gene, downregulates angiotensin II type 1 receptor expression in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 28, 1263–1269.
Mollnau, H., Wendt, M., Szocs, K., Lassegue, B., Schulz, E., Oelze, M., Li, H., Bodenschatz, M., August, M., Kleschyov, A. L., Tsilimingas, N., Walter, U., Forstermann, U., Meinertz, T., Griendling, K., and Munzel, T. (2002). Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ. Res. 90, E58–E65.
Mukai, Y., Shimokawa, H., Higashi, M., Morikawa, K., Matoba, T., Hiroki, J., Kunihiro, I., Talukder, H. M., and Takeshita, A. (2002). Inhibition of renin-angiotensin system ameliorates endothelial dysfunction associated with aging in rats. Arterioscler. Thromb. Vasc. Biol. 22, 1445–1450.
Munzel, T., Daiber, A., Ullrich, V., and Mulsch, A. (2005). Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler. Thromb. Vasc. Biol. 25, 1551–1557.
Najjar, S. S., Scuteri, A., and Lakatta, E. G. (2005). Arterial aging: is it an immutable cardiovascular risk factor? Hypertension 46, 454–462.
Nakashima, M., Mombouli, J. V., Taylor, A. A., and Vanhoutte, P. M. (1993). Endothelium-dependent hyperpolarization caused by bradykinin in human coronary arteries. J. Clin. Invest. 92, 2867–2871.
Nevado, J., Vallejo, S., El-Assar, M., Peiro, C., Sanchez-Ferrer, C. F., and Rodriguez-Manas, L. (2006). Changes in the human peritoneal mesothelial cells during aging. Kidney Int. 69, 313–322.
Newaz, M. A., Yousefipour, Z., and Oyekan, A. (2006). Oxidative stress-associated vascular aging is xanthine oxidase-dependent but not NAD(P)H oxidase-dependent. J. Cardiovasc. Pharmacol. 48, 88–94.
Ogami, M., Ikura, Y., Ohsawa, M., Matsuo, T., Kayo, S., Yoshimi, N., Hai, E., Shirai, N., Ehara, S., Komatsu, R., Naruko, T., and Ueda, M. (2004). Telomere shortening in human coronary artery diseases. Arterioscler. Thromb. Vasc. Biol. 24, 546–550.
Ota, H., Akishita, M., Eto, M., Iijima, K., Kaneki, M., and Ouchi, Y. (2007). Sirt1 modulates premature senescence-like phenotype in human endothelial cells. J. Mol. Cell Cardiol. 43, 571–579.
Ota, H., Eto, M., Ako, J., Ogawa, S., Iijima, K., Akishita, M., and Ouchi, Y. (2009). Sirolimus and everolimus induce endothelial cellular senescence via sirtuin 1 down-regulation: therapeutic implication of cilostazol after drug-eluting stent implantation. J. Am. Coll. Cardiol. 53, 2298–2305.
Ota, H., Eto, M., Kano, M. R., Ogawa, S., Iijima, K., Akishita, M., and Ouchi, Y. (2008). Cilostazol inhibits oxidative stress-induced premature senescence via upregulation of Sirt1 in human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 28, 1634–1639.
Ota, H., Eto, M., Ogawa, S., Iijima, K., Akishita, M., and Ouchi, Y. (2010). SIRT1/eNOS axis as a potential target against vascular senescence, dysfunction and atherosclerosis. J. Atheroscler. Thromb. 17, 431–435.
Pacher, P., Beckman, J. S., and Liaudet, L. (2007). Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 87, 315–424.
Pang, C. Y., Ma, Y. S., and Wei, Y. U. (2008). MtDNA mutations, functional decline and turnover of mitochondria in aging. Front. Biosci. 13, 3661–3675.
Panza, F., D’Introno, A., Colacicco, A. M., Capurso, C., Parigi, A. D., Capurso, S. A., Caselli, R. J., Pilotto, A., Scafato, E., Capurso, A., and Solfrizzi, V. (2006). Cognitive frailty: predementia syndrome and vascular risk factors. Neurobiol. Aging 27, 933–940.
Pascoal, I. F., and Umans, J. G. (1996). Effect of pregnancy on mechanisms of relaxation in human omental microvessels. Hypertension 28, 183–187.
Pasquier, D., and Dias, S. (2010). Endothelial progenitor cells: hope beyond controversy. Curr. Cancer Drug Targets 10, 914–921.
Patschan, S., Chen, J., Polotskaia, A., Mendelev, N., Cheng, J., Patschan, D., and Goligorsky, M. S. (2008). Lipid mediators of autophagy in stress-induced premature senescence of endothelial cells. Am. J. Physiol. Heart. Circ. Physiol. 294, H1119–H1129.
Patschan, S., and Goligorsky, M. S. (2008). Autophagy: the missing link between non-enzymatically glycated proteins inducing apoptosis and premature senescence of endothelial cells? Autophagy 4, 521–523.
Potente, M., and Dimmeler, S. (2008). NO targets SIRT1: a novel signaling network in endothelial senescence. Arterioscler. Thromb. Vasc. Biol. 28, 1577–1579.
Rajagopalan, S., Brook, R., Mehta, R. H., Supiano, M., and Pitt, B. (2002). Effect of losartan in aging-related endothelial impairment. Am. J. Cardiol. 89, 562–566.
Rajagopalan, S., Kariisa, M., Dellegrottaglie, S., Bard, R. L., Kehrer, C., Matlow, S., Daley, W., Pitt, B., and Brook, R. (2006). Angiotensin receptor blockade improves vascular compliance in healthy normotensive elderly individuals: results from a randomized double-blind placebo-controlled trial. J. Clin. Hypertens. (Greenwich) 8, 783–790.
Rajapakse, A. G., Yepuri, G., Carvas, J. M., Stein, S., Matter, C. M., Scerri, I., Ruffieux, J., Montani, J. P., Ming, X. F., and Yang, Z. (2011). Hyperactive S6K1 mediates oxidative stress and endothelial dysfunction in aging: inhibition by resveratrol. PLoS One 6, e19237.
Resch, T., Pircher, A., Kähler, C. M., Pratschke, J., and Hilbe, W. (2011). Endothelial progenitor cells: current issues on characterization and challenging clinical applications. Stem Cell Rev.. [Epub ahead of print].
Rippe, C., Lesniewski, L., Connell, M., LaRocca, T., Donato, A. J., and Seals, D. (2010). Short-term calorie restriction reverses vascular endothelial dysfunction in old mice by increasing nitric oxide and reducing oxidative stress. Aging Cell 9, 304–312.
Rodriguez-Mañas, L., El-Assar, M., Vallejo, S., Lopez-Doriga, P., Solis, J., Petidier, R., Montes, M., Nevado, J., Castro, M., Gomez-Guerrero, C., Peiro, C., and Sanchez-Ferrer, C. F. (2009). Endothelial dysfunction in aged humans is related with oxidative stress and vascular inflammation. Aging Cell 8, 226–238.
Rosso, A., Balsamo, A., Gambino, R., Dentelli, P., Falcioni, R., Cassader, M., Pegoraro, L., Pagano, G., and Brizzi, M. F. (2006). p53 Mediates the accelerated onset of senescence of endothelial progenitor cells in diabetes. J. Biol. Chem. 281, 4339–4347.
Sagach, V., Bondarenko, A., Bazilyuk, O., and Kotsuruba, A. (2006). Endothelial dysfunction: possible mechanisms and ways of correction. Exp. Clin. Cardiol. 11, 107–110.
Sanchez-Guijo, F. M., Oterino, E., Barbado, M. V., Carrancio, S., Lopez-Holgado, N., Muntion, S., Hernandez-Campo, P., Sanchez-Abarca, L. I., Perez-Simon, J. A., San Miguel, J. F., Briñon, J. G., and del Cañizo, M. C. (2010). Both CD133(+) cells and monocytes provide significant improvement for hindlimb ischemia, although they do not transdifferentiate into endothelial cells. Cell Transplant. 19, 103–112.
Santhanam, L., Christianson, D. W., Nyhan, D., and Berkowitz, D. E. (2008). Arginase and vascular aging. J. Appl. Physiol. 105, 1632–1642.
Santhanam, L., Lim, H. K., Miriel, V., Brown, T., Patel, M., Balanson, S., Ryoo, S., Anderson, M., Irani, K., Khanday, F., Di Costanzo, L., Nyhan, D., Hare, J. M., Christianson, D. W., Rivers, R., Shoukas, A., and Berkowitz, D. E. (2007). Inducible NO synthase dependent S-nitrosylation and activation of arginase1 contribute to age-related endothelial dysfunction. Circ. Res. 101, 692–702.
Scalera, F., Martens-Lobenhoffer, J., Bukowska, A., Lendeckel, U., Tager, M., and Bode-Boger, S. M. (2008). Effect of telmisartan on nitric oxide–asymmetrical dimethylarginine system: role of angiotensin II type 1 receptor gamma and peroxisome proliferator activated receptor gamma signaling during endothelial aging. Hypertension 51, 696–703.
Schiffrin, E. L. (2008). Oxidative stress, nitric oxide synthase, and superoxide dismutase: a matter of imbalance underlies endothelial dysfunction in the human coronary circulation. Hypertension 51, 31–32.
Schrage, W. G., Eisenach, J. H., and Joyner, M. J. (2007). Ageing reduces nitric-oxide- and prostaglandin-mediated vasodilatation in exercising humans. J. Physiol. (Lond.) 579, 227–236.
Schulze, F., Maas, R., Freese, R., Schwedhelm, E., Silberhorn, E., and Boger, R. H. (2005). Determination of a reference value for N(G), N(G)-dimethyl-L-arginine in 500 subjects. Eur. J. Clin. Invest. 35, 622–626.
Seo, B., Oemar, B. S., Siebenmann, R., Von Segesser, L., and Luscher, T. F. (1994). Both ETA and ETB receptors mediate contraction to endothelin-1 in human blood vessels. Circulation 89, 1203–1208.
Shi, Y., Man, R. Y., and Vanhoutte, P. M. (2008). Two isoforms of cyclooxygenase contribute to augmented endothelium-dependent contractions in femoral arteries of 1-year-old rats. Acta Pharmacol. Sin. 29, 185–192.
Simons, L. A., Von Konigsmark, M., Simons, J., Stocker, R., and Celermajer, D. S. (1999). Vitamin E ingestion does not improve arterial endothelial dysfunction in older adults. Atherosclerosis 143, 193–199.
Soucy, K. G., Ryoo, S., Benjo, A., Lim, H. K., Gupta, G., Sohi, J. S., Elser, J., Aon, M. A., Nyhan, D., Shoukas, A. A., and Berkowitz, D. E. (2006). Impaired shear stress-induced nitric oxide production through decreased NOS phosphorylation contributes to age-related vascular stiffness. J. Appl. Physiol. 101, 1751–1759.
Stegbauer, J., and Coffman, T. M. (2011). New insights into angiotensin receptor actions: from blood pressure to aging. Curr. Opin. Nephrol. Hypertens. 20, 84–88.
Stewart, K. G., Zhang, Y., and Davidge, S. T. (2000). Aging increases PGHS-2-dependent vasoconstriction in rat mesenteric arteries. Hypertension 35, 1242–1247.
Sun, D., Huang, A., Yan, E. H., Wu, Z., Yan, C., Kaminski, P. M., Oury, T. D., Wolin, M. S., and Kaley, G. (2004). Reduced release of nitric oxide to shear stress in mesenteric arteries of aged rats. Am. J. Physiol. Heart Circ. Physiol. 286, H2249–H2256.
Taddei, S., Virdis, A., Mattei, P., Ghiadoni, L., Fasolo, C. B., Sudano, I., and Salvetti, A. (1997). Hypertension causes premature aging of endothelial function in humans. Hypertension 29, 736–743.
Takamiya, M., Okigaki, M., Jin, D., Takai, S., Nozawa, Y., Adachi, Y., Urao, N., Tateishi, K., Nomura, T., Zen, K., Ashihara, E., Miyazaki, M., Tatsumi, T., Takahashi, T., and Matsubara, H. (2006). Granulocyte colony-stimulating factor-mobilized circulating c-Kit+/Flk-1+ progenitor cells regenerate endothelium and inhibit neointimal hyperplasia after vascular injury. Arterioscler. Thromb. Vasc. Biol. 26, 751–757.
Tang, E. H., and Vanhoutte, P. M. (2008). Gene expression changes of prostanoid synthases in endothelial cells and prostanoid receptors in vascular smooth muscle cells caused by aging and hypertension. Physiol. Genomics 32, 409–418.
Thijssen, D. H., Rongen, G. A., Van Dijk, A., Smits, P., and Hopman, M. T. (2007). Enhanced endothelin-1-mediated leg vascular tone in healthy older subjects. J. Appl. Physiol. 103, 852–857.
Toda, N. (2012). Age-related changes in endothelial function and blood flow regulation. Pharmacol. Ther. 133, 159–176.
Touyz, R. M. (2003). Reactive oxygen species in vascular biology: role in arterial hypertension. Expert Rev. Cardiovasc. Ther. 1, 91–106.
Tsukahara, H., Ende, H., Magazine, H. I., Bahou, W. F., and Goligorsky, M. S. (1994). Molecular and functional characterization of the non-isopeptide-selective ETB receptor in endothelial cells. Receptor coupling to nitric oxide synthase. J. Biol. Chem. 269, 21778–21785.
Ungvari, Z., Buffenstein, R., Austad, S. N., Podlutsky, A., Kaley, G., and Csiszar, A. (2008). Oxidative stress in vascular senescence: lessons from successfully aging species. Front. Biosci. 13, 5056–5070.
Ungvari, Z., Kaley, G., De Cabo, R., Sonntag, W. E., and Csiszar, A. (2010a). Mechanisms of vascular aging: new perspectives. J. Gerontol. A Biol. Sci. Med. Sci. 65, 1028–1041.
Ungvari, Z., Sonntag, W. E., and Csiszar, A. (2010b). Mitochondria and aging in the vascular system. J. Mol. Med. 88, 1021–1027.
Upmacis, R. K., Deeb, R. S., and Hajjar, D. P. (2006). Oxidative alterations of cyclooxygenase during atherogenesis. Prostaglandins Other Lipid Mediat. 80, 1–14.
Urakami-Harasawa, L., Shimokawa, H., Nakashima, M., Egashira, K., and Takeshita, A. (1997). Importance of endothelium-derived hyperpolarizing factor in human arteries. J. Clin. Invest. 100, 2793–2799.
Valko, M., Leibfritz, D., Moncol, J., Cronin, M. T., Mazur, M., and Telser, J. (2007). Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44–84.
van Bussel, B. C., Schouten, F., Henry, R. M., Schalkwijk, C. G., De Boer, M. R., Ferreira, I., Smulders, Y. M., Twisk, J. W., and Stehouwer, C. D. (2011). Endothelial dysfunction and low-grade inflammation are associated with greater arterial stiffness over a 6-year period. Hypertension 58, 588–595.
van der Loo, B., Labugger, R., Skepper, J. N., Bachschmid, M., Kilo, J., Powell, J. M., Palacios-Callender, M., Erusalimsky, J. D., Quaschning, T., Malinski, T., Gygi, D., Ullrich, V., and Luscher, T. F. (2000). Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 192, 1731–1744.
Van Guilder, G. P., Westby, C. M., Greiner, J. J., Stauffer, B. L., and Desouza, C. A. (2007). Endothelin-1 vasoconstrictor tone increases with age in healthy men but can be reduced by regular aerobic exercise. Hypertension 50, 403–409.
Vanhoutte, P. M., Feletou, M., and Taddei, S. (2005). Endothelium-dependent contractions in hypertension. Br. J. Pharmacol. 144, 449–458.
Vasa, M., Breitschopf, K., Zeiher, A. M., and Dimmeler, S. (2000). Nitric oxide activates telomerase and delays endothelial cell senescence. Circ. Res. 87, 540–542.
Vasquez-Vivar, J., Kalyanaraman, B., and Martasek, P. (2003). The role of tetrahydrobiopterin in superoxide generation from eNOS: enzymology and physiological implications. Free Radic. Res. 37, 121–127.
Virdis, A., Ghiadoni, L., Giannarelli, C., and Taddei, S. (2010). Endothelial dysfunction and vascular disease in later life. Maturitas 67, 20–24.
Volpato, S., Guralnik, J. M., Ferrucci, L., Balfour, J., Chaves, P., Fried, L. P., and Harris, T. B. (2001). Cardiovascular disease, interleukin-6, and risk of mortality in older women: the women’s health and aging study. Circulation 103, 947–953.
Walker, A. E., Seibert, S. M., Donato, A. J., Pierce, G. L., and Seals, D. R. (2010). Vascular endothelial function is related to white blood cell count and myeloperoxidase among healthy middle-aged and older adults. Hypertension 55, 363–369.
Wang, C. Y., Kim, H. H., Hiroi, Y., Sawada, N., Salomone, S., Benjamin, L. E., Walsh, K., Moskowitz, M. A., and Liao, J. K. (2009). Obesity increases vascular senescence and susceptibility to ischemic injury through chronic activation of Akt and mTOR. Sci. Signal 2, ra11.
Wang, M., Takagi, G., Asai, K., Resuello, R. G., Natividad, F. F., Vatner, D. E., Vatner, S. F., and Lakatta, E. G. (2003). Aging increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension 41, 1308–1316.
Wang, M., Zhang, J., Jiang, L. Q., Spinetti, G., Pintus, G., Monticone, R., Kolodgie, F. D., Virmani, R., and Lakatta, E. G. (2007). Proinflammatory profile within the grossly normal aged human aortic wall. Hypertension 50, 219–227.
Watson, N. L., Sutton-Tyrrell, K., Youk, A. O., Boudreau, R. M., Mackey, R. H., Simonsick, E. M., Rosano, C., Hardy, S. E., Windham, B. G., Harris, T. B., Najjar, S. S., Lakatta, E. G., Atkinson, H. H., Johnson, K. C., Bauer, D. C., and Newman, A. B. (2011). Arterial stiffness and gait speed in older adults with and without peripheral arterial disease. Am. J. Hypertens. 24, 90–95.
Weiss, E. P., and Fontana, L. (2011). Caloric restriction: powerful protection for the aging heart and vasculature. Am. J. Physiol. Heart Circ. Physiol. 301, H1205–H1219.
Williamson, K., Stringer, S. E., and Alexander, M. Y. (2012). Endothelial progenitor cells enter the aging arena. Front. Physiol. 3:30.
Wong, S. L., Leung, F. P., Lau, C. W., Au, C. L., Yung, L. M., Yao, X., Chen, Z. Y., Vanhoutte, P. M., Gollasch, M., and Huang, Y. (2009). Cyclooxygenase-2-derived prostaglandin F2alpha mediates endothelium-dependent contractions in the aortae of hamsters with increased impact during aging. Circ. Res. 104, 228–235.
Wray, D. W., Nishiyama, S. K., Harris, R. A., Zhao, J., Mcdaniel, J., Fjeldstad, A. S., Witman, M. A., Ives, S. J., Barrett-O’keefe, Z., and Richardson, R. S. (2012). Acute reversal of endothelial dysfunction in the elderly after antioxidant consumption. Hypertension 59, 818–824.
Yamagishi, S., and Matsui, T. (2011). Nitric oxide, a janus-faced therapeutic target for diabetic microangiopathy-friend or foe? Pharmacol. Res. 64, 187–194.
Yanagisawa, M., Kurihara, H., Kimura, S., Tomobe, Y., Kobayashi, M., Mitsui, Y., Yazaki, Y., Goto, K., and Masaki, T. (1988). A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 332, 411–415.
Yang, Y. M., Huang, A., Kaley, G., and Sun, D. (2009a). eNOS uncoupling and endothelial dysfunction in aged vessels. Am. J. Physiol. Heart Circ. Physiol. 297, H1829–H1836.
Yang, Z., Huang, X., Jiang, H., Zhang, Y., Liu, H., Qin, C., Eisner, G. M., Jose, P. A., Rudolph, L., and Ju, Z. (2009b). Short telomeres and prognosis of hypertension in a Chinese population. Hypertension 53, 639–645.
Yu, B. P., and Chung, H. Y. (2006). Adaptive mechanisms to oxidative stress during aging. Mech. Ageing Dev. 127, 436–443.
Yuan, Q., Peng, J., Liu, S. Y., Wang, C. J., Xiang, D. X., Xiong, X. M., Hu, C. P., and Li, Y. J. (2010). Inhibitory effect of resveratrol derivative BTM-0512 on high glucose-induced cell senescence involves dimethylaminohydrolase/asymmetric dimethylarginine pathway. Clin. Exp. Pharmacol. Physiol. 37, 630–635.
Keywords: endothelial dysfunction, aging, cardiovascular disease, nitric oxide, oxidative stress, inflammation, senescence
Citation: El Assar M, Angulo J, Vallejo S, Peiró C, Sánchez-Ferrer CF and Rodríguez-Mañas L (2012) Mechanisms involved in the aging-induced vascular dysfunction. Front. Physio. 3:132. doi: 10.3389/fphys.2012.00132
Received: 01 February 2012; Accepted: 20 April 2012;
Published online: 28 May 2012.
Edited by:
Elisabet Vila, Autonomous University of Barcelona, SpainReviewed by:
Anthony Donato, University of Utah, USAJunichi Sadoshima, University of Medicine and Dentistry of New, USA
Maria Alvarez De Sotomayor, Universidad de Sevilla, Spain
Anna Csiszar, University of Oklahoma, USA
Copyright: © 2012 El Assar, Angulo, Vallejo, Peiró, Sánchez-Ferrer and Rodríguez-Mañas. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Leocadio Rodríguez-Mañas, Jefe de Servicio de Geriatría del Hospital Universitario de Getafe, Ctra de Toledo, Km 12,500, 28905, GETAFE, Madrid, España. e-mail:bHJvZHJpZ3Vlei5odWdmQHNhbHVkLm1hZHJpZC5vcmc=