 Elisa Passini1*
Elisa Passini1* Oliver J. Britton1
Oliver J. Britton1 Hua Rong Lu2Jutta Rohrbacher2An N. Hermans2David J. Gallacher2Robert J. H. Greig3
Hua Rong Lu2Jutta Rohrbacher2An N. Hermans2David J. Gallacher2Robert J. H. Greig3 Alfonso Bueno-Orovio1
Alfonso Bueno-Orovio1 Blanca Rodriguez1
Blanca Rodriguez1- 1Computational Cardiovascular Science Group, Department of Computer Science, University of Oxford, Oxford, United Kingdom
- 2Global Safety, Pharmacology, Discovery Sciences, Janssen Research and Development, Janssen Pharmaceutica NV, Beerse, Belgium
- 3Oxford Computer Consultants Ltd., Oxford, United Kingdom
Early prediction of cardiotoxicity is critical for drug development. Current animal models raise ethical and translational questions, and have limited accuracy in clinical risk prediction. Human-based computer models constitute a fast, cheap and potentially effective alternative to experimental assays, also facilitating translation to human. Key challenges include consideration of inter-cellular variability in drug responses and integration of computational and experimental methods in safety pharmacology. Our aim is to evaluate the ability of in silico drug trials in populations of human action potential (AP) models to predict clinical risk of drug-induced arrhythmias based on ion channel information, and to compare simulation results against experimental assays commonly used for drug testing. A control population of 1,213 human ventricular AP models in agreement with experimental recordings was constructed. In silico drug trials were performed for 62 reference compounds at multiple concentrations, using pore-block drug models (IC50/Hill coefficient). Drug-induced changes in AP biomarkers were quantified, together with occurrence of repolarization/depolarization abnormalities. Simulation results were used to predict clinical risk based on reports of Torsade de Pointes arrhythmias, and further evaluated in a subset of compounds through comparison with electrocardiograms from rabbit wedge preparations and Ca2+-transient recordings in human induced pluripotent stem cell-derived cardiomyocytes (hiPS-CMs). Drug-induced changes in silico vary in magnitude depending on the specific ionic profile of each model in the population, thus allowing to identify cell sub-populations at higher risk of developing abnormal AP phenotypes. Models with low repolarization reserve (increased Ca2+/late Na+ currents and Na+/Ca2+-exchanger, reduced Na+/K+-pump) are highly vulnerable to drug-induced repolarization abnormalities, while those with reduced inward current density (fast/late Na+ and Ca2+ currents) exhibit high susceptibility to depolarization abnormalities. Repolarization abnormalities in silico predict clinical risk for all compounds with 89% accuracy. Drug-induced changes in biomarkers are in overall agreement across different assays: in silico AP duration changes reflect the ones observed in rabbit QT interval and hiPS-CMs Ca2+-transient, and simulated upstroke velocity captures variations in rabbit QRS complex. Our results demonstrate that human in silico drug trials constitute a powerful methodology for prediction of clinical pro-arrhythmic cardiotoxicity, ready for integration in the existing drug safety assessment pipelines.
Introduction
Cardiotoxicity is one of the main causes of withdrawal during drug development, and identifying at early stages drugs that may cause adverse effects in specific human sub-populations is still a major challenge (Stevens and Baker, 2009; Laverty et al., 2011). Adverse effects can potentially lead to lethal arrhythmias, and are therefore a major cause of concern.
Before clinical testing, drugs undergo a thorough pipeline of preclinical testing, including identification of drug effects on cardiac ion channels (and particularly hERG), as well as in a variety of animal experiments (Leishman et al., 2012; Vargas et al., 2015). Animal models are good for predicting QT interval prolongation (Vargas et al., 2015), and some of them, including rabbit wedge preparations, rabbit isolated hearts and the in vivo atrioventricular block dog, have shown sensitive predictions of drug-induced Torsade de Pointes (TdP) (Valentin et al., 2004; Liu et al., 2006; Sugiyama, 2008). However, most of these studies only consider a small set of drugs. In fact, independent assessment against a larger number of compound (in the order of magnitude of those tested in silico in this contribution) highlight a prediction accuracy of 75% (Lawrence et al., 2008).
More recently, in silico and in vitro tests are considered as potentially important human-based tools for safety pharmacology evaluation, through the use of computational multiscale human modeling and human stem cell-derived cardiomyocytes (Bass et al., 2015; Rodriguez et al., 2016). Their profile has also been raised by the Comprehensive in vitro Proarrhythmia Assay (CiPA) initiative promoted by the pharmaceutical industries, the United States Food and Drug Administration (FDA), the Health and Environmental Sciences Institute and the Cardiac Safety Research Consortium (Sager et al., 2014; Colatsky et al., 2016).
The widespread translation of in silico modeling from academia to industrial and regulatory settings requires increasing the credibility of the models, understanding of their predictive power through comparison with existing experimental methods, and facilitating their uptake through the provision of software that can reduce the technical barriers of in silico methods for non-specialist users.
The aim of this study is to evaluate the ability of in silico drug trials using human ventricular model populations to predict the risk of drug-induced adverse cardiac events, based on ion channel information, and to identify ionic profiles underlying a higher risk of repolarization abnormalities. In silico drug trials were run for a large set of reference compounds with cardiac effects, and simulation results were analyzed to extract several biomarkers of drug pro-arrhythmic cardiotoxicity and compared against clinical reports of TdP arrhythmias. Because in silico drug trials are likely to be embedded in existing safety pharmacology pipelines and thus combined with experimental methodologies, it is important to evaluate their consistency with experimental recordings. Therefore, the outputs of the in silico drug trials for a sub-set of 15 reference compounds with varied modes of action were compared against the well-established electrocardiogram (ECG) recordings from isolated rabbit wedge preparations Lu et al. (2016) as well as the more recently considered technique of Ca2+-transient (CT) recordings from human induced pluripotent stem cell-derived cardiomyocytes (hiPS-CMs) (Lu et al., 2015; Zeng et al., 2016), even if with still controversial advantages (Abi-Gerges et al., 2017).
Materials and Methods
Control Population of Human Ventricular Action Potential (AP) Models
All the in silico drug trials presented in this study were performed in a population of 1,213 human ventricular control models, built using the O'Hara-Rudy dynamic (ORd) model (O'Hara et al., 2011) as baseline and the methodology described by Britton et al. (2013) and further discussed by Muszkiewicz et al. (2016). The ORd human ventricular AP model was chosen for this study because of: (i) the large number of human ventricular experimental data obtained from more than 140 hearts used in its construction and evaluation; (ii) its ability to reproduce and probe pro-arrhythmic mechanisms, including repolarization abnormalities and APD alternans, as shown in multiple studies and reviewed by Britton et al. (2017); (iii) its choice within the CiPA initiative (Sager et al., 2014; Colatsky et al., 2016).
Ionic conductances were sampled in the [0–200]% range of the baseline model values, to include both healthy and potentially abnormal ionic current profiles (with low/high ion channel densities corresponding to loss/gain-of-function of specific ionic channels due to e.g., genetic mutations), but still with a healthy-looking AP. These are important as they have been implicated in increased pro-arrhythmic risk (Sanguinetti and Tristani-Firouzi, 2006; Itoh et al., 2016; Wang et al., 2016). Nine ionic conductances were considered: fast and late Na+ current (GNa and GNaL respectively), transient outward K+ current (Gto), rapid and slow delayed rectifier K+ current (GKr and GKs), inward rectified K+ current (GK1), Na+-Ca2+ exchanger (GNCX), Na+-K+ pump (GNaK), and the L-type Ca2+ current (GCaL).
Only the AP models exhibiting a phenotype in agreement with human experimental data from undiseased hearts (Britton et al., 2017) were selected for the control population, which consists of 1,213 models. A more detailed description of the control population used in this study is included in the Supplementary Material, together with the experimental AP biomarker ranges used for the calibration process (Table S1) and the scaling factors of the ionic conductances for the 1,213 models (Table S2).
In silico Drug Assay Design
A total of 62 reference compounds were considered in this study. The list includes antiarrhythmic drugs in Classes I, III, and IV, as well as other drugs used for different purposes but with known effects on cardiac ion channels. Most of these drugs have a multichannel action, which makes prediction and interpretation of cardiotoxicity challenging. Drugs were selected to include all the ones in Kramer et al. (2013), as well as 15 compounds widely used as reference compounds, which were characterized in more depth both in simulations and experiments and are listed in Table 1.
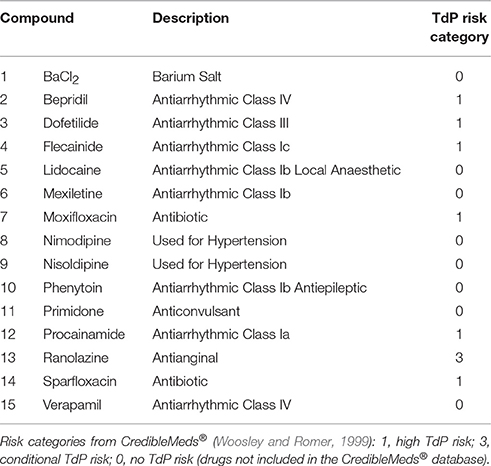
Table 1. List of the 15 compounds considered for in silico drug assays comparison against in vitro hiPS-CMs and ex vivo rabbit wedge preparations, including a short description and the clinical TdP Risk category based on CredibleMeds® (Woosley and Romer, 1999).
Each drug was assigned to a TdP risk category, based on the classification by CredibleMeds® (Woosley and Romer, 1999), available on www.crediblemeds.org (as of July 2017): 1 (high risk), the drug prolongs the QT interval and is clearly associated with a known TdP risk, even when taken as recommended; 2 (possible risk), the drug prolongs the QT interval, but there is a lack of evidence of TdP risk when taken as recommended; 3 (conditional risk), the drug is associated with TdP but only under certain circumstances, e.g., excessive dose or interaction with other drugs; NC (not classified), the drug was reviewed by CredibleMeds® but the evidence available was not enough to assign it to any of the previous categories, and therefore no action was taken. Of the 62 compounds, 24 are classified as high risk and 13 as potential/conditional risk, for a total of 37 drugs associated with TdP risk. Verapamil (classified as NC) and the remaining 24 compounds (not listed) are considered as TdP category 0 (no TdP risk) for the purpose of this study.
Drug effects were simulated using a simple pore-block model consistent with data available for drug/ion channel interactions, consisting of IC50 and Hill coefficient (h) for each drug/ion channel. Up to 7 ion channels were considered for this study: fast Na+ current (INa), rapid/slow delayed rectified K+ current (IKr/IKs), transient outward K+ current (Ito), L-type Ca2+ current (ICaL), inward rectifier K+ current (IK1), and late Na+ current (INaL). The experimental IC50 and h used for the drug assays were collected mainly from three different sources: (i) our internal database, measured with either manual or automated patch-clamp techniques (when the IC50 concentration was not reached in the experiments, an estimate was computed from the percentage of block at the maximum tested concentration, with h equal to 1); (ii) (Kramer et al., 2013), data acquired with automated patch-clamp; (iii) (Crumb et al., 2016), data acquired with manual patch-clamp.
For the compounds that were included in more than one of these datasets, multiple inhibitory profiles were considered to investigate the impact of variability in drug characterization. Each IC50 and h set was simulated separately, resulting in 87 different drug trials: each trial is referred to with the name of the compound together with a roman numeral, to differentiate multiple entries (e.g., Bepridil I, Bepridil II, Bepridil III).
Multiple concentrations were investigated for each compound, chosen to match those used in the experimental drug assays, as well as to explore different multiples of the maximal effective free therapeutic concentration (EFTPCmax), up to 100-fold. The EFTPCmax values were taken from literature, mainly from Kramer et al. (2013) or Crumb et al. (2016). When multiple values were found for the same compound, the higher one was considered for simulations.
The full list of compounds, together with the IC50, Hill coefficient and the EFTPCmax used for in silico drug trials is provided in Table S3.
Simulations and Simulated Data Analysis
All the simulations presented in this study were conducted using Virtual Assay (v.1.3.640 2014 Oxford University Innovation Ltd. Oxford, UK), a user-friendly C++ based software package with a graphical user interface for in silico drug assays, to facilitate its use by non-experts in computational modeling, and available upon request. Virtual Assay uses the ordinary differential equation solver CVODE, part of the open-source Sundials suite (Hindmarsh et al., 2005; Serban and Hindmarsh, 2005), implementing time adaptive Backward Differentiation Formulas with relative and absolute tolerance equal to 1e-5 and 1e-7, respectively. Our results could therefore be replicated using other software products, as Matlab (Mathworks Inc. Natwick, MA, USA) or Chaste (Pitt-Francis et al., 2008). As an example, a comparison of simulations obtained with Virtual Assay and with the Matlab solver ode15s (Shampine and Reichelt, 1997) is shown in the Supplemental Material, Figure S2. Simulation results were analyzed both in Virtual Assay and in Matlab.
Following drug application, all models were stimulated at 1 Hz for 150 beats, and the last AP trace in each simulation was analyzed. AP biomarkers were extracted, including: AP duration at 40, 50, and 90% of repolarization (APD40, APD50, APD90); APD90 dispersion, defined as the difference between the maximum and minimum value of APD90 in the population (ΔAPD90), AP triangulation, defined as the difference between APD90 and APD40 (Tri90−40); maximum upstroke velocity (dV/dtMAX); peak voltage (Vpeak); resting membrane potential (RMP), computed as in Britton et al. (2017). Drug-induced changes in those biomarkers are presented as percentage change in median with drug, compared to control (no drug).
All AP traces were automatically checked for repolarization and depolarization abnormalities (RA and DA, respectively). RA were defined as the presence of a positive derivative of the membrane potential (Vm) 150 ms after the AP peak (representative of early after-depolarizations, EADs), or when the membrane potential did not reach the resting condition following an AP upstroke (Vm > −40 mV) by the end of the beat. DA were defined as AP traces in which the upstroke phase was compromised, i.e., when the max upstroke Vm was lower than 0 mV, or when the time needed to reach 0 mV was longer than 100 ms.
Drugs were classified as risky when RA occurred in the population of models at different concentrations, based on: true positives (drug with reports of TdP risk classified as risky); true negatives (drug with no reports of TdP risk classified as safe), false positives (drug with no reports of TdP risk, classified as risky); false negatives (drugs with reports of TdP risk, classified as safe). The performances of the classification were evaluated based on: sensitivity, defined as the number of true positives divided by the sum of true positives and false negatives; specificity, defined the number of true negatives divided by the sum of the true negatives and false positives; accuracy, defined as the sum of true positives and true negatives divided by the total number of drugs. Classification results based on RA were compared against the ones obtained for APD prolongation at 10x EFTPCmax. APD90 prolongation threshold to define risk was fixed to 6%, considering the correspondence between QTc and APD90, and the current guidelines suggesting QTc prolongation >20 ms (which correspond to 5.7% for a normal QT of 350 ms) as a definite risk factor for TdP (Salvi et al., 2010). Results for the population of models were also compared against the same results obtained with the single ORd model.
A scoring system was developed by integrating RA occurrence at multiple concentrations. The fraction of models developing RA was multiplied by a factor inversely related to the drug concentration at which those RA occur (e.g., 1/100 for RA occurring at 100x EFTPCmax). Contributions from all the different concentrations were added together, and the total score was normalized, according to the following formula (where nRAi is the number of models showing RA at the tested concentration i, wi = EFTPCmax / i is the weight inversely related to the tested concentration i, and nmod is the total number of models in the population).
The TdP score thus obtained varies between 0 and 1, where 0 corresponds to a drug with no RA, and 1 to a drug which develops 100% of RA at every concentration. By using the proposed score, RA are considered more severe when occurring at low concentrations and/or affecting a high fraction of the population of models.
Experimental Drug Assays
In silico results were compared with properties computed from ECGs in rabbit wedge preparations and CT recordings from hiPS-CMs. Experimental data were acquired for the 15 compounds listed in Table 1 at multiple concentrations, as described below.
Recordings of ECGs from left ventricular rabbit wedge preparations have been previously described and partly published in Lu et al. (2016). The biomarkers extracted include QRS complex and QT interval duration, defined as the time from the onset of the QRS complex to the point at which the final downslope of the T wave crossed the isoelectric line.
CT recordings from hiPS-CMs (Cor 4U) were acquired as part of this study on pre-plated preparations from Axiogenesis (Cologne, Germany). Full method details are included in the Supplementary Material. Quantified biomarkers included CT beat rate (CTBR) and duration at 90% of the initial base value (CTD90), known to be correlated with APD (Gauthier et al., 2012; Spencer et al., 2014), similarly to other studies (Lu et al., 2015; Zeng et al., 2016).
All experimental results are presented as median percentage changes with respect to the baseline. Drug-induced changes in experimental values need to be compared against the effect measured without drugs, i.e., with vehicle, defining cut-off values. In the rabbit wedge, the changes measured with the vehicle were always quite small: <5% for QT and <3% for QRS. On the other hand, in CT assays using hiPS-CMs, the lower and upper limit were 19% and 24% of the baseline, with 95% confidence interval (n = 222 vehicle controls). Therefore, only CTD90 prolongations >25% were considered relevant for this assay. In silico, no biomarker changes are observed when the drug effect is not included, since the models are paced until steady state: therefore, the cut-off value is equal to 0%. Statistical analysis was performed with the Wilcoxon-Mann-Whitney Test by using R Project for Statistical Computing, and p < 0.05 was considered as significant. Very small p-values (e.g., p < 1e-6) were obtained in all simulation results due to the high number of models considered, and therefore we focus on the magnitude of the differences observed (White et al., 2014).
Results
In silico Drug Assays: Drug-Induced Changes in Action Potential (AP) Biomarkers
In silico drug trials for a total of 62 reference compounds were performed in the control population of 1,213 human ventricular AP control models, based on the ORd model (O'Hara et al., 2011) and constructed as described in Methods. Before drug application, all the models exhibit a healthy-looking AP phenotype, in agreement with human experimental recordings from un-diseased hearts (Figure S1A). When including drug effect for concentration up to 100-fold the EFTPCmax, each model responds in a different way, depending on its underlying ionic properties. We first evaluated drug-induced changes in the AP biomarkers.
Figure 1 shows APD90 and dV/dtMAX distributions from the in silico population for 5 compounds (Dofetilide I, Flecainide I, Nimodipine, Ranolazine I, and Verapamil II) at multiple concentrations. Extended results for additional compounds, including all AP biomarkers, are shown in Figures S3–S31.
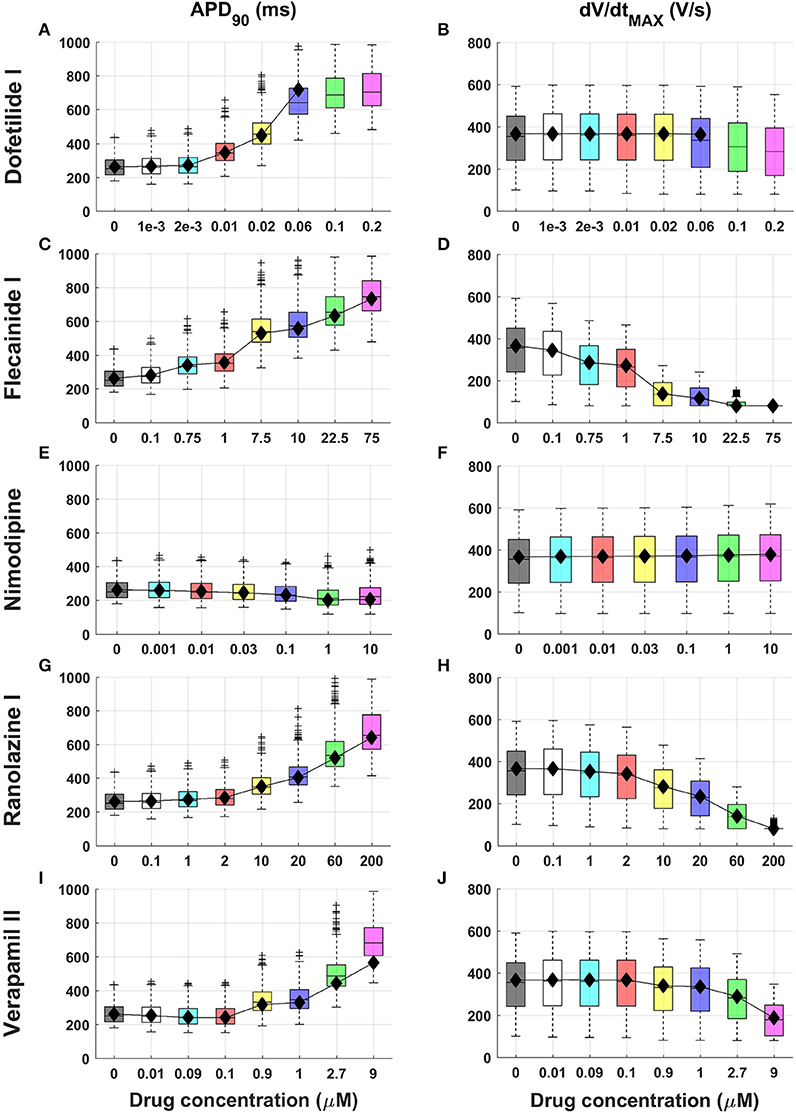
Figure 1. Explanatory examples of in silico drug trial results in a population of human computational models, showing drug-induced changes on APD90 and dV/dtMAX (left and right column, respectively) for 5 compounds (Dofetilide I, Flecainide I, Nimodipine, Ranolazine I and Verapamil II). Results are presented as boxplots of AP biomarkers for the population of human ventricular models at increasing concentrations (A–J). Results for the single ORd model are shown as black diamonds. On each box, the central mark is the median of the population, box limits are the 25 and 75th percentiles, and whiskers extend to the most extreme data points not considered outliers, plotted individually as separate crosses. Extended results for the selected 15 reference compounds, including all the AP biomarkers, are available in the Supplementary Material, Figures S3–S31.
Most drugs (all except Nimodipine and Nisoldipine) result in APD prolongation at 40, 50, and 90% of repolarization, as well as increased AP triangulation (Tri90−40), mainly as a result of hERG channel block (e.g., Figure 1, left column and A–C in Figures S3–S31). For 30x EFTPCmax dose, Flecainide III, Bepridil I, and Dofetilide III showed the largest APD90 prolongations (+180%, +175%, and +157%, respectively).
APD prolongation caused by BaCl2 (mainly due to IK1 block) is stronger in APD90 compared to APD40 and APD50 (Figures S3A–C). As a secondary effect of IK1 block, BaCl2 leads to a decrease in RMP (Figure S3H), which also exhibits larger variability, while for all the other compounds it remains almost constant (H in Figures S3–S31).
Consistent with their expected mode of action, class I anti-arrhythmic drugs (Procainamide, Lidocaine, Mexiletine, Phenytoin, Flecainide) as well as other drugs affecting Na+ channels as secondary effect (e.g., Bepridil) show a strong decrease of upstroke velocity (e.g., Figures 1D,H), together with a decrease of Vpeak (F, G in Figures S3–S31).
Verapamil II represents an interesting example of the combined block of IKr and ICaL (Figure 1I). For low concentrations (0.01–0.1 μM) the Ca2+ block is predominant, and APD90 is slightly decreased (−1 and −4% respectively), whereas IKr block compensates its effects for higher concentrations (>0.5 μM), resulting in a clear APD prolongation (e.g., +38% for 1 μM). Verapamil II also leads to slower AP upstroke for high concentrations (Figure 1J).
Results obtained using the baseline ORd model (Figure 1 and Figures S3–S31, black diamonds) are in overall agreement with the range of AP biomarkers in the population of human models. This is with the exception of cases in which the baseline ORd yields abnormal APs for high doses of certain drugs (e.g., Dofetilide I 0.1 and 0.2 μM, Figures 1A,B). In those cases, the human population of models still allows exploration of the full concentration range for each compound.
In silico Characterization of Drug-Induced Phenotypic Variability
Drug action resulted in an increase in the phenotypic variability yielded by the human ventricular population, as illustrated in Figure 2 for Moxifloxacin III (Figure 2A) and Dofetilide I (Figure 2B), mainly blocking IKr, and for Flecainide I (Figure 2C), inhibiting both IKr and INa. Following drug application, some models in the in silico population display normal but prolonged APs (gray traces) while a fraction develop repolarization abnormalities (RA, pink traces). Due to its strong effect on INa, Flecainide I (Figure 2C) also cause an overall reduction of dV/dtMAX, visible in the upstroke phase of the AP, and depolarization abnormalities (DA, green traces) in specific models.
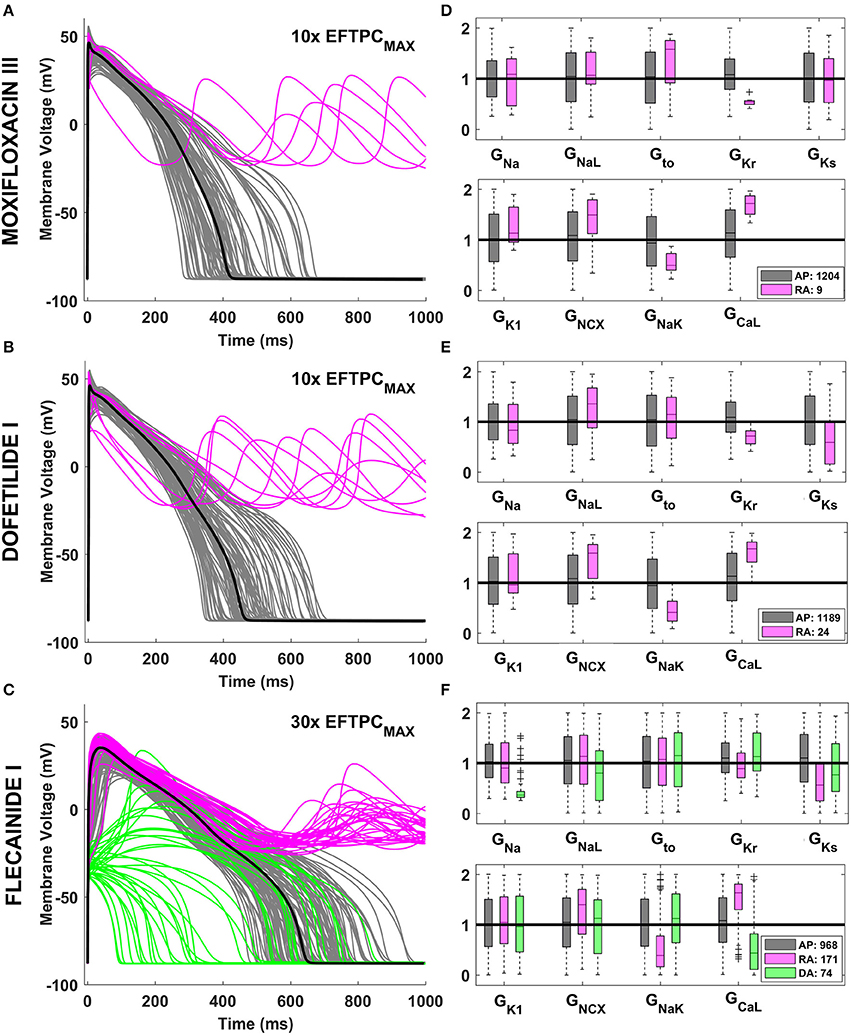
Figure 2. Explanatory examples of variability in drug response in the in silico population of human AP models, with the underlying ionic mechanisms. Representative AP traces of different drug-induced AP phenotypes are shown on the left side for Moxifloxacin III (A), Dofetilide I (B), and Flecainide I (C) at selected concentrations. Models with a normal AP are shown in gray, while models displaying RA and DA are shown in pink and green, respectively. In each panel, the baseline ORd model is highlighted in black. The distribution of ionic conductances for the different AP phenotypes is shown on the right side (D–F), by using boxplots of the corresponding scaling factors, and with the same color code. For each conductance, the values shown (between 0 and 2) represent the scaling factors of the models in the population compared to the baseline ORd model, which had all the scaling factors equal to 1. Boxplots description as in Figure 1.
Quantitative analysis of the underlying ionic mechanisms reveals consistency on the mechanisms underlying RA and DA across different drugs and concentrations. Models displaying RA are characterized by low GKr, and GNaK, and high GCaL and GNCX, i.e., a reduced repolarization reserve (Figures 2D–F, pink vs. gray boxplots). Low GKs also plays a role when a larger fraction of the population displays RA (Figures 2D,E). Models displaying DA are characterized mainly by low GNa/GCaL/GNaL, i.e. the net inward current in the initial phase of the AP is reduced (Figure 2F, green vs. gray boxplots).
Repolarization Abnormalities Occurrence Predicts TdP Risk
We hypothesized that occurrence of RA following drug application in the human population would be predictive of in vivo TdP, given the potential mechanistic link between them (El-sherif et al., 1990; Dutta et al., 2016). In silico drug trial predictions were evaluated against clinical reports of TdP using the TdP risk categories provided by CredibleMeds® (Woosley and Romer, 1999) and further described in Methods. When multiple inhibitory profiles were simulated for the same compound, the worse scenario was considered, i.e., the higher occurrence of RA, and the larger APD90 prolongation.
Figure 3 shows the classification results for the 49 compounds with either high or no TdP risk (Figure 3A), and for the full set of 62 compounds (Figure 3B) based on RA occurrence up to 100x EFTPCmax and APD90 prolongation >6% at 10x EFTPCmax for both the population of models (top row), and the single ORd model (bottom row). Accuracy reached 96% for the classification of high vs. no TdP risk compounds using the RA-based classification with the in silico population of models, compared to 80% based on APD prolongation (Figure 3A). Using the single ORd model, the higher accuracy was 76% and was obtained using APD prolongation as biomarker.
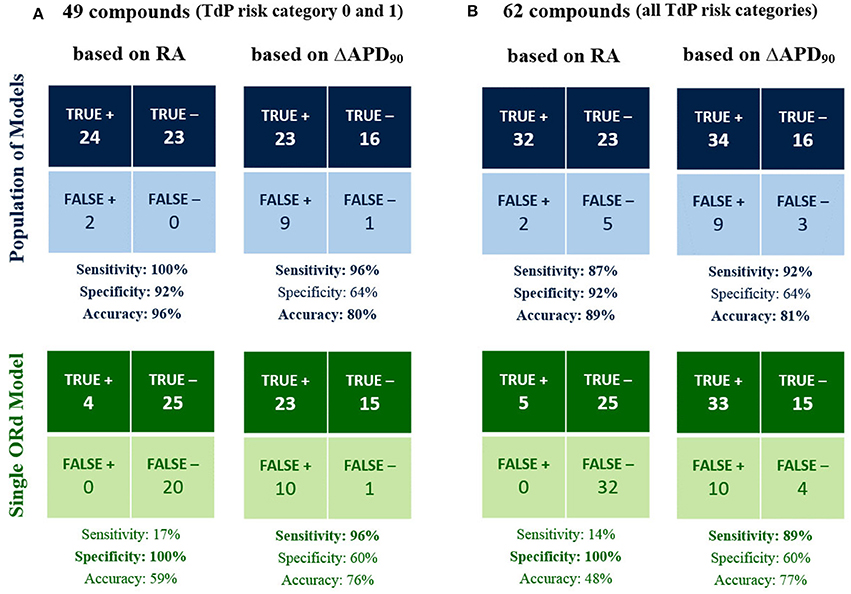
Figure 3. In silico prediction of in vivo TdP risk for the 49 compounds belonging to TdP risk category 0 and 1 (A) and for all the 62 tested compounds (B). In each panel, predictions based on the occurrence of RA in any of the model at 1x, 10x, 30x, and 100x EFTPCmax (1st column) are compared against predictions based on APD90 prolongation >6% at 10x EFTPCmax (2nd column). Results obtained using the population of models (top half) are compared against the ones for the baseline ORd model (bottom half). High sensitivity/specificity/accuracy (>80%) are highlighted in bold.
When including also compounds with possible/conditional risk (Figure 3B), accuracy with the RA-based classification for the in silico population was 89%, compared to 81% based on APD prolongation (48 and 77% based on RA and APD prolongation, respectively, for the single ORd model).
In summary, all drugs with high TdP risk (category 1) were correctly identified as risky with the RA-based classification in the in silico population, and only 5 drugs with possible/conditional TdP risk (category 2 and 3) resulted as false negatives: Clozapine, Dasatinib, Paroxetine, Saquinavir, Voriconazole. It is worth noting that drugs with possible/conditional TdP risk lack evidence of pro-arrhythmic risk when taken as recommended: TdP reports are usually related to excessive dose or interaction with other drugs.
Overall, classification based on APD prolongation exhibited high sensitivity, but low specificity. Indeed, many compounds prolong APD without being associated with TdP risk, e.g., Verapamil, thus resulting in false positives. On the contrary, false positives are rare in the RA-based classification (Lidocaine and Mexiletine), and they only develop RA at the maximum tested concentration (100x EFTPCmax). Indeed, both Lidocaine and Mexiletine are Class Ib anti-arrhythmic drugs, which have been associated with cardiotoxicity in case of overdose (Denaro and Benowitz, 1989).
RA-based classification is dependent on the maximum tested concentration: a higher concentration is more likely to provoke RA in the in silico population, thus increasing sensitivity but possibly decreasing specificity, since even safe drugs might lead to RA at very high doses. We reported here the classification results obtained for concentrations up to 100x EFTPCmax, and a comparison between results for 30x and 100x EFTPCmax is included in the Supplementary Material (Figure S32).
A New Scoring System to Evaluate In vivo Risk of Drug-Induced TdP
Figure 4 shows all the tested compounds classified using the TdP score computed from the in silico drug trials using the fraction of models displaying RA at each tested concentration, as described in Methods. The TdP score varies from 0 to 1, and is higher when RA occur at low concentrations and/or affecting a high fraction of the population of models.
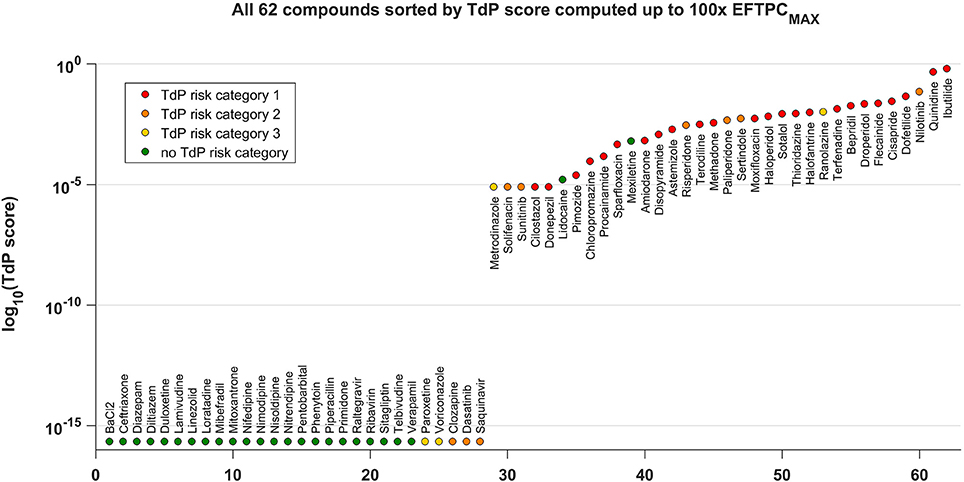
Figure 4. TdP score for all the 62 compounds, based on the occurrence of RA in the population of human AP models at 1x, 10x, 30x, and 100x EFTPCmax. The TdP score, which varies between 0 and 1, was computed by taking into account both the fraction of models displaying RA and the concentrations at which RA occur, as described in Methods. The logarithmic scale was considered to maximize the visual separation between safe and risky drugs, and log10(0) was approximated with the machine precision (10−16). All compounds with no report of TdP risk (in green) have 0 as TdP score (left side), except for Lidocaine and Mexiletine. All high risk compounds (in red) have a high TdP score (right side). Most of the compounds with possible or conditional TdP risk (in orange and yellow, respectively) have a TdP score >0, except for Paroxetine, Voriconazole, Clozapine, Dasatinib, and Saquinavir.
The distribution of compounds in the safe zone (TdP equal to 0, left side) and risky zone (TdP > 0, right side) reflects the classification results summarized in the confusion matrix with the higher (89%) accuracy (Figure 3). All safe compounds (no reported TdP risk, green dots) have a TdP score equal to 0, except Lidocaine and Mexiletine. All compounds with known risk of TdP (TdP risk category 1, red dots) have a positive score, and tend to be distributed toward the right end of the plot. Most of the compounds with possible or conditional TdP risk (TdP risk category 2 or 3, orange and yellow dots, respectively) have a positive score, except Clozapine, Dasatinib, Paroxetine, Saquinavir, Voriconazole. The TdP score is also dependent on the maximum considered concentrations. Again, higher concentrations lead to an increase in sensitivity while decreasing specificity. A comparison between the TdP scores computed up to 30x and 100x EFTPCmax is included in the Supplementary Material (Figure S33).
In silico Drug Assays are in Agreement with Rabbit Wedge and hiPs-CM Experimental Recordings
In silico drug trials are likely to be used as an additional tool for drug safety assessment in combination with experimental methods. It is therefore important to evaluate the consistency between in silico results and experimental data. Thus, simulation results for the 15 reference compounds with varied actions on ion channels (Table 1) were compared against recordings obtained using rabbit wedge and hiPS-CM preparations, as two techniques considered in safety pharmacology. In Figure 5, changes in QT interval duration in rabbit wedge, CTD90 in hiPS-CMs and in silico APD90 (from red to green, left side) were compared to evaluate drug-induced changes in repolarization. Changes in QRS complex duration in rabbit wedge and in silico dV/dtMAX were quantified to evaluate drug effects on depolarization (Figure 5, from purple to blue, right side). Negative variations in dV/dtMAX are considered as positive changes in depolarization time (opposite sign), to facilitate the comparison.
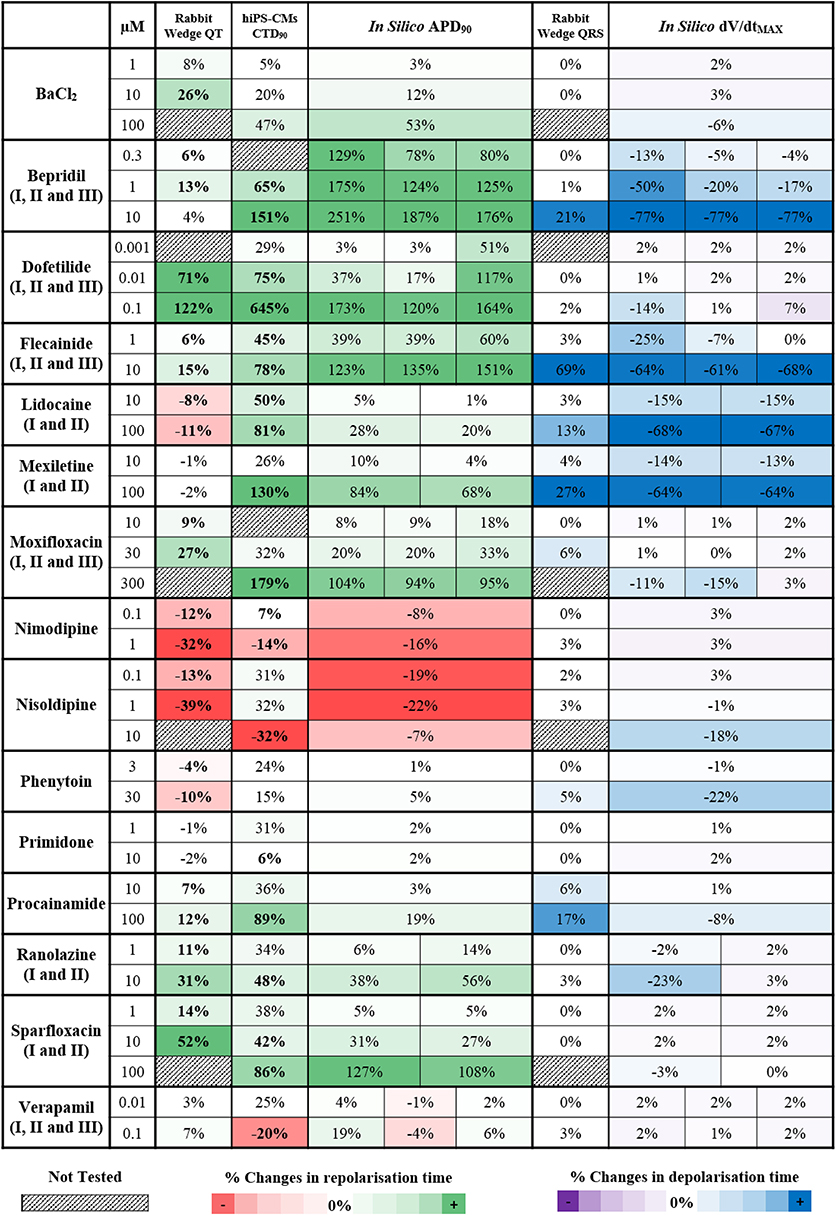
Figure 5. Qualitative and quantitative comparison of in silico drug trial results in the population of human ventricular AP models against ECG from rabbit wedge preparations and Ca2+ transient recordings from hiPS-CMs, for 15 reference compounds. On the left side (from red to green) are shown the drug-induced changes in the biomarkers related to the repolarization phase: QT interval in rabbit wedge, CTD90 in hiPS-CMs and APD90 in silico. On the right side (from purple to blue) are shown the drug-induced changes in the biomarkers related to the depolarization phase: QRS interval from rabbit wedge and dV/dtMAX in silico. For each assay, colors are scaled to span from the 15th to the 85th percentiles of the % changes observed in the biomarkers when considering drug effects, compared to no drug, and with respect to the cut-off values (3 and 5% for rabbit wedge QRS and QT, 25% for hiPS-CMs CTD90, and 0% for in silico APD90 and dV/dtMAX). To facilitate comparison, negative variations in dV/dtMAX were considered as positive changes in the depolarization time, and vice versa. When multiple combinations of IC50 and h were tested in simulation for the same compound, the corresponding in silico result sections consist of multiple sub-columns. Statistically significant changes in experiments have been highlighted in bold.
Drug-induced effects on biomarkers are in overall agreement for all three methodologies. Figure 5 presents consistent increase/decrease of QT, CTD90 and APD90, as well as consistency between positive changes in QRS and reduction of dV/dtMAX. Variations are generally larger in the in silico APD90 than in QT interval in rabbit wedge, indicating higher sensitivity of the in silico assay, and the wider range of ionic scenarios evaluated in the virtual population than in the limited number of experiments.
For Verapamil at 0.1 μM, small changes were observed in both QT interval in rabbit wedge and simulated APD, whereas CTD90 in hiPS-CMs was reduced. Such a reduction in CTD90 in the hiPS-CMs is also accompanied by a significant increase in beating rate (CTBR +61%), which does not occur in silico and in the rabbit wedge experiments as these are paced externally.
For Lidocaine and Mexiletine, their main effect is fast INa block, which results in a decreased dV/dtMAX in the in silico models, and a wider QRS complex in the rabbit wedge ECG. Furthermore, both in vitro CTD90 and in silico APD90 are prolonged, whereas QT interval decreases slightly (Lidocaine) or remains unchanged (Mexiletine) in rabbit wedge, suggesting that in vitro CT and in silico AP are more prone to display prolongation for non-selective Class I anti-arrhythmic drugs. It is worth noticing that the AP prolongation in silico is reduced when inhibition of the INaL current is taken into account in the simulations, corresponding to Lidocaine II and Mexiletine II (APD90 +84% vs. +68%, Mexiletine I vs. Mexiletine II, 100 μM). The tendency for prolongation under Na+ block of in silico AP and hiPS-CMs CT is confirmed also for another class I anti-arrhythmic drug (Phenytoin), which causes negligible changes in both CTD90 and in silico APD90, and a decrease in QT interval in rabbit wedge.
Bepridil constitutes a good example of multichannel block, intended to block ICaL, but also affecting IKr and INa. In the rabbit wedge, the QT interval is relatively prolonged at low concentrations (0.3–1 μM), compared to more selective Ca2+ blockers (e.g., Nimodipine and Nisoldipine), and it goes back to normal (+4%) at 10 μM. Both hiPS-CMs CT and in silico AP are prolonged in a dose-dependent manner, confirming once again the higher sensitive to IKr block of these techniques.
Interestingly, BaCl2 effects are of smaller magnitude in hiPS-CMs compared to rabbit wedge preparation and in silico models: relevant CTD90 prolongation was detected only at 100 μM (+47%), while up to 10 μM (more than 2-fold BaCl2 IC50 for IK1) the drug-induced effects on CT were negligible. This may be due to differences in IK1 expression between the cell types considered (Liang et al., 2013; Kim et al., 2015).
In silico results for compounds with multiple sets of IC50 and h are in overall agreement with each other, even if the magnitude of drug-induced changes may vary. As an example, the decrease in dV/dtMAX for the three variations of Flecainide is almost the same at 10 μM (−64, −61, and −68%, respectively), while for lower concentrations the differences between the three inhibition profiles are more noticeable (e.g., −25, −7, and 0% at 1 μM, respectively).
Discussion
In silico human electrophysiology drug trials using a population of human AP models were conducted for 62 compounds with varied electrophysiological profiles to evaluate their ability to predict clinical pro-arrhythmic risk and their consistency with electrophysiological recordings currently considered in safety assessment.
The main findings of this simulation study are:
1. RA occurrence in populations of human models proves to be more predictive of clinical TdP risk than APD prolongation and standard biomarkers obtained with the single ORd model. Accuracy of 96% and specificity of 92% was obtained in the classification of high risk vs. safe drugs, compared to 80 and 64%, based on APD prolongation. 100% sensitivity was achieved considering RA in the population compared to 17% with the single ORd model.
2. Human virtual cardiomyocytes exhibiting RA in in silico drug trials displayed low repolarization reserve, caused by low IKr/IKs/INaK and high ICaL/INCX, which is consistent with the prevalence of cardiotoxicity in patients with disease conditions such as myocardial ischaemia and heart failure. Low depolarization reserve caused by weak INa/INaL/ICaL was associated with DA under INa block.
3. A TdP risk score was developed to translate the high accuracy of RA abnormalities for risky drug classification into a non-binary system considering both data for multiple concentrations and the frequency of RA. This metric is higher when RA occur in a large fraction of the population of models, and at lower concentrations, thus informing on the likelihood of drug-induced adverse cardiac events in the population.
4. Drug-induced APD changes in silico are consistent with the ones measured in QT interval from rabbit wedge ECGs and CTD recorded from hiPS-CMs. Drugs affecting the depolarization phase provoke a decrease of dV/dtMAX in silico and a widening of QRS interval rabbit wedge ECGs. To show a qualitative and quantitative agreement between in silico drug trial results and two experimental models commonly used in safety pharmacology, is fundamental to build confidence in the integration of computer models for cardiotoxicity assessment.
Our results support the potential of RA in the in silico human population as a good predictor of clinical TdP risk, with sensitivity, specificity and accuracy higher or comparable to the ones obtained through animal studies (Valentin et al., 2009). RA-based classification for all 62 compounds reached 89% of accuracy, compared to 75% obtained for 64 compounds in rabbit isolated Langendorff heart model (Lawrence et al., 2006; Valentin et al., 2009). The in vivo atrioventricular block dog model showed sensitive predictions of drug-induced TdP, but in a limited set of 13 compounds (Sugiyama, 2008). Animal studies accuracy is higher when predicting QT prolongation rather that in vivo TdP risk: 85 and 79% for 19 compounds based on QT prolongation in in vivo dog studies and hERG assays, respectively (Valentin et al., 2009; Wallis, 2010); 90% accuracy for 40 compounds based on non-rodent QT prolongation (Vargas et al., 2015). However, “it is generally known that the sensitivity and the specificity of the QT interval prolongation as a surrogate marker of TdP is rather poor: only in 46% of the cases the TQT study results were concordant with the TdP risk classification, and 89% of drugs prolonging QT interval in thorough QT studies were approved by the FDA” (Wiśniowska et al., in press). This is confirmed in our study by the fact that RA-based predictions in the population of human models have higher accuracy compared to the ones based on APD prolongation, due to a higher specificity (92 vs. 64% for 62 compounds).
Our methodology also offers mechanistic insights into sub-populations at higher risk, which is a key advantage with respect to previous in silico and in vitro studies (Kramer et al., 2013; Lancaster and Sobie, 2016). We identify human in silico cardiomyocytes with high propensity to develop RA as those with low INaK/IKs/IKr and high INCX/ICaL. The ionic profile is consistent with ionic remodeling in cardiac specific diseases such as heart failure (Carmeliet, 1999; Coppini et al., 2013; Coronel et al., 2013), suggesting disease modeling as crucial when investigating cardiotoxicity in response to drug action (Walmsley et al., 2013; Gomez et al., 2014; Elshrif et al., 2015; Dutta et al., 2016; Passini et al., 2016).
The range of concentrations considered for drug trials plays an important role in risk prediction. Expanding the concentration up to 100x EFTPCmax allows to account for possible overdose, but most importantly for inter-subject variability in protein binding and metabolism, which can lead to important different in blood concentrations in patients taking the same drug dose, or even hormones which might change the effect of the drug on ion channels (Shuba et al., 2001). As an example, Amiodarone is a very controversial drug, considered safe by most clinicians but at the same time known to be associated with TdP risk (Jurado Román et al., 2012), and indeed belonging to TdP risk category 1. Amiodarone is almost completely bound to plasma proteins: reported values in literature range from 95.6% (Lalloz et al., 1984; Latini et al., 1984) to 99.98% (Veronese et al., 1988). In addition, absorption following oral administration is erratic and unpredictable (Latini et al., 1984). This can lead to EFTPCmax variation of more than 100-fold, with a big impact on drug-induced ion channels block.
An additional consideration about in silico trials concerns variability of recorded IC50 values. We show one possible way to consider this variability, by evaluating its implications in the in silico human population. In most cases, results obtained with different IC50 values were in overall agreement, thus building confidence in the answer provided. Should these results disagree, leading to contrasting scenarios, new ion channel recordings and experiments might be required for further drug characterization and refined in silico predictions. This may incorporate for example more detailed models of ion channel structure, based on the most recent crystallographic studies on human ion channels (Sun and MacKinnon, 2017; Wang and MacKinnon, 2017). Other in silico tools are also available at the ion channel level, to evaluate potential drug effects on the hERG channel, using ligand-based (Durdagi et al., 2011; Braga et al., 2015; Chemi et al., 2017) or receptor-based (Brindisi et al., 2014; Dempsey et al., 2014) approaches.
Indeed, in silico results are strictly dependent on the quality and consistency of the data used as inputs, which include ion channel assays costing time and money. In silico trials are a cheap complement to experimental methods following ion channel screening, which for some channels is already routine (hERG).
When fully integrated in the early stages of drug development, in silico methods provide predictions to partly replace animal experiments, thus reducing the corresponding costs. Therefore, in silico drug trials are likely to play soon a major role in drug development, identifying drug cardiotoxicity in the pre-clinical phase, thus improving the quality of new candidate drugs and reducing drug failure at later stages.
Our results are obtained using experimentally-calibrated population of human models for in silico drug trials. The wide range of conductances considered includes extreme up- or down-regulation of ion channels, which can be linked to specific mutations or diseased conditions known to be pro-arrhythmic (Sanguinetti and Tristani-Firouzi, 2006; Itoh et al., 2016; Wang et al., 2016). Previous studies have also considered aspects of population variability for gender and age, mostly by changing cell volume and area (rather than ionic conductances) using a commercial software (Polak et al., 2012). The same software was also recently used to investigate potential drug-induced arrhythmias for 12 drugs (Abbasi et al., 2017). In that study, variability was taken into account by using different AP models (Ten Tusscher, 2003; Ten Tusscher and Panfilov, 2006; O'Hara et al., 2011) and different cell types (endo-, epi-, and mid-myocardium). However, their results were presented only for a single model (mid-) since it was the one most prone to develop drug-induced APD prolongation and EADs.
Our results also demonstrate consistency between human-based in silico simulations and recordings obtained from experimental models traditionally used in safety pharmacology, including rabbit wedge ECGs (Lu et al., 2016) and hiPS-CMs CT recordings. Previous in silico studies have focused on predictions of QT prolongation in human (Mirams et al., 2014; Lancaster and Sobie, 2016) and animal models (Bottino et al., 2006; Davies et al., 2012; Beattie et al., 2013). Evaluating the in silico results against experimental data is important as in silico tools are likely to be used in combination with experimental recordings for validation and identification of potential unknown effects. Importantly, our results also identifies discrepancies between in silico results and experimental and clinical data, when considering compounds with strong multichannel action, leading to large AP prolongation in silico and only a moderate increase of rabbit wedge QT. The potential causes of such discrepancies include: (i) differences in the balance of inward/outward currents in human adult cardiomyocytes as represented in silico with respect to rabbit wedge preparations, which may lead to higher sensitivity to hERG block. Indeed, it has been shown that APD prolongation due to IKr block is more pronounced in human, compared to rabbit (Bányász et al., 2011; O'Hara and Rudy, 2012); (ii) the fact that in silico results in our study are focused on single cell electrophysiology, as opposed to tissue (rabbit wedge) or whole heart, where coupling and other mechanisms may act to modulate AP duration, as supported by the fact that both in silico APD and hiPS-CMs CTD show larger prolongation compared to rabbit QT; (iii) the IC50 values were often estimated based on current blocks measured at low drug concentrations, while simulations explored much higher ones. This can therefore lead to an overestimation of current blocks, producing a larger AP increase than expected. Further work could address these important factors.
To conclude, this study demonstrates that in silico drug trials in populations of human cardiomyocyte models constitute a powerful methodology to predict clinical risk of arrhythmias based on ion channel information. This study also highlights ionic profiles that have a higher risk of developing drug-induced abnormalities. This methodology is therefore ready for its integration into the existing pipeline for drug cardiotoxicity assessment, and contribute to the reduction of animal experiments in the near future.
Author Contributions
All the authors conceived and designed the study; EP performed the in silico drug assays, analyzed the data, prepared the figures and drafted the manuscript; HL, JR, and AH performed experimental drug assays; RG, OB, and EP developed the software; EP, OB, AB, and BR interpreted the results; all the authors edited and revised the manuscript.
Funding
EP, OB, AB, and BR are supported by BR's Wellcome Trust Senior Research Fellowship in Basic Biomedical Sciences (100246/Z/12/Z), an Engineering and Physical Sciences Research Council Impact Acceleration Award (EP/K503769/1), the CompBioMed project (European Commission grant agreement No 675451), the NC3Rs Infrastructure for Impact award (NC/P001076/1) and Project Grant (NC/P00122X/1), the Oxford British Heart Foundation Centre of Research Excellence (RE/08/004/23915, RE/13/1/30181) and the TransQST project (Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 116030, receiving support from the European Union's Horizon 2020 research and innovation programme and EFPIA).
Conflict of Interest Statement
HL, JR, AH, and DG are employees of Janssen Pharmaceutica NV. RG is an employee of Oxford Computer Consultants, Oxford, UK.
The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fphys.2017.00668/full#supplementary-material
Abbreviations
AP, Action potential; APDXX, Action potential duration at XX% of repolarization; CiPA, Comprehensive in vitro Proarrhythmia Assay; CT, Ca2+-transient; CTBR, Ca2+-transient beat rate; CTDXX, Ca2+-transient duration at XX% of the initial base value; DA, Depolarization abnormalities; dV/dtMAX, Maximum upstroke velocity; EADs, Early after-depolarizations; EFTPCmax, Maximal effective therapeutic free concentration; GX, IX conductance; h, Hill coefficient; hiPS-CMs, Human induced pluripotent stem cell-derived cardiomyocytes; IC50, Concentration for 50% channel inhibition; ICaL, L-type Ca2+ current; IK1, Inward rectifier K+ current; IKr, Rapid delayed rectifier K+ current; IKs, Slow delayed rectifier K+ current; INa, Fast Na+ current; INaK, Na+-K+ pump current; INaL, Late Na+ current; INCX, Na+-Ca2+ exchanger current; Ito, Transient outward K+ current; ORd, O'Hara-Rudy dynamic human ventricular model; RA, Repolarization abnormalities; RMP, Resting membrane potential; TdP, Torsade de Pointes; Tri90−40, AP triangulation; Vm, Membrane potential; Vpeak, Peak voltage.
References
Abbasi, M., Small, B. G., Patel, N., Jamei, M., and Polak, S. (2017). Early assessment of proarrhythmic risk of drugs using the in vitro data and single-cell-based in silico models: proof of concept. Toxicol. Mech. Methods 27, 88–99. doi: 10.1080/15376516.2016.1256460
Abi-Gerges, N., Pointon, A., Oldman, K. L., Brown, M. R., Pilling, M. A., Sefton, C. E., et al. (2017). Assessment of extracellular field potential and Ca2+ transient signals for early QT/pro-arrhythmia detection using human induced pluripotent stem cell-derived cardiomyocytes. J. Pharmacol. Toxicol. Methods 83, 1–15. doi: 10.1016/j.vascn.2016.09.001
Bányász, T., Bárándi, L., Harmati, G., Virág, L., Szentandrássy, N., Márton, I., et al. (2011). Mechanism of reverse rate-dependent action of cardioactive agents. Curr. Med. Chem. 18, 3597–3606. doi: 10.2174/092986711796642355
Bass, A. S., Hombo, T., Kasai, C., Kinter, L. B., and Valentin, J.-P. (2015). “A historical view and vision into the future of the field of safety pharmacology,” in Principles of Safety Pharmacology, Handbook of Experimental Pharmacology Vol. 229, eds M. K. Pugsley and M. J. Curtis (Berlin; Heidelberg: Springer), 3–45. doi: 10.1007/978-3-662-46943-9_1
Beattie, K. A., Luscombe, C., Williams, G., Munoz-Muriedas, J., Gavaghan, D. J., Cui, Y., et al. (2013). Evaluation of an in silico cardiac safety assay: using ion channel screening data to predict QT interval changes in the rabbit ventricular wedge. J. Pharmacol. Toxicol. Methods 68, 88–96. doi: 10.1016/j.vascn.2013.04.004
Bottino, D., Penland, R. C., Stamps, A., Traebert, M., Dumotier, B., Georgieva, A., et al. (2006). Preclinical cardiac safety assessment of pharmaceutical compounds using an integrated systems-based computer model of the heart. Prog. Biophys. Mol. Biol. 90, 414–443. doi: 10.1016/j.pbiomolbio.2005.06.006
Braga, R. C., Alves, V. M., Silva, M. F. B., Muratov, E., Fourches, D., Lião, L. M., et al. (2015). Pred-hERG: a novel web-accessible computational tool for predicting cardiac toxicity. Mol. Inform. 34, 698–701. doi: 10.1002/minf.201500040
Brindisi, M., Butini, S., Franceschini, S., Brogi, S., Trotta, F., Ros, S., et al. (2014). Targeting dopamine D3 and serotonin 5-HT1A and 5-HT2A receptors for developing effective antipsychotics: synthesis, biological characterization, and behavioral studies. J. Med. Chem. 57, 9578–9597. doi: 10.1021/jm501119j
Britton, O. J., Bueno-Orovio, A., Van Ammel, K., Lu, H. R., Towart, R., Gallacher, D. J., et al. (2013). Experimentally calibrated population of models predicts and explains intersubject variability in cardiac cellular electrophysiology. Proc. Natl. Acad. Sci. U.S.A. 110, E2098–E2105. doi: 10.1073/pnas.1304382110
Britton, O. J., Bueno-Orovio, A., Virág, L., Varró, A., and Rodriguez, B. (2017). The Electrogenic Na+/K+ pump is a key determinant of repolarization abnormality susceptibility in human ventricular cardiomyocytes: a population-based simulation study. Front. Physiol. 8:278. doi: 10.3389/fphys.2017.00278
Carmeliet, E. E. (1999). Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol. Rev. 79, 917–1017.
Chemi, G., Gemma, S., Campiani, G., Brogi, S., Butini, S., and Brindisi, M. (2017). Computational tool for fast in silico evaluation of hERG K+ channel affinity. Front. Chem. 5:7. doi: 10.3389/fchem.2017.00007
Colatsky, T., Fermini, B., Gintant, G., Pierson, J. B., Sager, P., Sekino, Y., et al. (2016). The comprehensive in vitro proarrhythmia assay (CiPA) initiative - Update on progress. J. Pharmacol. Toxicol. Methods 81, 15–20. doi: 10.1016/j.vascn.2016.06.002
Coppini, R., Ferrantini, C., Yao, L., Fan, P., Del Lungo, M., Stillitano, F., et al. (2013). Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 127, 575–584. doi: 10.1161/CIRCULATIONAHA.112.134932
Coronel, R., Wilders, R., Verkerk, A. O., Wiegerinck, R. F., Benoist, D., and Bernus, O. (2013). Electrophysiological changes in heart failure and their implications for arrhythmogenesis. Biochim. Biophys. Acta 1832, 2432–2441. doi: 10.1016/j.bbadis.2013.04.002
Crumb, W. J., Vicente, J., Johannesen, L., and Strauss, D. G. (2016). An evaluation of 30 clinical drugs against the comprehensive in vitro proarrhythmia assay (CiPA) proposed ion channel panel. J. Pharmacol. Toxicol. Methods 81, 251–262. doi: 10.1016/j.vascn.2016.03.009
Davies, M. R., Mistry, H. B., Hussein, L., Pollard, C. E., Valentin, J.-P., Swinton, J., et al. (2012). An in silico canine cardiac midmyocardial action potential duration model as a tool for early drug safety assessment. AJP Hear. Circ. Physiol. 302, H1466–H1480. doi: 10.1152/ajpheart.00808.2011
Dempsey, C. E., Wright, D., Colenso, C. K., Sessions, R. B., and Hancox, J. C. (2014). Assessing hERG pore models as templates for drug docking using published experimental constraints: the inactivated state in the context of drug block. J. Chem. Inf. Model. 54, 601–612. doi: 10.1021/ci400707h
Denaro, C. P., and Benowitz, N. L. (1989). Poisoning due to class 1B antiarrhythmic drugs: lignocaine, mexiletine and tocainide. Med. Toxicol. Adv. Drug Exp. 4, 412–428. doi: 10.1007/BF03259923
Durdagi, S., Duff, H. J., and Noskov, S. Y. (2011). Combined receptor and ligand-based approach to the universal pharmacophore model development for studies of drug blockade to the hERG1 pore domain. J. Chem. Inf. Model. 51, 463–474. doi: 10.1021/ci100409y
Dutta, S., Mincholé, A., Zacur, E., Quinn, T. A., Taggart, P., and Rodriguez, B. (2016). Early afterdepolarizations promote transmural reentry in ischemic human ventricles with reduced repolarization reserve. Prog. Biophys. Mol. Biol. 120, 236–248. doi: 10.1016/j.pbiomolbio.2016.01.008
El-sherif, N., Craelius, W., Boutjdir, M., and Gough, W. B. (1990). Early afterdepolarizations and arrhythmogenesis. J. Cardiovasc. Electrophysiol. 1, 157–160. doi: 10.1111/j.1540-8167.1990.tb01057.x
Elshrif, M. M., Shi, P., and Cherry, E. M. (2015). Representing variability and transmural differences in a model of human heart failure. IEEE J. Biomed. Heal. Inform. 19, 1308–1320. doi: 10.1109/JBHI.2015.2442833
Gauthier, L. D., Greenstein, J. L., and Winslow, R. L. (2012). Toward an integrative computational model of the guinea pig cardiac myocyte. Front. Physiol. 3:244. doi: 10.3389/fphys.2012.00244
Gomez, J. F., Cardona, K., Romero, L., Ferrero, J. M., and Trenor, B. (2014). Electrophysiological and structural remodeling in heart failure modulate arrhythmogenesis. 1D simulation study. PLoS ONE 9:e106602. doi: 10.1371/journal.pone.0106602
Hindmarsh, A. C., Brown, P. N., Grant, K. E., Lee, S. L., Serban, R., Shumaker, D. E., et al. (2005). SUNDIALS: suite of nonlinear and differential/algebraic equation solvers. ACM Trans. Math. Softw. 31, 363–396. doi: 10.1145/1089014.1089020
Itoh, H., Crotti, L., Aiba, T., Spazzolini, C., Denjoy, I., Fressart, V., et al. (2016). The genetics underlying acquired long QT syndrome: impact for genetic screening. Eur. Heart J. 37, 1456–1464. doi: 10.1093/eurheartj/ehv695
Jurado Román, A., Rubio Alonso, B., Martín Asenjo, R., Salguero Bodes, R., López Gil, M., and Arribas Ynsaurriaga, F. (2012). Proarrhythmic potential of amiodarone: an underestimated risk? Rev. Española Cardiol. 65, 292–294. doi: 10.1016/j.rec.2011.05.018
Kim, J. J., Yang, L., Lin, B., Zhu, X., Sun, B., Kaplan, A. D., et al. (2015). Mechanism of automaticity in cardiomyocytes derived from human induced pluripotent stem cells. J. Mol. Cell. Cardiol. 81, 81–93. doi: 10.1016/j.yjmcc.2015.01.013
Kramer, J., Obejero-Paz, C. A., Myatt, G., Kuryshev, Y. A., Bruening-Wright, A., Verducci, J. S., et al. (2013). MICE models: superior to the HERG model in predicting torsade de pointes. Sci. Rep. 3:2100. doi: 10.1038/srep02100
Lalloz, M. R. A., Byfield, P. G. H., Greenwood, R. M., and Himsworth, R. L. (1984). Binding of amiodarone by serum proteins and the effects of drugs, hormones and other interacting ligands. J. Pharm. Pharmacol. 36, 366–372. doi: 10.1111/j.2042-7158.1984.tb04400.x
Lancaster, M. C., and Sobie, E. A. (2016). Improved prediction of drug-induced torsades de pointes through simulations of dynamics and machine learning algorithms. Clin. Pharmacol. Ther. 100, 371–379. doi: 10.1002/cpt.367
Latini, R., Tognoni, G., and Kates, R. E. (1984). Clinical pharmacokinetics of amiodarone. Clin. Pharmacokinet. 9, 136–156. doi: 10.2165/00003088-198409020-00002
Laverty, H. G., Benson, C., Cartwright, E. J., Cross, M. J., Garland, C., Hammond, T., et al. (2011). How can we improve our understanding of cardiovascular safety liabilities to develop safer medicines? Br. J. Pharmacol. 163, 675–693. doi: 10.1111/j.1476-5381.2011.01255.x
Lawrence, C. L., Bridgland-Taylor, M. H., Pollard, C. E., Hammond, T. G., and Valentin, J.-P. (2006). A rabbit Langendorff heart proarrhythmia model: predictive value for clinical identification of Torsades de Pointes. Br. J. Pharmacol. 149, 845–860. doi: 10.1038/sj.bjp.0706894
Lawrence, C. L., Pollard, C. E., Hammond, T. G., and Valentin, J.-P. (2008). In vitro models of proarrhythmia. Br. J. Pharmacol. 154, 1516–1522. doi: 10.1038/bjp.2008.195
Leishman, D. J., Beck, T. W., Dybdal, N., Gallacher, D. J., Guth, B. D., Holbrook, M., et al. (2012). Best practice in the conduct of key nonclinical cardiovascular assessments in drug development: current recommendations from the Safety Pharmacology Society. J. Pharmacol. Toxicol. Methods 65, 93–101. doi: 10.1016/j.vascn.2011.08.006
Liang, P., Lan, F., Lee, A. S., Gong, T., Sanchez-Freire, V., Wang, Y., et al. (2013). Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation 127, 1677–1691. doi: 10.1161/CIRCULATIONAHA.113.001883
Liu, T., Brown, B. S., Wu, Y., Antzelevitch, C., Kowey, P. R., and Yan, G. X. (2006). Blinded validation of the isolated arterially perfused rabbit ventricular wedge in preclinical assessment of drug-induced proarrhythmias. Hear. Rhythm 3, 948–956. doi: 10.1016/j.hrthm.2006.04.021
Lu, H. R., Gallacher, D. J., and Yan, G. X. (2016). Assessment of drug-induced proarrhythmia: the importance of study design in the rabbit left ventricular wedge model. J. Pharmacol. Toxicol. Methods 81, 151–160. doi: 10.1016/j.vascn.2016.06.006
Lu, H. R., Whittaker, R., Price, J. H., Vega, R., Pfeiffer, E. R., Cerignoli, F., et al. (2015). High throughput measurement of Ca++ dynamics in human stem cell-derived cardiomyocytes by kinetic image cytometery: a cardiac risk assessment characterization using a large panel of cardioactive and inactive compounds. Toxicol. Sci. 148, 503–516. doi: 10.1093/toxsci/kfv201
Mirams, G. R., Davies, M. R., Brough, S. J., Bridgland-Taylor, M. H., Cui, Y., Gavaghan, D. J., et al. (2014). Prediction of thorough QT study results using action potential simulations based on ion channel screens. J. Pharmacol. Toxicol. Methods 70, 246–254. doi: 10.1016/j.vascn.2014.07.002
Muszkiewicz, A., Britton, O. J., Gemmell, P., Passini, E., Sanchez, C., Zhou, X., et al. (2016). Variability in cardiac electrophysiology: using experimentally-calibrated populations of models to move beyond the single virtual physiological human paradigm. Prog. Biophys. Mol. Biol. 120, 115–127. doi: 10.1016/j.pbiomolbio.2015.12.002
O'Hara, T., and Rudy, Y. (2012). Quantitative comparison of cardiac ventricular myocyte electrophysiology and response to drugs in human and nonhuman species. AJP Hear. Circ. Physiol. 302, H1023–H1030. doi: 10.1152/ajpheart.00785.2011
O'Hara, T., Virág, L., Varró, A., and Rudy, Y. (2011). Simulation of the undiseased human cardiac ventricular action potential: Model formulation and experimental validation. PLoS Comput. Biol. 7:e1002061. doi: 10.1371/journal.pcbi.1002061
Passini, E., Mincholé, A., Coppini, R., Cerbai, E., Rodriguez, B., Severi, S., et al. (2016). Mechanisms of pro-arrhythmic abnormalities in ventricular repolarisation and anti-arrhythmic therapies in human hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 96, 72–81. doi: 10.1016/j.yjmcc.2015.09.003
Pitt-Francis, J., Bernabeu, M. O., Cooper, J., Garny, A., Momtahan, L., Osborne, J., et al. (2008). Chaste: using agile programming techniques to develop computational biology software. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 366, 3111–3136. doi: 10.1098/rsta.2008.0096
Polak, S., Fijorek, K., Glinka, A., Wisniowska, B., and Mendyk, A. (2012). Virtual population generator for human cardiomyocytes parameters: in silico drug cardiotoxicity assessment. Toxicol. Mech. Methods 22, 31–40. doi: 10.3109/15376516.2011.585477
Rodriguez, B., Carusi, A., Abi-Gerges, N., Ariga, R., Britton, O. J., Bub, G., et al. (2016). Human-based approaches to pharmacology and cardiology: an interdisciplinary and intersectorial workshop. Europace 18, 1287–1298. doi: 10.1093/europace/euv320
Sager, P. T., Gintant, G., Turner, J. R., Pettit, S., and Stockbridge, N. (2014). Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am. Heart J. 167, 292–300. doi: 10.1016/j.ahj.2013.11.004
Salvi, V., Karnad, D. R., Panicker, G. K., and Kothari, S. (2010). Update on the evaluation of a new drug for effects on cardiac repolarization in humans: issues in early drug development. Br. J. Pharmacol. 159, 34–48. doi: 10.1111/j.1476-5381.2009.00427.x
Sanguinetti, M. C., and Tristani-Firouzi, M. (2006). hERG potassium channels and cardiac arrhythmia. Nature 440, 463–469. doi: 10.1038/nature04710
Serban, R., and Hindmarsh, A. C. (2005). “CVODES: the sensitivity-enabled ODE solver in SUNDIALS,” in Volume 6: 5th International Conference on Multibody Systems, Nonlinear Dynamics, and Control, Parts A, B, and C (Long Beach, CA: ASME), 257–269. doi: 10.1115/DETC2005-85597
Shampine, L. F., and Reichelt, M. W. (1997). The MATLAB ODE Suite. SIAM J. Sci. Comput. 18, 1–22. doi: 10.1137/S1064827594276424
Shuba, Y. M., Degtiar, V. E., Osipenko, V. N., Naidenov, V. G., and Woosley, R. L. (2001). Testosterone-mediated modulation of HERG blockade by proarrhythmic agents. Biochem. Pharmacol. 62, 41–49. doi: 10.1016/S0006-2952(01)00611-6
Spencer, C. I., Baba, S., Nakamura, K., Hua, E. A., Sears, M. A. F., Fu, C. C., et al. (2014). Calcium transients closely reflect prolonged action potentials in iPSC models of inherited cardiac arrhythmia. Stem Cell Rep. 3, 269–281. doi: 10.1016/j.stemcr.2014.06.003
Stevens, J. L., and Baker, T. K. (2009). The future of drug safety testing: expanding the view and narrowing the focus. Drug Discov. Today 14, 162–167. doi: 10.1016/j.drudis.2008.11.009
Sugiyama, A. (2008). Sensitive and reliable proarrhythmia in vivo animal models for predicting drug-induced torsades de pointes in patients with remodelled hearts. Br. J. Pharmacol. 154, 1528–1537. doi: 10.1038/bjp.2008.240
Sun, J., and MacKinnon, R. (2017). Cryo-EM structure of a KCNQ1/CaM complex reveals insights into congenital long QT syndrome. Cell 169, 1042–1050.e9. doi: 10.1016/j.cell.2017.05.019
Ten Tusscher, K. H. W. J. (2003). A model for human ventricular tissue. AJP Hear. Circ. Physiol. 286, H1573–H1589. doi: 10.1152/ajpheart.00794.2003
Ten Tusscher, K. H. W. J., and Panfilov, A. V. (2006). Cell model for efficient simulation of wave propagation in human ventricular tissue under normal and pathological conditions. Phys. Med. Biol. 51, 6141–6156. doi: 10.1088/0031-9155/51/23/014
Valentin, J.-P., Bialecki, R., Ewart, L., Hammond, T., Leishmann, D., Lindgren, S., et al. (2009). A framework to assess the translation of safety pharmacology data to humans. J. Pharmacol. Toxicol. Methods 60, 152–158. doi: 10.1016/j.vascn.2009.05.011
Valentin, J.-P., Hoffmann, P., De Clerck, F., Hammond, T. G., and Hondeghem, L. (2004). Review of the predictive value of the Langendorff heart model (Screenit system) in assessing the proarrhythmic potential of drugs. J. Pharmacol. Toxicol. Methods 49, 171–181. doi: 10.1016/j.vascn.2004.03.008
Vargas, H. M., Bass, A. S., Koerner, J., Matis-Mitchell, S., Pugsley, M. K., Skinner, M., et al. (2015). Evaluation of drug-induced QT interval prolongation in animal and human studies: a literature review of concordance. Br. J. Pharmacol. 172, 4002–4011. doi: 10.1111/bph.13207
Veronese, M., McLean, S., and Hendriks, R. (1988). Plasma protein binding of amiodarone in a patient population: measurement by erythrocyte partitioning and a novel glass–binding method. Br. J. Clin. Pharmacol. 26, 721–731. doi: 10.1111/j.1365-2125.1988.tb05311.x
Wallis, R. M. (2010). Integrated risk assessment and predictive value to humans of non-clinical repolarization assays. Br. J. Pharmacol. 159, 115–121. doi: 10.1111/j.1476-5381.2009.00395.x
Walmsley, J., Rodriguez, J. F., Mirams, G. R., Burrage, K., Efimov, I. R., and Rodriguez, B. (2013). mRNA expression levels in failing human hearts predict cellular electrophysiological remodeling: a population-based simulation study. PLoS ONE 8:e56359. doi: 10.1371/journal.pone.0056359
Wang, H.-G., Zhu, W., Kanter, R. J., Silva, J. R., Honeywell, C., Gow, R. M., et al. (2016). A novel NaV1.5 voltage sensor mutation associated with severe atrial and ventricular arrhythmias. J. Mol. Cell. Cardiol. 92, 52–62. doi: 10.1016/j.yjmcc.2016.01.014
Wang, W., and MacKinnon, R. (2017). Cryo-EM structure of the open human Ether-à-go-go-Related K+ channel hERG. Cell 169, 422–430.e10. doi: 10.1016/j.cell.2017.03.048
White, J. W., Rassweiler, A., Samhouri, J. F., Stier, A. C., and White, C. (2014). Ecologists should not use statistical significance tests to interpret simulation model results. Oikos 123, 385–388. doi: 10.1111/j.1600-0706.2013.01073.x
Wiśniowska, B., Tylutki, Z., and Polak, S. (in press). Thorough QT (TQT) studies: concordance with torsadogenesis and an evolving cardiac safety testing paradigm. Drug Discov. Today. doi: 10.1016/j.drudis.2017.04.017
Woosley, R., and Romer, K. (1999). www.crediblemeds.org, QTdrugs List. AZCERT, Inc. 1822 Innov. Park Dr., Oro Val. AZ 85755. Available online at: www.crediblemeds.org (Accessed July 1, 2017).
Zeng, H., Roman, M. I., Lis, E., Lagrutta, A., and Sannajust, F. (2016). Use of FDSS/μCell imaging platform for preclinical cardiac electrophysiology safety screening of compounds in human induced pluripotent stem cell-derived cardiomyocytes. J. Pharmacol. Toxicol. Methods 81, 217–222. doi: 10.1016/j.vascn.2016.05.009
Keywords: in silico drug trials, drug safety, drug cardiotoxicity, Torsade de Pointes, computer models, human ventricular action potential
Citation: Passini E, Britton OJ, Lu HR, Rohrbacher J, Hermans AN, Gallacher DJ, Greig RJH, Bueno-Orovio A and Rodriguez B (2017) Human In Silico Drug Trials Demonstrate Higher Accuracy than Animal Models in Predicting Clinical Pro-Arrhythmic Cardiotoxicity. Front. Physiol. 8:668. doi: 10.3389/fphys.2017.00668
Received: 01 July 2017; Accepted: 23 August 2017;
Published: 12 September 2017.
Edited by:
Ovidiu Constantin Baltatu, Anhembi Morumbi University, BrazilReviewed by:
David Christini, Weill Cornell Medical College, United StatesSimone Brogi, University of Siena, Italy
Copyright © 2017 Passini, Britton, Lu, Rohrbacher, Hermans, Gallacher, Greig, Bueno-Orovio and Rodriguez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisa Passini, ZWxpc2EucGFzc2luaUBjcy5veC5hYy51aw==