 Li-Yen R. Hu
Li-Yen R. Hu Aikaterini Kontrogianni-Konstantopoulos
Aikaterini Kontrogianni-Konstantopoulos- Department of Biochemistry and Molecular Biology, University of Maryland School of Medicine, Baltimore, MD, United States
Obscurin is a giant cytoskeletal protein with structural and regulatory roles encoded by the OBSCN gene. Recently, mutations in OBSCN were associated with the development of different forms of cardiomyopathies, including hypertrophic cardiomyopathy (HCM). We previously reported that homozygous mice carrying the HCM-linked R4344Q obscurin mutation develop arrhythmia by 1-year of age under sedentary conditions characterized by increased heart rate, frequent incidents of premature ventricular contractions, and episodes of spontaneous ventricular tachycardia. In an effort to delineate the molecular mechanisms that contribute to the observed arrhythmic phenotype, we subjected protein lysates prepared from left ventricles of 1-year old R4344Q and wild-type mice to comparative proteomics analysis using tandem mass spectrometry; raw data are available via ProteomeXchange with identifier PXD017314. We found that the expression levels of proteins involved in cardiac function and disease, cytoskeletal organization, electropotential regulation, molecular transport and metabolism were significantly altered. Moreover, phospho-proteomic evaluation revealed changes in the phosphorylation profile of Ca2+ cycling proteins, including sAnk1.5, a major binding partner of obscurin localized in the sarcoplasmic reticulum; notably, this is the first report indicating that sAnk1 undergoes phosphorylation. Taken together, our findings implicate obscurin in diverse cellular processes within the myocardium, which is consistent with its multiple binding partners, localization in different subcellular compartments, and disease association.
Introduction
Muscle contraction is a highly regulated process that depends on the coordinated assembly of the sarcomeric cytoskeleton and internal membrane systems regulating Ca2+ cycling. As such, giant scaffolding proteins guide the incorporation of myofilament and accessory proteins into striated structures and contribute to the sarcomeric alignment of the sarcoplasmic reticulum (SR) and transverse tubules (t-tubules) to form functional couplons where excitation-contraction (EC) takes place (Wang et al., 2018). Given that EC is an energy demanding process, enzymes supplying adequate levels of metabolites are also targeted to the mature sarcomere in order to sustain repetitive contractions, and thus the ability of the heart to beat incessantly (Hu et al., 2015).
Obscurin, encoded by the OBSCN gene, is a giant scaffolding protein surrounding sarcomeric Z-disks and M-bands. It is highly modular consisting of 68 immunoglobulin (Ig) and 3 fibronectin-III (Fn-III) domains in addition to an array of signaling motifs that include an isoleucine glutamine (IQ) motif that binds calmodulin, a Src homology 3 (SH3) domain, a Rho-guanine nucleotide exchange factor (Rho-GEF) domain in tandem with a pleckstrin homology (PH) domain, and two active Ser/Thr kinase domains (Wang et al., 2018). Given its unique position at the periphery of Z-disks and M-bands, obscurin mediates binding to structural, regulatory and membrane-associated proteins residing in different subcellular compartments via its adhesion and signaling motifs (Bagnato et al., 2003; Kontrogianni-Konstantopoulos et al., 2003; Bowman et al., 2007; Fukuzawa et al., 2008; Ackermann et al., 2009; Ford-Speelman et al., 2009; Hu and Kontrogianni-Konstantopoulos, 2013; Randazzo et al., 2013; Qadota et al., 2016; Shriver et al., 2016). Through these binding interactions, obscurin plays key roles in the proper incorporation of myosin thick filaments into A-bands and the stability and maintenance of A- and M-bands (Kontrogianni-Konstantopoulos et al., 2006; Ackermann et al., 2009), the subsarcolemmal organization of the microtubule network and dystrophin into costameres (Randazzo et al., 2013), cell adhesion (Hu and Kontrogianni-Konstantopoulos, 2013), stress/stretch and growth responses (Ackermann et al., 2017; Randazzo et al., 2017), the organization and cytoskeletal anchoring of the SR membranes (Bagnato et al., 2003; Kontrogianni-Konstantopoulos et al., 2003; Lange et al., 2009), and Ca2+ homeostasis (Spooner et al., 2012; Hu et al., 2017).
Recent studies have further highlighted the essential roles of obscurin in cardiac pathophysiology as missense and frameshift mutations in the OBSCN gene were shown to co-segregate with hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM) and left ventricular non-compaction (LVNC) (Grogan and Kontrogianni-Konstantopoulos, 2018). Although the etiologies that underlie the pathogenicity of the majority of these mutations are elusive, work from our group has demonstrated that homozygous knock-in female mice carrying the HCM-linked R4344Q mutation located in Ig58 develop ventricular arrhythmia by 1-year of age under sedentary conditions, compensatory HCM in the presence of mild operational stress, and a DCM-like phenotype following exertion of sustained mechanical stress (Hu et al., 2017). Our biochemical and electrophysiological studies further showed that Ca2+ deregulation underlies at least in part these phenotypic manifestations as mutant obscurin (containing the R4344Q substitution) exhibits enhanced binding to phospholamban (PLN), the major regulator of the sarco/endoplasmic reticulum Ca2+ adenosine triphosphatase 2 (SERCA2), resulting in increased SR Ca2+ load and contractility kinetics, likely due to sequestration of PLN and disinhibition of SERCA2 (Hu et al., 2017).
Given the cardiac pathology that the R4344Q knock-in animals develop, we sought insights into the molecular pathways and cellular processes that are altered in the mutant myocardium. To this end, we performed an in-depth proteomics analysis coupled with evaluation of post-translational alterations using protein lysates prepared from left ventricles of sedentary, 1-year old female knock-in and wild-type animals. Our studies reveal alterations in the expression levels and post-translational regulation via phosphorylation of proteins involved in major pathophysiological processes and highlight the involvement of obscurin in their modulation.
Materials and Methods
All animal care, experimental procedures, and methodologies were performed in accordance with the protocols approved by the Institutional Animal Care and Use Committees of the University of Maryland School of Medicine.
Sample Preparation
Isogenic 1-year old female wild-type (n = 3) and obscurinR4344Q knock-in (n = 3) mice (Hu et al., 2017) were perfused with phosphate buffered saline (PBS) supplemented with a complete Mini Protease Inhibitor Cocktail (Roche, Indianapolis, IN, United States). The hearts were excised, and flash-frozen in liquid nitrogen. The left ventricles were isolated and used for generation of protein lysates with freshly prepared urea-thiourea buffer containing 8M urea, 2M thiourea, 0.05M Tris-HCl, pH 6.8, 75mM dithiothreitol, 3% (w/v) sodium dodecyl sulfate, 10% (v/v) glycerol supplemented with Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific, Waltham, MA, United States) and Halt Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). Hundred μg of protein lysates from each left ventricle were reduced with 4.5 mM tris-(2-carboxyethyl) phosphine (TCEP) (Thermo Fisher Scientific) and subjected to alkylation with 4.3 mM methyl methanethiosuphonate (Thermo Fisher Scientific). The alkylated lysates were precipitated with 10% (v/v) trichloroacetic acid in acetone (Sigma-Aldrich, St. Louis, MO, United States). The precipitated protein samples were reconstituted in 100 μl of 100 mM tri-ethyl-ammonium bicarbonate (TEAB), pH 8, and digested with 4 μg of trypsin/LysC mixture (V5071, Promega, Madison, WI, United States) at 37°C overnight. Peptides were labeled with 0.8 mg Tandem Mass Tag (TMT) 10-plex Isobaric Label Reagent (Thermo Fisher Scientific) in 100 mM TEAB, according to the manufacturer’s instructions. The isobaric nature of TMT allows the same peptides collected from different samples to co-migrate in liquid chromatography but be resolved by mass spectrometry. Thus, this approach allows quantification of the relative abundance of peptides from multiple samples, and enhances the signal-to-noise ratio for the identification of a particular peptide (Thompson et al., 2003).
Basic Reverse Phase Fractionation
One third of combined TMT 10-plex labeled tryptic peptides from each sample (i.e., wild-type and obscurinR4344Q knock-in) was reconstituted in 2 ml of buffer A (10 mM TEAB in H20, pH 8) and subjected to basic Reverse Phase (bRP) fractionation (Johns Hopkins University Mass Spectrometry and Proteomic Facility). The injection rate of the samples was set at 250 μl/min over 8 min. Peptides were fractionated on a 5 μm/2.1 × 100 mm XBridge C18 Column (Waters, Milford, MA, United States) using a high-performance liquid chromatography (HPLC) system (Agilent, Santa Clara, CA, United States). Fractionation was carried out by a linear gradient between buffer A and buffer B (10 mM TEAB in 90% acetonitrile) with the flow rate set at 250 μl/min. Eighty-four (84) fractions of 225 μl were collected between 20 and 95 min with 0–100% of buffer B. The 84 fractions were re-combined in 24 fractions that were analyzed by liquid chromatography/tandem mass spectrometry.
Liquid Chromatography/Tandem Mass Spectrometry
Each of the 24 bRP fractions was reconstituted in 28 μl of 2% acetonitrile/0.1% formic acid. Five hundred nanograms (500 ng) per fraction were subjected to reverse phase chromatography in a 2–90% acetonitrile/0.1% formic acid gradient over 90 min at 300 nl/min on a 5 μm/120 Å/75 μm × 150 mm ProntoSIL-120-5-C18 H column (Bischoff Chromatography, Munich, Germany), and subsequently sprayed on a nano-LC-Q Exactive HF (Thermo Fisher Scientific) through 1 μm emitter tip (Scientific Instrument Services Inc, Ringoes, NJ, United States) at 2.0 kV interfaced with the nanoAcquity LC system (Waters). Survey scans (including full mass spectra) were acquired on Orbi-trap within 350–1700 Da m/z using the Data dependent Top 15 method with dynamic exclusion of 20 s. Precursor ions were individually isolated with 1.0 Da and fragmented using Higher energy Collision activated Dissociation (HCD) activation collision energy of 32. Precursor and fragment ions were analyzed at 120,000 resolution, 3xe6 Automatic Gain Control (AGC) target and 50 ms maximum Injection Time (max IT), and at 60,000 resolution, 1xe5 AGC target and 200 ms max IT, respectively.
Data Analysis
MS/MS spectra were analyzed with the Proteome Discoverer (PD) software (v1.4 Thermo Fisher Scientific) using 3Nodes; common PD nodes with spectra were extracted, charge state deconvoluted, and deisotoped using Xtract option at resolution 100K at 400Da in an MS2 processor. MS/MS spectra from 3Nodes were analyzed with Mascot (v2.5.1, Matrix Science, London, United Kingdom) with the RefSeq2015 Complete Database (Mus musculus species, trypsin as an enzyme, mc2). We performed two searches for post-translational modifications. Both searches accounted for TMT10plex on the NH2-terminus and carbamidomethylation on cysteine as static modifications, and TMT on lysine, oxidation on methionine and deamidation on QN as variable modifications. In addition, the first search included phosphorylation on serine and threonine, and the phosphorylation sites were identified with both Mascot and PhosphoRS4.1. The second search included O-GlcNAcylation on serine and threonine, and glutathionylation on cysteine. Peptides identified from Mascot searches were processed with PD to match to corresponding proteins with 0.01% threshold of False Discover Rate (FDR) based on a concatenated decoy database search to calculate protein and peptide ratios. The mass spectra reporter ion intensities ascertained from PD provided multiple spectra values for all the detected modified peptides of all samples. The median signal value of all intensities for each peptide’s modification class, e.g., phosphorylated or dephosphorylated, was calculated and converted to log2 notation (with 0.0 values excluded as nulls) to represent that peptide and its modification state for subsequent analysis. To minimize any potential experimental artifact effect due to differential sample processing and/or loading, these peptide values were quantile-normalized across samples so that all samples’ modified peptide values had the same median and distributions. A two-tailed t-test using a one-way ANOVA model was applied to evaluate the adjusted modified peptides between wild-type and R4344Q LV samples using the Partek Genomics Suite 6.6 analytic platform (Partek Inc. St. Louis, MO, United States). The 95% confidence interval of the linear fold-change provided by the Partek Genomics Suite analytic platform was converted to relative ratio change. Functions and pathways of proteins with altered expression levels were analyzed with Ingenuity Pathway Analysis (IPA) (Qiagen, Hilden, Germany).
Data Repository
The raw mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (Perez-Riverol et al., 2019) with the dataset identifier PXD017314. In addition, all proteins identified from the mass spec analysis along with the relative statistics (i.e., knock-in vs. wild-type) regardless of whether they were deemed significantly altered or not are included in Supplementary File S1.
Results and Discussion
The R4344Q obscurin mutation was originally identified via genetic screening in a young adult Japanese patient presenting with HCM (Arimura et al., 2007). The patient’s mother was also diagnosed with HCM, and although she was not screened for the mutation, it was postulated that the index patient most likely inherited the mutation from her since the father did not carry it and did not develop HCM (Arimura et al., 2007).
To study the disease pathogenesis due to the R4344Q mutation, we recently generated a knock-in mouse model carrying the mutation (Hu et al., 2017). Examination of female homozygous knock-in mice indicated that they exhibit increased heart rate, frequent incidents of premature ventricular contractions, and episodes of spontaneous ventricular tachycardia at 1-year of age under sedentary conditions (Hu et al., 2017); of note, heterozygous knock-in mice did not show any cardiac pathology. Exertion of sustained mechanical stress induced via transaortic constriction (TAC) surgery in young R4344Q knock-in mice resulted in the development of a DCM-like phenotype, which was associated with increased fibrosis and calcification (Hu et al., 2017). Interestingly, sham knock-in animals subjected to surgery but not TAC developed HCM, possibly as an adaptive response (Hu et al., 2017). Taken together, these observations indicated that the presence of the R4344Q mutation results in cardiac remodeling and dysfunction in response to the physiological process of aging and following mild or sustained pathological stress in the young.
In the current study, we focused on the molecular alterations taking place in the R4344Q myocardia as a function of aging under sedentary conditions. To identify differences in the relative abundance and post-translational regulation of key proteins contributing to the observed arrhythmic phenotype, we performed an unbiased, in-depth, comparative proteomics analysis using left ventricles (LV) from 1-year old isogenic wild-type (n = 3) and R4344Q knock-in (n = 3) homozygous female mice. We identified 4,343 proteins via our proteomic screening, 181 of which exhibited significantly altered expression (Supplementary File S1). In addition to changes in the expression levels of these 181 proteins, we found alterations in the post-translational regulation of select ones, with phosphorylation being the major modification.
Cardiac function and disease development, electropotential regulation, cytoskeletal organization, molecular transport, and metabolism were the main cellular processes that were significantly affected in the R4344Q female myocardium compared to wild-type (Figure 1). Among the 181 proteins whose expression levels were significantly altered in these cellular processes, some are well known to regulate cardiac homeostasis while others are currently understudied (Tables 1–6). Herein, we discuss the identified altered cellular processes and key relevant proteins aiming to provide a mechanistic interpretation of the arrhythmic phenotype developed by the R4344Q female animals in response to the physiological and irreversible process of aging. We followed three rules in the selection of proteins or families of proteins to discuss: (1) they are involved in several processes and/or sub-processes, (2) alterations in their expression levels or post-translational modifications have been linked to cardiac pathology, and (3) they directly or indirectly associate with obscurin. Of note, proteins that are involved in many processes and/or sub-processes are discussed under the ones for which more information regarding their role(s) is known.
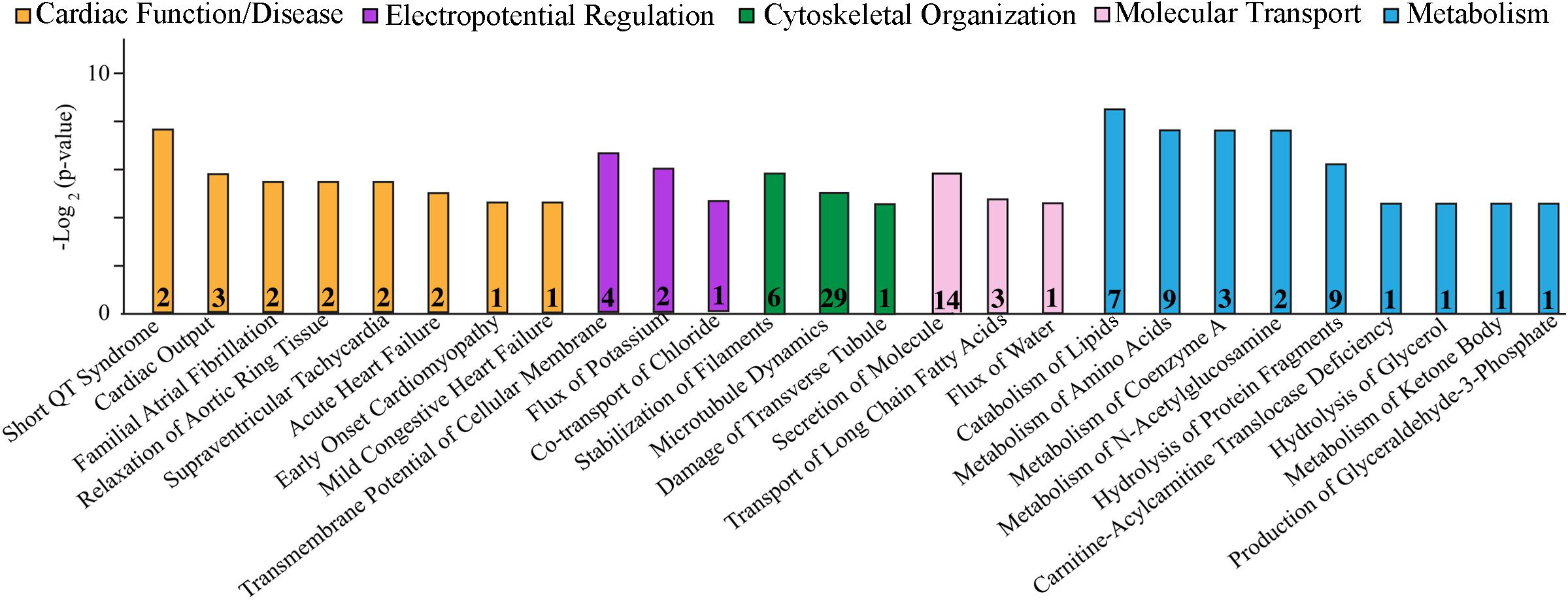
Figure 1. Cellular process/disease annotation of proteins with altered expression in left ventricles of 1-year-old R4344Q knock-in female mice. The expression levels of proteins involved in cardiac function and disease (orange), electropotential regulation (purple), cytoskeleton organization (green), molecular transport (pink), and metabolism (cyan) are significantly altered in 1-year old R4344Q myocardia (n = 3) compared to isogenic gender- and age-matched control animals (n = 3). The p-value for each process is represented by the height of the respective bar, after being transformed into the negative logarithmic value with the base of 2. The number of proteins whose expression levels are altered in each process are denoted in the relevant columns.
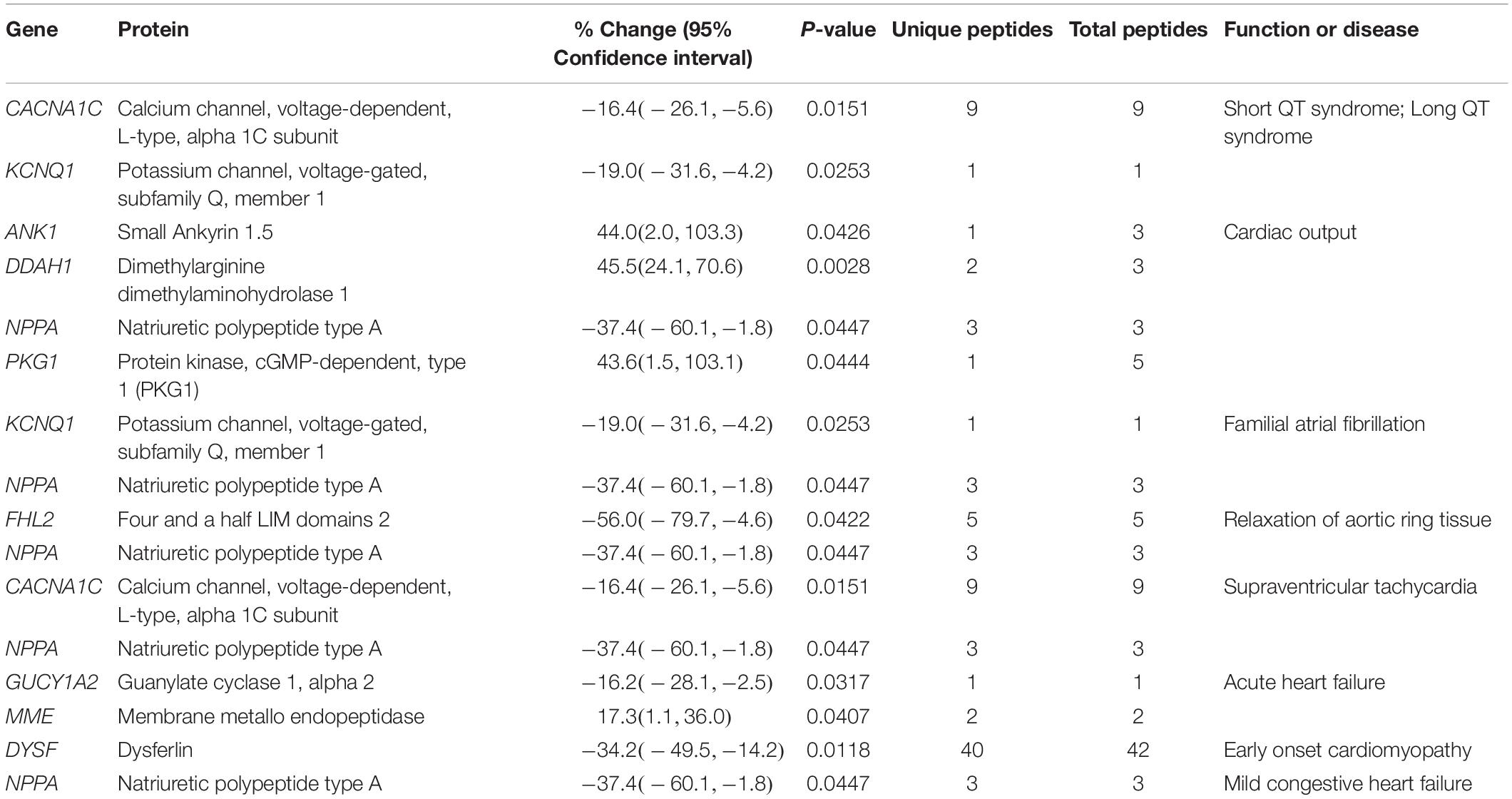
Table 1. Altered expression levels of proteins involved in cardiac function and disease.
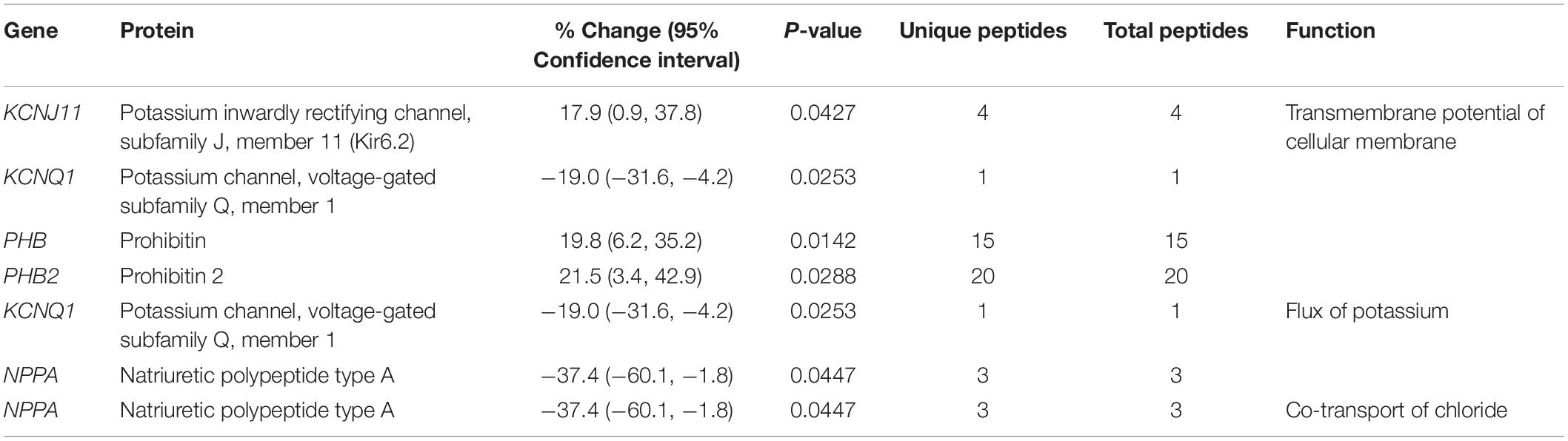
Table 2. Altered expression levels of proteins involved in electropotential regulation.
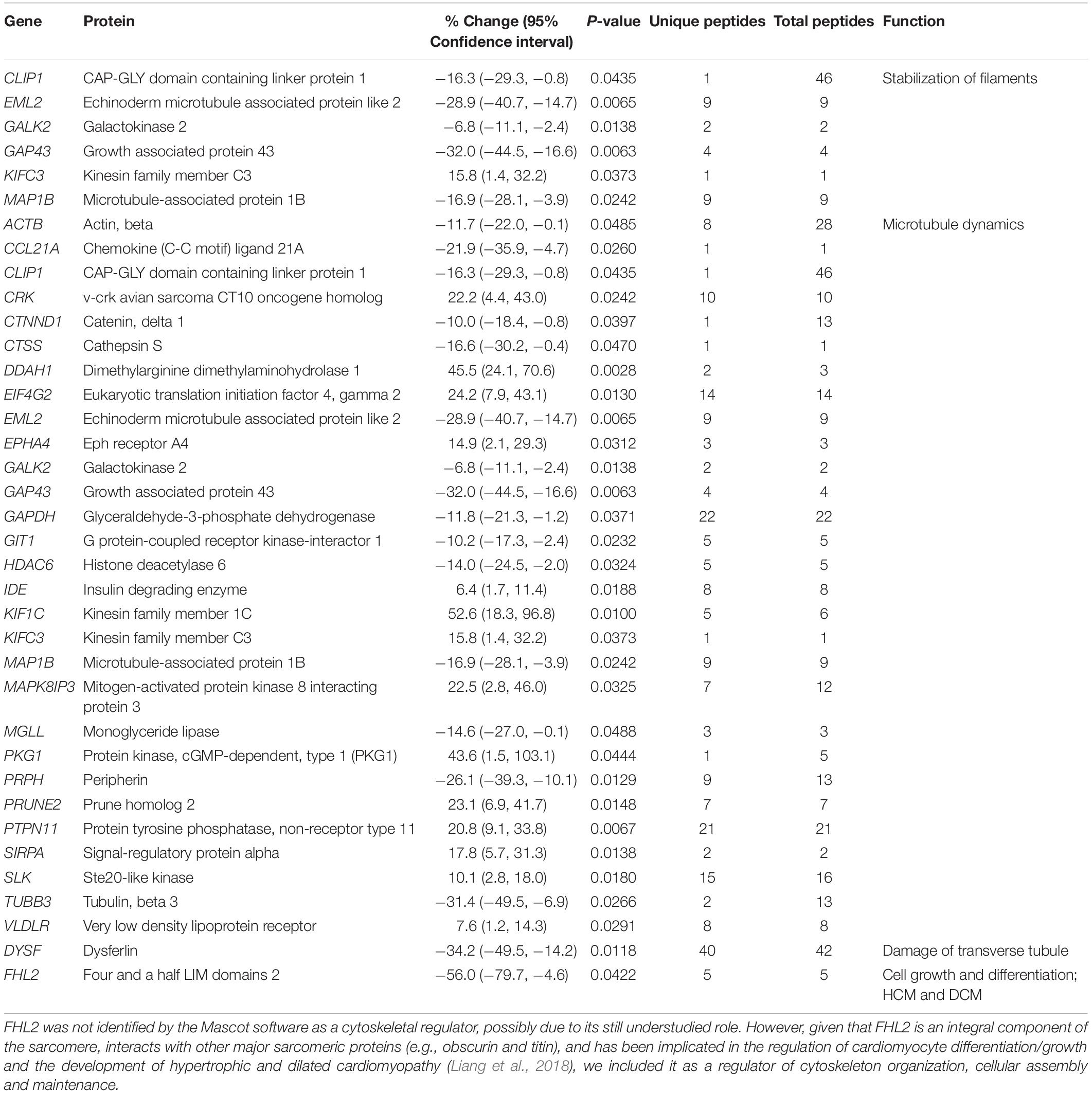
Table 3. Altered expression levels of proteins involved in cytoskeleton organization, cellular assembly and maintenance.
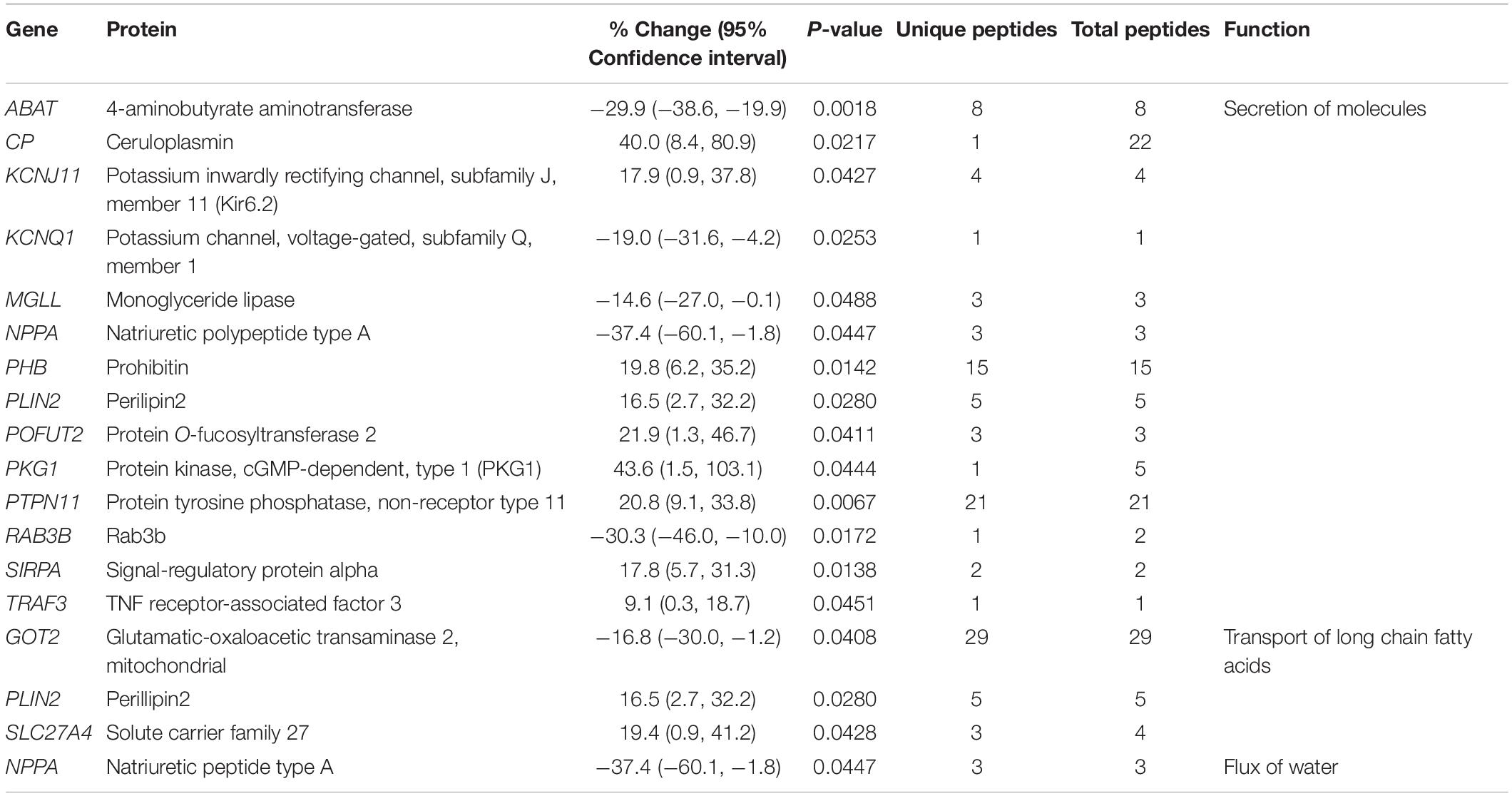
Table 4. Altered expression levels of proteins involved in molecular transport.
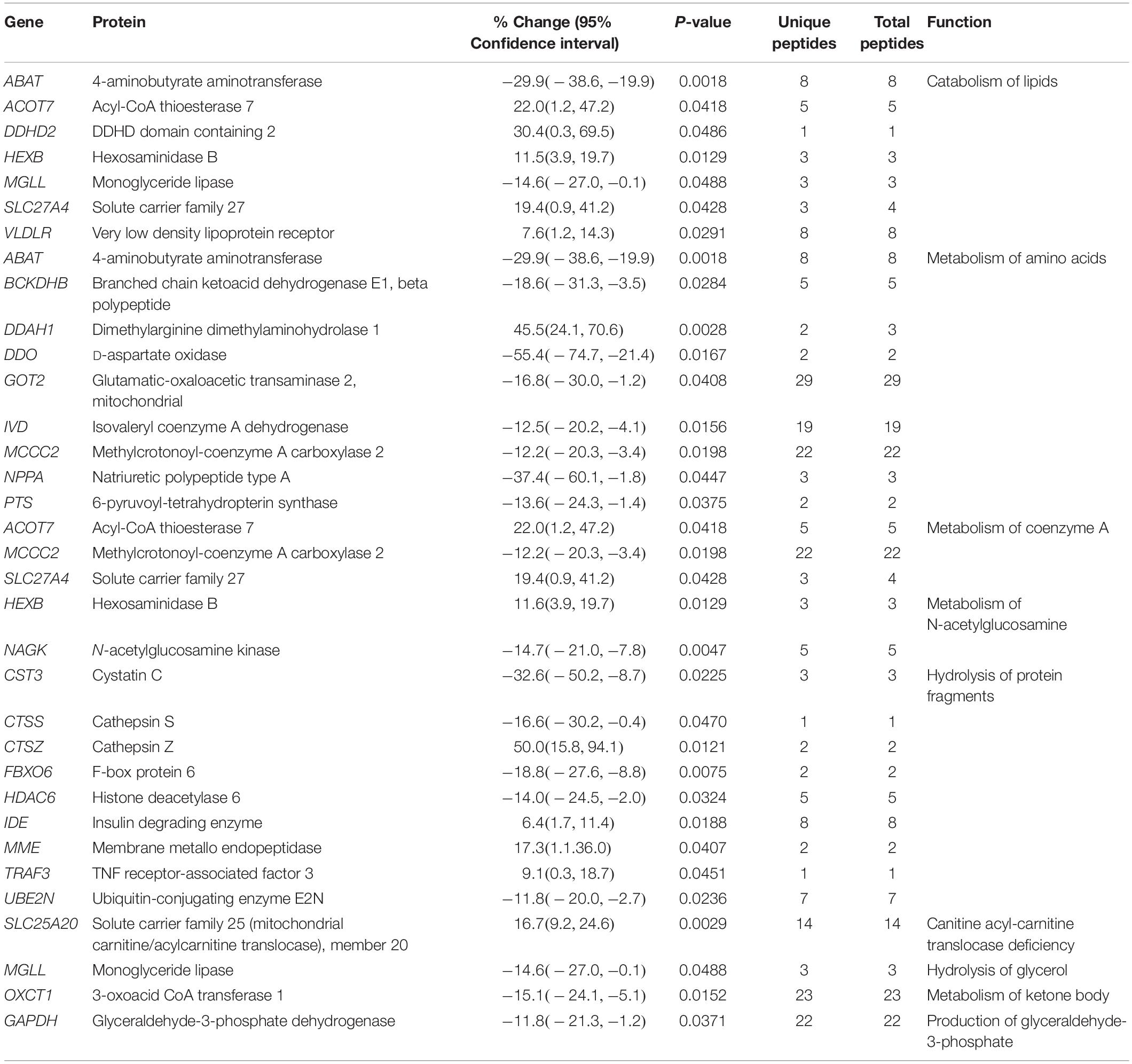
Table 5. Altered expression levels of proteins involved in metabolism.
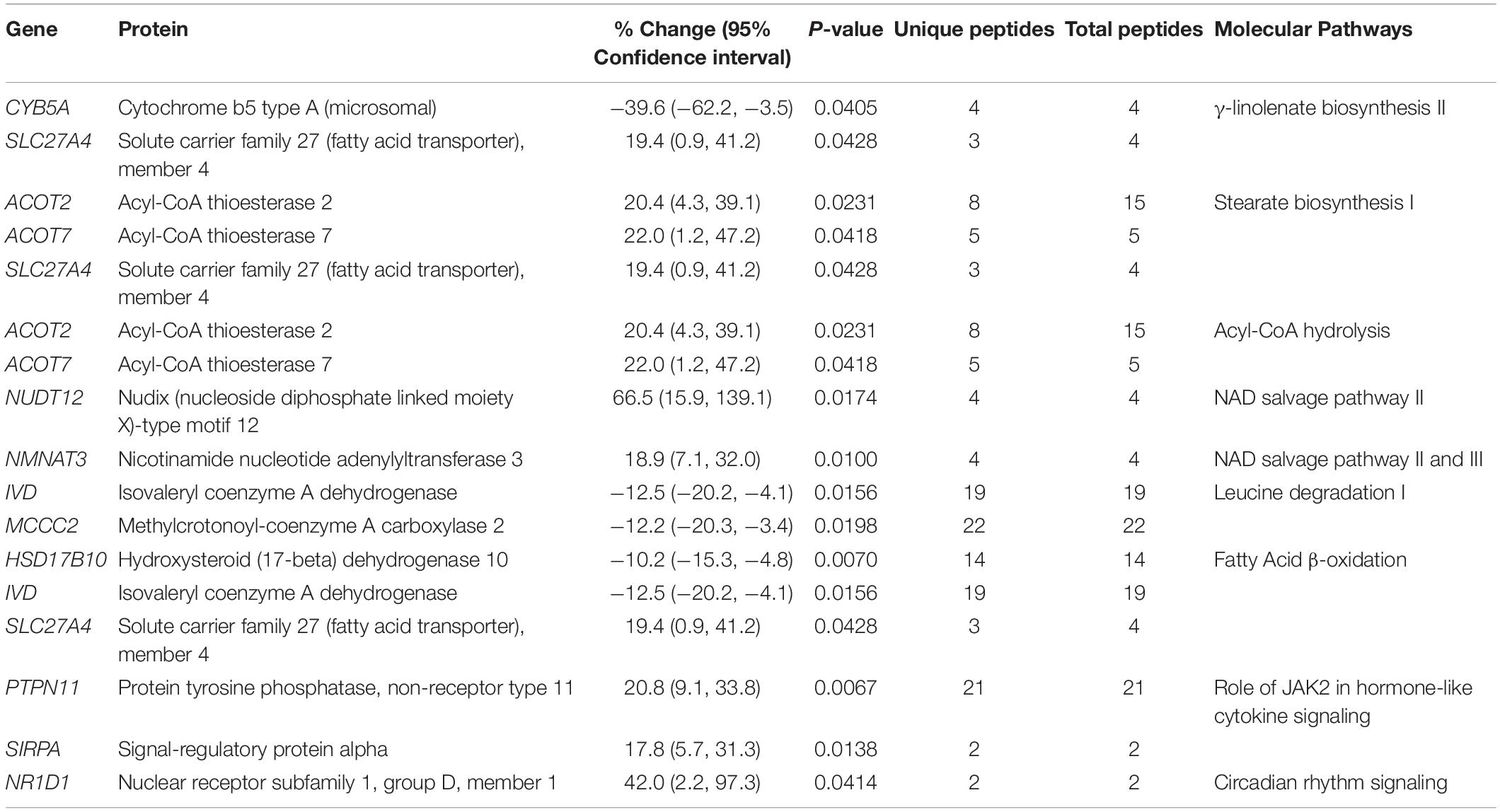
Table 6. Altered expression levels of proteins involved in various molecular pathways.
Cardiac Function and Disease
Consistent with the myopathic and arrhythmic phenotype that the 1-year old female R4344Q mice develop, the expression levels of several proteins involved in cardiac output and relaxation or the development of short and long QT syndrome, atrial fibrillation, supraventricular tachycardia, hypertrophy, and heart failure are significantly altered. These include ion channels, enzymes, secreted peptides, membrane and sarcomeric proteins (Table 1). Given that cardiac function and disease is broad and all-encompassing, we will discuss key identified proteins in the respective (sub)processes they are involved.
Ca2+-Handling Proteins
The expression levels of two Ca2+-handling proteins were significantly altered in the 1-year old female R4344Q left ventricles, the L-type voltage-dependent Ca2+ channel alpha 1C subunit (Cacna1C) and small ankyrin 1.5 (sAnk1.5) (Table 1). In particular, the expression levels of Cacna1C were decreased (∼16%), whereas the expression levels of sAnk1 were increased (44%) compared to wild-type (Table 1). Activation of the L-type voltage-dependent Ca2+ channel by membrane depolarization is essential to induce Ca2+ release from the sarcoplasmic reticulum (SR) membranes primarily via the ryanodine receptor (RyR) (Grunnet, 2010). Assuming that the open-probability of the Cacna1C L-type Ca2+ channel remains unaltered in the R4344Q myocardia, its reduced expression levels would potentially result in delayed Ca2+ release from the SR. Interestingly, we did not observe such an effect in isolated R4344Q cardiomyocytes (Hu et al., 2017), suggesting that the ∼16% decrease in the expression levels of Cacna1C was not sufficient to notably delay SR Ca2+ release or that other compensatory mechanisms alleviated such an effect.
Moreover, sAnk1.5 is a transmembrane protein residing in the SR with its COOH-terminus extending in the myoplasm where it binds to the extreme COOH-terminus of obscurin-A at two sites with distinct affinities (Bagnato et al., 2003; Kontrogianni-Konstantopoulos et al., 2003; Busby et al., 2010). Loss-of-function studies have postulated that sAnk1.5 is essential for the assembly and sarcomeric alignment of the SR through its interaction with obscurin-A (Kontrogianni-Konstantopoulos et al., 2006; Lange et al., 2009). Contrary to our proteomics analysis, we did not find the expression levels of sAnk1.5 statistically increased in LV from 1-year old R4344Q female mice compared to wild-type via immunoblotting, although we did observe a clear trend (Hu et al., 2017). This discrepancy may be due to the different methods used, i.e., the qualitative nature and low sensitivity of immunoblotting vs. the quantitative nature and high sensitivity of proteomics (Aebersold et al., 2013), and/or the commonly observed inter-individual variability among isogenic animals of the same genotype that may obscure the identification of statistical differences.
In addition to binding obscurin-A, sAnk1.5 interacts directly with the SarcoEndoplasmic Reticulum Ca2+ ATPase 1 (SERCA1) and its major regulator sarcolipin (SLN) (Desmond et al., 2015), which are preferentially expressed in skeletal muscles (Shaikh et al., 2016); importantly, SLN is also present in healthy atrial cells and SERCA1 is upregulated in atrial cells in pathological states (Shaikh et al., 2016). sAnk1 reduces the apparent Ca2+ affinity of SERCA1, albeit to a lesser extent compared to SLN (Desmond et al., 2015), and ablates the inhibitory effect of SLN to SERCA1 activity (Desmond et al., 2017). Whether sAnk1.5 may play a similar regulatory role in the myocardium, by directly binding to SERCA2 and/or its major regulator phospholamban (PLN) is currently unknown, yet intriguing. Our previous work demonstrated that wild-type obscurin binds to PLN modestly, but this interaction is markedly enhanced in the presence of the R4344Q mutation (Hu et al., 2017). We therefore proposed that mutant obscurin might sequester PLN leading to SERCA2 disinhibition (Hu et al., 2017). If sAnk1.5 is a regulator of SERCA2, too, its up-regulation may contribute to the increased SERCA2 activity (∼40%) that we observed in the R4344Q myocardium in two ways: (1) through direct binding to PLN and ablation of its inhibitory effect on SERCA2 (similar to mutant obscurin) or (2) by being a less potent inhibitor of SERCA2 activity, and competing PLN away due to its excess amounts in the R4344Q myocardium. Experiments in our lab are currently under way to test these possibilities.
Electropotential Regulation
Our proteomics studies also revealed alterations in the expression levels of proteins involved in electropotential regulation. Specifically, the expression levels of two potassium (K+) channels, K+ channel voltage-gated subfamily Q member 1 (Kcnq1) and K+ inwardly rectifying channel subfamily J member 11 (Kir6.2) were significantly decreased (∼19%) and increased (∼18%), respectively (Table 2). In the healthy myocardium, Kcnq1 is responsible for K+ extrusion during the repolarization phase (Finlay et al., 2017). Loss-of-function mutations in KCNQ1 have been associated with long QT syndrome, ventricular tachycardia, ventricular fibrillation, extrasystoles and atrial flutter (Table 1) (Tester and Ackerman, 2014), while gain-of-function mutations have been linked to short QT syndrome, cardiac arrest, and atrial arrhythmia including atrial flutter and atrial fibrillation (Table 1) (Lee et al., 2017). Conversely, the KATP channel Kir6.2 is predominantly closed, but opens in response to cardiac stress (e.g., hypoxia or ischemia) to permit K+ efflux (Foster and Coetzee, 2016). The opening of Kir6.2 due to stress has been regarded as cardioprotective, since it leads to shortening of action potentials and suppression of Ca2+ entry into cardiomyocytes resulting in inhibition of contractility and reduced energy consumption (Foster and Coetzee, 2016). Alterations in the expression levels or open probability of K+ channels have been linked to deregulation of the electrochemical gradient of cardiomyocytes resulting in abnormal or asynchronous beating (Finlay et al., 2017). Given that the expression of Kir6.2 is increased by ∼18% in the R4344Q LV, if its open probability remains unaltered, it may at least partially account for the development of tachycardia (Hu et al., 2017) due to the shortened duration of action potentials.
In addition to K+ channels, proteins interacting with ion channels may also contribute to electropotential regulation. Prohibitin and prohibitin 2, interacting with the large-conductance Ca2+-activated Big K+ (BK) channel in chicken cochleae (Sokolowski et al., 2011), are up-regulated by ∼20% in the R4344Q obscurin model (Table 2). Their expression profiles are interdependent, as alterations in the levels of one are followed by the other. They form a multimeric complex consisting of 12–16 pairs of heterodimers in the inner mitochondrial membrane where they are responsible for organizing the mitochondrial genome and maintaining mitochondrial morphology. In addition to mitochondria, prohibitin and prohibitin 2 localize in the nucleus and the plasma membrane, serving as a transcriptional activator (prohibitin), regulator of chromatin morphology (prohibitin 2), and signal transducers (both proteins), whereas not much is known about their plasma membrane localization (Osman et al., 2009). In cardiomyocytes, overexpression of prohibitin protected cardiomyocytes from apoptosis against oxidative stress, and ameliorated diabetic cardiomyopathy in a type-2 diabetic rat model (Dong et al., 2016). Consistent with its protective role, the expression levels of prohibitin were decreased in a rat cardiac hypertrophy model induced by isoproterenol (Chowdhury et al., 2013). Given that knockdown of prohibitin in epithelial cells resulted in inhibition of Complex I of the mitochondrial electron transport chain and increased production of reactive oxygen species (ROS) (Schleicher et al., 2008), it is plausible that upregulation of prohibitin (and possibly prohibitin 2) in the R4344Q obscurin model is an adaptive mechanism of the mutant myocardium promoting cardiomyocyte survival by improving cellular respiration and diminishing ROS production.
Natriuretic polypeptide type A (NppA) is involved in electropotential regulation (Table 2) in addition to other processes (Tables 1, 4, 5), and its levels are significantly decreased (∼37%) in the R4344Q LV compared to wild-type controls. It indirectly inhibits the Na+/K+/2Cl– co-transport in cardiomyocytes by stimulating guanylyl cyclase (GC) and increasing cytoplasmic cGMP levels, therefore reducing cell volume (Clemo and Baumgarten, 1995). Moreover, NppA has anti-proliferative and anti-hypertrophic effects in the heart by inhibiting the Mitogen Activated Protein Kinase (MAPK) axis (Song et al., 2015). Secretion of NppA from cardiac muscle to plasma is regulated by the Kir6.2 ATP-sensitive K+ channel, as Kir6.2-deficient atrial myocytes release higher levels of NppA post-mechanical stretch than wild-type cells (Saegusa et al., 2005). In accord with the decreased Nppa levels (∼37%) in R4344Q LV, Kir6.2 expression is increased (∼18%; Table 2). Given that the expression NppA is affected in the R4344Q LV alongside with its direct involvement in several pathophysiological cellular processes, it is not surprising that our proteomics analysis found many of them altered.
Cytoskeletal Organization, Cellular Assembly and Maintenance
As obscurin is a bona fide cytoskeletal protein, it is not surprising that the proteomics analysis indicated alterations in several proteins involved in cytoskeletal organization (Table 3). Proteins involved in filament stabilization and microtubule dynamics were the main two subgroups that were significantly altered. Given that the expression profile and pathophysiological role of the identified proteins involved in filament stabilization are currently understudied in the heart, we will discuss select proteins involved in microtubule dynamics.
The expression levels of 23 proteins regulating microtubule dynamics were altered. As such, the levels of G protein-coupled receptor kinase-interactor 1 (Git1) and catenin-delta1 (Ichii and Takeichi, 2007; Cernohorska et al., 2016) that promote microtubule assembly were decreased ∼10% (Table 3). Conversely, the levels of Prune homolog 2 (Prune2), an interacting partner of Microtubule Associated Protein 6 (MAP6), shown to suppress microtubule stability in response to cold conferred by MAP6 (Arama et al., 2012), are increased by ∼23% (Table 3). While down-regulation of Git1 and catenin-delta1 and up-regulation of Prune2 suggest that the R4344Q myocardium favors microtubule disassembly, alterations in the expression levels of enzymes involved in the post-translational regulation of microtubules suggest a contrary scenario. Specifically, the levels of Histone deacetylase 6 (HDAC6) are reduced by ∼14%, whereas the levels of Protein tyrosine phosphatase non-receptor type 11 (Ptpn11) are increased by ∼21% (Table 3). Since HDAC6-mediated deacetylation of α-tubulin promotes microtubule destabilization (Hubbert et al., 2002) and gain-of-function of Ptpn11 reduces HDAC6-mediated microtubule deacetylation (Tien and Chang, 2014), the coincident down-regulation of HDAC6 and up-regulation of Ptpn11 in the R4344Q myocardium likely results in increased acetylation of α-tubulin and thus microtubule stabilization. In view of the pivotal role of microtubules in mechanotransduction in the healthy heart and their potential as a novel therapeutic target for the “stiff” heart (Chaldakov, 2018), characterization of microtubule structure and dynamics in the R4344Q myocardium is of high pathophysiological relevance, which our lab plans to investigate.
Tethered at the Z/I junction through its interaction with titin, Four and a Half Lim domains protein 2 (FHL2) interacts with cytoskeletal (including obscurin) and signaling proteins as well as metabolic enzymes (Liang et al., 2018). FHL2 has been postulated to suppress cardiac hypertrophy by inhibiting the MAPK/ERK and calcineurin/nuclear factor of activated T-cells (NFAT) signaling cascades (Liang et al., 2018). As such, mutations in FHL2 are associated with the development of HCM and DCM (Friedrich et al., 2014). In human patients with HCM, the mRNA and protein levels of FHL2 are reduced by >50% (Friedrich et al., 2014). In agreement with this, FHL2 is down-regulated by ∼56% (Table 3) in the R4344Q LV. Although the R4344Q myocardia do not undergo obvious hypertrophy under sedentary conditions by 1-year of age, they are arrhythmic, and develop hypertrophy at 3-months of age due to mild stress and dilation following sustained mechanical stress (Hu et al., 2017). It is therefore possible that the reduced levels of FHL2 contribute to these phenotypic manifestations although it is currently unknown whether up-regulation of the MAPK/ERK pathway takes place in the R4344Q hearts.
Moreover, dysferlin levels were notably reduced by ∼34% in the obscurin R4344Q myocardium (Table 3). Given that dysferlin is essential in maintaining and/or repairing sarcolemmal and transverse tubule integrity in striated muscle cells (McElhanon and Bhattacharya, 2018), and dysferlin-null mice develop early onset cardiomyopathy when subjected to stress exercise, it is possible that cardiomyocytes containing mutant obscurin are more susceptible to mechanical stress and pathological remodeling of the plasma membrane and internal membrane systems.
Molecular Transport
The expression levels of several proteins involved in molecular transport are also altered in the obscurin R4344Q LV (Table 4). In addition to K+ channels and Nppa discussed above, proteins involved in iron and fatty acid transport and secretion of extracellular proteases may impact cellular metabolism and homeostasis. Ceruloplasmin is essential for converting ferrous (Fe2+) to ferric (Fe3+) to promote its transport and compartmentalization in the body. Up-regulation of ceruloplasmin by ∼40% (Table 4) may thus enhance iron transport in the obscurin R4344Q mice. Additionally, ceruloplasmin may carry out other physiological functions, such as scavenging of free radicals and copper transport (Wang and Wang, 2018). It was previously shown that up-regulation of ceruloplasmin may confer cardiac protection to isolated rat hearts subjected to ischemia and reperfusion (Atanasiu et al., 1995). Consistent with this, the expression levels of ceruloplasmin are up-regulated in heart failure patients, potentially as a compensatory response (Cabassi et al., 2014). Thus, up-regulation of ceruloplasmin may be an adaptive mechanism developed by the R4344Q model to ameliorate the cardiomyopathic pathology.
The expression levels of three proteins involved in the transport of long chain fatty acids are altered, too, including glutamic-oxaloacetic transaminase 2 (Got2), perilipin 2 (Plin2), and solute carrier family 27 (Table 4). Got2 and Plin2 upregulation in the heart has been shown to lead to accumulation of lipid droplets (Holloway et al., 2011; Ueno et al., 2017). Interestingly, the expression levels of solute carrier family 27 have been suggested to positively correlate with fatty acid uptake (Milger et al., 2006), but also lipid oxidation (Jeppesen et al., 2012). In the R4344Q myocardium, Got2 expression was down-regulated by 16.8%, whereas Plin2 and solute carrier family 27 expression was up-regulated by 16.5 and 19.4%, respectively (Table 4). Given that no prominent lipid droplet formation was observed in the hearts of these animals (our unpublished observations), it is likely that the altered expression levels of these proteins are not sufficient to lead to detectable lipid droplet accumulation by 1-year of age. Follow-up studies with older animals will assess the possibility of lipid accumulation as a function of aging in the R4344Q model.
In addition to the proteins discussed above, others involved in molecular transport have been shown to confer cardiac protection or exacerbate cardiac pathology. Among those, cardiac-specific overexpression of signal-regulatory protein alpha (Sirpα) has been suggested to attenuate pressure overload-induced cardiac hypertrophy in mice (Jiang et al., 2014), whereas mice treated with a pharmacological inhibitor of monoglyceride lipase (Mgll) exhibit increased infarct size and worsened cardiac function post-myocardial infarction (Schloss et al., 2019). Interestingly, while Sirpα is up-regulated by 17.8%, Mgll is down-regulated by 14.6% in the R4344Q myocardium (Table 4). Moreover, mice with cardiac-specific deletion of TNF receptor-associated factor 3 (TRAF3) showed significantly reduced cardiac hypertrophy and dysfunction 4-weeks after aortic banding, whereas transgenic mice overexpressing TRAF3 developed severe cardiac hypertrophy in response to pressure overload (Jiang et al., 2015); notably, TRAF3 is modestly up-regulated (9.1%) in the R4344Q myocardia. Thus, the absence of apparent cardiac remodeling, yet the development of arrhythmia in 1-year old R4344Q animals (Hu et al., 2017), appears to be modulated in a complex manner by the concomitantly altered expression of several positive and negative regulators of cardiac (mal)adaptation.
Metabolism
Proteins involved in the regulation of metabolic pathways are affected in the R4344Q myocardia, too (Table 5). With the exception of 4-aminobutyrate aminotransferase (Abat) and Mgll whose expression levels are down-regulated, the remaining proteins involved in lipid catabolism are up-regulated (Table 5). Mgll contributes to the catabolism of triglycerides and monoglycerides, and has been implicated in suppressing the inflammatory response post-myocardial infarction (Schloss et al., 2019). Consistent with this, pharmacological inhibition of Mgll exacerbates cardiac pathology post myocardial infarction, as discussed above (Schloss et al., 2019). In contrast to a general up-regulation of proteins involved in lipid catabolism, proteins involved in amino acid metabolism are down-regulated, with the exception of dimethylarginine dimethylaminohydrolase 1 (Ddah1), which was significantly up-regulated (45.5%; Table 5). Ddah1 degrades asymmetric dimethylarginine (ADMA), which inhibits nitric oxide synthase (NOS) and may serve as a marker of cardiovascular disease (Aldamiz-Echevarria and Andrade, 2012). In response to myocardial ischemia, Ddah1 is up-regulated in a canine model, along with reduced ADMA levels in the myocardial interstitial fluid (Zhang et al., 2017). Mice with cardiac-specific deletion of Ddah1 exhibited exacerbated LV hypertrophy, increased ventricular fibrosis, and reduced ejection fraction and fractional shortening in response to transverse aortic constriction and acute myocardial infarction (Xu et al., 2017; Hou et al., 2018). Moreover, Ddah1-null cardiomyocytes showed increased apoptosis when subjected to oxidative stress (Hou et al., 2018). Consistent with this, over-expression of Ddah1 in heart transplants of recipient mice led to attenuation of oxidative stress and suppression of graft coronary artery disease (Tanaka et al., 2005). Thus, up-regulation of Ddah1 may be a compensatory mechanism preventing cardiomyocyte hypertrophy and/or apoptosis in the R4344Q model.
In addition to the alterations in major cellular processes discussed above, we also identified affected molecular pathways. As such, biosynthesis and/or degradation pathways of fatty acids and amino acids, cytokine signaling, circadian rhythm, and the NAD salvage pathway were identified (Figure 2 and Table 6). Notably, with the exception of Cytochrome b5 type A (Cyb5a), Nudix (nucleoside diphosphate linked moiety X)-type motif 12 (Nudt12), Nicotinamide nucleotide adenylyltransferase (Nmnat3) and Nuclear receptor subfamily 1 group D (Nr1d1), all other proteins participating in these pathways contribute to additional cellular processes, discussed in Tables 1–5. Although minimal information is available regarding the function of Cyb5a, Nudt12, and Nr1d1 in cardiac pathophysiology, Nmnat3 was previously suggested to confer anti-hypertrophic effects in response to angiotensin II induced cardiac hypertrophy (Yue et al., 2016). Thus, up-regulation of Nmnat3 (∼19%) may protect the R4344Q knock-in mice from developing hypertrophy under sedentary conditions.
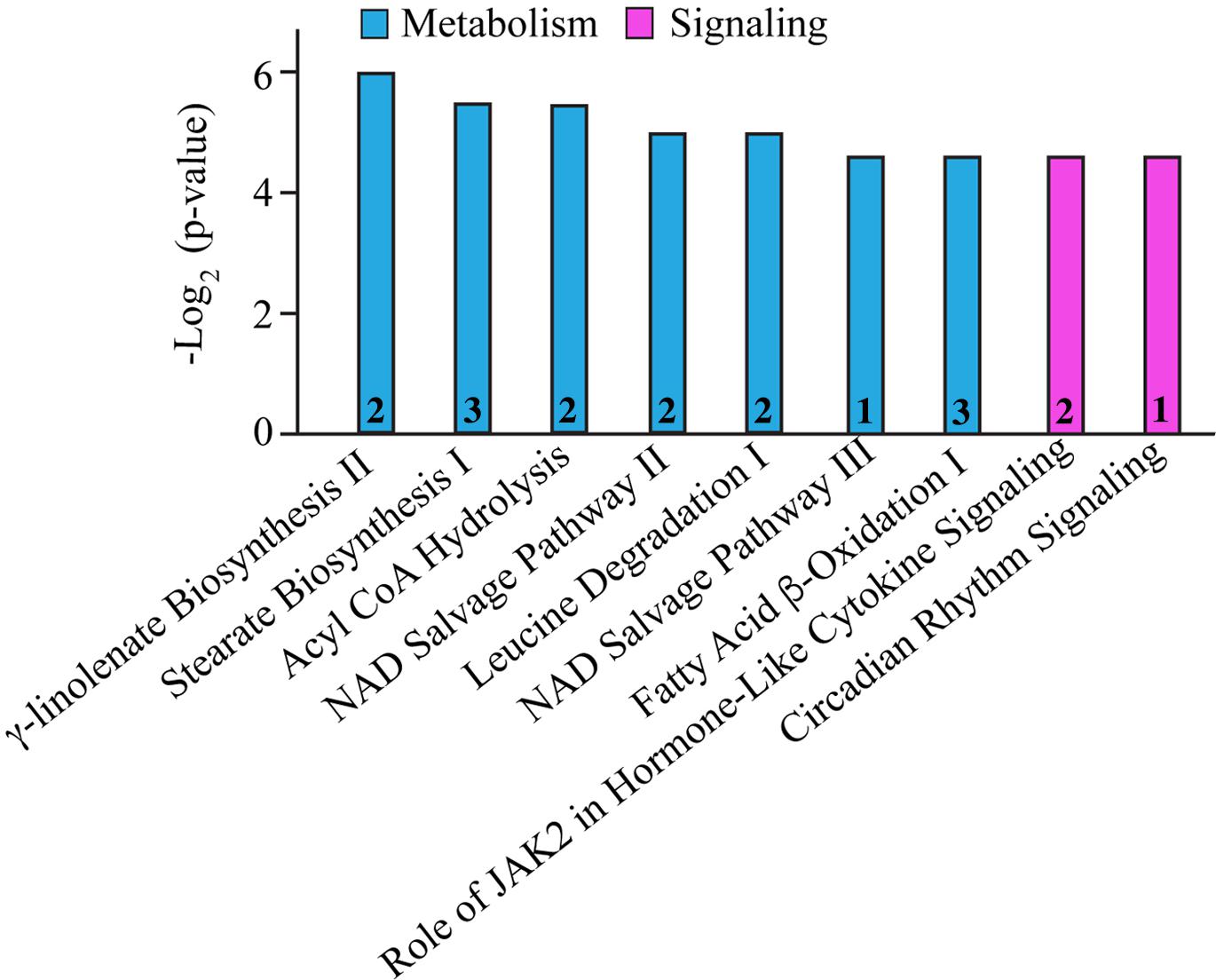
Figure 2. Molecular pathway annotation of proteins with altered expression levels in left ventricles of 1-year-old knock-in female animals. The expression levels of proteins involved in cellular metabolism (cyan) and signaling (pink) pathways are significantly altered in 1-year-old R4344Q myocardia (n = 3) compared to isogenic gender- and age-matched control animals (n = 3). The p-value for each molecular pathway is represented by the height of the respective bar after being transformed into the negative logarithmic value with the base of 2. The number of proteins whose expression levels are altered in each pathway are denoted in the relevant columns.
Altered Phosphorylation Levels
Moreover, we also examined whether the R4344Q LV exhibited alterations in protein post-translational modifications. Although several modifications were tested (e.g., glycosylation and glutathionylation), alterations in phosphorylation were the only significant ones. Two protein kinases (protein kinase cGMP-dependent type 1, PKG1, and Ste20-like kinase, Slk) and one protein phosphatase (protein tyrosine phosphatase, non-receptor type 11, Ptpn11) exhibited increased expression levels in the R4344Q myocardium (Tables 1, 3, 4, 6). PKG1 phosphorylates multiple substrates in the heart including cardiac myosin binding protein-C (cMyBP-C; Thoonen et al., 2015), transient receptor potential canonical channel 6 (TRPC6; Koitabashi et al., 2010), regulator of G-protein signaling subtype 4 (RGS4; Tokudome et al., 2008), and the BK channel, enhancing its activity (Frankenreiter et al., 2017). Although no direct phosphorylation on the L-type voltage-gated Ca2+ channel, Cacna1C, by PKG1 has been reported, it has been suggested that a PKG1-mediated pathway may suppress its open probability (Schroder et al., 2003). Thus, PKG1 may modulate cardiac contractility and output via the coordinated regulation of the activity of multiple ion channels in the heart. Consistent with this, PKG1 appears to have a cardioprotective role in the heart by attenuating hypertrophy and decreasing the extent of fibrosis via inhibition of phosphodiesterase 5 (PDE5) (Nishida et al., 2010). Given the ∼40% upregulation of PKG1 in the R4344Q LV, we speculate that it is one of the compensatory responses of the mutant myocardium contributing to preserved morphology and accounting for the presence of mild fibrosis (Hu et al., 2017). Slk, the second kinase whose expression is altered in the R4344Q myocardium, regulates microtubule organization and focal adhesion turnover through phosphorylation of dynactin and paxillin, respectively (Zhapparova et al., 2013). Up-regulated by 10%, Slk may modulate cytoskeletal organization in the R4344Q myocardium, alongside with other proteins involved in microtubule dynamics that exhibit altered expression. Moreover, the only phosphatase that is altered in the presence of the R4344Q mutation, Ptpn11, is also up-regulated by 20.8%. In cardiac development, Ptpn11 is required for maintenance of cardiac progenitor cells, as inhibition of Ptpn11 prevents cardiac differentiation (Langdon et al., 2007). Overexpression of wild-type Ptpn11 in neonatal rat ventricular cardiomyocytes (NRVM) failed to increase cell length in response to leukemia inhibitory factor (LIF) known to cause hypertrophy, whereas overexpression of a dominant-negative form of Ptpn11 led to attenuated cell elongation in response to LIF (Nakaoka et al., 2010). Moreover, Ptpn11 constitutively dephosphorylates focal adhesion kinase (FAK) and prevents cardiac hypertrophy, as down-regulation of Ptpn11 in NRVM prevented association of Ptpn11 with FAK leading to hypertrophy (Marin et al., 2008). Thus, the ∼20% upregulation of Ptpn11 that we observed in the R4344Q myocardium may aid along with PKG1 in the prevention of a hypertrophic maladaptive response in the R4344Q myocardium under sedentary conditions.
Our proteomics screening identified alterations in the phosphorylation profile of sarcomeric and Ca2+ associated proteins, too (Table 7). As such, myosin heavy chain 6 (Myh6) showed increased phosphorylation levels (∼25%) on Ser1309 in the R4344Q LV compared to wild-type. Ser1309 is located in the coiled-coil region of the myosin tail and has been previously reported as a phosphosite in the murine heart downstream of the β-adrenergic receptor pathway; however, the responsible kinase and functional significance of this modification are still uncharacterized (Lundby et al., 2013). The Ca2+ associated proteins sAnk1.5 and junctophilin 2 (Jph2) exhibited alterations in their phosphorylation status, too. sAnk1.5 residing in the SR membranes displayed a ∼38% increase in the phosphorylation levels of Ser55 (NP_001297366.1; Figure 3). To our knowledge, this is the first study reporting regulation of sAnk1.5 via phosphorylation. Although the impact of Ser55 phosphorylation is currently elusive, it is possible that it may regulate the dynamic binding of sAnk1.5 to obscurin, SERCA and/or SLN/PLN (Bagnato et al., 2003; Kontrogianni-Konstantopoulos et al., 2003; Desmond et al., 2017) (please see above). Contrary to sAnk1.5, Jph2 that links the SR with the transverse tubules (Beavers et al., 2014) exhibited reduced phosphorylation (38%) at Ser597. Ser597 has been previously reported as a phosphosite of Jph2 (Lundby et al., 2013) although neither the responsible kinase nor the biological importance of this modification has been identified. Recently, striated preferentially expressed gene (SPEG) kinase, a homolog of the obscurin kinase isoform, was found to directly interact with Jph2 (Quick et al., 2017). Conditional loss of SPEG in murine hearts resulted in reduced overall Ser phosphorylation levels of Jph-2, but the affected phosphosites are not known (Quick et al., 2017). Lastly, the phosphorylation levels of Kelch domain containing 7A (Klhdc7a) and ubiquitin-associated protein 2-like (Ubap2l) proteins were drastically altered in R4344Q LV with Klhdc7a exhibiting a ∼195% increase in the phosphorylation levels of Ser361 and Ubap2l displaying a ∼53% decrease in the phosphorylation levels of Ser487 compared to wild-type; of note, both of these phospho-sites have been previously reported in the literature (Klhdc7a1 and Ubap212). Currently, no study has addressed the role(s) of Klhdc7a. On the other hand, there are several reports on Ubap2l implicating it in cancer progression, and specifically in cell proliferation and epithelial to mesenchymal transition. Although the role of Ubap2l in the mammalian heart remains to be elucidated, it is reasonable to postulate that it might be involved in growth and/or hypertrophic responses. Given the up-regulation of Pkrg1 and Slk kinases, it is tempting to speculate that they may directly or indirectly mediate at least some of these phosphorylation events, a hypothesis that we plan to address.
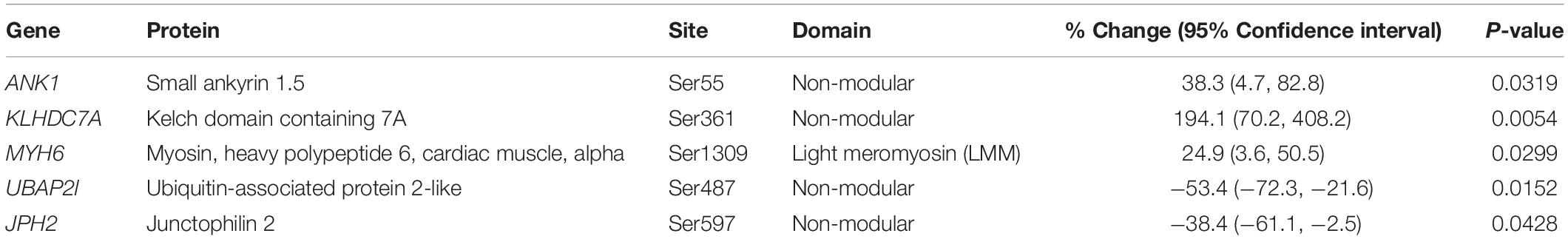
Table 7. Altered phosphorylation levels of identified proteins.
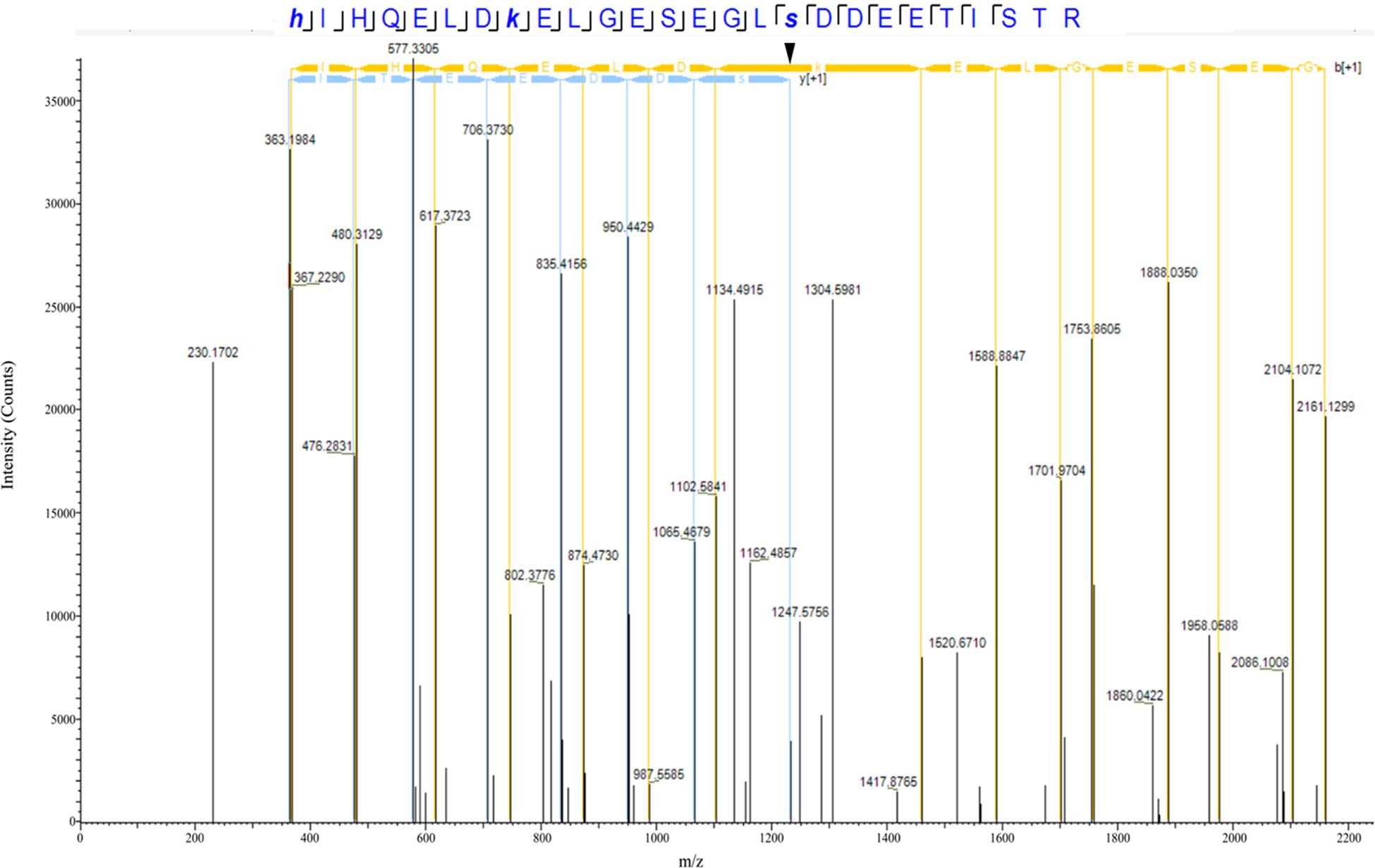
Figure 3. Identification of a novel phosphorylation site in sAnk1. The spectrum of the identified sAnk1 phosphorylated peptide is shown with the phospho-Ser55 peak denoted with an arrowhead.
Taken together, our detailed proteomics study revealed alterations in the expression levels of major proteins involved in cardiac homeostasis, Ca2+ handling, cytoskeletal organization, ion and molecular transport, and metabolism. Given the presence of a single amino acid substitution (R4344Q) in a protein that may contain 6,600–8,000 amino acids depending on the isoform, it is simultaneously striking and perplexing to consider the identified molecular (this study) and functional (Hu et al., 2017) consequences. Although obscurin has been implicated in several cellular processes (Wang et al., 2018), its unequivocal contribution to the organization and cytoskeletal anchoring of the SR membranes and Ca2+ regulation (Bagnato et al., 2003; Kontrogianni-Konstantopoulos et al., 2003; Lange et al., 2009; Hu et al., 2017; Randazzo et al., 2017) strongly suggest that alterations in the expression and phosphorylation levels of Ca2+ handling proteins, ion channels and relevant enzymes may underlie the arrhythmic phenotype observed as a function of aging and the cardiac remodeling in response to stress (Hu et al., 2017). However, given the identified alterations in proteins involved in additional cellular processes for which there is some (e.g., microtubule dynamics; (Randazzo et al., 2013) or no (e.g., metabolism) evidence for the involvement of obscurin, it is appealing to hypothesize that obscurin may have new (previously unidentified) roles in the myocardium. Further work will assess this intriguing possibility, especially given the escalating number of OBSCN missense mutations associated with different forms of congenital cardiomyopathy (Grogan and Kontrogianni-Konstantopoulos, 2018).
Although our comprehensive proteomics analysis yielded a number of important and novel findings, we are cognizant of potential limitations of our study, including: (1) the relatively small number of animals that was analyzed per genotype, (2) the lack of relevant human biopsies, and (3) the examination of homozygous, rather than heterozygous, R4344Q animals, given that the mutation exhibits a dominant inheritance in humans.
Variability in isogenic animals is indeed a major concern for “omics” studies, but also a reality, whether a small or a large cohort is used. Although the source of variability is not well-understood, studies suggest that it might originate from random genetic variations despite extensive inbreeding, environmental influences such as intrauterine position of embryos or feeding hierarchy and maternal care of newborns as well as stochastic events affecting molecular and cellular processes (Loos et al., 2015). As such, there has been a recent tendency to perform individualized sample analysis, especially when human biopsies are used, refraining from collating and averaging data, a tactic that has likely obscured important idiosyncratic alterations. At this time, we are not aware of available cardiac biopsies that carry the obscurin R4344Q mutation or the disease progression and life-span of the affected patients, therefore evaluation of our preclinical mouse model appears to be the most optimal approach.
Moreover, our earlier (Hu et al., 2017) and current studies have focused on the homozygous population of the R4344Q model although the monoallelic presence of the mutation in humans is linked to HCM. Examination of the heterozygous R4344Q population under sedentary conditions did not reveal any cardiac pathology. This is not an uncommon phenomenon, where disease severity manifests to different extents between humans and the respective preclinical models, likely due to the phylogenetic distance of humans and rodents, species-specific differences, and the presence of non-controlled environmental influences and stresses that humans are subjected to (Camacho et al., 2016).
Notwithstanding the aforementioned limitations, our comprehensive proteomics analysis provides important insights about the molecular and cellular alterations that take place in the R4344Q myocardium by confirming some obvious ones and unraveling some new and unexpected ones to be interrogated in the future.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committees of the University of Maryland School of Medicine.
Author Contributions
AK-K conceived the study. L-YH and AK-K designed the study and wrote the manuscript. L-YH performed the experiments and analyzed the data.
Funding
This work was supported by NIH/NIAMS (2T32AR7592-16 to L-YH) and the American Heart Association (16GRNT31290010 to AK-K).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Drs. Robert Cole and Tatiana Boronina for their technical assistance with tandem mass-tagged mass spectrometry (Mass Spectrometry and Proteomics Facility, Johns Hopkins University School of Medicine), and Mr. Conover Talbot (Transcriptomics and Deep Sequencing Core/Core Analysis Unit, Johns Hopkins University School of Medicine) for his assistance with the biostatistical analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2020.00478/full#supplementary-material
FILE S1 | Relative abundance of proteins identified in the R4344Q-obscurin myocardia compared to wild-type.
Footnotes
References
Ackermann, M. A., Hu, L. Y., Bowman, A. L., Bloch, R. J., and Kontrogianni-Konstantopoulos, A. (2009). Obscurin interacts with a novel isoform of MyBP-C slow at the periphery of the sarcomeric M-band and regulates thick filament assembly. Mol. Biol. Cell 20, 2963–2978. doi: 10.1091/mbc.e08-12-1251
Ackermann, M. A., King, B., Lieberman, N. A. P., Bobbili, P. J., Rudloff, M., Berndsen, C. E., et al. (2017). Novel obscurins mediate cardiomyocyte adhesion and size via the PI3K/AKT/mTOR signaling pathway. J. Mol. Cell Cardiol. 111, 27–39. doi: 10.1016/j.yjmcc.2017.08.004
Aebersold, R., Burlingame, A. L., and Bradshaw, R. A. (2013). Western blots versus selected reaction monitoring assays: time to turn the tables? Mol. Cell Proteomics. 12, 2381–2382. doi: 10.1074/mcp.E113.031658
Aldamiz-Echevarria, L., and Andrade, F. (2012). Asymmetric dimethylarginine, endothelial dysfunction and renal disease. Int. J. Mol. Sci. 13, 11288–11311. doi: 10.3390/ijms130911288
Arama, J., Boulay, A. C., Bosc, C., Delphin, C., Loew, D., Rostaing, P., et al. (2012). Bmcc1s, a novel brain-isoform of Bmcc1, affects cell morphology by regulating MAP6/STOP functions. PLoS One 7:e35488. doi: 10.1371/journal.pone.0035488
Arimura, T., Matsumoto, Y., Okazaki, O., Hayashi, T., Takahashi, M., Inagaki, N., et al. (2007). Structural analysis of obscurin gene in hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 362, 281–287. doi: 10.1016/j.bbrc.2007.07.183
Atanasiu, R., Dumoulin, M. J., Chahine, R., Mateescu, M. A., and Nadeau, R. (1995). Antiarrhythmic effects of ceruloplasmin during reperfusion in the ischemic isolated rat heart. Can. J. Physiol. Pharmacol. 73, 1253–1261. doi: 10.1139/y95-177
Bagnato, P., Barone, V., Giacomello, E., Rossi, D., and Sorrentino, V. (2003). Binding of an ankyrin-1 isoform to obscurin suggests a molecular link between the sarcoplasmic reticulum and myofibrils in striated muscles. J. Cell Biol. 160, 245–253. doi: 10.1083/jcb.200208109
Beavers, D. L., Landstrom, A. P., Chiang, D. Y., and Wehrens, X. H. (2014). Emerging roles of junctophilin-2 in the heart and implications for cardiac diseases. Cardiovasc. Res. 103, 198–205. doi: 10.1093/cvr/cvu151
Bowman, A. L., Kontrogianni-Konstantopoulos, A., Hirsch, S. S., Geisler, S. B., Gonzalez-Serratos, H., Russell, M. W., et al. (2007). Different obscurin isoforms localize to distinct sites at sarcomeres. FEBS Lett. 581, 1549–1554. doi: 10.1016/j.febslet.2007.03.011
Busby, B., Willis, C. D., Ackermann, M. A., Kontrogianni-Konstantopoulos, A., and Bloch, R. J. (2010). Characterization and comparison of two binding sites on obscurin for small ankyrin 1. Biochemistry 49, 9948–9956. doi: 10.1021/bi101165p
Cabassi, A., Binno, S. M., Tedeschi, S., Ruzicka, V., Dancelli, S., Rocco, R., et al. (2014). Low serum ferroxidase I activity is associated with mortality in heart failure and related to both peroxynitrite-induced cysteine oxidation and tyrosine nitration of ceruloplasmin. Circ. Res. 114, 1723–1732. doi: 10.1161/CIRCRESAHA.114.302849
Camacho, P., Fan, H., Liu, Z., and He, J. Q. (2016). Small mammalian animal models of heart disease. Am. J. Cardiovasc. Dis. 6, 70–80.
Cernohorska, M., Sulimenko, V., Hajkova, Z., Sulimenko, T., Sladkova, V., Vinopal, S., et al. (2016). GIT1/betaPIX signaling proteins and PAK1 kinase regulate microtubule nucleation. Biochim. Biophys. Acta 1863, 1282–1297. doi: 10.1016/j.bbamcr.2017.01.002
Chaldakov, G. N. (2018). Colchicine, a microtubule-disassembling drug, in the therapy of cardiovascular diseases. Cell Biol. Int. 42, 1079–1084. doi: 10.1002/cbin.10988
Chowdhury, D., Tangutur, A. D., Khatua, T. N., Saxena, P., Banerjee, S. K., and Bhadra, M. P. (2013). A proteomic view of isoproterenol induced cardiac hypertrophy: prohibitin identified as a potential biomarker in rats. J. Transl. Med. 11:130. doi: 10.1186/1479-5876-11-130
Clemo, H. F., and Baumgarten, C. M. (1995). cGMP and atrial natriuretic factor regulate cell volume of rabbit atrial myocytes. Circ. Res. 77, 741–749. doi: 10.1161/01.res.77.4.741
Desmond, P. F., Labuza, A., Muriel, J., Markwardt, M. L., Mancini, A. E., Rizzo, M. A., et al. (2017). Interactions between small ankyrin 1 and sarcolipin coordinately regulate activity of the sarco(endo)plasmic reticulum Ca(2+)-ATPase (SERCA1). J. Biol. Chem. 292, 10961–10972. doi: 10.1074/jbc.M117.783613
Desmond, P. F., Muriel, J., Markwardt, M. L., Rizzo, M. A., and Bloch, R. J. (2015). Identification of small ankyrin 1 as a novel sarco(endo)plasmic reticulum Ca2+-ATPase 1 (SERCA1) regulatory protein in skeletal muscle. J. Biol. Chem. 290, 27854–27867. doi: 10.1074/jbc.M115.676585
Dong, W. Q., Chao, M., Lu, Q. H., Chai, W. L., Zhang, W., Chen, X. Y., et al. (2016). Prohibitin overexpression improves myocardial function in diabetic cardiomyopathy. Oncotarget. 7, 66–80. doi: 10.18632/oncotarget.6384
Finlay, M., Harmer, S. C., and Tinker, A. (2017). The control of cardiac ventricular excitability by autonomic pathways. Pharmacol. Ther. 174, 97–111. doi: 10.1016/j.pharmthera.2017.02.023
Ford-Speelman, D. L., Roche, J. A., Bowman, A. L., and Bloch, R. J. (2009). The rho-guanine nucleotide exchange factor domain of obscurin activates rhoA signaling in skeletal muscle. Mol. Biol. Cell 20, 3905–3917. doi: 10.1091/mbc.e08-10-1029
Foster, M. N., and Coetzee, W. A. (2016). KATP channels in the cardiovascular system. Physiol. Rev. 96, 177–252.
Frankenreiter, S., Bednarczyk, P., Kniess, A., Bork, N. I., Straubinger, J., Koprowski, P., et al. (2017). cGMP-Elevating compounds and ischemic conditioning provide cardioprotection against ischemia and reperfusion injury via cardiomyocyte-specific BK channels. Circulation 136, 2337–2355. doi: 10.1161/CIRCULATIONAHA.117.028723
Friedrich, F. W., Reischmann, S., Schwalm, A., Unger, A., Ramanujam, D., Munch, J., et al. (2014). FHL2 expression and variants in hypertrophic cardiomyopathy. Basic Res. Cardiol. 109:451. doi: 10.1007/s00395-014-0451-8
Fukuzawa, A., Lange, S., Holt, M., Vihola, A., Carmignac, V., Ferreiro, A., et al. (2008). Interactions with titin and myomesin target obscurin and obscurin-like 1 to the M-band: implications for hereditary myopathies. J. Cell Sci. 121, 1841–1851. doi: 10.1242/jcs.028019
Grogan, A., and Kontrogianni-Konstantopoulos, A. (2018). Unraveling obscurins in heart disease. Pflugers Arch. 471:735. doi: 10.1007/s00424-018-2191-3
Grunnet, M. (2010). Repolarization of the cardiac action potential. Does an increase in repolarization capacity constitute a new anti-arrhythmic principle? Acta Physiol. 198(Suppl. 676), 1–48. doi: 10.1111/j.1748-1716.2009.02072.x
Holloway, G. P., Snook, L. A., Harris, R. J., Glatz, J. F., Luiken, J. J., and Bonen, A. (2011). In obese Zucker rats, lipids accumulate in the heart despite normal mitochondrial content, morphology and long-chain fatty acid oxidation. J. Physiol. 589, 169–180. doi: 10.1113/jphysiol.2010.198663
Hou, L., Guo, J., Xu, F., Weng, X., Yue, W., and Ge, J. (2018). Cardiomyocyte dimethylarginine dimethylaminohydrolase1 attenuates left-ventricular remodeling after acute myocardial infarction: involvement in oxidative stress and apoptosis. Basic Res. Cardiol. 113:28. doi: 10.1007/s00395-018-0685-y
Hu, L. R., Ackermann, M. A., Hecker, P. A., Prosser, B. L., King, B., O’Connell, K. A., et al. (2017). Deregulated Ca(2+) cycling underlies the development of arrhythmia and heart disease due to mutant obscurin. Sci. Adv. 3:e1603081. doi: 10.1126/sciadv.1603081
Hu, L. Y., Ackermann, M. A., and Kontrogianni-Konstantopoulos, A. (2015). The sarcomeric M-region: a molecular command center for diverse cellular processes. Biomed Res. Int. 2015:714197. doi: 10.1155/2015/714197
Hu, L. Y., and Kontrogianni-Konstantopoulos, A. (2013). The kinase domains of obscurin interact with intercellular adhesion proteins. FASEB J. 27, 2001–2012. doi: 10.1096/fj.12-221317
Hubbert, C., Guardiola, A., Shao, R., Kawaguchi, Y., Ito, A., Nixon, A., et al. (2002). HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458. doi: 10.1074/jbc.M109.042754
Ichii, T., and Takeichi, M. (2007). p120-catenin regulates microtubule dynamics and cell migration in a cadherin-independent manner. Genes Cells 12, 827–839. doi: 10.1111/j.1365-2443.2007.01095.x
Jeppesen, J., Jordy, A. B., Sjoberg, K. A., Fullekrug, J., Stahl, A., Nybo, L., et al. (2012). Enhanced fatty acid oxidation and FATP4 protein expression after endurance exercise training in human skeletal muscle. PLoS One 7:e29391. doi: 10.1371/journal.pone.0029391
Jiang, D. S., Zhang, X. F., Gao, L., Zong, J., Zhou, H., Liu, Y., et al. (2014). Signal regulatory protein-alpha protects against cardiac hypertrophy via the disruption of toll-like receptor 4 signaling. Hypertension 63, 96–104. doi: 10.1161/HYPERTENSIONAHA.113.01506
Jiang, X., Deng, K. Q., Luo, Y., Jiang, D. S., Gao, L., Zhang, X. F., et al. (2015). Tumor necrosis factor receptor-associated factor 3 is a positive regulator of pathological cardiac hypertrophy. Hypertension 66, 356–367. doi: 10.1161/HYPERTENSIONAHA.115.05469
Koitabashi, N., Aiba, T., Hesketh, G. G., Rowell, J., Zhang, M., Takimoto, E., et al. (2010). Cyclic GMP/PKG-dependent inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation Novel mechanism of cardiac stress modulation by PDE5 inhibition. J. Mol. Cell Cardiol. 48, 713–724. doi: 10.1016/j.yjmcc.2009.11.015
Kontrogianni-Konstantopoulos, A., Catino, D. H., Strong, J. C., Sutter, S., Borisov, A. B., Pumplin, D. W., et al. (2006). Obscurin modulates the assembly and organization of sarcomeres and the sarcoplasmic reticulum. FASEB J. 20, 2102–2111. doi: 10.1096/fj.06-5761com
Kontrogianni-Konstantopoulos, A., Jones, E. M., Van Rossum, D. B., and Bloch, R. J. (2003). Obscurin is a ligand for small ankyrin 1 in skeletal muscle. Mol. Biol. Cell 14, 1138–1148. doi: 10.1091/mbc.e02-07-0411
Langdon, Y. G., Goetz, S. C., Berg, A. E., Swanik, J. T., and Conlon, F. L. (2007). SHP-2 is required for the maintenance of cardiac progenitors. Development 134, 4119–4130. doi: 10.1242/dev.009290
Lange, S., Ouyang, K., Meyer, G., Cui, L., Cheng, H., Lieber, R. L., et al. (2009). Obscurin determines the architecture of the longitudinal sarcoplasmic reticulum. J. Cell Sci. 122, 2640–2650. doi: 10.1242/jcs.046193
Lee, H. C., Rudy, Y., Liang, H., Chen, C. C., Luo, C. H., Sheu, S. H., et al. (2017). Pro-arrhythmogenic Effects of the V141M KCNQ1 mutation in short QT syndrome and its potential therapeutic targets: insights from modeling. J. Med. Biol. Eng. 37, 780–789. doi: 10.1007/s40846-017-0257-x
Liang, Y., Bradford, W. H., Zhang, J., and Sheikh, F. (2018). Four and a half LIM domain protein signaling and cardiomyopathy. Biophys. Rev. 10, 1073–1085. doi: 10.1007/s12551-018-0434-3
Loos, M., Koopmans, B., Aarts, E., Maroteaux, G., van der Sluis, S., Neuro, B. M. P. C., et al. (2015). Within-strain variation in behavior differs consistently between common inbred strains of mice. Mamm Genome 26, 348–354. doi: 10.1007/s00335-015-9578-7
Lundby, A., Andersen, M. N., Steffensen, A. B., Horn, H., Kelstrup, C. D., Francavilla, C., et al. (2013). In vivo phosphoproteomics analysis reveals the cardiac targets of beta-adrenergic receptor signaling. Sci. Signal. 6:rs11. doi: 10.1126/scisignal.2003506
Marin, T. M., Clemente, C. F., Santos, A. M., Picardi, P. K., Pascoal, V. D., Lopes-Cendes, I., et al. (2008). Shp2 negatively regulates growth in cardiomyocytes by controlling focal adhesion kinase/Src and mTOR pathways. Circ. Res. 103, 813–824. doi: 10.1161/CIRCRESAHA.108.179754
McElhanon, K. E., and Bhattacharya, S. (2018). Altered membrane integrity in the progression of muscle diseases. Life Sci. 192, 166–172. doi: 10.1016/j.lfs.2017.11.035
Milger, K., Herrmann, T., Becker, C., Gotthardt, D., Zickwolf, J., Ehehalt, R., et al. (2006). Cellular uptake of fatty acids driven by the ER-localized acyl-CoA synthetase FATP4. J. Cell Sci. 119, 4678–4688. doi: 10.1242/jcs.03280
Nakaoka, Y., Shioyama, W., Kunimoto, S., Arita, Y., Higuchi, K., Yamamoto, K., et al. (2010). SHP2 mediates gp130-dependent cardiomyocyte hypertrophy via negative regulation of skeletal alpha-actin gene. J. Mol. Cell Cardiol. 49, 157–164. doi: 10.1016/j.yjmcc.2010.03.001
Nishida, M., Watanabe, K., Sato, Y., Nakaya, M., Kitajima, N., Ide, T., et al. (2010). Phosphorylation of TRPC6 channels at Thr69 is required for anti-hypertrophic effects of phosphodiesterase 5 inhibition. J. Biol. Chem. 285, 13244–13253. doi: 10.1074/jbc.M109.074104
Osman, C., Merkwirth, C., and Langer, T. (2009). Prohibitins and the functional compartmentalization of mitochondrial membranes. J. Cell Sci. 122, 3823–3830. doi: 10.1242/jcs.037655
Perez-Riverol, Y., Csordas, A., Bai, J., Bernal-Llinares, M., Hewapathirana, S., Kundu, D. J., et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450. doi: 10.1093/nar/gky1106
Qadota, H., Mayans, O., Matsunaga, Y., McMurry, J. L., Wilson, K. J., Kwon, G. E., et al. (2016). The SH3 domain of UNC-89 (obscurin) interacts with paramyosin, a coiled-coil protein, in Caenorhabditis elegans muscle. Mol. Biol. Cell 27, 1606–1620. doi: 10.1091/mbc.E15-09-0675
Quick, A. P., Wang, Q., Philippen, L. E., Barreto-Torres, G., Chiang, D. Y., Beavers, D., et al. (2017). SPEG (Striated muscle preferentially expressed protein kinase) is essential for cardiac function by regulating junctional membrane complex activity. Circ. Res. 120, 110–119. doi: 10.1161/CIRCRESAHA.116.309977
Randazzo, D., Blaauw, B., Paolini, C., Pierantozzi, E., Spinozzi, S., Lange, S., et al. (2017). Exercise-induced alterations and loss of sarcomeric M-line organization in the diaphragm muscle of obscurin knockout mice. Am. J. Physiol. Cell Physiol. 312, C16–C28. doi: 10.1152/ajpcell.00098.2016
Randazzo, D., Giacomello, E., Lorenzini, S., Rossi, D., Pierantozzi, E., Blaauw, B., et al. (2013). Obscurin is required for ankyrinB-dependent dystrophin localization and sarcolemma integrity. J. Cell Biol. 200, 523–536. doi: 10.1083/jcb.201205118
Saegusa, N., Sato, T., Saito, T., Tamagawa, M., Komuro, I., and Nakaya, H. (2005). Kir6.2-deficient mice are susceptible to stimulated ANP secretion: K(ATP) channel acts as a negative feedback mechanism? Cardiovasc. Res. 67, 60–68. doi: 10.1016/j.cardiores.2005.03.011
Schleicher, M., Shepherd, B. R., Suarez, Y., Fernandez-Hernando, C., Yu, J., Pan, Y., et al. (2008). Prohibitin-1 maintains the angiogenic capacity of endothelial cells by regulating mitochondrial function and senescence. J. Cell Biol. 180, 101–112. doi: 10.1083/jcb.200706072
Schloss, M. J., Horckmans, M., Guillamat-Prats, R., Hering, D., Lauer, E., Lenglet, S., et al. (2019). 2-Arachidonoylglycerol mobilizes myeloid cells and worsens heart function after acute myocardial infarction. Cardiovasc. Res. 115, 602–613. doi: 10.1093/cvr/cvy242
Schroder, F., Klein, G., Fiedler, B., Bastein, M., Schnasse, N., Hillmer, A., et al. (2003). Single L-type Ca(2+) channel regulation by cGMP-dependent protein kinase type I in adult cardiomyocytes from PKG I transgenic mice. Cardiovasc. Res. 60, 268–277. doi: 10.1016/s0008-6363(03)00546-7
Shaikh, S. A., Sahoo, S. K., and Periasamy, M. (2016). Phospholamban and sarcolipin: are they functionally redundant or distinct regulators of the Sarco(Endo)plasmic reticulum calcium ATPase? J. Mol. Cell Cardiol. 91, 81–91. doi: 10.1016/j.yjmcc.2015.12.030
Shriver, M., Marimuthu, S., Paul, C., Geist, J., Seale, T., Konstantopoulos, K., et al. (2016). Giant obscurins regulate the PI3K cascade in breast epithelial cells via direct binding to the PI3K/p85 regulatory subunit. Oncotarget 7, 45414–45428. doi: 10.18632/oncotarget.9985
Sokolowski, B., Orchard, S., Harvey, M., Sridhar, S., and Sakai, Y. (2011). Conserved BK channel-protein interactions reveal signals relevant to cell death and survival. PLoS One 6:e28532. doi: 10.1371/journal.pone.0028532
Song, W., Wang, H., and Wu, Q. (2015). Atrial natriuretic peptide in cardiovascular biology and disease (NPPA). Gene 569, 1–6. doi: 10.1016/j.gene.2015.06.029
Spooner, P. M., Bonner, J., Maricq, A. V., Benian, G. M., and Norman, K. R. (2012). Large isoforms of UNC-89 (obscurin) are required for muscle cell architecture and optimal calcium release in Caenorhabditis elegans. PLoS One 7:e40182. doi: 10.1371/journal.pone.0040182
Tanaka, M., Fedoseyeva, E. V., and Robbins, R. C. (2005). Graft coronary artery disease in murine cardiac allografts: proposal to meet the need for standardized assessment. J. Heart Lung. Transplant. 24, 316–322. doi: 10.1016/j.healun.2003.12.011
Tester, D. J., and Ackerman, M. J. (2014). Genetics of long QT syndrome. Methodist. Debakey Cardiovasc. J. 10, 29–33.
Thompson, A., Schafer, J., Kuhn, K., Kienle, S., Schwarz, J., Schmidt, G., et al. (2003). Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904. doi: 10.1021/ac0262560
Thoonen, R., Giovanni, S., Govindan, S., Lee, D. I., Wang, G. R., Calamaras, T. D., et al. (2015). Molecular screen identifies cardiac myosin-binding protein-c as a protein kinase G-Ialpha Substrate. Circ. Heart Fail 8, 1115–1122. doi: 10.1161/CIRCHEARTFAILURE.115.002308
Tien, S. C., and Chang, Z. F. (2014). Oncogenic Shp2 disturbs microtubule regulation to cause HDAC6-dependent ERK hyperactivation. Oncogene 33, 2938–2946. doi: 10.1038/onc.2013.241
Tokudome, T., Kishimoto, I., Horio, T., Arai, Y., Schwenke, D. O., Hino, J., et al. (2008). Regulator of G-protein signaling subtype 4 mediates antihypertrophic effect of locally secreted natriuretic peptides in the heart. Circulation 117, 2329–2339. doi: 10.1161/CIRCULATIONAHA.107.732990
Ueno, M., Suzuki, J., Hirose, M., Sato, S., Imagawa, M., Zenimaru, Y., et al. (2017). Cardiac overexpression of perilipin 2 induces dynamic steatosis: prevention by hormone-sensitive lipase. Am. J. Physiol. Endocrinol. Metab. 313, E699–E709. doi: 10.1152/ajpendo.00098.2017
Wang, B., and Wang, X. P. (2018). Does ceruloplasmin defend against neurodegenerative diseases? Curr. Neuropharmacol. 7, 539–549. doi: 10.2174/1570159X16666180508113025
Wang, L., Geist, J., Grogan, A., Hu, L. R., and Kontrogianni-Konstantopoulos, A. (2018). Thick Filament Protein Network. Functions, and disease association. Compr. Physiol. 8, 631–709. doi: 10.1002/cphy.c170023
Xu, X., Zhang, P., Kwak, D., Fassett, J., Yue, W., Atzler, D., et al. (2017). Cardiomyocyte dimethylarginine dimethylaminohydrolase-1 (DDAH1) plays an important role in attenuating ventricular hypertrophy and dysfunction. Basic Res. Cardiol. 112:55. doi: 10.1007/s00395-017-0644-z
Yue, Z., Ma, Y., You, J., Li, Z., Ding, Y., He, P., et al. (2016). NMNAT3 is involved in the protective effect of SIRT3 in Ang II-induced cardiac hypertrophy. Exp. Cell Res. 347, 261–273. doi: 10.1016/j.yexcr.2016.07.006
Zhang, P., Fassett, J. T., Zhu, G., Li, J., Hu, X., Xu, X., et al. (2017). Repetitive ischemia increases myocardial dimethylarginine dimethylaminohydrolase 1 expression. Vasc. Med. 22, 179–188. doi: 10.1177/1358863X16681215
Keywords: obscurin, cardiomyopathy, tandem mass-tagged mass spectrometry, phosphoproteomics, cytoskeleton, Ca2+ cycling, metabolism, ion and molecular transport
Citation: Hu L-YR and Kontrogianni-Konstantopoulos A (2020) Proteomic Analysis of Myocardia Containing the Obscurin R4344Q Mutation Linked to Hypertrophic Cardiomyopathy. Front. Physiol. 11:478. doi: 10.3389/fphys.2020.00478
Received: 13 December 2019; Accepted: 20 April 2020;
Published: 18 May 2020.
Edited by:
Julian Stelzer, Case Western Reserve University, United StatesReviewed by:
Steven Baxter Marston, Imperial College London, United KingdomJohn Jeshurun Michael, Cornell University, United States
Aldrin V. Gomes, University of California, Davis, United States
Copyright © 2020 Hu and Kontrogianni-Konstantopoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aikaterini Kontrogianni-Konstantopoulos, YWtvbnRyb2dpYW5uaUBzb20udW1hcnlsYW5kLmVkdQ==; YWtvbnMwMDFAdW1hcnlsYW5kLmVkdQ==