 Kristin Wenzel1,2
Kristin Wenzel1,2 Elisabeth Krämer3,4Birgit Geertz3,4
Elisabeth Krämer3,4Birgit Geertz3,4 Lucie Carrier3,4Stephan B. Felix1,2
Lucie Carrier3,4Stephan B. Felix1,2 Stephanie Könemann1,2
Stephanie Könemann1,2 Saskia Schlossarek3,4*
Saskia Schlossarek3,4*- 1Department of Internal Medicine B, University Medicine Greifswald, Greifswald, Germany
- 2German Centre for Cardiovascular Research (DZHK), partner site Greifswald, Greifswald, Germany
- 3Institute of Experimental Pharmacology and Toxicology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 4German Centre for Cardiovascular Research (DZHK), partner site Hamburg/Kiel/Lübeck, Hamburg, Germany
The ubiquitin-proteasome system (UPS) and the autophagy-lysosomal pathway (ALP) are the main proteolytic systems involved in cellular homeostasis. Since cardiomyocytes, as terminally differentiated cells, lack the ability to share damaged proteins with their daughter cells, they are especially reliant on these protein degradation systems for their proper function. Alterations of the UPS and ALP have been reported in a wide range of cardiac diseases, including cardiomyopathies. In this study, we determined whether the UPS and ALP are altered in a mouse model of eccentric left ventricular (LV) hypertrophy expressing both cyclin T1 and Gαq under the control of the cardiac-specific α-myosin heavy chain promoter (double transgenic; DTG). Compared to wild-type (WT) littermates, DTG mice showed higher end-diastolic (ED) LV wall thicknesses and diameter with preserved ejection fraction (EF). The cardiomyopathic phenotype was further confirmed by an upregulation of the fetal gene program and genes associated with fibrosis as well as a downregulation of genes involved in Ca2+ handling. Likewise, higher NT-proBNP levels were detected in DTG mice. Investigation of the UPS showed elevated steady-state levels of (poly)ubiquitinated proteins without alterations of all proteasomal activities in DTG mice. Evaluation of ALP key marker revealed a mixed pattern with higher protein levels of microtubule-associated protein 1 light chain 3 beta (LC3)-I and lysosomal-associated membrane protein-2, lower protein levels of beclin-1 and FYVE and coiled-coil domain-containing protein 1 (FYCO1) and unchanged protein levels of p62/SQSTM1 in DTG mice when compared to WT. At transcriptional level, a > 1.2-fold expression was observed for Erbb2, Hdac6, Lamp2, Nrg1, and Sqstm1, while a < 0.8-fold expression was revealed for Fyco1 in DTG mice. The results related to the ALP suggested overall a repression of the ALP during the initiation process, but an induction of the ALP at the level of autophagosome-lysosome fusion and the delivery of ubiquitinated cargo to the ALP for degradation.
Introduction
The adult heart undergoes distinct remodeling processes in response to acute or chronic insults, which involve myocardial hypertrophy, ventricular wall thickening and dilatation as well as cardiomyocyte apoptosis and the development of fibrosis (Frey and Olson, 2003; Dorn and Force, 2005). Cardiac myocytes are terminally differentiated and maintain their cellular homeostasis by activation of degradation programs. The two main proteolytic systems are the ubiquitin-proteasome system (UPS) and the autophagy-lysosomal pathway (ALP). The highly selective degradation process by the UPS is ATP-dependent and involves the polyubiquitination of a target protein through a series of enzymatic reactions and the subsequent degradation of this (poly)ubiquitinated protein by the 26S proteasome (Ciechanover, 2007). During the proteolytic process of ALP small portions of the cytoplasm or complete organelles are enclosed by a phagophore to form a double−membrane vesicle, termed autophagosome, which subsequently fuses with a lysosome to form an autolysosome, in which lysosomal proteases degrade the autophagosomal content (Mizushima et al., 2008). Several lines of evidence indicate that alterations of the UPS and the ALP may be involved in cardiac diseases, such as dilated and hypertrophic cardiomyopathies (Mearini et al., 2008; Zheng and Wang, 2010; Day, 2013; Zech et al., 2020). A marked accumulation of (poly)ubiquitinated proteins has been reported as a common feature of cardiac disorders (Weekes et al., 2003; Birks et al., 2008; Predmore et al., 2010), whereas the proteasomal activities have been shown to be higher or lower depending on the status of the cardiac disease (Depre et al., 2006; Tsukamoto et al., 2006; Birks et al., 2008; Predmore et al., 2010; Schlossarek et al., 2012; Thottakara et al., 2015). Likewise, an altered autophagic flux and dysregulated expression of ALP key marker have been observed in a wide range of cardiac diseases such as desmin-related cardiomyopathy (Maloyan et al., 2010; Pattison et al., 2011; Bhuiyan et al., 2013), dilated cardiomyopathy (Choi et al., 2012; Ramos et al., 2012) and hypertrophic cardiomyopathy with reduced ejection fraction (EF; Schlossarek et al., 2012; Singh et al., 2017). The role of the ALP in the development of cardiac hypertrophy and its progression to heart failure is being discussed controversially (Zhu et al., 2007; Rothermel and Hill, 2008). Currently, a basal ALP activity is thought to be important to maintain normal cardiac function, whereas a decrease or an increase in ALP activity could mediate the adaption of the heart during stress conditions (Lavandero et al., 2013).
Traditionally, the UPS and ALP have been considered to act separately, but recent data suggest that they functionally cooperate with each other to maintain cellular homeostasis (Wang et al., 2008; Korolchuk et al., 2010; Lamark and Johansen, 2010; Singh et al., 2020). In the present study, we therefore aimed to investigate alterations of both the UPS and the ALP in a mouse model of eccentric left ventricular (LV) hypertrophy with preserved EF expressing both cyclin T1 and Gαq under the control of the cardiac-specific α-myosin heavy chain promoter.
Materials and Methods
Ethics Statement
All experimental procedures were conducted in compliance with the national guidelines of the German animal protective law for the use of laboratory animals. All animal procedures conform to the guidelines of Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. The protocols used were approved by the local animal care committee (Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei Mecklenburg-Vorpommern; LALLF-MV, 7221.3-1-061/15).
Experimental Animals
Double transgenic (DTG) α-myosin heavy chain (αMHC)-cyclin T1 × αMHC-Gαq mice on a Friend leukaemia virus strain (FVB) background of either sex were obtained by crossbreeding heterogeneous αMHC-cyclin T1 mice, kindly provided by Prof. Michael D. Schneider (Imperial College London, United Kingdom), with αMHC-Gαq overexpressing mice, courtesy of Prof. Gerald W. Dorn 2nd (Washington University, St. Louis, United States). The heart-specific activation of Cdk9 via forced expression of cyclin T1 induces LV hypertrophy by phosphorylation of the RNA polymerase II (Sano et al., 2004). Gαq overexpression further enhances the transcription elongation, thereby serving as an additional hypertrophic stimulus (D’Angelo et al., 1997). Gαq overexpression results in cardiac hypertrophy, defined as a conserved program of fetal gene expression, increased heart weight, and increased cardiomyocyte size. Thus, the DTG mice show a global increase in RNA synthesis leading to myocyte enlargement and hypertrophic growth and further development of fibrosis as well as apoptosis.
Wild-type (WT) littermates were used as healthy controls and both genotypes were investigated at the age of 6–8 weeks.
Study Design and Tissue Processing
Cardiac function of 6–8-weeks-old mice was assessed via magnetic resonance imaging (MRI). Afterward, the mice were sacrificed while still being under isoflurane anesthesia. Hearts were harvested and approximately 500 μL whole blood was collected. Organs were washed in PBS and subsequently frozen at −80°C. Blood was transferred to an Eppendorf tube containing 0.3 μM EDTA (10 μL/200 μL blood) and centrifuged at 300 g for 15 min at room temperature. Plasma was transferred to a new Eppendorf tube and stored at −20°C.
Magnetic Resonance Imaging Measurement
Anesthesia was induced using 4% isoflurane in 100% oxygen and maintained with 2–3% isoflurane in 100% oxygen at a flow rate of 1.0 L/min using a face mask during the procedure. The depth of anesthesia was monitored using a pressure sensor for respiration. Breathing rates were maintained at 30–40 breaths per minute. Body temperature was continuously measured via a rectal thermal probe and kept at 35°C using a flowing-water heating blanket. Heart function was measured using a 7.1 Tesla MR system (ClinScan 70/30 30 Bruker, Ettlingen, Germany) with 290 mT/m gradients field strength. For signal excitation a head coil was used, while signal reception was realized using a four-channel rat brain array. After three localizer sequences and a fast single shot fast low angle shot (FLASH) single shot axial sequence, as 2 chamber view (2 CHV) and as 4 chamber view (4 CHV) were acquired. In addition, prospective electrocardiogram triggered and respiration gated CINE sequences were generated in 2 CHV [repetition time (TR): 5.8 ms, echo time (TE) 2.37 ms, field-of-view (FoV): 35 × 35 mm, FoV Phase: 100%, flip angle: 20°, slice thickness 0.7 mm] and 4 CHV (TR: 5.8 ms, TE: 2.37 ms, FoV: 35 × 35 mm, FoV Phase: 100%, flip angle: 20°, slice thickness 0.7 mm) were obtained. Afterward, five short axis CINE (flash) sequences of the left ventricle (TR: 5.7 ms, TE: 2.25 ms, FoV: 35 × 35 mm, FoV Phase: 100%, flip angle: 25°, slice thickness: 1.0 mm) were acquired. The results were analyzed using the software Segment, version 1.9 R3510 (Medviso and Osirix, version 5.8.2 32-bit, Lund, Sweden). The endocardium and epicardium were delineated in all five short axes in both end-diastole and end-systole, which the software used to calculate the specific area in μm2. Papillary muscles were included in the delineation of the endocardium. Followed by a conversion in a specific volume (in μm3), utilizing a slice thickness of 1 mm, left ventricular end-diastolic volume (LVEDV) was acquired by using the volume from each slice. The end-diastole was determined as being the phase with the largest area of the LV cavity and the end-systole being the one with the smallest area in each slice. The EF was calculated with the help of LVEDV and left ventricular end-systolic volume (LVESV) (EF = [(LVEDV − LVESV)/LVEDV] × 100). Fractional shortening (FS) was calculated using the left mid-ventricular diameter (FS = [(LVEDD − LVESD)/LVEDD] × 100). The LV mass was computed by multiplying the LV volume by the specific density of a healthy (assuming the lack of infarcted regions in the heart) myocardium of 1.05 g/ml.
ELISA
Mouse plasma N-terminal pro-brain natriuretic peptide (NT-proBNP) concentrations were measured using ELISA kits (SEA485Mu, Cloud Clone Corp., Houston, TX, United States) according to the instructions of the manufacturer.
RNA Extraction and Expression Analysis With the NanoString nCounter® Elements
Total RNA was extracted from powdered whole heart tissue samples using the SV Total RNA isolation kit (Promega, Madison, WI, United States) according to the manufacturer‘s instructions.
For gene expression analysis, a customized NanoString’s nCounter Elements TagSet panel was used. About 50 ng of each sample were hybridized to the target-specific capture and reporter probes at 67°C overnight (16 h) according to manufacturer’s instructions. Samples were cooled down to 4°C, supplemented with 15 μl H2O, and loaded into the NanoString cartridge. Afterward, the nCounter Gene Expression Assay was started immediately. Raw data were analyzed with the nCounter Sprint Profiler. Transcript levels were determined with the nSolver Data Analysis Software including background subtraction using negative controls and normalization to six housekeeping genes (Abcf1, Actb, Cltc, Gapdh, Pgk1, and Tubb5). Data represent the mean of normalized counts and are expressed as fold change over WT.
Analysis of Protein Degradation Pathways UPS and ALP
Sample Preparation
Tissue samples were powdered, and protein extraction was performed in two steps. First, the organ powder (about 30 mg) was dissolved in 150 μl water with a protease inhibitor cocktail (complete miniTM, Roche Diagnostics, Rotkreuz, Switzerland). After three freeze-thaw-cycles the tissue was homogenized by using Tissue Lyser (2 × 30 s at 20 Hz) and centrifuged at 4°C, full speed for 30 min in a table-top centrifuge. The supernatant was kept as the cytosolic fraction. Secondly, the pellet of the first step was homogenized in 240 μl SDS-buffer (3% SDS, 30 mM Tris-base, pH 8.8, 5 mM EDTA, 30 mM NaF, 10% glycerol and 1 mM DTT) and centrifuged at room temperature, full speed for 10 min in a table-top centrifuge. The supernatant was kept as the membrane-enriched fraction.
Western Blot
Proteins of the membrane-enriched fraction were loaded on acrylamide/bisacrylamide (29:1) gels and electrotransferred to nitrocellulose membranes, except for LC3 analysis, for which proteins were electrotransferred to polyvinylidene fluoride membranes. Antibodies against the following proteins were used for western blot analysis: Beclin-1 (Cell Signaling Technology, 3738), FYCO1 (Novus Biologicals, NBP1-47266), LAMP-2 (Abcam, ab13524), LC3 (Cell Signaling Technology, 2775), p62 (Sigma Aldrich, P0067), ribosomal protein S6 (Cell Signaling Technology, 2217), and ubiquitinated proteins (Enzo Life Sciences, BML-PW8810). Signals were revealed with the Clarity Western ECL substrate (Bio-Rad) and acquired with the ChemiDoc Touch Imaging System (Bio-Rad). Signals were quantified with the Image Lab Software (Bio-Rad).
Determination of the 20S and 26S Proteasomal Activities
The activities of the 20S and 26S proteasome were assessed in the cytosolic protein fraction. For determination of the activity, 30 μg of protein were diluted in incubation buffer (20 mM HEPES, 0.5 mM EDTA, 5 mM MgCl2, 1 mM DTT) to a final volume of 50 μl. Samples were pre-incubated in this buffer for 2 h at 4°C. Following pre-incubation, the synthetic fluorogenic substrates Suc-LLVY-AMC (Enzo Life Sciences, BML-P802), Z-LLE-AMC (Enzo Life Sciences, BML-ZW9345) and Ac-RLR-AMC (Enzo Life Sciences, BML-AW9785) were added to the samples at a final concentration of 60 μM, 45 μM and 40 μM in the presence and absence of 28, 14, and 14 μM ATP for chymotrypsin-like, caspase-like and trypsin-like activity, respectively. After incubation in the dark for 1 h at 37°C, the fluorescence of the released AMC reporters was measured using the TECAN Safire2 microplate reader at an excitation wavelength of 380 nm and an emission wavelength of 460 nm. Each sample was measured in duplicate. The mean of the blank (incubation buffer only) was subtracted from the mean of each sample duplicate.
Data Analysis
Data are presented as mean ± SEM. Statistical analyses were performed using the unpaired Student’s t-test. All analyses were realized using GraphPad Prism 8. A value of p < 0.05 was considered statistically significant.
Results
DTG Mice Exhibit Eccentric LV Hypertrophy With a Preserved Systolic Function
Representative MRI images of WT and DTG hearts are shown in Supplementary Figure 1. MRI measurements of the DTG mice showed significantly higher LVED anterior and posterior wall thicknesses (Figures 1A,B), LVED and LVES diameters (Figures 1C,D), and LVED volumes (Figure 1E) than WT mice, whereas LVES did not differ (Figure 1F). In line with this, the LV mass (Figure 1G), but not the body weight (Figure 1H), and LV mass/body weight ratio (Figure 1I) were higher in DTG than WT mice. In contrast, no difference in EF and FS was observed between the two groups (Figures 1J,K). The stroke volume was higher in DTG than WT mice (Figure 1L). Thus, DTG mice showed eccentric LV hypertrophy with preserved systolic function.
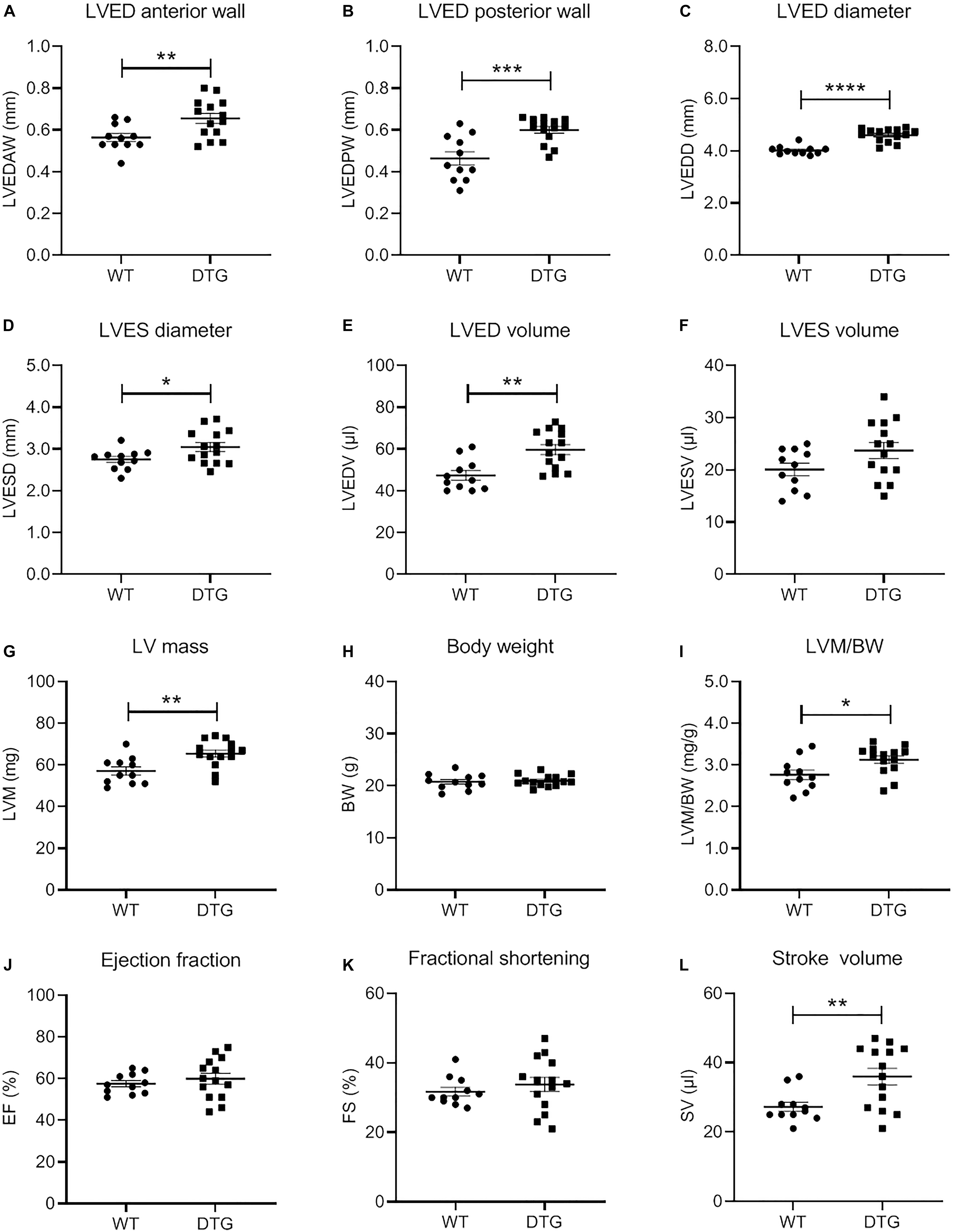
Figure 1. Evaluation of morphological and functional parameters of the heart of DTG and WT mice. Scatter plots show the difference in (A) left ventricular (LV) end-diastolic (ED) anterior wall thickness (LVEDAW), (B) LVED posterior wall thickness (LVEDPW), (C) LVED diameter (LVEDD), (D) LV end-systolic (ES) diameter (LVESD), (E) LVED volume (LVEDV), (F) LVES volume (LVESV), (G) LV mass (LVM), (H) body weight (BW), (I) LVM/BW, (J) ejection fraction (EF), (K) fractional shortening (FS), and (L) stroke volume (SV) of DTG and WT mice. Data are presented as mean ± SEM with *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 vs. WT, unpaired Student’s t-test. N = 11–14.
DTG Mice Show Typical Gene Expression Changes Associated With Pathological Cardiac Hypertrophy
To further examine the disease state of the DTG mice, the expression of several genes related to cardiac hypertrophy, fetal gene program, fibrosis, apoptosis and Ca2+ handling was evaluated by using the NanoString’s nCounter Elements technology (Table 1). While expression levels of genes related to hypertrophy (Fhl1 and Rcan1) were not significantly higher, the expression of Fhl2, which has been shown to be down-regulated during hypertrophy, was markedly lower in DTG mice. The expression level of genes associated with the reactivation of the fetal gene program (Nppa, Nppb, and Myh7) was significantly higher in DTG than WT mice. Genes related to fibrosis (Col1a1, p < 0.001 and Col3a1, p = 0.06) and genes specific for fibroblasts (Ctgf, S100a4 and Postn) were up-regulated in DTG mice. Furthermore, the expression of Bcl2, a key regulator of apoptosis, and Vwf, a marker of endothelial dysfunction, was higher in DTG mice. In contrast, genes involved in Ca2+ handling (Atp2a2, Pln, and Ryr2) were downregulated in DTG mice.
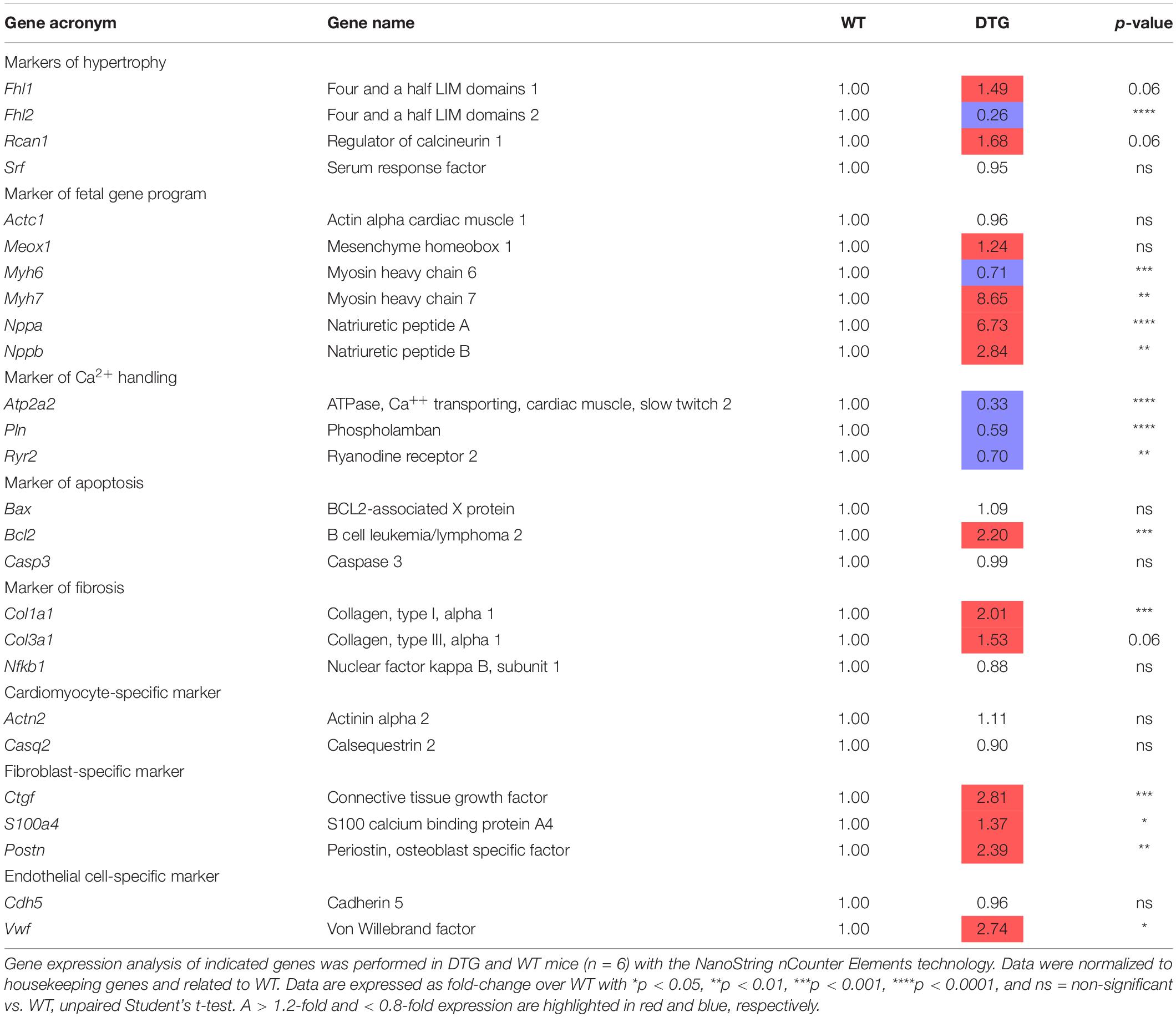
Table 1. Gene expression analysis in DTG and WT mice.
DTG Mice Express Higher Level of NT-proBNP
NT-proBNP is used in clinical routine as a prognostic biomarker for LV hypertrophy and heart failure. The NT-proBNP level was ∼13-fold higher in DTG than in WT mice (Figure 2). This is in line with the results of the gene expression analysis for the natriuretic peptides Nppa and Nppb which were higher expressed in DTG mice than in healthy littermates.
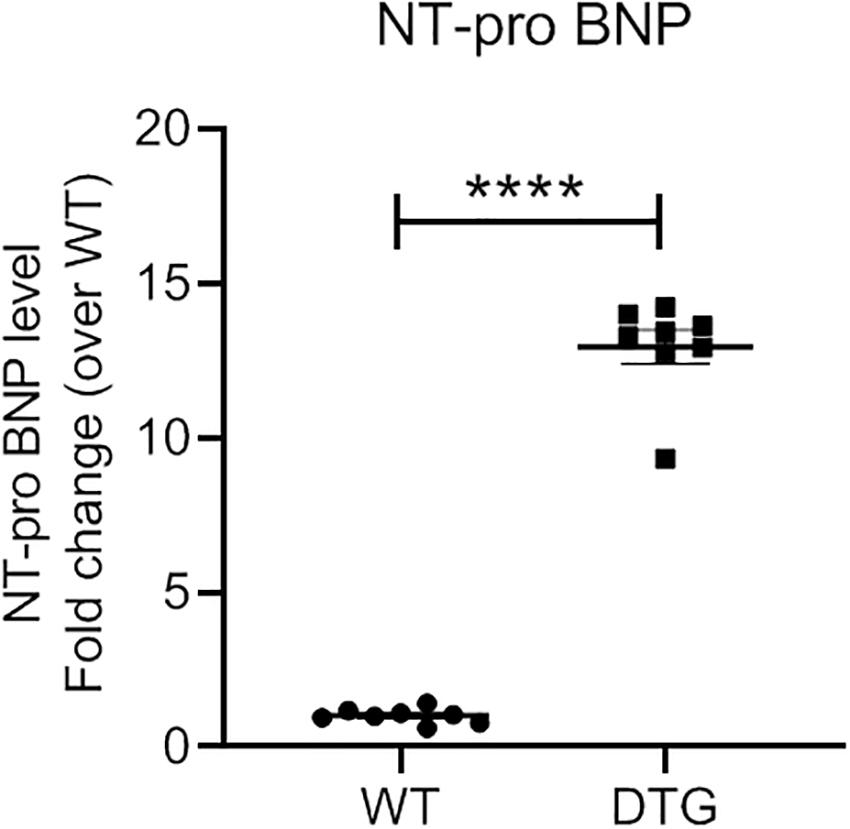
Figure 2. Plasma NT-proBNP level in DTG and WT mice. Data are presented as mean ± SEM with ****p < 0.0001 vs. WT, unpaired Student’s t-test. N = 8.
DTG Mice Display Accumulation of (Poly)ubiquitinated Proteins Without Alteration of Proteasomal Activities
Since accumulation of (poly)ubiquitinated proteins and altered proteasomal activities have been reported in cardiac diseases, we determined the steady-state levels of (poly)ubiquitinated proteins by Western blot and measured the 20S (ATP-independent) and 26S (ATP-dependent) proteasomal activities by using synthetic fluorogenic substrates in heart tissue of DTG and WT mice. The steady-state level of polyubiquitinated proteins was 1.5-fold higher in DTG than in WT mice (Figure 3A). Additionally, cardiac sections have been stained with a monoclonal antibody directed against ubiquitinated proteins. In agreement with the Western blot analysis, the number of dots of (poly)ubiquitinated proteins was significantly higher in DTG than in WT mice (Supplementary Figure 2 and Supplementary Table 1). In contrast, the 20S and 26S chymotrypsin-like, caspase-like and trypsin-like activities did not differ between DTG and WT mice (Figure 3B).
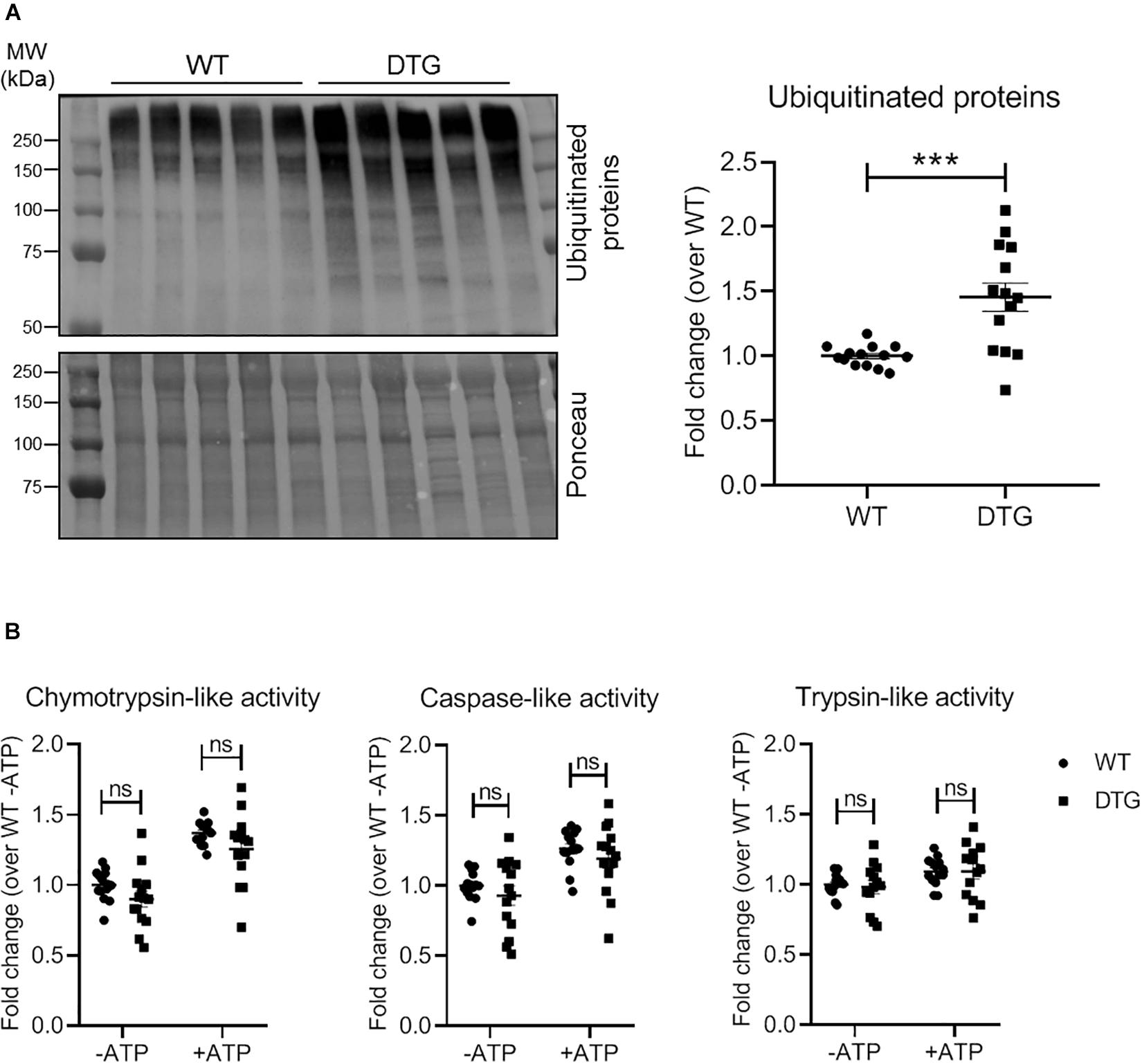
Figure 3. Steady-state levels of ubiquitinated proteins and proteasomal activities in DTG and WT mice. (A) Representative Western blot and quantification of the steady-state levels of ubiquitinated proteins normalized to Ponceau and related to WT. (B) 20S (ATP-independent) and 26S (ATP-dependent) chymotrypsin-like, caspase-like and trypsin-like activities were measured by using specific fluorogenic substrates. Data are presented as mean ± SEM with ***p < 0.001 vs. WT and ns = non-significant, unpaired Student’s t-test. N = 13–14.
DTG Mice Show an Altered ALP
To identify the regulation of autophagic processes in the disease pattern of cardiac hypertrophy, several markers involved in the process of the ALP were determined using the NanoString’s nCounter Elements technology (Table 2) and Western blot analysis (Figure 4).
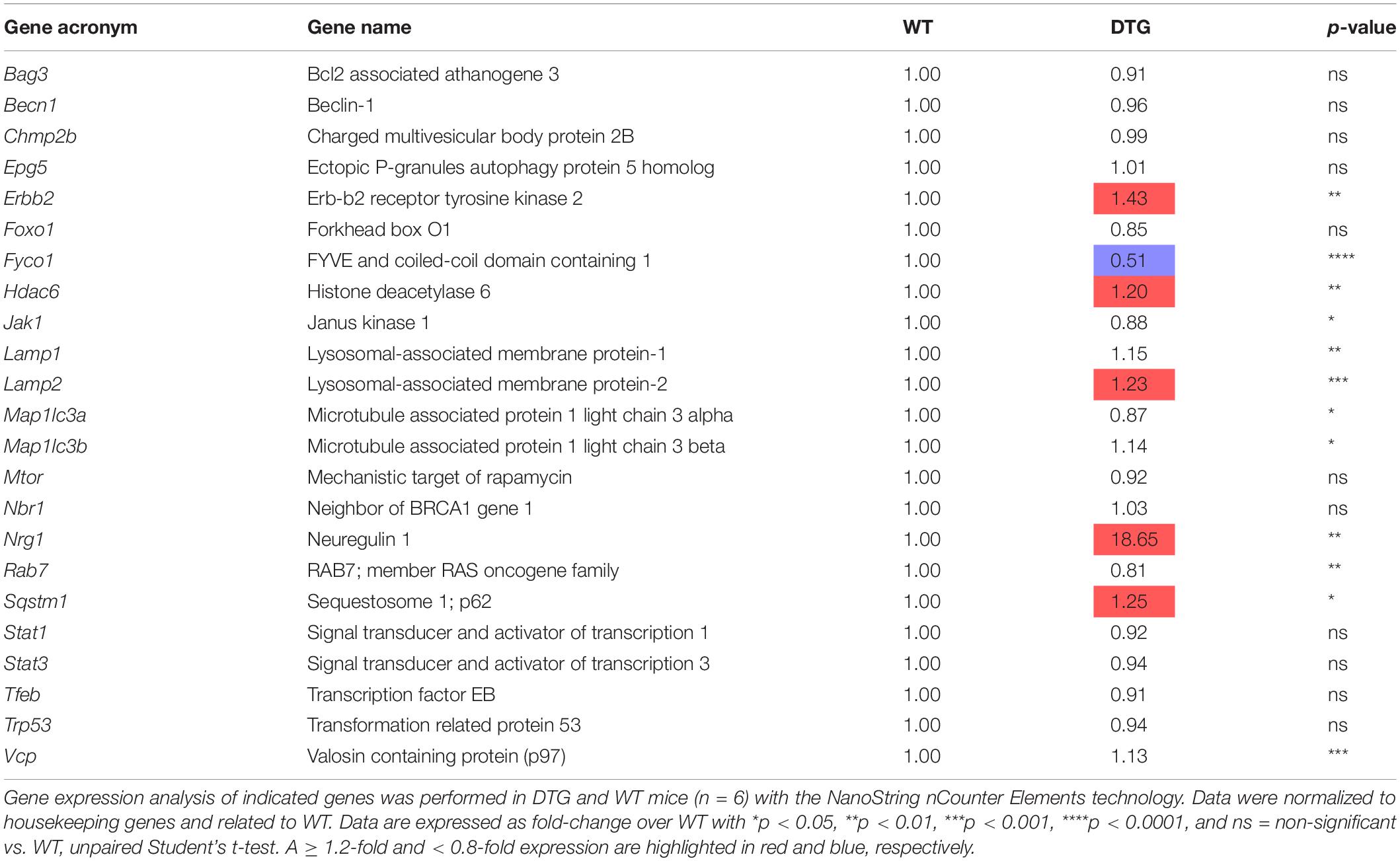
Table 2. Autophagy-lysosomal pathway gene expression analysis in DTG and WT mice.
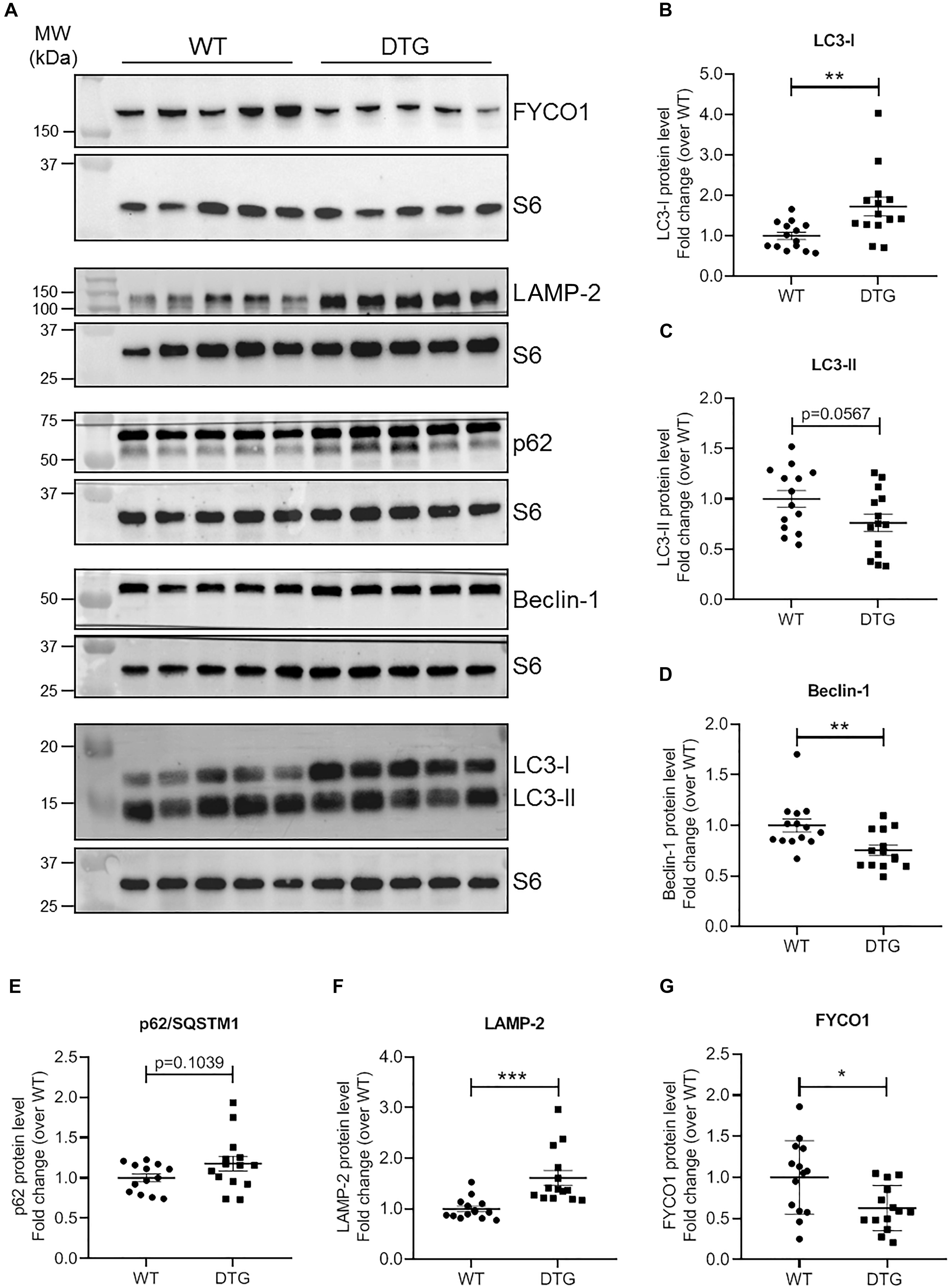
Figure 4. ALP marker proteins in DTG and WT mice. (A) Representative Western blots of indicated proteins. S6 ribosomal protein was used as loading control. Quantification of (B) LC3-I, (C) LC3-II, (D) Beclin-1, (E) p62/SQSTM1, (F) LAMP-2, and (G) FYCO1 protein levels normalized to S6 ribosomal protein and related to WT. Data are presented as mean ± SEM with *p < 0.05, **p < 0.01 and ***p < 0.001 vs. WT, unpaired Student’s t-test. N = 13–14.
Microtubule-associated protein 1 light chain 3 beta (hereafter referred to as LC3) is a central protein in the autophagy pathway and exists in a soluble form termed LC3-I and a lipidated form referred to as LC3-II. LC3-II is incorporated into the autophagosomal membrane and is important for autophagosome formation and maturation, shuttling and fusion with the lysosomes (Zech et al., 2020). Whereas Map1lc3b transcript and LC3-I protein levels were higher in DTG mice, LC3-II protein levels tended to be lower in DTG than WT mice (Table 2 and Figures 4A–C). Next, we evaluated beclin-1, which is involved in the phagophore structure initiation, an early step of autophagy (Zech et al., 2020). Beclin-1 protein levels were lower in DTG than WT mice, while beclin-1 mRNA levels did not differ between the groups (Figures 4A,D and Table 2). Furthermore, the protein levels of p62/SQSTM1, which acts as a shuttle protein that binds ubiquitinated proteins and LC3-II and directs ALP-mediated degradation of ubiquitinated proteins (Pankiv et al., 2007), did not differ between DTG and WT mice, whereas the expression of Sqstm1 was induced on transcriptional level in DTG mice (Figures 4A,E and Table 2). The expression of the lysosomal-associated membrane protein-2 (LAMP-2) was higher on both mRNA and protein levels in DTG than WT mice (Table 2 and Figures 4A,F). In contrast, the expression of FYVE and coiled-coil domain-containing protein 1 (FYCO1), which forms an adaptor protein complex with LC3 and Rab7 (Pankiv et al., 2010), was lower on both mRNA and protein levels in DTG than WT mice (Table 2 and Figures 4A,G). A > 1.2-fold expression was further observed for Erbb2, Hdac6 and Nrg1 at transcriptional level in DTG mice (Table 2).
Discussion
In the present study we investigated alterations of the two major proteolytic systems namely UPS and ALP in a mouse model of eccentric LV hypertrophy with preserved EF. The major findings of the present study are: (1) accumulation of (poly)ubiquitinated proteins, (2) no alteration of the 20S and 26S proteasomal activities, (3) higher protein levels of LC3-I and LAMP-2, (4) lower protein levels of beclin-1 and FYCO1, (5) > 1.2-fold expression for Erbb2, Hdac6, Lamp2, Nrg1, and Sqstm1, and (6) < 0.8-fold expression for Fyco1 at transcriptional level in DTG mice. These findings indicate an altered autophagic clearance of damaged and/or dysfunctional proteins in a mouse model of eccentric LV hypertrophy.
In recent years, the UPS was found to play a fundamental role in several biological processes including cell proliferation, adaption to stress and cell death (Rock et al., 1994; King et al., 1996; Breitschopf et al., 2000). Various studies found that activation or impairment of the UPS is associated with different cardiac diseases (Mearini et al., 2008; Su and Wang, 2010; Schlossarek and Carrier, 2011; Day, 2013). An accumulation of (poly)ubiquitinated proteins was documented in failing human hearts (Weekes et al., 2003; Birks et al., 2008; Predmore et al., 2010). In our transgenic mouse model showing eccentric LV hypertrophy with preserved EF the classical signature of a hypertrophic response including the up-regulation of the genes encoding BNP (Nppb), ANP (Nppa) and α-myosin heavy chain (Myh7) was detected together with higher levels of (poly)ubiquitinated proteins and without alterations of proteasomal activities.
The UPS and ALP have long been considered to act separately, but recent data suggest that they work as a consortium (Wang et al., 2008; Korolchuk et al., 2010; Lamark and Johansen, 2010; Singh et al., 2020). Perturbations in the degradation process of either pathway have been reported to affect the activity of the other one (Korolchuk et al., 2010; Pan et al., 2020). For instance, impairment of the UPS has been shown to trigger autophagy (Iwata et al., 2005; Pandey et al., 2007), suggesting autophagic upregulation as a compensatory mechanism. Evaluation of the ALP key marker LC3 showed that LC3-II protein levels tended to be lower in DTG than WT mice, whereas Map1lc3b transcript and LC3-I protein levels were higher in DTG mice. Lower LC3-II protein level could indicate an alteration in the lipidation process from LC3-I to LC3-II (Kabeya et al., 2000). On the other hand, LC3-II is a substrate of the ALP as well and lower levels of LC3-II protein could as well indicate a rapid degradation via the ALP. The elevated Map1lc3b transcript and LC3-I protein levels support more the second scenario suggesting an activated or at least proper working ALP in the DTG mice. Further support for an activated ALP is the increased expression of LAMP-2 on both mRNA and protein levels in the DTG mice, indicating an intact autophagosome-lysosome fusion and an enhanced lysosomal activity. Autophagosome-lysosome fusion occurs in the vicinity of the centrosome. To get to the perinuclear region, autophagic vesicles have to move along the microtubules and this interaction with the microtubules is mediated by motor proteins (Mackeh et al., 2013). So-called plus-end-directed motor proteins transport autophagic vesicles toward the cellular periphery and minus-end-directed motor proteins mediate the transport to the perinuclear region (Hirokawa et al., 2009). The balance between active plus-end- and minus-end-directed motor proteins on the surface of autophagic vesicles determines the directionality of their intracellular movement (Mackeh et al., 2013). FYCO1 forms an adaptor protein complex with LC3 and Rab7 and promotes the plus-end-directed transport of autophagic vesicles (Pankiv et al., 2010). We observed markedly reduced levels of FYCO1 on both transcript and protein levels in DTG mice, suggesting that the directionality of the intracellular movements of autophagic vesicles is maybe shifted toward the perinuclear region.
Protein levels of p62/SQSTM1 did not differ between DTG and WT mice. This protein serves as a linker between ubiquitinated proteins and LC3-II thereby directing ALP-mediated degradation of ubiquitinated proteins (Pankiv et al., 2007) and is as well degraded by the ALP (Bjorkoy et al., 2005). The induced expression of Sqstm1 at transcriptional level may indicate an increased demand for p62/SQSTM1. Unaltered p62/SQSTM1 protein level in spite of elevated transcript level could indicate a proper or even induced working ALP degrading its substrate. Another protein serving as a linker between ubiquitinated cargo and the ALP is the histone deacetylase 6 (HDAC6). HDAC6 binds ubiquitin and interacts directly with dynein motors, thereby ensuring efficient delivery of substrates to the ALP for degradation (Kawaguchi et al., 2003). A protective effect of compensatory upregulation of autophagy during proteasomal inhibition has been proposed to be dependent on HDAC6 (Iwata et al., 2005; Pandey et al., 2007). We did not determine the protein levels, but found as well elevated Hdac6 transcript levels in the DTG mice suggesting similar to p62/SQSTM1 an induced expression to deal with the higher amounts of ubiquitinated proteins. Interestingly, beclin-1 protein levels were lower in DTG than WT mice, while Becn1 mRNA levels did not differ between the groups. Beclin-1 is involved in the phagophore structure initiation (Zech et al., 2020), and lower protein level suggest, in contrast to our other data, rather a repression of the ALP, at least during the initiation process. Bcl-2 (B-cell leukemia/lymphoma 2), which was significantly up-regulated at the transcriptional level in the DTG mice, is directly linked to the autophagy process. Bcl-2 interacts with beclin-1 by inhibiting the formation of the beclin-1/Vsp34 PI3K complex and therefore the beclin-1-dependent autophagy (Pattingre et al., 2005). Pattingre et al. (2005) proposed that this anti-autophagic action of Bcl-2 may help to maintain autophagy at an optimal level for cell survival rather than cell death. In addition, Bcl-2 partly mediates the anti-autophagic effects of neuregulin-1 (Nrg-1) (An et al., 2013). Nrg-1 and its corresponding receptor ErbB2 (Erb-b2 receptor tyrosine kinase 2) were significantly up-regulated at the transcriptional level in the hearts of the DTG mice. Nrg-1 induces a number of cellular responses, such as regulation of cell proliferation and differentiation and plays a critical role in cardiovascular development and maintenance of heart function by promoting cardiac myocyte survival and maintenance of Ca2+ homeostasis (De Keulenaer et al., 2019). Although there was a marked increase in Nrg1 expression in the DTG mice, almost all genes involved in Ca2+ handling were significantly down-regulated. Nevertheless, the increase in Nrg1 and its receptor Erbb2 suggests an induction of survival, structural and functional maintenance and anti-autophagic processes, probably similar to Bcl-2 to maintain autophagy at an optimal level.
Many studies have shown that activation of G-protein-coupled receptors can regulate autophagy (Wauson et al., 2014). Liu et al. (2017) investigated the ALP in a transgenic mouse model of cardiac-specific inducible Gαq expressing a constitutively active GαqQ209L fused to a modified hormone-binding domain of the estrogen receptor (Liu et al., 2017). In contrast to the DTG mice, which exhibit a preserved ejection function, the transgenic GαqQ209L mice showed a reduced FS after 7 days of treatment with tamoxifen. At this time point, Liu et al. reported an activated autophagy in the GαqQ209L mice including elevated protein levels of beclin-1, p62, LC3-II, LAMP-1, and LAMP-2 (Liu et al., 2017).
In conclusion, evaluation of the ALP in the DTG αMHC-cyclin T1 × αMHC-Gαq mice revealed a mixed pattern in that the results suggested a repression of the ALP during the initiation process, but an induction of the ALP at the level of autophagosome-lysosome fusion and the delivery of ubiquitinated cargo to the ALP for degradation.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by the Landesamt für Landwirtschaft, Lebensmittelsicherheit und Fischerei Mecklenburg-Vorpommern.
Author Contributions
KW contributed to the conception and design of the study, management of the mouse cohorts, execution of experiments, analysis and interpretation of data, figure preparation, and drafting of the manuscript. EK and BG contributed to the execution of experiments. LC and SK contributed to the conception and design of the study, analysis and interpretation of data, and drafting of the manuscript. SF contributed to the conception and design of the study and drafting of the manuscript. SS contributed to the conception and design of the study, execution of experiments, analysis and interpretation of data, figure preparation, and drafting of the manuscript. All authors critically discussed the results, and reviewed and approved the manuscript before submission.
Funding
This study was supported by the German Centre for Cardiovascular Research (DZHK; Grant B15-072).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Ellen Orthey for her technical assistence.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2021.614878/full#supplementary-material
References
An, T., Huang, Y., Zhou, Q., Wei, B. Q., Zhang, R. C., Yin, S. J., et al. (2013). Neuregulin-1 attenuates doxorubicin-induced autophagy in neonatal rat cardiomyocytes. J. Cardiovasc. Pharmacol. 62, 130–137. doi: 10.1097/FJC.0b013e318291c094
Bhuiyan, M. S., Pattison, J. S., Osinska, H., James, J., Gulick, J., Mclendon, P. M., et al. (2013). Enhanced autophagy ameliorates cardiac proteinopathy. J. Clin. Invest. 123, 5284–5297. doi: 10.1172/JCI70877
Birks, E. J., Latif, N., Enesa, K., Folkvang, T., Luong Le, A., Sarathchandra, P., et al. (2008). Elevated p53 expression is associated with dysregulation of the ubiquitin-proteasome system in dilated cardiomyopathy. Cardiovasc. Res. 79, 472–480. doi: 10.1093/cvr/cvn083
Bjorkoy, G., Lamark, T., Brech, A., Outzen, H., Perander, M., Overvatn, A., et al. (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614. doi: 10.1083/jcb.200507002
Breitschopf, K., Zeiher, A. M., and Dimmeler, S. (2000). Ubiquitin-mediated degradation of the proapoptotic active form of bid. A functional consequence on apoptosis induction. J. Biol. Chem. 275, 21648–21652. doi: 10.1074/jbc.M001083200
Choi, J. C., Muchir, A., Wu, W., Iwata, S., Homma, S., Morrow, J. P., et al. (2012). Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci. Transl. Med. 4:144ra102. doi: 10.1126/scitranslmed.3003875
Ciechanover, A. (2007). Intracellular protein degradation from a vague idea through the lysosome and the ubiquitin-proteasome system and on to human diseases and drug targeting: nobel Lecture, December 8, 2004. Ann. N. Y. Acad. Sci. 1116, 1–28. doi: 10.1196/annals.1402.078
D’Angelo, D. D., Sakata, Y., Lorenz, J. N., Boivin, G. P., Walsh, R. A., Liggett, S. B., et al. (1997). Transgenic Galphaq overexpression induces cardiac contractile failure in mice. Proc. Natl. Acad. Sci. U. S. A. 94, 8121–8126. doi: 10.1073/pnas.94.15.8121
Day, S. M. (2013). The ubiquitin proteasome system in human cardiomyopathies and heart failure. Am. J. Physiol. Heart Circ. Physiol. 304, H1283–93. doi: 10.1152/ajpheart.00249.2012
De Keulenaer, G. W., Feyen, E., Dugaucquier, L., Shakeri, H., Shchendrygina, A., Belenkov, Y. N., et al. (2019). Mechanisms of the Multitasking Endothelial Protein NRG-1 as a Compensatory Factor During Chronic Heart Failure. Circ. Heart Fail. 12:e006288. doi: 10.1161/CIRCHEARTFAILURE.119.006288
Depre, C., Wang, Q., Yan, L., Hedhli, N., Peter, P., Chen, L., et al. (2006). Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation 114, 1821–1828.
Dorn, G. W. II., and Force, T. (2005). Protein kinase cascades in the regulation of cardiac hypertrophy. J. Clin. Invest. 115, 527–537. doi: 10.1172/JCI24178
Frey, N., and Olson, E. N. (2003). Cardiac hypertrophy: the good, the bad, and the ugly. Annu. Rev. Physiol. 65, 45–79. doi: 10.1146/annurev.physiol.65.092101.142243
Hirokawa, N., Noda, Y., Tanaka, Y., and Niwa, S. (2009). Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 10, 682–696. doi: 10.1038/nrm2774
Iwata, A., Riley, B. E., Johnston, J. A., and Kopito, R. R. (2005). HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 280, 40282–40292. doi: 10.1074/jbc.M508786200
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., et al. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728. doi: 10.1093/emboj/19.21.5720
Kawaguchi, Y., Kovacs, J. J., Mclaurin, A., Vance, J. M., Ito, A., and Yao, T. P. (2003). The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727–738. doi: 10.1016/S0092-8674(03)00939-5
King, R. W., Deshaies, R. J., Peters, J. M., and Kirschner, M. W. (1996). How proteolysis drives the cell cycle. Science 274, 1652–1659.
Korolchuk, V. I., Menzies, F. M., and Rubinsztein, D. C. (2010). Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 584, 1393–1398. doi: 10.1016/j.febslet.2009.12.047
Lamark, T., and Johansen, T. (2010). Autophagy: links with the proteasome. Curr. Opin. Cell Biol. 22, 192–198. doi: 10.1016/j.ceb.2009.11.002
Lavandero, S., Troncoso, R., Rothermel, B. A., Martinet, W., Sadoshima, J., and Hill, J. A. (2013). Cardiovascular autophagy Concepts, controversies, and perspectives. Autophagy 9, 1455–1466. doi: 10.4161/auto.25969
Liu, S., Jiang, Y. P., Ballou, L. M., Zong, W. X., and Lin, R. Z. (2017). Activation of Galphaq in Cardiomyocytes Increases Vps34 Activity and Stimulates Autophagy. J. Cardiovasc. Pharmacol. 69, 198–211. doi: 10.1097/FJC.0000000000000461
Mackeh, R., Perdiz, D., Lorin, S., Codogno, P., and Pous, C. (2013). Autophagy and microtubules - new story, old players. J. Cell Sci. 126, 1071–1080. doi: 10.1242/jcs.115626
Maloyan, A., Sayegh, J., Osinska, H., Chua, B. H., and Robbins, J. (2010). Manipulation of death pathways in desmin-related cardiomyopathy. Circ. Res. 106, 1524–1532. doi: 10.1161/CIRCRESAHA.109.212639
Mearini, G., Schlossarek, S., Willis, M. S., and Carrier, L. (2008). The ubiquitin-proteasome system in cardiac dysfunction. Biochim. Biophys. Acta 1782, 749–763. doi: 10.1016/j.bbadis.2008.06.009
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451, 1069–1075. doi: 10.1038/nature06639
Pan, B., Li, J., Parajuli, N., Tian, Z. W., Wu, P. L., Lewno, M. T., et al. (2020). The Calcineurin-TFEB-p62 Pathway Mediates the Activation of Cardiac Macroautophagy by Proteasomal Malfunction. Circ. Res. 127, 502–518. doi: 10.1161/Circresaha.119.316007
Pandey, U. B., Nie, Z. P., Batlevi, Y., Mccray, B. A., Ritson, G. P., Nedelsky, N. B., et al. (2007). HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863. doi: 10.1038/nature05853
Pankiv, S., Alemu, E. A., Brech, A., Bruun, J. A., Lamark, T., Overvatn, A., et al. (2010). FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 188, 253–269. doi: 10.1083/jcb.200907015
Pankiv, S., Clausen, T. H., Lamark, T., Brech, A., Bruun, J. A., Outzen, H., et al. (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145. doi: 10.1074/jbc.M702824200
Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., et al. (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939. doi: 10.1016/j.cell.2005.07.002
Pattison, J. S., Osinska, H., and Robbins, J. (2011). Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ. Res. 109, 151–160. doi: 10.1161/CIRCRESAHA.110.237339
Predmore, J. M., Wang, P., Davis, F., Bartolone, S., Westfall, M. V., Dyke, D. B., et al. (2010). Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 121, 997–1004. doi: 10.1161/CIRCULATIONAHA.109.904557
Ramos, F. J., Chen, S. C., Garelick, M. G., Dai, D. F., Liao, C. Y., Schreiber, K. H., et al. (2012). Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 4:144ra103. doi: 10.1126/scitranslmed.3003802
Rock, K. L., Gramm, C., Rothstein, L., Clark, K., Stein, R., Dick, L., et al. (1994). Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78, 761–771. doi: 10.1016/s0092-8674(94)90462-6
Rothermel, B. A., and Hill, J. A. (2008). Autophagy in load-induced heart disease. Circ. Res. 103, 1363–1369. doi: 10.1161/CIRCRESAHA.108.186551
Sano, M., Wang, S. C., Shirai, M., Scaglia, F., Xie, M., Sakai, S., et al. (2004). Activation of cardiac Cdk9 represses PGC-1 and confers a predisposition to heart failure. EMBO J. 23, 3559–3569. doi: 10.1038/sj.emboj.7600351
Schlossarek, S., and Carrier, L. (2011). The ubiquitin-proteasome system in cardiomyopathies. Curr. Opin. Cardiol. 26, 190–195. doi: 10.1097/HCO.0b013e32834598fe
Schlossarek, S., Englmann, D. R., Sultan, K. R., Sauer, M., Eschenhagen, T., and Carrier, L. (2012). Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Basic Res. Cardiol. 107, 1–13. doi: 10.1007/s00395-011-0235-3
Singh, S. R., Meyer-Jens, M., Alizoti, E., Bacon, W. C., Davis, G., Osinska, H., et al. (2020). A high-throughput screening identifies ZNF418 as a novel regulator of the ubiquitin-proteasome system and autophagy-lysosomal pathway. Autophagy 27, 1–16. doi: 10.1080/15548627.2020.1856493
Singh, S. R., Zech, A. T. L., Geertz, B., Reischmann-Dusener, S., Osinska, H., Prondzynski, M., et al. (2017). Activation of Autophagy Ameliorates Cardiomyopathy in Mybpc3-Targeted Knockin Mice. Circ. Heart Fail. 10:e004140. doi: 10.1161/CIRCHEARTFAILURE.117.004140
Su, H., and Wang, X. (2010). The ubiquitin-proteasome system in cardiac proteinopathy: a quality control perspective. Cardiovasc. Res. 85, 253–262. doi: 10.1093/cvr/cvp287
Thottakara, T., Friedrich, F. W., Reischmann, S., Braumann, S., Schlossarek, S., Kramer, E., et al. (2015). The E3 ubiquitin ligase Asb2beta is downregulated in a mouse model of hypertrophic cardiomyopathy and targets desmin for proteasomal degradation. J. Mol. Cell Cardiol. 87, 214–224. doi: 10.1016/j.yjmcc.2015.08.020
Tsukamoto, O., Minamino, T., Okada, K., Shintani, Y., Takashima, S., Kato, H., et al. (2006). Depression of proteasome activities during the progression of cardiac dysfunction in pressure-overloaded heart of mice. Biochem. Biophys. Res. Commun. 340, 1125–1133. doi: 10.1016/j.bbrc.2005.12.120
Wang, X., Su, H., and Ranek, M. J. (2008). Protein quality control and degradation in cardiomyocytes. J. Mol. Cell Cardiol. 45, 11–27. doi: 10.1016/j.yjmcc.2008.03.025
Wauson, E. M., Dbouk, H. A., Ghosh, A. B., and Cobb, M. H. (2014). G protein-coupled receptors and the regulation of autophagy. Trends Endocrinol. Metab. 25, 274–282. doi: 10.1016/j.tem.2014.03.006
Weekes, J., Morrison, K., Mullen, A., Wait, R., Barton, P., and Dunn, M. J. (2003). Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics 3, 208–216. doi: 10.1002/pmic.200390029
Zech, A. T. L., Singh, S. R., Schlossarek, S., and Carrier, L. (2020). Autophagy in cardiomyopathies. Biochim. Biophys. Acta Mol. Cell Res. 1867:118432. doi: 10.1016/j.bbamcr.2019.01.013
Zheng, Q., and Wang, X. (2010). Autophagy and the ubiquitin-proteasome system in cardiac dysfunction. Panminerva Med. 52, 9–25.
Keywords: autophagy-lysosomal pathway, cardiomyopathy, left ventricular hypertrophy, protein degradation, ubiquitin-proteasome system
Citation: Wenzel K, Krämer E, Geertz B, Carrier L, Felix SB, Könemann S and Schlossarek S (2021) A Transgenic Mouse Model of Eccentric Left Ventricular Hypertrophy With Preserved Ejection Fraction Exhibits Alterations in the Autophagy-Lysosomal Pathway. Front. Physiol. 12:614878. doi: 10.3389/fphys.2021.614878
Received: 07 October 2020; Accepted: 29 March 2021;
Published: 22 April 2021.
Edited by:
Xuejun Wang, University of South Dakota, United StatesReviewed by:
Zhenlin Li, Université Pierre et Marie Curie, FranceZhiqun Zhou, University of Miami, United States
Copyright © 2021 Wenzel, Krämer, Geertz, Carrier, Felix, Könemann and Schlossarek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saskia Schlossarek, cy5zY2hsb3NzYXJla0B1a2UuZGU=