 Michael Clerx
Michael Clerx Gary R. Mirams
Gary R. Mirams Albert J. Rogers2
Albert J. Rogers2 Sanjiv M. Narayan
Sanjiv M. Narayan Wayne R. Giles
Wayne R. Giles- 1Centre for Mathematical Medicine and Biology, School of Mathematical Sciences, University of Nottingham, Nottingham, United Kingdom
- 2Department of Medicine and Cardiovascular Institute, Stanford University, Stanford, CA, United States
- 3Department of Physiology and Pharmacology, University of Calgary, Calgary, AB, Canada
Although plasma electrolyte levels are quickly and precisely regulated in the mammalian cardiovascular system, even small transient changes in K+, Na+, Ca2+, and/or Mg2+ can significantly alter physiological responses in the heart, blood vessels, and intrinsic (intracardiac) autonomic nervous system. We have used mathematical models of the human atrial action potential (AP) to explore the electrophysiological mechanisms that underlie changes in resting potential (Vr) and the AP following decreases in plasma K+, [K+]o, that were selected to mimic clinical hypokalemia. Such changes may be associated with arrhythmias and are commonly encountered in patients (i) in therapy for hypertension and heart failure; (ii) undergoing renal dialysis; (iii) with any disease with acid-base imbalance; or (iv) post-operatively. Our study emphasizes clinically-relevant hypokalemic conditions, corresponding to [K+]o reductions of approximately 1.5 mM from the normal value of 4 to 4.5 mM. We show how the resulting electrophysiological responses in human atrial myocytes progress within two distinct time frames:
(i) Immediately after [K+]o is reduced, the K+-sensing mechanism of the background inward rectifier current (IK1) responds. Specifically, its highly non-linear current-voltage relationship changes significantly as judged by the voltage dependence of its region of outward current. This rapidly alters, and sometimes even depolarizes, Vr and can also markedly prolong the final repolarization phase of the AP, thus modulating excitability and refractoriness.
(ii) A second much slower electrophysiological response (developing 5–10 minutes after [K+]o is reduced) results from alterations in the intracellular electrolyte balance. A progressive shift in intracellular [Na+]i causes a change in the outward electrogenic current generated by the Na+/K+ pump, thereby modifying Vr and AP repolarization and changing the human atrial electrophysiological substrate.
In this study, these two effects were investigated quantitatively, using seven published models of the human atrial AP. This highlighted the important role of IK1 rectification when analyzing both the mechanisms by which [K+]o regulates Vr and how the AP waveform may contribute to “trigger” mechanisms within the proarrhythmic substrate. Our simulations complement and extend previous studies aimed at understanding key factors by which decreases in [K+]o can produce effects that are known to promote atrial arrhythmias in human hearts.
Introduction
Detailed knowledge of plasma electrolyte (e.g., K+, Na+, Ca2+, and Mg2+) levels, and an understanding of the consequences of even small changes in one or more of them, are needed for optimal clinical management of various chronic diseases (Franse et al., 2000; Ahmed et al., 2007; Barrett Bowling et al., 2010; El-Sherif and Turitto, 2011; Alderman et al., 2012; Wang et al., 2013; Collins et al., 2017; Krogager et al., 2020). Examples of the translational implications of changed electrolyte levels can be found in renal physiology/pathophysiology and dialysis (Genovesi et al., 2008; Krueger et al., 2011; Zimmerman et al., 2012; Vincenti et al., 2014), in case management of hypertension (Wrong, 1961; Krogager et al., 2020), and in clinical cardiac arrhythmia management (Weaver and Burchell, 1960; Zaza, 2009; Goyal et al., 2012). During in-hospital acute patient care, plasma levels of Mg2+, Ca2+, K+, and Na+ are monitored closely, and plasma K+ levels ([K+]o) can provide essential information for diagnoses and guidance for treatment (Macdonald and Struthers, 2004; Mehta and Emmett, 2017).
Regulatory mechanisms for plasma electrolytes are both robust and precise; and even small changes in divalent cation levels or in [K+]o can have significant physiological and clinical consequences. Indeed, large-scale studies have linked such electrolyte changes to fluid imbalances and resulting congestion, reduced muscle cramping, and lowered incidence of cardiac arrhythmias (Barrett Bowling et al., 2010; El-Sherif and Turitto, 2011; Collins et al., 2017) including atrial fibrillation (AF; Krijthe et al., 2012; Buiten et al., 2014). As a result, close monitoring of electrolyte levels is recommended. Hypokalemia is known to occur in 20 to 30% of hospitalized patients (Paice et al., 1986; Collins et al., 2017), but whether or not prevention or correction of abnormal plasma potassium levels has a beneficial impact on morbidity and mortality has yet to be shown in a large-scale trial (Mehta and Emmett, 2017). A computational review focusing on plasma electrolyte changes, and in particular on hemodialysis effects, was published by Passini et al. (2014).
In this study we focus on one potentially important plasma electrolyte alteration; the electrophysiological consequences of small, but significant, decreases in plasma K+ levels (hypokalemia) in the human atrium. It is well established that small reductions (1–2 mM) in [K+]o can occur during renal dialysis (Zimmerman et al., 2012; Vincenti et al., 2014), treatment for hypertension (Krogager et al., 2020), diuretic therapies (Osadchii, 2010), or after intense or chronic exercise (Paterson, 1997). Our goal is to gain a detailed understanding of the short- and long-term effects of moderate hypokalemia on human atrial electrophysiological parameters including the resting potential (Vr), action potential (AP) waveform, refractory period dynamics and conduction velocity (CV). Previously, we have studied some of the effects of hyperkalemia on human atrial electrophysiological properties (Nygren and Giles, 2000) and reviewed hypokalemia in the ventricle (Trenor et al., 2018).
Before discussing our methods and results, we briefly review the normal atrial [K+]o level, the critical [K+]o-sensing mechanisms in atrial myocytes, and some important past experimental results on hyper- and hypo-kalemic effects on atrial electrophysiology.
Normal or Physiological Levels of [K+]o
In most in vitro physiological experimental settings, [K+]o is set by the superfusate at 5.4 mM. In contrast, a recent detailed electrolyte analysis of plasma from healthy adult human establishes the mean of this important electrolyte parameter to be around 4.2 mM (Collins et al., 2017). Analyzing both a control group (n = 339,297) and a large and geographically varied cohort of patients (n = 911,689) with heart failure, chronic kidney disease, and/or diabetes mellitus this study found mortality was lowest in the group with potassium levels from 4 to 5 mM (Collins et al., 2017; Mehta and Emmett, 2017). This was in agreement with an earlier study (Macdonald and Struthers, 2004) that reported lowest mortality for [K+]o levels of 4.5 to 5.5 mM. Indeed, the paper by Collins et al. defines “normal” levels as 4–4.5 mM, with “moderate hypokalemia” and “moderate hyperkalemia” given as 3.5–4 mM and 4.5–5 mM, respectively. Based on this information, it appears that many experimental studies of cardiovascular physiology (and pathophysiology) have been carried out under somewhat hyperkalemic conditions (5.4 mM instead of 4–4.5 mM). One reason for this is that much more stable electrophysiological recordings can be obtained in 5.4 than in 4 mM [K+]o.
[K+]o-Sensing Mechanisms in Human Atrial Myocytes
Inwardly Rectifying K+ Channels
This distinction between 5.4 and 4 mM [K+]o is important, since all skeletal muscles and most cardiac tissues exhibit a mechanism for sensing and then rapidly transducing changes in [K+]o into electrophysiological responses (altered Vr or AP waveform). This mechanism is finely tuned to [K+]o levels between 3 and 8 mM (Bailly et al., 1998; Bouchard et al., 2004; Burns et al., 2004; Trenor et al., 2018). These very quickly developing [K+]o-dependent changes arise mainly from altered current flow through a subset of the inwardly rectifying K+ channels, Kir2.1, 2.2, and 2.3 (Melnyk et al., 2002; Anumonwo and Lopatin, 2010) that exhibit a highly non-linear current-voltage relationship (Dhamoon and Jalife, 2005; Hibino et al., 2010). The corresponding transmembrane K+ current, denoted IK1 in cardiac myocytes, can strongly regulate the resting potential and alter the final phase of repolarization of the AP (Shimoni et al., 1992; Anumonwo and Lopatin, 2010; Weiss et al., 2017).
Na+/K+ Pump-Mediated Regulation of Electrophysiology and Contraction
However, it is also well known that the changes in [K+]o that are frequently encountered in clinical settings can significantly alter Na+/K+ pump current (Glitsch, 2001; Burns et al., 2004). K+ binding at the Na+/K+ external (plasma) site has a dissociation constant, Kd, (or half-maximal concentration, K0.5) of approximately 1.5 to 2 mM in both mammalian ventricular tissue (Nakao and Gadsby, 1989; Glitsch, 2001) and in human atrial myocytes (Workman et al., 2003), with the response saturating at approximately 8 mM. Thus, even relatively small changes in [K+]o can significantly alter the electrogenic current INaK (Blanco and Mercer, 1998). In cardiac myocytes, in most physiological settings, this outwardly directed current can hyperpolarize Vr by approximately 5 mV (Workman et al., 2003) and altered Na+/K+ pump activity may also produce changes in the AP waveform or duration (Glitsch, 2001; Workman et al., 2003; Sanchez et al., 2012). In addition, and perhaps more importantly, maintained and relatively long-term changes in Na+/K+ pump activity can significantly alter intracellular Na+ concentration, [Na+]i (Glitsch, 2001).
Electrophysiology of Human Atrial Myocytes
The availability of human atrial tissue (usually excised segments of the right atrial appendage) from open heart surgical procedures has enabled detailed electrophysiological studies of these preparations in both healthy and diseased conditions (Gelband et al., 1972; Ten Eick and Singer, 1979). A number of well-known papers provide important background on human atrium responses to both hypo- and hyper-kalemia for our study. Significant findings include:
i The resting potential in atrial myocytes is largely determined by IK1, a current which is highly sensitive to changes in [K+]o. IK1 is sensitive to [K+]o via three distinct mechanisms: a conductance decrease with decreased [K+]o (Sakmann and Trube, 1984); a shift in the reversal potential; and a change in the voltage-dependence of rectification (see Lu, 2004 for a review).
ii The resting potential is also modulated by the atrial Na+/K+ pump current (Rasmussen et al., 1984, 1986; Workman et al., 2003), which is highly sensitive to [K+]o and [Na+]i.
iii Although hyperkalemic increases above 5 mM are known to strongly depolarize Vr, very few studies in human atrium have investigated this effect at [K+]o levels in the hypokalemic range. A tissue study by Gelband et al. (1972) shows a sharp change in the [K+]o dependence at this point, while Ten Eick and Singer (1979) even show depolarization at very low [K+]o (see Supplementary Figure 1).
iv The resting potential, Vr, at the [K+]o level that is considered “normal” in in vitro atrial experimental settings (5.4 mM) is somewhat more depolarized than in ventricular myocytes, with values in the –80 to –75 mV range commonly reported (Ten Eick and Singer, 1979; Mary-Rabine et al., 1983; Workman et al., 2001).
v Even moderate changes in Vr affect sodium current availability, thereby significantly altering excitability and CV (Whalley et al., 1994; Skibsbye et al., 2016), so that [K+]o changes may alter the effectiveness of drugs that target the fast sodium current (Singh and Vaughan Williams, 1971).
vi Electrophysiological properties of the human atrial myocyte change or remodel significantly under long lasting or chronic AF conditions. Two prominent features of the new phenotype are a marked reduction in atrial AP duration and a significant hyperpolarization of Vr (Workman et al., 2001).
These findings inspired three questions, which we address using mathematical models of the atrial AP: (i) What is the role of the non-linear (inwardly rectifying) background K+ current IK1 in regulating the resting potential and AP in hypokalemia? (ii) How do the two outward currents in human atrium that are strongly modulated by [K+]o (IK1 and INaK) interact in settings that mimic clinical hypokalemia? (iii) What are the main functional changes in human atrial tissue strands that result from short- or long-term hypokalemia?
To address these questions and relate our findings to previous publications that have studied similar problems (Passini et al., 2014; Vincenti et al., 2014), the first part of our mathematical modeling study focuses on the almost immediate changes in the AP, one second after a sudden change in [K+]o. This initial part is expected to reveal the immediate effects due to changes in IK1. Part two consists of a similar analysis done under quasi steady-state conditions (after 15 min), when changes due to the Na+/K+ pump have developed. In part three, these insights regarding [K+]o-induced effects, are applied to assess excitability and conduction dynamics in a simulated strand of human atrial myocytes. Previous work by Passini et al. (2014) has shown that replicating electrolyte changes is challenging for human atrial myocyte models. We attempted to mitigate this by performing a multi-model study. Although we restrict ourselves to a single model in the main manuscript, we will refer to supplementary results from the other models throughout.
Materials and Methods
Simulations were performed using models of single human atrial myocytes and human atrial tissue strands. In total, our study used seven published mathematical models of the human AP, as listed in Table 1. All models and scripts that were used are available for download at: https://github.com/CardiacModelling/AtrialLowK.
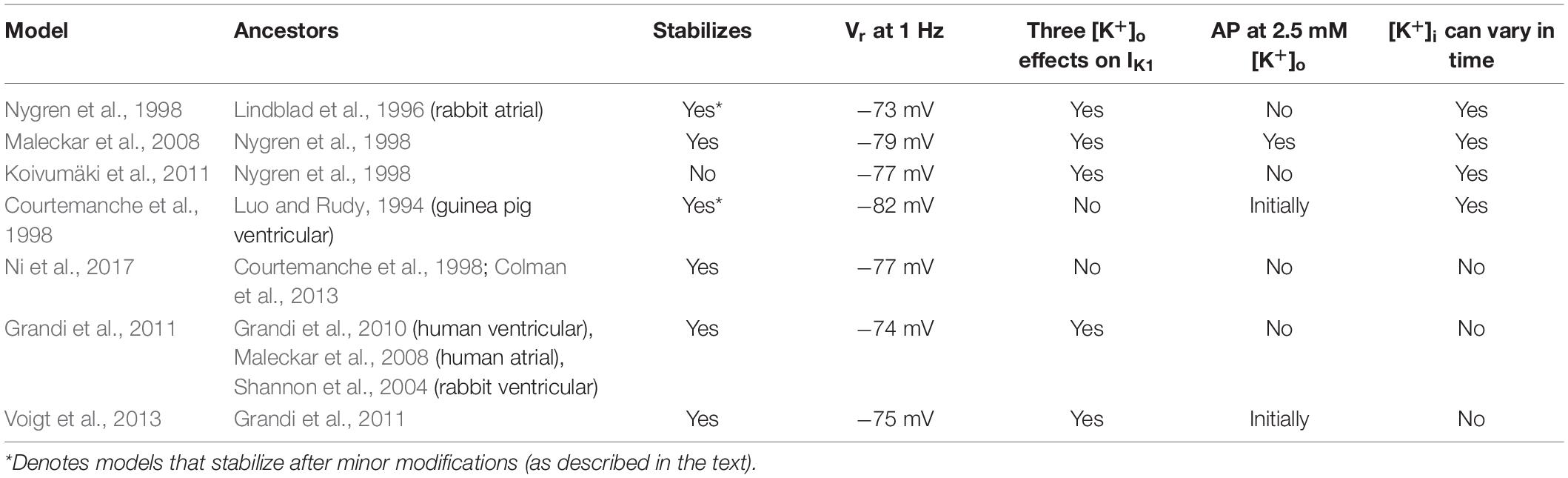
Table 1. Summary of the mathematical models of the human atrial action potential and resting potential that form the basis of this study.
Mathematical Models of the Human Atrial Action Potential
A number of different mathematical models of the human atrial AP and resting potential have been published (for reviews see Wilhelms et al., 2013; Heijman et al., 2016; Vagos et al., 2018). While some of these models include detailed subcellular spatial modeling of intracellular Ca2+ homeostasis/buffering, we have used less complex models that have a limited number of “compartments” representing average concentration in the cytoplasm, sarcoplasmic reticulum, and dyadic space, etc.
As the first step in this study, the seven published models that are listed in Table 1 were implemented. CellML 1.0 (Hedley et al., 2001) implementations of the models by Courtemanche et al. (1998); Nygren et al. (1998), and Maleckar et al. (2008) were downloaded from the Physiome Model Repository (Yu et al., 2011) and then converted to Myokit “mmt” format (Clerx et al., 2016). Novel Myokit and CellML implementations were created for the Grandi et al. (2011); Koivumäki et al. (2011), and Ni et al. (2017) models based on code kindly provided by the authors. These three “re-implementations” were verified by comparing the calculated state derivatives of the original implementations with our versions at the default initial conditions. A Myokit implementation of the Voigt et al. (2013) model was provided by the original authors. Myokit and CellML files for all models used in this study are provided in the online repository accompanying paper.
All figures were made using an approach similar to that described by Cooper et al. (2011), where essential variables in each model were annotated with labels and unit information so that simulations could be written in a model-agnostic manner. In some models a number of different currents (e.g., ICaL) are coded in such a way that the total current is divided into components according to their ion selectivity or subcellular localization. When plotting these currents, we show the sum of all components. The Nygren et al. and Maleckar et al. models both include a “restricted diffusion” or “cleft” space immediately outside the cell membrane, in which the [K+]o profile can differ from the bulk [K+]o. In simulations with these models, any effects of restricted diffusion within these cleft spaces were removed by assigning the same [K+]o values for both the bulk extracellular (or superfusate) volume and the cleft space.
Single-Cell Simulation Procedures
Single human atrial myocyte AP simulations were performed using Myokit’s CVODE-based (Hindmarsh et al., 2005) simulation engine. All simulations were run with a tolerance setting of 10–8 for both absolute and relative errors. All single myocyte models were paced at 1 Hz using a 0.5 ms stimulus. The first stimulus was applied 50 ms after the start of the simulated experiment, the second stimulus at 1,050 ms, and so on.
Before running experiments, the models were conditioned by: (i) setting the stimulus amplitude to twice threshold value required to elicit an AP (determined at baseline [K+]o); and (ii) pacing each model until the AP was stable from beat to beat. Specifically, each model was paced until the maximum relative change in any state variable was less than 0.001% from beat to beat: maxi|xi[j]−xi[j−1]|/ri < 10−5. where xi[j] is the value of the i-th state variable at t = j⋅1,000ms, and ri is the range (maximum – minimum) of the i-th state variable during the first AP. For most models this “stable” condition was reached within a few hundred paces, although >10,000 paces were needed for the Grandi-Pandit-Voigt et al. model. Our implementation of the Koivumäki et al. model never stabilized enough to meet this criterion, so was instead used with a state obtained after > 10,000 paces.
Minor modifications had to be made to the models of Courtemanche et al. and Nygren et al. to ensure stabilization. In both models, the equations for the internal K+ concentration [K+]i were updated to include the influence of the stimulus current, which has become a standard approach to ensure conservation of charge (Jacquemet, 2007). The Nygren et al. (1998) model contains a constant term ΦNa,en introduced to approximately balance the charge influx corresponding to the stimulus. This extra term is not required when stimulus charges are explicitly accounted for in [K+]i, and so ΦNa,en was set to zero (Jacquemet, 2007).
Na+/K+ Pump I–V Relationships
Some figures show simulated APs along with I–V curves for INaK. These I–V curves were calculated by varying membrane potential but holding other variables affecting INaK (e.g., the internal sodium concentration) fixed at their values just before the graph was made (i.e., corresponding to the left-most point of the shown AP).
Simulations of Human Atrial AP Conduction in Simulated Strands
To investigate the effects of low [K+]o conditions on AP refractoriness and CV, simulations were carried out based on 1–D strands consisting of electrotonically coupled atrial myocytes. This approach was implemented to approximate the experimental procedure used by Smeets et al. (1986). Simulations were run using a forward-Euler integration scheme implemented in Myokit (Clerx et al., 2016), using a step size of 0.001 ms and a Rush-Larsen approximation for Hodgkin-Huxley gate variables. At each selected time, t, the intercellular current flow between any two neighboring atrial myocytes (denoted i and j) was calculated as Iij(t) = g(Vi–Vj) and used to evaluate the state variable derivatives in the model for each cell (with conductance g determined as explained below). In this study, 200 myocytes were included, leading to a strand length of 2 cm (assuming a myocyte length of 0.1 mm). Excitation and AP propagation were initiated at 1 Hz by delivering 2 ms stimuli, adjusted to be 2 times the diastolic threshold. These stimuli were applied only to the left-most 5 myocytes of each simulated strand.
To determine the cell-to-cell conductance g, all 200 atrial myocyte models were set to the same initial conditions that were used in single myocyte simulations (i.e., the steady-state conditions for a single cell at 1 Hz). Simulations were run from this starting point, and g in each strand was varied until a CV of 80 cm/s was obtained (Spach et al., 2007). CV was calculated by estimating the start time of the AP in cells 50 through to 150, and using linear least squares (with the myocyte dimensions given above).
After the stimulus parameters and cell-to-cell conductance were estimated using the procedure above, each simulated strand was preconditioned by: (i) using a [K+]o of 5.4 mM, applying the single-cell steady-state to each of the connected cell models; and (ii) simulating 50 APs at a frequency of 1 Hz. This approach was adopted to mimic commonly-used experimental conditions at a heart rate of a healthy subject at rest. To check whether this procedure led to a sufficiently stable system, we then simulated one further AP and calculated the relative change in all state variables before and after this beat. This was found to be in the range of 0.01 to 0.2% in all models (see Supplementary Section 6).
Short- and Long-Term Effects of Hypokalemia on the Refractory Period and Conduction Velocity
In both the single myocyte and the tissue strand components of this study, the immediate and longer-term effects of step changes to different levels of [K+]o were evaluated by analyzing changes in AP waveform, Vr, and non-linear I–V characteristics at a time point either 1 s or 15 min after the [K+]o change.
In the human atrial strand simulations, a standard S1/S2 paired stimulus protocol was used to determine the functional refractory period (FRP). By convention, the FRP was defined as the shortest time between the two successive stimuli that successfully elicited a second propagating AP with a CV of at least 40 cm/s. Values for the wavelength (WL) of conduction or “wavelength of the cardiac impulse” parameter (Smeets et al., 1986) were obtained as the product of FRP and CV.
Results
Immediate Effects
Although several computational models of the human atrial AP and resting potential have been published, none of them have been formulated or validated using data obtained at low (or perhaps even normal) [K+]o, and predictions for low plasma electrolyte levels from published models are known to vary qualitatively and quantitatively (Passini et al., 2014). We therefore started our study by evaluating seven different published mathematical models of human atrial electrophysiology. A feature-by-feature comparison of these models is given in Table 1, and baseline APs and calcium transients are shown in Supplementary Figure 2. A comparison of the currents active during the AP and in the diastolic phase is shown in Supplementary Figure 3 while Supplementary Section 2.3 compares the models’ restitution characteristics. A detailed description of the IK1 and INaK equations in each model is provided in Supplementary Section 3.
We inspected each model:
i to check whether each exhibited stable resting potential values and characteristic AP waveforms at a 1 Hz heart rate;
ii to ensure a sufficiently strong Na+ current to sustain propagated AP responses in our atrial strand simulations; and
iii perhaps most importantly, to determine whether each model could accurately reproduce the three-part dependence of IK1 on [K+]o, via its reversal potential, maximum conductance, and most importantly its rectification.
From the models that fit these criteria, we selected the one by Voigt et al. (2013) for use in the main text – but we will refer to the Supplementary Material throughout for complementary results in the other models.
In Figure 1, we compare the Voigt-Heijman et al. model’s output with that of its predecessor, the Grandi-Pandit-Voigt et al. model. The three currents that were modified in the Voigt-Heijman et al. model are highlighted in bold on the y-axis labels. Both simulations were run at baseline conditions and paced at a 1 Hz frequency. The AP and Vr data in Figure 1A confirm that the resulting two AP waveforms are very similar. The values of the resting potential, Vr, both fall within the range that has been established in human atrial tissue (Gelband et al., 1972; Mary-Rabine et al., 1983; Workman et al., 2001).
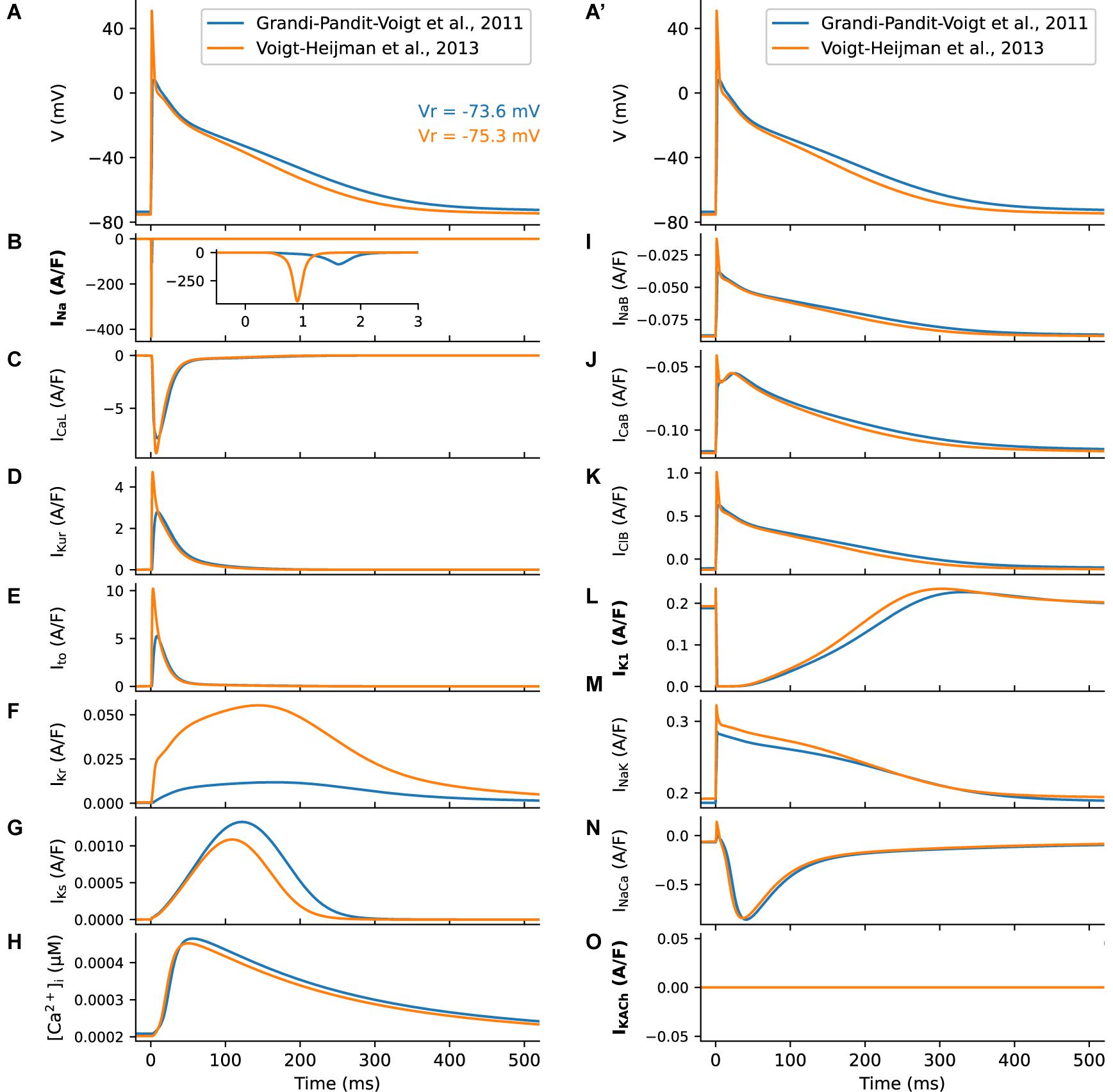
Figure 1. Summary of the human atrial membrane action potential and underlying transmembrane ionic currents. The AP traces (A, A’) and underlying currents were computed using the original Grandi et al. (2011) model (blue) and the Voigt et al. (2013) modification. Where the underlying model formulation differs, labels for the respective transmembrane currents have been highlighted in bold. (B) Shows marked differences in the Na+ currents that generate the AP upstroke, caused by the changed Na+ current formulation. (C) shows L-type Ca2+ currents; (D) illustrates the rapidly activating delayed rectifier K+ current, IKur; and (E) denotes the Ca2+-independent transient outward current, Ito. (F, G) show the fast and slow time- and voltage-dependent delayed rectifier K+ currents, IKr (F) and IKs (G). (H), illustrates model generated changes in intracellular [Ca2+]i that take place during the AP. In both models there are three time-independent or background currents: the Na+ current is shown in (I); the background Ca2+ current in (J) and the background Cl– current in (K). Data depicting the inwardly rectifying K+ current, IK1 is shown in (L). Although the equations for this current differ between models, only modest differences between the produced current traces are observed. The electrogenic current generated by the Na+/K+ pump is shown in (M). A second distinct electrogenic current produced by the Na+/Ca2+ exchanger is illustrated in (N). In the Voigt et al. (2013) model it is also possible to simulate one of the effects of acetylcholine by utilizing a second distinct background K+ current, IKACh. In this study, we did not make use of this feature and thus this current was turned off as illustrated in (O). Note that the two superimposed APs in (A, A’) are identical; this was done to more clearly illustrate the sizes and time courses of the underlying currents in each column relative to the AP waveform.
Compared to its predecessor, the Voigt-Heijman et al. model produces a much larger transient Na+ current, as shown in Figure 1B. This difference is important: It is essential if this model is to be used in our simulations of AP propagation (summarized in Figure 5). A similar modification was used in previous studies using the Grandi-Pandit-Voigt model in a tissue context (Deo et al., 2013; Martins et al., 2014).
The Voigt-Heijman et al. model also generates significantly larger K+ currents than those in the Grandi-Pandit-Voigt et al. model (Figures 1D–G), despite the equations for these currents being identical in both models. Thus, the Voigt et al. (2013) model may have a larger safety factor for repolarization, or repolarization reserve (Varró and Baczkó, 2011). As seen in Figure 1M the Na+/K+ currents are very similar in these two models of the human atrial AP.
One drawback of this model, and of most other human atrial AP models, is that intracellular K+ ([K+]i) is a fixed parameter. In principle, this choice could limit the model’s ability to accurately reproduce long-term effects of hypokalemia due to slow intracellular concentration changes. However, none of the simulated experiments in this study would be expected to significantly alter [K+]i. This is confirmed in Supplementary Figures 15–17, showing at most 2 mM deviation in [K+]i for the three models with time-varying [K+]i and [K+]o-sensitive IK1 rectification.
An essential property of the biophysical behavior of a strong inward rectifier such as IK1 is the effect of [K+]o–-sensitivity on its conductance and rectification properties, which counteract the effect of hypokalemia on its electrochemical driving force. The magnitude and direction of these three effects is plotted independently in Supplementary Figures 7, 9, while Supplementary Figure 8 compares the [K+]o-sensitivity of IK1 rectification in the used models.
Figure 2 shows a side-by-side comparison of predicted electrophysiological changes after 1 s of exposure to an altered [K+]o in the Courtemanche et al. and Voigt-Heijman et al. models. In Figure 2A, the respective baseline AP waveforms and Vr levels, and the responses of both to selected changes in [K+]o in the range 2.5 to 8 mM, are shown. Note that in the Voigt-Heijman et al. model at [K+]o concentrations of less than approximately 4 mM, Vr moves in the depolarizing direction and the AP lengthens. In contrast, in the Courtemanche et al. model the same changes in [K+]o produce a progressive hyperpolarization of Vr and shortening of the AP. Information concerning the ionic basis for these changes in Vr and the atrial AP waveforms is presented in Figure 2B, where the I–V relationships for the background inwardly rectifying K+ current, IK1, are shown. It is apparent that the qualitative behavior is very different for the two models, although IK1 is significantly affected by [K+]o in both. Finally, the I–V curves in Figure 2C show that there are immediate effects on INaK (via Vr), but that these are moderate compared to the dramatic changes in IK1.
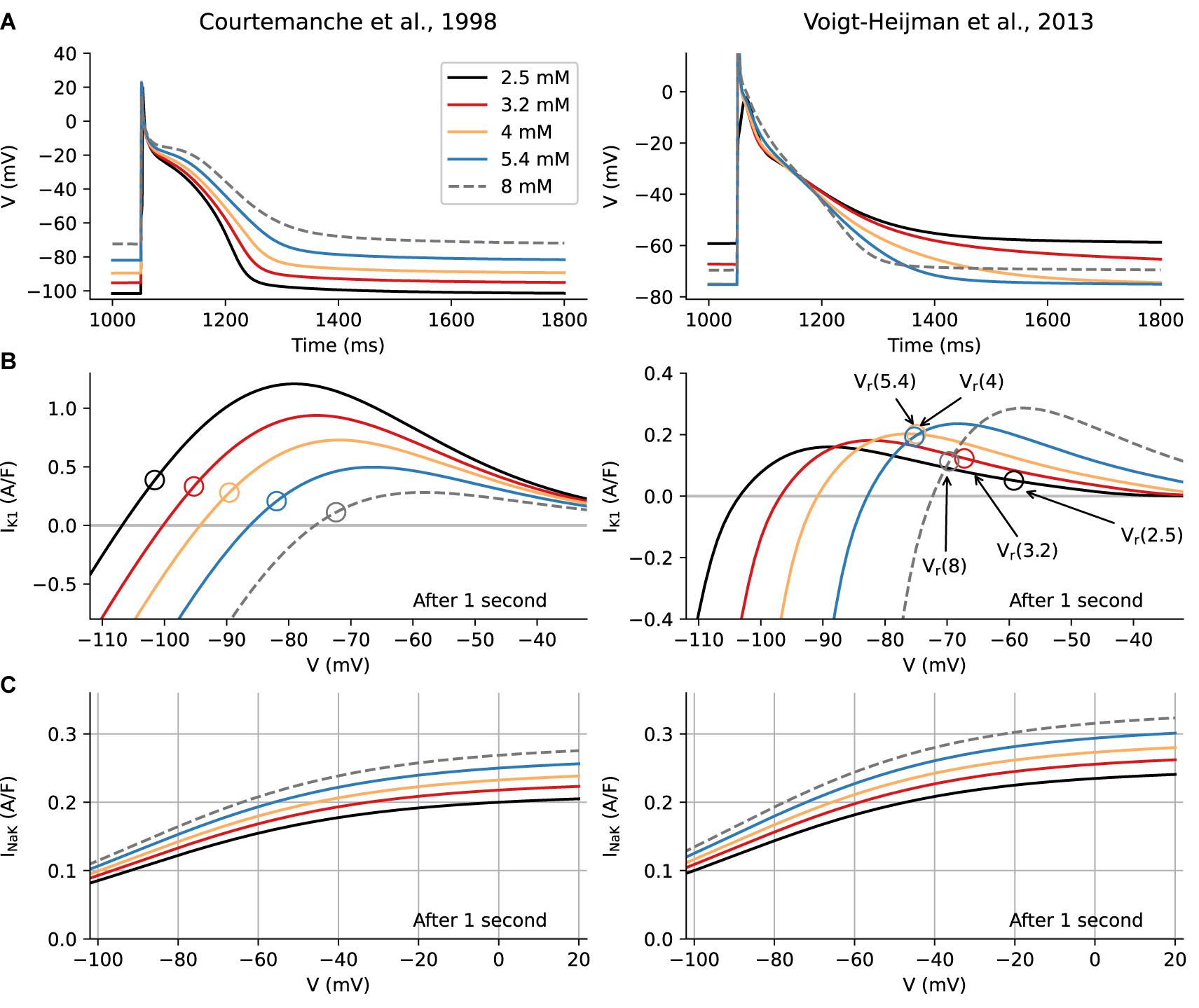
Figure 2. Immediate effects of alterations in [K+]o levels on human atrial Action Potential (AP), Resting Potential (Vr), and underlying ionic currents. Computational results for the Courtemanche et al. model are shown in the left column, and analogous results from the Voigt-Heijman et al. model are shown on the right. Each panel in Row A shows five superimposed AP waveforms that illustrate how [K+]o-induced changes in repolarization affect the AP waveform and the resting potential, Vr, 1 s after a step change in [K+]o. Note that in both models, hyperkalemia depolarizes Vr, but hypokalemic effects are model-dependent. In the Courtemanche et al. model the waveforms for the [K+]o levels representing clinical hypokalemia (3.2 and 2.5 mM) show a pronounced hyperpolarization and a shortening of the AP, while in the Voigt-Heijman et al. model hypokalemia depolarizes Vr and prolongs the AP. The origins of this difference can be seen in Row B, which depicts the I-V curve for IK1 at the selected [K+]o levels. Note that these are color-coded to match the AP traces in Row A, while open circles denote the resting potential of the myocyte in the corresponding [K+]o condition. In the Courtemanche et al. model, the only effect of lowering [K+]o is an increase in electrochemical driving force for IK1 (which is more larger at more depolarized potentials, causing the appearance of a peak current that moves “left and up”). In the Voigt-Heijman et al. this effect is still present, but the [K+]o -sensitivity of IK1 rectification and conductance counteracts this effect. Importantly, as described in section “Results” and Supplementary Section 3, the [K+]o dependence of IK1 gives rise to a complex pattern of changes in the non-linear inwardly rectifying I–V relation for IK1. The superimposed I–V curves in each panel of Row C illustrate the corresponding I–V curve of electrogenic current, INaK, generated by the Na+/K+ pump, in each of the [K+]o levels that were used for this analysis.
The significantly different pattern of results shown in the two columns of Figures 2A,B should be considered in conjunction with well-known molecular features and biophysical properties of the K+ channels that generate IK1 in human atrium (see section “Introduction”). In response to decreases in [K+]o the outward component of the non-linear I–V curves for IK1 not only translates in the hyperpolarizing direction (as would be expected from the increased electrochemical driving force for K+) but also “flattens.” While the hyperpolarizing translation leads to an increased current at Vr (as seen in the Courtemanche model), the flattening effect is dominant, so that a smaller outward current is generated in low [K+]o (Shimoni et al., 1992; Trenor et al., 2018). The superimposed I-V curves in Figure 2B also reveal that, when the effects on rectification and maximum conductance are taken into account, changes in [K+]o result in the outward component of the I–V curves exhibiting “cross over” within a range of membrane potentials (–80 to –60 mV). Importantly, this voltage range falls within the normal resting potential and the mid-to-final repolarization phases of the human atrial AP (Bouchard et al., 2004; Dhamoon and Jalife, 2005; Trenor et al., 2018). It is mainly for this reason that in 4 to 2.5 mM [K+]o, although the electrochemical driving force for K+ fluxes has increased, the size of the outward current component decreases substantially. Thus, very soon after lowering [K+]o, this very rapid change in IK1 is the main reason for a depolarization of Vr, and significantly lengthens the final phase of repolarization in the ventricle (Shimoni et al., 1992). In marked contrast, when this essential feature of IK1 is not incorporated into the model, as in the Courtemanche et al. model, Vr progressively hyperpolarizes and the AP shortens in low [K+]o.
In Supplementary Figure 10, we show the full Vr-[K+]o relationship predicted by the seven models, and compare it with the observations by Gelband et al. (1972) and Ten Eick and Singer (1979). This analysis shows that all models with [K+]o-sensitive IK1 predict a depolarization of Vr at severely hypokalemic levels, although the point where the depolarization sets in varies from 4 to 5 mM in the Voigt-Heijman, Grandi-Pandit-Voigt, and Nygren et al. models, to the much lower value of 2 mM in the model by Koivumäki et al. The 2014 paper by Passini et al., recommended using a modified version of the Courtemanche et al. model, in which Vr starts depolarizing at around 3 mM [K+]o.
Delayed Effects
The computational results presented in Figure 2 revealed rapid changes in the background K+ current, IK1, in response to decreases in [K+]o. As noted in the Introduction, we analyze the range of [K+]o known to be present in clinical situations, e.g., as a consequence of diuretic treatment for hypertension or heart failure as well as during renal dialysis procedures (Zaza, 2009; Zimmerman et al., 2012). To begin to gain an improved understanding of the ionic basis for atrial arrhythmias associated with hypokalemia, and to relate our findings to previous detailed studies of low [K+]o conditions in experimental and clinical settings (Aronsen et al., 2015; Weiss et al., 2017), it was necessary to do corresponding AP simulations at times 10–15 min after altering [K+]o. This is because electrophysiological changes due to the Na+/K+ pump would be expected to develop very slowly compared to the ion channel-mediated mechanisms associated with IK1. Accordingly, in Parts II and III of our study, electrophysiological changes in both single myocyte and strand models were assessed and compared almost immediately (1 s) after changing [K+]o levels, and also 15 min later (as an approximation for quasi steady-state conditions). Our rationale was that, in principle, comparison of these two time points could reveal rapidly-developing consequences of hypokalemia (which might serve as a localized “trigger” for atrial rhythm disturbances) and more slowly-developing and long-lasting alterations to the atrial substrate (that may contribute to proarrhythmia; Zhang et al., 2005; Krogh-Madsen et al., 2012; Pandit and Jalife, 2013).
Figure 3 is based on these two sets of calculations, each done using the Voigt-Heijman et al., model, paced at 1 Hz for either 1 s or for 15 min after a sudden [K+]o change, as described in Methods. Figure 3A (1 s) and Figure 3B (15 min) show the resulting changes in AP waveforms and in Vr at the selected values of [K+]o. Figures 3C,D show ionic currents due to IK1 (continuous lines) and the Na+/K+ pump current, INaK (broken lines).
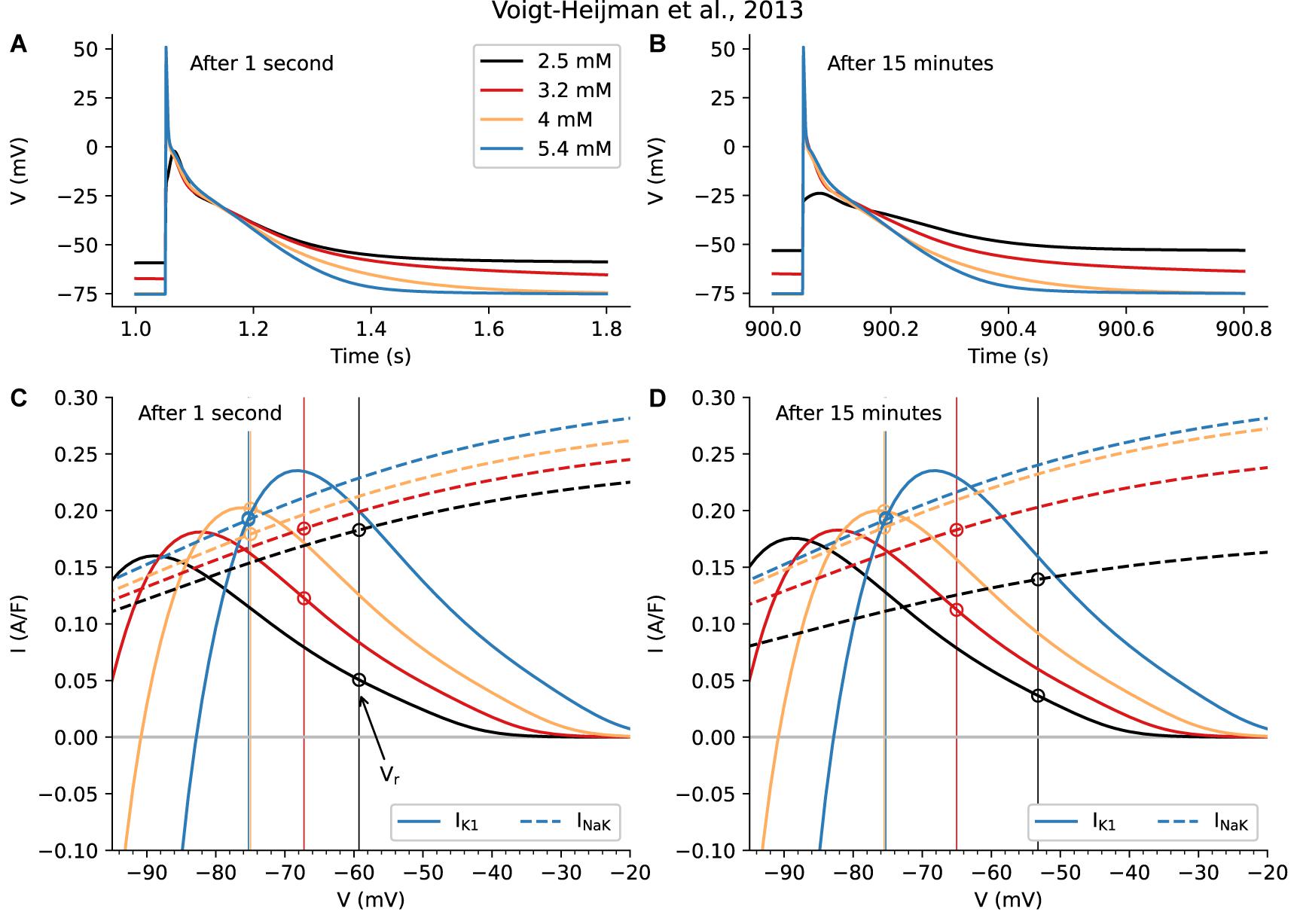
Figure 3. Evaluation of the Short- and Long-Term Physiological Effects of Changes in [K+]o on the Background K+ Current, IK1, and the Na+/K+ Pump Current, IP. Data in the left column (A, C) was obtained 1 s after changing from 5.4 mM to the shown levels of [K+]o; the results in the right column (B, D) are 15 min after changes in [K+]o. The superimposed AP traces in (A) are similar to those in Figure 2 and are included here to provide a direct comparison of very rapid short-term changes in [K+]o with those that develop in a time-dependent fashion, shown in (C). Note that the overall patterns of changes in Vr and AP waveforms are similar when captured at 1 s vs. at 15 min. However, at 15 min in hypokalemic conditions (2.5 and 3.2 mM [K+]o) Vr depolarizes slightly more and the final phase of AP repolarization is slowed markedly. Note also the very small depolarization observed for 2.5 mM after 15 min. (C, D) provide a direct comparison of the size and voltage-dependence of IK1 and INaK. Side-by-side comparison of these two components reveals that, as expected, there is a time-dependent decrease in INaK in the two hypokalemic [K+]o conditions (3.2 and 2.5 mM). This, and the decrease in the outward current due to IK1, are primary reasons that Vr depolarizes to a new stable level. By contrast, the initial [K+]o induced reduction in INaK seen when switching from 5.4 to 4 mM is almost fully restored after 15 min.
As shown in Figure 3C, when [K+]o is decreased, net outward INaK exhibits an immediate downward shift, confirming that its affinity for [K+]o is a significant regulatory variable for Na+/K+ pump current in human atrium (Workman et al., 2003). The KD for [K+]o in the Voigt et al. (2013) model is set at 1.5 mM in accordance with classical experimental analyses (Glitsch, 2001).
The more slowly-developing, time-dependent changes shown in Figure 3D reveal that, for a small change from 5.4 to 4 mM, the initial downward shift (decrease) of INaK is partially recovered. This recovery occurs as a result of the well-known sensitivity of the Na+/K+ pump to changes in [Na+]i (Glitsch, 2001). For larger decreases in [K+]o, to 3.2 or 2.5 mM, the Na+/K+ pump appears not to recover, but instead INaK is further decreased.
The superimposed data sets in Figures 3C,D can be used to address the fundamental question: What are the differences in the sizes of the low [K+]o-induced changes in IK1 vs. those due to the electrogenic Na+/K+ pump current, INaK? Beginning with the current changes that occur in the range of the resting membrane potential (−80 to −50 mV), one can see from Figure 3C that the very rapid (within 1 s) changes in IK1 are at least as large as those due to the changes in INaK. Thus, very soon after [K+]o is decreased, IK1 is an important regulator of membrane potential and action potential duration (APD). In combination, the changes are likely to contribute to the “triggering mechanism” for atrial flutter/fibrillation. However, after 15 min the changes in INaK become more significant.
Second, and equally importantly, in the range of membrane potential that corresponds to the mid- to final repolarization phases of the AP (approx. −30 to −65 mV), the immediate current changes due to IK1 are larger than those due to INaK. This reveals that the highly non-linear change in IK1 is the predominant factor in regulating atrial AP duration in the short term. However, after 15 min the change in INaK has also become a significant regulatory factor. It is important to recall that both AP duration and the time course of repolarization strongly regulate Ca2+ fluxes through L-type Ca2+ channels in cardiac preparations having approximately triangular shaped APs (Clark et al., 1996; Sah et al., 2003) such as the human atrial myocardium. In addition, even small changes in Vr during the diastolic interval can markedly alter electrogenic current generated by the Na+/Ca+ exchanger, and thus strongly modulate [Ca2+]i (Clark et al., 1996).
Complementary results in the other models are shown in Supplementary Figures 11–17. For the models with [K+]o-sensitive IK1 rectification, they show a similar pattern of results, where the effects of small decreases in [K+]o on INaK are compensated, while larger changes are exacerbated over time.
Slowly-developing effects (minutes) in cell electrophysiology settings may occur through changes in intracellular electrolyte balances. Since the Na+/K+ pump is a determinant of the internal sodium concentration ([Na+]i) as well as being sensitive to small [Na+]i changes, we investigated the role of [Na+]i in the observed long-term changes. Our results are shown in Figure 4, which illustrates the time-dependent changes during the first 8 min after the change in [K+]o. At the end of this interval the system was seen to stabilize.
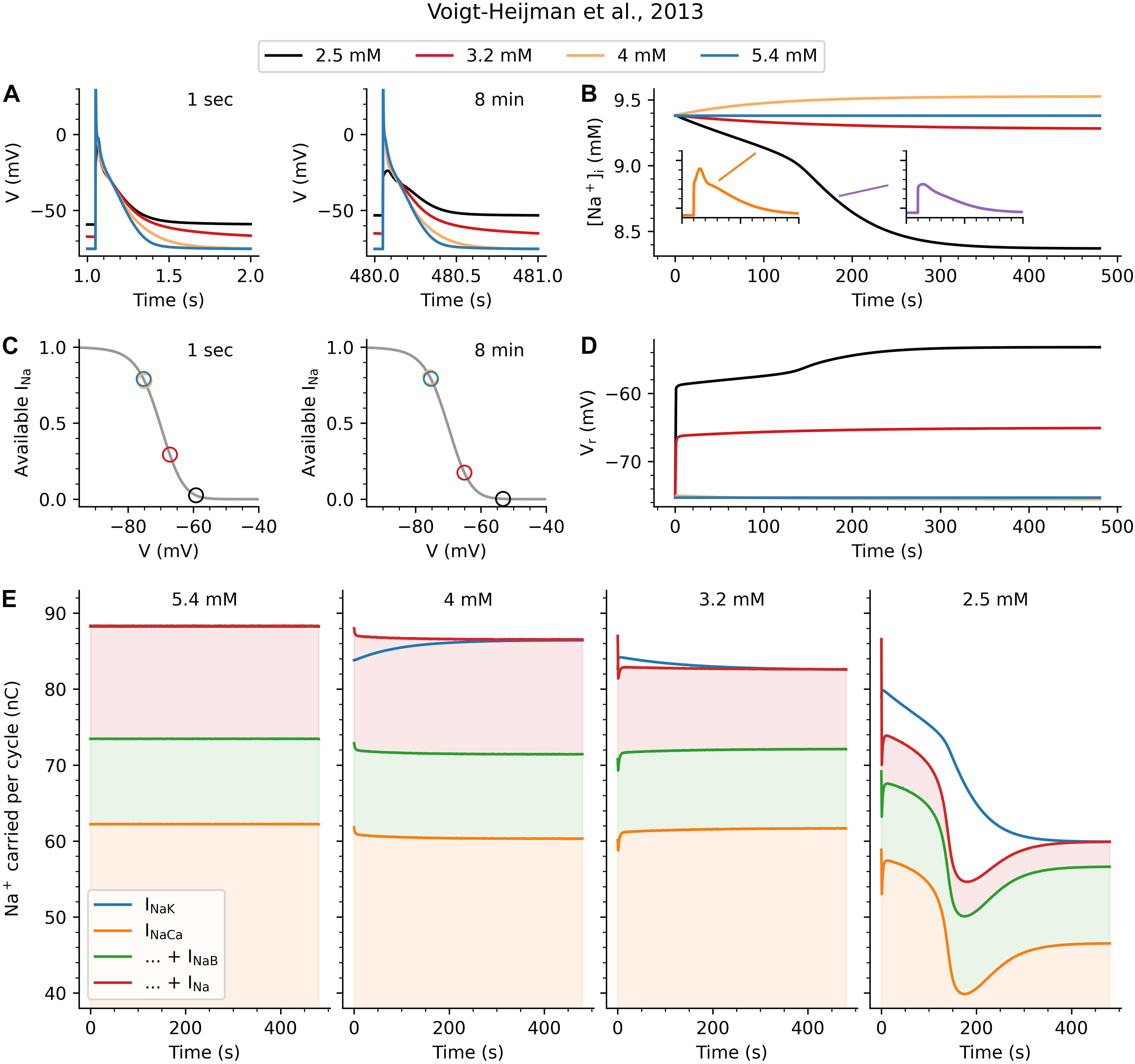
Figure 4. Mechanism underlying longer-term effects of [K+]o on Vr and AP. (A) shows the AP either 1 s (left) or 8 min (right) after a change in [K+]o from its baseline value of 5.4 mM. The same pattern of effects as in Figure 3 is observed here at 8 min, indicating that the system has stabilized. (B) shows how the [K+]o changes can cause either a [Na+]i increase (4 mM) or decrease (3.2 and 2.5 mM) over time. While the changes in [Na+]i start gradually in all cases, the 2.5 mM hypokalemic condition reveals a secondary effect occurring after around 140 beats. During this rapid transition phase, [Na+]i steeply declines and the AP waveform undergoes a qualitative change, from a triangular waveform to something resembling a subthreshold response. (C) is based on steady-state inactivation characteristics (gray lines) for INa. It shows the amount of recovered (available) INa at Vr after 1 s or after 8 min (colored circles) for all [K+]o conditions (note that the circles for 5.4 mM and 4 mM overlap). (D) shows how the changes to Vr develop over time, again showing a dramatic increase at around 140 s for 2.5 mM. (E) shows the charge carried into and out of the cell by Na+ ions during each pacing cycle (AP and diastolic interval). The blue line in each graph indicates INaK, the only outward Na+ flux. The orange, green, and red shaded areas shows the influx due to INaCa, INaB, and INa, respectively. The left-most graph illustrates the stable 5.4 mM situation, in which the system is in balance and the sum of inward fluxes equals the outward flux through INaK. The second panel (4 mM) shows an instantaneous reduction in inward flux (through reduced INaCa), and a greater reduction in outward flux (through reduced INaK). Over time, this causes the [Na+]i changes seen in (B) until the system restabilizes. The dominant instantaneous effect in the third graph (3.2 mM) is a decrease in Na+ influx through INa. Again the Na + /K + pump adjusts to compensate and rebalance the system, but now at a reduced [Na+]i and somewhat depolarized Vr. Finally, in the (2.5) case the gradual depolarization of Vr grows until no INa is available for an INa-driven regenerative AP. These mechanisms are described in further detail in the text and in Figure 6.
Action potentials either 1 s or 8 min after the [K+]o change are shown in Figure 4A. The changes in Vr are very similar to those illustrated in earlier figures. Figure 4B shows the changes in [Na+]i during the 1 Hz AP train, from the start of the simulation (including the very first AP before the graph in Figure 4A). There is a qualitative difference between the response to a small decrease (5.4 to 4 mM), where [Na+]i increases, and the larger decreases (to 3.2 and 2.5 mM), where [Na+]i decreases. A secondary change is seen to occur around 140 beats, when the rate of the progressive decrease in [Na+]i suddenly accelerates. The insets in Figure 4B show an AP before and after this time point, indicating a transition from a “normal” AP morphology to an small depolarization, resembling a “subthreshold response.” The basis for this sudden and significant change can be seen in the next panels. Figure 4C consists of two plots of the steady-state voltage-dependence of INa inactivation (gray lines). These curves are the same in both plots, and were obtained by multiplying the corresponding relationships for slow and fast inactivation together. The color-coded circles indicate the location of Vr, 1 s and 8 min after the change to [K+]o, showing whether INa is available or inactivated at rest. As can be seen, INa availability is almost entirely unaffected for the change to 4 mM. In contrast, the depolarized Vr at lower [K+]o values causes a significant reduction in the amount of available INa. As Figure 4D shows, the progressively depolarizing Vr for these hypokalemic conditions leads to a slowly decreasing amount of available INa. In the 2.5 mM [K+]o experiment, this ultimately results in a loss of inward INa current and a failure to depolarize or generate an AP.
Finally, Figure 4E shows the effects of the changes in [Na+]i and INaK on the Na+ homeostasis in the atrial myocyte. The left-most column shows the situation when [K+]o is unchanged and the system is in balance. The orange, green, and red shaded areas show the charges carried into the myocyte by Na+ influx during each pacing cycle (AP and diastolic interval) for the Na+/Ca2+ exchanger (INaCa), the background Na+ current (INaB), and the fast Na+ current (INa). At steady-state, the sum of these three Na+ influxes is balanced by the efflux of Na+ through INaK almost exactly (the model contains a separate slowly activating Na+ current, INaL, omitted here for clarity) so that the blue line for INaK and the red line for the sum of the inward fluxes overlap.
The second column in Figure 4E shows effects of a sudden [K+]o change from 5.4 to 4 mM, to which the Na+/K+ pump can adequately respond: While the immediate effect is a reduction of INaK (the blue line dips), this triggers a feedback response wherein reduced INaK causes an [Na+]i accumulation which increases INaK until the system is once again in balance (the blue and red lines overlap).
A perhaps more unexpected effect is seen in the third column of Figure 4E, which illustrates the restoration of Na+ balance after a step change to 3.2 mM [K+]o. Recall from Figure 3 that the immediate effects of this step-change include a reduction of IK1 and a depolarization of Vr. In this setting, the dominant and immediate effect is the reduction of Na+ influx through INa, due to the reduced INa availability produced by the depolarization of Vr. Again the Na+/K+ pump, acting as one element in a [Na+] homeostasis feedback mechanism, reacts to restore the balance. In this setting, reducing pump current and Na+ efflux, leads to the slow progressive increase in Vr seen in Figure 4D.
Finally, the fourth column of Figure 4E shows the effects of a reduction to 2.5 mM [K+]o. Initially, effects are similar to the 3.2 mM case: IK1 decreases, leading to Vr depolarization and a large and rapid decrease in Na+ influx through INa; followed by a steady decrease in [Na+]i as the Na+/K+ pump reduces its efflux to match. In this scenario, however, the resulting Vr depolarization is so large that AP excitation fails, causing the significant shifts seen starting around 100 beats. Although Na+ balance is eventually restored the atrial myocyte has lost its excitability.
While we did not repeat this detailed analysis for all seven models, very similar patterns of results can be seen in the other models in Supplementary Figures 11–17. Changes to [Na+]i differ in direction according to the size of the [K+]o decrease, and there is often a sudden qualitative change a few minutes into the experiment. As expected, the models without [K+]o-sensitive IK1 rectification show a very different, monophasic [Na+]i-response. This monophasic response is also shown by the model by Maleckar et al., which hyperpolarizes at 3.2 and 2.5 mM [K+]o (see Supplementary Figure 10).
Strand Simulations
The third part of this study consisted of a series of computations aimed at revealing some of the important contributing factors for either (i) triggering atrial arrhythmias or (ii) producing long-lasting electrophysiological changes in multicellular atrial syncytium (often referred to as the substrate) in the setting of hypokalemia. It is known that both hyper- and hypo-kalemia are associated with initiation or prolongation of rhythm disturbances in both the ventricles and atria of mammalian hearts (Gettes et al., 1962; Weiss et al., 2017). For these studies, a model system (described in detail in section “Materials and Methods”) was formulated on the basis of a one-dimensional strand of 200 identical Voigt et al. (2013) human atrial myocyte models. Stimulus protocols (described in section “Materials and Methods”) and analyses were applied to determine the dynamics of the FRP, alterations in CV and related changes in conduction WL. Simulations were stopped at either 1 s or 15 min following changes to the same levels of [K+]o that were used previously in single myocyte studies (Figures 1–4). A 1 Hz stimulus train was applied.
The design of these studies and most aspects of the data analysis were guided by an extensive experimental study concerning the effects of [K+]o on rabbit atrial tissues by Smeets et al., 1986. Results obtained at 1 s and 15 min after [K+]o changes are illustrated in Figure 5. In Figure 5A the Vr values and AP waveforms formed in response to selected [K+]o levels are shown as measured in the first myocyte in the simulated human atrial tissue strand. The vertical lines indicate the FRP at each [K+]o level. At 2.5 mM [K+]o the atrial tissue strand generates only a very small regenerative AP. It fails to support propagation/conduction, so that no FRP can be shown. The reasons for this discrepancy were not pursued in detail. The complete results of these simulations are summarized in Figures 5B–F, in terms of: the resting potential (5B), CV (5C), and (where possible) the APD (5D), FRP (5E), and WL (5F). In agreement with the Smeets et al. study, the CV is decreased at lower [K+]o levels, in both the 1 s and 15 min settings. However, the observed elongation of the APD in these simulations leads to a much increased FRP at low [K+]o, resulting in an increased WL of conduction. This finding is in contrast to the decrease in WL reported by Smeets et al.
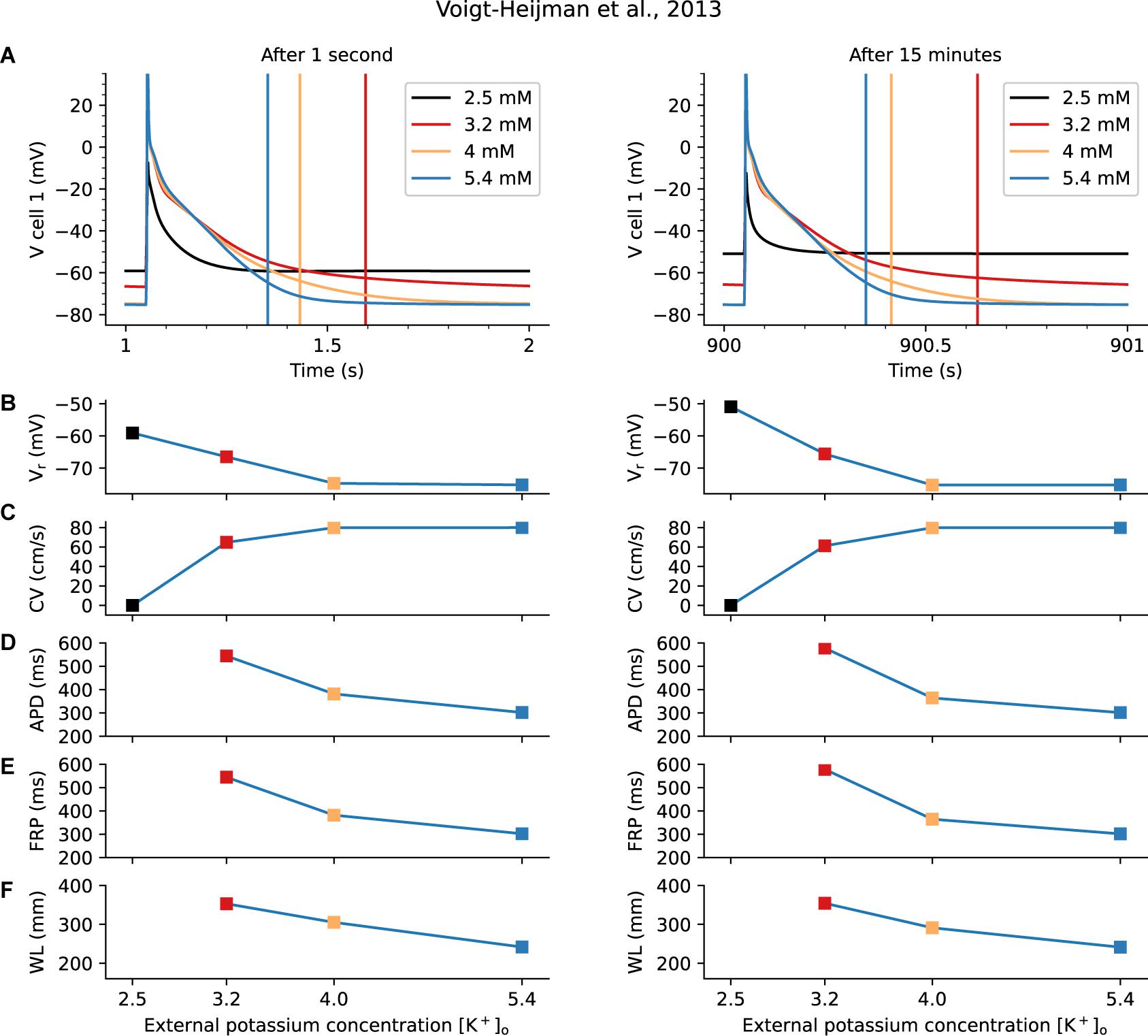
Figure 5. Human Atrial Tissue “Strand” Model for Evaluation of Effects of Hypokalemia on Excitability, Conduction Velocity and Wave Length of Conduction. The Voigt-Heijman et al. model of the human atrial myocyte was used as the basis for constructing a 1-D, 200 myocyte atrial strand. Standard steady-state pacing and extra stimulus protocols (S1–S2) were applied, and the results are shown either immediately (1 s) after altering [K+]o or under quasi steady-state conditions (after 15 min). The separate panels in Row A show individual AP waveforms in the first myocyte in the simulated strand, at the [K+]o levels that were the focus of this study. The vertical lines in these graphs indicate the functional refractory periods (FRP) that were estimated from the tissue strand response after either 1 s (left) or 15 min (right). These results, and the corresponding Vr, CV, APD, and WL values are presented in Rows B–F below. Note that in 2.5 mM [K+]o the small AP failed to support conduction, and so no APD, FRP, or WL could be determined. See section “Results” for more details.
Complementary results in generated by the other models are shown in Supplementary Section 6. Since the Voigt-Heijman et al. model results after 15 min did not differ qualitatively from the results after 1 s, we did not run the computationally expensive 15 min fiber simulations for all models. As before, the results differ dramatically for the models with and without [K+]o-dependent IK1 rectification, which show decreases in both APD and FRP. We note, however, that none of the models tested showed the expected prolonged APD with FRP shortening. Finally, results relating to single-cell excitability are shown in Supplementary Section 7.
Discussion
The computational work in this paper has illustrated how hypokalemia can lead to an increased deviation of Vr from the potassium reversal potential, even resulting in depolarization at lower [K+]o. To produce this effect, models of the AP and Vr under hyper- or hypokalemic conditions must include the three [K+]o effects on the inwardly rectifying background K+ current, IK1, which is expressed in both the left and right atrium of mammalian hearts (Voigt et al., 2010). In the human atrium, IK1, is generated mainly by the K+ channel transcripts, Kir2.1 and Kir2.3 (Melnyk et al., 2002). These K+ transcripts, alone or in combination, can sense and react to changes in [K+]o with high affinity and very rapid response times. Recognition and understanding of these properties provides the basis for understanding the direct and indirect effects of clinically-relevant hypokalemia (Macdonald and Struthers, 2004; Krijthe et al., 2012; Mehta and Emmett, 2017). Specifically, validated mathematical models must be able to accurately reproduce the [K+]o-dependent modulation of the highly non-linear I-V relationship (voltage-dependent rectification) generated by “strong inward rectifier” transcripts such as Kir2.1/2.3 (Lu, 2004; Dhamoon and Jalife, 2005; Anumonwo and Lopatin, 2010; Hibino et al., 2010) in all myocytes from human atrium (Figure 2) and ventricles (Bouchard et al., 2004; Trenor et al., 2018).
Based on our application of five models that include [K+]o-dependent IK1 rectification, we saw that even small decreases in [K+]o can very rapidly produce a significant lengthening of the AP duration. This effect occurs almost immediately (within 1 s or 1 heartbeat), and is mediated mostly through IK1 and Vr. At both the single myocyte level and in our 200 myocyte 1-D strand of atrial tissue, this hypokalemia-induced change in the AP waveform significantly lengthened the refractory period and the WL for conduction (Figure 5). A further insight gained from our simulations is that even small decreases in [K+]o, e.g., 4 to 3.2 mM; or 3.2 to 2.5 mM can alter the outward electrogenic current produced by the Na+/K+ pump, and that this interacts with Vr, [Na+]i, and INa in ways that are predictable in the short term (seconds), but much more complex in the longer term (minutes). This was explored in detail in Figure 4. Notably, qualitatively different effects on the AP and Vr were observed when changing [K+]o from 5.4 mM to 4, 3.2, and 2.5 mM, as summarized in Figure 6. While the analysis in Figures 4, 6 is more strongly model-dependent than our previous results, we believe they show a pattern of changes that is shared by other models that include [K+]o-dependent IK1 rectification, as can be seen in Supplementary Figures 11–17.
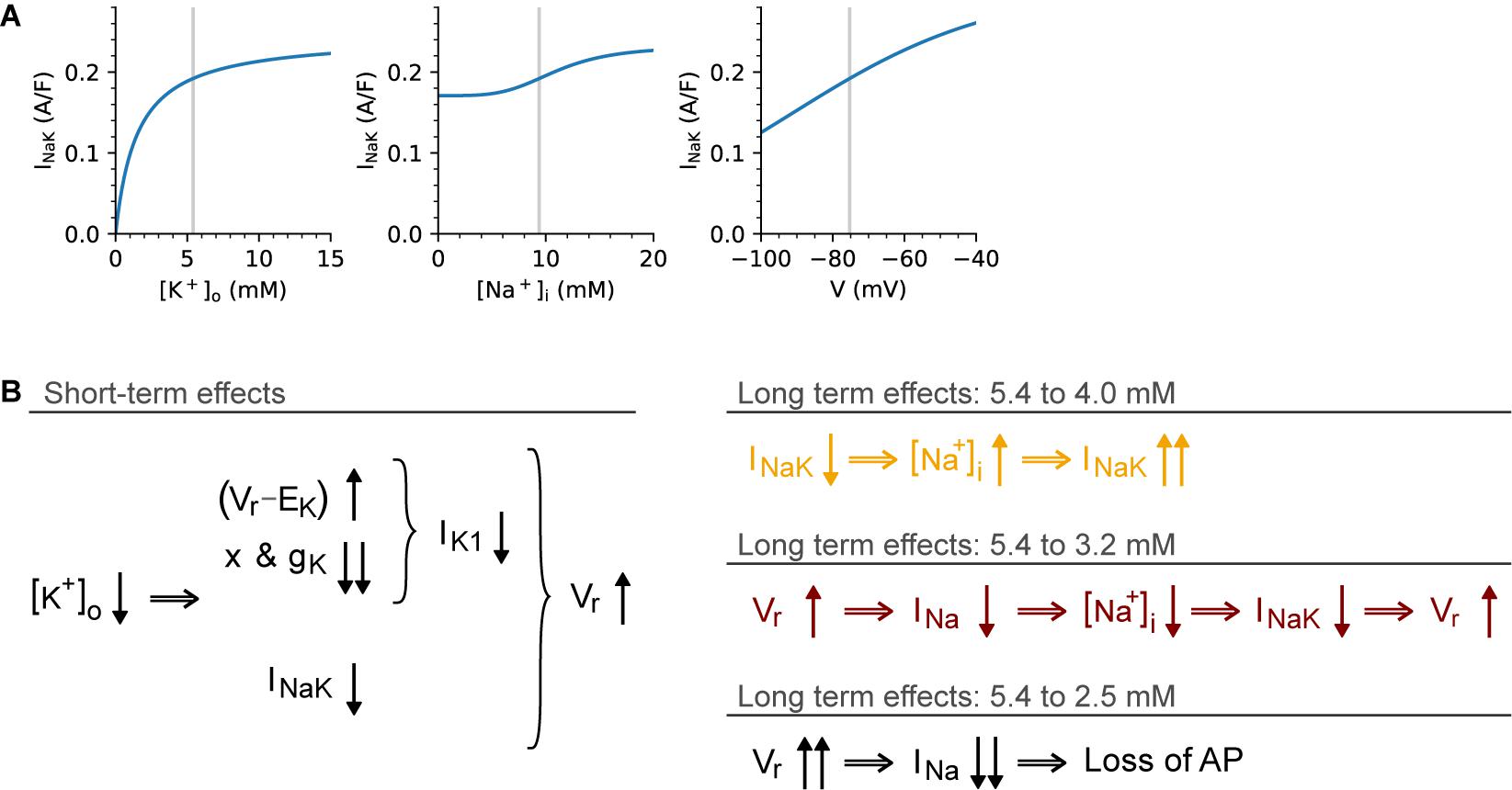
Figure 6. Qualitatively different long-term effects of different [K+]o changes. This diagram summarizes the origins of the contrasting short- and long-term effects seen in Figure 4. (A) illustrates the sensitivity of INaK to [K+]o (left), [Na+]i (center), and Vr (right), as determined from the Voigt-Heijman model under baseline conditions. The schematic in (B) shows the immediate (left) and delayed (right) effects of [K+]o changes. The short-term effects are similar in all cases: a step change in [K+]o causes an increased electrochemical driving force for IK1 at potentials near Vr, this causes an increase in IK1. However, this effect is overshadowed by the IK1 decrease due to the effects of [K+]o on IK1 rectification and maximum conductance. In response to lower [K+]o, INaK shows an instantaneous decrease, and together these effects cause a depolarization of Vr in all tested [K+]o levels. In the long-term, however, the effect is size-dependent: For small decreases (5.4 to 4 mM), the effect is dominated by the initial reduction in INaK, which leads to an increased Na+ influx. This, in turn, causes [Na+]i to rise progressively and INaK to increase until Na+ balance is restored. For larger decreases in [K+]o (5.4 to 3.2 mM) the effects of Vr depolarization on INa “availability” become dominant. The depolarized Vr reduces INa availability, so that both peak current and Na+ influx through INa is decreased, causing a reduction in [Na+]i. This in turn further reduces INaK until its Na+ efflux once again matches the (reduced) influx. In this case, balance is restored, but at a depolarized Vr level, which markedly reduces excitability. Finally, for the largest reduction in [K+]o studied (5.4 to 2.5 mM), this same pattern develops, but at a faster rate until the depolarization of Vr reaches a point where INa availability is too low to support an INa-driven depolarization.
Finally, we showed that these effects persist in strands of connected myocyte models (Figure 5), although we did not observe the reduced refractory period seen with prolonged APD by, e.g., Smeets et al. (1986).
Relationship to Previous Basic Science Studies
The significant physiological and pathophysiological effects in the mammalian heart of changes in plasma K+ are the basis of previous mechanism-oriented studies of both hypo- and hyperkalemia (Gettes et al., 1962; Kléber, 1983; for review see Weiss et al., 2017). We briefly discuss how our work fits in with earlier work, starting with the relationship between Vr and [K+]o.
A common line of reasoning holds that the resting potential in atrial myocytes is dictated by the reversal potential for potassium ions, EK, and is strongly regulated IK1. But it has also been well established that Vr increasingly deviates from EK as [K+]o is lowered from hyperkalemic to normo- and hypokalemic conditions. For example, Figure 4 in a study by Vaughan Williams (1959) shows an increasing deviation from EK in rabbit ventricle, starting at values as high as 6 or 7 mM. Similar examples in human atria can be seen in Figure 2 of Gelband et al. (1972) and Table 1 of McCullough et al. (1987). Ten Eick and Singer (1979) showed depolarization at 2 mM for healthy human atrial cells, an effect which was already present at 4 mM for diseased (partially depolarized) cells. Figures 2, 3 in McCullough et al. (1990) show a depolarization when lowering [K+]o from 7 to 4 mM in cells from diseased human ventricle, and perform experiments to show that these cells can exist at one of two stable states (with a Vr of either −80 or −45 mV). Recent work by Ma et al. (2011) provides further review of what they call “paradoxical depolarization,” and later work by the same group links this to Kir channels (Zuo et al., 2017).
In our Supplementary Figure 10 we show predictions from all seven models for the relationship between Vr and [K+]o. We demonstrate that all human atrial models that incorporate [K+]o-dependent IK1 rectification predict this effect – although at a variety of potassium levels, with most models predicting a depolarization starting at approximately 4 mM. Howewver, the model by Maleckar et al. stays closer to EK until approximately 1.5 mM. Interestingly, the models predict a relatively smooth Vr-[K+]o relationship after 1 s of hypokalemia, but after 15 min most models display a sharp cut-off instead, corresponding to the observation of two stable Vr levels in, e.g., Gelband et al. (1972) and McCullough et al. (1990). This variability in model predictions may be expected based on Supplementary Figure 3, which displays the relative contribution of ionic currents at Vr. In most models, this consists of IK1 and INaK (outward), balanced by the (inward) Na+/Ca2+-current INaCa and several “background currents.” Compared to, e.g., IKr and INa these currents have perhaps not received as much attention as they warrant from the modeling community, which is further corroborated by the fact that both the IK1 and INaK formulations in all models used in this study date back to the early or mid 1990s. This has been partly ascribed to the difficulty of measuring these relatively small “diastolic” currents (Bueno-Orovio et al., 2014). In this respect, it would be useful to perform new experiments measuring Vr under different hypokalemic conditions, e.g., in right-atrial appendage samples and perhaps in the presence of pharmaceutical blockers such as Ouabain. Such experiments could resolve uncertainties regarding the Vr-[K+]o relationship we highlighted in this study, but also have the potential to add to the relatively sparse data record on the balance between INaK and other diastolic currents.
Many previous studies have interpreted both short- and long-term effects of low [K+]o on cardiac muscle mainly in terms of its established inhibitory effects on the Na+/K+ pump (Aronsen et al., 2015; Weiss et al., 2017). This interpretation is logical since it is known that an essential regulatory site on the external surface of this integral membrane protein complex binds K+ with high affinity (Glitsch, 2001). Thus, a decrease in [K+]o to 3.2 mM from 4 or 5.4 mM can significantly reduce the activity of the Na+/K+ pump, and therefore result in a gradual increase in [Na+]i. This change in the electrochemical driving force for Na+ alters Na+/Ca2+ exchange activity and can produce an increase in [Ca2+]i (Pogwizd et al., 2001; Matchkov et al., 2007). Maintained increases in [Ca2+]i can contribute to a proarrhythmic substrate by a variety of ionic mechanisms. These are likely to include: (i) inhibitory effects of [Ca2+]i on Na+ channels (Chadda et al., 2017; Huang, 2017) and/or (ii) on the background K+ current, IK1 (Nagy et al., 2013) in the mammalian heart. Changes in [Ca2+]i can also alter myocyte-to-myocyte electrotonic communication by changing the conductance of the connexins in gap junctions (Matchkov et al., 2007; Varró et al., 2020). It is also important to note that changes in Vr in response to alterations in [K+]o can strongly modulate the Na+/Ca+ exchange current and thus alter the AP waveform (Baczkó et al., 2003) and myocyte contractility (Ng et al., 1987; White and Terrar, 1991).
In contrast, not all of the prominent low [K+]o effects that we have identified can be fully explained by Na+/K+ pump-based mechanisms. For example, after the change to low [K+]o, the AP lengthening occurs almost instantaneously. Therefore, this effect can not be due to a partial inhibition Na+/K+ pump activity. Based on our computational analysis, we agree, however, that at later times after small reductions in [K+]o the electrogenic current generated by the Na+/K+ pump does indeed have an important effect on excitability and perhaps on CV in atrial myocytes and tissue (see Figure 5). As mentioned, this effect is due to a small, but significant, increase in [Na+]i. The exceptional sensitivity of all known isoforms of the Na+/K+ pump to changes in [Na+]i within the physiological range (approximately 9–11 mM) can alter the associated electrogenic current, and thus change diastolic membrane potential, Vr (Glitsch, 2001). This effect appears to be quite prominent in human atrium (Sanchez et al., 2012).
We also acknowledge that in accordance with an essential part of the published working hypothesis for the effects of hypokalemia, there would be expected to be an increase in [Ca2+]i. However, additional and more detailed analysis of Ca2+ homeostasis in the human atrial myocyte is needed to reveal whether this is due to the IK1-induced broadening of the AP waveform and related effects on dynamics of the L-type Ca2+ current (Clark et al., 1996; Sah et al., 2003; Bouchard et al., 2004); altered release of Ca2+ from the sarcoplasmic reticulum (Koivumäki et al., 2011); a change in the ability of the Na+/Ca2+ exchanger to extrude Ca2+ (Pogwizd et al., 2001; Baczkó et al., 2003); or a combination of these effects.
Several studies have addressed the effects of [K+]o on CV, or on the maximum upstroke velocity V̇max which is known to correlate with CV. Early studies observed a “biphasic” relationship between [K+]o and CV, in which CV peaked at around 9–10 mM in mammalian ventricular cells (Kagiyama et al., 1982; Buchanan et al., 1985) and near 4 mM in sheep Purkinje cells (Dominguez and Fozzard, 1970). Similarly, V̇max was found to decrease with hyperkalemia, but remained relatively unchanged in hypokalemic conditions. All three studies reported a hyperpolarization of Vr with reduced [K+]o. Although effects of hyperkalemia could be explained quite readily by the depolarization of Vr and resulting inactivation of INa; the effects of lowering [K+]o were more of a puzzle. In this respect it is interesting to note the work of Kishida et al. (1979), who showed that the effects of [K+]o are partially voltage-independent, and the follow-up work by Whalley et al. (1994) who demonstrated that the effects of [K+]o on CV can be altered significantly by application of sub millimolar concentrations of BaCl2, a well-known and selective blocker of IK1 (Whalley et al., 1994).
Consistent with this literature, our strand simulations show a decrease in CV with hyperpolarization. Previous work by Nygren and Giles (2000) also showed a peak in CV at around 8 mM. Single-cell results on V̇max are shown in Supplementary Figure 21. In contrast to the experimental studies, these show a “biphasic” response similar to the CV experiments. Interestingly, this effect occurs regardless of whether the models hyperpolarize or depolarize at lowered [K+]o. The Courtemanche et al. and Ni et al. models both hyperpolarize at low [K+]o, leading to a very slight increase in INa availability (Supplementary Figure 23) but their lack of [K+]o-dependent IK1 rectification also causes a significant increase in IK1 (Supplementary Figures 13, 14) which as expected reduces both CV and V̇max. Conversely, the four models with [K+]o-dependent IK1 rectification that depolarize at low [K+]o all show a strong reduction in available INa, leading to a reduced CV and V̇max in spite of the reduced IK1. The most interesting model in this respect is the one by Maleckar et al., which has [K+]o-dependent IK1 rectification but still hyperpolarizes and so is most similar to the ventricular data sets described above. In this model, the slight hyperpolarization causes a small increase in INa availability only when stepping from 5.4 to 4 mM [K+]o (Supplementary Figure 24), while IK1 increases further at 3.2 and 2.5 mM (Supplementary Figure 16). This is likely to be part of the reason why, in this model, although CV keeps decreasing at more negative potentials (Supplementary Figure 20) the V̇max response flattens below 4 mM.
Relationship to Previous Clinical Findings
Our simulations illustrate how even moderate hypokalemia leads to a reduction in IK1, and a resulting increase in APD. This reduction of the atrial myocyte’s ability to stabilize at Vr implies an increased susceptibility to variability in the late stages of repolarization and therefore an increased susceptibility to variability in APD. In human atrium (Maleckar et al., 2009b) and in most other mammalian cardiac preparations, variability in APD is known to contribute to initiation and maintenance of atrial rhythm disturbances (Hondeghem et al., 2001; Kim et al., 2002; Fabritz et al., 2003). In fact, APD alternans often precedes initiation of atrial rhythm disturbances in patients (Narayan et al., 2011; Huang, 2017). This reduction in stability at Vr is further explored in Supplementary Section 8 where we show a reduction in [K+]o leads to an increase in membrane resistance at rest and in the late stages of repolarization, so that small fluctuations in current at this stage can cause large changes in the membrane potential (and thereby in the APD).
There is a general consensus that the [K+]o levels in a healthy adult human are closer to 4 to 4.5 mM than the 5.4 mM value typically used in wet lab and simulation experiments (Macdonald and Struthers, 2004; Mehta and Emmett, 2017) and that quite small changes (±1 to 2 mM) can confer a proarrhythmic phenotype in mammalian atria (Zaza, 2009; Krijthe et al., 2012). The associated hypo- or hyperkalemic phenotypes are observed quite frequently in clinical settings and must be monitored and managed effectively. A primary example is the need for optimization of the use of diuretics, and related transient or maintained alterations in [K+]o. In clinical settings, such as management of hypertension this often results in hypokalemia and related atrial rhythm disturbances (Franse et al., 2000; Zaza, 2009; Wang et al., 2013; Krogager et al., 2020). In addition, patients in the postoperative period following coronary bypass, or in the setting of heart failure, can also present with hypokalemia and associated electrophysiological instability of their atria and ventricles (Ahmed et al., 2007; Barrett Bowling et al., 2010; Severi et al., 2010; Collins et al., 2017). Perhaps the most common situation in which plasma K+ is actively manipulated in clinical settings and when it can decrease to hypokalemic levels is during renal dialysis (Genovesi et al., 2008; Zimmerman et al., 2012). It is recognized that some aspects of atrial rhythm disturbances are strongly associated with clinical hypokalemia (Vincenti et al., 2014).
Basic and clinical cardiac electrophysiologists have long attempted to detect and understand [K+]o-induced rhythm disturbances in mammalian heart. This has been done by monitoring refractoriness, CV and AP duration (Smeets et al., 1986; Nygren and Giles, 2000; Killeen et al., 2007b). Analysis often involves relating these factors in terms of the WL for conduction-that is, the product of CV and AP duration (Jacquemet et al., 2005; King et al., 2013a). Quite recently, based on detailed monophasic AP recordings from mouse hearts, this analysis paradigm (changes in WL for conduction and its restitution) has been updated and placed in a semi quantitative context (Sabir et al., 2007, 2008; Matthews et al., 2013). In brief, this comprehensive analysis provides a relationship between conduction WL restitution and AP duration alternans in the setting of hypokalemia (Killeen et al., 2007a,b). An important underlying physiological principle is that alterations in (i) the amount of inactivation (availability) of the Na+ current, (ii) its reactivation during the diastolic period, and (iii) a progressive inhibitory effect on INa of increases in [Ca2+]i (King et al., 2013b; Chadda et al., 2017; Huang, 2017) combine to contribute to a proarrhythmic substrate.
The dynamics of INa are particularly important in mammalian atria, given that its resting potential is relatively depolarized (e.g., −75 mV) compared to that in the ventricle. As a result, much of this essential rapidly activating inward current is not available or inactivated at diastolic membrane potentials (Sakakibara et al., 1992; Whalley et al., 1994). In fact, a recent study of [K+]o-induced atrial rhythm disturbances concludes that alterations in INa that result in its repetitive activation is a major proarrhythmic mechanism (Tazmini et al., 2020). In addition, Pandit and Jalife (2013), when reviewing mechanisms that underlie fibrillation, have suggested that functional linkages of IK1 and INa are likely to be essential for rotor formation and dynamics. Recent work by Sossalla et al. (2010) has shown a significant decrease in peak INa (but an increase in its late component) in isolated atrial myocytes from patients with AF, which could be further exacerbated by [K+]o-dependent INa decrease.
As mentioned previously, increases in [Ca2+]i have also been reported to be able to decrease K+ currents that are carried by IK1 (Nagy et al., 2013). In fact, much earlier studies of the relationship between [K+]o and INa had also drawn attention to K+-induced changes in membrane potential and related alterations in the availability and dynamics of the Na+ current in atrial tissue (Whalley et al., 1994; Spach et al., 2007).
Future Considerations
This study was performed to address a specific question and therefore has only a quite limited scope. To put our findings in a broader context, it is interesting to consider other published findings that also provide insights into how IK1 can change the human atrial electrophysiological substrate. For example, it is now known that mammalian atrial myocytes include a functional transverse tubule system (Richards et al., 2011; Tazmini et al., 2020). This finding, coupled with previous demonstrations that the K+ channels that are responsible for IK1 show prominent expression in the transverse tubule system (Clark et al., 2001), give rise to the possibility that in the failing heart (Lichter et al., 2014) a fraction of IK1 channel expression is lost due to disease-related decreases in T-tubule density. By analogy to what is known about skeletal muscle, this decrease in IK1 density could destabilize the resting potential and perhaps also alter excitability (Riisager et al., 2014). This change may be particularly relevant during relatively high frequency firing that is characteristic of chronic AF (Fraser et al., 2011, c.f. Tazmini et al., 2020).
Functionally significant, reversible changes in [K+]o have also been demonstrated in the mammalian cardiovascular system in the setting of maintained exercise (Paterson, 1997). One of the factors responsible for the association of exercise for endurance training with the incidence of AF may be explained by the striking sensitivity and rapid responsiveness of IK1 to small changes in [K+]o, and related effects on AP waveform and refractoriness that are revealed by our simulations (Figures 1, 5).
The antiarrhythmic drug flecainide has also been shown to enhance a background K+ current that is generated by a subset of the Kir2.X isoforms that produce IK1 (Caballero et al., 2010). This effect may be a significant factor in some of the proarrhythmic actions of flecainide both in ventricle and in supraventricular tissues. Somewhat similarly, nitric oxide can enhance IK1 expression in cardiac tissue (Gómez et al., 2009). Thus, changes in intrinsic tissue metabolism, including alterations in intracellular phospholipid levels such as PIP2 (Lopatin and Nichols, 1996) may be an important cofactor to consider in future studies concerning the [K+]o dependence of atrial physiology and pathophysiology (Gómez et al., 2009). In particular, details of the negative slope region of the IK1 I–V curve will need to be studied in detail (Baronas and Kurata, 2014).
Initiation and maintenance of atrial rhythm disturbances is known to be modulated by enhanced production of free radicals that are generated, e.g., during a sterile inflammatory response (Coppini et al., 2019; Varró et al., 2020). This finding may be related to the pattern of results revealed by our computations in two ways: (i) It is known that Na+/K+ pump activity is reduced significantly by this type of “redox challenge” (Figtree et al., 2012) and to free radical production and in particular enhanced generation of hydrogen peroxide can increase the slowly inactivating or late component of the Na+ current (Ward et al., 2006; Pezhouman et al., 2018). Both of these effects would be expected to destabilize the resting potential, and may be proarrhythmic through their ability to reduce the net outward current in diastole.
Finally, it is important to recognize that atrial tissue does not consist only or even mainly of myocytes. Rather, both atria in mature mammalian hearts include very significant populations of fibroblasts/myofibroblasts that connect, one to another, to form a syncytium. Fibroblasts and myofibroblasts also form connexin-mediated electrotonic interactions with immediately adjacent atrial myocytes. These functional interactions, especially when combined with the fact that these fibroblasts express the background K+ current, IK1 (Chilton et al., 2005) have important consequences for our main findings and conclusions. Thus, detailed consideration of electrophysiological effects due to altered plasma K+ should also consider changes in the atrial myocyte resting potential or AP waveform that are generated in part through electrotonic interactions with fibroblasts (Maleckar et al., 2009a). Consideration of fibroblast/myocyte electrotonic interactions also requires recognition of the possibility that progressive fibrosis as occurs during healthy aging; or localized changes in the strength of myocyte-to-myocyte coupling could contribute to functional alterations and important electrophysiological changes in the human atrium (Spach et al., 2007; Maleckar et al., 2009a; Huang, 2017; Varró et al., 2020).
Limitations and Future Directions
Atrial rhythm disturbances are the most common cardiac arrhythmias in the aged population (c.f. Tazmini et al., 2020). Accordingly, a number of different approaches have been developed for attempting to detect, manage and possibly even predict episodes of AF. Mathematical simulations of AF are quite advanced, and have already given rise to interesting insights and controversies. Recent reviews summarize available mathematical modeling approaches and also provide opinions concerning “what’s next” (Wilhelms et al., 2013; Vagos et al., 2018; Grandi et al., 2019). Very informative studies of underlying mechanisms and development and refinement of related mathematical models for AF that are directly relevant to the present study have been published (Severi et al., 2010; Krueger et al., 2011; Passini et al., 2014). With further refinement and validation, computational platforms for simulating atrial rhythm disturbances are beginning to make useful contributions Examples include: drug discovery and repurposing (Courtemanche et al., 1999; Koivumäki et al., 2014; Ni et al., 2017, 2020); detailed study of the genetic and molecular causes of arrhythmias (Pandit and Jalife, 2013; Colman et al., 2017; Zhang et al., 2020); and safety pharmacology initiatives (Mirams et al., 2012; Yang et al., 2020). Our findings provide important insights and criteria for selection of robust methods and rational approaches for ongoing or future computational studies of atrial rhythm disturbances.
However, this study also highlights several issues with numerical modeling of atrial cell and tissue electrophysiology which will need to be considered in more detail. In the short-term (1 s) simulations we identified strong interactions between opposing effects: in IK1, increases in electrochemical driving force are balanced by changes to its conductance and the voltage-dependence of its rectification. Such a situation, where large opposing effects partially cancel each other to create a smaller effect, requires a highly accurate parameterization. As a result, even small errors in modeling and parameterization of IK1 can result in large effects. At diastole, Vr is determined by the precise balance between IK1, INaK, INaCa, and several “background currents.” Formulations for these currents are typically inherited from older models or added without too much justification, making this an area of AP modeling that requires additional attention.
The longer term effects of hypokalemia were dominated by the interplay of IK1, INaK, INa, Vr, and [Na+]i. Importantly, even though the changes in [K+]o were small, three qualitatively different results were seen. This adds to the functional significance of this study, because it shows that such results are possible as even a result of very small changes in [K+]o. However, predicting exactly which effects will occur at which [K+]o level requires a model that has precisely the right balance between ionic current conductances and that has accurate sensitivities to internal and external electrolytes. Earlier studies have shown that human atrial AP models fall short in this respect (Passini et al., 2014). We have attempted to remedy model-specific effects by repeating the main results in multiple models. However, given the importance of IK1 and INaK in this respect it is worth noting that the seven AP models all inherit these formulations from the same sources, so that one could argue we only really compare 2 or 3 models. Finally, it may not be possible to get the balance between the opposing diastolic currents exactly right without measuring several quantities simultaneously in the same cell – which is technically highly challenging (Whittaker et al., 2020). The complexity of the low [K+]o response offers a challenging but also a promising perspective as a relatively simple but highly revealing experimental maneuver to be used in experimental characterization and model building.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://github.com/CardiacModelling/AtrialLowK.
Author Contributions
MC was responsible for implementation, evaluation, and updates in all of the models of the human atrial action potential that were utilized in this study and carried out all of the simulations and constructed figures. AR and SN provided guidance concerning current clinical investigations of atrial fibrillation, as well as manuscript editing. MC, GM, and WG were involved in the design of the project and wrote the manuscript. All authors have reviewed and approved the final version of this manuscript.
Funding
This work was supported by the National Institutes of Health (grant no. NIH RO1 HL149134 “Machine Learning in Atrial Fibrillation,” and grant no. NIH RO1 HL83359 “Dynamics of Human Atrial Fibrillation”); the Wellcome Trust (grant no. 212203/Z/18/Z); and the Biotechnology and Biological Sciences Research Council (grant no. BB/P010008/1). SN acknowledges support from the National Institutes of Health. GM and MC acknowledge support from the Wellcome Trust via a Wellcome Trust Senior Research Fellowship to GM and MC acknowledges support from a BBSRC project grant. This research was funded in whole, or in part, by the Wellcome Trust [212203/Z/18/Z]. For the purpose of open access, the author has applied a CC-BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr Dominic Whittaker for insightful discussions, and Dr Eleonora Grandi, Dr Haibo Ni, Dr Jussi Koivumäki, Dr Jordi Heijman, and Dr Henry Sutanto for providing their model codes.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2021.651162/full#supplementary-material
References
Ahmed, A., Zannad, F., Love, T. E., Tallaj, J., Gheorghiade, M., Ekundayo, O. J., et al. (2007). A propensity matched study of the association of low serum potassium levels and mortality in chronic heart failure. Eur. Heart J. 28, 1334–1343. doi: 10.1093/eurheartj/ehm091
Alderman, M. H., Piller, L. B., Ford, C. E., Probstfield, J. L., Oparil, S., Cushman, W. C., et al. (2012). Clinical significance of incident hypokalemia and hyperkalemia in treated hypertensive patients in the antihypertensive and lipid-lowering treatment to prevent heart attack trial. Hypertension 59, 926–933. doi: 10.1161/HYPERTENSIONAHA.111.180554
Anumonwo, J. M., and Lopatin, A. N. (2010). Cardiac strong inward rectifier potassium channels. J Mol. Cell.Cardiol. 48, 45–54. doi: 10.1016/j.yjmcc.2009.08.013
Aronsen, J. M., Skogestad, J., Lewalle, A., Louch, W. E., Hougen, K., Stokke, M. K., et al. (2015). Hypokalemia induces Ca2+ overload and Ca2+ waves in ventricular myocytes by reducing NKA α2 activity. J. Physiol. 593, 1509–1521. doi: 10.1113/jphysiol.2014.279893
Baczkó, I., Giles, W. R., and Light, P. E. (2003). Resting membrane potential regulates Na+-Ca2+ exchange-mediated Ca2+ overload during hypoxia-reoxygenation in rat ventricular myocytes. J. Physiol. 550, 889–898. doi: 10.1113/jphysiol.2003.043372
Bailly, P., Mouchoniere, M., Benitah, J. P., Camilleri, L., Vassort, G., and Lorente, P. (1998). Extracellular K+ dependence of inward rectification kinetics in human left ventricular cardiomyocytes. Circulation 98, 2753–2759. doi: 10.1161/01.cir.98.24.2753
Baronas, V. A., and Kurata, H. T. (2014). Inward rectifiers and their regulation by endogenous polyamines. Front. Physiol. 5:325. doi: 10.3389/fphys.2014.00325
Barrett Bowling, C., Pitt, B., Ahmed, M. I., Aban, I. B., Sanders, P. W., Mujib, M., et al. (2010). Hypokalemia and outcomes in patients with chronic heart failure and chronic kidney disease: findings from propensity-matched studies. Circ. Heart Fail. 3, 253–260. doi: 10.1161/CIRCHEARTFAILURE.109.899526
Blanco, G., and Mercer, R. W. (1998). Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am. J. Physiol. Renal Physiol. 275, F633–F650. doi: 10.1152/ajprenal.1998.275.5.F633
Bouchard, R., Clark, R. B., Juhasz, A. E., and Giles, W. R. (2004). Changes in extracellular K+ concentration modulate contractility of rat and rabbit cardiac myocytes via the inward rectifier K+ current IK1. J. Physiol. 556, 773–790. doi: 10.1113/jphysiol.2003.058248
Buchanan, J. W. Jr., Saito, T., and Gettes, L. S. (1985). The effects of antiarrhythmic drugs, stimulation frequency, and potassium-induced resting membrane potential changes on conduction velocity and dV/dtmax in guinea pig myocardium. Circ. Res. 56, 696–703. doi: 10.1161/01.RES.56.5.696
Bueno-Orovio, A., Sánchez, C., Pueyo, E., and Rodriguez, B. (2014). Na/K pump regulation of cardiac repolarization: insights from a systems biology approach. Pflügers Arch. Eur. J. Physiol. 466, 183–193. doi: 10.1007/s00424-013-1293-1
Buiten, M. S., de Bie, M. K., Rotmans, J. I., et al. (2014). The dialysis procedure as a trigger for atrial fibrillation: new insights in the development of atrial fibrillation in dialysis patients. Heart 100, 685–690. doi: 10.1136/heartjnl-2013-305417
Burns, W. R., Cohen, K. D., and Jackson, W. F. (2004). K+-induced dilation of hamster cremasteric arterioles involves both the Na+/K+-ATPase and inward rectifier K+ channels. Microcirculation 11, 279–293. doi: 10.1080/10739680490425985
Caballero, R., Dolz-Gaitón, P., Gómez, R., Amorós, I., Barana, A., González de la Fuente, M., et al. (2010). Flecainide increases Kir2.1 currents by interacting with cysteine 311, decreasing the polyamine-induced rectification. Proc. Natl. Acad. Sci. U.S.A. 107, 15631–15636. doi: 10.1073/pnas.1004021107
Chadda, K. R., Jeevaratnam, K., Lei, M., and Huang, C. L.-H. (2017). Sodium channel biophysics, late sodium current and genetic arrhythmic syndromes. Pflugers Arch. Eur. J. Physiol. 469, 629–641. doi: 10.1007/s00424-017-1959-1
Chilton, L., Ohya, S., Freed, D., George, E., Drobic, V., Shibukawa, Y., et al. (2005). K+ currents regulate the resting membrane potential, proliferation and contractile responses in ventricular fibroblasts and myofibroblasts. Am. J. Physiol. Heart Circ. Physiol. 288, H2931–H2939. doi: 10.1152/ajpheart.01220.2004
Clark, R., Bouchard, R., and Giles, W. R. (1996). Action potential duration modulates calcium influx, Na+-Ca2+ exchange and intracellular calcium release in rat ventricular myocytes. Ann. N. Y. Acad. Sci. 779, 417–429. doi: 10.1111/j.1749-6632.1996.tb44817.x
Clark, R. B., Tremblay, A., Melnyk, P., Allen, B. G., Giles, W. R., and Fiset, C. (2001). T-tubule localization of the inward-rectifier K+ channel in mouse ventricular myocytes: a role in K+ accumulation. J. Physiol. 537, 979–992. doi: 10.1111/j.1469-7793.2001.00979.x
Clerx, M., Collins, P., de Lange, E., and Volders, P. G. (2016). Myokit: a simple interface to cardiac cellular electrophysiology. Prog. Biophys. Mol. Biol. 120, 100–114. doi: 10.1016/j.pbiomolbio.2015.12.008
Collins, A. J., Pitt, B., Reaven, N., Funk, S., McGaughey, K., Wilson, D., et al. (2017). Association of serum potassium with all-cause mortality in patients with and without heart failure, chronic kidney disease and/or diabetes. Am. J. Nephrol. 46, 213–221. doi: 10.1159/000479802
Colman, M. A., Aslanidi, O. V., Kharche, S., Boyett, M. R., Garratt, C., Hancox, J. C., et al. (2013). Pro-arrhythmogenic effects of atrial fibrillation-induced electrical remodelling: insights from the three-dimensional virtual human atria. J. Physiol. 591, 4249–4272. doi: 10.1113/jphysiol.2013.254987
Colman, M. A., Ni, H., Liang, B., Schmitt, N., and Zhang, H. (2017). In silico assessment of genetic variation in kcna5 reveals multiple mechanisms of human atrial arrhythmogenesis. PLoS Comp. Biol. 13:e1005587. doi: 10.1371/journal.pcbi.1005587
Cooper, J., Mirams, G. R., and Niederer, S. A. (2011). High-throughput functional curation of cellular electrophysiology models. Prog. Biophys. Mol. Biol. 107, 11–20. doi: 10.1016/j.pbiomolbio.2011.06.003
Coppini, R., Santini, L., Palandri, C., Sartiani, L., Cerbai, E., and Raimondi, L. (2019). Pharmacological inhibition of serine proteases to reduce cardiac inflammation and fibrosis in atrial fibrillation. Front. Pharmacol. 10:1420. doi: 10.3389/fphar.2019.01420
Courtemanche, M., Ramirez, R. J., and Nattel, S. (1998). Ionic mechanisms underlying human atrial action potential properties: insights from a mathematical model. Am. J. Physiol. Heart Circ. Physiol. 275, H301–H321. doi: 10.1152/ajpheart.1998.275.1.H301
Courtemanche, M., Ramirez, R. J., and Nattel, S. (1999). Ionic targets for drug therapy and atrial fibrillation-induced electrical remodeling: insights from a mathematical model. Cardiovasc. Res. 42, 477–489. doi: 10.1016/s0008-6363(99)00034-6
Deo, M., Ruan, Y., Pandit, S. V., Shah, K., Berenfeld, O., Blaufox, A., et al. (2013). KCNJ2 mutation in short QT syndrome 3 results in atrial fibrillation and ventricular proarrhythmia. Proc. Natl. Acad. Sci. U.S.A. 110, 4291–4296. doi: 10.1073/pnas.1218154110
Dhamoon, A. S., and Jalife, J. (2005). The inward rectifier current IK1 controls cardiac excitability and is involved in arrhythmogenesis. Heart Rhythm. 2, 316–324. doi: 10.1016/j.hrthm.2004.11.012
Dominguez, G., and Fozzard, H. A. (1970). Influence of extracellular K+ concentration on cable properties and excitability of sheep cardiac Purkinje fibers. Circ. Res. 26, 565–574. doi: 10.1161/01.RES.26.5.565
El-Sherif, N., and Turitto, G. (2011). Electrolyte disorders and arrhythmogenesis. Cardiol. J. 18, 233–245.
Fabritz, I., Kirchhof, P., Franz, M. R., Eckardt, L., Mönnig, G., Milberg, P., et al. (2003). Prolonged action potential durations, increased dispersion of repolarization, and polymorphic ventricular tachycardia in a mouse model of proarrhythmia. Basic Res. Cardiol. 98, 25–32. doi: 10.1007/s00395-003-0386-y
Figtree, G. A., Keyvan Karimi, G., and Rasmussen, H. H. (2012). Oxidative regulation of the Na+-K+ pump in the cardiovascular system. Free Radic. Biol. Med. 53, 2263–2268. doi: 10.1016/j.freeradbiomed.2012.10.539
Franse, L. V., Pahor, M., Di Bari, M., Somes, G. W., Cushman, W. C., and Applegate, W. B. (2000). Hypokalemia associated with diuretic use and cardiovascular events in the systolic hypertension in the elderly program. Hypertension 35, 1025–1030. doi: 10.1161/01.hyp.35.5.1025
Fraser, J. A., Huang, C. L. H., and Pedersen, T. H. (2011). Relationships between resting conductances, excitability, and t-system ionic homeostasis in skeletal muscle. J. Gen. Physiol. 138, 95–116. doi: 10.1085/jgp.201110617
Gelband, H., Bush, H. L., Rosen, M. R., Myerburg, R. J., and Hoffman, B. F. (1972). Electrophysiologic properties of isolated preparations of human atrial myocardium. Circ. Res. 30, 293–300. doi: 10.1161/01.res.30.3.293
Genovesi, S., Dossi, C., Vigano, M. R., Galbiati, E., Prolo, F., Stella, A., et al. (2008). Electrolyte concentration during haemodialysis and QT interval prolongation in uraemic patients. Europace 10, 771–777. doi: 10.1093/europace/eun028
Gettes, L. S., Surawicz, B., and Shiue, J. C. (1962). Effect of high K, low K, and quinidine on QRS duration and ventricular action potential. Am. J. Physiol. 203, 1135–1140. doi: 10.1152/ajplegacy.1962.203.6.1135
Glitsch, H. G. (2001). Electrophysiology of the sodium-potassium ATPase in cardiac cells. Physiol. Rev. 81, 1791–1826. doi: 10.1152/physrev.2001.81.4.1791
Gómez, R., Caballero, R., Barana, A., Amorós, I., Calvo, E., López, J. A., et al. (2009). Nitric oxide increases cardiac IK1 by nitrosylation of cysteine 76 of Kir2.1 channels. Circ. Res. 105, 383–392. doi: 10.1161/CIRCRESAHA.109.197558
Goyal, A., Spertus, J. A., Gosch, K., Venkitachalam, L., Jones, P. G., Van den Berghe, G., et al. (2012). Serum potassium levels and mortality in acute myocardial infarction. JAMA 307, 157–164. doi: 10.1001/jama.2011.1967
Grandi, E., Dobrev, D., and Heijman, J. (2019). Computational modeling: what does it tell us about atrial fibrillation therapy? Int. J. Cardiol. 287, 155–161. doi: 10.1016/j.ijcard.2019.01.077
Grandi, E., Pandit, S. V., Voigt, N., Workman, A. J., Dobrev, D., Jalife, J., et al. (2011). Human atrial action potential and Ca2+ model: sinus rhythm and chronic atrial fibrillation. Circ. Res. 109, 1055–1066. doi: 10.1161/CIRCRESAHA.111.253955
Grandi, E., Pasqualini, F. S., and Bers, D. M. (2010). A novel computational model of the human ventricular action potential and Ca transient. J. Mol. Cell Cardiol. 48, 112–121. doi: 10.1016/j.yjmcc.2009.09.019
Hedley, W. J., Nelson, M. R., Bellivant, D. P., and Nielsen, P. F. (2001). A short introduction to CellML. Phil. Trans. Roy. Soc. Lond. Series A Math. Phys. Eng. Sci. 359, 1073–1089. doi: 10.1098/rsta.2001.0817
Heijman, J., Erfanian Abdoust, P., Voigt, N., Nattel, S., and Dobrev, D. (2016). Computational models of atrial cellular electrophysiology and calcium handling, and their role in atrial fibrillation. J. Physiol. 594, 537–553. doi: 10.1113/JP271404
Hibino, H., Inanobe, A., Furutani, K., et al. (2010). Inwardly rectifying potassium channels: their structure, function and physiological roles. Physiol. Rev. 90, 291–366. doi: 10.1152/physrev.00021.2009
Hindmarsh, A. C., Brown, P. N., Grant, K. E., Lee, S. L., Serban, R., Shumaker, D. E., et al. (2005). SUNDIALS: suite of nonlinear and differential/algebraic equation solvers. ACM Trans. Math. Softw. 31, 363–396. doi: 10.1145/1089014.1089020
Hondeghem, L. M., Carlsson, L., and Duker, G. (2001). Instability and triangulation of the action potential predict serious proarrhythmia, but action potential duration prolongation is antiarrhythmic. Circulation 103, 2004–2013. doi: 10.1161/01.cir.103.15.2004
Huang, C. L. (2017). Murine electrophysiological models of cardiac arrhythmogenesis. Physiol. Rev. 97, 283–409. doi: 10.1152/physrev.00007.2016
Jacquemet, V. (2007). Steady-state solutions in mathematical models of atrial cell electrophysiology and their stability. Math. Biosci. 208, 241–269. doi: 10.1016/j.mbs.2006.10.007
Jacquemet, V., Virag, N., and Kappenberger, L. (2005). Wavelength and vulnerability to atrial fibrillation: insights from a computer model of human atria. Europace 7, S83–S92. doi: 10.1016/j.eupc.2005.03.017
Kagiyama, Y., Hill, J. L., and Gettes, L. S. (1982). Interaction of acidosis and increased extracellular potassium on action potential characteristics and conduction in guinea pig ventricular muscle. Circ. Res. 51, 614–623. doi: 10.1161/01.RES.51.5.614
Killeen, M. J., Gurung, I. S., Thomas, G., et al. (2007a). Separation of early afterdepolarizations from arrhythmogenic substrate in the isolated perfused hypokalaemic murine heart through modifiers of calcium homeostasis. Acta Physiol. 191, 43–58. doi: 10.1111/j.1748-1716.2007.01715.x
Killeen, M. J., Thomas, G., Gurung, I. S., et al. (2007b). Arrhythmogenic mechanisms in the isolated perfused hypokalaemic murine heart. Acta Physiol. 189, 33–46. doi: 10.1111/j.1748-1716.2006.01643.x
Kim, B.-S., Kim, Y.-H., Hwang, G.-S., Pak, H.-N., Lee, S. C., Shim, W. J., et al. (2002). Action potential duration restitution kinetics in human atrial fibrillation. J. Am. Col. Cardiol. 39, 1329–1326. doi: 10.1016/s0735-1097(02)01760-6
King, J., Huang, C. L. H., and Fraser, J. A. (2013a). Determinants of myocardial conduction velocity: implications for arrhythmogenesis. Front. Physiol. 4:154. doi: 10.3389/fphys.2013.00154
King, J., Zhang, Y., Lei, M., et al. (2013b). Atrial arrhythmia, triggering events and conduction abnormalities in isolated murine RyR2-P2328S hearts. Acta Physiol. 207, 308–323. doi: 10.1111/apha.12006
Kishida, H., Surawicz, B. O. R. Y. S., and Fu, L. T. (1979). Effects of K+ and K+-induced polarization on (dV/dt) max, threshold potential, and membrane input resistance in guinea pig and cat ventricular myocardium. Circ. Res. 44, 800–814. doi: 10.1161/01.RES.44.6.800
Kléber, A. G. (1983). Resting membrane potential, extracellular potassium activity, and intracellular sodium activity during acute global ischemia in isolated perfusion guinea pig hearts. Circ. Res. 52, 442–450. doi: 10.1161/01.res.52.4.442
Koivumäki, J. T., Korhonen, T., and Tavi, P. (2011). Impact of sarcoplasmic reticulum calcium release on calcium dynamics and action potential morphology in human atrial myocytes: a computational study. PLoS Comput. Biol. 7:e1001067. doi: 10.1371/journal.pcbi.1001067
Koivumäki, J. T., Seemann, G., Maleckar, M. M., and Tavi, P. (2014). In silico screening of the key cellular remodeling targets in chronic atrial fibrillation. PLoS Comput. Biol. 10:e1003620. doi: 10.1371/journal.pcbi.1003620
Krijthe, B. P., Heeringa, J., Kors, J. A., Hofman, A., Franco, O. H., Witteman, J. C. M., et al. (2012). Serum potassium levels and the risk of atrial fibrillation. The Rotterdam study. Int. J. Cardiol. 168, 5411–5415. doi: 10.1016/j.ijcard.2013.08.048
Krogager, M. L., Mortensen, R. N., Lund, P. E., Boggild, H., Hansen, S. M., Kragholm, K., et al. (2020). Risk of developing hypokalemia in patients with hypertension treated with combination antihypertensive therapy. Hypertension 75, 966–972. doi: 10.1161/HYPERTENSIONAHA.119.14223
Krogh-Madsen, T., Abbott, G. W., and Christini, D. J. (2012). Effects of electrical and structural remodeling on atrial fibrillation maintenance: a simulation study. PLoS Comput. Biol. 8:e10002390. doi: 10.1371/journal.pcbi.1002390
Krueger, M. W., Severi, S., Rhode, K., Genovesi, S., Weber, F. M., Vincenti, A., et al. (2011). Alterations of atrial electrophysiology related to hemodialysis session: insights from a multiscale computer model. J. Electrocardiol. 44, 176–183. doi: 10.1016/j.jelectrocard.2010.11.016
Lichter, J. G., Carruth, E., Mitchell, C., Barth, A. S., Aiba, T., Kass, D. A., et al. (2014). Remodeling of the sarcomeric cytoskeleton in cardiac ventricular myocytes during heart failure and after cardiac resynchronization therapy. J. Mol. Cell Cardiol. 72, 186–195. doi: 10.1016/j.yjmcc.2014.03.012
Lindblad, D. S., Murphey, C. R., Clark, J. W., and Giles, W. R. (1996). A model of the action potential and underlying membrane currents in a rabbit atrial cell. Am. J. Physiol. Heart Circ. Physiol. 271, H1666–H1696. doi: 10.1152/ajpheart.1996.271.4.H1666
Lopatin, A. N., and Nichols, C. G. (1996). [K+] dependence of polyamine-induced rectification in inward rectifier potassium channels (IRK1, Kir2.1). J. Gen. Physiol. 108, 105–113. doi: 10.1085/jgp.108.2.105
Lu, Z. (2004). Mechanism of rectification in inward-rectifier K+ channels. Annu. Rev. Physiol. 66, 103–129. doi: 10.1146/annurev.physiol.66.032102.150822
Luo, C. H., and Rudy, Y. (1994). A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ. Res. 74, 1071–1096. doi: 10.1161/01.RES.74.6.1071
Ma, L., Zhang, X., and Chen, H. (2011). TWIK-1 two-pore domain potassium channels change ion selectivity and conduct inward leak sodium currents in hypokalemia. Sci. Signal. 4:ra37. doi: 10.1126/scisignal.2001726
Macdonald, J. E., and Struthers, A. D. (2004). What is the optimal serum potassium level in cardiovascular patients? J. Am. Coll. Cardiol. 43, 155–161. doi: 10.1016/j.jacc.2003.06.021
Maleckar, M. M., Greenstein, J. L., Giles, W. R., and Trayanova, N. A. (2008). Mathematical simulations of ligand-gated and cell-type specific effects on the action potential of human atrium. Prog. Biophys. Mol. Biol. 98, 161–170. doi: 10.1016/j.pbiomolbio.2009.01.010
Maleckar, M. M., Greenstein, J. L., Giles, W. R., and Trayanova, N. A. (2009a). Electrotonic coupling between human atrial myocytes and fibroblasts alters myocyte excitability and repolarization. Biophys. J. 97, 2179–2190. doi: 10.1016/j.bpj.2009.07.054
Maleckar, M. M., Greenstein, J. L., Giles, W. R., and Trayanova, N. A. (2009b). K+ current changes account for the rate dependence of the action potential in the human atrial myocyte. Am. J. Physiol. Heart Circ. Physiol. 297, H1398–H1410. doi: 10.1152/ajpheart.00411.2009
Martins, R. P., Kaur, K., Hwang, E., Ramirez, R. J., Willis, B. C., Filgueiras-Rama, D., et al. (2014). Dominant frequency increase rate predicts transition from paroxysmal to long-term persistent atrial fibrillation. Circulation 129, 1472–1482. doi: 10.1161/CIRCULATIONAHA.113.004742
Mary-Rabine, L., Albert, A., Pham, T. D., Hordof, A., Feoglio, J. J., Malm, J. R., et al. (1983). The relationship of human atrial cellular electrophysiology to clinical function and ultrastructure. Circ. Res. 52, 188–199. doi: 10.1161/01.res.52.2.188
Matchkov, V. V., Gustafsson, H., Rahman, A., Briggs Boedtkjer, D. M., Gorintin, S., Hansen, A. K., et al. (2007). Interaction between Na+/K+-pump and Na+/Ca2+-exchanger modulates intercellular communication. Circ. Res. 100, 1026–1035. doi: 10.1161/01.RES.0000262659.09293.56
Matthews, G. D. K., Guzadhur, L., Sabir, I., Grace, A., and Huang, C. L. H. (2013). Action potential wavelength restitution predicts alternans and arrhythmia in murine Scn5a+\- hearts. J. Physiol. 591, 4167–4188. doi: 10.1113/jphysiol.2013.254938
McCullough, J. R., Baumgarten, C. M., and Singer, D. H. (1987). Intra-and extracellular potassium activities and the potassium equilibrium potential in partially depolarized human atrial cells. J. Mol. Cell. Cardiol. 19, 477–486. doi: 10.1016/S0022-2828(87)80399-1
McCullough, J. R., Chua, W. T., Rasmussen, H. H., Ten Eick, R. E., and Singer, D. H. (1990). Two stable levels of diastolic potential at physiological K+ concentrations in human ventricular myocardial cells. Circ. Res. 66, 191–201. doi: 10.1161/01.RES.66.1.191
Mehta, A., and Emmett, M. (2017). The importance of a ‘just right’ serum potassium level. Am. J. Nephrol. 46, 210–212. doi: 10.1159/000479803
Melnyk, P., Zhang, L., Shrier, A., and Nattell, S. (2002). Differential distribution of Kir2.1 and Kir2.3 subunits in canine atrium and ventricle. Am. J. Physiol. Heart Circ. Physiol. 283, H1123–H1133. doi: 10.1152/ajpheart.00934.2001
Mirams, G. R., Davies, M. R., Cui, Y., Kohl, P., and Noble, D. (2012). Application of cardiac electrophysiology simulations to pro-arrhythmic safety testing. Br. J. Pharmacol. 167, 932–945. doi: 10.1111/j.1476-5381.2012.02020.x
Nagy, N., Acsai, K., Kormos, A., et al. (2013). [Ca2+]i-induced augmentation of the inward rectifier potassium current (IK1) in canine and human ventricular myocardium. Pflugers Archiv. Eur. J. Physiol. 465, 1621–1635. doi: 10.1007/s00424-013-1309-x
Nakao, M., and Gadsby, D. C. (1989). [Na] and [K] dependence of the Na/K pump current-voltage relationship in guinea pig ventricular myocytes. J. Gen. Physiol. 94, 539–565. doi: 10.1085/jgp.94.3.539
Narayan, S. M., Franz, M. R., Clopton, P., Pruvot, E. J., and Krummen, D. E. (2011). Repolarization alternans reveals vulnerability to human atrial fibrillation. Circulation 123, 2922–2930. doi: 10.1161/CIRCULATIONAHA.110.977827
Ng, Y. C., Hume, J. R., and Akera, A. T. (1987). Paradoxical positive inotropic effect of K+ in the rat heart. Am. J. Physiol. Heart. Circ. Physiol. 21, H1005–H1015. doi: 10.1152/ajpheart.1987.252.5.H1005
Ni, H., Fogli Iseppe, A., Giles, W. R., Narayan, S. M., Zhang, H., Edwards, A. G., et al. (2020). Populations of in silico myocytes and tissues reveal synergy of multiatrial-predominant K+-current block in atrial fibrillation. Br. J. Pharmacol. 177, 4497–4515. doi: 10.1111/bph.15198
Ni, H., Whittaker, D. G., Wang, W., Giles, W. R., Narayan, S. M., and Zhang, H. (2017). Synergistic anti-arrhythmic effects in human atria with combined use of sodium blockers and acacetin. Front. Physiol. 8:946. doi: 10.3389/fphys.2017.00946
Nygren, A., Fiset, C., Firek, L., Clark, J. W., Lindblad, D. S., Clark, R. B., et al. (1998). Mathematical model of an adult human atrial cell: the role of K+ currents in repolarization. Circ. Res. 82, 63–81. doi: 10.1161/01.res.82.1.63
Nygren, A., and Giles, W. R. (2000). Mathematical simulation of slowing of cardiac conduction velocity by elevated extracellular [K+] in a human atrial strand. Ann. Biomed. Eng. 28, 951–957. doi: 10.1114/1.1308489
Osadchii, O. E. (2010). Mechanisms of hypokalemia-induced ventricular arrhythmogenicity. Fund. Clin.Pharmacol. 24, 547–559. doi: 10.1111/j.1472-8206.2010.00835.x
Paice, B. J., Paterson, K. R., Onyanga-Omara, F., Donnelly, T., Gray, J. M., and Lawson, D. H. (1986). Record linkage study of hypokalaemia in hospitalized patients. Postgrad. Med. J. 62, 187–191. doi: 10.1136/pgmj.62.725.187
Pandit, S. V., and Jalife, J. (2013). Rotors and the dynamics of cardiac fibrillation. Circ. Res. 112, 849–862. doi: 10.1161/CIRCRESAHA.111.300158
Passini, E., Genovesi, S., and Severi, S. (2014). Human atrial cell models to analyse haemodialysis-related effects on cardiac electrophysiology: work in progress. Comp. Math. Meth. Med. 2014:291598. doi: 10.1155/2014/291598
Paterson, D. J. (1997). Potassium and breathing in exercise. Sports Med. 23, 149–163. doi: 10.2165/00007256-199723030-00002
Pezhouman, A., Cao, H., Fishbein, M. C., Belardinelli, L., Weiss, J. N., and Karagueuzian, H. S. (2018). Atrial fibrillation initiated by early afterdepolarization-mediated triggered activity during acute oxidative stress: efficacy of late sodium current blockade. J. Heart Health 4:0.16966/2379-769X.146. doi: 10.16966/2379-769X.146
Pogwizd, S. M., Schlotthauer, K., Li, L., Yuan, W., and Bers, D. M. (2001). Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current and residual beta-adrenergic responsiveness. Circ. Res. 88, 1159–1167. doi: 10.1161/hh1101.091193
Rasmussen, H. H., Singer, D. H., and Ten Eick, R. E. (1986). Characterization of a sodium pump-induced hyperpolarization in isolated human atrium. Am. J. Physiol. Heart Circ. Physiol. 20, H331–H339. doi: 10.1152/ajpheart.1986.251.2.H331
Rasmussen, H. H., Ten Eick, R. E., McCullough, J. R., and Singer, D. H. (1984). Evidence for electrogenic Na+ pumping in human atrial myocardium. J. Mol. Cell. Cardiol. 16, 771–773. doi: 10.1016/s0022-2828(84)80660-4
Richards, M. A., Clarke, J. D., Saravanan, P., Voigt, N., Dobrev, D., Eisner, D. A., et al. (2011). Transverse tubules are a common feature in large mammalian atrial myocytes including human. Am. J. Physiol. Heart Circ. Physiol. 301, H1996–H2005. doi: 10.1152/ajpheart.00284.2011
Riisager, A., Duehmke, R., Nielsen, O. B., Huang, C. L., and Pedersen, T. H. (2014). Determination of cable parameters in skeletal muscle fibres during repetitive firing of action potentials. J. Physiol. 592, 4417–4429. doi: 10.1113/jphysiol.2014.280529
Sabir, I. N., Fraser, J. A., Cass, T. R., Grace, A. A., and Huang, C. L. H. (2007). A quantitative analysis of the effect of cycle length on arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts. Pflugers Arch. 454, 925–936. doi: 10.1007/s00424-007-0255-x
Sabir, I. N., Li, L. M., Grace, A. A., and Huang, C. L. H. (2008). Restitution analysis of alternans and its relationship to arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts. Pflugers Arch. 455, 653–666. doi: 10.1007/s00424-007-0327-y
Sah, R., Ramirez, R. J., Oudit, G., Gidrewicz, D., Trivieri, M. G., Zobel, C., et al. (2003). Regulation of cardiac excitation-contraction coupling by action potential repolarization: role of the transient outward potassium current Ito. J. Physiol. 546, 5–18. doi: 10.1113/jphysiol.2002.026468
Sakakibara, Y., Wasserstrom, A., Furukawa, T., Jia, H., Arentzen, C. E., Hartz, R. S., et al. (1992). Characterization of the sodium current in single human atrial myocytes. Circ. Res. 71, 535–546. doi: 10.1161/01.res.71.3.535
Sakmann, B., and Trube, G. (1984). Conductance properties of single inwardly rectifying potassium channels in ventricular cells from guinea-pig heart. J. Physiol. 347, 641–657. doi: 10.1113/jphysiol.1984.sp015088
Sanchez, C., Corrias, A., Bueno-Orovio, A., et al. (2012). The Na+K+ pump is an important modulator of refractoriness and rotor dynamics in human atrial tissue. Am. J. Physiol. Heart Circ. Physiol. 302, H1146–H1159. doi: 10.1152/ajpheart.00668.2011
Severi, S., Pogliani, D., Fantini, G., Fabbrini, P., Vigano, M. R., Galbiati, E., et al. (2010). Alterations of atrial electrophysiology induced by electrolyte variations: combined computational and P-wave analysis. Europace 12, 842–849. doi: 10.1093/europace/euq042
Shannon, T. R., Wang, F., Puglisi, J., Weber, C., and Bers, D. M. (2004). A mathematical treatment of integrated Ca2+ dynamics within the ventricular myocyte. Biophys. J. 87, 3351–3371. doi: 10.1529/biophysj.104.047449
Shimoni, Y., Clark, R., and Giles, W. R. (1992). Role of an inwardly rectifying potassium current in rabbit ventricular action potential. J. Physiol. 448, 709–727. doi: 10.1113/jphysiol.1992.sp019066
Singh, B. N., and Vaughan Williams, E. M. (1971). Effect of altering potassium concentration on the action of lidocaine and diphenylhydantoin on rabbit atrial and ventricular muscle. Circ. Res. 29, 286–295. doi: 10.1161/01.res.29.3.286
Skibsbye, L., Jespersen, T., Christ, T., Maleckar, M. M., van den Brink, J., Tavi, P., et al. (2016). Refractoriness in human atria: time and voltage dependence of sodium channel availability. J. Mol. Cell. Cardiol. 101, 26–34. doi: 10.1016/j.yjmcc.2016.10.009
Smeets, J. L. R. M., Allessie, M. A., Lammers, W. J. E. P., Bonke, F. I. M., and Hollen, J. (1986). The wavelength of the cardiac impulse and reentrant arrhythmias in isolated rabbit atrium. Circ. Res. 58, 96–108. doi: 10.1161/01.res.58.1.96
Sossalla, S., Kallmeyer, B., Wagner, S., Mazur, M., Maurer, U., Toischer, K., et al. (2010). Altered Na+ currents in atrial fibrillation: effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J. Am. Coll. Cardiol. 55, 2330–2342. doi: 10.1016/j.jacc.2009.12.055
Spach, K. S., Heidlage, J. F., Dolber, P. C., and Barr, R. C. (2007). Mechanism of origin of conduction disturbances in aging human atrial bundles: experimental and model study. Heart Rhythm. 4, 175–185. doi: 10.1016/j.hrthm.2006.10.023
Tazmini, K., Frist, M., Lewalle, A., Laasmaa, M., Morotti, S., Lipsett, D. B., et al. (2020). Hypokalemia promotes arrhythmia by distinct mechanisms in atrial and ventricular myocytes. Circ. Res. 126, 889–906. doi: 10.1161/CIRCRESAHA.119.315641
Ten Eick, R. E., and Singer, D. H. (1979). Electrophysiological properties of diseased human atrium. Circ. Res. 44, 545–557. doi: 10.1161/01.res.44.4.545
Trenor, B., Cardona, K., Romero, L., Gomez, J., Saiz, J., Rajamani, S., et al. (2018). Proarrhythmic effects of low plasma K+ in human ventricle: an illustrated review. Trends Cardiovas. Med. 28, 233–242. doi: 10.1016/j.tcm.2017.11.002
Vagos, M. R. S. S., van Herck, I. G. M., Sundnes, J., Arevalo, H. J., Edwards, A. G., and Koivumäki, J. T. (2018). Computational modeling of electrophysiology and pharmacotherapy of atrial fibrillation: recent advances and future challenges. Front. Physiol. 9:1221. doi: 10.3389/fphys.2018.01221
Varró, A., and Baczkó, I. (2011). Cardiac ventricular repolarization reserve: a principle for understanding drug-related proarrhythmic risk. Br. J. Pharmacol. 164, 14–36. doi: 10.1111/j.1476-5381.2011.01367.x
Varró, A., Tomek, J., Nagy, N., Virág, L., Passini, E., Rodriguez, B., et al. (2020). Cardiac transmembrane ion channels and action potentials: cellular physiology and arrhythmogenic behaviour. Physiol. Rev. [Epub ahead of print]. doi: 10.1152/physrev.00024.2019
Vaughan Williams, E. M. (1959). The effect of changes in extracellular potassium concentration on the intracellular potentials of isolated rabbit atria. J. Physiol. 146, 411–427. doi: 10.1113/jphysiol.1959.sp006201
Vincenti, A., Passini, E., Fabbrini, P., Luise, M. C., Severi, S., and Genovesi, S. (2014). Recurrent intradialytic paroxysmal atrial fibrillation: hypotheses on onset mechanisms based on clinical data and computational analysis. Eruopace 16, 396–404. doi: 10.1093/europace/eut346
Voigt, N., Heijman, J., Trausch, A., Mintert-Jancke, E., Pott, L., Ravens, U., et al. (2013). Impaired Na+-dependent regulation of acetylcholine-activated inward-rectifier K+ current modulates action potential rate dependence in patients with chronic atrial fibrillation. J. Mol. Cell. Cardiol. 61, 142–152. doi: 10.1016/j.yjmcc.2013.03.011
Voigt, N., Trausch, A., Knaut, M., Matschke, K., Varró, A., Van Wagoner, D. R., et al. (2010). Left-to-right atrial inward rectifier potassium current gradients in patients with paroxysmal versus chronic atrial fibrillation. Circ. Arrhythm. Electrophysiol. 3, 472–480. doi: 10.1161/CIRCEP.110.954636
Wang, H.-H., Hung, C.-C., Hwang, D.-Y., Kuo, M.-C., Chiu, Y.-W., Chang, J. M., et al. (2013). Hypokalemia, its contributing factors and renal outcomes in patients with chronic kidney disease. PLoS One 8:e67140. doi: 10.1371/journal.pone.0067140
Ward, C. A., Bazazi, H., Clark, R. B., Nygren, A., and Giles, W. R. (2006). Actions of emigrated neutrophils on Na+ and K+ currents in rat ventricular myocytes. Prog. Biophys, Mol. Biol. 90, 249–269. doi: 10.1016/j.pbiomolbio.2005.07.003
Weaver, W. F., and Burchell, H. B. (1960). Serum potassium and the electrocardiogram in hypokalemia. Circulation 21, 505–521. doi: 10.1161/01.cir.21.4.505
Weiss, J. N., Qu, Z., and Shivkumar, K. (2017). Electrophysiology of hypokalemia and hyperkalemia. Circ. Arrhythm. Electrophysiol. 10:e004667. doi: 10.1161/CIRCEP.116.004667
Whalley, D. W., Wendt, D. J., Starmer, C. F., Rudy, Y., and Grant, A. O. (1994). Voltage-independent effects of extracellular K+ on the Na+ current and phase 0 of the action potential in isolated cardiac myocytes. Circ. Res. 75, 491–502. doi: 10.1161/01.res.75.3.491
White, E., and Terrar, D. A. (1991). Action potential duration and the inotropic response to reduced extracellular potassium in guinea-pig ventricular myocytes. Exp. Physiol. 76, 705–716. doi: 10.1113/expphysiol.1991.sp003537
Whittaker, D. G., Clerx, M., Lei, C. L., Christini, D. J., and Mirams, G. R. (2020). Calibration of ionic and cellular cardiac electrophysiology models. Wiley Interdiscip. Syst. Biol. Med. 12:e1482. doi: 10.1002/wsbm.1482
Wilhelms, M., Hettmann, H., Maleckar, M. M. C., Koivumäki, J. T., Dössel, O., and Seemann, G. (2013). Benchmarking electrophysiological models of human atrial myocytes. Front. Physiol. 3:487. doi: 10.3389/fphys.2012.00487
Workman, A. J., Kane, K. A., and Rankin, A. C. (2001). The contribution of ionic currents to changes in refractoriness of human atrial myocytes associated with chronic atrial fibrillation. Cardiovasc. Res. 52, 226–235. doi: 10.1016/s0008-6363(01)00380-7
Workman, A. J., Kane, K. A., and Rankin, A. C. (2003). Characterization of the Na,K pump current in atrial cells from patients with and without chronic atrial fibrillation. Cardiovasc. Res. 59, 593–602. doi: 10.1016/s0008-6363(03)00466-8
Wrong, O. (1961). Incidence of hypokalaemia in severe hypertension. Br. Med. J. 2, 419–421. doi: 10.1136/bmj.2.5249.419
Yang, P. C., DeMarco, K. R., Aghasafari, P., Jeng, M. T., Dawson, J. R. D., Bekker, S., et al. (2020). A computational pipeline to predict cardiotoxicity. Circ. Res. 126, 947–964. doi: 10.1161/CIRCRESAHA.119.316404
Yu, T., Lloyd, C. M., Nickerson, D. P., Cooling, M. T., Miller, A. K., Garny, A., et al. (2011). The physiome model repository 2. Bioinformatics 27, 743–744. doi: 10.1093/bioinformatics/btq723
Zaza, A. (2009). Serum potassium and arrhythmias. Europace 11, 421–422. doi: 10.1093/europace/eup005
Zhang, H., Garratt, C. J., Zhu, J., and Holden, A. V. (2005). Role of up-regulation of IK1 in action potential shortening associated with atrial fibrillation in humans. Cardiovas. Res. 66, 493–502. doi: 10.1016/j.cardiores.2005.01.020
Zhang, H., Zhang, S., Wang, W., Wang, K., and Shen, W. (2020). A mathematical model of the mouse atrial myocyte with inter-atrial electrophysiological heterogeneity. Front. Physiol. 11:972. doi: 10.3389/fphys.2020.00972
Zimmerman, D., Sood, M. M., Rigatto, C., Holden, R. M., Hiremath, H. S., and Clase, C. M. (2012). Systematic review and meta-analysis of incidence, prevalence and outcomes of atrial fibrillation in patients on dialysis. Nephrol. Dial. Transpl. 27, 3816–3822. doi: 10.1093/ndt/gfs416
Keywords: hypokalemia, mathematical modeling, inwardly rectifying K+ current, sodium potassium (Na+/K+-ATPase) pump, action potential repolarization, atrial fibrillation (AF), renal dialysis, plasma potassium levels
Citation: Clerx M, Mirams GR, Rogers AJ, Narayan SM and Giles WR (2021) Immediate and Delayed Response of Simulated Human Atrial Myocytes to Clinically-Relevant Hypokalemia. Front. Physiol. 12:651162. doi: 10.3389/fphys.2021.651162
Received: 08 January 2021; Accepted: 22 March 2021;
Published: 26 May 2021.
Edited by:
Axel Loewe, Karlsruhe Institute of Technology (KIT), GermanyReviewed by:
Beatriz Trenor, Universitat Politècnica de València, SpainSteve Poelzing, Virginia Tech, United States
Copyright © 2021 Clerx, Mirams, Rogers, Narayan and Giles. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Clerx, TWljaGFlbC5DbGVyeEBub3R0aW5naGFtLmFjLnVr