Abstract
Hypertension (HTN) affects more than one-third of the US population and remains the top risk factor for the development of cardiovascular disease (CVD). Identifying the underlying mechanisms for developing HTN are of critical importance because the risk of developing CVD doubles with ∼20 mmHg increase in systolic blood pressure (BP). Endothelial dysfunction, especially in the resistance arteries, is the primary site for initiation of sub-clinical HTN. Furthermore, inflammation and reactive oxygen and nitrogen species (ROS/RNS) not only influence the endothelium independently, but also have a synergistic influence on each other. Together, the interplay between inflammation, ROS and vascular dysfunction is referred to as the vascular health triad, and affects BP regulation in humans. While the interplay of the vascular health triad is well established, new underlying mechanistic targets are under investigation, including: Inducible nitric oxide synthase, hydrogen peroxide, hydrogen sulfide, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and nuclear factor activated T cells. This review outlines the role of these unusual suspects in vascular health and function in humans. This review connects the dots using these unusual suspects underlying inflammation, ROS and vascular dysfunction especially in individuals at risk of or with diagnosed HTN based on novel studies performed in humans.
Introduction
Cardiovascular disease (CVD) is the leading cause of morbidity and mortality worldwide, and the prevalence of CVD increases with aging (Virani et al., 2021). The increase in blood pressure (BP) or diagnosed hypertension (HTN) is widely accepted as the primary precursor to CVD; the risk of CVD is assumed to increase in a linear fashion as BP increases. In general, the risk of CVD doubles when there is an approximately 20 mmHg increase in systolic BP and 10 mmHg increase in diastolic BP (Lewington et al., 2002). Further, BP rises substantially as humans age (Guzik and Touyz, 2017); however, the rise in BP with age is linear among men but rises in a differential pattern among women. Sex differences in HTN and their underlying mechanisms have been reviewed in detail previously (Ji et al., 2020).
In 2017, the American College of Cardiology (ACC) and American Heart Association (AHA) redefined the classifications of HTN diagnosis (Whelton et al., 2018). In the revised categorizations, the BP threshold has been lowered for stage 1 HTN and prehypertension (now elevated BP). Stage 1 HTN is defined as resting systolic BP between 130 and 139 mmHg or diastolic BP between 80 and 89 mmHg, and elevated BP is now defined as systolic BP between 120 and 129 mmHg and diastolic BP below 80 mmHg. Elevated BP is a strong predictor of late life HTN and CVD (Whelton et al., 2018). The revised categorizations have drastically increased the prevalence of HTN to about 46% of Americans (Virani et al., 2021), and have highlighted the importance of preclinical and clinical research in humans to identify therapeutic targets for interventions that extend the health of aging humans (Whelton et al., 2018). Moreover, identification of novel drug treatment based targets is important as medication adherence with traditional antihypertensives (diuretics, angiotensin converting enzyme inhibitors, calcium channel blockers, etc.) remains a significant issue.
The main site for vascular resistance, and thus a critical component of BP regulation, are the arterioles in the microcirculation (Nowroozpoor et al., 2021). The arterioles have similar anatomical layers as larger arteries; however, the lumen size of the arterioles (10–150 μm diameter) creates a substantial resistance to the blood flow and thus BP responses. In addition to the lumen size, the smooth muscle tone being normally in a state of contraction makes the resistance vessels an important site for BP control. Vascular resistance within the arteries is controlled by a complex interplay between local vasodilators, sympathetic modulation, and endocrine (paracrine) driven changes. Not only is the microvascular bed the first site to present with dysfunction, but it can also experience dysfunction without displaying any evidence in the macrovasculature or feed arteries (Nowroozpoor et al., 2021). Well established causes of endothelial dysfunction include imbalances in inflammation and/or reactive oxygen species (ROS). This is of particular importance because endothelial dysfunction can further potentiate imbalances in inflammation and ROS, leading to a “never ending cycle (or triad)” (Figure 1). The cumulative effects of these minor insults on the vasculature lead to a pro-hypertensive environment. In this mini-review we will first present aspects of the vascular health triad and then novel mechanisms that induce imbalances within the system, leading to HTN in humans. Table 1 summarizes the methods utilized to elucidate mechanisms underlying hypertension-associated vascular dysfunction A comprehensive discussion of the renal and sympathetic contributions to HTN is beyond the scope of this review, but have been recently reviewed (Grassi et al., 2018, 2019; Fonkoue et al., 2019; Holwerda et al., 2019; Keir et al., 2020).
FIGURE 1
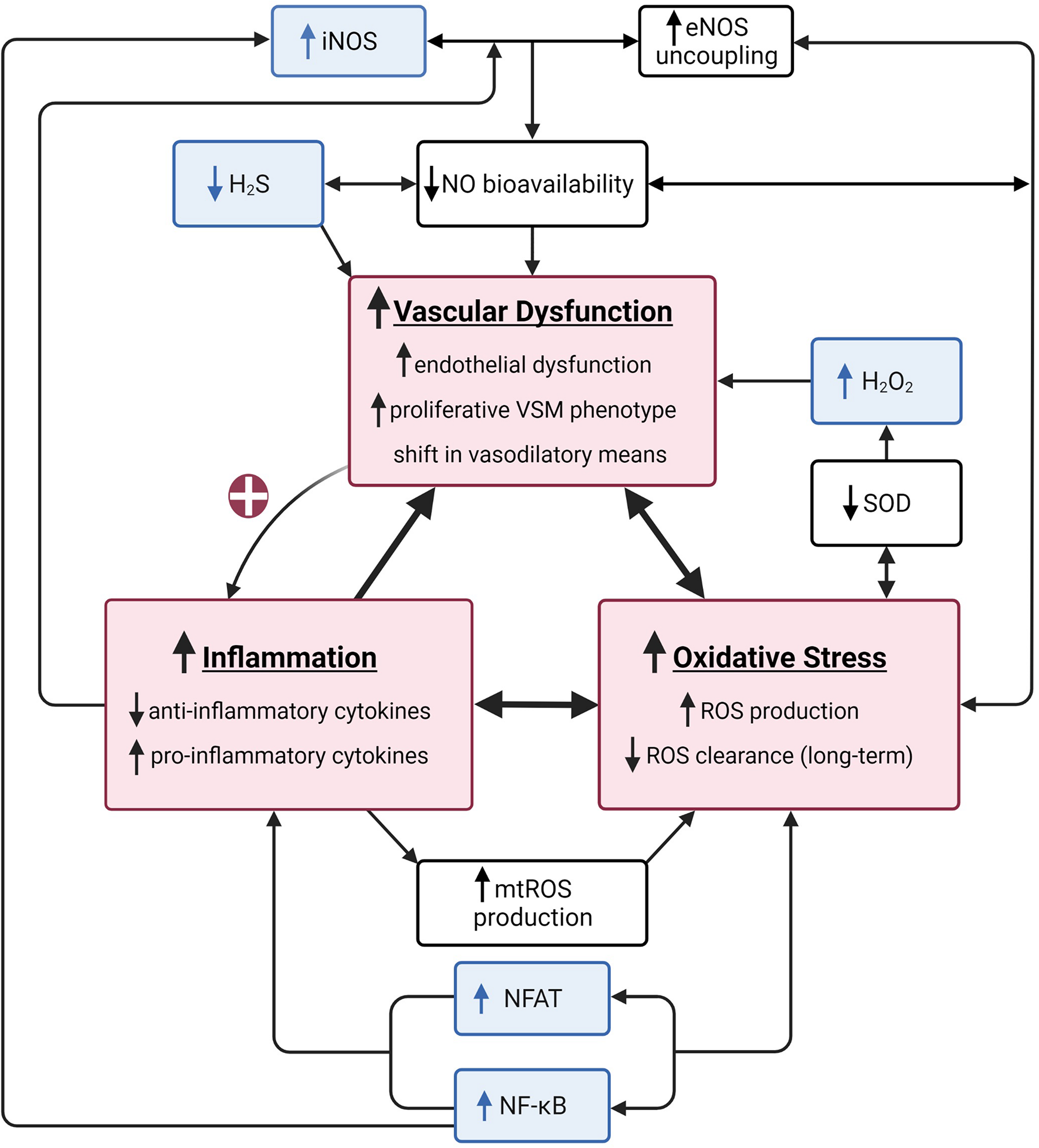
A summary of the vascular health triad. The vascular health triad is composed of oxidative stress, inflammation, and vascular dysfunction (red boxes). These outcomes are synergistically interdependent as their underlying mechanisms directly (e.g., high inflammatory state causes a decrease in nitric oxide (NO) bioavailability resulting in increased vascular dysfunction) or indirectly [e.g., vascular dysfunction increasing inflammation via positive feedback loop (red plus sign)] interact, resulting in a vicious cycle of increased cardiovascular disease risk. Well-established underlying mechanisms of the triad include decreased NO bioavailability via increased endothelial nitric oxide synthase (eNOS) uncoupling, decreased superoxide dismutase (SOD), and increased mitochondrial reactive oxygen species (mtROS) production (white boxes). The unusual suspects include inducible nitric oxide synthase (iNOS), hydrogen sulfide (H2S), hydrogen peroxide (H2O2), nuclear factor kappa-light-chain-enhancer of active B cells (NF-κB), and nuclear factor of activated T cells (NFAT; blue boxes). Created with BioRender.com.
TABLE 1
| Method | References | Pathway | |
| In vivo | |||
|
Flow-mediated Dilation + Doppler Ultrasound |
|||
| Gokce et al., 2001; Benjamin et al., 2004; Juonala et al., 2004; McGowan et al., 2006; Buus et al., 2007; Yang et al., 2010; Broxterman et al., 2019; Figueiredo et al., 2012 | Endothelium-dependent dilation | ||
| Ratchford et al., 2019a | Endothelium-dependent dilation | ||
| Oxidative stress (acute antioxidant cocktail) | |||
| Sapp et al., 2021 | Endothelium-dependent dilation | ||
| Inflammation (influenza vaccine) | |||
| Craighead et al., 2020 | Endothelium-dependent dilation, Endothelium in-dependent dilation (sublingual NTG), Oxidative stress (Vitamin C) | ||
|
Venous Occlusion Plethysmography |
|||
| On et al., 2002 | Endothelium-dependent (ACh) | ||
| Bottino et al., 2015 | Endothelium-dependent dilation, Endothelium in-dependent (sublingual NTG) | ||
| Endothelium-dependent (ACh, Bradykinin) | |||
| Hingorani et al., 2000 | Endothelium in-dependent (NTG, verapamil) | ||
| Inflammation (salmonella typhi vaccine) | |||
|
Arterial Infusions + Doppler Ultrasound |
|||
| Takase et al., 2006; Broxterman et al., 2019 | Endothelium-dependent dilation (ACh) | ||
| Gokce et al., 2001; McGowan et al., 2006; Buus et al., 2007 | Endothelium in-dependent dilation (NTG) | ||
|
Iontophoresis + Laser Doppler Flowmetry |
|||
| Cupisti et al., 2000 | Endothelium-dependent dilation (ACh) Endothelium-independent dilation (SNP) | ||
| Farkas et al., 2004 | |||
| Lindstedt et al., 2006 | Endothelium-dependent dilation (ACh; local heating) Endothelium in-dependent dilation (SNP) | ||
|
Intradermal Microdialysis + Laser Doppler Flowmetry |
|||
| Smith et al., 2011 | Endothelium-dependent dilation (ACh, local heating) NO (L-NAME) iNOS (1400W) nNOS (NPLA) | ||
| Smith et al., 2013 | Vasoconstriction (local cooling, yohimbine + proprananol), Rho/Rho-Kinase (fasudil) | ||
| Bruning et al., 2015 | Endothelium-dependent dilation (local heating), NO (L-NAME) | ||
| Craighead et al., 2017 | Endothelium-dependent dilation (ACh, local heating), NO (L-NAME), iNOS (1400w) | ||
| HTN Greaney et al., 2017 | Endothelium-dependent dilation (ACh), NO (L-NAME), H2S (Na2S, AOAA) | ||
| Craighead et al., 2018 | Endothelium independent dilation (SNP), NE-induced vasoconstriction, Lysyl Oxidation (BAPN) | ||
| Dillon et al., 2020 | Endothelium-dependent (ACh), NO (L-NAME) | ||
|
Sublingual NTG + Doppler Ultrasound |
|||
| McGowan et al., 2006 | Endothelium in-dependent dilation | ||
|
Non-invasive Single Limb Exercise + Doppler Ultrasound |
|||
| Ratchford et al., 2019b | BF response to exercise (IHG) | ||
| McGowan et al., 2006 | BF response to exercise (static IHG, dynamic knee extension) | ||
|
Single Limb Exercise + NIRS-derived TSI |
|||
| Sprick et al., 2019 | BF response to exercise (IHG) | ||
|
Ex vivo / in vitro Circulating concentrations |
|||
| Hingorani et al., 2000 | IL-1β, IL-1Ra IL-6, TNF-α | ||
| Bottino et al., 2015 | IL-1, IL-6, OxLDL, sICAM, sVCAM, sE-Selectin, TNF-α | ||
| Junqueira et al., 2018 | Adiponectin, CRP, endothelin, ICAM, VCAM | ||
| Craighead et al., 2020 | IL-6, OxLDL, TNF-α, total antioxidant status, hsCRP | ||
|
PBMCs |
|||
| Huang et al., 2016 | IL-6, NFAT, TNF-α | ||
|
Arterial Biopsy |
|||
| Phillips et al., 2007 | NO (L-NAME) | ||
| Oxidative stress (H2O2) | |||
| Donato et al., 2007 | Oxidative stress (nitrotyrosine, NFκB) | ||
| Migrino et al., 2011 | Endothelial-dependent dilation (ACh), Endothelial in-dependent dilation (papaverine), Oxidative stress (SOD, BH4, mitoquinone, gp91ds-tat) | ||
| Beyer et al., 2014 | Oxidative stress (H2O2, mtROS, tempol) | ||
| Beyer et al., 2017 | Oxidative stress (H2O2, mtROS) | ||
| Kadlec et al., 2017 | H2O2, NO, PGC-1α | ||
| Hughes et al., 2021b | NO (L-NAME, c-PTIO), H2O2 (peg-cat) Oxidative stress (rotenone) | ||
| Hader et al., 2019 | |||
| Hughes et al., 2021a | H2O2 | ||
|
Venous Endothelial Cell Biopsy |
|||
| Donato et al., 2007 | Oxidative stress (NADPH oxidase p47phox, SOD, NFκB) | ||
| Pierce et al., 2009 | Inflammation (NFκB, TNF-α) | ||
| Oxidative stress (NADPH oxidase p47phox) | |||
| Craighead et al., 2020 | Oxidative stress (NADPH, MnSOD), Inflammation (NFkB) | ||
|
Cutaneous Biopsies |
|||
| Smith et al., 2011 | eNOS, iNOS, nNOS, pVASP | ||
| Smith et al., 2013 | ROCK activity and expression | ||
| HTN Greaney et al., 2017 | H2S (CSE, 3-MPST) | ||
|
Myograph |
|||
| Buus et al., 2007 | “small artery relaxation with ACh” | ||
Summary of methodology for mechanisms mediating vascular function in hypertensive adults.
3-MPST, 3-mercaptopyruvate sulfurtransferase; ACh, acetylcholine; BAPN, β-aminopropionitrile; CRP, C-reactive protein; CSE, cystathionine gamma-lyase; H2S, hydrogen sulfide; hsCRP, high-sensitivity C-reactive protein; IHG, intermittent handgrip; IL, interleukin; iNOS, inducible nitric oxide synthase; L-NAME, NG-nitro-l-arginine methyl ester; MnSOD, manganese superoxide dismutase; NADPH, nicotinamide adenine dinucleotide phosphate; NE, norepinephrine; NFkB, nuclear factor kappa-light-chain-enhancer of activated B cells; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; NPLA, N(ω)-propyl-L-arginine; NTG, nitroglycerin; oxLDL, oxidized low-density lipoprotein; PBMCs, peripheral blood mononuclear cells; ROCK, rho-associated protein kinase; ROS, reactive oxygen species; sICAM, soluble inter-cellular adhesion molecule; SNP, sodium nitroprusside; sVCAM, soluble vascular cell adhesion molecule; TNF-α, tumor necrosis factor alpha.
Vascular Health—Role of Inflammation and Reactive Oxygen and Nitrogen Species—“The Usual Suspects”
In young, healthy adults, inflammation and reactive oxygen and nitrogen species (ROS/RNS) serve a critical, positive physiological role in vascular homeostasis. The maintenance of vascular health is the complex relationship between vasoprotective factors, such as the nitric oxide system, and other pathways that impair these mechanisms, including both inflammation and ROS. “Normal” vascular function is often characterized by the ability to efficiently vasodilate or vasoconstrict in response to a stimulus. On the contrary, vascular dysfunction is characterized by the loss of efficiency in the vasodilatory component even in the presence of a stimulus. Inflammation is one such stimulus (and a cornerstone of the vascular health triad) which influences vascular function acutely and chronically—in a temporal fashion. Inflammation is heightened as a natural defense mechanism to tissue injury, infection, or pathogen infiltration. The initial inflammatory cascade is characterized by heightened pro-inflammatory cytokine release, immune cell movement to the site of invasion or injury, and the release of local chemoattractants, notably, adhesion molecules (Wadley et al., 2013). This acute inflammation (influx of inflammatory cytokines) can transiently impair vascular function (8–32 h), through an acute impairment in nitric oxide (NO) bioavailability, even in young otherwise healthy individuals (Hingorani et al., 2000). The second component of the vascular health triad is ROS, which are free radicals, such as superoxide or peroxynitrite, that are integral to cellular signaling (El Assar et al., 2013; Wadley et al., 2013). ROS are produced via oxidative metabolism and proteins, such as NADPH oxidase, xanthine oxidase and via endothelial nitric oxide synthase (eNOS) uncoupling (Jacobi et al., 2005; Ding et al., 2007; El Assar et al., 2013; Wadley et al., 2013). Endogenous antioxidants, such as superoxide dismutase (SOD), glutathione, and NO, clear ROS enzymatically, or through direct chemical reaction (Selemidis et al., 2007; El Assar et al., 2013; Wadley et al., 2013). The aspect of mitochondrial ROS is covered in detail in an excellent review by Kirkman et al. (2021).
In young, otherwise healthy adults, there is (I) redundancy in the vasodilatory pathways with abundant bioavailability of endothelium-derived vasodilating substances (e.g., NO, prostaglandins, EDHF) (II) low basal concentrations of vasoconstrictive substances and (III) low basal concentrations of inflammatory cytokines and ROS. However, while there is ample data indicating impaired endothelial function in human subject cohorts who are at risk for the development of HTN later in life, including those with a family history of HTN (Greaney et al., 2015; Matthews et al., 2017), and non-traditional CVD risk factors (Martens et al., 2016; Greaney et al., 2019; Katulka et al., 2019), there is lack of data in individuals who are young and otherwise healthy but have undiagnosed HTN. Moreover, due to this redundancy it is difficult to unravel these interrelated mechanisms using gross measures of endothelial function in conduit arteries.
Our laboratory recently induced acute inflammation using an influenza vaccine in vivo and in vitro in young, healthy African American and Caucasian American individuals (Sapp et al., 2021). Although the vaccine stimulus did not impact conduit artery function (as measured by flow-mediated dilation), there were decreases in eNOS messenger RNA in the African American group, coinciding with race-specific changes in intracellular and extracellular microRNAs (miR) related to inflammation (miR-221-3p, 222-3p, and 150-5p) (Sapp et al., 2021). Circulating miRs are novel biomarkers of acute and chronic inflammation (Mi et al., 2013; Benz et al., 2016). miRs play mechanistic roles in endothelial activation, inflammation, and dysfunction; thus, initiating events in HTN (Cheng et al., 2014; Shi et al., 2015; Fernández-Hernando and Suárez, 2018). Furthermore, certain miRs [miR-146-protective (Wang et al., 2017); miR-34a- pro-inflammatory (Badi et al., 2018); miR-570-3p- pro-senescence (Baker et al., 2019); miR-217- pro-inflammatory (Zhang et al., 2019)] regulate ROS mechanisms via sirtuin-1 (Yamakuchi, 2012; Yamakuchi and Hashiguchi, 2018). Circulating miRs are taken up by endothelial cells, where they affect endothelial cell function, promote inflammation, and increase ROS (Njock and Fish, 2017; Zhong et al., 2018). Furthermore, the flow patterns within the arterial tree, i.e., turbulent flow near bifurcations or atherosclerotic lesions which can accentuate endothelial permeability or in contrast laminar flow which can exert beneficial effects on the vessel wall tend to trigger a number of miRs (Marin et al., 2013; Schmitz et al., 2019). Case in point, laminar shear stress (could be exercise induced) can upregulate miR-126 which is generally accepted as an anti-inflammatory and anti-atherogenic miR (Sapp et al., 2017). Therefore, in recent years circulating miRs have emerged as novel molecules mediating cell-to-cell communication in physiological processes (Njock and Fish, 2017; Bär et al., 2019). Mechanistically, emerging data in humans indicates significant cross-talk between inflammation and ROS within the endothelial cells in young, otherwise healthy adults, even before measurable conduit artery or macrovascular dysfunction plausibly through miRs (Sapp et al., 2021).
Vascular Health Triad—Aging or Diseased States
In aging and diseased states, the vascular health triad becomes a positive feedback loop of heightened inflammation, oxidative stress, and vascular dysfunction. Chronic overproduction of inflammatory mediators, such as inflammatory cytokines, adhesion molecules, and inflammatory proteins, result in systemic low-grade inflammation initiating the complex cascade of heightened NADPH oxidase activity, eNOS uncoupling, and mitochondrial dysfunction, all of which result in heightened ROS production. Early in aging or disease pathogenesis, heightened antioxidant concentrations accommodate excessive ROS production. Following long-term exposure to increased ROS and inflammation, antioxidant defense mechanisms are reduced. Previous studies have interrogated these mechanisms with the acute administration of antioxidant cocktails (including ascorbic acid and alpha-tocopherol) in older adults (Eskurza et al., 2004a,b; Crecelius et al., 2010; Wray et al., 2012; Richards et al., 2015; Trinity et al., 2016), post-menopausal females (Ozemek et al., 2016), and individuals with heart failure with preserved ejection fraction (Ratchford et al., 2019a). However, more recent studies have focused on targeted pharmacological agents to identify specific ROS molecules within the triad (Alexander et al., 2013; Hurr et al., 2018; Martens et al., 2018; Park et al., 2018; Rossman et al., 2018). One emerging link in this triad is through the inducible nitric oxide synthase (iNOS) pathway. iNOS activity is increased during inflammation and NO is produced in toxic concentrations to prevent cell death by clearing excessive ROS (Aktan, 2004). NO reacts with superoxide (), a main type of ROS produced during heightened inflammation, to form peroxynitrite (ONOO−). Concurrently, superoxide dismutase (SOD) reacts with superoxide () to form hydrogen peroxide (H2O2). Interestingly H2O2 becomes the main contributing vasodilatory substance when NO bioavailability is decreased (Phillips et al., 2007; Beyer et al., 2014; Kadlec et al., 2017). Initially with acute inflammatory/ROS stimuli, other vasodilatory substances, such as prostaglandins and H2O2, can compensate for the impairment in NO-mediated vasodilation (Beyer et al., 2017). However, if the allostatic load presented by inflammation and ROS persists, not only do the compensatory mechanisms fail, but eventually deleterious vascular remodeling occurs. Therefore, with aging and/or disease progression the healthy functioning of the vasculature is disrupted due to (I) loss of redundant vasodilatory pathways, with limited bioavailability of vasodilating substances (especially NO), (II) increased circulating vasoconstrictive substances and (III) increased concentrations of inflammatory cytokines and ROS.
Typically, when considering vascular dysfunction, the role of NO bioavailability/scavenging due to eNOS uncoupling, decreased superoxide dismutase (SOD), and increased mitochondrial reactive oxygen species (mtROS) production are of significant importance (Figure 1). Therefore, in the following sections we have presented some of the newer and not widely discussed factors which can also have role in decreased NO bioavailability and/or increased NO scavenging, leading to vascular dysfunction.
Vascular Health Triad—“The Unusual Suspects”
Role of Inducible Nitric Oxide Synthase
During heightened inflammation and oxidative stress, there are various intersecting pathways that contribute to impaired NO bioavailability and, eventually, vascular dysfunction. Increased iNOS expression is stimulated by nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), interleukin-6 (IL-6), and ROS producers, such as the p47phox subunit of NADPH oxidases (Hemmrich et al., 2003; Wu et al., 2008; Li et al., 2015). With chronic low-grade inflammation, iNOS activity increases, resulting in increased iNOS-derived NO production. To pharmacodissect the NOS balance in humans and prominent role of iNOS, we performed a unique bed to bench experiment evaluating endothelial-dependent microvascular function in individuals with HTN during iNOS, neuronal NOS (nNOS) and non-selective-NOS inhibition (Smith et al., 2011). Interestingly, the attenuated endothelial-dependent vasodilation in hypertensive adults was restored with iNOS inhibition, suggesting a prominent role of iNOS in hypertension-induced microvascular dysfunction. Even though the eNOS expression was similar between normotensives and hypertensives adults, iNOS expression in biopsy samples from hypertensive subjects was significantly greater as compared to age-matched normotensives (Smith et al., 2011). Thus, the NO produced during inflammation with HTN adopts a scavenging and cell-preserving role, as opposed to an active vasodilatory role.
Hydrogen Peroxide (H2O2) and H2O2-Mediated Primary Vasodilatory Mechanism
SOD is a primary antioxidant defense system with three known forms: SOD1 (cytosolic), SOD2 (mitochondrial), and SOD3 (circulating) (Zelko et al., 2002). SOD scavenges O2– radicals to form H2O2. During states of heightened inflammation and ROS, NADPH oxidase activity and eNOS uncoupling subsequently increase, resulting in increased O2– production (El Assar et al., 2013; Risbano and Gladwin, 2013; Wadley et al., 2013). In early disease initiation and progression, SOD activity increases to clear excessively produced O2–, subsequently increasing the production of H2O2 during both acute and chronic increases in inflammation and oxidative stress (El Assar et al., 2013). Recent studies from the Gutterman laboratory have suggested a novel theory of shifts in vasodilatory pathways during vascular inflammatory conditions, especially HTN (Migrino et al., 2011; Beyer et al., 2014, 2017; Kadlec et al., 2017). The NO-mediated vasodilatory pathway is vasoprotective, as it helps maintain normal BP in young, healthy individuals. However, with increased intraluminal pressure, as seen in HTN and reduced bioavailability of NO, there is a shift in microvascular vasodilatory pathways toward H2O2-dependent mechanisms, even though the total magnitude of the vasodilation to a given shear stimulus remains the same (Beyer et al., 2014). Hughes et al. (2021b) examined the effect of transient increases in intraluminal pressure in resistance arterioles of hypertensive individuals with and without coronary artery disease were evaluated. In this model, there was a compensatory switch to the H2O2-mediated vasodilatory pathway following increased intraluminal pressure, suggesting diseased and healthy aged adults have similar shifts in primary vasodilatory mechanisms, but along a different time course (Hughes et al., 2021b). Furthermore, in the presence of transient increases in intraluminal pressure, even in isolated arterioles of healthy individuals, H2O2 is typically mitochondria derived (Beyer et al., 2014). These ex-vivo studies have also led to potential targets, such as autophagy and extranuclear telomerase (Hughes et al., 2021a). Taken together, these studies suggests an important switch in the physiological mechanism of vasodilation and heightened ROS production, specifically production of O2–, H2O2, and ONOO−as propagating a positive feedback loop that promotes vascular dysfunction.
Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells and Nuclear Factor From Activated T Cells
Investigating upstream transcriptional molecular targets underlying endothelial and vascular smooth muscle cell dysfunction in humans has yielded significant understanding of the complexity of these signaling mechanisms. Despite the complexity and multiple downstream effects, these studies are necessary for the development of targeted treatment and prevention strategies for HTN in humans. It is well established that NF-κB are an important intracellular mediator of inflammation and vascular dysfunction (specifically, NO-mediated mechanisms) (Donato et al., 2007, 2009; Pierce et al., 2009; Lee et al., 2014). In an elegant series of preclinical and clinical studies, Donato et al. (2009) established that activation of NF-κB mediates age-related vascular dysfunction. These studies interrogated NF-κB functionality in human subjects using a short-term (4 days) high-dose oral salsalate approach (Pierce et al., 2009). Salsalate, a non-acytlated salicylate, inhibits NF-κB translocation to the nucleus, reducing ROS synthesis through NADPH oxidases (Kopp and Ghosh, 1994; Pierce et al., 1996). They demonstrated that oral salsalate reduces endothelial cell NF-κ p65 expression by ∼25%, total nitrotrysine, a global marker of oxidative stress, and NADPH oxidase p47phox expression by 25 and 30%, respectively. Importantly, inhibition of NF-κB functionally resulted in improved NO-dependent vasodilation. Additionally, when comparing inactive to habitually active older adults, active adults had reduced NF-κB p65 expression, reduced nitrotyrosine, and endothelial function similar to their younger counterparts (Walker et al., 2014).
A growing body of literature suggests that the family of Ca2+/calcineurin-sensitive transcriptional factors of nuclear factor from activated T-cells (NFAT) may play an essential role as a molecular switch that initiates dysfunction in both the endothelium (Cockerill et al., 1995; Boss et al., 1998; Armesilla et al., 1999; Bochkov et al., 2002; Gonzalez Bosc et al., 2004; Zetterqvist et al., 2015; Huang et al., 2016) and vascular smooth muscle (Suzuki et al., 2002; Lipskaia et al., 2003; Amberg et al., 2004; Nilsson et al., 2006; Nieves-Cintrón et al., 2007; Orr et al., 2009; Pang and Sun, 2009; Nilsson-Berglund et al., 2010; Berglund et al., 2012; Shiny et al., 2016; Soudani et al., 2016; Govatati et al., 2019). NFATs regulate multiple downstream mechanisms that initiate vascular dysfunction. Specifically, NFATs (I) impair endothelial function through NO-dependent mechanisms (Armesilla et al., 1999; Bochkov et al., 2002; Johnson et al., 2003; Norata et al., 2007; Garcia-Vaz et al., 2020; Wang et al., 2020), (II) increase the expression of inflammatory mediators in the arterial wall promoting atherosclerosis (Pierce et al., 1996; Pang and Sun, 2009; Nilsson-Berglund et al., 2010; Berglund et al., 2012; Zetterqvist et al., 2014; Weng et al., 2017), and (III) initiate pathogenic VSM proliferation (Suzuki et al., 2002; Lipskaia et al., 2003; Nilsson et al., 2006; Nieves-Cintrón et al., 2007; Donato et al., 2009; Orr et al., 2009; Pang and Sun, 2009; Nilsson-Berglund et al., 2010; Yan et al., 2015; Shiny et al., 2016; Soudani et al., 2016; Govatati et al., 2019). In preclinical models, inhibition of NFAT has prevented the activation of inflammatory cytokines (Kiani et al., 2000; Zetterqvist et al., 2014; Bretz et al., 2015; Huang et al., 2016), enhanced eNOS expression (Smith et al., 2011), increased NO bioavailability (Friedman et al., 2014; Zetterqvist et al., 2014, 2015; Garcia-Vaz et al., 2020), prevented VSM proliferation (Lipskaia et al., 2003; Berglund et al., 2012; Shiny et al., 2016), lowered BP (Garcia-Vaz et al., 2020), and reduced total atherosclerotic load (Norata et al., 2007; Nilsson-Berglund et al., 2010; Zetterqvist et al., 2014). Currently, these investigations/findings are limited to cellular and animal models. There are promising approaches, including examining NFATs in the skin microcirculation and in peripheral blood mononuclear cells, for investigating the role of NFATs in humans (Huang et al., 2016). Elucidation of the role of NFATs and their putative upstream contributions to the vascular health triad in humans is still needed.
Hydrogen Sulfide
As one of the three gasotransmitters ubiquitously synthesized in mammalian systems, hydrogen sulfide (H2S) is emerging as a critical component of vascular homeostasis (Polhemus and Lefer, 2014). HTN-associated microvascular dysfunction is characterized by a loss of endothelium-dependent signaling pathways, including hydrogen sulfide (H2S) (Cupisti et al., 2000; Lindstedt et al., 2006; Smith et al., 2011; Craighead et al., 2017; Greaney et al., 2017). Similar to NO, H2S exerts several beneficial physiological effects, including inhibiting inflammatory markers and leukocyte adhesion molecules, enhancing anti-inflammatory markers, and acting as an antioxidant (Polhemus and Lefer, 2014). NO and H2S vasodilatory pathways are synergistically interdependent. Both exogenous and endogenous enzymatic H2S synthesis helps to maintain NO bioavailability by stabilizing the eNOS dimer and improving tetrahydrobiopterin (BH4) bioavailability (Zhao et al., 2001; Coletta et al., 2012; Altaany et al., 2013; Polhemus and Lefer, 2014; Greaney et al., 2017). Exogenous administration of H2S improves NO-dependent vasodilation, while blockade of the H2S producing enzyme, cystahionine y-layase (CSE), impairs NO-dependent vasodilation (Coletta et al., 2012; Altaany et al., 2013). Reciprocally, blockade of NO synthesis also reduces H2S-dependent vasodilation (Zhao et al., 2001; Coletta et al., 2012). We reported that the H2S-dependent contribution to endothelium-dependent vasodilation is functionally absent in naïve-to-therapy hypertensive adults (Greaney et al., 2017). This was partially due to reduced endogenous enzymatic synthesis of H2S, as expression and activity of H2S producing enzymes, including cystathione-γ-lyase and 3-mercaptopyruvate transulferase, were reduced in hypertensive compared to normotensive adults (Greaney et al., 2017). However, vascular responsiveness to exogenous H2S donors remained intact in hypertensive adults. In preclinical models, treatment with an H2S donating antihypertensive agents improves endothelial function and normalizes BP (Bucci et al., 2014; Ji et al., 2014; Al-Magableh et al., 2015; Xue et al., 2015). These improvements were partially mediated by increased NO bioavailability (Bucci et al., 2014; Ji et al., 2014; Al-Magableh et al., 2015; Xue et al., 2015), demonstrating the synergistic nature of these gasotransmitter pathways. Various nutraceutical intervention studies suggest that allicin, the bioactive component of garlic, improves vascular function, specifically through the H2S enzymatic pathway (Cui et al., 2020). Due to the ubiquitous nature of H2S, there are several clinical trials evaluating the impact of H2S donating pharmacologics in a variety of disease states. At present, the H2S synthetic pathway remains an underexplored therapeutic target in human HTN and other CVDs, including heart failure.
Potential Targets
In humans, the potential targets to mitigate the vascular health triad and improve the vascular function have been pursued from a global, holistic approach of exercise training to more specific, targeted treatments. In humans, ascorbic acid and folic acid supplementation have been shown to improve vascular function in populations with inflammatory diseases (Alexander et al., 2013; Karbach et al., 2014; Stanhewicz et al., 2015; Stanhewicz and Kenney, 2017). BH4 precursors and antioxidants are the main therapeutic targets facilitating eNOS coupling and reducing eNOS-derived ROS production. BH4 in the form of saproterin is an orphan drug used in the treatment of certain genetic variants of phenylketonuria. Saproterin has improved eNOS function and NO-dependent vasodilation in aged and hypercholesterolemic human subjects in both acute and interventional studies. Specifically, saproterin (or, BH4) supplementation works through eNOS coupling mechanisms and not simply through its moderate antioxidant capacity. Folic acid (and its active metabolite 5-MTHF) is a cost-effective strategy for improving BH4 bioavailability through BH2 recycling. Thus, reducing eNOS uncoupling is an attractive, accessible, and affordable intervention for improving the vascular health triad in aging and diseased states.
Similarly, mitochondrial ROS targeted interventions have shown promise in the past decade. Nicotinamide riboside (NR) is a sirtuin-1 (SIRT-1) precursor that has gained recent popularity in vascular intervention treatment research. NR is a precursor to NAD+, and SIRT1 is a NAD+-dependent deacetylase. Similar to resveratrol and Mito Q, NR supplementation has had vastly positive impacts on vascular function and oxidative stress in pre-clinical animal models (Yoshino et al., 2011; Mills et al., 2016). NR has been shown to improve vascular function and reduce oxidative stress in aged mice. However, in humans, these findings have not been replicated. NR has shown small impacts on various measures of vascular function (Martens et al., 2018). Thus, there is a knowledge gap in the positive benefits of long-term NR supplementation.
Conclusion
In summary, there is ample evidence that the shift from healthy endothelial function to dysfunction, typically preceding HTN and CVD, is driven by a cross-talk between inflammation and ROS. There are numerous “players” that have been recently identified to be responsible for this abnormal shift toward dysfunction. Therefore, it is crucial to investigate these key players in human experimental models to further understand and improve human vascular function with the ultimate goal of preventing CVD risk.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
SR and LA conceived the research review. SR, GD, SM, and LA designed the research review, drafted, revised, and approved the final version of the manuscript. GD and SM prepared figure and table. All authors contributed to the article and approved the submitted version.
Funding
Support for this work was provided by National Institute of Health (NIH) grant T32-5T32-AG049676 to GD, NIH grant R01-HL-093238 to LA, and University of Maryland (UMD) Tier 1 (pilot grant) to SR.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Aktan F. (2004). iNOS-mediated nitric oxide production and its regulation.Life Sci.75639–653. 10.1016/j.lfs.2003.10.042
2
Alexander L. M. Kutz J. L. Kenney W. L. (2013). Tetrahydrobiopterin increases NO-dependent vasodilation in hypercholesterolemic human skin through eNOS-coupling mechanisms.Am. J. Physiol. Regul. Integr. Comp. Physiol.304R164–R169. 10.1152/ajpregu.00448.2012
3
Al-Magableh M. R. Kemp-Harper B. K. Hart J. L. (2015). Hydrogen sulfide treatment reduces blood pressure and oxidative stress in angiotensin II-induced hypertensive mice.Hypertens. Res.3813–20. 10.1038/hr.2014.125
4
Altaany Z. Yang G. Wang R. (2013). Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells.J. Cell. Mol. Med.17879–888. 10.1111/jcmm.12077
5
Amberg G. C. Rossow C. F. Navedo M. F. Santana L. F. (2004). NFATc3 regulates Kv2.1 expression in arterial smooth muscle.J. Biol. Chem.27947326–47334. 10.1074/jbc.M408789200
6
Armesilla A. L. Lorenzo E. Gomez del Arco P. Martinez-Martinez S. Alfranca A. Redondo J. M. (1999). Vascular endothelial growth factor activates nuclear factor of activated T cells in human endothelial cells: a role for tissue factor gene expression.Mol. Cell Biol.192032–2043. 10.1128/MCB.19.3.2032
7
Badi I. Mancinelli L. Polizzotto A. Ferri D. Zeni F. Burba I. et al (2018). miR-34a Promotes Vascular Smooth Muscle Cell Calcification by Downregulating SIRT1 (Sirtuin 1) and Axl (AXL Receptor Tyrosine Kinase).Arterioscler. Thromb. Vascul. Biol.382079–2090. 10.1161/ATVBAHA.118.311298
8
Baker J. R. Vuppusetty C. Colley T. Hassibi S. Fenwick P. S. Donnelly L. E. et al (2019). MicroRNA-570 is a novel regulator of cellular senescence and inflammaging.FASEB J.331605–1616. 10.1096/fj.201800965R
9
Bär C. Thum T. de Gonzalo-Calvo D. (2019). Circulating miRNAs as mediators in cell-to-cell communication.Epigenomics11111–113. 10.2217/epi-2018-0183
10
Benjamin E. J. Larson M. G. Keyes M. J. Mitchell G. F. Vasan R. S. Keaney J. F. Jr. et al (2004). Clinical correlates and heritability of flow-mediated dilation in the community: the Framingham Heart Study.Circulation109613–619. 10.1161/01.CIR.0000112565.60887.1E
11
Benz F. Roy S. Trautwein C. Roderburg C. Luedde T. (2016). Circulating MicroRNAs as Biomarkers for Sepsis.Int. J. Mol. Sci.17:78. 10.3390/ijms17010078
12
Berglund L. M. Kotova O. Osmark P. Grufman H. Xing C. Lydrup M. L. et al (2012). NFAT regulates the expression of AIF-1 and IRT-1: yin and yang splice variants of neointima formation and atherosclerosis.Cardiovasc. Res.93414–423. 10.1093/cvr/cvr309
13
Beyer A. M. Durand M. J. Hockenberry J. Gamblin T. C. Phillips S. A. Gutterman D. D. (2014). An acute rise in intraluminal pressure shifts the mediator of flow-mediated dilation from nitric oxide to hydrogen peroxide in human arterioles.Am. J. Physiol. Heart Circ. Physiol.307H1587–H1593. 10.1152/ajpheart.00557.2014
14
Beyer A. M. Zinkevich N. Miller B. Liu Y. Wittenburg A. L. Mitchell M. et al (2017). Transition in the mechanism of flow-mediated dilation with aging and development of coronary artery disease.Basic Res. Cardiol.112:5. 10.1007/s00395-016-0594-x
15
Bochkov V. N. Mechtcheriakova D. Lucerna M. Huber J. Malli R. Graier W. F. et al (2002). Oxidized phospholipids stimulate tissue factor expression in human endothelial cells via activation of ERK/EGR-1 and Ca(++)/NFAT.Blood99199–206. 10.1182/blood.V99.1.199
16
Boss V. Wang X. Koppelman L. F. Xu K. Murphy T. J. (1998). Histamine induces nuclear factor of activated T cell-mediated transcription and cyclosporin A-sensitive interleukin-8 mRNA expression in human umbilical vein endothelial cells.Mol. Pharmacol.54264–272. 10.1124/mol.54.2.264
17
Bottino D. A. Lopes F. G. de Oliveira F. J. Mecenas A. S. Clapauch R. Bouskela E. (2015). Relationship between biomarkers of inflammation, oxidative stress and endothelial/microcirculatory function in successful aging versus healthy youth: a transversal study.BMC Geriatr.15:41. 10.1186/s12877-015-0044-x
18
Bretz C. A. Savage S. R. Capozzi M. E. Suarez S. Penn J. S. (2015). NFAT isoforms play distinct roles in TNFalpha-induced retinal leukostasis.Sci. Rep.5:14963. 10.1038/srep14963
19
Broxterman R. M. Witman M. A. Trinity J. D. Groot H. J. Rossman M. J. Park S. Y. et al (2019). Strong Relationship Between Vascular Function in the Coronary and Brachial Arteries.Hypertension74208–215. 10.1161/HYPERTENSIONAHA.119.12881
20
Bruning R. S. Kenney W. L. Alexander L. M. (2015). Altered skin flowmotion in hypertensive humans.Microvascular. Res.9781–87. 10.1016/j.mvr.2014.01.001
21
Bucci M. Vellecco V. Cantalupo A. Brancaleone V. Zhou Z. Evangelista S. et al (2014). Hydrogen sulfide accounts for the peripheral vascular effects of zofenopril independently of ACE inhibition.Cardiovasc. Res.102138–147. 10.1093/cvr/cvu026
22
Buus N. H. Jørgensen C. G. Mulvany M. J. Sørensen K. E. (2007). Large and small artery endothelial function in patients with essential hypertension–effect of ACE inhibition and beta-blockade.Blood Press.16106–113. 10.1080/08037050701343688
23
Cheng H. S. Njock M. S. Khyzha N. Dang L. T. Fish J. E. (2014). Noncoding RNAs regulate NF-κB signaling to modulate blood vessel inflammation.Front. Genet.5:422. 10.3389/fgene.2014.00422
24
Cockerill G. W. Bert A. G. Ryan G. R. Gamble J. R. Vadas M. A. Cockerill P. N. (1995). Regulation of granulocyte-macrophage colony-stimulating factor and E-selectin expression in endothelial cells by cyclosporin A and the T-cell transcription factor NFAT.Blood862689–2698. 10.1182/blood.V86.7.2689.2689
25
Coletta C. Papapetropoulos A. Erdelyi K. Olah G. Modis K. Panopoulos P. et al (2012). Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation.Proc. Natl. Acad. Sci. U S A.1099161–9166. 10.1073/pnas.1202916109
26
Craighead D. H. Freeberg K. A. Seals D. R. (2020). Vascular Endothelial Function in Midlife/Older Adults Classified According to 2017 American College of Cardiology/American Heart Association Blood Pressure Guidelines.J. Am. Heart Associat.9:e016625. 10.1161/JAHA.120.016625
27
Craighead D. H. Smith C. J. Alexander L. M. (2017). Blood pressure normalization via pharmacotherapy improves cutaneous microvascular function through NO-dependent and NO-independent mechanisms.Microcirculation24:12382. 10.1111/micc.12382
28
Craighead D. H. Wang H. Santhanam L. Alexander L. M. (2018). Acute lysyl oxidase inhibition alters microvascular function in normotensive but not hypertensive men and women.Am. J. Physiol. Heart Circ. Physiol.314H424–H433. 10.1152/ajpheart.00521.2017
29
Crecelius A. R. Kirby B. S. Voyles W. F. Dinenno F. A. (2010). Nitric oxide, but not vasodilating prostaglandins, contributes to the improvement of exercise hyperemia via ascorbic acid in healthy older adults.Am. J. Physiol. Heart Circ. Physiol.299H1633–H1641. 10.1152/ajpheart.00614.2010
30
Cui T. Liu W. Chen S. Yu C. Li Y. Zhang J. Y. (2020). Antihypertensive effects of allicin on spontaneously hypertensive rats via vasorelaxation and hydrogen sulfide mechanisms.Biomed. Pharmacother.128:110240. 10.1016/j.biopha.2020.110240
31
Cupisti A. Rossi M. Placidi S. Fabbri A. Morelli E. Vagheggini G. et al (2000). Responses of the skin microcirculation to acetylcholine in patients with essential hypertension and in normotensive patients with chronic renal failure.Nephron85114–119. 10.1159/000045643
32
Dillon G. A. Greaney J. L. Shank S. Leuenberger U. A. Alexander L. M. (2020). AHA/ACC-defined stage 1 hypertensive adults do not display cutaneous microvascular endothelial dysfunction.Am. J. Physiol. Heart Circ. Physiol.319H539–H546. 10.1152/ajpheart.00179.2020
33
Ding H. Aljofan M. Triggle C. R. (2007). Oxidative stress and increased eNOS and NADPH oxidase expression in mouse microvessel endothelial cells.J. Cell Physiol.212682–689. 10.1002/jcp.21063
34
Donato A. J. Eskurza I. Silver A. E. Levy A. S. Pierce G. L. Gates P. E. et al (2007). Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-kappaB.Circ. Res.1001659–1666. 10.1161/01.RES.0000269183.13937.e8
35
Donato A. J. Pierce G. L. Lesniewski L. A. Seals D. R. (2009). Role of NFkappaB in age-related vascular endothelial dysfunction in humans.Aging1678–680. 10.18632/aging.100080
36
El Assar M. Angulo J. Rodriguez-Manas L. (2013). Oxidative stress and vascular inflammation in aging.Free Radic. Biol. Med.65380–401. 10.1016/j.freeradbiomed.2013.07.003
37
Eskurza I. Monahan K. D. Robinson J. A. Seals D. R. (2004a). Ascorbic acid does not affect large elastic artery compliance or central blood pressure in young and older men.Am. J. Physiol. Heart Circ. Physiol.286H1528–H1534. 10.1152/ajpheart.00879.2003
38
Eskurza I. Monahan K. D. Robinson J. A. Seals D. R. (2004b). Effect of acute and chronic ascorbic acid on flow-mediated dilatation with sedentary and physically active human ageing.J. Physiol.556(Pt 1), 315–324. 10.1113/jphysiol.2003.057042
39
Farkas K. Kolossváry E. Járai Z. Nemcsik J. Farsang C. (2004). Non-invasive assessment of microvascular endothelial function by laser Doppler flowmetry in patients with essential hypertension.Atherosclerosis17397–102. 10.1016/j.atherosclerosis.2003.11.015
40
Fernández-Hernando C. Suárez Y. (2018). MicroRNAs in endothelial cell homeostasis and vascular disease.Curr. Opin. Hematol.25227–236. 10.1097/MOH.0000000000000424
41
Figueiredo V. N. Yugar-Toledo J. C. Martins L. C. Martins L. B. de Faria A. P. de Haro Moraes C. et al (2012). Vascular stiffness and endothelial dysfunction: Correlations at different levels of blood pressure.Blood Press.2131–38. 10.3109/08037051.2011.617045
42
Fonkoue I. T. Le N. A. Kankam M. L. DaCosta D. Jones T. N. Marvar P. J. et al (2019). Sympathoexcitation and impaired arterial baroreflex sensitivity are linked to vascular inflammation in individuals with elevated resting blood pressure.Physiol. Rep.7:e14057. 10.14814/phy2.14057
43
Friedman J. K. Nitta C. H. Henderson K. M. Codianni S. J. Sanchez L. Ramiro-Diaz J. M. et al (2014). Intermittent hypoxia-induced increases in reactive oxygen species activate NFATc3 increasing endothelin-1 vasoconstrictor reactivity.Vascul. Pharmacol.6017–24. 10.1016/j.vph.2013.11.001
44
Garcia-Vaz E. McNeilly A. D. Berglund L. M. Ahmad A. Gallagher J. R. Dutius Andersson A. M. et al (2020). Inhibition of NFAT Signaling Restores Microvascular Endothelial Function in Diabetic Mice.Diabetes69424–435. 10.2337/db18-0870
45
Gokce N. Holbrook M. Duffy S. J. Demissie S. Cupples L. A. Biegelsen E. et al (2001). Effects of race and hypertension on flow-mediated and nitroglycerin-mediated dilation of the brachial artery.Hypertension381349–1354. 10.1161/hy1201.096575
46
Gonzalez Bosc L. V. Wilkerson M. K. Bradley K. N. Eckman D. M. Hill-Eubanks D. C. Nelson M. T. (2004). Intraluminal pressure is a stimulus for NFATc3 nuclear accumulation: role of calcium, endothelium-derived nitric oxide, and cGMP-dependent protein kinase.J. Biol. Chem.27910702–10709. 10.1074/jbc.M312920200
47
Govatati S. Pichavaram P. Janjanam J. Zhang B. Singh N. K. Mani A. M. et al (2019). NFATc1-E2F1-LMCD1-Mediated IL-33 Expression by Thrombin Is Required for Injury-Induced Neointima Formation.Arterioscler. Thromb. Vascul. Biol.391212–1226. 10.1161/ATVBAHA.119.312729
48
Grassi G. Biffi A. Seravalle G. Trevano F. Q. Dell’Oro R. Corrao G. et al (2019). Sympathetic Neural Overdrive in the Obese and Overweight State.Hypertension74349–358. 10.1161/HYPERTENSIONAHA.119.12885
49
Grassi G. Pisano A. Bolignano D. Seravalle G. D’Arrigo G. Quarti-Trevano F. et al (2018). Sympathetic Nerve Traffic Activation in Essential Hypertension and Its Correlates: Systematic Reviews and Meta-Analyses.Hypertension72483–491. 10.1161/HYPERTENSIONAHA.118.11038
50
Greaney J. L. Kutz J. L. Shank S. W. Jandu S. Santhanam L. Alexander L. M. (2017). Impaired Hydrogen Sulfide-Mediated Vasodilation Contributes to Microvascular Endothelial Dysfunction in Hypertensive Adults.Hypertension69902–909. 10.1161/HYPERTENSIONAHA.116.08964
51
Greaney J. L. Matthews E. L. Wenner M. M. (2015). Sympathetic reactivity in young women with a family history of hypertension.Am. J. Physiol. Heart Circ. Physiol.308H816–H822. 10.1152/ajpheart.00867.2014
52
Greaney J. L. Saunders E. F. H. Santhanam L. Alexander L. M. (2019). Oxidative Stress Contributes to Microvascular Endothelial Dysfunction in Men and Women With Major Depressive Disorder.Circ. Res.124564–574. 10.1161/CIRCRESAHA.118.313764
53
Guzik T. J. Touyz R. M. (2017). Oxidative Stress, Inflammation, and Vascular Aging in Hypertension.Hypertension70660–667. 10.1161/HYPERTENSIONAHA.117.07802
54
Hader S. N. Zinkevich N. Norwood Toro L. E. Kriegel A. J. Kong A. Freed J. K. et al (2019). Detrimental effects of chemotherapy on human coronary microvascular function.Am. J. Physiol. Heart Circ. Physiol.317H705–H710. 10.1152/ajpheart.00370.2019
55
Hemmrich K. Suschek C. V. Lerzynski G. Kolb-Bachofen V. (2003). iNOS activity is essential for endothelial stress gene expression protecting against oxidative damage.J. Appl. Physiol.951937–1946. 10.1152/japplphysiol.00419.2003
56
Hingorani A. D. Cross J. Kharbanda R. K. Mullen M. J. Bhagat K. Taylor M. et al (2000). Acute systemic inflammation impairs endothelium-dependent dilatation in humans.Circulation102994–999. 10.1161/01.CIR.102.9.994
57
Holwerda S. W. Luehrs R. E. DuBose L. Collins M. T. Wooldridge N. A. Stroud A. K. et al (2019). Elevated Muscle Sympathetic Nerve Activity Contributes to Central Artery Stiffness in Young and Middle-Age/Older Adults.Hypertension731025–1035. 10.1161/HYPERTENSIONAHA.118.12462
58
Huang S.-S. He S.-L. Zhang Y.-M. (2016). The effects of telmisartan on the nuclear factor of activated T lymphocytes signalling pathway in hypertensive patients.J. Renin Angiotensin Aldosterone Syst.17:1470320316655005. 10.1177/1470320316655005
59
Hughes W. E. Zinkevich N. Gutterman D. D. Beyer A. M. (2021b). Hypertension preserves the magnitude of microvascular flow-mediated dilation following transient elevation in intraluminal pressure.Physiol. Rep.9:e14507. 10.14814/phy2.14507
60
Hughes W. E. Chabowski D. S. Ait-Aissa K. Fetterman J. L. Hockenberry J. Beyer A. M. et al (2021a). Critical Interaction Between Telomerase and Autophagy in Mediating Flow-Induced Human Arteriolar Vasodilation.Arterioscler. Thromb. Vascul. Biol.41446–457. 10.1161/ATVBAHA.120.314944
61
Hurr C. Patik J. C. Kim K. Christmas K. M. Brothers R. M. (2018). Tempol augments the blunted cutaneous microvascular thermal reactivity in healthy young African Americans.Exp. Physiol.103343–349. 10.1113/EP086776
62
Jacobi J. Kristal B. Chezar J. Shaul S. M. Sela S. (2005). Exogenous superoxide mediates pro-oxidative, proinflammatory, and procoagulatory changes in primary endothelial cell cultures.Free Radic. Biol. Med.391238–1248. 10.1016/j.freeradbiomed.2005.06.010
63
Ji H. Kim A. Ebinger J. E. Niiranen T. J. Claggett B. L. Bairey Merz C. N. et al (2020). Sex Differences in Blood Pressure Trajectories Over the Life Course.JAMA Cardiol.519–26. 10.1001/jamacardio.2019.5306
64
Ji W. Liu S. Dai J. Yang T. Jiang X. Duan X. et al (2014). Hydrogen sulfide defends against the cardiovascular risk of Nw-nitro-L-argininemethyl ester-induced hypertension in rats via the nitric oxide/endothelial nitric oxide synthase pathway.Chin. Med. J.1273751–3757.
65
Johnson E. N. Lee Y. M. Sander T. L. Rabkin E. Schoen F. J. Kaushal S. et al (2003). NFATc1 mediates vascular endothelial growth factor-induced proliferation of human pulmonary valve endothelial cells.J. Biol. Chem.2781686–1692. 10.1074/jbc.M210250200
66
Junqueira C. L. C. Magalhães M. E. C. Brandão A. A. Ferreira E. Junqueira A. S. M. Neto J. F. N. et al (2018). Evaluation of endothelial function by VOP and inflammatory biomarkers in patients with arterial hypertension.J. Hum. Hypertens.32105–113. 10.1038/s41371-017-0024-z
67
Juonala M. Viikari J. S. Laitinen T. Marniemi J. Helenius H. Rönnemaa T. et al (2004). Interrelations between brachial endothelial function and carotid intima-media thickness in young adults: the cardiovascular risk in young Finns study.Circulation1102918–2923. 10.1161/01.CIR.0000147540.88559.00
68
Kadlec A. O. Chabowski D. S. Ait-Aissa K. Hockenberry J. C. Otterson M. F. Durand M. J. et al (2017). PGC-1alpha (Peroxisome Proliferator-Activated Receptor gamma Coactivator 1-alpha) Overexpression in Coronary Artery Disease Recruits NO and Hydrogen Peroxide During Flow-Mediated Dilation and Protects Against Increased Intraluminal Pressure.Hypertension70166–173. 10.1161/HYPERTENSIONAHA.117.09289
69
Karbach S. Wenzel P. Waisman A. Munzel T. Daiber A. (2014). eNOS uncoupling in cardiovascular diseases–the role of oxidative stress and inflammation.Curr. Pharm. Des.203579–3594. 10.2174/13816128113196660748
70
Katulka E. K. Hirt A. E. Kirkman D. L. Edwards D. G. Witman M. A. H. (2019). Altered vascular function in chronic kidney disease: evidence from passive leg movement.Physiol. Rep.7:e14075. 10.14814/phy2.14075
71
Keir D. A. Badrov M. B. Tomlinson G. Notarius C. F. Kimmerly D. S. Millar P. J. et al (2020). Influence of Sex and Age on Muscle Sympathetic Nerve Activity of Healthy Normotensive Adults.Hypertension76997–1005. 10.1161/HYPERTENSIONAHA.120.15208
72
Kiani A. Rao A. Aramburu J. (2000). Manipulating immune responses with immunosuppressive agents that target NFAT.Immunity12359–372. 10.1016/S1074-7613(00)80188-0
73
Kirkman D. L. Robinson A. T. Rossman M. J. Seals D. R. Edwards D. G. (2021). Mitochondrial contributions to vascular endothelial dysfunction, arterial stiffness, and cardiovascular diseases.Am. J. Physiol. Heart Circ. Physiol.320H2080–H2100. 10.1152/ajpheart.00917.2020
74
Kopp E. Ghosh S. (1994). Inhibition of NF-kappa B by sodium salicylate and aspirin.Science265956–959. 10.1126/science.8052854
75
Lee K. S. Kim J. Kwak S. N. Lee K. S. Lee D. K. Ha K. S. et al (2014). Functional role of NF-κB in expression of human endothelial nitric oxide synthase.Biochem. Biophys. Res. Communicat.448101–107. 10.1016/j.bbrc.2014.04.079
76
Lewington S. Clarke R. Qizilbash N. Peto R. Collins R. (2002). Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies.Lancet3601903–1913. 10.1016/S0140-6736(02)11911-8
77
Li L. Sapkota M. Kim S. W. Soh Y. (2015). Herbacetin inhibits inducible nitric oxide synthase via JNK and nuclear factor-κB in LPS-stimulated RAW264.7 cells.Eur. J. Pharmacol.765115–123. 10.1016/j.ejphar.2015.08.032
78
Lindstedt I. H. Edvinsson M. L. Edvinsson L. (2006). Reduced responsiveness of cutaneous microcirculation in essential hypertension–a pilot study.Blood Press.15275–280. 10.1080/08037050600996586
79
Lipskaia L. Pourci M. L. Delomenie C. Combettes L. Goudouneche D. Paul J. L. et al (2003). Phosphatidylinositol 3-kinase and calcium-activated transcription pathways are required for VLDL-induced smooth muscle cell proliferation.Circ. Res.921115–1122. 10.1161/01.RES.0000074880.25540.D0
80
Marin T. Gongol B. Chen Z. Woo B. Subramaniam S. Chien S. et al (2013). Mechanosensitive microRNAs-role in endothelial responses to shear stress and redox state.Free Radic. Biol. Med.6461–68. 10.1016/j.freeradbiomed.2013.05.034
81
Martens C. R. Denman B. A. Mazzo M. R. Armstrong M. L. Reisdorph N. McQueen M. B. et al (2018). Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults.Nat. Commun.9:1286. 10.1038/s41467-018-03421-7
82
Martens C. R. Kirkman D. L. Edwards D. G. (2016). The Vascular Endothelium in Chronic Kidney Disease: A Novel Target for Aerobic Exercise.Exerc. Sport Sci. Rev.4412–19. 10.1249/JES.0000000000000065
83
Matthews E. L. Greaney J. L. Wenner M. M. (2017). Rapid onset pressor response to exercise in young women with a family history of hypertension.Exp. Physiol.1021092–1099. 10.1113/EP086466
84
McGowan C. L. Levy A. S. Millar P. J. Guzman J. C. Morillo C. A. McCartney N. et al (2006). Acute vascular responses to isometric handgrip exercise and effects of training in persons medicated for hypertension.Am. J. Physiol. Heart Circ. Physiol.291H1797–H1802. 10.1152/ajpheart.01113.2005
85
Mi S. Zhang J. Zhang W. Huang R. S. (2013). Circulating microRNAs as biomarkers for inflammatory diseases.Microrna263–71. 10.2174/2211536611302010007
86
Migrino R. Q. Truran S. Gutterman D. D. Franco D. A. Bright M. Schlundt B. et al (2011). Human microvascular dysfunction and apoptotic injury induced by AL amyloidosis light chain proteins.Am. J. Physiol. Heart Circ. Physiol.301H2305–H2312. 10.1152/ajpheart.00503.2011
87
Mills K. F. Yoshida S. Stein L. R. Grozio A. Kubota S. Sasaki Y. et al (2016). Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice.Cell Metab.24795–806. 10.1016/j.cmet.2016.09.013
88
Nieves-Cintrón M. Amberg G. C. Nichols C. B. Molkentin J. D. Santana L. F. (2007). Activation of NFATc3 down-regulates the beta1 subunit of large conductance, calcium-activated K+ channels in arterial smooth muscle and contributes to hypertension.J. Biol. Chem.2823231–3240. 10.1074/jbc.M608822200
89
Nilsson J. Nilsson L. M. Chen Y. W. Molkentin J. D. Erlinge D. Gomez M. F. (2006). High glucose activates nuclear factor of activated T cells in native vascular smooth muscle.Arterioscler. Thromb. Vascul. Biol.26794–800. 10.1161/01.ATV.0000209513.00765.13
90
Nilsson-Berglund L. M. Zetterqvist A. V. Nilsson-Ohman J. Sigvardsson M. Gonzalez Bosc L. V. Smith M. L. et al (2010). Nuclear factor of activated T cells regulates osteopontin expression in arterial smooth muscle in response to diabetes-induced hyperglycemia.Arterioscler. Thromb. Vascul. Biol.30218–224. 10.1161/ATVBAHA.109.199299
91
Njock M. S. Fish J. E. (2017). Endothelial miRNAs as Cellular Messengers in Cardiometabolic Diseases.Trends Endocrinol. Metab.28237–246. 10.1016/j.tem.2016.11.009
92
Norata G. D. Grigore L. Raselli S. Redaelli L. Hamsten A. Maggi F. et al (2007). Post-prandial endothelial dysfunction in hypertriglyceridemic subjects: molecular mechanisms and gene expression studies.Atherosclerosis193321–327. 10.1016/j.atherosclerosis.2006.09.015
93
Nowroozpoor A. Gutterman D. Safdar B. (2021). Is microvascular dysfunction a systemic disorder with common biomarkers found in the heart, brain, and kidneys? - A scoping review.Microvasc. Res.134:104123. 10.1016/j.mvr.2020.104123
94
On Y. K. Kim C. H. Sohn D. W. Oh B. H. Lee M. M. Park Y. B. et al (2002). Improvement of endothelial function by amlodipine and vitamin C in essential hypertension.Korean J. Intern. Med.17131–137. 10.3904/kjim.2002.17.2.131
95
Orr A. W. Lee M. Y. Lemmon J. A. Yurdagul A. Gomez M. F. Bortz P. D. S. et al (2009). Molecular Mechanisms of Collagen Isotype-Specific Modulation of Smooth Muscle Cell Phenotype.Arterioscl. Throm. Vas.29225–231. 10.1161/ATVBAHA.108.178749
96
Ozemek C. Hildreth K. L. Groves D. W. Moreau K. L. (2016). Acute ascorbic acid infusion increases left ventricular diastolic function in postmenopausal women.Maturitas.92154–161. 10.1016/j.maturitas.2016.08.007
97
Pang X. Sun N. L. (2009). Calcineurin-NFAT signaling is involved in phenylephrine-induced vascular smooth muscle cell proliferation.Acta Pharmacol. Sin.30537–544. 10.1038/aps.2009.28
98
Park S. Y. Kwon O. S. Andtbacka R. H. I. Hyngstrom J. R. Reese V. Murphy M. P. et al (2018). Age-related endothelial dysfunction in human skeletal muscle feed arteries: the role of free radicals derived from mitochondria in the vasculature.Acta physiol.222:12893. 10.1111/apha.12893
99
Phillips S. A. Hatoum O. A. Gutterman D. D. (2007). The mechanism of flow-induced dilation in human adipose arterioles involves hydrogen peroxide during CAD.Am. J. Physiol. Heart Circ. Physiol.292H93–H100. 10.1152/ajpheart.00819.2006
100
Pierce G. L. Lesniewski L. A. Lawson B. R. Beske S. D. Seals D. R. (2009). Nuclear factor-{kappa}B activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans.Circulation1191284–1292. 10.1161/CIRCULATIONAHA.108.804294
101
Pierce J. W. Read M. A. Ding H. Luscinskas F. W. Collins T. (1996). Salicylates inhibit I kappa B-alpha phosphorylation, endothelial-leukocyte adhesion molecule expression, and neutrophil transmigration.J. Immunol.1563961–3969.
102
Polhemus D. J. Lefer D. J. (2014). Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease.Circ. Res.114730–737. 10.1161/CIRCRESAHA.114.300505
103
Ratchford S. M. Clifton H. L. Gifford J. R. LaSalle D. T. Thurston T. S. Bunsawat K. et al (2019a). Impact of acute antioxidant administration on inflammation and vascular function in heart failure with preserved ejection fraction.Am. J. Physiol. Regul. Integr. Comp. Physiol.317R607–R614. 10.1152/ajpregu.00184.2019
104
Ratchford S. M. Broxterman R. M. La Salle D. T. Kwon O. S. Park S. Y. Hopkins P. N. et al (2019b). Salt restriction lowers blood pressure at rest and during exercise without altering peripheral hemodynamics in hypertensive individuals.Am. J. Physiol. Heart Circ. Physiol.317H1194–H1202.
105
Richards J. C. Crecelius A. R. Larson D. G. Dinenno F. A. (2015). Acute ascorbic acid ingestion increases skeletal muscle blood flow and oxygen consumption via local vasodilation during graded handgrip exercise in older adults.Am. J. Physiol. Heart Circ. Physiol.309H360–H368.
106
Risbano M. G. Gladwin M. T. (2013). Therapeutics targeting of dysregulated redox equilibrium and endothelial dysfunction.Handb. Exp. Pharmacol.218315–349. 10.1007/978-3-662-45805-1_13
107
Rossman M. J. Santos-Parker J. R. Steward C. A. C. Bispham N. Z. Cuevas L. M. Rosenberg H. L. et al (2018). Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults.Hypertension711056–1063. 10.1161/HYPERTENSIONAHA.117.10787
108
Sapp R. M. Chesney C. A. Springer C. B. Laskowski M. R. Singer D. B. Eagan L. E. et al (2021). Race-specific changes in endothelial inflammation and microRNA in response to an acute inflammatory stimulus.Am. J. Physiol. Heart Circ. Physiol.320H2371–H2384. 10.1152/ajpheart.00991.2020
109
Sapp R. M. Shill D. D. Roth S. M. Hagberg J. M. (2017). Circulating microRNAs in acute and chronic exercise: more than mere biomarkers.J. Appl. Physiol.122702–717. 10.1152/japplphysiol.00982.2016
110
Schmitz B. Breulmann F. L. Jubran B. Rolfes F. Thorwesten L. Kruger M. et al (2019). A three-step approach identifies novel shear stress-sensitive endothelial microRNAs involved in vasculoprotective effects of high-intensity interval training (HIIT).Oncotarget103625–3640. 10.18632/oncotarget.26944
111
Selemidis S. Dusting G. J. Peshavariya H. Kemp-Harper B. K. Drummond G. R. (2007). Nitric oxide suppresses NADPH oxidase-dependent superoxide production by S-nitrosylation in human endothelial cells.Cardiovasc. Res.75349–358. 10.1016/j.cardiores.2007.03.030
112
Shi L. Liao J. Liu B. Zeng F. Zhang L. (2015). Mechanisms and therapeutic potential of microRNAs in hypertension.Drug Discov. Today201188–1204. 10.1016/j.drudis.2015.05.007
113
Shiny A. Regin B. Mohan V. Balasubramanyam M. (2016). Coordinated augmentation of NFAT and NOD signaling mediates proliferative VSMC phenotype switch under hyperinsulinemia.Atherosclerosis246257–266. 10.1016/j.atherosclerosis.2016.01.006
114
Smith C. J. Santhanam L. Alexander L. M. (2013). Rho-Kinase activity and cutaneous vasoconstriction is upregulated in essential hypertensive humans.Microvascul. Res.8758–64. 10.1016/j.mvr.2013.02.005
115
Smith C. J. Santhanam L. Bruning R. S. Stanhewicz A. Berkowitz D. E. Holowatz L. A. (2011). Upregulation of inducible nitric oxide synthase contributes to attenuated cutaneous vasodilation in essential hypertensive humans.Hypertension58935–942. 10.1161/HYPERTENSIONAHA.111.178129
116
Soudani N. Ghantous C. M. Farhat Z. Shebaby W. N. Zibara K. Zeidan A. (2016). Calcineurin/NFAT Activation-Dependence of Leptin Synthesis and Vascular Growth in Response to Mechanical Stretch.Front. Physiol.7:433. 10.3389/fphys.2016.00433
117
Sprick J. D. Downey R. M. Morison D. L. Fonkoue I. T. Li Y. DaCosta D. et al (2019). Functional sympatholysis is impaired in end-stage renal disease.Am. J. Physiol. Regul. Integr. Comp. Physiol.316R504–R511. 10.1152/ajpregu.00380.2018
118
Stanhewicz A. E. Alexander L. M. Kenney W. L. (2015). Folic acid supplementation improves microvascular function in older adults through nitric oxide-dependent mechanisms.Clin. Sci.129159–167. 10.1042/CS20140821
119
Stanhewicz A. E. Kenney W. L. (2017). Role of folic acid in nitric oxide bioavailability and vascular endothelial function.Nutrit. Rev.7561–70. 10.1093/nutrit/nuw053
120
Suzuki E. Nishimatsu H. Satonaka H. Walsh K. Goto A. Omata M. et al (2002). Angiotensin II induces myocyte enhancer factor 2- and calcineurin/nuclear factor of activated T cell-dependent transcriptional activation in vascular myocytes.Circ. Res.901004–1011. 10.1161/01.RES.0000017629.70769.CC
121
Takase B. Hamabe A. Satomura K. Akima T. Uehata A. Matsui T. et al (2006). Comparable prognostic value of vasodilator response to acetylcholine in brachial and coronary arteries for predicting long-term cardiovascular events in suspected coronary artery disease.Circ. J.7049–56. 10.1253/circj.70.49
122
Trinity J. D. Wray D. W. Witman M. A. Layec G. Barrett-O’Keefe Z. Ives S. J. et al (2016). Ascorbic acid improves brachial artery vasodilation during progressive handgrip exercise in the elderly through a nitric oxide-mediated mechanism.Am. J. Physiol. Heart Circ. Physiol.310H765–H774. 10.1152/ajpheart.00817.2015
123
Virani S. S. Alonso A. Aparicio H. J. Benjamin E. J. Bittencourt M. S. Callaway C. W. et al (2021). Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association.Circulation143e254–e743. 10.1161/CIR.0000000000000950
124
Wadley A. J. Veldhuijzen van Zanten J. J. Aldred S. (2013). The interactions of oxidative stress and inflammation with vascular dysfunction in ageing: the vascular health triad.Age35705–718. 10.1007/s11357-012-9402-1
125
Walker A. E. Kaplon R. E. Pierce G. L. Nowlan M. J. Seals D. R. (2014). Prevention of age-related endothelial dysfunction by habitual aerobic exercise in healthy humans: possible role of nuclear factor κB.Clin. Sci.127645–654. 10.1042/CS20140030
126
Wang Q. Li D. Han Y. Ding X. Xu T. Tang B. (2017). MicroRNA-146 protects A549 and H1975 cells from LPS-induced apoptosis and inflammation injury.J. Biosci.42637–645. 10.1007/s12038-017-9715-4
127
Wang Y. Hu J. Liu J. Geng Z. Tao Y. Zheng F. et al (2020). The role of Ca(2+)/NFAT in Dysfunction and Inflammation of Human Coronary Endothelial Cells induced by Sera from patients with Kawasaki disease.Sci. Rep.10:4706. 10.1038/s41598-020-61667-y
128
Weng J. Wang C. Zhong W. Li B. Wang Z. Shao C. et al (2017). Activation of CD137 Signaling Promotes Angiogenesis in Atherosclerosis via Modulating Endothelial Smad1/5-NFATc1 Pathway.J. Am. Heart Assoc.6:e004756. 10.1161/JAHA.116.004756
129
Whelton P. K. Carey R. M. Aronow W. S. Casey D. E. Jr. Collins K. J. Dennison Himmelfarb C. et al (2018). 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines.J. Am. Coll. Cardiol.71e127–e248. 10.1161/HYP.0000000000000076
130
Wray D. W. Nishiyama S. K. Harris R. A. Zhao J. McDaniel J. Fjeldstad A. S. et al (2012). Acute reversal of endothelial dysfunction in the elderly after antioxidant consumption.Hypertension59818–824. 10.1161/HYPERTENSIONAHA.111.189456
131
Wu F. Tyml K. Wilson J. X. (2008). iNOS expression requires NADPH oxidase-dependent redox signaling in microvascular endothelial cells.J. Cell Physiol.217207–214. 10.1002/jcp.21495
132
Xue H. Zhou S. Xiao L. Guo Q. Liu S. Wu Y. (2015). Hydrogen sulfide improves the endothelial dysfunction in renovascular hypertensive rats.Physiol. Res.64663–672. 10.33549/physiolres.932848
133
Yamakuchi M. (2012). MicroRNA Regulation of SIRT1.Front. Physiol.3:68. 10.3389/fphys.2012.00068
134
Yamakuchi M. Hashiguchi T. (2018). Endothelial Cell Aging: How miRNAs Contribute?J. Clin. Med.7:170. 10.3390/jcm7070170
135
Yan J. Yin Y. Zhong W. Wang C. Wang Z. (2015). CD137 Regulates NFATc1 Expression in Mouse VSMCs through TRAF6/NF-κB p65 Signaling Pathway.Mediat. Inflammat.2015:639780. 10.1155/2015/639780
136
Yang P. Liu Y. F. Yang L. Wei Q. Zeng H. (2010). Mechanism and clinical significance of the prothrombotic state in patients with essential hypertension.Clin. Cardiol.33E81–E86. 10.1002/clc.20719
137
Yoshino J. Mills K. F. Yoon M. J. Imai S. (2011). Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice.Cell Metab.14528–536. 10.1016/j.cmet.2011.08.014
138
Zelko I. N. Mariani T. J. Folz R. J. (2002). Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression.Free Radic. Biol. Med.33337–349. 10.1016/S0891-5849(02)00905-X
139
Zetterqvist A. V. Berglund L. M. Blanco F. Garcia-Vaz E. Wigren M. Duner P. et al (2014). Inhibition of nuclear factor of activated T-cells (NFAT) suppresses accelerated atherosclerosis in diabetic mice.PLoS One8:e65020. 10.1371/journal.pone.0065020
140
Zetterqvist A. V. Blanco F. Öhman J. Kotova O. Berglund L. M. de Frutos Garcia S. et al (2015). Nuclear factor of activated T cells is activated in the endothelium of retinal microvessels in diabetic mice.J. Diabet. Res.2015:428473. 10.1155/2015/428473
141
Zhang L. Chen J. He Q. Chao Z. Li X. Chen M. (2019). MicroRNA-217 is involved in the progression of atherosclerosis through regulating inflammatory responses by targeting sirtuin 1.Mol. Med. Rep.203182–3190. 10.3892/mmr.2019.10581
142
Zhao W. Zhang J. Lu Y. Wang R. (2001). The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener.EMBO J.206008–6016. 10.1093/emboj/20.21.6008
143
Zhong L. Simard M. J. Huot J. (2018). Endothelial microRNAs regulating the NF-kappaB pathway and cell adhesion molecules during inflammation.FASEB J.324070–4084. 10.1096/fj.201701536R
Summary
Keywords
blood pressure, endothelium, inflammation, oxidative stress, reactive oxygen species
Citation
Ranadive SM, Dillon GA, Mascone SE and Alexander LM (2021) Vascular Health Triad in Humans With Hypertension—Not the Usual Suspects. Front. Physiol. 12:746278. doi: 10.3389/fphys.2021.746278
Received
23 July 2021
Accepted
25 August 2021
Published
01 October 2021
Volume
12 - 2021
Edited by
Jochen Steppan, Johns Hopkins University, United States
Reviewed by
Eduardo Damasceno Costa, Federal University of Minas Gerais, Brazil; Yu Huang, China University of Geosciences Wuhan, China; Deepesh Pandey, Johns Hopkins University, United States
Updates
Copyright
© 2021 Ranadive, Dillon, Mascone and Alexander.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sushant M. Ranadive, ranadive@umd.edu
This article was submitted to Vascular Physiology, a section of the journal Frontiers in Physiology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.