 Jose M. Ferrero
Jose M. Ferrero Ana Gonzalez-Ascaso1,2
Ana Gonzalez-Ascaso1,2 Jose F. Rodriguez Matas
Jose F. Rodriguez Matas- 1Centro de Investigacion e Innovacion en Bioingenieria, Universitat Politecnica de Valencia, Valencia, Spain
- 2Dipartimento di Chimica, Materiali e Ingegneria Chimica ”Giulio Natta”, Politecnico di Milano, Milan, Italy
Acute myocardial ischemia induces hyperkalemia (accumulation of extracellular potassium), a major perpetrator of lethal reentrant ventricular arrhythmias. Despite considerable experimental efforts to explain this pathology in the last decades, the intimate mechanisms behind hyperkalemia remain partially unknown. In order to investigate these mechanisms, we developed a novel computational model of acute myocardial ischemia which couples a) an electrophysiologically detailed human cardiomyocyte model that incorporates modifications to account for ischemia-induced changes in transmembrane currents, with b) a model of cardiac tissue and extracellular K+ transport. The resulting model is able to reproduce and explain the triphasic time course of extracellular K+ concentration within the ischemic zone, with values of
1 Introduction
Globally, cardiovascular disease (CVD) remains the leading cause of death and disability, accounting for around 18.5 million deaths every year, approximately one-third of all deaths globally (Roth et al., 2020). Among CVD, atherosclerotic coronary artery disease is the most common pathology. This disease may cause a partial or complete occlusion of a coronary artery which, in turn, causes myocardial ischemia and infarction. If ischemia is regional (the most common case) and affects only a part of the myocardium, it introduces abnormal heterogeneity in the tissue. Indeed, resting membrane potential, action potential (AP) duration and effective refractory period, among others, may differ from one site to another. This ischemia-induced heterogeneity provides an important pro-arrhythmic substrate (Kléber, 1984; Janse and Wit, 1989; Coronel, 1994).
The acute phase of myocardial ischemia corresponds to the first 30–60 min after coronary artery occlusion and is associated with a high incidence of arrhythmic events (Smith et al., 1995; Yan et al., 2004; Cascio et al., 2005). During the first 30 min, the period in which this study will focus, ischemia is characterized by important metabolic changes inducing profound electrophysiological alterations in the behavior of affected cardiomyocytes. The main metabolic changes include a reduction in intracellular adenosine triphosphate (ATP), changes alterations in intracellular adenosine diphosphate (ADP), a reduction of tissue pH, and an increase in lysophosphatidylcholine (LPC) (Daleau, 1999; Sakamoto et al., 2000; Terkildsen et al., 2007). These changes cause alterations in the cell resting membrane potential, which becomes less negative, and modulate the inward and outward transmembrane currents during the AP, leading to hyperkalemia (an increase in extracellular potassium concentration,
The increase in extracellular potassium is related to alterations of the electrical behavior of ischemic cardiomyocytes (Janse and Wit, 1989). Specifically, resting membrane potential increases (which reduces cell excitability), action potential duration shortens and conduction velocity diminishes, among other alterations. These electrophysiological changes provide a potential substrate for the generation of arrhythmic events leading to ventricular fibrillation and sudden cardiac death (Harris et al., 1954; Smith et al., 1995). The evidence comes from experiments involving isolated whole hearts subject to regional acute myocardial ischemia (Harris et al., 1954; Janse et al., 1980; Janse and Kléber, 1981; Weiss and Shine, 1981; Weiss and Shine, 1982b; Coronel et al., 1989; Janse and Wit, 1989; Coronel, 1994; Weiss et al., 2017). In these experiments, ischemia (and hyperkalemia in particular) was found to promote unidirectional block and reentry due to the partial or complete loss of cell excitability provoked by extracellular potassium accumulation. Also, the appearance of an “injury current” flowing from the ischemic zone to the normal zone has been hypothesised to induce a source-sink imbalance which could induce ectopic activity that would act as a trigger for reentry (Janse et al., 1980; Janse and van Capelle, 1982; Coronel et al., 1991; Coronel, 1994). Computer simulations have also highlighted the arrhythmogenic effects of ischemia-induced hyperkalemia in virtual 2D tissues (Ferrero et al., 2003; Tice et al., 2007; Trénor et al., 2007; Romero et al., 2009) and 3D virtual bi-ventricular heart models (Mena Tobar et al., 2018; Carpio et al., 2022).
Despite the importance of this phenomenon, and the number of experimental studies trying to elucidate the ionic and tissue related mechanisms behind the increase in
Indeed, using solely experimental means to elucidate the intimate mechanisms responsible for
Understanding the mechanisms that lead to hyperkalemia would enable a better understanding of its arrhythmogenic effects and the development of more solid approaches to its treatment. With this in mind, this work attempts to study, with the aid of computer simulations, the intimate mechanisms underlying the increase in
2 Materials and methods
2.1 Virtual tissue description
In this work, “0D simulations” corresponds to a virtual single isolated ventricular cardiomyocyte, and “1D simulations” to a virtual myocardial one-dimensional strand 4 cm in length. In the latter case, in order to simulate regional ischemia, the first 2 cm correspond to normal cells (“normoxic tissue”), whereas the rest of the fiber was subject to ischemic conditions (“altered tissue”), as described below.
2.2 Action potential model
Unless otherwise stated, the simulations were carried out using a modified version of the O’Hara model (O’Hara et al., 2011). This model is widely accepted to simulate APs and the underlying ionic currents in human ventricular cardiomyocytes, and was adopted by an expert group of the FDA as the starting point for developing an in silico model suitable for regulatory decision making (Colatsky et al., 2016). The model includes mathematical descriptions of fifteen transmembrane currents flowing through ion channels, pumps and exchangers, as well as the calcium transients involved in the calcium-induced calcium release process, and calcium buffering in the intracellular medium. In particular, the model includes seven different ionic currents that carry potassium ions, namely the rapid delayed rectifier potassium current (IKr), the slow delayed rectifier potassium current (IKs), the transient outward potassium current (Ito), the inward rectifier potassium current (IK1), the background potassium current (IKb), the potassium component of the L-type calcium current (ICaL) and the sodium/potassium (NaK) pump (INaK).
To obtain realistic values of AP upstroke velocity and propagation velocity, the formulation of INa and INaL were modified as in Carpio et al. (2019) [which, in turn, includes the modifications by Dutta et al. (2017)].
2.3 Cellular model of acute ischemia
To simulate the effects of acute myocardial ischemia at the cell membrane level, more profound changes were made to the O’Hara model [as in Carpio et al. (2022)]. First, a model of the ATP-sensitive potassium current (IK(ATP)), which is absent in the O’Hara model, was formulated using the model by Ferrero et al. (1996) adapted to human cardiomyocytes using data from Babenko et al. (1998) by changing the maximum conductance and the sensitivity to intracellular ATP and ADP concentrations ([ATP]i and [ADP]i, respectively). Secondly, the effects of [ATP]i and [ADP]i on ionic pumps were modeled using data from Cortassa et al. (2006) and Terkildsen et al. (2007) by introducing different [ATP]i and [ADP]i dependent scaling factors affecting the NaK pump (INaK), the sarcolemmal calcium pump (IpCa)and the SERCA pump (Iup). Thirdly, we introduced the effects of extracellular and intracellular acidosis in the model by applying different multiplicative factors that depend on pHo and/or pHi to several pH-dependent currents. Specifically, we used data from Saegusa et al. (2011) to model the effects of acidosis in ICaL; data from Murphy et al. (2011); Watson and Gold (1995) for both INa and INaL; data from Doering et al. (1996); Egger and Niggli (2000) for the sodium/calcium exchanger (ICaNa); and data from Friedrich et al. (1996) to model the effects of acidosis on INaK. Finally, the effects of LPC) on INa and INaL were modeled following Gautier et al. (2008).
2.4 Simulation of progressive acute ischemia
Each simulation corresponds to 5 min of normoxia followed by 30 min of acute ischemia. During this 30 min period, hypoxia and acidosis are progressive, and the time evolution of the ischemic parameters is shown in Figures 1A–D. For the [ATP]i (Figure 1A), the time course reported by Sakamoto et al. (2000) for guinea pig was adopted with a normoxic value of [ATP]i = 10 mmol/L as in O’Hara et al. (2011) and Cao et al. (2018). Regarding pH (Figure 1C), the time course of pHi reported by Sakamoto et al. (2000) for guinea pig was assumed. The same time course was adopted for pHo, with a normoxic value of pHo = 7.4 measured in guinea pig (Vaughan-Jones et al., 2009). For [LPC]i, a linear time course was assumed with a normoxic value of [LPC]i = 2 μmol/L and a concentration of [LPC]i = 20 μmol/L at 30 min from the onset of ischemia, estimated from the works by Sobel et al. (1978) and Daleau (1999). As for free intracellular ADP, due to its low basal concentrations, direct measurements of [ADP]i are not possible and must be estimated using mass-action kinetics principles. In this regard, different profiles of ADP have been derived (Allen and Orchard, 1987; Weiss et al., 1992; Terkildsen et al., 2007). In this work, the ADP profile derived by Terkildsen et al. (2007) has been adopted.
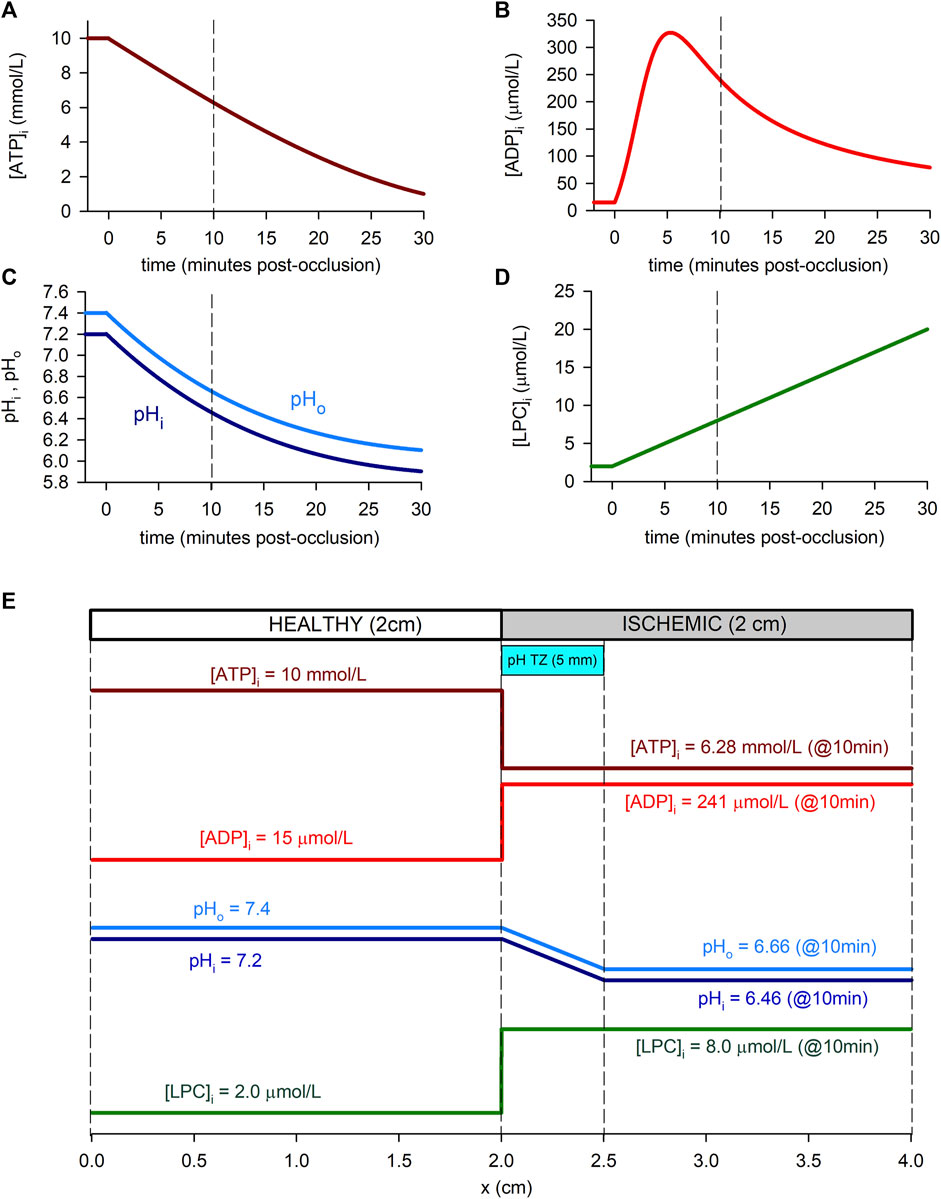
FIGURE 1. Time course of ischemia-related parameters in the simulated cell (“0D simulations”, panels (A–D) and spatial profiles in the simulated tissue (“1D simulations”, panel (E). (A): Intracellular ATP concentration. We adopted the time course reported by Sakamoto et al. (2000) for guinea-pig (at 37°C) with the normoxic value of O’Hara et al. (2011) and Cao et al. (2018). (B): Intracellular ADP concentration, assuming a time course as reported by Terkildsen et al. (2007), based on Befroy et al. (1999) for guinea-pig at 37°C. (C): Intracellular and extracellular pH, adopting the time course for guinea-pig reported by Sakamoto et al. (2000) (at 37°C) and a normoxic value for pHo by Vaughan-Jones et al. (2009). (D): Intracellular LPC concentration, assumed linear and estimated using data from Gautier et al. (2008) in rat at 22°C and Daleau (1999) for guinea-pig at 30°C. (E): Spatial profiles of the ischemic parameters in relation to the simulated 1D tissue. As an example, values within the ischemic part of the strand correspond to 10 min post-occlusion [dashed lines in panels (A–D)]. A pH transition zone (pH TZ) was assumed to be 0.5 cm wide Coronel et al. (1988). pH was assumed to vary linearly within that zone.
In the 1D simulations, these dynamic changes affected only the “altered tissue”, while in the “normoxic tissue” the values of the parameters remained normal ([ATP]i = 10 mmol/L [ADP]i = 15 μmol/L, pHi = 7.2, pHo = 7.4, and [LPC]i = 2 μmol/L). In the case of [ATP]i [ADP]i and [LPC]i, these dynamic changes have been imposed in a stepwise manner at the transition between the normal and ischemic tissue, as indicated in the study from Walfridsson et al. (1985) that reports the anoxic border to develop in less than 1 mm. On the contrary, a transition zone of 0.5 cm has been considered for pHi and pHo following the model by Ferrero et al. (2003) which was inspired in the studies from Coronel et al. (1988). Figure 1E shows the spatial transition for the cyanotic and the acidosis border used in the simulations.
Due to the large variability of the data present in the literature, additional simulations considering different time-courses and baseline values for the different metabolites were performed for completeness. Results from these simulations are available as part of the Supplementary Figures S1, S3 and will be discussed later.
2.5 Stimulation protocol
The cell (in the “0D simulations”) and the first (leftmost) cell of the “normoxic tissue” (in the “1D simulations”) were stimulated during 35 min with a train of pulses 0.5 milliseconds in width and twice normoxic diastolic threshold in amplitude, with a frequency of 1 Hz (unless otherwise stated). The frequency of 1Hz was chosen since most of the experimental studies found in the literature were conducted at this frequency. However, to test the sensitivity of the model to heart rate, additional simulations were carried out in which the cell was paced at different frequencies: i) a quiescent cell (0 beats per minute, bpm), ii) a bradycardic situation (30 bpm), iii) our control heart rate (60 bpm), iv) a mild tachycardia (120 bpm), and v) a severe tachycardia (180 bpm).
2.6 Tissue electrophysiology equations
In the 1D simulations, cardiac tissue was modeled as a continuous fiber with a length L of 4 cm. The transmembrane potential (Vm), and the extracellular potential (Vo), were computed using the bidomain model in two steps (Keller et al., 2010). First, Vm was obtained as the solution of the 1D reaction-diffusion equation:
where DV is the effective tissue conductivity, Cm is the specific membrane capacitance, Iion is the transmembrane ionic current density (sum of the transmembrane currents included in the O’Hara model), and Istm is the stimulation current density. The extracellular potential is governed by the following partial differential equation
where λ is the extracellular to intracellular conductivity ratio. Eqs 1, 2 are subject to non-conduction boundary conditions at both cable ends.
In the “0D simulations”, Eq 1 without the diffusion term was used:
2.7 Computation of the injury current
One of the interests of this study was to investigate on the characteristics of the “injury current” which flows between the altered and normal tissue during acute ischemia, and to evaluate its potential role in arrhythmogenesis during acute myocardial ischemia (Janse and Kléber, 1981; Coronel et al., 1991). For this purpose the intracellular injury current,
This equation is computed as a postprocessing step in the simulations.
2.8 Potassium transport equations
To model the dynamic changes in extracellular potassium concentration
where DK is the extracellular potassium diffusion coefficient, F is the Faraday’s constant, R is the gas constant, T is the absolute temperature, Vo is the extracellular potential, Ac is the cell surface, vo is the volume of the extracellular compartment, ∑IKx the total transmembrane potassium current (IKr + IKs + Ito + IK1 + IK(ATP) + IKb + ICaK - 2INaK),
In the “0D simulations”, Eq. 7 without the diffusion term was used:
2.9 Computation of potassium flux rates
One of the objectives of this study was to compute the contribution of each individual sarcolemmal current to extracellular potassium accumulation. For this purpose, we define the potassium flux rate (KFRx) generated by a particular potassium current IKx as the rate of increase of
From Eq. 9 (particularized for a specific potassium current IKx), the value of KFRx may be computed as follows:
where BCL (Basic Cycle Length) is the stimulation period (1,000 milliseconds, unless otherwise stated).
The total potassium flux rate (KFRT) is given by:
2.10 Numerical and computational methods
The “0D simulations” were performed with a custom-made script in MATLAB (MathWorks, Natick, MA) using an adaptive time step method. On the contrary, for the “1D simulations”, Eqs 1–7 were solved with an in-house C code using the operator splitting numerical scheme together with the explicit Euler method. Eq. 2 was integrated with custom-made software routines in MATLAB as a postprocessing step assuming a value of λ = 3.647 from Niederer et al. (2011). These simulation codes are available upon request.
For the “1D simulations”, a constant time step of Δt = 0.02 m and a space discretization of Δx = 0.25 mm were used. The parameters used in the simulations are shown in Table 1. The effective tissue conductivity was assumed to be DV = 0.0026 m that gives a conduction velocity of 70 cm/s in normoxic tissue (Taggart et al., 2000). The extracellular potassium diffusion coefficient DK = 1.5 ⋅ 10–8 cm2/s was taken from Niederer (2013). However, additional simulations were conducted at different values of DV and DK to test the influence that electrotonic coupling and
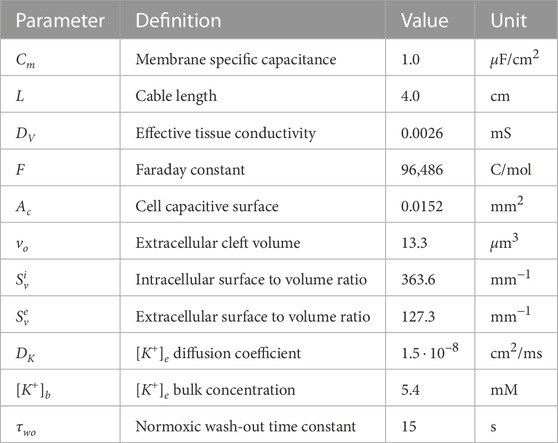
TABLE 1. Model parameters for the electrophysiology and transport equations.
The accuracy of the 1D numerical simulations was verified against simulations that employed smaller space and time discretizations of Δx = 0.125 mm and Δt = 0.01 m respectively. Results indicate that reducing the space discretization and time step to these values lead to an increase in the conduction velocity of about 2.0%. However, major results, and main conclusions, of this study obtained with these smaller time and space discretizations are still valid as discussed in the following section and reported in the Supplementary Figure S5.
3 Results
3.1 Single cell simulations
This section describes the main results related to the “0D simulations”, corresponding to an isolated ventricular cardiac myocyte.
3.1.1 Extracellular potassium accumulation
Figure 2 summarizes some of the main findings corresponding to the “0D simulations”. The solid line in Figure 2A shows the time course of
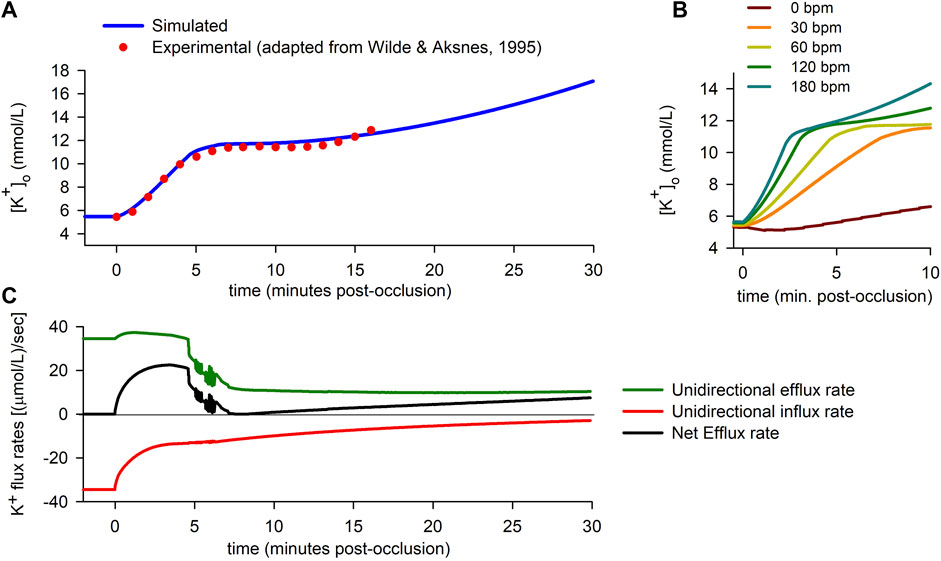
FIGURE 2. Extracellular potassium accumulation and transmembrane potassium fluxes. (A): Extracellular potassium concentration
To test the validity of the simulation, results were compared to experimental data published elsewhere (Wilde and Aksnes, 1995). The dots in Figure 2A correspond to adapted experimental data of extracellular potassium accumulation obtained in rabbit hearts subject to acute global ischemia. The data shown in the figure have been shifted +0.9 mmol/L to account for the difference between the normoxic value of
In order to test the soundness and sensitivity of the results to the specific time courses of intracellular ATP and intra/extra-cellular pH, we carried out alternative simulations using different time courses and/or initial normoxic values and/or final ischemic values for said ischemic parameters. The results, which can be found in Supplementary Figure S2, show that, despite of small quantitative differences, the qualitative features of the extracellular K+ behavior remain unaltered. As for the effects of different possible time courses of intracellular ADP, its effects are presented and discussed below.
Finally, the effect of heart rate is shown in Figure 2B. The results show that the rate of rise of extracellular potassium increases with heart rate. In the particular case of a quiescent cell, the rise in
3.1.2 Potassium flux rates
The increase in
During the normoxic period, efflux and influx are equal in magnitude [34.5 (μmol/L)/s], so there is no net potassium efflux. As soon as ischemia begins, an imbalance between the potassium efflux and influx quickly arises, with a rapid decrease in the magnitude of the influx rate being coupled to a lesser increase in the efflux rate. The resulting net efflux rise (Figure 2C) during the first ≈4 min post-occlusion provokes the first rising phase in
Just before the fifth minute of ischemia, potassium influx has almost stabilized [at ≈ 13 (μmol/L)/s]. However, the efflux rate suffers an abrupt drop, just prior to showing notorious oscillations [which are due to alternans in the action potential duration (APD), as explained below]. In the following 2 minutes, the efflux keeps decreasing until it almost equals the magnitude of the influx. As a result, the net potassium efflux falls to near zero values, which in turn gives rise to the plateau in
To better understand the causes of the dynamic changes in potassium efflux and influx, the individual contributions of the different potassium transmembrane currents were computed using Eq. (10) at selected time instants depicted by the short vertical lines in Figure 3A. The results are depicted in panels B to F in Figure 3. The coloured bars represent efflux rates (when positive) or influx rates (when negative). The continuous data of the time course of the flux rates corresponding to the individual contributions of all the potassium currents is shown in Supplementary Figure S3.
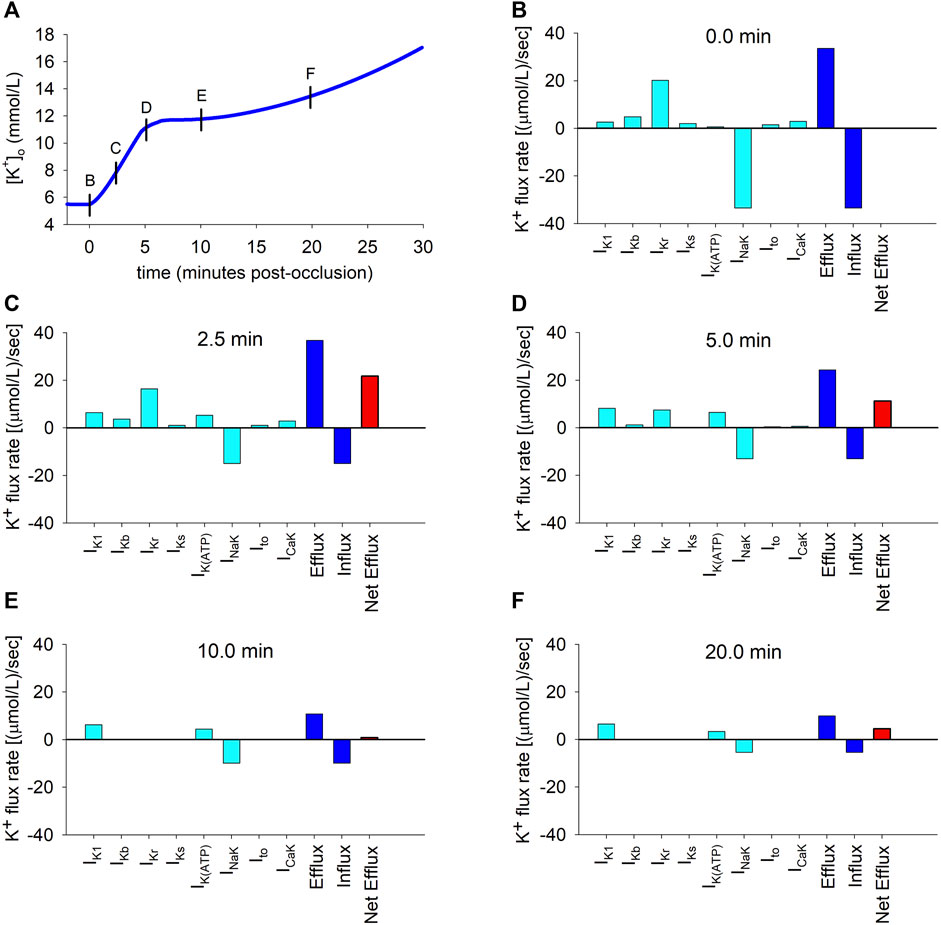
FIGURE 3. Potassium transmembrane fluxes carried by individual ionic currents. (A): Simulated ischemic
As shown in Figure 3B, which corresponds to the normoxic situation just before the onset of ischemia, the current carrying the highest potassium efflux is IKr, with other currents playing a minor role. Unsurprisingly, the NaK pump is the only current that generates a noticeable potassium influx. At this time point, influx and efflux are balanced.
Two and a half minutes later (Figure 3C), the most notorious change is the decrease in the magnitude of the influx rate [from 34.5 to 15.0 (μmol/L)/s] caused by the reduction in the current transported by the NaK pump. Concomitantly, the efflux rates through IK1 and IK(ATP) increase considerably, counteracting the decrease in the IKr efflux rate, with changes in other potassium currents being negligible. Thus, the unidirectional potassium efflux increases (from 34.5 to 37.2 (μmol/L)/s). The overall result is an increase in the net efflux [from 0 to 22.2 (μmol/L)/s], which provokes the primary rise in
Five minutes post-occlusion (Figure 3D), the efflux rates through various ion channels have considerably changed. Most noticeably, the efflux generated by IKr has strongly diminished and the only currents generating an appreciable potassium efflux are now IKr, IK1 and IK(ATP). Even if the influx through the NaK pump has very slightly decreased, this important reduction in the efflux provokes a strong decrease in the net efflux rate [now 11.2 (μmol/L)/s]. This is responsible for the decrease in the rate of rise in
Ten minutes post-occlusion (Figure 3E), when
This situation is similar after 20 min of ischemia (Figure 3F), except that the NaK pump influx is further reduced, again generating an imbalance between influx and efflux that gives rise to a net efflux of 4.5 (μmol/L)/s and a secondary slow rise in
3.1.3 Ionic currents
To further investigate the causes of these changes in efflux and influx as ischemia develops, we plotted the time course of APs and selected potassium currents at the time instants discussed above. The top row in Figure 4 shows how APs change as ischemia progresses. Resting membrane potential becomes increasingly less negative (a direct consequence of the increase in
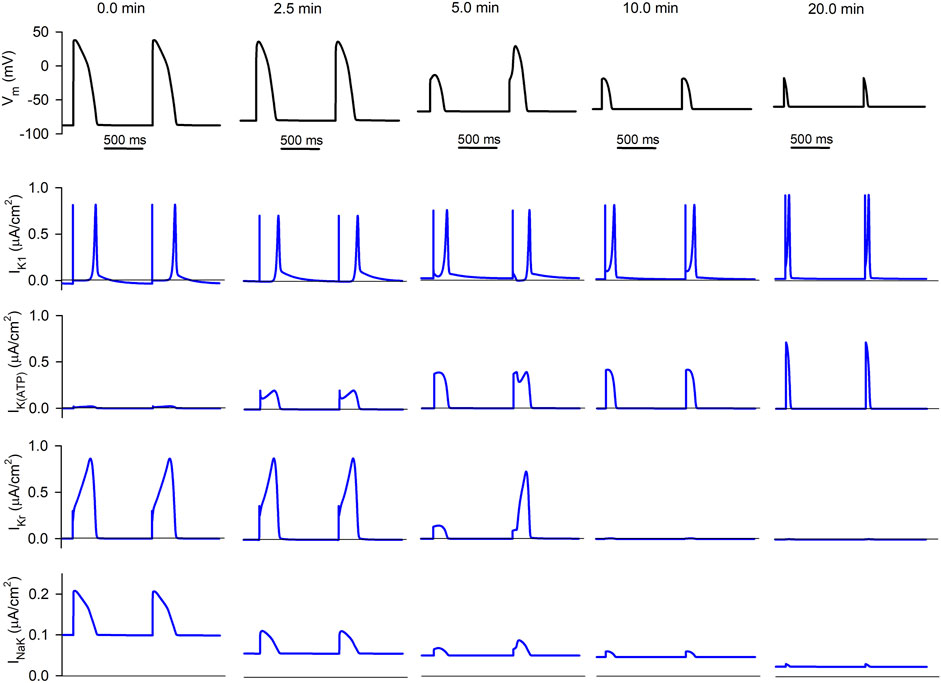
FIGURE 4. Simulated APs and selected ionic currents in an acutely ischemic isolated ventricular cardiomyocyte. Top row (black traces): consecutive APs 0.0, 2.5, 5.0, 10.0, and 20.0 min after the onset of progressive ischemia. Lower rows (blue traces): inward rectifier potassium current (IK1), ATP-sensitive potassium current (IK(ATP)), rapid delayed rectifier potassium current (IKr), and sodium/potassium pump (INaK), respectively.
The second row shows the progressive evolution of IK1. Basically, its diastolic value keeps increasing as ischemia develops due to the hump in its current-voltage relationship that implies a higher current at membrane potentials moderately higher than the potassium Nernst potential (EK). This causes the increase in its associated efflux seen in Figures 3D–F.
The third row represents the evolution of IK(ATP). While it is almost non-existent in normoxia, its amplitude keeps increasing in ischemia (due to the decrease in the intracellular ATP/ADP ratio). Although its duration decreases with time (due to the reduction in APD), the area below the curve (proportional to the efflux generated by the current) is almost constant.
The fourth row shows the progression of IKr (which generates the highest potassium efflux in normoxia) with ischemia. Its duration has decreased after 2.5 min of ischemia, thus reducing the efflux, and it has almost vanished 10 min after the onset of ischemia (due to the cell being almost non-excitable at this point). Five minutes post-occlusion, AP alternans cause an alternation in the amplitude and duration of IKr, which is very low in the short AP compared to the long one. Further effects of AP alternans in the time course of
Finally, the lower row corresponds to the NaK pump current. The decrease in intracellular ATP levels continuously reduces the magnitude of the current, thus reducing the influx of potassium as was apparent in Figures 3B–F.
3.1.4 Action potential alternans and their effect in potassium accumulation
Once we identified the causes of the triphasic nature of extracellular potassium accumulation in regards to potassium fluxes, we further analyzed the influence of the dynamically changing AP morphology during ischemia on the time course of
The data depicted in red in the lower graph in Figure 5A represents the time course of the APD at 90% repolarization (APD90) during the 30 min period of simulated ischemia at the same time scale as
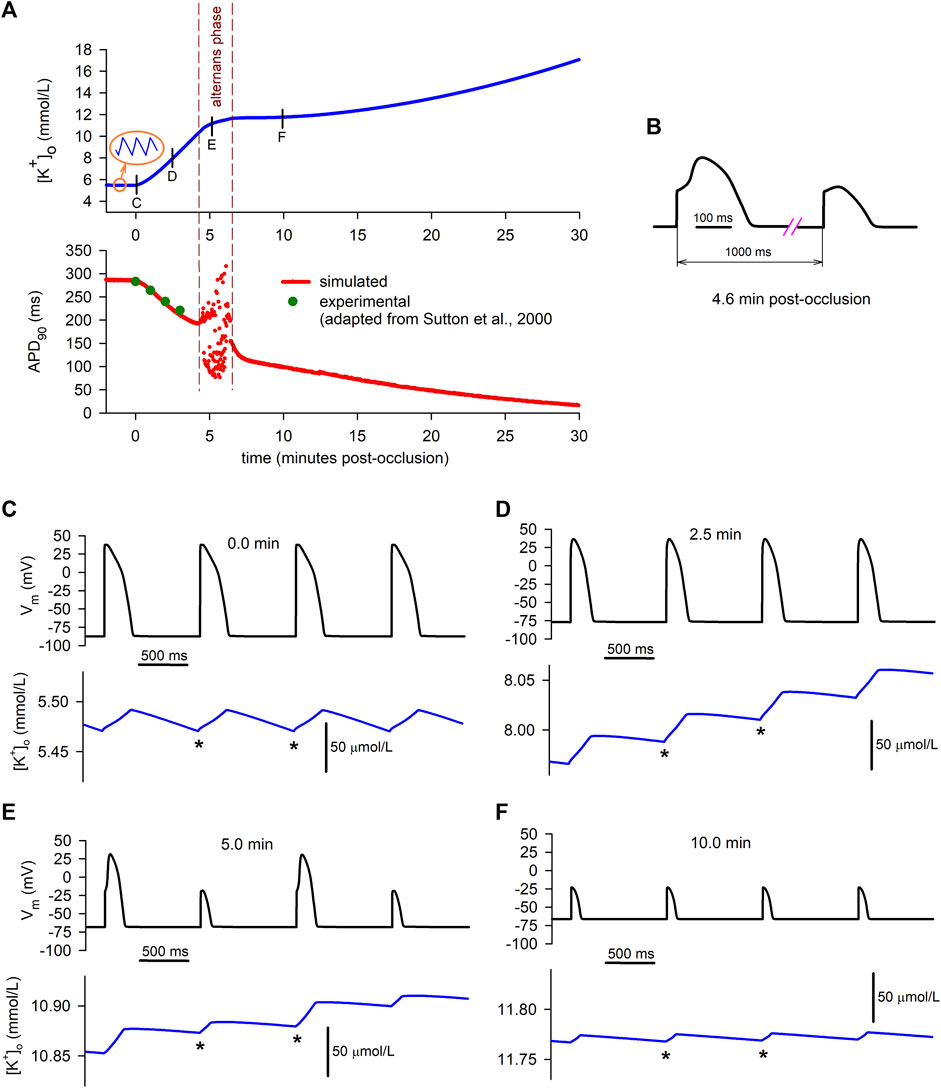
FIGURE 5. Relationship between simulated extracellular potassium concentration and APs. (A): time course of
As shown in the lower graph of Panel A, between the fourth and the sixth minute of ischemia (approximately), APD90 values begin to oscillate, indicating that the cell is developing electrical alternans. This alternans phase is delimited by the dashed vertical lines. Figure 5B shows the detail of the waveform of two consecutive alternating APs obtained in our simulations (at the 4.6 min mark). At the beginning of this phase, alternans with a 2:2 pattern (i.e., long–short–ong–short) are predominant, but shortly afterwards more complex patterns arise (e.g., long–short–short–long–long–short–long).
As the dashed vertical lines indicate, the period of occurrence of alternans matches the smooth transition from the primary rising phase of
During normoxia (Figure 5C), APs do not alternate at all, and potassium currents are balanced in a way that the diastolic minimum potassium levels (DmKL, indicated by the * signs) reach the same value at the end of each diastolic period. This is consistent with the normoxic stable behaviour of the “macro”
On the contrary, at the 2.5 min mark in the ischemic cell (Figure 5D), the positive net potassium efflux seen in Figures 2C, Figure 3C is related to a continuous increase in the DmKL from one AP to the following one. Consequently, the “macro”
Finally, 10 min after the onset of ischemia (Figure 5F), only short APs are present in the cell due to its partial loss of excitability provoked by an already elevated value of
3.2 Tissue simulations
This section describes the main results related to the “1D simulations”, corresponding to a virtual 4 cm strand subject to normal conditions in its first 2 cm (the “normoxic tissue”) and to progressive acutely ischemic conditions in the last 2 cm (the “altered tissue”).
3.2.1 Extracellular potassium accumulation
Figure 6A shows the spatial profiles of
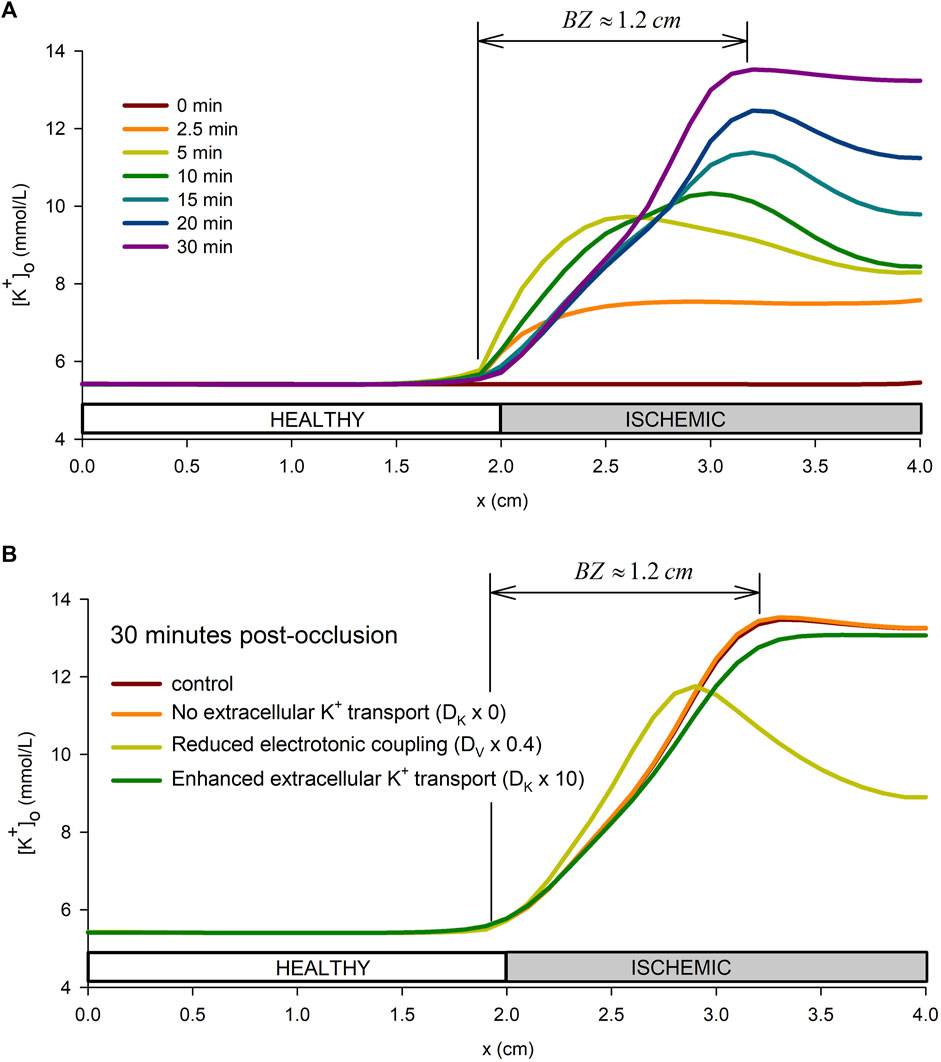
FIGURE 6. Spatial profile of extracellular potassium accumulation. (A): spatial profile in the tissue preparation at different times from the onset of ischemia. (B): influence of the tissue conductivity and extracellular potassium diffusion coefficient on the extracellular potassium spatial profile at 30 min from the onset of ischemia.
To test the sensitivity of the results to the time-course of [ADP]i and the spatial extension of the pH transition zone, simulations considering the ADP time-course proposed by Weiss et al. (1992) were performed for three different pH transition zone sizes (0 cm, 0.5 cm, and 1 cm). Results from the simulations are shown in Supplementary Figure S4. The results show that the ADP data from Weiss et al. (1992) leads to a lower rate of rise of
Furthermore, to better understand the mechanisms behind the formation of the BZ, a sensitivity analysis of the effect of the tissue conductivity and the potassium diffusion coefficient was performed. Together with the control simulation, the following combination of parameters were considered: i) The tissue conductivity, DV, was reduced by 20%, by 60%, and by 80% down to 0.0005 m (conduction velocity of 30 cm/s) while keeping the extracellular potassium diffusion coefficient, DK, at the control value in Table 1; ii) the extracellular potassium diffusion coefficient, DK, was multiplied by 0 (no extracellular potassium transport i.e., DK = 0.0), by 10, and by 100 while keeping the effective tissue conductivity to the value in Table 1. Results of these simulations are show in Supplementary Figure S6, whereas Figure 6B shows only results for four representative cases. Results show that increasing DK reduces the value of
To better understand the role of extracellular potassium transport in the
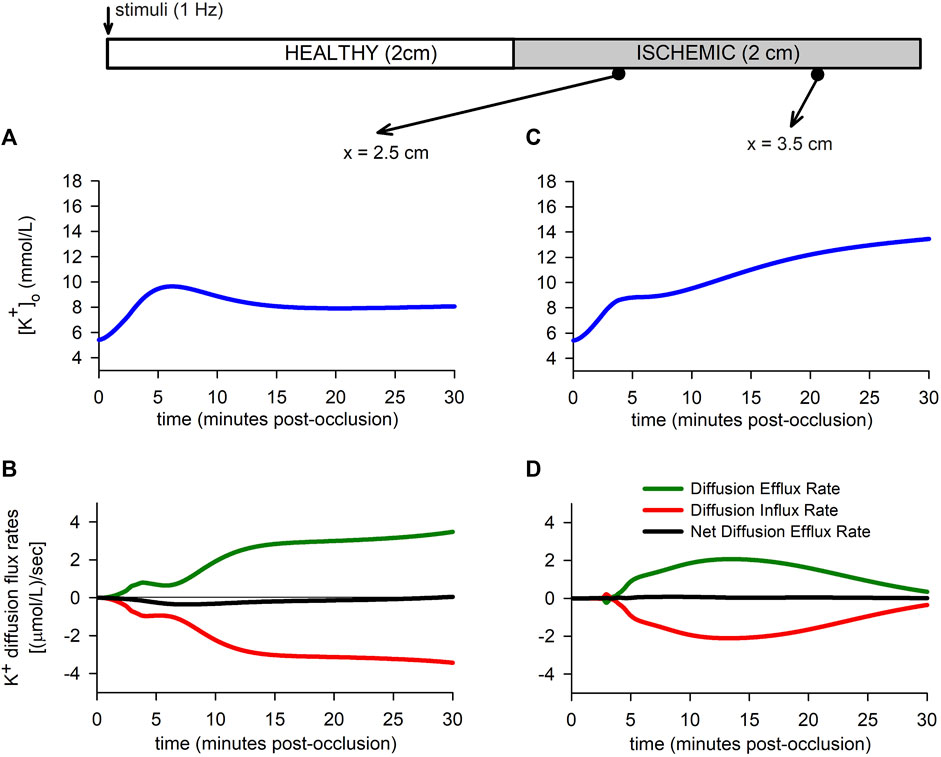
FIGURE 7. Extracellular potassium flux rates. (A,B): Extracellular potassium accumulation [Panel (A)] and extracellular potassium diffusion flux rate [Panel (B)] for a point located in the BZ. (C,D): Extracellular potassium accumulation [Panel (C)] and extracellular potassium diffusion flux rate [Panel (D)] for a point located in the CIZ.
3.2.2 Electrophysiological manifestations of the extracellular potassium accumulation in tissue
Figure 8 shows the
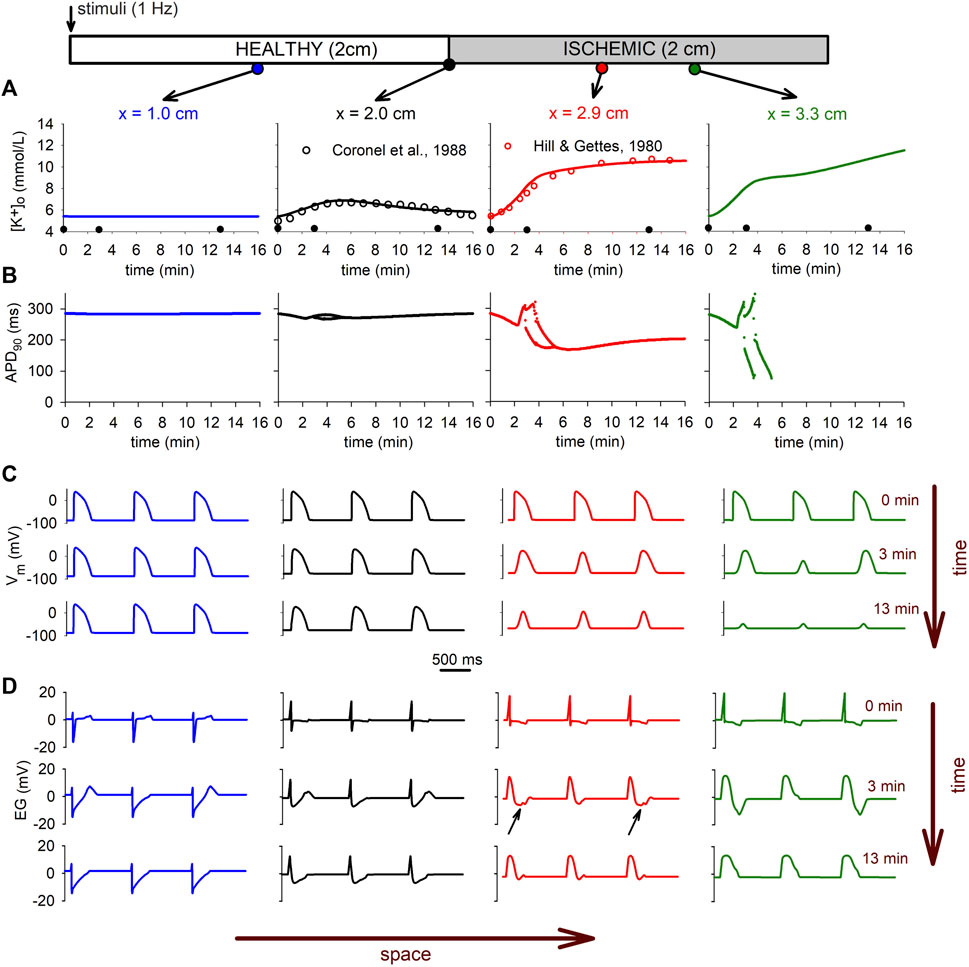
FIGURE 8. Electrophysiological manifestations of the extracellular potassium accumulation in tissue. (A): time course of the
The heterogeneity observed in the
These electrophysiologic changes are clearly reflected in the electrograms depicted in Figure 8D for different positions in the tissue. The electrograms show the typical depression in the ST segment due to ischemia in the altered region (small black arrows in Figure 8D), whereas the alternans in the AP are reflected in alternans in the T-wave (see Figure 8D right panel). The conduction block in the CIZ at minute 13 is evident by the total suppression in of the T-wave in all electrograms, and the monophasic electrogram at the CIZ (x = 3.3 cm).
3.2.3 Injury current during acute regional ischemia
The hypothesis that during acute ischemia, a current flowing between the ischemic and the normal tissue, a current of injury, may cause depolarization of the normal cells near the border zone has been proposed in several studies (Janse et al., 1980; Coronel et al., 1991). To explore this hypothesis more in detail by means of numerical simulations, the injury current in the tissue at 3 min and 4 min after the onset of ischemia were computed. The upper panel in Figure 9 show the AP at three position in the tissue located at the normal zone (x = 0.9 mm), the border zone (x = 2.3 mm), and the central ischemic zone (x = 3.2 mm), whereas panel B shows the electrograms at the same positions. The instant tA has been chosen in correspondence with the end of repolarization of the cell in the normal zone, coincident with the point of deep negative T wave in the electrogram recorded in the ischemic zone. On the contrary, tB corresponds to an instant of complete repolarization in the three positions. Panel C shows the depolarization current density for the stimulations at 3 and 4 min after the onset of ischemia, at the instant when the propagation front is entering the altered region (x = 2 cm). Panel D depicts the intracellular injury currents at instants tA and tB at 3 min and 4 min after the onset of ischemia.
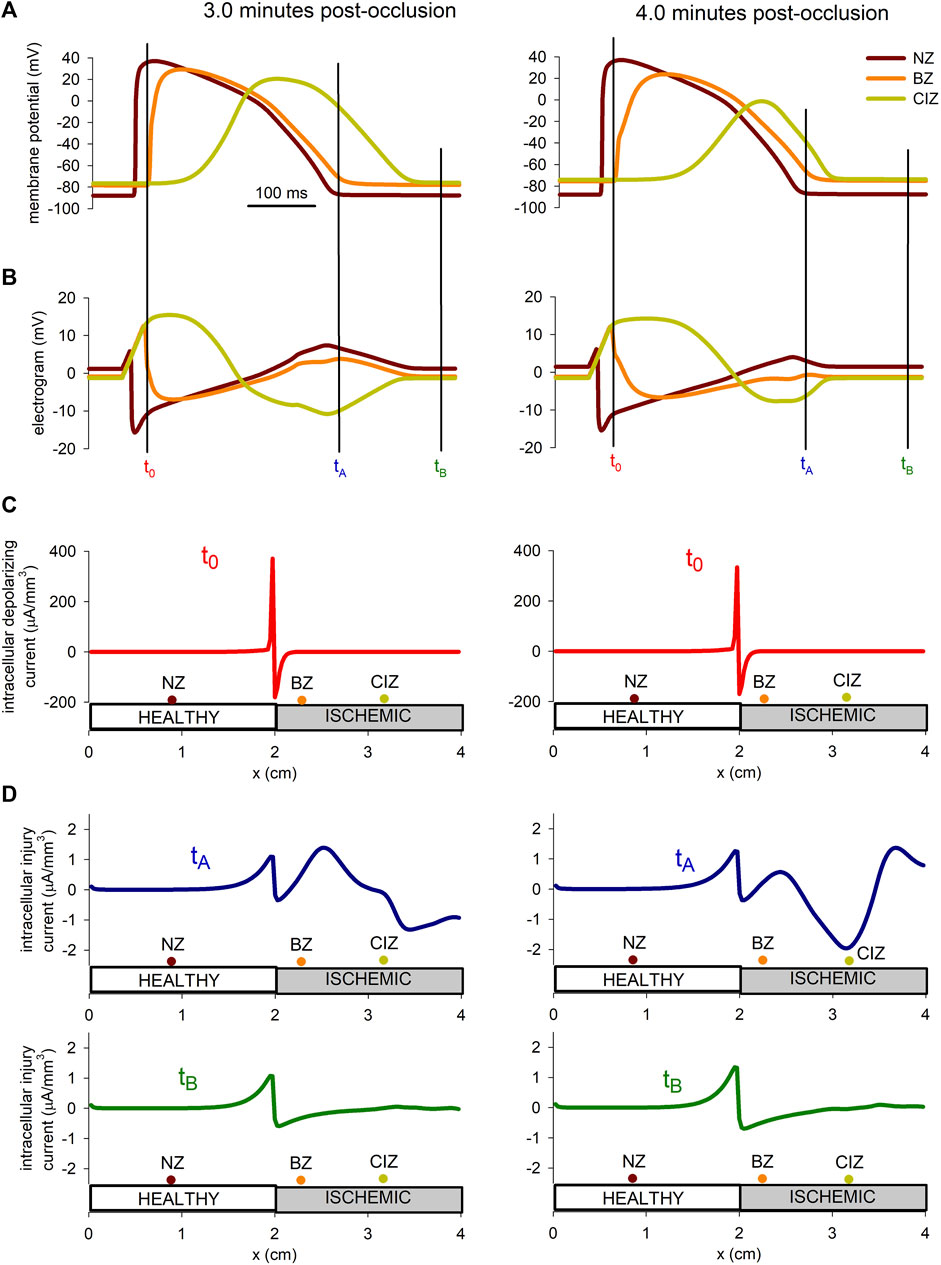
FIGURE 9. Injury current during acute regional ischemia. (A): Action potentials at three different locations within the cable at two instances after the onset of ischemia. (B): Simulated electrograms at the same positions and time instants as in panel (B). (C) Intracellular depolarizing current at the moment the depolarizing front enters the ischemic region. (D) Intracellular injury current at the moment of deepest negative T-wave in the ischemic zone and during the diastolic phase.
The results in Figure 9 show that the magnitude of the depolarization current, required to generate an ectopic beat, is about eight times larger than the magnitude of the injury current flowing from the ischemic to the normal zone.
4 Discussion
4.1 Main findings
The main findings of our work can be summarized as follows.
1. Ischemia-induced extracellular potassium accumulation and its characteristic triphasic time course are an intrinsic feature of the isolated ischemic cell (Figure 2). Though modulated by diffusion of potassium within the tissue, they are not directly caused by it.
2. The primary (initial) rising phase of
3. The smooth transition to the plateau phase of
4. Once the alternating period is over, the plateau phase of
5. The secondary rise in
6. The results obtained in a regionally ischemic tissue explain the progressive appearance of a potassium BZ of approximately 1.2 cm within the proximal end of the ischemic zone.
7. The simulations suggest that the
8. Results from the simulations suggest that the injury current associated with the deep negative T-wave in the ischemic zone may not be sufficient to depolarize healthy cells in the normal zone. In addition, part of the outward injury current occurs inside the altered tissue, where the cells are still within the effective refractory period.
While findings 1, 2, 4, 6, and 8 confirm previous hypothesis and results from previous modeling works regarding the extracellular K+ accumulation during acute ischemia, findings 3, 4, 5, and 7 identify novel mechanisms behind the time course of K+ accumulation and its spatial heterogeneity during acute ischemia. These findings are discussed in detail in the following sections.
4.2 Comparison with experimental data
The results obtained with our model show a very high degree of agreement with experimental data. This applies both to the time course of
Experimental evidence shows that the rate of rise of
As for the evolution of the APD, data from the literature obtained in ischemic human hearts is scarce because it is very difficult to obtain. The data from Sutton et al. (2000), plotted in Figure 5A (lower panel), was recorded during 3 min from a single left ventricular epicardial site in patients undergoing coronary artery surgery during cardiopulmonary bypass. To the best of our knowledge, they are the only available MAP data for comparison purposes in human in-vivo. Our results are in very close agreement with these data (see Figure 5A).
A very close agreement with experimental data is also found in the tissue simulations. Indeed, the two middle panels of Figure 8A show a direct comparison of the time course of
As for the time course of the net efflux rate resulting from our simulations, it is also in accordance with experimental measurements in animals (Wilde et al., 1990). In this experiment, a rapid increase in the net efflux rate, followed by a decrease to almost zero and an eventual slower increase, was found in globally ischemic mammalian hearts. The values they obtained for the net efflux rate are in the range of our simulated values.
Unfortunately, to our knowledge, the rise in
As for the dispersion of
According to our simulations, the injury current during the negative T wave, corresponding to a potential gradient of almost 60 mV accross the BZ (see Figure 9) was of 1.5 μA/mm3 at 3 min post-occlusion and 2 μA/mm3 at 4 min post-occlusion. These values are found to be in good agreement with the 2 μA/mm3 reported by Janse et al. (1980) in canine hearts at 4 min after LAD occlusion. Further, we found that this current was about eight times smaller than the excitatory current provided by the traveling front. This result is higher than the results from Janse et al. (1980) who reported an excitatory current as twice as large as the injury current. However, they computed the current with electrodes spaced a distance between 1.5 and 4 mm which lead to an underestimation of the excitatory current as demonstrated in Coronel et al. (2000). On the contrary, the intracellular depolarizing current computed by our model at the metabolic border was found to be in good agreement with the experiments from Coronel et al. (2000) on porcine heart, and the simulation results from Potse et al. (2007a).
In summary, the good agreement between experimental evidences and the results obtained with our model, both in the single cell and tissue levels, enables us to suggest new hypothesis regarding the intimate mechanisms of ischemia-induced hyperkalemia, as will be explained in the next sections.
4.3 The mechanisms of extracellular potassium accumulation
Experimental evidence shows that extracellular potassium concentration in the interstitium of acutely ischemic myocardial tissue shows a triphasic time pattern, with an initial rapid rise being followed by a plateau phase and a secondary slower increase. Regardless of the intimate mechanisms involved, dynamic changes in
4.3.1 Primary rising phase
Due to the difficulties related to the direct measurement of the individual contributions to the rapid primary rise of
Regarding the partial inhibition of the NaK pump, due to the decrease in the intracellular ATP/ADP ratio, this effect is included in our model. Indeed, the last row in Figure 4 shows the decrease in magnitude of the current carried by the pump. Moreover, the bars in Figure 3 (and also the results shown in Supplementary Figure S3) show that the magnitude of the influx rate through the pump rapidly decreases during the primary rising phase of
Regarding the enhancement of the ATP-sensitive potassium current, Our results show that, indeed, this factor is not as essential as the depression of the NaK pump in the primary rise of
Regarding the modification in sodium influx and its role in the primary phase of potassium accumulation, the classical hypothesis is that the increase in potassium efflux is a passive consequence of an increased sodium influx to maintain electroneutrality (Wilde and Aksnes, 1995; Shivkumar et al., 1997). Further, there is evidence that
Finally, a shrinkage of the interstitial extracellular space provoked by the ischemia-induced alteration of osmotic pressure has also been hypothesized to play a role in the increase in
4.3.2 Transition to the potassium plateau
One of the most intriguing and novel result from our simulations refers to the causes of the smooth transition to the extracellular potassium plateau and its relationship with AP alternans. As explained in the Results section, the model predicts the appearance of electrical alternans between the fourth and the sixth minute of ischemia in the isolated cell model (Figures 5B,E). Moreover, the tissue simulations also show AP alternans in the distal altered zone (Figure 8B). This is in correspondence with experimental results in animals. Indeed, these type of alternans, occurring at normal/low stimulation frequencies (1 Hz in our case) around 5 min post-occlusion (or even earlier), are a well-known feature of acute ischemia and have been observed in AP or MAP recordings and/or in electrograms (Kléber et al., 1978; Nakashima et al., 1978; Janse, 1990; Martišienė et al., 2015; Watanabe and Gettes, 2016). Moreover, the experimentally-obtained waveforms of the alternating APs are very similar to our simulated results shown in Figure 5B. The “long” AP has the upstroke divided in two phases, whereas the “short” one lacks the secondary phase. These exact features were found in the APs directly recorded in pig hearts 5 min after clamping the LAD coronary artery in pig hearts [see Figure 2 in Kléber et al. (1978); Figure 4 in Janse (1990)]. The morphology of the AP alternans seen in Figure 1 in Downar et al. (1977) are very similar to those found in our tissue simulations (Figure 8).
In our “0D simulations” (isolated cell), the onset of the
According to our simulations, the flattening of the
Different experimental results have been published in which APD and
The mechanisms that give rise to the
Indeed, in our “0D simulations”, the net potassium efflux rate is almost zero in the plateau around the 10 min mark. It stays below 2.2 (μmol/L)/s (10% of its peak value) from the 6.8 to the 13.4 min mark. During this period, influx and efflux rates are almost balanced with a value of ≈10 (μmol/L)/s, which is much smaller than in normoxia (≈34.5 (μmol/L)/s). On the one hand, influx rate is smaller than in normoxia because of the progressive partial inhibition of the NaK pump. On the other hand, efflux rate is also smaller due to the short duration of the AP plateau, which practically eliminates all potassium currents except for IK1 and IK(ATP). The former is not AP plateau-dependent (due to its inward rectification nature), while the latter has a high peak due to the low value of the ATP/ADP ratio at this point in time. The overall result is a new stable
A further proof of the influence on APD on the plateau phase is given in Supplementary Figure S9. As explained above, artificial interventions were made to eliminate AP alternans. The lack of a plateau phase in this separate simulation is because such interventions did not only abolish alternans but also maintained APD90 levels high compared to the control simulation.
Another classical explanation of the potassium plateau, different to considerations on APD, is related to the behaviour of the NaK pump. This has been argued both from experimental observations (see Wilde and Aksnes. (1995) for a review) and from previous computational modeling studies (Rodríguez et al., 2002; Terkildsen et al., 2007). Simulations carried out by Terkildsen et al. (2007) showed that the time course of INaK is triphasic and, according to them, the
4.3.3 Secondary rising phase
The prior hypotheses regarding the causes of the secondary slower rise in potassium accumulation are scarce. Classically, this phase has been related to cell irreversible damage (Weiss and Shine, 1982b). It is not completely clear if the cause is related to an increase in potassium efflux provoked by the loss of sarcolemmal integrity or by a further depletion of NaK pump activity (Weiss and Shine, 1986). According to a previous modeling study, the latter could be the main cause (Terkildsen et al., 2007). From our results, a complementary hypothesis may be established. Cell membrane damage was not included in our model, so our results discard the relevance of this factor. According to our simulations, potassium efflux rate almost plateaus during the secondary rise in
This phenomenon may be alternatively explained from a different perspective. The rate of rise in the secondary
4.3.4 The potassium border zone
The potassium gradient that is established during acute regional ischemia near the metabolic border, the BZ, is believed to be related with the extracellular potassium transport (Coronel et al., 1988; Coronel et al., 1989). However, our simulations indicate the size of the BZ to be related to the electrotonic coupling between cells close to the metabolic border, with a minor role of the extracellular potassium transport in modulating the BZ (see Figure 6B). Our results indicate that, an effective modulation of the BZ by potassium transport requires a ten-fold increase in the potassium diffusion coefficient (see Figure 6B). These findings are in agreement with the work from Potse et al. (2007b) who conclude that the mechanism that leads to a smooth distribution of
4.4 Limitations of the study
Our study presents several potential limitations. The first one is that the simulation of progressive acute ischemia relies on the chosen time courses of the ischemic parameters which are imposed to the model (see Figures 1A–D). They have been taken from experimental measurements or calculations reported in the literature (Weiss et al., 1992; Daleau, 1999; Sakamoto et al., 2000; Terkildsen et al., 2007; Gautier et al., 2008). None of them correspond to measurements in human hearts (due to the lack of data) but rather belong to different species (guinea-pig and rabbit) and were obtained in different laboratories and contexts. However, our additional simulations shown in Supplementary Figures S1, S3, as already discussed, indicate that the results obtained with different ischemic parameter time courses are qualitatively similar, so the main conclusions of our study are not hampered by this potential limitation.
Another potential limitation is related to the fact that our results have been obtained with a particular AP model (O’Hara et al. (2011), so the results may be model-dependent. We preferred to use the model by O’Hara et al. because it has been widely used in the past decade and it was adopted by an expert group of the Federal Drug administration (FDA) as the starting point for developing an in silico model suitable for regulatory decision making (Colatsky et al., 2016). However, to asses the impact on the chosen model on the results, we carried out another separate simulation using the more recent human ventricular cell AP model by Tomek et al. (2019). The result is shown in Supplementary Figure S10. Again, the results with both models are very similar and our main conclusions regarding the potassium triphasic time course and the mechanisms responsible for the plateau are still valid.
Our tissue simulations have been performed with a 1D model, which may represent a limitation since the effect of electrotonic coupling is enhanced with spatial dimensionality. However, if the stimulation consists in a planar wave in 2D or 3D this effect is greatly reduced and differences in the results between 1D, 2D or 3D simulations are small. In this regard, our future work will simulate 30 min of dynamic acute ischemia in a real 3D geometry of a biventricular heart considering electrophysiological variability and tissue structure. We hope, in this case to reproduce a realistic border zone, but most importantly, this new set of simulations may provide further insights about the importance of electrotonic coupling in
One of the main limitations of the study is that our model does not consider the mechano-sensitivity of K (ATP) channels (Van Wagoner, 1993; Kohl et al., 2006; Peyronnet et al., 2016; Quinn and Kohl, 2021). Taking into account that stretch modulates the channel sensitivity to ATP and ADP, this could lead to further shortening of the AP duration and in turn this could affect the results regarding extracellular potassium accumulation and the formation of the BZ. Some models in the literature have considered this effect [e.g., (Li et al., 2006)] by simply scaling the maximum conductance of K (ATP) channels to account for stretch. We chose not to do so due to the lack of data regarding the stretch levels in the CIZ and the BZ. However, we acknowledge that this could affect the main results of this work. For instance, our main findings 1 and 2 could be hampered by the lack of mechano-electric feedback in the model. Further simulations with a model that includes the mechano-sensitivity of K (ATP) channels, as well as other stretch-activated non-specific channels, would be needed to asses its impact on the results, which could be related to possible depolarizing effects in the BZ that could eventually lead to triggered activity. However, such simulations were beyond the scope of the present work.
Moreover, the cellular ischemic model lacks a description of the effect of other ischemia-related parameters, such as cathecolamines, adenosine, fatty acid metabolites, etc., which could affect the outcome of the model. Also, the tissue model was one-dimensional and represented an electrophysiology homogeneous (though metabolically heterogeneous) cardiac strand. While it was useful to study the effects of regional ischemia on extracellular potassium accumulation, it lacks a proper anatomical and structural description of realistic three-dimensional ventricles. In particular, transmural and apex-to-base differences in APs, as well as anisotropy, could not be included in the model. Finally, inter-subject variability was not considered in our study. To do so, a set of population models should be carried out in which some relevant parameters (e.g., maximum conductances of ionic currents) were randomly varied within predetermined ranges. Our results thus correspond to an “average” cell and tissue.
5 Conclusion
A model of cardiac tissue electrophysiology to investigate the spatio-temporal evolution of
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author. The MATLAB and C codes used in the simulations are available upon request to the corresponding author.
Author contributions
JF and JRM conceived and designed the study. Computational simulations were performed by JF, JRM, and AG-A. The analysis/interpretation of the results was performed by JF and JRM, with contributions of AG-A. JF and JRM contributed to draft the manuscript, and all authors revised and approved the submitted version.
Funding
This work was supported by the “Programa Salvador de Madariaga 2018” of the Spanish Ministry of Science, Innovation and Universities (Grant Reference PRX18/00489), by Grant PID 2019-104356RB-C41 funded by the Spanish Ministry of Science and Innovation, and Italian Ministry of Education, University and Research, Grant number 1613 FISR2019_03221, CECOMES.
Acknowledgments
The authors would like to thank Dr. Ruben Coronel, Dr. Veronique Meijborg and Dr. Bas Boukens, from the Academic Medical Center in Amsterdam (The Netherlands), for their helpful discussions, comments and inputs on our work. Also, we acknowledge the help of Mireia Garcia-Daras and Patricia Olcina, from the Polytechnic University of Valencia (Spain), with the preliminary simulations that where the embryo of this work. Finally, we want to thank Marta Girones-Sanguesa, from the Polytechnic University of Valencia (Spain), for preliminary simulations with the Tomek et al. model.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2023.1074160/full#supplementary-material
References
Allen, D. G., and Orchard, C. H. (1987). Myocardial contractile function during ischemia and hypoxia. Circulation Res. 60, 153–168. doi:10.1161/01.RES.60.2.153
Babenko, A. P., Gonzalez, G., Aguilar-Bryan, L., and Bryan, J. (1998). Reconstituted human cardiac KATP channels. Circulation Res. 83, 1132–1143. doi:10.1161/01.RES.83.11.1132
Befroy, D. E., Powell, T., Radda, G. K., and Clarke, K. (1999). Osmotic shock: Modulation of contractile function, phi, and ischemic damage in perfused Guinea pig heart. Am. J. Physiology-Heart Circulatory Physiology 276, H1236–H1244. doi:10.1152/ajpheart.1999.276.4.H1236
Bersohn, M. M., Philipson, K. D., and Fukushima, J. Y. (1982). Sodium-calcium exchange and sarcolemmal enzymes in ischemic rabbit hearts. Am. J. Physiology-Cell Physiology 242, C288–C295. doi:10.1152/ajpcell.1982.242.5.C288
Cao, F., Zervou, S., and Lygate, C. A. (2018). The creatine kinase system as a therapeutic target for myocardial ischaemia–reperfusion injury. Biochem. Soc. Trans. 46, 1119–1127. doi:10.1042/BST20170504
Carpio, E. F., Gomez, J. F., Rodríguez-Matas, J. F., Trenor, B., and Ferrero, J. M. (2022). Analysis of vulnerability to reentry in acute myocardial ischemia using a realistic human heart model. Comput. Biol. Med. 141, 105038. doi:10.1016/j.compbiomed.2021.105038
Carpio, E. F., Gomez, J. F., Sebastian, R., Lopez-Perez, A., Castellanos, E., Almendral, J., et al. (2019). Optimization of lead placement in the right ventricle during cardiac resynchronization therapy. a simulation study. Front. Physiology 10, 74. doi:10.3389/fphys.2019.00074
Cascio, W. E., Yang, H., Muller-Borer, B. J., and Johnson, T. A. (2005). Ischemia-induced arrhythmia: The role of connexins, gap junctions, and attendant changes in impulse propagation. J. Electrocardiol. 38, 55–59. doi:10.1016/j.jelectrocard.2005.06.019
Colatsky, T., Fermini, B., Gintant, G., Pierson, J. B., Sager, P., Sekino, Y., et al. (2016). The comprehensive in vitro proarrhythmia assay (cipa) initiative — Update on progress. J. Pharmacol. Toxicol. Methods 81, 15–20. doi:10.1016/j.vascn.2016.06.002
Coronel, R., Fiolet, J. W., Wilms-Schopman, F. J., Schaapherder, A. F., Johnson, T. A., Gettes, L. S., et al. (1988). Distribution of extracellular potassium and its relation to electrophysiologic changes during acute myocardial ischemia in the isolated perfused porcine heart. Circulation 77, 1125–1138. doi:10.1161/01.CIR.77.5.1125
Coronel, R., Fiolet, J. W., Wilms-Schopman, J. G., Opthof, T., Schaapherder, A. F., and Janse, M. J. (1989). Distribution of extracellular potassium and electrophysiologic changes during two-stage coronary ligation in the isolated, perfused canine heart. Circulation 80, 165–177. doi:10.1161/01.CIR.80.1.165
Coronel, R. (1994). Heterogeneity in extracellular potassium concentration during early myocardial ischaemia and reperfusion: Implications for arrhythmogenesis. Cardiovasc. Res. 28, 770–777. doi:10.1093/cvr/28.6.770
Coronel, R., Wilms-Schopman, F. J., De Groot, J. R., Janse, M. J., Van Capelle, F. J., and De Bakker, J. M. (2000). Laplacian electrograms and the interpretation of complex ventricular activation patterns during ventricular fibrillation. J. Cardiovasc. Electrophysiol. 11, 1119–1128. doi:10.1111/j.1540-8167.2000.tb01758.x
Coronel, R., Wilms-Schopman, F. J., Opthof, T., van Capelle, F. J., and Janse, M. J. (1991). Injury current and gradients of diastolic stimulation threshold, tq potential, and extracellular potassium concentration during acute regional ischemia in the isolated perfused pig heart. Circulation Res. 68, 1241–1249. doi:10.1161/01.RES.68.5.1241
Cortassa, S., Aon, M. A., O’Rourke, B., Jacques, R., Tseng, H.-J., Marbán, E., et al. (2006). A computational model integrating electrophysiology, contraction, and mitochondrial bioenergetics in the ventricular myocyte. Biophysical J. 91, 1564–1589. doi:10.1529/biophysj.105.076174
Daleau, P. (1999). Lysophosphatidylcholine, a metabolite which accumulates early in myocardium during ischemia, reduces gap junctional coupling in cardiac cells. J. Mol. Cell. Cardiol. 31, 1391–1401. doi:10.1006/jmcc.1999.0973
Doering, A. E., Eisner, D. A., and Lederer, W. J. (1996). Cardiac Na − Ca exchange and pH. Ann. N. Y. Acad. Sci. 779, 182–198. doi:10.1111/j.1749-6632.1996.tb44786.x
Downar, E., Janse, M. J., and Durrer, D. (1977). The effect of acute coronary artery occlusion on subepicardial transmembrane potentials in the intact porcine heart. Circulation 56, 217–224. doi:10.1161/01.CIR.56.2.217
Dutta, S., Mincholé, A., Quinn, T. A., and Rodriguez, B. (2017). Electrophysiological properties of computational human ventricular cell action potential models under acute ischemic conditions. Prog. Biophysics Mol. Biol, 129, 40–52. doi:10.1016/j.pbiomolbio.2017.02.007
Egger, M., and Niggli, E. (2000). Paradoxical block of the Na+ − Ca2+ exchanger by extracellular protons in Guinea-pig ventricular myocytes. J. Physiology 523, 353–366. doi:10.1111/j.1469-7793.2000.t01-1-00353.x
Ferrero, J. M., Sáiz, J., Ferrero, J. M., and Thakor, N. V. (1996). Simulation of action potentials from metabolically impaired cardiac myocytes. Role of ATP-sensitive K+ current. Circulation Res. 79, 208–221. doi:10.1161/01.RES.79.2.208
Ferrero, J., Trenor, B., Saiz, J., Montilla, F., and Hernandez, V. (2003). Electrical activity and reentry in acute regional ischemia: Insights from simulations. Proceedings of the 25th annual international conference of the IEEE engineering in medicine and biology society (IEEE cat. No.03CH37439) 11, 17–20. doi:10.1109/IEMBS.2003.1279483
Friedrich, R., Hirche, H., Kebbel, U., Zylka, V., and Bissig, R. (1981). Changes of extracellular Na+, K+, Ca2+ and H+ of the ischemic myocardium in pigs. Basic Res. Cardiol. 76, 453–456. doi:10.1007/BF01908341
Friedrich, T., Bamberg, E., and Nagel, G. (1996). Na+, K+ − ATPase pump currents in giant excised patches activated by an atp concentration jump. Biophysical J. 71, 2486–2500. doi:10.1016/S0006-3495(96)79442-0
Gadsby, D. C. (1980). Activation of electrogenic Na+K+ exchange by extracellular K+ in canine cardiac purkinje fibers. Proc. Natl. Acad. Sci. 77, 4035–4039. doi:10.1073/pnas.77.7.4035
Gautier, M., Zhang, H., and Fearon, I. M. (2008). Peroxynitrite formation mediates lpc-induced augmentation of cardiac late sodium currents. J. Mol. Cell. Cardiol. 44, 241–251. doi:10.1016/j.yjmcc.2007.09.007
Glitsch, H. G., Kampmann, W., and Pusch, H. (1981). Activation of active na transport in sheep purkinje fibres by external K or Rb ions. Pflügers Arch. 391, 28–34. doi:10.1007/BF00580690
Harper, J. R., Johnston, T. A., Engle, C. L., Martin, D. G., Eleet, W., and Gettes, L. S. (1993). Effect of rate on changes in conduction velocity and extracellular potassium concentration during acute ischemia in the in situ pig heart. J. Cardiovasc. Electrophysiol. 4, 661–671. doi:10.1111/j.1540-8167.1993.tb01252.x
Harris, A. S., Bisteni, A., Russell, R. A., Brigham, J. C., and Firestone, J. E. (1954). Excitatory factors in ventricular tachycardia resulting from myocardial ischemia. potassium a major excitant. Science 119, 200–203. doi:10.1126/science.119.3085.200
Hill, J. L., and Gettes, L. S. (1980). Effect of acute coronary artery occlusion on local myocardial extracellular k+ activity in swine. Circulation 61, 768–778. doi:10.1161/01.CIR.61.4.768
Hirche, H., Franz, C., Bös, L., Bissig, R., Lang, R., and Schramm, M. (1980). Myocardial extracellular K+ and H+ increase and noradrenaline release as possible cause of early arrhythmias following acute coronary artery occlusion in pigs. J. Mol. Cell. Cardiol. 12, 579–593. doi:10.1016/0022-2828(80)90016-4
Hotokebuchi, N., Yano, T., Nishizono, Y., and Nishi, K. (1987). Changes in intra- and extracellular potassium and intracellular sodium activities induced by repetitive stimulation and their relation to membrane potential in Guinea-pig papillary muscle. Jpn. J. Physiology 37, 797–819. doi:10.2170/jjphysiol.37.797
Janse, M. J. (1990). Electrical activity immediately following myocardial infarction. New York: Futura, 739–753.
Janse, M. J., and Kléber, A. G. (1981). Electrophysiological changes and ventricular arrhythmias in the early phase of regional myocardial ischemia. Circulation Res. 49, 1069–1081. doi:10.1161/01.RES.49.5.1069
Janse, M. J., and van Capelle, F. J. (1982). Electrotonic interactions across an inexcitable region as a cause of ectopic activity in acute regional myocardial ischemia. a study in intact porcine and canine hearts and computer models. Circulation Res. 50, 527–537. doi:10.1161/01.RES.50.4.527
Janse, M. J., van Capelle, F. J., Morsink, H., Kléber, A. G., Wilms-Schopman, F., Cardinal, R., et al. (1980). Flow of ”injury” current and patterns of excitation during early ventricular arrhythmias in acute regional myocardial ischemia in isolated porcine and canine hearts. evidence for two different arrhythmogenic mechanisms. Circulation Res. 47, 151–165. doi:10.1161/01.RES.47.2.151
Janse, M. J., and Wit, A. L. (1989). Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol. Rev. 69, 1049–1169. doi:10.1152/physrev.1989.69.4.1049
Kanda, A., Watanabe, I., Williams, M. L., Engle, C. L., Li, S., Koch, G. G., et al. (1997). Unanticipated lessening of the rise in extracellular potassium during ischemia by pinacidil. Circulation 95, 1937–1944. doi:10.1161/01.CIR.95.7.1937
Kazbanov, I. V., Clayton, R. H., Nash, M. P., Bradley, C. P., Paterson, D. J., Hayward, M. P., et al. (2014). Effect of global cardiac ischemia on human ventricular fibrillation: Insights from a multi-scale mechanistic model of the human heart. PLOS Comput. Biol. 10, 10038911–e1003915. doi:10.1371/journal.pcbi.1003891
Keller, D., Weber, F. M., Seemann, G., and Dössel, O. (2010). Ranking the influence of tissue conductivities on forward-calculated ecgs. IEEE Trans. Biomed. Eng. 57, 1568–1576. doi:10.1109/TBME.2010.2046485
Kléber, A. G. (1984). Extracellular potassium accumulation in acute myocardial ischemia. J. Mol. Cell. Cardiol. 16, 389–394. doi:10.1016/S0022-2828(84)80610-0
Kléber, A. G., Janse, M. J., van Capelle, F. J., and Durrer, D. (1978). Mechanism and time course of s-t and t-q segment changes during acute regional myocardial ischemia in the pig heart determined by extracellular and intracellular recordings. Circulation Res. 42, 603–613. doi:10.1161/01.RES.42.5.603
Kléber, A. G. (1983). Resting membrane potential, extracellular potassium activity, and intracellular sodium activity during acute global ischemia in isolated perfused Guinea pig hearts. Circulation Res. 52, 442–450. doi:10.1161/01.RES.52.4.442
Kléber, A., Riegger, C. B., and Janse, M. J. (1987). Extracellular K+ and H+ shifts in early ischemia: Mechanisms and relation to changes in impulse propagation. J. Mol. Cell. Cardiol. 19, 35–44. doi:10.1016/S0022-2828(87)80608-9
Kohl, P., Bollensdorff, C., and Garny, A. (2006). Effects of mechanosensitive ion channels on ventricular electrophysiology: Experimental and theoretical models. Exp. Physiol. 91, 307–321. doi:10.1113/expphysiol.2005.031062
Kunze, D. L. (1977). Rate-dependent changes in extracellular potassium in the rabbit atrium. Circulation Res. 41, 122–127. doi:10.1161/01.RES.41.1.122
Li, W., Kohl, P., and Trayanova, N. (2006). Myocardial ischemia lowers precordial thump efficacy: An inquiry into mechanisms using three-dimensional simulations. Heart rhythm. 3, 179–186. doi:10.1016/j.hrthm.2005.10.033
Luo, C. H., and Rudy, Y. (1994). A dynamic model of the cardiac ventricular action potential. i. simulations of ionic currents and concentration changes. Circulation Res. 74, 1071–1096. doi:10.1161/01.RES.74.6.1071
Martišienė, I., Jurevičius, J., Vosyliutė, R., Navalinskas, A., Treinys, R., Mačianskienė, R., et al. (2015). Evolution of action potential alternans in rabbit heart during acute regional ischemia. BioMed Res. Int. 2015, 951704. doi:10.1155/2015/951704
Mena Tobar, A., Ferrero, J. M., Migliavacca, F., and Rodriguez Matas, J. F. (2018). Vulnerability in regionally ischemic human heart. effect of the extracellular potassium concentration. J. Comput. Sci. 24, 160–168. doi:10.1016/j.jocs.2017.11.009
Mitani, A., and Shattock, M. J. (1992). Role of Na-activated K channel, Na − K − Cl cotransport, and Na − K pump in [K]e changes during ischemia in rat heart. Am. J. Physiology-Heart Circulatory Physiology 263, H333–H340. doi:10.1152/ajpheart.1992.263.2.H333
Murphy, L., Renodin, D., Antzelevitch, C., Di Diego, J. M., and Cordeiro, J. M. (2011). Extracellular proton depression of peak and late Na+ current in the canine left ventricle. Am. J. Physiology-Heart Circulatory Physiology 301, H936–H944. doi:10.1152/ajpheart.00204.2011
Nakashima, M., Hashimoto, H., Kanamura, M., Nagaya, T., Hashizume, M., and Oishi, H. (1978). Experimental studies and clinical report on the electrical alternans of ST segment during myocardial ischemia. Jpn. Heart J. 19, 396–408. doi:10.1536/ihj.19.396
Niederer, S. A., Kerfoot, E., Benson, A. P., Bernabeu, M. O., Bernus, O., Bradley, C., et al. (2011). Verification of cardiac tissue electrophysiology simulators using an N-version benchmark. Philosophical Trans. R. Soc. A Math. Phys. Eng. Sci. 369, 4331–4351. doi:10.1098/rsta.2011.0139
Niederer, S. (2013). Regulation of ion gradients across myocardial ischemic border zones: A biophysical modelling analysis. PLOS ONE 8, 603233–e60421. doi:10.1371/journal.pone.0060323
O’Hara, T., Virág, L., Varró, A., and Rudy, Y. (2011). Simulation of the undiseased human cardiac ventricular action potential: Model formulation and experimental validation. PLOS Comput. Biol. 7, 10020611–e1002129. doi:10.1371/journal.pcbi.1002061
Parker, J. O., Chiong, M. A., West, R. O., and Case, R. B. (1970). The effect of ischemia and alterations of heart rate on myocardial potassium balance in man. Circulation 42, 205–217. doi:10.1161/01.CIR.42.2.205
Peyronnet, R., Nerbonne, J. M., and Kohl, P. (2016). Cardiac mechano-gated ion channels and arrhythmias. Circulation Res. 118, 311–329. doi:10.1161/CIRCRESAHA.115.305043
Potse, M., Coronel, R., LeBlanc, A.-R., and Vinet, A. (2007a). “Modeling transport of interstitial potassium in regional myocardial ischemia: Effect on the injury current,” in 2007 29th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, 6330–6333. doi:10.1109/IEMBS.2007.4353803
Potse, M., Coronel, R., LeBlanc, A. R., and Vinet, A. (2007b). The role of extracellular potassium transport in computer models of the ischemic zone. Med. Biol. Eng. Comput. 45, 1187–1199. doi:10.1007/s11517-007-0276-9
Quinn, T. A., and Kohl, P. (2021). Cardiac mechano-electric coupling: Acute effects of mechanical stimulation on heart rate and rhythm. Physiol. Rev. 101, 37–92. doi:10.1152/physrev.00036.2019
Rodríguez, B., Ferrero, J. M., and Trénor, B. (2002). Mechanistic investigation of extracellular K+ accumulation during acute myocardial ischemia: A simulation study. Am. J. Physiology-Heart Circulatory Physiology 283, H490–H500. doi:10.1152/ajpheart.00625.2001
Romero, L., Trénor, B., Alonso, J., Tobón, C., Saiz, J., and Ferrero, J. (2009). The relative role of refractoriness and source–sink relationship in reentry generation during simulated acute ischemia. Ann. Biomed. Eng. 37, 1560–1571. doi:10.1007/s10439-009-9721-2
Roth, G. A., Mensah, G. A., and Fuster, V. (2020). The global burden of cardiovascular diseases and risks: A compass for global action. J. Am. Coll. Cardiol. 76, 2980–2981. doi:10.1016/j.jacc.2020.11.021
Saegusa, N., Moorhouse, E., Vaughan-Jones, R. D., and Spitzer, K. W. (2011). Influence of pH on Ca2+ current and its control of electrical and Ca2+ signaling in ventricular myocytes. J. General Physiology 138, 537–559. doi:10.1085/jgp.201110658
Sakamoto, K., Ishikawa, M., Koga, K., Urushidani, T., and Nagao, T. (2000). Energy preserving effect of l-cis diltiazem in isolated ischemic and reperfused Guinea pig hearts:a 31P − NMR study. Jpn. J. Pharmacol. 83, 225–232. doi:10.1254/jjp.83.225
Shivkumar, K., Deutsch, N. A., Lamp, S. T., Khuu, K., Goldhaber, J. I., and Weiss, J. N. (1997). Mechanism of hypoxic K loss in rabbit ventricle. J. Clin. Investigation 100, 1782–1788. doi:10.1172/JCI119705
Smith, W. T., Fleet, W. F., Johnson, T. A., Engle, C. L., and Cascio, W. E. (1995). The Ib phase of ventricular arrhythmias in ischemic in situ porcine heart is related to changes in cell-to-cell electrical coupling. Experimental Cardiology Group, University of North Carolina. Circulation 92, 3051–3060. doi:10.1161/01.CIR.92.10.3051
Sobel, B. E., Corr, P. B., Robison, A. K., Goldstein, R. A., Witkowski, F. X., and Klein, M. S. (1978). Accumulation of lysophosphoglycerides with arrhythmogenic properties in ischemic myocardium. J. Clin. Investigation 62, 546–553. doi:10.1172/JCI109159
Sutton, P. M. I., Taggart, P., Opthof, T., Coronel, R., Trimlett, R., Pugsley, W., et al. (2000). Repolarisation and refractoriness during early ischaemia in humans. Heart 84, 365–369. doi:10.1136/heart.84.4.365
Taggart, P., Sutton, P. M., Opthof, T., Coronel, R., Trimlett, R., Pugsley, W., et al. (2000). Inhomogeneous transmural conduction during early ischaemia in patients with coronary artery disease. J. Mol. Cell. Cardiol. 32, 621–630. doi:10.1006/jmcc.2000.1105
Terkildsen, J. R., Crampin, E. J., and Smith, N. P. (2007). The balance between inactivation and activation of the Na+ − K+ pump underlies the triphasic accumulation of extracellular K+ during myocardial ischemia. Am. J. Physiology-Heart Circulatory Physiology 293, H3036–H3045. doi:10.1152/ajpheart.00771.2007
Tice, B. M., Rodríguez, B., Eason, J., and Trayanova, N. (2007). Mechanistic investigation into the arrhythmogenic role of transmural heterogeneities in regional ischaemia phase 1A. EP Eur. 9, vi46–vi58. doi:10.1093/europace/eum204
Tomek, J., Bueno-Orovio, A., Passini, E., Zhou, X., Minchole, A., Britton, O., et al. (2019). Development, calibration, and validation of a novel human ventricular myocyte model in health, disease, and drug block. eLife 8, e48890. doi:10.7554/eLife.48890
Trénor, B., Romero, L., Ferrero, J., Sáiz, J., Moltó, G., and Alonso, J. (2007). Vulnerability to reentry in a regionally ischemic tissue: A simulation study. Ann. Biomed. Eng. 35, 1756–1770. doi:10.1007/s10439-007-9353-3
Van Wagoner, D. R. (1993). Mechanosensitive gating of atrial atp-sensitive potassium channels. Circulation Res. 72, 973–983. doi:10.1161/01.RES.72.5.973
Vaughan-Jones, R. D., Spitzer, K. W., and Swietach, P. (2009). Intracellular ph regulation in heart. J. Mol. Cell. Cardiol. 46, 318–331. doi:10.1016/j.yjmcc.2008.10.024
Walfridsson, H., Odman, S., and Lund, N. (1985). Myocardial oxygen pressure across the lateral border zone after acute coronary occlusion in the pig heart. Adv. Exp. Med. Biol. 191, 203–210. doi:10.1007/978-1-4684-3291-6_20
Watanabe, I., and Gettes, L. S. (2018). Effects of verapamil and pinacidil on extracellular K+, pH, and the incidence of ventricular fibrillation during 60 minutes of ischemia. Int. Heart J. 59, 589–595. doi:10.1536/ihj.17-175
Watanabe, I., and Gettes, L. S. (2016). Initial and secondary ST − T alternans during acute myocardial ischemia in the in-situ pig heart. Int. Heart J. 57, 327–335. doi:10.1536/ihj.15-337
Watson, C. L., and Gold, M. R. (1995). Effect of intracellular and extracellular acidosis on sodium current in ventricular myocytes. Am. J. Physiology-Heart Circulatory Physiology 268, H1749–H1756. doi:10.1152/ajpheart.1995.268.4.H1749
Weiss, J. N., Lamp, S. T., and Shine, K. I. (1989). Cellular K+ loss and anion efflux during myocardial ischemia and metabolic inhibition. Am. J. Physiology-Heart Circulatory Physiology 256, H1165–H1175. doi:10.1152/ajpheart.1989.256.4.H1165
Weiss, J. N., Qu, Z., and Shivkumar, K. (2017). Electrophysiology of hypokalemia and hyperkalemia. Circulation Arrhythmia Electrophysiol. 10, e004667. doi:10.1161/CIRCEP.116.004667
Weiss, J. N., Venkatesh, N., and Lamp, S. T. (1992). ATP-sensitive K+ channels and cellular K+ loss in hypoxic and ischaemic mammalian ventricle. J. Physiology 447, 649–673. doi:10.1113/jphysiol.1992.sp019022
Weiss, J., and Shine, K. I. (1986). Effects of heart rate on extracellular K+ accumulation during myocardial ischemia. Am. J. Physiology-Heart Circulatory Physiology 250, H982–H991. doi:10.1152/ajpheart.1986.250.6.H982
Weiss, J., and Shine, K. I. (1982a). Extracellular K+ accumulation during myocardial ischemia in isolated rabbit heart. Am. J. Physiology-Heart Circulatory Physiology 242, H619–H628. doi:10.1152/ajpheart.1982.242.4.H619
Weiss, J., and Shine, K. I. (1981). Extracellular potassium accumulation during myocardial ischemia: Implications for arrhythmogenesis. J. Mol. Cell. Cardiol. 13, 699–704. doi:10.1016/0022-2828(81)90277-7
Weiss, J., and Shine, K. I. (1982b). [K+]o accumulation and electrophysiological alterations during early myocardial ischemia. Am. J. Physiology-Heart Circulatory Physiology 243, H318–H327. doi:10.1152/ajpheart.1982.243.2.H318
Wilde, A. A., and Aksnes, G. (1995). Myocardial potassium loss and cell depolarisation in ischaemia and hypoxia. Cardiovasc. Res. 29, 1–15. doi:10.1016/S0008-6363(96)88539-7
Wilde, A. A., Escande, D., Schumacher, C. A., Thuringer, D., Mestre, M., Fiolet, J. W., et al. (1990). Potassium accumulation in the globally ischemic mammalian heart. a role for the ATP-sensitive potassium channel. Circulation Res. 67, 835–843. doi:10.1161/01.RES.67.4.835
Yan, G.-X., Joshi, A., Guo, D., Hlaing, T., Martin, J., Xu, X., et al. (2004). Phase 2 reentry as a trigger to initiate ventricular fibrillation during early acute myocardial ischemia. Circulation 110, 1036–1041. doi:10.1161/01.CIR.0000140258.09964.19
Keywords: hyperkalemia, myocardial ischemia, injury current, computational model, alternans, potassium loss
Citation: Ferrero JM, Gonzalez-Ascaso A and Matas JFR (2023) The mechanisms of potassium loss in acute myocardial ischemia: New insights from computational simulations. Front. Physiol. 14:1074160. doi: 10.3389/fphys.2023.1074160
Received: 19 October 2022; Accepted: 13 February 2023;
Published: 27 February 2023.
Edited by:
Mark Potse, UMR5251 Institut de mathématiques de Bordeaux (IMB), FranceReviewed by:
Mary (Molly) Maleckar, Simula Research Laboratory, NorwayBreanne A. Cameron, University Heart Center Freiburg, Germany
Steven Alexander Niederer, King’s College London, United Kingdom
Copyright © 2023 Ferrero, Gonzalez-Ascaso and Matas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jose M. Ferrero, Y2ZlcnJlcm9AY2kyYi51cHYuZXM=