Abstract
Duchenne muscular dystrophy (DMD) is a severe, progressive, and ultimately fatal disease of skeletal muscle wasting, respiratory insufficiency, and cardiomyopathy. The identification of the dystrophin gene as central to DMD pathogenesis has led to the understanding of the muscle membrane and the proteins involved in membrane stability as the focal point of the disease. The lessons learned from decades of research in human genetics, biochemistry, and physiology have culminated in establishing the myriad functionalities of dystrophin in striated muscle biology. Here, we review the pathophysiological basis of DMD and discuss recent progress toward the development of therapeutic strategies for DMD that are currently close to or are in human clinical trials. The first section of the review focuses on DMD and the mechanisms contributing to membrane instability, inflammation, and fibrosis. The second section discusses therapeutic strategies currently used to treat DMD. This includes a focus on outlining the strengths and limitations of approaches directed at correcting the genetic defect through dystrophin gene replacement, modification, repair, and/or a range of dystrophin-independent approaches. The final section highlights the different therapeutic strategies for DMD currently in clinical trials.
1 Genetic basis and clinical presentation of DMD
Duchenne muscular dystrophy (DMD) is a severe X-linked recessive disorder caused by mutations in the dystrophin gene and consequent complete loss of dystrophin protein expression (Hoffman et al., 1987). The incidence of DMD is estimated at 1:5,000 boys worldwide, making it one of the most common recessive disorders in humans. The DMD gene that encodes dystrophin is located at position Xp21.1 of the X chromosome. DMD is the largest known gene in the human body, spanning 2.5 MB of DNA with 79 exons and 78 introns (Cohn and Campbell, 2000). The corresponding 14 kb mRNA transcript generates a 427 kDa protein with 3,685 amino acids (Hoffman et al., 1987; Emery, 2002).
Due to its massive size, the mutation rate for the dystrophin gene is quite high, with about one-third of mutations occurring de novo and the remaining caused by germline mosaicism or inheritance from a carrier mother (Nallamilli et al., 2021). More than 7,000 different dystrophin mutations have been identified in patients with DMD or Becker muscular dystrophy (BMD) (Bladen et al., 2015). Approximately 60% of mutations in patients with DMD are deletions, and 11% are duplications, with the remainder being small mutations affecting the coding sequence and splice sites (Bladen et al., 2015). Deletions and duplications occur predominantly in two hotspots of the DMD gene, located at exons 3–9 and 45–55 (Min et al., 2019a). The high mutation rate, along with the many different ways in which a frameshift could occur, raises significant challenges for the development of gene correction therapies applicable to patients with different dystrophin mutations.
DMD mutations preventing the production of dystrophin hold dire consequences for protection against muscle membrane stress. Dystrophin forms a mechanical link between cytoskeletal actin and the extracellular matrix (ECM), which protects the sarcolemma from the force associated with stretch and contraction (Campbell and Kahl, 1989; Boland et al., 1996; Houang et al., 2018). Secondary to deficient membrane stability is the disorganization of numerous proteins associated with dystrophin, as well as impairment of their distribution and cell signaling, both leading to severe and progressive muscle wasting (Emery, 1993; Jungbluth et al., 2018). Women act as carriers, and although affected men appear normal at birth, steady muscle weakness and wasting are first apparent in proximal limb muscles before spreading to more distal muscles (Mercuri et al., 2019), leading to the disease being typically diagnosed in the first few years of life (Tyler, 2003; Birnkrant et al., 2018). The earliest symptoms such as a waddling gait, frequent falls, difficulties standing up from a sitting position (Gower’s sign), and struggling to climb stairs can be seen between ages 2 and 3 (Emery, 2002). Affected children exhibit delayed gross motor development, and at ages 10–12, most patients require a wheelchair (Tsuda, 2018). As muscle weakness progresses, scoliosis and joint contractures develop, which promote restrictive lung disease (Bushby et al., 2010). Assisted ventilation is necessary by about 15–20 years of age, and the majority of DMD patients die from cardiac/respiratory failure between 20 and 30 years of age, even when optimal care has been administered (Mendell et al., 2012; Mercuri et al., 2019). Additionally, the corresponding immobility hastens the decline in bone density and increases the risk of fracture (McDonald et al., 2002).
The cardiac muscle must withstand significant mechanical stress to pump blood throughout the body, and the consequences of this stress are greatly exacerbated in DMD patients. Membrane instability leads to gradual cardiac myocyte death, resulting in fibrosis and secondary fatty infiltration (Townsend et al., 2011; Houang et al., 2018; Tsuda, 2018). This process decreases diastolic/systolic function, which plays a role in the atrophy and remodeling that gives rise to heterogeneous pathological alterations to the ventricular myocardium (Amin et al., 2002; Desguerre et al., 2009; Spurney, 2011a; Lee et al., 2014). Life-threatening consequences of cardiac muscle dystrophin deficiency include dilated cardiomyopathy, arrhythmias, and congestive heart failure (Kamdar and Garry, 2016; McNally and Wyatt, 2017; Buddhe et al., 2019; Meyers and Townsend, 2019). Along with these debilitating effects on cardiac function, the pulmonary ramifications of DMD are a key co-contributor to patient mortality (Emery, 2002). Weakened respiratory muscles, particularly the diminishment of diaphragm function, cause severe ventilatory insufficiency (Benditt and Boitano, 2005; Bushby et al., 2010). Additionally, incidences of otherwise benign upper respiratory tract infections can lead to serious acute respiratory failure in DMD, as patients cannot cough effectively. Diminished cough capacity causes airway secretions to be retained that in turn increases the risk of pneumonia and atelectasis (Bushby et al., 2010). Reduction in vital capacity and elevated residual volume due to impaired expiratory muscle function is also exhibited, which further necessitates assisted ventilation later in the disease progression (Bach et al., 1997; Benditt and Boitano, 2005; Bushby et al., 2010).
Although dystrophin deficiency in muscle tissues is the primary cause of the life-limiting issues associated with DMD, a lack of brain dystrophin has consequences as well (Duan et al., 2021). DMD patients reportedly have a higher susceptibility to seizures as compared to dystrophin replete individuals. Additionally, cognitive, neuropsychological, and neurobehavioral abnormalities are more likely to occur in DMD patients (Boyce et al., 1991; Kim et al., 1995). The general intelligence of affected boys is one standard deviation below the normal mean IQ, with 34.8% of patients reported to have intellectual disabilities (Cotton et al., 2001). Moreover, ADHD, autism spectrum disorder, and moderate-to-severe reading problems were exhibited in 11.7%, 3.1%, and 20% of patients, respectively (Hendriksen and Vles, 2006; Hendriksen and Vles, 2008).
The survival of patients with DMD has improved over time. With optimal care, patients with DMD can survive to live in their thirties and forties, mainly owing to the development of guidelines for care and management and improved treatment for cardiopulmonary dysfunction (Birnkrant et al., 2018).
2 Dystrophin function
Dystrophin is an essential cytoskeletal protein in muscle that localizes to the inner surface of the muscle cell membrane and is enriched at the costameres and sites of cell–cell contact (Ervasti and Campbell, 1993). Dystrophin is the connection point joining the actin cytoskeleton to the inner surface of the sarcolemma (Hoffman et al., 1987; Ahn and Kunkel, 1993). Dystrophin has the characteristic biochemical properties of a membrane cytoskeletal protein and is recognized as a major member of the spectrin-type superfamily of actin-binding proteins (Petrof et al., 1993; Straub et al., 1997). In muscle cells, dystrophin plays an important role in maintaining membrane integrity and preventing membrane rupture (Emery, 2002; Gao and McNally, 2015).
The dystrophin protein has four main functional domains: 1) an N-terminal region with homologies to the actin-binding domains of α-actinin that anchors the protein to the cytoskeleton; 2) a central rod domain containing 24 spectrin-like repeats separated by four hinge regions; 3) a cysteine-rich domain that anchors dystrophin to the muscle membrane via interaction with the transmembrane protein β-dystroglycan; and 4) a C-terminal domain that binds dystrobrevin and syntrophins, mediating sarcolemma localization (Ervasti, 2013). The N-terminal domain includes two calponin homology domains (CH1 and CH2). This conventional CH-actin binding domain binds directly to F actin, anchoring dystrophin to the cytoskeleton (Rybakova et al., 1996). The spectrin-like repeats in the central rod of dystrophin indicate that dystrophin works as a shock absorber, helping to resist repeated rounds of muscle contraction and relaxation. The rod domain also harbors a second actin-binding motif (ABD2). ABD2 is found near the middle of the rod and works in concert with ABD1 to form a strong lateral association with actin filaments (Straub et al., 1992). The rod domain also mediates dystrophin interaction with microtubules via spectrin-like repeats 20–23 and is required for the organization of the microtubule lattice in skeletal muscle cells. Furthermore, the rod forms a flexible linker between the amino- and carboxy-termini. Four short proline-rich spacers, called “hinges,” are interspersed in the mid-rod domain providing elasticity to the protein (Koenig et al., 1988). Hinge 4 is at the end of the rod domain and contains a WW domain, a domain implicated in protein–protein interactions (Koenig and Kunkel, 1990; Straub et al., 1992). The WW domain, along with two neighboring EF-hands, binds the carboxy-terminus of β-dystroglycan, anchoring dystrophin at sarcolemma (Koenig et al., 1988). In dystrophin, the two EF-hands are located in the cysteine-rich domain, which resides between the central rod and the C-terminus. The cysteine-rich domain also contains a zinc finger (ZZ) domain (Ishikawa-Sakurai et al., 2004). The carboxy-terminal (CT) domain is α-helical and provides binding sites for dystrobrevin and syntrophins, mediating their sarcolemma localization.
Connection is achieved through the dystrophin-glycoprotein complex (DGC), which consists of extracellular (α-dystroglycan), transmembrane (sarcoglycans, β-dystroglycan, sarcospan), and cytoplasmic proteins (dystrophin, syntrophins, neuronal nitric oxide synthase, dystrobrevin), providing a link between the intracellular cytoskeleton and the ECM (Campbell and Kahl, 1989; Ervasti and Campbell, 1991).
It has been shown that α-dystroglycan resides on the extracellular surface of the sarcolemma and functions as a receptor for extracellular ligands such as laminin (Montanaro et al., 1999). Here, α-dystroglycan is tightly associated with β-dystroglycan, a transmembrane protein that also interacts with dystrophin. At the sarcolemma, β-dystroglycan is tightly associated with the sarcoglycan subcomplex, which is composed of four single-pass transmembrane proteins: α-sarcoglycan, β-sarcoglycan, γ-sarcoglycan, and δ-sarcoglycan (Ervasti and Campbell, 1993). The sarcoglycan complex is also tightly associated with sarcospan, a small transmembrane protein. On the cytoplasmic side of the sarcolemma, dystrophin maintains its membrane localization by interacting with β-dystroglycan. Dystrophin binds to the intracellular actin network to link the cytoskeleton to DGC, which in turn connects to basal lamina by interacting with ECM ligands. Other cytoplasmic components of DGC include α-dystrobrevin, syntrophins, and neuronal nitric oxide synthase (nNOS) (Ervasti, 2013). The α-dystrobrevin/syntrophin triplet associates with dystrophin and anchors nNOS to the sarcolemma (Guiraud et al., 2015).
3 Pathophysiological mechanisms of Duchenne muscular dystrophy
3.1 Membrane instability
In striated muscle, force is generated through cycles of contraction and relaxation of muscle fibers as driven via the sarcomere (Gordon et al., 2000). As discussed previously, in healthy muscle, the integrity of the sarcolemma is maintained through interactions between the sarcolemma, cytoskeleton, and ECM through the DGC and the integrin complex (Houang et al., 2018; Duan et al., 2021). However, in DMD, due to the lack of dystrophin and its associated proteins, the physical connection between the sarcolemma, cytoskeleton, and ECM is lost, causing the sarcolemma to become leaky and highly susceptible to injury (Guiraud et al., 2015; Houang et al., 2018; Duan et al., 2021). This mechanism is supported by several observations. Previous work has identified hallmarks of muscle injury in DMD patients in the form of sarcolemmal tears (so-called “delta” lesions) (Mokri and Engel, 1975; Pestronk et al., 1982; Duan et al., 2021), increased levels of creatine kinase in the serum, and the uptake of large proteins including albumin and Evans blue dye (Houang et al., 2018; Duan et al., 2021). As levels of muscle damage are directly linked to levels of muscle stress, those muscles that undergo higher levels of stress are more likely to incur higher levels of damage. This can be seen in skeletal muscles involved in the exercise, specifically those muscles undergoing lengthening contractions, where it has been shown that repetitive contractions lead to increases in sarcolemmal damage (Petrof et al., 1993; Allen et al., 2016). This can also be seen in the diaphragm, where increased levels of injury are seen in DMD patients (Stedman et al., 1991). Taken together, these works demonstrate the need for the dystrophin complex to maintain the structural integrity of the muscle sarcolemma and highlight the devastating consequences of muscle fiber membrane stability in DMD.
3.2 Calcium dysregulation
A major consequence of the increased levels of membrane instability and leakiness in DMD muscles is an increase in intracellular calcium levels and disrupted calcium homeostasis within the muscle (Hopf et al., 1996; Mareedu et al., 2021). There is some debate over the mechanisms leading to increased intracellular calcium in the DMD muscle. Previous work has suggested that calcium can enter the muscle fibers through membrane tears (Yasuda et al., 2005; Houang et al., 2018), through a number of different calcium channels, including store-operated, voltage-gated, or stretch-activated (Mareedu et al., 2021), or through sodium channels (Mareedu et al., 2021).
Although the mechanism of calcium entry is not fully resolved, it is clear that increased calcium levels in DMD muscles have a significant negative impact on function. This can be seen through the myriad of calcium-signaling pathways in the cell, including calmodulin (Pertille et al., 2010), calpains (Gailly et al., 2007), and dysferlin (Bansal et al., 2003; Houang et al., 2018), all of which can negatively impact calcium cycling and muscle repair. Another possible mechanism of calcium dysfunction in DMD that has become a recent area of focus is the sarcoplasmic reticulum (SR) (Duan et al., 2021). In DMD muscle fibers, it has been shown that there is an increase in passive calcium leak from the SR, and therefore sarco-endoplasmic reticulum calcium ATPase (SERCA) function is reduced, leading to an increase in cytosolic calcium levels (Duan et al., 2021; Mareedu et al., 2021). It has been suggested that this process may occur through SR crosstalk with reactive oxygen species (ROS) (Papa and Skulachev, 1997; Houang et al., 2018; Mareedu et al., 2021). Further damage in the muscle can also be the result of calcium-mediated changes in mitochondrial function, which can lead to a reduction in ATP production and loss of membrane potential (Kyrychenko et al., 2015; Houang et al., 2018; Kang et al., 2018; Mareedu et al., 2021). Both of these mechanisms are discussed in more detail as follows. It is evident in dystrophic muscle fibers that the number of ways calcium-signaling can negatively impact muscle function and the increased levels of cytosolic calcium in DMD muscle is unmistakably a driving force in the pathology of the disease.
3.3 Inflammation
Immune cell infiltration is a main feature of DMD and is strongly associated with disease severity (Rosenberg et al., 2015; Villalta et al., 2015; Saclier et al., 2021). In DMD patients, inflammation onset precedes the onset of clinical symptoms (Pescatori et al., 2007). The involvement of the immune system after DMD onset, when there is already marked muscle damage, is well established (Vidal et al., 2008; Burzyn et al., 2013; Mojumdar et al., 2014; Mázala et al., 2020; Saclier et al., 2021). However, little is known about the role of inflammation in the earliest phase of DMD prior to the clinical onset (Evans et al., 2009).
Skeletal muscle is host to a heterogeneous combination of endothelial, stem, and immune cells, including macrophages, natural killer (NK) cells, T cells, B cells, and neutrophils (Tidball and Villalta, 2010). Immune cells play an important role in skeletal muscle homeostasis, repair, and regeneration (Tidball and Villalta, 2010; Castiglioni et al., 2015; Sciorati et al., 2016). The unique immune-privileged status of skeletal muscle (Sciorati et al., 2016) relies on the absence of constitutive MHCI expression and the lower number of antigen-presenting cells and pro-inflammatory cells in homeostasis, resulting in less necrosis and a lower capacity to generate abscesses (Evans et al., 2009; Tibdall et al., 2018). In the setting of acute muscle injury, neutrophils and pro-inflammatory macrophages are early players and are essential in clearing damaged fibers. They are then gradually replaced by anti-inflammatory pro-regenerative macrophages to stimulate fibers regeneration and fibrosis (Evans et al., 2009; Tidball and Villalta, 2010; Castiglioni et al., 2015; Tidball et al., 2018; Gehlert and Jacko, 2019). A distinct population of regulatory T cells (Foxp3+ Treg) drives the transition to a pro-regenerative state and potentiates muscle regeneration and repair (Burzyn et al., 2013).
As detailed previously, membrane instability is a primary defect in DMD. In healthy muscles, intense exercise causes membrane damage and leakage of cytoplasmic content into the extracellular compartment (Sorichter et al., 1998; Sorichter et al., 1999). This is associated with increased circulating levels of muscle proteins such as fatty acid binding protein and muscle-specific creatine kinase that can be used as biomarkers to detect and evaluate these injuries (Sorichter et al., 1998). Due to the muscle’s unique immune privilege, rapid membrane repair mechanisms, and membrane stability conferred by intact dystrophin, this inflammatory response is limited, controlled, and quickly resolved in healthy muscle (Sciorati et al., 2016). In DMD, the muscle loses its immune privilege. Membrane instability caused by dystrophin deficiency leads to the continuous release of cytoplasmic content, in particular, damage-associated molecular patterns (DAMPs) that are ligands to toll-like receptors (TLRs), purinoceptors (P2RX7), and other pattern recognition receptors (PRRs) on muscle and immune cells (Henriques-Pons et al., 2014; Giordano et al., 2015; Sinadinos et al., 2015). Upon activation, PRR downstream signaling cascades initiate the innate immune response. Moreover, pro-inflammatory cytokines induce the expression of MHCI and MHCII on muscle fibers, thus breaking down the muscle immune privilege. In addition to membrane instability, intracellular calcium overload and increased oxidative stress further induce tissue damage, contributing to triggering the inflammatory cascade in dystrophin-deficient muscles (Wehling et al., 2001; Henriques-Pons et al., 2014).
The immune cell infiltration in young DMD patients is predominantly formed by macrophages and T cells (Evans et al., 2009). Similar findings were revealed in dystrophin-deficient murine muscles (Lozanoska-Ochser et al., 2018). Early in the DMD time course, degenerated muscle fibers are cleared and repaired by regeneration (Tidball and Villalta, 2010; Tidball et al., 2018). The co-existence of lesions of different ages and the overlapping of pro-inflammatory and anti-inflammatory signaling prevent the full resolution of inflammation and sustain a chronic inflammatory response, which further progresses muscle damage. As the disease progresses, the impaired regenerative capacity and the chronic inflammatory environment of dystrophic muscle lead to fibrosis and adipose tissue accumulation (Vidal et al., 2008; Evans et al., 2009; Rosenberg et al., 2015; Mázala et al., 2020).
Macrophages play a key role in the disease progression of DMD (Rosenberg et al., 2015). Genetic ablation of CCR2 in a dystrophin-deficient mouse model (mdx), which prevents the monocytes from exiting the bone marrow, reduces the pro-inflammatory macrophage infiltration into the dystrophin-deficient muscle and is shown to improve muscle histology and function (Mojumdar et al., 2014). Bhattarai et al. (2022) showed, in the necrotic phase, a TLR4-dependent alteration of the inflammatory phenotype of the mdx mice bone-marrow-derived macrophages, with implications in major remodeling of the macrophage metabolic and epigenetic landscape. These changes correspond to characteristics of trained immunity and indicate that trained immunity can play an important role in DMD pathogenesis and progression (Bhattarai et al., 2022). T cells also play a crucial role in DMD pathogenesis (Lozanoska-Ochser et al., 2018; Tidball et al., 2018). CD4+ helper T cells promote the immune response by producing inflammatory cytokines (Spencer et al., 2001). CD8+ T-cell perforin-mediated cytotoxicity and fatty acid synthetase-induced cytotoxicity cause muscle fiber apoptosis (Spencer et al., 1997; Cai et al., 2000; Spencer et al., 2001). Effector T cells were recently shown to be one of the earliest cell types infiltrating the dystrophic skeletal muscle (in 2-week-old mdx mice) via a protein kinase C θ- (PKCθ-) dependent mechanism (Lozanoska-Ochser et al., 2018). The absence of PKCθ, a key regulator of effector T-cell activation, significantly reduced T-cell infiltration and the innate immune cell infiltrate in mdx/Prkcq−/− muscle (mdx lacking Prkcq, the PKCθ-coding gene), and improved the muscle phenotype (Lozanoska-Ochser et al., 2018).
3.4 Mitochondrial energetics
Mitochondria play a pivotal role in the bioenergetics of muscle fibers via oxidative phosphorylation. The electron transport chain oxidizes NADH or FADH2 through Complexes I and II in the matrix of the mitochondria and generates a proton gradient across the inner membrane of the mitochondria while transporting the electrons (Ahmad et al., 2022). The ATP synthase utilizes this proton gradient to synthesize ATP from ADP and inorganic phosphate. Fine orchestration of the electron transport chain complexes is essential for normal bioenergetics. DMD results in an abnormal environment that is not favorable for mitochondrial homeostasis (Cole et al., 2002; Allen et al., 2016). Instability in the DMD muscle membrane triggers extracellular Ca2+ inflow, leading to mitochondrial Ca2+ overload (Turner et al., 1988; Mareedu et al., 2021; Garcia-Castaneda et al., 2022). Although Ca2+ buffering is another key function of the mitochondria in the muscle through the mitochondrial Ca2+ uniporter (MCU), pathogenically high levels of cytoplasmic [Ca2+] can lead to Ca2+ overload in the mitochondria and trigger the opening of the mitochondrial permeability transition pore (mPTP) (Hurst et al., 2017; Briston et al., 2019).
Morphologically, mitochondria in dystrophin-deficient muscle display decreased size and sparse cristae formation, which are attributed to an increased mitochondrial fission/fusion cycle (Pant et al., 2015; Kang et al., 2018; Moore et al., 2020). Furthermore, diminished protein expression involved in PINK1/PARKIN mitophagy also causes structural damage and mitochondrial dysfunction in dystrophic cardiac muscles (Pant et al., 2015). These observations combined suggest that increased mitochondrial dynamics and suppressed mitophagy in dystrophin-deficient muscle also contribute to mitochondrial dysfunction (Pant et al., 2015).
ATP synthesis capacity has also been found to be disrupted in dystrophin-deficient muscle, and this is shown by a reduction in mitochondrial density and maximal ATP synthesis rate (Percival et al., 2013), as well as mitochondrial respiration (Passaquin et al., 2002; Onopiuk et al., 2009; Schuh et al., 2012). Interestingly, mitochondrial dysfunction and metabolic abnormalities were found to precede the onset of myofiber necrosis in young mdx mice (Townsend et al., 2011; Moore et al., 2020).
3.5 Reactive oxygen species dysregulation
In healthy muscle, a basal level of reactive oxygen species (ROS) is produced, and this is required for optimal cellular function, such as contraction (Reid et al., 1992; Reid, 2001; Powers et al., 2011). Muscles from animal models and patients with DMD produce significantly more free radicals than normal muscle, making the dystrophic muscle more vulnerable than healthy muscle to oxidative stress. Electrons leaked from the electron transport chain in mitochondria are one of the major sources of ROS production in muscle fiber cells (Fridovich, 2004). Specifically, electrons leaked from Complexes I and III combine with oxygen to form superoxide and eventually hydrogen peroxide by superoxide dismutase (e− + O2 → O2− → H2O2) at the mitochondrial matrix and cytosol (Chance et al., 1979). Within a healthy cell, ROS and antioxidants maintain a delicate balance, referred to as the optimal redox state. However, an uncontrolled amount of ROS production in pathology that exceeds the capacity of innate antioxidants generates redox unbalance leading to pathological dysfunction and even cell death (Powers et al., 2011).
Within the muscle fiber, mitochondria form a reticular network that surrounds the contractile apparatus, providing a short distance for the ATP supply within the healthy muscle (Glancy et al., 2015). In pathology, however, this proximity becomes deleterious for the redox-sensitive contractile proteins. Myofilament structures, including troponin, tropomyosin, myosin, and actin, contain a thiol group that is sensitive to redox balance, and exposure to an excessive amount of ROS disrupts myofilament Ca2+ sensitivity and cross-bridge kinetics causing muscle contraction dysfunction (Andrade et al., 1998; Terrill et al., 2013).
DMD is noted for pathologically elevated ROS production from the mitochondria in muscle. Specifically, Complex I-supported maximal H2O2 production has been found to be markedly elevated in skeletal muscle from young mdx mice (Godin et al., 2012; Hughes et al., 2019). Furthermore, skeletal muscles from mdx mice display decreased Ca2+ retention capacity, leading to susceptibility to Ca2+-induced mitochondrial permeability transition pore (mPTP) opening, as discussed previously. Interestingly, mdx skeletal and cardiac muscle have an adaptive elevation in ROS buffering capacity that counteracts the elevated ROS production from the mitochondria (Godin et al., 2012; Hughes et al., 2019; Hughes et al., 2020), and although this can temporarily dampen ROS, it still leads to an increase in oxidative damage as the disease progresses. Chronic inflammation due to sarcolemma damage and myofiber necrosis in dystrophin-deficient cardiac and skeletal muscle is a central feature of DMD, and this contributes to elevated ROS production from mitochondria (Terrill et al., 2016; Tulangekar and Sztal, 2021). Lastly, pathological conditions, including DMD, that impact mitochondrial function can cause more severe effects on the diaphragm than on limb muscle due to the relatively greater ROS emission rates per mass and mitochondrial content (Hahn et al., 2019). This suggests that the mitochondria as a crucial therapeutic target when considering the involvement of diaphragm weakness and wasting leads to cardiorespiratory failure in DMD patients (Mead et al., 2014).
NADPH oxidase 2 (NOX2) also has a considerable role in the pathology of DMD muscle as a source of ROS (Williams and Allen, 2007; Jung et al., 2008; Whitehead et al., 2010). NOX2 is activated by the microtubule-associated protein Rac1 upon mechanical stretching of the muscle, leading to ROS production. This process is greatly increased in DMD as the microtubule lattice becomes disorganized and dense due to the lack of dystrophin-microtubule interaction (Khairallah et al., 2012). NOX2 in phagocytic cells produces superoxide during respiratory bursts for host immunity against bacteria and infections. In DMD, continuous contraction-induced muscle membrane damage also leads to NOX2 activation (Moe et al., 2011; Zhao et al., 2015). Furthermore, biopsies from DMD patients showed greater gene expression of NOX2 compared to age-matched controls (Petrillo et al., 2017), and inhibition of NOX2 expression improved muscle function in mdx mice (Whitehead et al., 2015). Another important source of free radicals in dystrophin-deficient muscles is reactive nitrogen species produced by the cytosolic activation of delocalized nNOS. Together, this free-radical production leads to increased DNA, protein, and lipid oxidation in dystrophin-deficient muscles (Kim et al., 2013; Grounds et al., 2020). Mitochondria and NOX2 have shown potential targets for ROS production.
Indeed, there have been several reports showing significant improvements in muscle function, as well as prevention of myofiber necrosis and membrane permeability from mdx skeletal muscle by N-acetylcysteine supplementation (Whitehead et al., 2008; Terrill et al., 2012; Pinniger et al., 2017).
3.6 Fibrosis
Fibrosis is a wound-healing process wherein an excessive amount of connective tissue is formed, leading to permanent scar tissue. The fraction of ECM in normal muscle is ∼5%, which can be drastically increased in diseased or injured muscles (Lieber and Ward, 2013). Repeated injury/regeneration cycles and chronic inflammation cause fibrosis and loss of function, and this is another phenotypic feature of DMD (Kharraz et al., 2014; Gallardo et al., 2021). In DMD, fibrosis decreases contractile function and reduces the amount of healthy muscle for therapy (Kharraz et al., 2014). Muscle membrane damage in DMD and the subsequent accumulation of acidic metabolites and inflammation amplification initiate fibrotic tissue production by extracellular fluid containing fibronectin, glycosaminoglycans, and proteoglycans (Klingler et al., 2012). Specifically, transforming growth factor-β (TGF-β) is released from the skeletal muscle in injury or inflammation, and this activates myofibroblasts, leading to excessive production of ECM dominated by type I and type III collagen (Zhou et al., 2006; Lieber and Ward, 2013; Meng et al., 2016). Myostatin, which is a negative regulator of muscle mass, also stimulates muscle fibroblasts to proliferate ECM proteins (Li et al., 2008). In addition, angiotensin II, collagen triple helix repeat-containing 1 (Cthrc1) protein, and Wnt signaling regulate fibrogenic turnovers of the muscle cells (Bedair et al., 2008; Li et al., 2011; Cisternas et al., 2014).
The ECM functions as the major source of passive load-bearing in muscles. When comparing the normalized stiffness of a single muscle fiber, fiber bundle (ECM intact), and fiber alone (ECM non-intact), muscle fiber bundles had much higher stress, and this demonstrated that the ECM provides a large portion of the load-bearing function (Meyer and Lieber, 2011). In this light, DMD skeletal muscle displays significantly greater stiffness (Lacourpaille et al., 2015; Lacourpaille et al., 2017; Yu et al., 2022). In animal models of DMD, the lower limbs of mdx mice displayed an increase in the amount of collagen, and this was reported to have a close relationship with increased stiffness in the muscle (Brashear et al., 2021).
Because the fibrotic process begins early in DMD, therapeutic approaches aiming to reduce fibrosis have gained increased interest as a potential strategy for DMD. Therapeutic approaches inhibiting fibrosis (collagen type 1) have been shown to increase the expression of utrophin and the number of revertant myofibers (Levi et al., 2015). Other therapeutic targets that have shown the ability to limit the accumulation of fibrosis in DMD models and in patients include the renin-angiotensin system, cytokines, and tyrosine kinase receptors (Kharraz et al., 2014), ACE inhibitors, ARBs, and mineralocorticoid receptor antagonists (Heier et al., 2019; Kim et al., 2019).
4 Therapeutic strategies for DMD
Despite major therapeutic advances over the past 30 years, there is no curative treatment for DMD (McNally and Pytel, 2007; Mercuri et al., 2019). Nonetheless, a multidisciplinary medical, surgical, and rehabilitative approach targeting the symptoms of DMD can alter the natural course of the disease, improving longevity and quality of life. Glucocorticosteroids, such as deflazacort or prednisone, are the current standard of care and the most widely used treatment for DMD patients (Bushby et al., 2004). The exact mechanism by which glucocorticosteroids delay disease progression in DMD is not fully understood. However, it has been suggested that glucocorticosteroids increase total muscle mass and strength in patients with DMD through the stimulation of insulin-like growth factors, decreased inflammation, decreased lymphocyte reaction and cytokine production, and enhanced myoblast proliferation (for more information on this topic, please see earlier reviews (Angelini and Peterle, 2012; McDonald et al., 2018; McDonald et al., 2020). Although glucocorticosteroids and many other pharmacological strategies can delay symptoms by targeting the secondary effects of DMD, many are only partially effective because they treat just one aspect of DMD pathogenesis. Nevertheless, long-term corticosteroid treatment has a variety of unfavorable side effects, including weight gain, short stature, puberty delay, behavioral issues, and pathologic bone fractures (Connolly et al., 2002). A promising oral glucocorticosteroid analog named VBP-15 was recently shown to improve muscle strength without side effects in the mdx mouse (Damsker et al., 2013; Heier et al., 2013), and a phase 1 clinical trial is currently underway on healthy human volunteers.
As reviewed before, there has been great interest in developing genetic approaches to treat DMD. Herein, we highlight some of the most promising therapeutic strategies that have been used to treat DMD, including gene therapy approaches via direct replacement of dystrophin and newer approaches involving the manipulation of the cellular machinery at the level of gene transcription, mRNA processing, or translation.
4.1 Microdystrophin viral gene therapy
Viral-mediated gene therapy has long been considered a potential therapeutic strategy to treat DMD based on its capability to restore the missing dystrophin by providing a functional copy of the dystrophin gene or repairing dystrophin, thereby restoring dystrophin throughout the body. The fact that DMD patients can be candidates for replacement gene therapy regardless of the underlying dystrophin mutation makes this a very attractive approach to correct DMD defects (Gregorevic et al., 2004; Chamberlain and Chamberlain, 2017; Duan, 2018a; Crudele and Chamberlain, 2019).
Implementing DMD gene therapy has turned out to be a very challenging endeavor, involving two main challenges. The first problem is the dystrophin gene’s immense size. The dystrophin gene, including its coding sequence, poses a problem for viral vector packaging (Roberts et al., 1993). The second hurdle involves systemic delivery to treat all the striated muscles in the body. Thus, a whole-body systemic therapy approach is required (Townsend et al., 2007; Chamberlain and Chamberlain, 2017). Numerous truncated dystrophin designs and body-wide systemic viral gene transfer approaches have been investigated in many labs to overcome these barriers (Gregorevic et al., 2004; Odom et al., 2007; Duan, 2016a; Duan, 2018b; Ramos et al., 2019).
The most common viral vectors implemented for neuromuscular disorders are lentiviruses and adeno-associated viruses (AAV). Table 1 provides a comparison of different vector systems for treating DMD. Among all the virus-based technologies in use for DMD clinical trials, adeno-associated viral (AAV) vectors encoding mini- or microdystrophins show encouraging treatment results in the preclinical setting (Kawecka et al., 2015; Duan, 2018b). The full-length dystrophin cDNA is far larger than the 4.7 kb packaging capacity of AAVs. Hence, these vectors are unable to deliver the full-length dystrophin cDNA. Two or three AAV vector (tri-AAV) in vivo recombination systems have been reported to deliver full-length dystrophin, albeit at very low efficiency (Lostal et al., 2014; Morgan and Muntoni, 2021). Studies have been directed to generate engineered AAVs with increased packaging capacity that induces efficient transduction associated with a modest immune response (Rogers et al., 2011). The currently used recombinant AAVs (rAAV) are erased of all viral genes, specifically the rep and cap genes that encode the nonstructural and structural proteins, and replaced by a sequence of interest, resulting in a maximum encapsidation capacity of around 5 kb (Dong et al., 1996). These vectors mostly remain episomal as extrachromosomal concatemers and rarely integrate into the genome. However, Schnepp et al. (2003) reported rAAV integration rates of about 0.5% in muscles from adult mice. AAV serotypes display a variety of cellular tropisms with a range of efficiency for targeting different muscles (Gao et al., 2004; Zincarelli et al., 2008).
TABLE 1
| Characteristics | Adenovirus | Lentivirus | AAV |
|---|---|---|---|
| Genome | dsDNA | ssRNA | ssDNA |
| Capacity (kb) | Up to 30 kb | Up to 10 kb | Up to 4.7 kb |
| Genome integration | NO | Yes | On rare events |
| Expression duration | Transient | Persistent | Intermediate |
| Non-dividing cell transduction | Yes | Yes | Yes |
| Ability to target satellite cells | NO | Yes | NO |
| Immune response | High | Low | Very low |
| Risk | Elicits viral-induced inflammation/cytotoxicity | Insertional mutagenesis/potential for replication of competent virus | High safety profile |
Viral vector comparison in terms of their applicability to DMD gene therapy.
Initial studies featuring direct AAV vector injection into muscle showed high-level and durable gene transfer (Kessler et al., 1996; Xiao et al., 1996), although this was limited to the injected muscles and represents a non-clinically feasible condition as a continuous injection would be necessary for a therapeutic treatment modality. A major advancement was achieved when the Chamberlain lab (Gregorevic et al., 2004) and Xiao lab (Wang et al., 2005) demonstrated the success of using effective systemic gene transfer to cause whole-body muscle transduction in rodents with AAV pseudotype-6 vectors. Subsequently, the Duan lab reported the success of using systemic AAV-9 transduction in canines (Yue et al., 2008). Currently, a broad variety of AAV capsids that are either isolated from nature or engineered in laboratories have been used to accomplish systemic delivery (Townsend et al., 2007; Kotterman and Schaffer, 2014; Duan, 2016b). For instance, in rodents and large animals, a single intravenous administration of AAV-6, AAV-8, and AAV-9 led to whole-body muscle transduction. Additionally, limiting the expression to tissues of interest can be achieved by utilizing cell type-specific promoters. These advances spurred the development of AAV as the vector of choice for muscle gene therapy (Salva et al., 2007; Wang et al., 2014; Duan, 2016b).
Because AAV vectors rarely integrate into the muscle fiber genome, it follows that with muscle turnover, the microdystrophin transgenes will be diluted and lose effectiveness over time. As AAV vectors do not effectively target satellite cells, upon muscle injury, the satellite cells that are recruited to take part in the repair process will not contain the therapeutic transgene, causing the transduced cells to become diluted and eventually disappear with time (Arnett et al., 2014).
AAV administration may also induce an innate or cellular immune response to the vector, including anti-AAV capsid-neutralizing antibody production. In addition, some patients already have pre-existing AAV-neutralizing antibodies, which exclude them from receiving this treatment (Calcedo et al., 2009; Boutin et al., 2010). Strategies to counter this antibody response, such as by plasmapheresis, alternative AAV serotype, or immune-modulating drugs, are being investigated. The high safety profile of AAVs is evident by the fact that the most severe side effect to date using various AAV serotypes in several clinical trials was a temporary tissue inflammation that did not result in any long-term damage (Büning and Schmidt, 2015). Several clinical trials are investigating the safety and tolerability of this promising therapy in DMD patients (for more details, see Clinical trials section).
As clinical trials continue to develop, other factors controlling the function and stability of the transgene are being evaluated. The Chamberlain group reported that AAV-mediated delivery of microdystrophin resulted in continued expression that was maintained for at least 27 months in both skeletal and cardiac muscles (Ramos et al., 2019). Conversely, Dupont et al. (2015) reported short-lived AAV-derived therapeutic mRNAs in the dystrophic milieu. This finding highlights the necessity for additional elements to be considered when rAAV-mediated gene therapy is being optimized. Designing improved gene therapy treatments for DMD will be made possible with continued research into vector development, optimization of expression cassettes, and further investigations, including immune modulation.
As previously indicated, the coding sequence of the muscle isoform of the dystrophin gene is approximately 11 kb long, and this length presents a significant difficulty in developing gene transfer therapy (Duan, 2018a). Observations in patients with BMD, typically a milder and less common form of muscular dystrophy, have suggested that the transfer of the full-length dystrophin open reading frame (ORF) may not be necessary for developing effective gene therapy. Thus, the disease phenotype may be alleviated with a smaller dystrophin gene construct. In BMD, mutations cause the loss of some exons but do not abolish dystrophin expression, as in the case of DMD (England et al., 1990).
The capacity to excise specific coding regions within the dystrophin gene while maximizing protein function has been made possible by the availability of the full-length sequence of dystrophin together with the knowledge of related protein domains (Jung et al., 1995; Rafael et al., 1996; Lai et al., 2009). Some domains of the dystrophin sequence are reported to be somewhat dispensable for protein function. Hence, a series of internally truncated dystrophin constructs lacking regions of the rod domain and/or the C-terminal domain were devised to be used in place of the full dystrophin (Yuasa et al., 1998; Harper et al., 2002; Goyenvalle et al., 2011; Chamberlain and Chamberlain, 2017). Microdystrophins are partially functional proteins that lack more than half of the typical amino acid sequence of dystrophin. Early versions of microdystrophin, which are about one-third the size of the human dystrophin cDNA (around 3.7 kb in size), lacked the C-terminal domain and up to 20 SRs. In pre-clinical studies, these were very successful at reducing necrosis in dystrophic mdx mouse muscles (Phelps et al., 1995; Gregorevic et al., 2006). They have also been successful in enhancing force generation in striated muscles of dystrophic mouse and canine models for DMD and in preserving the sarcolemma from the injury induced by contraction (Harper et al., 2002; Watchko et al., 2002; Abmayr and Chamberlain, 2006; Le Guiner et al., 2017). These characteristics are attained via micro-dys binding to γ-actin filaments and β-dystroglycan through the N-terminal actin-binding domain and the cystine-rich β-dystroglycan binding domain, respectively. Thus, microdystrophin can form a mechanically robust connection between the subsarcolemmal cytoskeleton and the ECM (Corrado et al., 1996; Rafael et al., 1996; Harper et al., 2002; Gardner et al., 2006; Ervasti, 2013).
Dystrophin structure-function studies have suggested that at least four SRs from the central rod domain are required to connect the crucial N-terminal and cysteine-rich domains. There are a variety of strategies to build such miniature dystrophins. Although it has been demonstrated that a variety of microdystrophins containing various SR combinations can alleviate the dystrophic pathophysiology, other SR arrangements have produced proteins with low or reduced functional capacity (Harper et al., 2002). For example, microdystrophin, ΔR4-R23/ΔCT (also known as mDysH2), one of the most used designs, reduces muscle necrosis and enhances muscle strength, but it has also been reported to cause ringbinden in some myofibers after myotendinous junction injury (Harper et al., 2002; Banks et al., 2008). Ringbinden was caused by a polyproline tract in hinge 2, which was corrected by replacing hinge 2 with hinge 3 (Banks et al., 2010). It is worth mentioning that Sarepta is testing this first-generation ΔR4-R23/ΔCT microdystrophin derived under the striated muscle-specific MHCK7 regulatory cassette in a human clinical trial (ClinicalTrials.gov: NCT03375164) (Figure 1) (Table 2) (Harper et al., 2002; Salva et al., 2007). (For more details, see Clinical trials section.)
FIGURE 1
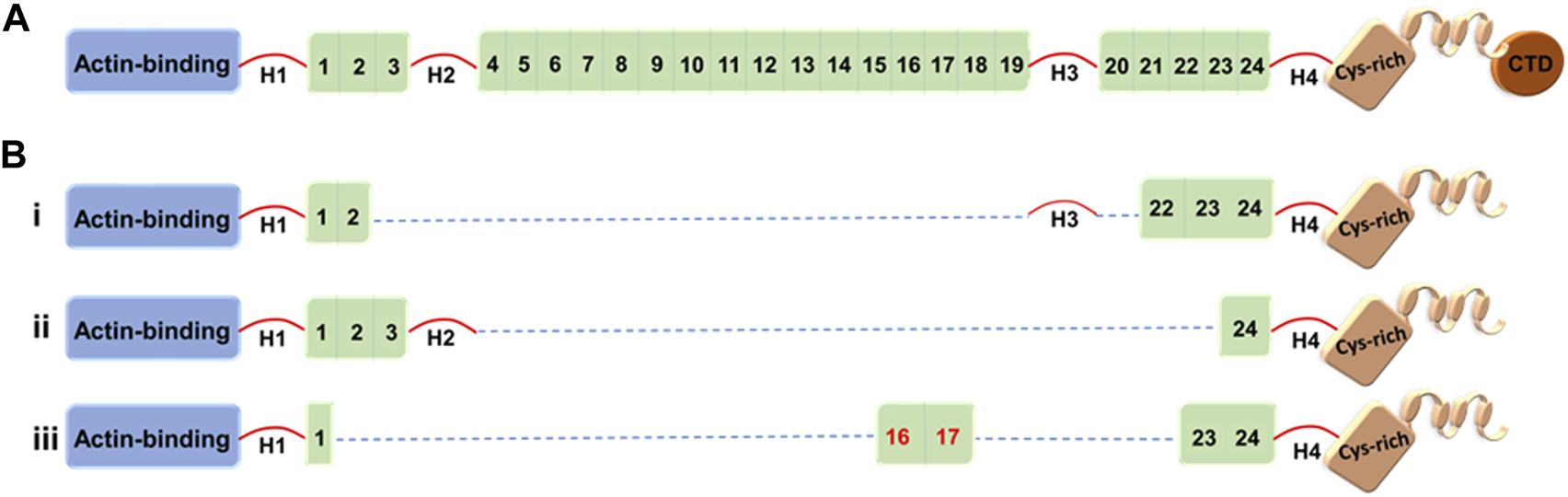
Full-length dystrophin and representative truncated dystrophins currently in use in clinical trials. (A) Full-length dystrophin versus(B) the truncated dystrophin constructs: i) mini-dystrophin Δ3990, ii) microdystrophin ΔR4-23/ΔCT (µDysH2) and iii) microdystrophin μDys-5R. Domains are identified as the N-terminal actin-binding domain, 1–24 spectrin-like repeats, four hinges (H1–H4), a cysteine-rich domain, and a C-terminal domain (CTD). The nNOS-binding domain is shown by the red numbers (repeats 16 and 17).
TABLE 2
| Company | Solid biosciences | Sarepta therapeutics | Pfizer | Nationwide Children’s hospital |
|---|---|---|---|---|
| Description | Genetic: SGT-001 | Genetic: rAAVrh74.MHCK7 microdystrophin | Genetic: PF-06939926 | Administration of rAAVrh74.MCK.GAL GT2 to DMD patients |
| Clinical trial number | NCT03368742 | NCT03375164 | NCT03362502 | NCT03333590 |
| Trial title | Microdystrophin gene transfer study in adolescents and children with DMD (IGNITE DMD) | A randomized, double-blind, placebo-controlled study of SRP-90001 | A study to evaluate the safety and tolerability of PF-06939926 gene therapy in DMD | Gene transfer clinical trial to deliver rAAVrh74.MCK.GALGT2 for DMD |
| Delivery route | Intravenous injection | Intravenous injection into peripheral arm vein | Intravenous injection | Intravascular |
| AAV serotype | AAV9 | AAVrh74 | AAV9 | AAVrh74 |
| Current stage | Active, but no longer recruiting participants | Active, but no longer recruiting participants | Active, but no longer recruiting participants | Active, not recruiting |
| Primary outcome | Safety | Safety | Safety and tolerability | Safety and tolerability |
Clinical trials using adeno-associated virus- (AAV-) mediated microdystrophin transfer for DMD therapy.
Several other mini-/microdystrophin designs with clinical potential have been generated and tested in animal models (Banks et al., 2007; 2010; Jørgensen et al., 2009; Lai et al., 2009; Koo et al., 2011; McCourt et al., 2015; Hakim et al., 2017; Le Guiner et al., 2017; Duan, 2018b; Nelson et al., 2018). Among these, Δ3990 (mini-dystrophin), ΔR4–23/ΔCT (microdystrophin with deletion of repeats 4–23 and deletion of the C-terminal domain), and R16/17-containing microdystrophin (μDys-5R) are particularly noteworthy given the high safety profile and potency of these constructs in the murine and canine models (Figure 1) (Wang et al., 2000; Harper et al., 2002; Hakim et al., 2017). Strategic design of these microgenes to increase therapeutic efficacy includes codon optimization—a modification that switches a particular DNA sequence by changing its codons to match the most prevalent tRNAs, resulting in a more efficient translation (Foster et al., 2008) and the inclusion of nNOS-binding domain (repeats 16 and 17) (Lai et al., 2009).
Although all microdystrophins currently in clinical trials include hinges H1 and H4 (Figure 1), Wasala et al. (2022) recently studied the importance of including hinge 1/4 in microdystrophin gene therapy. They reported that hinge 1 is essential for the correct sarcolemmal localization of microdystrophin, restores the DGC, and enhances specific muscle force significantly compared to hinge 4. However, the construct including H1 and H4 outperformed the H1-only or H4-only microdystrophins. The Chamberlain group has been a leader in the field of testing microdystrophins with different structural configurations, including modification with central rod and/or various hinge domains (Harper et al., 2002; Wang et al., 2012). Here, vectors were evaluated in mdx4cv mice and delivered using recombinant AAV6 vector. When compared to first-generation microdystrophins, two of the new microdystrophins were found to improve force generation while also localizing neuronal nitric oxide synthase to the sarcolemma, namely, μDys5 (ΔR2-R15/ΔR18-R22/ΔCT) and μDys7 (ΔH2-R15 + H3/ΔR18-R23/ΔCT) (Ramos et al., 2019).
It is worth noting that studies are needed to fully investigate the stability, function, and proper localization of the several different micro/mini-dystrophin constructs, as it is not entirely clear if the proteins will localize correctly within the cytoskeletal network.
Additional research is required to assess how functional these microdystrophins are in humans, including their therapeutic effectiveness. A functional improvement in DMD patients may also be more challenging to predict, given that the DMD animal models (where preclinical studies are tested) frequently exhibit a milder skeletal and cardiac muscle phenotype than humans. Additionally, because microdystrophins are shorter than the typical length of truncated dystrophins in BMD patients, microdystrophin therapy in DMD patients may not fully achieve the goal of attaining a BMD-like phenotype (Mercuri and Muntoni, 2013). Accordingly, the loss of functionality owing to the deletion of specific dystrophin protein subdomains continues to be problematic in terms of treatment effectiveness. It will also be crucial to achieve significant therapeutic efficacy in the heart since treating skeletal muscle without also treating the associated cardiomyopathy could paradoxically add more strain on the myocardium, due to increased physical activity, and hasten the development of heart disease (Townsend et al., 2008; 2009). This may be a general concern with treatments that more specifically target skeletal muscles without also addressing heart tissue.
4.2 Exon-skipping therapy
Exon skipping has been proposed and studied as a possible treatment for DMD for more than two decades. The goal of exon skipping therapy is to restore the disrupted open reading frame (ORF) of the dystrophin gene transcripts in DMD patients, allowing them to make a Becker’s muscular dystrophy-like protein (Figure 2) (Niks and Aartsma-Rus, 2017). It is based on manipulating the pre-mRNA splicing of dystrophin transcripts to bypass a defective exon from the splicing machinery, restoring the reading frame and producing a partially functional, internally deleted dystrophin (Figure 2) (Schneider and Aartsma-Rus, 2021). The resulting dystrophin proteins are usually larger than the majority of mini- and microdystrophin gene therapy constructs currently under investigation.
FIGURE 2
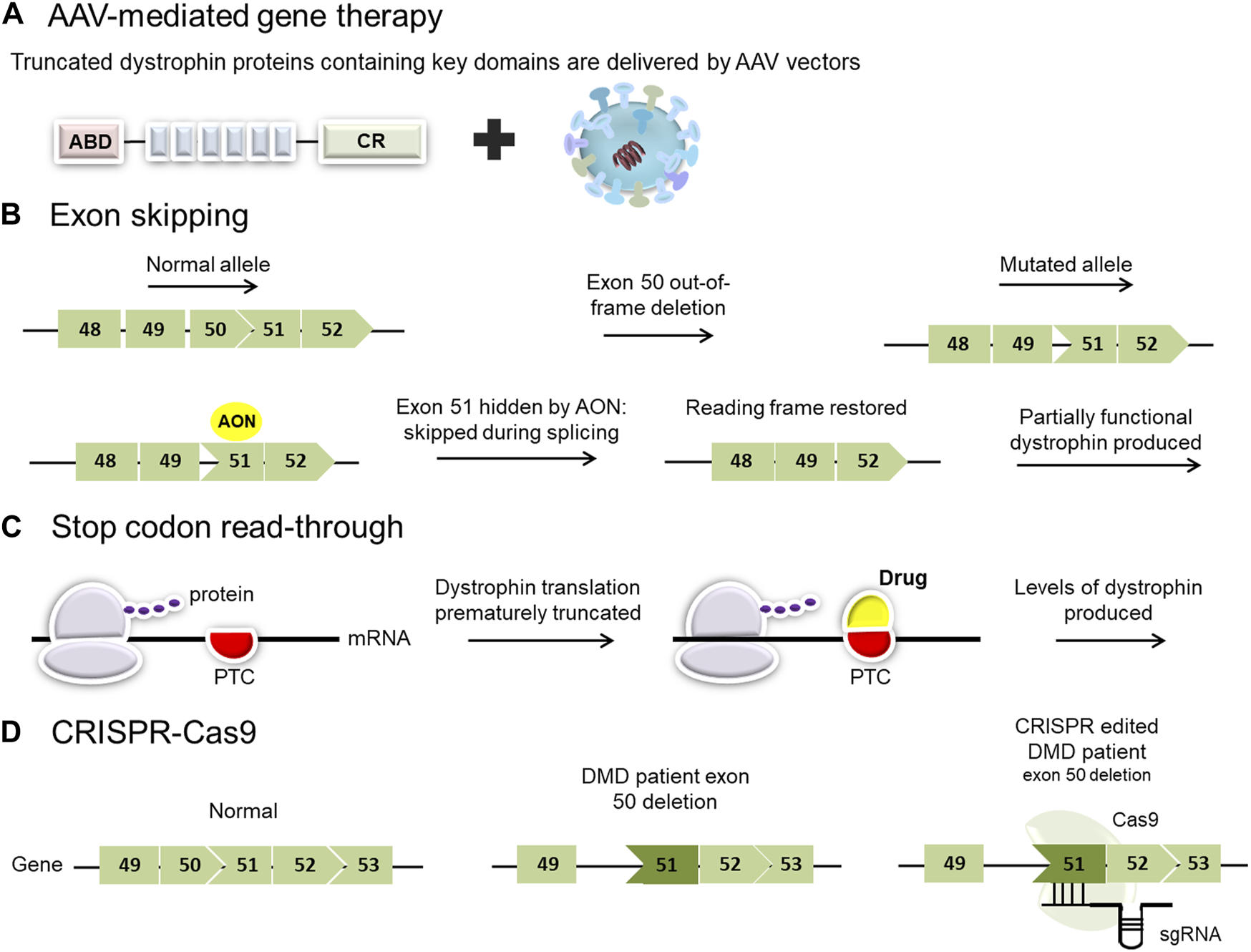
Overview of current and proposed experimental therapies for Duchenne muscular dystrophy (DMD). (A) AAV-mediated gene therapy employs viral vectors to deliver micro- or mini-dystrophin genes. Clinical trials using different adeno-associated virus (AAV) serotypes have shown promise for the treatment of patients with DMD. (B) Exon-skipping strategies seek to mask exons adjacent to others that have been deleted. This results in the restoration of the reading frame and permits the translation of a slightly smaller dystrophin product. (C) Stop codon read-through is a small molecule therapy aimed at reducing ribosomal sensitivity to mRNA stop codons, thus promoting ongoing dystrophin translation in those patients with nonsense mutations. (PTC: premature termination codon). (D) Genome editing, employing a CRISPR/Cas9 platform, has the potential to target specific pathogenic variants in the DMD gene but carries a risk of off-target effects.
Exon-skipping entails three experimental approaches: CRISPR DNA editing system, U7 snRNP-mediated splice blocking, and antisense oligonucleotides (AONs) (Takeda et al., 2021). CRISPR DNA editing techniques work at the DNA level to change myofiber genomic DNA in converting a DMD out-of-frame deletion into a BMD-like in-frame deletion. CRISPR relies on viral vectors to deliver the DNA editing technology (for more information on CRISPR-mediated exon skipping, see CRISPR-Cas9 mediated gene editing strategies). U7 snRNP-mediated inhibition of splicing is another approach for exon-skipping where modified U7 snRNP genes are delivered via AAV vectors (Vulin et al., 2004; Gadgil and Raczyńska, 2021). In this instance, an antisense sequence that targets the exon takes the place of the typical antisense portion that hybridizes to histone RNA. This does not target mRNA but pre-mRNA (similar to the mechanism of AON) (Gushchina et al., 2021). The third approach to accomplishing exon skipping is using oligonucleotide drugs that bind to the pre-mRNA (prior to splicing) to modulate RNA splicing. These oligonucleotide techniques have used a variety of drug chemistry with variable success, and this approach will be the focus of the following section.
4.2.1 AONs-mediated exon-skipping therapy
AONs are small 20–30 nucleotides of modified DNA or RNA homologs that specifically bind to their target exon prior to pre-mRNA splicing in a Watson-Crick-like manner, hiding the target exon from the splicing machinery so that it is spliced out with its flanking introns blocking its incorporation to the mature mRNA (Schneider and Aartsma-Rus, 2021). Antisense-mediated exon skipping is a method that targets a specific mutation (Bladen et al., 2015). For the reading frame to be restored, different exons have to be skipped depending on the location and size of the mutations (Aartsma-Rus et al., 2009; Bladen et al., 2015). Accordingly, different dystrophin proteins will be generated after skipping different exons for different mutations. Hence, it is crucial to have a precise genetic diagnosis of the disease to be able to design effective AONs (Arechavala-Gomeza et al., 2007).
For AON therapy, the majority of DMD patients would have one or more exons deleted. Overall, in DMD patients, 70% of the reading frame can be restored by single exon skipping, whereas another 8% would benefit from double exon skipping to restore the reading frame (Bladen et al., 2015). DMD deletions frequently cluster in hotspots between exons 45 and 55; therefore, skipping target exons in this area could be of benefit to a large group of patients. Certain exons have been reported to have the highest applicability for skipping. Namely, exon 51 could be applied for (13%–14%) of patients, exon 45 and exon 53 both could be skipped for another 8%–10% of patients, and exon 44 could be skipped for an additional 6% of the patients (Mann et al., 2001; Van Deutekom et al., 2001).
Different chemical modifications have been tested to give the AONs drug-like characteristics in trying to enhance their bioavailability, improving their resistance to exonucleases and endonucleases, and increasing their affinity to the target RNA transcripts (Saleh et al., 2012). 2ʹ-O-methyl phosphorothioate (2OMePS) AONs and phosphorodiamidate morpholino oligomers (PMOs) are used among other chemistries for DMD exon skipping clinical development approaches (Verma, 2018; Schneider and Aartsma-Rus, 2021).
A major challenge for the success of AON approaches centers on achieving adequate delivery to the myofiber nuclei so that the drug can hit its pre-mRNA target in the nucleus (prior to splicing) and prevent the splicing of the targeted exon (Takeda et al., 2021). Three different techniques have been used for the delivery of the oligonucleotides to the myofiber, namely, cell transfections, in vivo intramuscular injections, and in vivo systemic delivery (intravenous, intraperitoneal, or subcutaneous). Depending on the delivery technique, various oligonucleotide chemistries exhibit different degrees of myofiber delivery efficacy and exon-skipping efficiency (Heemskerk et al., 2009; Hoffman et al., 2011; Takeda et al., 2021).
The use of nanoparticles as a delivery vehicle has been investigated for enhancing AON uptake and reducing possible immune reactions. Polymethyl methacrylate (PMMA) nanoparticles include two subgroups: T1 and ZM2 have been tested for AON therapy in mdx mice (Schneider and Aartsma-Rus, 2021). T1 nanoparticles were shown to have slow biodegradability; however, they formed aggregates that precluded their use via intravenous administration and raised concerns about possible adverse effects (Rimessi et al., 2009). Regarding ZM2-encapsulated AONs, intraperitoneal delivery was accompanied by higher dystrophin restoration levels in skeletal muscle and hearts as compared to naked 2OMePs AONs (Rimessi et al., 2009; Ferlini et al., 2010). Moreover, the effect lasted for 90 days after the course of treatment (7 weeks) (Bassi et al., 2012). Still, more studies are needed to better understand the pharmacodynamics, pharmacokinetics, and any related side effects of this type of delivery.
2OMePS molecules possess a negative charge. They can effectively transfect cells in vitro and can be absorbed via intramuscular injection. Unfortunately, their use in clinical trials has been stopped, as no detectable dystrophin production in patient muscle was observed (Mann et al., 2001; Lu et al., 2003). Morpholinos (PMO) are uncharged compounds that are challenging to introduce into cells in vitro. They show little or no systemic distribution to normal myofibers but exhibit high levels of systemic delivery and efficient exon skipping in dystrophic muscle (Heemskerk et al., 2009; Hoffman et al., 2011; Novak et al., 2017). The effective delivery of PMOs was limited to the inflammatory foci and active muscle-regenerating regions of the dystrophic muscle tissue, where the macrophages seem to play a role in their delivery (Novak et al., 2017). Morpholinos have shown an acceptable safety profile at very large systemic doses in mouse, dog, and human studies (Yokota et al., 2009; Wu et al., 2010; Clemens et al., 2020; Komaki et al., 2020). Nevertheless, as the underlying mutations that cause DMD disease remain and due to transcript and protein turnover, repeated drug injection is required. Overall, the highest levels of dystrophin restoration have been achieved via morpholino chemistry using systemic delivery (Schneider and Aartsma-Rus, 2021).
Several strategies have been proposed to increase the effectiveness of AONs and their uptake by skeletal muscles and in the heart for multi-exons skipping or enhancing their delivery using muscle-homing peptide conjugation (e.g., arginine-rich peptide) or via nanoparticles. Multi-exon skipping approaches, which permit the skipping of several adjacent exons at once, are under investigation in order to broaden the range of mutations eligible for treatment (Goyenvalle et al., 2012). Cocktails of antisense oligonucleotides have been developed to target two or more neighboring exons within a particular area to induce multi-exon skipping, such as the major mutational hotspots (exons 45–55) (Taglia et al., 2015). Another favorable approach is conjugating AONs to arginine-rich cell-penetrating peptides (CPPs) to enhance cellular uptake. The conjugated PMO-based AONs are termed peptide phosphorodiamidate morpholino oligomers (PPMO). In mdx mice and canine X-linked muscular dystrophy model of DMD, PPMOs were shown to be safe and permitted a longer duration of effective exon skipping in the tissues evaluated, including the heart and diaphragm (Jearawiriyapaisarn et al., 2008; Wu et al., 2008; Echigoya et al., 2017).
Despite the attractiveness and applicability of single or multiple exon(s) skipping techniques and the fact that four AONs have already been approved for clinical trials, it is becoming clear that their delivery efficiency is not satisfactory (Sheikh and Yokota, 2021). This could be concluded from the low levels of dystrophin restoration in skeletal muscle biopsies and the lower levels observed in heart muscle (Heemskerk et al., 2009; Wu et al., 2009; Heemskerk et al., 2010; Jearawiriyapaisarn et al., 2010; Wu et al., 2010; Malerba et al., 2011; Yin et al., 2011). Therefore, ultimate clinical effectiveness will necessitate repeated injections. For exon skipping to reach clinical milestones, drug-induced dystrophin needs to be produced at higher levels in both skeletal and heart muscles. Hence, it is crucial to understand the underlying pathogenesis to maximize the treatment benefit. Epigenetic modulators that enhance or abolish the inhibitory effect of gene expression need to be considered with this treatment to retain and stabilize dystrophin transcripts and protein at multiple levels.
Taken together, the exon-skipping technique has advanced from in vitro proof of concept research of the clinical trial stage to FDA approval (Eser and Topaloğlu, 2022). Only single exon skipping has been approved for clinical trials (for more details, see Clinical trials section). The multiple exon-skipping approach is still facing hurdles (e.g., higher drug doses required, differential uptake and efficiency of the used ASOs) (Mitrpant et al., 2009; Yokota et al., 2009; Aartsma-Rus et al., 2014).
4.3 Stop codon read-through
Around 10%–15% of patients with DMD have a nonsense mutation that induces a premature termination codon (PTC) in the mRNA, causing the ribosome to terminate translation and failing to synthesize the remainder of the protein (Flanigan, 2014; Bladen et al., 2015). The principle of stop codon read-through is to induce the ribosome to continue translating the mRNA through the premature stop codon and continue through the rest of the transcript. The drug works by binding to the ribosome and its partners, disrupting its ability to recognize the premature stop codon, leading to the transformation of the PTC into a correct codon, thereby resulting in the continuation of the translation (Figure 2) (Bordeira-Carriço et al., 2012). This mechanism ideally needs to be specific to premature stop codons, without read-through of bona fide stop codons, to efficiently enable partial restoration of dystrophin expression in muscle (Welch et al., 2007). A number of drugs can induce stop codon read-through, and their efficacies rely on the nature of the nonsense mutation and the surrounding nucleotide sequences. These drugs can be divided into two main groups: aminoglycoside antibiotics (e.g., gentamycin; amikacin, tobramycin, and paromomycin) or chemical compounds without known analogs (e.g., ataluren or negamycin) (Bidou et al., 2012). Although gentamycin demonstrated efficacy in inducing stop codon read-through for numerous pathologies, including DMD (Barton-Davis et al., 1999; Wagner et al., 2001; Politano et al., 2003; Malik et al., 2010), toxicity remains a major concern limiting gentamycin from clinical considerations (Prayle and Smyth, 2010; Beringer and Winter 2011). In the past few years, several studies have aimed to select molecules capable of promoting mRNA translation despite the presence of a PTC. Ataluren (Translarna TM, previously known as PTC124) is a small oral molecule developed by PTC Therapeutics as a potentially safer alternative to induce stop codon read-through (Welch et al., 2007).
4.4 CRISPR-Cas9 mediated gene editing strategies
Over the past decade, gene editing has moved to the forefront with the identification of mechanisms of DNA repair (Stephenson and Flanigan, 2021). The CRISPR-Cas system has made it possible for research labs all around the world to successfully incorporate gene editing into their work (Jinek et al., 2012; Mali et al., 2013; Perez-Pinera et al., 2013). The two main components of the CRISPR/Cas9 system are the CRISPR-associated (Cas) endonuclease and a single guide RNA (sgRNA), which directs Cas9 to a particular ∼20-nucleotide region in the genome that contains the complementary sequence (Figure 2).
Numerous engineered Cas endonucleases have been generated using naturally occurring Cas enzymes as guides. Targeted modifications have improved enzyme fidelity, reduced off-target mutations, increased editing efficiency, and used a more dispensable PAM sequence (Mali et al., 2013; Makarova et al., 2020). Engineered Cas proteins have extended to include nickase Cas9 (nCas9) (Ran et al., 2013) and deactivated Cas9 (dCas9) (Qi et al., 2013) that have been engineered to retain their programmable DNA-binding ability while decreasing or abolishing the Cas9 endonuclease activity, respectively. This has increased the safety of the CRISPR/Cas9 system owing to the absence of any DSBs (Dominguez et al., 2016). These may be combined with other elements, such as precise base editors (Komor et al., 2016; Yuan et al., 2018) or transcriptional activators (Qi et al., 2013; Wojtal et al., 2016), for alternative therapeutic strategies (Chen et al., 2022). Thus, the toolkit of CRISPR gene-editing systems has advanced, providing a growing number of options for CRISPR-based therapies.
The potential treatment of DMD using CRISPR/Cas9-mediated therapeutic techniques and the difficulties of CRISPR/Cas9-mediated therapeutic genome editing will be the main topics of this section.
4.4.1 CRISPR technology as a gene-editing therapeutic for DMD
A single infusion of CRISPR genome-editing components can theoretically treat DMD by correcting genetic mutations at the genome level utilizing CRISPR/Cas programmable nucleases (Olson, 2021; Chen et al., 2022), a concept that represents a promising therapeutic strategy for the long-term correction of genetic illnesses (Knott and Doudna, 2018). Several of the disease characteristics of DMD make gene editing an exciting treatment modality. First, owing to the relative dispensability of certain dystrophin domains, it is possible to remove mutant exons from the rod domain of the gene and restore the ORF yielding partially functional truncated dystrophins, as discussed previously. Second, a genetic repair of even a small percentage of muscle nuclei could enable the generation of dystrophin and its dissemination throughout the myofibers as skeletal fibers are multinucleated. Moreover, because the dystrophin gene is located on the X chromosome, affected boys only have one mutant allele that needs to be repaired. Therefore, unintentionally altering a wild-type copy of the gene is obviated (Long et al., 2014).
Both in vitro and in vivo studies using CRISPR-mediated gene editing for DMD have been conducted. Therapeutic effects have been reported in human cells and mice, rats, dogs, and piglets with various DMD mutations (Ousterout et al., 2015; Young et al., 2016; Amoasii et al., 2018; Duchene et al., 2018; Min et al., 2019a; Moretti et al., 2020; Pickar-Oliver et al., 2021; Szabo et al., 2021; Zhang et al., 2021). Attaining greater insights using sequence-humanized animal models will be required prior to any clinical translation. This would include providing crucial information such as the required dose, delivery vehicle, route of administration, and the percentage levels required for functional dystrophin recovery (Aartsma-Rus and van Putten, 2020; Olson, 2021). Off-target effects are also a concern and must be addressed prior to human trials.
4.4.2 Exon deletion
Exon excision, exon skipping, exon reframing, and exon knock-in are the main approaches for the CRISPR-mediated therapeutic repair of DMD mutations. Additionally, newly engineered CRISPR system technologies like the base or prime editors have been used to enable more accurate gene editing.
The majority (∼60%) of DMD patients have a deletion of one or more exons. Deleting the out-of-frame exon to restore the ORF is a typical method for achieving single or multiple exon deletions. This can be accomplished by double-cut exon deletion to one exon or over the entire hotspot region. To cut out single or multiple mutant exons, two sgRNAs flanking these sites can be targeted with Cas9, and this will cause the ORF to be restored by splicing the neighboring in-frame exons together (Ousterout et al., 2015; Maggio et al., 2016a; Young et al., 2016). Importantly, because this approach is less mutation-specific and permits the ablation of many exons in a mutational hotspot, it is applicable to a higher percentage of DMD patients. However, this is at the expense of shorter dystrophins compared to other correction strategies. This method, though appealing, has a number of drawbacks, including the increased risk of off-target mutations and a sharp decline in editing efficiency simply because two directed DSBs must occur at the same time for the edit/correction to be successful. Added to this issue is the challenge of delivering the two sgRNAs simultaneously.
DMD mutational hot spot deletion has the potential to cure around 60% of DMD patients, based on BMD patients with more mild symptoms, at least in skeletal muscles (Aartsma-Rus et al., 2006; Doo et al., 2012; Ousterout et al., 2015). Ousterout et al. (2015) designed multiplexed sgRNAs to restore the dystrophin reading frame by targeting the mutational hotspot in ∆Ex48-50 DMD patient-derived myoblasts. Here, dystrophin expression was restored in vitro following gene editing. However, this correction was not as efficient as deleting exon 51 alone, and this raised concerns about the editing efficiency for deleting a larger DNA fragment. Other groups targeted this same region in vitro and demonstrated a successful restoration of dystrophin in multiple DMD cell lines (Maggio et al., 2016b; Young et al., 2016; Duchene et al., 2018). In vivo studies have also been conducted in the humanized DMD del45/mdx mouse model. Exons 45–55 were successfully deleted, and dystrophin expression was observed after gene correction (Young et al., 2017).
In vitro and in vivo studies using the double-cut gene-editing approach have been used to target single-exon deletions (Maggio et al., 2016b; Long et al., 2016; Tabebordbar et al., 2016; Zhang et al., 2017; Long et al., 2018; Matre et al., 2019; Moretti et al., 2020). Although removing a smaller region lessens some of the problems with the double-cut method, it also eliminates the multi-exon deletion advantages noted previously. Postnatal single exon deletion editing has been studied in the mdx mouse model. Here, dystrophin expression was partially restored in cardiac and skeletal muscle and was shown to last up to 1 year in some studies after a single intravenous injection of an adeno-associated virus that encodes the CRISPR cassette (Nelson et al., 2016; Nelson et al., 2019).
Exon duplication mutations, which affect 5% of DMD patients, can also be corrected using exon deletion techniques. One duplicate exon can be targeted with a single sgRNA designed against the intron region next to the duplicated exon. In the presence of Cas9, sgRNA will produce two cuts and eliminate one of the duplicate exons. Thus, in principle, the dystrophin gene ORF can be restored, resulting in the production of full-length dystrophin protein identical to normal dystrophin (Long et al., 2018). However, exon duplication excision is presently challenging to investigate in vivo owing to the lack of animal models with this mutation (Bladen et al., 2015). CRISPR/Cas9 technology was recently used to develop a mouse model with a muti-exon tandem duplication of exon 50. This duplication mutation was then corrected using the sgRNA CRISPR/Cas9 technique. This method removed the duplication mutation, restored the expression of full-length dystrophin, and improved muscle functionality in vivo (Maino et al., 2021). Thus, this approach has the potential to treat duplication mutations in DMD.
4.4.3 Exon skipping by gene editing
In comparison to AON-based exon skipping therapy that modulates dystrophin mRNA, CRISPR gene editing corrects the underlying mutation in the genome. Thus, it is regarded as a permanent genomic correction. The conservative CRISPR-based exon skipping approach using only one sgRNA, instead of two sgRNAs flanking a mutant exon, to abolish either the splice acceptor site or splice donor site of the out-of-frame exon is the commonly used method for exon skipping (Amoasii et al., 2018). The sequence encoding the exon splice acceptor or donor site is then deleted by NHEJ induced via CRISPR system-triggered DSB, leading to out-of-frame exon skipping and splicing to the next accessible exon. Compared to utilizing two sgRNAs to flank the exon for removal, a single-sgRNA method for exon skipping increases editing efficiency (Long et al., 2018).
Several in vitro and in vivo studies have been conducted to evaluate this approach. In vitro, single-cut exon skipping has been used to skip exons 43, 45, 51, and 53 by NHEJ-induced disruption of splice acceptor sites, and successful skipping was demonstrated in iPSCs-derived skeletal muscle myoblasts isolated from DMD patients. Dystrophin restoration and functional improvement were observed in the gene-edited cell lines (Maggio et al., 2016b; Long et al., 2018; Min et al., 2019b). This approach has also been tested in vivo in both mouse (Amoasii et al., 2017; Min et al., 2019b; Wei et al., 2020) and canine models (Amoasii et al., 2018). Targeting the splice acceptor site of exon 51 results in skipping exon 51 and restoration of the dystrophin ORF. Dystrophin expression restoration and improved muscle function were demonstrated in both the DMD mouse and canine models that lack exon 50 (Amoasii et al., 2017; Amoasii et al., 2018). Exon 45 skipping via CRISPR/Cas9 has also been demonstrated to restore the reading frame in the ∆Ex44 mouse model, allowing exon 43 to splice to exon 46, thus restoring the dystrophin reading frame (Nelson et al., 2017; Min et al., 2019b).
4.4.4 Exon reframing by gene editing
Another strategy to restore the dystrophin ORF involves Cas9 induction of a single NHEJ (Li et al., 2015; Iyombe-Engembe et al., 2016; Amoasii et al., 2017; Long et al., 2018). This form of gene editing is designed to “reframe” the ORF of the dystrophin transcript by introducing small insertions and deletions (INDELS) via NHEJ of double-stranded DNA breaks generated by CRISPR-Cas9. When using a sgRNA to induce NHEJ in an out-of-frame exon, the generated indels result in a targeted frameshift to restore the dystrophin gene back in frame (Min et al., 2018). This method is often referred to as single-cut myoediting because only one sgRNA is used to direct Cas9 editing, requiring only a single cut to restore the dystrophin ORF in targeted muscle cells. A number of groups have shown efficient restoration of the dystrophin ORF through exon reframing in human iPSC-derived cardiac myocytes, mouse models, and large animal models of DMD (Ousterout et al., 2015; Amoasii et al., 2017; 2018; Bengtsson et al., 2017; Kyrychenko et al., 2017; Zhang et al., 2017; Long et al., 2018; Yuan et al., 2018). Exon reframing produces small indels during the repair and serves as an efficient strategy to preserve a large portion of the dystrophin genomic sequence while bypassing the DMD mutation. This strategy offers the possibility of permanently correcting specific DMD mutations.
4.4.5 Exon knock-in
In contrast to NHEJ-mediated exon deletion, skipping or reframing HDR-mediated exon knock-in can restore the full-length dystrophin gene expression. This approach incorporates a DNA donor template with the appropriate sequence as a part of the editing components. HDR-mediated genome editing has been demonstrated in small and large animal models of DMD (Long et al., 2014; Bengtsson et al., 2017; Zhang et al., 2017; Zhu et al., 2017; Mata López et al., 2020). An example of this strategy is the correction of exon 23 in the germline of mdx mice via SpCas9 with a 180-nt single-stranded DNA oligonucleotide template. Correction rates ranged from 2% to 100% correction of the Dmd gene in the resultant mosaic mice (Long et al., 2014). HDR is not active in quiescent or G1-arrested cells, rendering it unsuitable in mature myofibers and cardiac myocytes. Additionally, the approach cannot be used to correct DMD deletion mutations because of the length restriction on the donor DNA template (Zhang et al., 2021).
Homology-independent targeted integration (HITI) is a gene editing method with a relatively high efficiency in postmitotic cells. HITI can accurately knock in a missing exon(s) at a specific locus using NHEJ, which bypasses the requirement of HDR. HITI has been developed to overcome the HDR-related challenges noted previously (Suzuki et al., 2016). Delivering a donor plasmid with two Cas9 cleavage sites flanking the desired donor sequence is a key component of HITI. Following the Cas9 cleavage of the targeted genomic DNA and the donor plasmid, the NHEJ repair pathway will then incorporate the donor sequence. Although this exon knock-in strategy to restore full-length dystrophin protein is promising, it has not yet been extensively evaluated in the setting of DMD. Pickar-Oliver et al. examined the HITI-mediated approach to insert the missing human exon 52 or a superexon encoding the human dystrophin cDNA sequence downstream of exon 51 into its corresponding position within the dystrophin gene in a humanized mouse model of DMD. The DMD model used contained an out-of-frame deletion of exon 52, and full-length dystrophin restoration in skeletal and cardiac muscles was demonstrated (Pickar-Oliver et al., 2021). This technique enables the entire restoration of full-length dystrophin, even though the insertion efficiencies were low, and would be applicable to about 20% of DMD patients.
4.4.6 Base editing
Roughly 25%–30% of patients with DMD have point mutations (Aartsma-Rus et al., 2006; Bladen et al., 2015). Base editing is a recently developed approach to expand the toolbox of gene editing strategies to treat DMD (Chemello et al., 2020). In base editing, there are two major categories of DNA base editors: cytosine base editors (CBEs), which convert the C:G base pair into a T:A base pair (Komor et al., 2016; Nishida et al., 2016; Gaudelli et al., 2017) and adenine base editors (ABEs), which convert A:T base pairs to G:C base pairs (Gaudelli et al., 2017). In this strategy, Cas9 nickase (nCas9) or deactivated Cas9 (dCas9) is fused to a cytidine deaminase or an engineered adenine deaminase protein, allowing precise single-base pair conversations without double-stranded breaks (Gaudelli et al., 2017; Huang et al., 2021). These RNA-guided nucleotide-specific base editors do not rely on the NHEJ repair pathway, and as a consequence, small indels through error-prone NHEJ at the target site are not produced. Furthermore, a donor DNA template for HDR is not required (Min et al., 2018). Recently, a CRISPR/Cas9 adenine base editor (ABE7.10) was used to substitute a single adenine with guanine in a DMD mouse model containing a nonsense mutation on exon 20 (Ryu et al., 2018). This strategy has also been used to induce exon skipping by mutating target DNA bases of splice motifs (Gapinske et al., 2018). In this context, a CBE base editor (hAID P182X) was implemented in various canonical intronic motifs to modulate the splicing of different genes (Yuan et al., 2018).
4.4.7 Prime editing
More recently, a new strategy, termed prime editing, has been developed, which has been added to the CRISPR techniques to treat DMD point mutations (Anzalone et al., 2019). Prime editing takes advantage of a catalytically inactive nCas9. As a result, no DSBs are produced. The nCas9 is coupled to an engineered reverse transcriptase and delivered with an extended sgRNA termed the “prime editing” guide RNA (pegRNA). Reverse transcriptase uses the pegRNA as a template to add a DNA alteration at the target location (Anzalone et al., 2020). This approach enables single-base transitions or transversions, as well as site-specific genomic insertions and deletions, without introducing DSBs or the need for an exogenous donor DNA as a template for HDR repair. With the aid of endogenous DNA repair pathways, this newly generated DNA flap is then incorporated into the genome. While size restrictions still pose a problem for in vivo distribution, prime editing provides the ability to correct a number of DMD-causing mutations. In a proof-of-concept study by Chemello et al. (2021), prime editing was tested in the context of DMD. The approach was shown to be capable of reframing the dystrophin ORF in DMD (∆Ex51) cardiac myocytes derived from human iPSCs (Chemello et al., 2021). Subsequently, the Tremblay group reported the correction of point mutations in exon 6 in human DMD myoblasts using the prime editing technique (Happi Mbakam et al., 2022b).
4.4.8 Delivery of CRISPR in vivo
An effective delivery mechanism is necessary to accomplish efficient in vivo postnatal genome editing. Cas9 and sgRNA, the two components of the CRISPR system, can be administered to the target organs via a variety of forms. DNA/DNA, mRNA/sgRNA, and protein/sgRNA are the possible nucleotide forms for Cas9 and sgRNA, respectively. The CRISPR delivery strategies frequently employed in DMD include viral and nonviral methods (Min et al., 2019a).
4.4.8.1 Viral delivery
For the delivery of CRISPR/Cas9 components, lentivirus, adenovirus, and adeno-associated virus (AAV) have been employed (Min et al., 2019a), and clinical trials using AAV for gene replacement therapy have been approved by the FDA (Mendell et al., 2017). AAV serotypes 1, 6, 8, 9, rh10, and rh74 have tropism for skeletal muscle and heart. These serotypes have been successfully used in numerous preclinical investigations to deliver CRISPR gene editing components for postnatal genome editing (Lau and Suh, 2017; Wang et al., 2017).
The SpCas9 ORF is about 4.2 kb in length, which is close to the AAV cargo limit. Thus, a second AAV vector that carries the donor template or sgRNA is required. Hepatocytes and muscle cells have been successfully edited in vivo using a dual-AAV approach (Yang et al., 2016). Staphylococcus aureus (Sa) Cas9, a smaller Cas9 protein encoded by a 3.2-kb cDNA, has been employed for gene editing in mdx mice to avoid the requirement for a dual-vector system (Nelson et al., 2016; Tabebordbar et al., 2016; Bengtsson et al., 2017). For more flexibility with promoter usage in DMD gene therapy, new AAV serotypes have been engineered, such as the MyoAAV 2A serotype, which appears more efficient than AAV9 in muscle fibers. This serotype can be employed with genome-specific modifiers, including base or prime editors, to maximize therapeutic potential (Mendell et al., 2020; Tabebordbar et al., 2021; Happi Mbakam et al., 2022a).
4.4.8.2 Nonviral delivery
Cas9 and sgRNAs can be delivered in vivo through nonviral delivery mechanisms in a variety of forms, including DNA, mRNA, and ribonucleoproteins (RNP). Electroporation has been used to deliver negatively charged DNA or mRNA into muscle cells (Xu et al., 2016). Direct delivery of Cas9 and sgRNA constructs into the skeletal muscle of mdx mice using this technique led to the restoration of dystrophin expression (Xu et al., 2016). Another delivery method for CRISPR components, such as RNP or mRNA/sgRNA, is via lipid-mediated nanoparticles (Zuris et al., 2015; Miller et al., 2017). The cationic lipid nanoparticles can enclose the complex, which can then be transported into cells via endocytosis and pinocytosis. This method is relatively inexpensive compared to other approaches (Kim et al., 2014). Gold nanoparticles coupled to DNA and complexed with cationic endosomal disruptive polymers have also been reported to successfully deliver CRISPR RNP to mdx mice (Lee et al., 2017). Even though it seems to be an appealing strategy for delivering the RNP complex, a major challenge is the systemic delivery requirement to reach the heart, diaphragm, and skeletal muscles through the body. This, together with issues of efficiency, needs to be addressed before effective clinical applications can be investigated.
4.4.9 Gene editing: clinical considerations
Theoretically, gene editing has the potential to cure the disease with a single treatment, “one and done,” by correcting the genetic defect causing the condition. Gene editing can enable the expression of normal or nearly normal dystrophin to maximize functional outcomes. The resultant dystrophin, post-gene editing, is anticipated to be larger than microdystrophins which are undergoing clinical trials. Physiological temporal-spatial expression is expected to be normal because the expression of dystrophin after gene editing will be driven under the control of the endogenous dystrophin gene locus. This is compared to therapeutic microdystrophin gene therapy, where expression is dependent on the cis-regulatory sequences designed and incorporated in the construct within the AAV vector (Min et al., 2019b).
4.4.10 Gene editing: future challenges
Despite recent advances, the gene-editing experimental approach is still in the early stages of development for DMD therapy, with several concerns that should be addressed. Notably, off-target mutation risk in vivo is a significant concern. Muscle-specific promoters have been used to drive the CRISPR cassette expression to reduce possible off-target effects in non-muscle tissues (Young et al., 2016; Miller et al., 2017). Other issues to be addressed are the delivery efficiency to all the affected skeletal muscles and the heart, in addition to issues related to the maintenance of long-term dystrophin expression. Cardiac myocytes are long-lived cells with minimal regeneration capacity; thus, upon adequate delivery of the editing cassette and precise correction, myocytes are anticipated to exhibit long-term dystrophin expression. However, skeletal muscle fibers have a remarkable regeneration capacity (Ma et al., 2022). Skeletal muscle cells are generated via satellite cell recruitment, the muscle stem cells that are not efficiently transduced by AAV (Arnett et al., 2014; Min et al., 2019b). Thus, the skeletal edited muscle cell proportion will be diluted with every regeneration cycle, ultimately reducing dystrophin expression in the fibers.
Furthermore, there are a number of immunological issues to take into account, such as immunogenicity toward Cas protein or the likelihood of immune reaction toward the replaced dystrophin, a possibility that remains with all gene replacement therapies. More research is required to fully address these concerns (Min et al., 2019b). Thus, further studies focusing on the safety and efficiency of the gene editing CRISPR system are necessary prior to clinical translation (Erkut and Yokota, 2022). The CRISPR system has recently been introduced into clinical trials for other diseases, such as cancer; allergy; and cardiovascular, immunological, and neurological disorders (Sharma et al., 2021). Even though the CRISPR system faces several challenges, it has undoubtedly created new possibilities for treating monogenic diseases that will certainly pave the way for many applications in the upcoming years.
4.5 Cell transplantation therapy
DMD cell-based therapeutics aim to transplant cells with functional dystrophin into dystrophin-deficient muscles. This is one of the earliest genetic approaches attempting to treat DMD (Smythe et al., 2000; Skuk et al., 2004; Skuk et al., 2006). However, multiple factors must be considered for successful muscle recovery. Autogenic cell transplantation, relying on genetic engineering of the host’s induced pluripotent stem cells (iPSCs) to restore functional dystrophin expression, is associated with lower immunogenic risk than allogenic cell transplantation (obtained from a healthy donor) requiring immunosuppression (Maffioletti et al., 2014). Ideally, the transplanted cells should be competent to cross the vascular wall (blood–muscle barrier) upon systemic delivery to target different striated muscle entities (heart, diaphragm, and limb muscles) to limit the requirement for multiple injection sites into individual muscles. Moreover, to provide a long-term effect and reduce the risks of inducing an immune response from repeated deliveries, the transplanted cells must have a high muscle engraftment rate, incorporate into the host myocytes, and persist through self-renewal (Wallace et al., 2008).
Various cell types are candidates for transplantation and are being studied for DMD therapy. Satellite cells (myoblasts) are quiescent muscle stem cells that reside beneath the basal lamina (Mauro, 1961; Moss and Leblond, 1971). Upon muscle damage, myoblasts are activated to proliferate, self-renew, and repair. Other cell types, notably bone marrow-derived cells, pericytes, mesoangioblasts, and mesenchymal stem cells, are also under investigation because of their ability to access the muscle compartment and their myogenic potential. However, the limited capacity of these cells to be expanded in vitro constitutes a limitation for wide therapeutic usage. Nonetheless, based on the early successes in preclinical studies, a clinical trial is underway to transplant skeletal muscle-derived mesoangioblasts into the circulation of patients with DMD. Induced pluripotent stem cells (iPSCs), featuring an infinite self-renewal capacity, can be differentiated into myogenic lineages. However, iPSCs-derived myogenic progenitors could carry the risk of unlimited growth and teratoma development in vivo (Rong et al., 2012).
One of the most challenging hurdles for cell-based clinical applications centers on delivery. Although preclinical studies show promise (Rouger et al., 2011), most systemic studies report only limited engraftment of dystrophin restoration (Briggs and Morgan, 2013; Hogrel et al., 2013). Myoblasts lack extravasation ability, and continuous efforts are being made to improve their trans-endothelial migration ability (Choi et al., 2022). As for extravasation-competent stem cells, only a small fraction of transplanted stem cells appears to reach the muscle upon systemic delivery (Dellavalle et al., 2007). Additionally, although local high-density myoblast injections (intra-muscular injections) produce some dystrophin restoration around the needle track (Skuk et al., 2004; 2006), this approach is only practical for superficial small muscles.
On another note, cardiosphere-derived stem cells offer a transplantable cell type with therapeutic potential. Preliminary results from the randomized, phase I/II HOPE-Duchenne clinical trial (NCT02485938) revealed that the intra-coronary infusion of cardiosphere-derived cells was well tolerated and suggested a positive effect on upper limb and heart function for up to 12 months (Taylor et al., 2019). Furthermore, phase II of the Hope-2 clinical trial (NCT04428476) recently revealed that intravenous delivery of cardiosphere-derived cells is safe and has beneficial effects in slowing the deterioration of muscle function in patients with late-stage DMD (McDonald et al., 2022).
5 Small molecule therapy for DMD: copolymers
Despite the numerous trials exploring potential therapeutics, there remains a dearth of effective treatments available for DMD patients. In this context, it is worth considering additional strategic approaches that target the primary defect of DMD: severe muscle membrane fragility. As discussed previously, as the primary pathophysiological defect in DMD is the marked susceptibility to contraction-induced membrane stress, a unique therapeutic approach is the use of synthetic membrane stabilizers to prevent muscle damage by directly stabilizing the dystrophin-deficient muscle membrane (Houang et al., 2015; 2017).
Poloxamers emerged from industrial applications in the 1940s. Recently, however, amphiphilic characteristics of poloxamer have shown a protective effect on the cell membrane. Poloxamer 188 (P188; PEO75-PPO30-PEO75, 8400 g/mol) is a non-ionic copolymer consisting of a triblock—a hydrophobic chain of poly(propylene oxide) (PPO) in the middle flanked by two hydrophilic poly(ethylene oxide) (PEO) chains on each side (Bates and Fredrickson, 1990). Studies using P188 have reported positive results in skeletal muscle fibers with electric injury (Lee et al., 1992; Lee et al., 1999; Collins et al., 2007), ischemia-reperfusion injuries (Hunter et al., 2010), heat injury (Merchant et al., 1998), radiation injury (Hannig et al., 1999), and sickle cell disease (Adams-Graves et al., 1997; Ballas et al., 2004). These results are due to the interactions between the phospholipid bilayer of the cells and the amphiphilic copolymer (Houang et al., 2017). Similar to these trials, DMD has been considered a potential candidate for the P188 treatment due to the fragile muscle cell membrane caused by dystrophin deficiency. In DMD, P188 is hypothesized to be beneficial in preventing cardiac myocyte damage induced by passive stretch and extracellular Ca2+ inflow triggered by micro-tears in the cell membrane (Yasuda et al., 2005) (Figure 3).
FIGURE 3
Copolymer-based muscle membrane stabilization of dystrophic muscle. Representation of copolymer-based stabilization of the damaged membrane via the interface of its hydrophobic PPO block (red) with stabilization by PEO blocks (blue) to prevent the entry of extracellular Ca2+ into the cell.
P188-treatment in isolated cardiac myocytes from mdx mice in vitro restored compliance and blocked susceptibility to stretch-induced Ca2+ overload (Yasuda et al., 2005). P188 delivery in vivo to mdx mice rapidly improved the geometry of the ventricles and suppressed the acute cardiac failure induced by a dobutamine stress test (Yasuda et al., 2005). P188 also prevented cardiomyopathy induced by isoproterenol in mdx mice (Spurney et al., 2011b). Similarly, systemic delivery of P188 for 8 weeks to dystrophic dogs (Golden retriever muscular dystrophy model) markedly reduced myocardial fibrosis, prevented left-ventricular remodeling, and inhibited the increase in cardiac Troponin I (TnI) and brain natriuretic peptide (BNP) in serum (Townsend et al., 2010). Furthermore, the direct application of P188 to isolated canine cardiac myocytes instantly restored poor myocyte compliance (Townsend et al., 2010).
For dystrophic skeletal muscle, the delivery method of P188 was found to be an important factor in the effectiveness of rescue of function. Interestingly, subcutaneous delivery of P188 showed protection against lengthening contraction-induced force loss, but this was not seen by intraperitoneal delivery. Furthermore, P188 significantly decreased baseline and lengthening contraction-induced membrane instability, as shown by reduced Evans Blue dye uptake in the mdx skeletal muscles (Houang et al., 2015). Skeletal muscles such as the diaphragm were also found to be protected by longer-term systemic delivery of P188 showing improved respiratory parameters, reduced centralized myonuclei, reduced variation in fiber size, and reduced collagen deposition (Markham et al., 2015).
Poloxamers can be manufactured in diverse forms by differing weight ratios of the PEO chain, molecular weight, and end group (Houang et al., 2018). Variations in hydrophilic and hydrophobic block lengths alter the hydrophobicity balance that determines the insertion of the molecule into the lipid bilayer of the cell and, thus, stabilization (Demina et al., 2005; Houang et al., 2017; Kim et al., 2017; Houang et al., 2018). For example, more hydrophobic poloxamers, such as P335 (PEO38–PPO54–PEO38), P333 (PEO20–PPO54–PEO20), and P181 (PEO2–PPO30–PEO2), insert into the liposomal membrane and eventually lyse the cell (Houang et al., 2018). Compared to P188, extended P188 (PEO140-PPO44-PEO140, 14,600 g/mol) also showed a robust protective effect at lower concentrations against the intracellular enzyme lactate dehydrogenase release induced by hypoosmotic stress (Houang et al., 2015). Furthermore, diblock copolymers (PEO75-PPO15) with a tert-butoxy end group (-C4) showed significant protection against lengthening contraction-induced force loss in mdx mice, whereas a hydroxyl end group (-H) did not show any protection (Houang et al., 2017). This finding showed that the hydrophobicity of the end group also directly influences interactions with the cell membrane and stabilization.
For membrane stabilizers in DMD, Phrixus Pharmaceuticals, Inc. has initiated a Phase 2 single-site, open-labeled trial for skeletal limb, cardiac, and respiratory muscle endpoints in non-ambulatory DMD patients (ClinicalTrials.gov Identifier: NCT03558958). Further research to determine the optimal structure-function of synthetic membrane-protecting copolymers will be useful in seeking a full therapeutic effect in DMD.
6 Non-dystrophin-based therapeutics
This section focuses on the genetic strategies related to the correction of or compensation for the lack of dystrophin via meaningful alternatives to direct dystrophin reconstitution. The advantage of dystrophin complementary approaches is that they may be applied to a number of MD patients, regardless of the type of mutation. Several pharmacological strategies to slow down the disease progress, such as those reducing inflammation or fibrosis, are also in development, and these have been reviewed by different groups (Spinazzola and Kunkel, 2016) and will not be covered in this review.
6.1 Utrophin modulation
Utrophin is a structural and functional autosomal paralog of dystrophin encoded by the UTRN gene (Love et al., 1989). This large cytoskeletal protein (395 kDa) shows sequence and structural similarity to dystrophin. Unlike dystrophin, utrophin is highly expressed in developing muscle and is localized at the sarcolemma in early human development (Love et al., 1989; Pons et al., 1993) when dystrophin is absent or present only at low levels. As the myofiber matures, utrophin levels decrease and become enriched at the neuromuscular junction (McNally, 2012).
Utrophin content is increased at the sarcolemma of skeletal muscle in DMD and BMD patients compared to healthy individuals (Weir et al., 2004; Arechavala et al., 2010; Mamchaoui, 2018). The baseline mild phenotype observed in the mdx mouse has been proposed to result from the efficient regeneration process in this model along with utrophin upregulation as a compensatory mechanism to mitigate the lack of dystrophin (Guiraud et al., 2015). This view is supported by the fact that mice deficient in both utrophin and dystrophin display a much more severe pathology compared to mdx mice (Deconinck et al., 1997). Furthermore, proof of principle that utrophin could play a role as a dystrophin surrogate comes from preclinical studies which established that an increase in utrophin protein levels in transgenic mdx mice prevented pathology, and this prevention was dependent on the amount of utrophin expressed (Tinsley et al., 1998). Studies in animal models have provided compelling evidence that utrophin functions in these scenarios to directly protect the muscle against dystrophin-deficient membrane instability. Because utrophin is an endogenous protein that can substitute for dystrophin, several approaches have been initiated to upregulate utrophin expression (Soblechero-Martín et al., 2021), including direct mechanisms, such as gene or protein replacement (Sonnemann et al., 2009), and indirect methods, such as transcriptional upregulation of the utrophin promoter or by stabilization of the protein or RNA (Michel et al., 2008).
Utrophin is expressed from at least two promoters, known as promoters A and B (Burton et al., 1999). Expression from promoter B is predominantly in endothelial cells in blood vessels, whereas expression from promoter A occurs in muscle and other tissues (Weir et al., 2002). Utrophin upregulation by stimulating promoter A activity is a promising pharmacological approach that has been extensively investigated. Thousands of small molecule candidates from drug libraries have been tested by high-throughput screening (HTS) assays to identify molecules acting at the utrophin A promotor site. Numerous small molecules, including heregulin (Krag et al., 2004), nabumetone (Moorwood et al., 2011), L-arginine (Voisin et al., 2005), and okadaic acid, have shown promising results at the preclinical level, with dose-dependent activation of the utrophin promoter. Ezutromid (SMTC1100) was the first orally bioavailable utrophin regulator that showed increased UTRN transcription (Soblechero-Martín et al., 2021). Pre-clinical results with the small molecule drug Ezutromid led Summit Therapeutics to initiate clinical trials as a potential treatment for DMD and BMD. Promising results from a Phase 1 healthy volunteer study showed that SMTC1100 is safe and well tolerated, with plasma levels achieved above those thought to be required to modulate utrophin (Ricotti et al., 2016). However, a Phase 2 clinical study (NCT02858362) failed to achieve both the primary (changes in leg muscle magnetic resonance parameters) and secondary (increased utrophin levels and decreased muscle damage) endpoints (Soblechero-Martín et al., 2021). Based on these results, Summit Therapeutics abandoned the development program of Ezutromid (Ricotti et al., 2016; Muntoni et al., 2019).
Direct utrophin protein replacement using recombinant full-length or truncated utrophin is another potential strategy to increase utrophin levels in vivo. The TAT protein transduction domain of the human immunodeficiency virus (HIV1) has been used to generate chimeric protein microutrophin (TAT-μUtr) and utrophin protein (TAT-Utr) (Sonnemann et al., 2009). Preclinical studies in mdx and dko mice were promising; however, further progress has not been reported. A similar pathway to micro-dys gene therapy has been pursued in the development of utrophin gene therapy. A number of preclinical studies using “micro-utrophin” (µUtrn) gene delivery have been reported recently, with some studies reporting restoration of the DGC, prevention of myofiber degeneration, normalization of serum CK levels, and improvement of muscle function (Ebihara et al., 2022). Furthermore, additional studies on double knockout (dko) mice and canine X-linked muscular dystrophy dogs have shown that µUtrn improves the severe pathological dystrophic phenotype in these models (Cerletti et al., 2003). One advantage to delivering utrophin is that it is less likely to elicit an immune response. It has been shown that viral gene delivery of dystrophin into DMD patients has been associated with an immune response, which limits the expression of dystrophin (Mendell et al., 2010). A protein such as utrophin, which is normally expressed in DMD patients, should not be associated with a similar immune response.
Utrophin, while highly similar to dystrophin, does not share all of the dystrophin binding domains. Therefore, it may not be able to fully substitute for dystrophin. Nonetheless, utrophin upregulation has been shown as a promising therapeutic approach, applicable to all DMD and BMD patients, irrespective of their dystrophin mutation. Many pathways involved in utrophin expression are currently being investigated. However, the amount of utrophin required by dystrophic patients to achieve a relevant clinical benefit remains to be determined. Further research is required before utrophin therapies can apply to DMD and BMD patients.
6.2 α7-Integrin upregulation
The α7β1 integrin protein is the predominant laminin-binding integrin in skeletal, cardiac, and vascular smooth muscle (Burkin and Kaufman, 1999). The integrin/laminin complex serves as a mechano-signaling anchor that binds laminin and links the ECM on the surface of muscle cells with the intracellular actin cytoskeleton (Hynes, 1992). α7 is present throughout the sarcolemma and is enriched at the myotendinous and neuromuscular junction. In particular, α7β1-integrin/laminin-211 plays a critical role in the functional integrity and maintenance of skeletal myoblasts and adult myofibers (Hodges et al., 1997; Mayer et al., 1997). α7 has been shown to be an important modifier of dystrophic symptoms, and defects in the components of this complex cause muscular dystrophy, illustrating the essential role of the α7 chain (Hodges et al., 1997; Guo et al., 2006). Interestingly, α7-integrin expression is increased at the sarcolemma in the mdx mouse and DMD patients (Hodges et al., 1997), demonstrating evidence that integrin upregulation may serve to functionally compensate for the lack of dystrophin. Moreover, knockout of both dystrophin and α7 integrin has been shown to produce a significantly more severe dystrophic phenotype, further supporting the evidence of a compensatory role for α7 integrin for dystrophin (Rooney et al., 2006).
Burkin et al. (2001, 2005) showed that transgenic overexpression of α7-integrin in dystrophin/utrophin double knockout mice (mdx/utrn−/−) alleviates pathology, extends viability and mobility, and reduces kyphosis. In addition, increased expression of α7 in mdx mice significantly protected against loss of force, reversed muscle pathology, and stabilized sarcolemmal integrity (Heller et al., 2013). Importantly, eightfold overexpression of α7-integrin does not demonstrate detectable toxicity or disruption to global gene expression profiles (Liu et al., 2008). Therefore, similar to utrophin, small compound screens for α7-integrin modulators appear to be a viable approach (Gurpur et al., 2009).
Laminin-111 is another potential candidate of interest (Rooney et al., 2009a). Injection of laminin-111 protein in the mdx mouse increased the expression of α7-integrin, stabilized the sarcolemma, and protected muscle from exercise-induced damage (Rooney et al., 2009a; 2009b). Conversely, transgenic expression of the laminin α1 chain to enhance heterotrimer formation of laminin-111 in the mdx mouse reported no improvement of the dystrophic symptoms (Gawlik et al., 2011), showing that further studies are necessary to validate the functionality of laminin-111 protein therapy in DMD.
6.3 Myostatin, follistatin, and other muscle growth strategies
Myostatin is a transforming growth factor-β-superfamily member that acts as a negative regulator of skeletal muscle growth (Mcpherron et al., 1997). The biological relevance of myostatin was addressed in mice through gene overexpression studies using normal or dominant-negative forms of the protein, systemic administration of myostatin protein or inactivating antibody, and gene inactivation (Zhu et al., 2000; Yang et al., 2001; Nishi et al., 2002; Grobet et al., 2003; Walsh and Celeste, 2005; Reisz-porszasz et al., 2022). Regardless of the approach used to induce myostatin inhibition, studies have shown functional improvement of the dystrophic muscle, such as increasing body weight, muscle mass, and muscle size, along with a significant decrease in muscle degeneration. Based on these studies and others carried out in cultured cells, the general consensus is that myostatin regulates postnatal muscle-fiber size by maintaining satellite cells in a quiescent state and inhibiting protein synthesis. Myostatin also regulates the number of muscle fibers during development by blocking the proliferation and differentiation of myoblasts (Lee, 2004; Tobin and Celeste, 2005).
However, some concerns have been raised about whether myostatin inhibition leads to a truly healthy muscle, as an exercise in myostatin-deficient cattle leads to early exhaustion. Moreover, it can even be deleterious, as disused muscle atrophy is markedly more severe in the context of myostatin deficiency (Mcmahon et al., 2022). Nonetheless, considering the effect of myostatin on muscle growth, inhibition of myostatin has been considered a therapeutic target in the treatment of muscle-degenerative and wasting conditions, such as muscular dystrophies, and clinical trials have been designed to increase muscle mass and strength in several of the most common forms of adult muscular dystrophy. Nevertheless, clinical trials in humans have been disappointing owing to a lack of improvement in muscle strength (Wagner et al., 2008) or adverse effects (Attie et al., 2013). Despite these negative results, other clinical trials based on blocking myostatin activity have been initiated (Mulivor et al., 2014; Pfizer, 2014).
7 Clinical trials and approved therapies
A growing number of pharmaceutical companies and startups have directed efforts toward DMD therapeutics. Many challenges remain, and the problem of delivery to all muscles of the body needs to be resolved. Nevertheless, there is little doubt that although a cure remains elusive, there has been a rapid expansion in the number of treatments entering clinical trials that have the potential to provide a significant clinical impact on the quality of life of DMD patients (ClinicalTrials.gov). Some of these approaches have received regulatory approval in the USA, Europe, and Japan (Mercuri et al., 2019). The increase in the number of patient registries and the ability to link natural histories significantly enhance opportunities to examine inter-individual differences to best evaluate disease progression/regression. These opportunities will greatly facilitate the emerging field of personalized/combinatory therapies, which represent the future of effective DMD treatments and will set the stage for other rare disease therapeutics.
Gene therapy has been elevated in the DMD field due to the promise that this approach could “stably” correct DMD defects via restoring dystrophin throughout the body (Gregorevic et al., 2006; Chamberlain and Chamberlain, 2017). Adeno-associated virus- (AAV) mediated gene therapies have advanced significantly since their clinical trial debut in 2003 (Kuzmin et al., 2021). Several studies have demonstrated the body-wide expression and therapeutic effect of this approach following intravascular AAV microdystrophin delivery (Mendell et al., 2021). Based on the many positive efficacy reports from experiments in the mdx mouse model and golden retrievers with muscular dystrophy (GRMD dog model) (Birch et al., 2023), four microdystrophin constructs have moved forward and are currently being investigated in clinical trials (Figure 1) (Table 2). The clinical trials were initiated in the United States in December 2017 and are now ongoing in Europe.
Clinical trial PF-06939926 is an AAV9-mediated transfer of microdystrophin currently tested in a Pfizer Phase 1b open-label clinical trial evaluating dose, safety, and tolerability of a single IV infusion of microdystrophin in ambulatory and non-ambulatory DMD patients (NCT03362502 and NCT04281485) (Table 2). PF-06939926 has been tested in men with DMD, ages 4 and older, at 21 sites, and two doses have been used: a low dose of 1 × 1014 vector genomes (vg)/kg) and a higher dose of 2 × 1014 vg/kg. Early data from five of six boys showed some evidence of improvement (or at least no decline) in the NorthStar Ambulatory Assessment (NSAA), compared with participants given a placebo during previous clinical trials. Side effects and serious adverse events were reported in some patients. Tragically, a death of a patient was reported, which led to the trial being put on hold due to questions about Pfizer’s potency tests. Recently, the FDA announced the removal of the clinical hold after the company had addressed the agency’s request regarding a potency assay and implementation of a protocol amendment.
SGT-001, an AAV9-mediated transfer of microdystrophin from Solid Biosciences, is also being evaluated in a phase I clinical trial, IGNITE DMD (NCT03368742) (Table 2). After a suspension due to a serious adverse effect in one patient, the study was recently re-activated with an amended clinical protocol and using SGT-001 manufactured with a second-generation process (Solid Bioscience). A clinical trial by Sarepta Therapeutics, Inc. is ongoing to investigate the safety and efficacy of the IV infusion of rAAVrh74.MHCK7 microdystrophin (SRP-9001), in a first open-label phase I/II trial (NCT03375164). Some evidence of 12-week dystrophin expression and a good safety profile was communicated from the first 11 participants enrolled in Study SRP-9001–103 (NCT04626674), another open-label Phase I study being conducted in partnership with Roche (Sarepta therapeutics).
Overall, to date, clinical trials have accrued preliminary results that have unfortunately demonstrated a lack of clinically meaningful success and missed milestones for efficacy (Mullard, 2021a). Thus far, the human trial data do not report the same success reported in animal works, indicating this promising approach requires significant further enhancement (Mullard, 2021b).
There is a clear need for caution as several serious treatment-related adverse events have been reported. Hepatic transaminitis, classed as severe, was reported in participants in trials in DMD, following SGT001 (Wagner et al., 2021) and rAAVrh74.MHCK7.microdystrophin (NCT03375164) (Mendell et al., 2020). One incidence of vomiting requiring hospital admission was reported as a severe adverse event with the PF-06939926 microdystrophin product (NCT03362502) (Belluscio et al., 2021). Moreover, two patients receiving rAAVrh74.MHCK7.microdystrophin (NCT03375164) experienced severe rhabdomyolysis (Today MDN, 2019; Novack et al., 2021). Significant issues remain to be considered, such as pre-existing DMD population immunity to certain AAV serotypes, the efficacy of the specific truncated micro- or mini-dystrophin variants used, and re-administration challenges and serotype switching considerations.
Consequently, other approaches aim to produce nearly full-length dystrophin by targeting only the region that encompasses the mutation in an effort to bypass the genetic defect. During the last decade, numerous approaches have been developed to directly correct genetic defects at the RNA level (Hanson et al., 2021), as discussed previously. In the case of dystrophin, such approaches are designed to either restore the expression of a full-length transcript or a nearly full-length DMD.
Ataluren (Translarna™, previously known as PTC124) is an orally bioavailable small molecule developed by PTC Therapeutics designed to enable the formation of a functioning protein in patients with genetic disorders caused by a nonsense mutation (Politano, 2021). Ataluren has demonstrated its efficacy in vivo using the mdx mouse model (Welch et al., 2007). However, laboratories failed to replicate this finding, questioning the specificity of Ataluren against stop codons (Auld et al., 2009; McElroy et al., 2013). Nonetheless, supported by evidence for functional improvement in a cell model of Hurler syndrome (Goldmann et al., 2012) and in mouse models of nonsense mutation-associated cystic fibrosis (Kerem et al., 2014), Ataluren advanced to clinical trials and was granted conditional marketing authorization by EMA in 2014 (https://www.ema.europa.eu/en/medicines/human/EPAR/translarna) for DMD ambulatory patients and those aged ≥2 years (Table 3). To date, two randomized, double-blind, placebo-controlled trials of Ataluren in DMD patients have been conducted: a Phase IIb trial (NCT00592553) on 174 randomized patients and a Phase III trial (NCT01826487) on 230 randomized patients (Table 3). In both trials, Ataluren was well-tolerated (Campbell et al., 2020). However, the trials failed to achieve the 48-week primary endpoint of improved distance walked in the 6-min walk test (SMWT) compared with patients who received placebo treatment. Nonetheless, these trials reported a trend of therapeutic efficacy, specifically a 29-m increase in the SMWT and an improvement in timed function tests in those who received Ataluren compared with placebo (Campbell et al., 2020; Politano, 2021). Unfortunately, very little evidence exists to inform the question of whether Ataluren displays any degree of efficiency or delivers a benefit in the heart.
TABLE 3
| Drug name | Description | Company | Delivery route | Current stage | Clinical trial number |
|---|---|---|---|---|---|
| Stop codon read-through | |||||
| Ataluren | Nonsense suppression | PTC Therapeutics | Oral | Phase III | NCT01557400 |
| Exon skipping | |||||
| Eteplirsen (AVI-4568) | PMO morpholino targeting exon 51 | Sarepta Therapeutics | Intravenous | Phase II | NCT03218995 |
| Phase II | NCT04179409 | ||||
| Phase III | NCT03992430 | ||||
| Phase III | NCT03985878 | ||||
| Golodirsen (SRP4053) | PMO morpholino targeting exon 53 | Sarepta Therapeutics | Intravenous | Phase II | NCT04179409 |
| Phase III | NCT02500381 | ||||
| Phase III | NCT03532542 | ||||
| Casimersen (SRP4053) | PMO morpholino targeting exon 45 | Sarepta Therapeutics | Intravenous | Phase II | NCT04179409 |
| Viltolarsen (NCNP-01) | PMO morpholino targeting exon 53 | Nippon Shinyaku Co. Ltd | Intravenous | Phase II | NCT03167255 |
| Drisapersen (PRO051) | 2′-O-methyl PS Targeting exon 51 | BioMarin Pharmaceutical Inc | Intravenous | Extension | NCT02636686 |
Clinical trials using stop codon read-through and exon skipping for DMD therapy in 2023.
A number of chemical versions of AONs exist to be applied for exon-skipping, including 2′-O-methyl-modified RNA, phosphorodiamidate morpholino oligomers (PMO), and tricycloDNA antisense. All of them have been used in vitro and in vivo in mouse and dog models and demonstrated efficient skipping that resulted in histology and functional improvements (Niks and Aartsma-Rus, 2017), as previously discussed. Several PMO compounds are in development to target the exons that represent the highest proportions of deletions amenable to exon-skipping. Eteplirsen (marketed under the trade name Exondys 51) is a morpholino ASO from Sarepta Therapeutics designed to mask a splice acceptor sequence in exon 51 of the dystrophin gene, thereby promoting exon skipping and restoration of the reading frame in the 13% of patients with amenable frame-shifting mutations. This was the first drug approved by the FDA as a specific DMD therapy. Patients’ muscle biopsies after approximately a year of systemic dosing showed modest efficacy in the production of dystrophin protein expression (∼1%), which is considered to be inadequate to confer significant clinical benefits (NCT01396239, NCT01540409, NCT02255552) (Table 3) (Mendell et al., 2016; Niks and Aartsma-Rus, 2017).
Regardless, the drug was approved, and efforts are underway to develop more effective oligonucleotide therapies. Tissue penetration and longevity of PMOs have remained an ongoing concern. To address these shortcomings, other delivery systems for AONs are currently being investigated to identify the modifications that can optimize cellular penetration and systemic safety, with a major focus on PPMOs (Nguyen and Yokota, 2019). Additionally, multiple other exon-skipping AONs are being actively developed, with the FDA recently giving approval for Golodirsen (SRP-4053) and Viltolarsen, designed to skip exon 53, and Casimersen (SRP-4045), designed to skip exon 45 (Table 3). So far, data collected from DMD patients show that PMO AONs are well tolerated and safe in DMD following weekly intravenous (IV) administration (Frank et al., 2020; Komaki et al., 2020; Wagner et al., 2021).
8 Conclusion
The identification of the dystrophin gene as the cause of DMD has led to improved diagnosis while providing deep insights into the biochemistry and cellular physiology of the striated muscle cytoskeleton-membrane-ECM interface. The preceding decades have been marked by careful mechanistic studies aiming to tease apart the molecular processes disrupted by dystrophin loss in an effort to understand and correct the underlying pathology of DMD. These insights have greatly impacted DMD diagnosis and care, including the opportunity to develop novel, personalized, and effective therapeutic strategies to prolong and improve the quality of life of patients with DMD.
9 Future directions
The future of heart/skeletal muscle therapy in DMD will likely require a combination of approaches to achieve optimal outcomes, including a therapeutic approach to correct the genetic defect and target the secondary effects caused by the lack of dystrophin. Advances in technology have made many of the problems with therapeutic approaches more tractable. Despite that, two crucial and challenging issues shared by all new drugs are delivery and targeting. Finding an appropriate vehicle is critical to consistently reach the proper drug target at minimal, effective dosing. This is especially significant considering the enormous healthcare costs associated with these proposed therapies. Nonetheless, there is little doubt that although a cure remains elusive, there has been a rapid expansion in the number of treatments entering clinical trials that have the potential to provide a significant clinical impact on the quality of life for DMD patients. This turning point in the development of DMD therapies marks the beginning of a new mission for correcting monogenic muscular disorders. In turn, these approaches set an example for research progress for other related disorders.
Statements
Author contributions
ABBA searched the literature, organized the article structure, prepared tables and figures, and wrote and guided the manuscript for this review. NH searched the literature, prepared tables and figures, and wrote the manuscript. HC, AM, DH, and JB searched the literature and wrote the manuscript. JM guided the manuscript, provided critical input, and revised the manuscript for this review. All authors contributed to the article and approved the submitted version.
Funding
National Institutes of Health (R01)—Title: Copolymer-based sarcolemma stabilization for protecting dystrophic skeletal muscles in vivo (1R01AR071349). National Institutes of Health (R01)—Title: Skeletal muscle sarcomere function in health and disease (R01AR079477). National Institutes of Health (R01)—Title: Targeted membrane integrity in cardiac ischemia and reperfusion (R01HL122323). Regenerative Medicine Minnesota—Title: Advancing a genetically edited biosensor platform in human induced pluripotent stem cell-derived cardiac muscle for personalized medicine and drug discovery. This work was supported by grants from the NIH, the American Heart Association, the Muscular Dystrophy Association, and Regenerative Medicine Minnesota.
Conflict of interest
JM is on the scientific advisory board of and holds shares in Phrixus Pharmaceuticals Inc., a company developing novel therapeutics for heart failure. The terms of this arrangement have been reviewed and approved by the University of Minnesota in accordance with its conflict-of-interest policies.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Aartsma-Rus A. Ferlini A. Goemans N. Pasmooij A. M. Wells D. J. Bushby K. et al (2014). Translational and regulatory challenges for exon skipping therapies. Hum. Gene Ther.25, 885–892. 10.1089/hum.2014.086
2
Aartsma-Rus A. Fokkema I. Verschuuren J. Ginjaar I. van Deutekom J. van Ommen G. J. et al (2009). Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat.30, 293–299. 10.1002/humu.20918
3
Aartsma-Rus A. van Putten M. (2020). The use of genetically humanized animal models for personalized medicine approaches. Dis. Model Mech.13, dmm041673. 10.1242/dmm.041673
4
Aartsma‐Rus A. Van Deutekom J. C. T. Fokkema I. F. Van Ommen G. B. Den Dunnen J. T. (2006). Entries in the leiden duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading‐frame rule. Muscle and Nerve Official J. Am. Assoc. Electrodiagn. Med.34, 135–144. 10.1002/mus.20586
5
Abmayr S. Chamberlain J. (2006). The structure and function of dystrophin. Molecular mechanisms of muscular dystrophies. Georget. Landes Biosci., 14–34.
6
Adams-Graves P. Kedar A. Koshy M. Steinberg M. Veith R. Ward D. et al (1997). RheothRx (poloxamer 188) injection for the acute painful episode of sickle cell disease: A pilot study. Blood90, 2041–2046. 10.1182/blood.V90.5.2041
7
Ahmad M. Wolberg A. Kahwaji C. I. (2022). “Biochemistry, electron transport chain,” in StatPearls (treasure island (FL).
8
Ahn A. H. Kunkel L. M. (1993). The structural and functional diversity of dystrophin. Nat. Genet.3 (4), 283–291. 10.1038/ng0493-283
9
Allen D. G. Whitehead N. P. Froehner S. C. (2016). Absence of dystrophin disrupts skeletal muscle signaling: Roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol. Rev.96, 253–305. 10.1152/physrev.00007.2015
10
Amin R. S. Kimball T. R. Bean J. A. Jeffries J. L. Paul Willging J. Cotton R. T. et al (2002). Left ventricular hypertrophy and abnormal ventricular geometry in children and adolescents with obstructive sleep apnea. Am. J. Respir. Crit. Care Med.165, 1395–1399. 10.1164/rccm.2105118
11
Amoasii L. Hildyard J. C. W. Li H. Sanchez-Ortiz E. Mireault A. Caballero D. et al (2018). Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science362, 86–91. 10.1126/science.aau1549
12
Amoasii L. Long C. Li H. Mireault A. A. Shelton J. M. Sanchez-Ortiz E. et al (2017). Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci. Transl. Med.9, eaan8081. 10.1126/scitranslmed.aan8081
13
Andrade F. H. Reid M. B. Allen D. G. Westerblad H. (1998). Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol.509 (2), 565–575. 10.1111/j.1469-7793.1998.565bn.x
14
Angelini C. Peterle E. (2012). Old and new therapeutic developments in steroid treatment in Duchenne muscular dystrophy. Acta Myol.31, 9–15.
15
Anzalone A. V. Koblan L. W. Liu D. R. (2020). Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol.38, 824–844. 10.1038/s41587-020-0561-9
16
Anzalone A. V. Randolph P. B. Davis J. R. Sousa A. A. Koblan L. W. Levy J. M. et al (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature576, 149–157. 10.1038/s41586-019-1711-4
17
Arechavala V. Kinali M. Feng L. Brown S. C. Sewry C. Morgan J. E. et al (2010). Immunohistological intensity measurements as a tool to assess sarcolemma-associated protein expression. Neuropathol. Appl. Neurobiol.36, 265–274. 10.1111/j.1365-2990.2009.01056.x
18
Arechavala-Gomeza V. Graham I. R. Popplewell L. J. Adams A. M. Aartsma-Rus A. Kinali M. et al (2007). Comparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mRNA splicing in human muscle. Hum. Gene Ther.18, 798–810. 10.1089/hum.2006.061
19
Arnett A. L. H. Konieczny P. Ramos J. N. Hall J. Odom G. Yablonka-Reuveni Z. et al (2014). Adeno-associated viral vectors do not efficiently target muscle satellite cells. Mol. Therapy-Methods Clin. Dev.1, 14038. 10.1038/mtm.2014.38
20
Attie K. M. Borgstein N. G. Yang Y. Condon C. H. Seehra J. A. S. S. Sherman M. L. (2013). A single ascending-dose study of muscle regulator ace-031 in healthy volunteers. 10.1002/mus.23539
21
Auld D. S. Thorne N. Maguire W. F. Inglese J. (2009). Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc. Natl. Acad. Sci. U. S. A.106, 3585–3590. 10.1073/pnas.0813345106
22
Bach J. R. Ishikawa Y. Kim H. (1997). Prevention of pulmonary morbidity for patients with Duchenne muscular dystrophy. Chest112, 1024–1028. 10.1378/chest.112.4.1024
23
Ballas S. K. Files B. Luchtman-Jones L. Benjamin L. Swerdlow P. Hilliard L. et al (2004). Safety of purified poloxamer 188 in sickle cell disease: phase I study of a non-ionic surfactant in the management of acute chest syndrome. Hemoglobin28, 85–102. 10.1081/hem-120035919
24
Banks G. B. Combs A. C. Chamberlain J. R. Chamberlain J. S. (2008). Molecular and cellular adaptations to chronic myotendinous strain injury in mdx mice expressing a truncated dystrophin. Hum. Mol. Genet.17, 3975–3986. 10.1093/hmg/ddn301
25
Banks G. B. Gregorevic P. Allen J. M. Finn E. E. Chamberlain J. S. (2007). Functional capacity of dystrophins carrying deletions in the N-terminal actin-binding domain. Hum. Mol. Genet.16, 2105–2113. 10.1093/hmg/ddm158
26
Banks G. B. Judge L. M. Allen J. M. Chamberlain J. S. (2010). The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS Genet.6, e1000958. 10.1371/journal.pgen.1000958
27
Bansal D. Miyake K. Vogel S. S. Groh S. Chen C. C. Williamson R. et al (2003). Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature423, 168–172. 10.1038/nature01573
28
Barton-Davis E. R. Cordier L. Shoturma D. I. Leland S. E. Sweeney H. L. (1999). Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Investigation104, 375–381. 10.1172/JCI7866
29
Bassi E. Falzarano S. Fabris M. Gualandi F. Merlini L. Vattemi G. et al (2012). Persistent dystrophin protein restoration 90 days after a course of intraperitoneally administered naked 2′ OMePS AON and ZM2 NP-AON complexes in mdx mice. J. Biomed. Biotechnol.2012, 897076. 10.1155/2012/897076
30
Bates F. S. Fredrickson G. H. (1990). Block copolymer thermodynamics: Theory and experiment. Annu. Rev. Phys. Chem.41, 525–557. 10.1146/annurev.pc.41.100190.002521
31
Bedair H. S. Karthikeyan T. Quintero A. Li Y. Huard J. (2008). Angiotensin II receptor blockade administered after injury improves muscle regeneration and decreases fibrosis in normal skeletal muscle. Am. J. Sports Med.36, 1548–1554. 10.1177/0363546508315470
32
Benditt J. O. Boitano L. (2005). Respiratory support of individuals with Duchenne muscular dystrophy: Toward a standard of care. Phys. Med. Rehabil. Clin. N. Am.16, 1125–1139. 10.1016/j.pmr.2005.08.017
33
Bengtsson N. E. Hall J. K. Odom G. L. Phelps M. P. Andrus C. R. Hawkins R. D. et al (2017). Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun.8, 14454–14510. 10.1038/ncomms14454
34
Beringer P. Winter M. E. (2011). “Aminoglycoside antibiotics,” in Basic clinical pharmacokineticsFifth Edition, 134–181. 10.1002/9783527678679.dg00403
35
Bhattarai S. Li Q. Ding J. Liang F. Gusev E. Lapohos O. et al (2022). TLR4 is a regulator of trained immunity in a murine model of Duchenne muscular dystrophy. Nat. Commun.13, 879. 10.1038/s41467-022-28531-1
36
Bidou L. Allamand V. Rousset J. P. Namy O. (2012). Sense from nonsense: Therapies for premature stop codon diseases. Trends Mol. Med.18, 679–688. 10.1016/j.molmed.2012.09.008
37
Birch S. M. Lawlor M. W. Conlon T. J. Guo L. J. Crudele J. M. Hawkins E. C. et al (2023). Assessment of systemic AAV-microdystrophin gene therapy in the GRMD model of Duchenne muscular dystrophy. Sci. Transl. Med.15, eabo1815. 10.1126/scitranslmed.abo1815
38
Birnkrant D. J. Bushby K. Bann C. M. Apkon S. D. Blackwell A. Colvin M. K. et al (2018). Diagnosis and management of duchenne muscular dystrophy, part 3: Primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol.17, 445–455. 10.1016/S1474-4422(18)30026-7
39
Bladen C. L. Salgado D. Monges S. Foncuberta M. E. Kekou K. Kosma K. et al (2015). The TREAT‐NMD DMD global database: Analysis of more than 7,000 duchenne muscular dystrophy mutations. Hum. Mutat.36, 395–402. 10.1002/humu.22758
40
Boland B. J. Silbert P. L. Groover R. V. Wollan P. C. Silverstein M. D. (1996). Skeletal, cardiac, and smooth muscle failure in Duchenne muscular dystrophy. Pediatr. Neurol.14, 7–12. 10.1016/0887-8994(95)00251-0
41
Bordeira-Carriço R. Pêgo A. P. Santos M. Oliveira C. (2012). Cancer syndromes and therapy by stop-codon readthrough. Trends Mol. Med.18, 667–678. 10.1016/j.molmed.2012.09.004
42
Boutin S. Monteilhet V. Veron P. Leborgne C. Benveniste O. Montus M. F. et al (2010). Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: Implications for gene therapy using AAV vectors. Hum. Gene Ther.21, 704–712. 10.1089/hum.2009.182
43
Boyce F. M. Beggs A. H. Feener C. Kunkel L. M. (1991). Dystrophin is transcribed in brain from a distant upstream promoter. Proc. Natl. Acad. Sci. U. S. A.88, 1276–1280. 10.1073/pnas.88.4.1276
44
Brashear S. E. Wohlgemuth R. P. Gonzalez G. Smith L. R. (2021). Passive stiffness of fibrotic skeletal muscle in mdx mice relates to collagen architecture. J. Physiol.599, 943–962. 10.1113/JP280656
45
Briggs D. Morgan J. E. (2013). Recent progress in satellite cell/myoblast engraftment - relevance for therapy. FEBS J.280, 4281–4293. 10.1111/febs.12273
46
Briston T. Selwood D. L. Szabadkai G. Duchen M. R. (2019). Mitochondrial permeability transition: A molecular lesion with multiple drug targets. Trends Pharmacol. Sci.40, 50–70. 10.1016/j.tips.2018.11.004
47
Buddhe S. Cripe L. Friedland-little J. Eghtesady P. Finder J. Hor K. et al (2019)., 142. Cardiac, S72–S81. 10.1542/peds.2018-0333ICardiac management of the patient with duchenne muscular dystrophyHHS Public Access
48
Büning H. Schmidt M. (2015). Adeno-associated vector toxicity—To be or not to be?Mol. Ther.23, 1673–1675. 10.1038/mt.2015.182
49
Burkin D. J. Kaufman S. J. (1999). The α 7 β 1 integrin in muscle development and disease, 183–190.
50
Burkin D. J. Wallace G. Q. Milner D. J. Chaney E. J. Mulligan J. A. Kaufman S. J. (2005). Transgenic expression of {alpha}7{beta}1 integrin maintains muscle integrity, increases regenerative capacity, promotes hypertrophy, and reduces cardiomyopathy in dystrophic mice. Increases Regen. Capacity , Promot. Hypertrophy , Reduces Cardiomyopathy Dystrophic Mice166, 253–263. 10.1016/s0002-9440(10)62249-3
51
Burkin D. J. Wallace G. Q. Nicol K. J. Kaufman D. J. Kaufman S. J. (2001). Enhanced expression of the alpha 7 beta 1 integrin reduces muscular dystrophy and restores viability in dystrophic miceDystrophy Restores Viability Dystrophic Mice152, 1207–1218. 10.1083/jcb.152.6.1207
52
Burton E. A. Tinsley J. M. Holzfeind P. J. Rodrigues N. R. Davies K. E. (1999). A second promoter provides an alternative target for therapeutic up-regulation of utrophin in Duchenne muscular dystrophy, 96.
53
Burzyn D. Kuswanto W. Kolodin D. Shadrach J. L. Cerletti M. Jang Y. et al (2013). A special population of regulatory T cells potentiates muscle repair. Cell155, 1282–1295. 10.1016/j.cell.2013.10.054
54
Bushby K. Finkel R. Birnkrant D. J. Case L. E. Clemens P. R. Cripe L. et al (2010). Diagnosis and management of duchenne muscular dystrophy, part 2: Implementation of multidisciplinary care. Lancet Neurol.9, 177–189. 10.1016/S1474-4422(09)70272-8
55
Bushby K. Muntoni F. Urtizberea A. Hughes R. Griggs R. (2004). Report on the 124th ENMC International Workshop. Treatment of Duchenne muscular dystrophy; defining the gold standards of management in the use of corticosteroids 2-4 April 2004, Naarden, The Netherlands. Neuromuscul. Disord.14, 526–534. 10.1016/j.nmd.2004.05.006
56
Cai B. Spencer M. J. Nakamura G. Tseng-Ong L. Tidball J. G. (2000). Eosinophilia of dystrophin-deficient muscle is promoted by perforin-mediated cytotoxicity by T cell effectors. Am. J. Pathol.156, 1789–1796. 10.1016/S0002-9440(10)65050-X
57
Calcedo R. Vandenberghe L. H. Gao G. Lin J. Wilson J. M. (2009). Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J. Infect. Dis.199, 381–390. 10.1086/595830
58
Campbell C. Barohn R. J. Bertini E. Chabrol B. Comi P. Darras B. T. et al (2020). Meta-analyses of ataluren randomized controlled trials in nonsense mutation Duchenne muscular dystrophy. J. Comp. Eff. Res.9, 973–984. 10.2217/cer-2020-0095
59
Campbell K. P. Kahl S. D. (1989). Association of dystrophin and an integral membrane glycoprotein. Nature338, 259–262. 10.1038/338259a0
60
Castiglioni A. Corna G. Rigamonti E. Basso V. Vezzoli M. Monno A. et al (2015). FOXP3+ T cells recruited to sites of sterile skeletal muscle injury regulate the fate of satellite cells and guide effective tissue regeneration. PLoS One10, e0128094. 10.1371/journal.pone.0128094
61
Cerletti M. Negri T. Cozzi F. Colpo R. Andreetta F. Croci D. et al (2003). Dystrophic phenotype of canine X-linked muscular dystrophy is mitigated by adenovirus-mediated utrophin gene transfer. Gene Ther.10, 750–757. 10.1038/sj.gt.3301941
62
Chamberlain J. R. Chamberlain J. S. (2017). Progress toward gene therapy for duchenne muscular dystrophy. Mol. Ther.25, 1125–1131. 10.1016/j.ymthe.2017.02.019
63
Chance B. Sies H. Boveris A. (1979). Hydroperoxide metabolism in mammalian organs. Physiol. Rev.59, 527–605. 10.1152/physrev.1979.59.3.527
64
Chemello F. Bassel-duby R. Olson E. N. (2020). Correction of muscular dystrophies by CRISPR gene editing, 130.
65
Chemello F. Chai A. C. Li H. Rodriguez-Caycedo C. Sanchez-Ortiz E. Atmanli A. et al (2021). Precise correction of Duchenne muscular dystrophy exon deletion mutations by base and prime editing. Sci. Adv.7, eabg4910. 10.1126/sciadv.abg4910
66
Chen G. Wei T. Yang H. Li G. Li H. (2022). CRISPR-based therapeutic gene editing for duchenne muscular dystrophy: Advances, challenges and perspectives. Cells11, 2964. 10.3390/cells11192964
67
Choi S. Ferrari G. Moyle L. A. Mackinlay K. Naouar N. Jalal S. et al (2022). Assessing and enhancing migration of human myogenic progenitors using directed iPS cell differentiation and advanced tissue modelling. EMBO Mol. Med.14, e14526. 10.15252/emmm.202114526
68
Cisternas P. Vio C. P. Inestrosa N. C. (2014). Role of Wnt signaling in tissue fibrosis, lessons from skeletal muscle and kidney. Curr. Mol. Med.14, 510–522. 10.2174/1566524014666140414210346
69
Clemens P. R. Rao V. K. Connolly A. M. Harper A. D. Mah J. K. Smith E. C. et al (2020). Safety, tolerability, and efficacy of viltolarsen in boys with duchenne muscular dystrophy amenable to exon 53 skipping: A phase 2 randomized clinical trial. JAMA Neurol.77, 982–991. 10.1001/jamaneurol.2020.1264
70
Cohn R. D. Campbell K. P. (2000). Molecular basis of muscular dystrophies. Muscle Nerve23, 1456–1471. 10.1002/1097-4598(200010)23:10<1456:AID-MUS2>3.0.CO;2-T
71
Cole M. A. Rafael J. A. Taylor D. J. Lodi R. Davies K. E. Styles P. (2002). A quantitative study of bioenergetics in skeletal muscle lacking utrophin and dystrophin. Neuromuscul. Disord.12, 247–257. 10.1016/s0960-8966(01)00278-4
72
Collins J. M. Despa F. Lee R. C. (2007). Structural and functional recovery of electropermeabilized skeletal muscle in-vivo after treatment with surfactant poloxamer 188. Biochim. Biophys. Acta1768, 1238–1246. 10.1016/j.bbamem.2007.01.012
73
Connolly A. M. Schierbecker J. Renna R. Florence J. (2002). High dose weekly oral prednisone improves strength in boys with Duchenne muscular dystrophy. Neuromuscul. Disord.12, 917–925. 10.1016/S0960-8966(02)00180-3
74
Corrado K. Rafael J. A. Mills P. L. Cole N. M. Faulkner J. A. Wang K. et al (1996). Transgenic mdx mice expressing dystrophin with a deletion in the actin-binding domain display a “‘mild becker’” phenotype. J. Cell Biol.134, 873–884. 10.1083/jcb.134.4.873
75
Cotton S. Voudouris N. J. Greenwood K. M. (2001). Intelligence and Duchenne muscular dystrophy: Full-scale, verbal, and performance intelligence quotients. Dev. Med. Child. Neurol.43, 497–501. 10.1017/s0012162201000913
76
Crudele J. M. Chamberlain J. S. (2019). AAV-based gene therapies for the muscular dystrophies. Hum. Mol. Genet.28, R102–R107. 10.1093/hmg/ddz128
77
Damsker J. M. Dillingham B. C. Rose M. C. Balsley M. A. Heier C. R. Watson A. M. et al (2013). VBP15, a glucocorticoid analogue, is effective at reducing allergic lung inflammation in mice. PLoS One8, e63871–e63879. 10.1371/journal.pone.0063871
78
Deconinck A. E. Rafael J. A. Skinner J. A. Brown S. C. Potter A. C. Metzinger L. et al (1997). Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell90, 717–727. 10.1016/S0092-8674(00)80532-2
79
Dellavalle A. Sampaolesi M. Tonlorenzi R. Tagliafico E. Sacchetti B. Perani L. et al (2007). Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat. Cell Biol.9, 255–267. 10.1038/ncb1542
80
Demina T. Grozdova I. Krylova O. Zhirnov A. Istratov V. Frey H. et al (2005). Relationship between the structure of amphiphilic copolymers and their ability to disturb lipid bilayers. Biochemistry.44, 4042–4054. 10.1021/bi048373q
81
Desguerre I. Christov C. Mayer M. Zeller R. Becane H. M. Bastuji-Garin S. et al (2009). Clinical heterogeneity of Duchenne muscular dystrophy (DMD): Definition of sub-phenotypes and predictive criteria by long-term follow-up. PLoS One4, e4347. 10.1371/journal.pone.0004347
82
Dominguez A. A. Lim W. A. Qi L. S. (2016). Beyond editing: Repurposing CRISPR-cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol.17, 5–15. 10.1038/nrm.2015.2
83
Dong J. Y. Fan P. D. Frizzell R. A. (1996). Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther.7, 2101–2112. 10.1089/hum.1996.7.17-2101
84
Doo K. H. Ryu H. W. Kim S. S. Lim B. C. Hwang H. Kim K. J. et al (2012). A case of Becker muscular dystrophy with early manifestation of cardiomyopathy. Korean J. Pediatr.55, 350–353. 10.3345/kjp.2012.55.9.350
85
Duan D. (2016a). Dystrophin gene replacement and gene repair therapy for duchenne muscular dystrophy in 2016: An interview. Hum. Gene Ther. Clin. Dev.27, 9–18. 10.1089/humc.2016.001
86
Duan D. Goemans N. Takeda S. Mercuri E. Aartsma-Rus A. (2021). Duchenne muscular dystrophy. Nat. Rev. Dis. Prim.7, 13. 10.1038/s41572-021-00248-3
87
Duan D. (2018a). Micro-dystrophin gene therapy goes systemic in duchenne muscular dystrophy patients. Hum. Gene Ther.29, 733–736. 10.1089/hum.2018.012
88
Duan D. (2018b). Systemic AAV micro-dystrophin gene therapy for duchenne muscular dystrophy. Mol. Ther.26, 2337–2356. 10.1016/j.ymthe.2018.07.011
89
Duan D. (2016b). Systemic delivery of adeno-associated viral vectors. Curr. Opin. Virol.21, 16–25. 10.1016/j.coviro.2016.07.006
90
Duchene B. L. Cherif K. Iyombe-Engembe J. P. Guyon A. Rousseau J. Ouellet D. L. et al (2018). CRISPR-induced deletion with SaCas9 restores dystrophin expression in dystrophic models in vitro and in vivo. Mol. Ther.26, 2604–2616. 10.1016/j.ymthe.2018.08.010
91
Dupont J. B. Tournaire B. Georger C. Marolleau B. Jeanson-Leh L. Ledevin M. et al (2015). Short-lived recombinant adeno-associated virus transgene expression in dystrophic muscle is associated with oxidative damage to transgene mRNA. Mol. Therapy-Methods Clin. Dev.2, 15010. 10.1038/mtm.2015.10
92
Ebihara S. Guibinga G. Gilbert R. Nalbantoglu J. Massie B. Karpati G. et al (2022). Differential effects of dystrophin and utrophin gene transfer in immunocompetent muscular dystrophy (mdx) mice, 133–144.
93
Echigoya Y. Nakamura A. Nagata T. Urasawa N. Lim K. R. Q. Trieu N. et al (2017). Effects of systemic multiexon skipping with peptide-conjugated morpholinos in the heart of a dog model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. U. S. A.114, 4213–4218. 10.1073/pnas.1613203114
94
Emery A. E. H. (1993). Duchenne muscular dystrophy-Meryon’s disease. Neuromuscul. Disord.3, 263–266. 10.1016/0960-8966(93)90018-F
95
Emery A. E. H. (2002). “The muscular dystrophies,” in Lancet (Elsevier Limited), 687–695. 10.1016/S0140-6736(02)07815-7
96
England S. B. Nicholson L. V. B. Johnson M. A. Forrest S. M. Love D. R. Zubrzycka-Gaarn E. E. et al (1990). Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature343, 180–182. 10.1038/343180a0
97
Erkut E. Yokota T. (2022). CRISPR therapeutics for duchenne muscular dystrophy. Int. J. Mol. Sci.23, 1832. 10.3390/ijms23031832
98
Ervasti J. M. Campbell K. P. (1993). A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol.122, 809–823. 10.1083/jcb.122.4.809
99
Ervasti J. M. Campbell K. P. (1991). Membrane organization of the dystrophin-glycoprotein complex. Cell66, 1121–1131. 10.1016/0092-8674(91)90035-W
100
Ervasti J. M. (2013). “Structure and function of the dystrophin-glycoprotein complex,” in Madame curie bioscience database. [Internet] (Landes Bioscience).
101
Eser G. Topaloğlu H. (2022). Current outline of exon skipping trials in duchenne muscular dystrophy. Genes (Basel)13, 1241. 10.3390/genes13071241
102
Evans N. P. Misyak S. A. Robertson J. L. Bassaganya-Riera J. Grange R. W. (2009). Dysregulated intracellular signaling and inflammatory gene expression during initial disease onset in duchenne muscular dystrophy. Am. J. Phys. Med. Rehabil.88, 502–522. 10.1097/PHM.0b013e3181a5a24f
103
Ferlini A. Sabatelli P. Fabris M. Bassi E. Falzarano S. Vattemi G. et al (2010). Dystrophin restoration in skeletal, heart and skin arrector pili smooth muscle of mdx mice by ZM2 NP-AON complexes. Gene Ther.17, 432–438. 10.1038/gt.2009.145
104
Flanigan K. M. (2014). Duchenne and becker muscular dystrophies. Neurol. Clin.32, 671–688. 10.1016/j.ncl.2014.05.002
105
Foster H. Sharp P. S. Athanasopoulos T. Trollet C. Graham I. R. Foster K. et al (2008). Codon and mRNA sequence optimization of microdystrophin transgenes improves expression and physiological outcome in dystrophic mdx mice following AAV2/8 gene transfer. Mol. Ther.16, 1825–1832. 10.1038/mt.2008.186
106
Frank D. E. Schnell F. J. Akana C. El-husayni S. H. (2020). Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. 10.1212/WNL.0000000000009233
107
Fridovich I. (2004). Mitochondria: Are they the seat of senescence?Aging Cell3, 13–16. 10.1046/j.1474-9728.2003.00075.x
108
Gadgil A. Raczyńska K. D. (2021). U7 snRNA: A tool for gene therapy. J. Gene Med.23, 1–15. 10.1002/jgm.3321
109
Gailly P. De Backer F. Van Schoor M. Gillis J. M. (2007). In situ measurements of calpain activity in isolated muscle fibres from normal and dystrophin-lacking mdx mice. J. Physiology582, 1261–1275. 10.1113/jphysiol.2007.132191
110
Gallardo F. S. Cordova-Casanova A. Brandan E. (2021). The linkage between inflammation and fibrosis in muscular dystrophies: The axis autotaxin-lysophosphatidic acid as a new therapeutic target?J. Cell Commun. Signal15, 317–334. 10.1007/s12079-021-00610-w
111
Gao G. Vandenberghe L. H. Alvira M. R. Lu Y. Calcedo R. Zhou X. et al (2004). Clades of Adeno-associated viruses are widely disseminated in human tissues. J. Virol.78, 6381–6388. 10.1128/JVI.78.12.6381-6388.2004
112
Gao Q. McNally E. (2015). The dystrophin complex: Structure, function, and implications for therapy. Compr. Physiol.5, 1223–1239. 10.1002/cphy.c140048
113
Gapinske M. Luu A. Winter J. Woods W. S. Kostan K. A. Shiva N. (2018). CRISPR-SKIP: Programmable gene splicing with single base editors, 1–11.
114
Garcia-Castaneda M. Michelucci A. Zhao N. Malik S. Dirksen R. T. (2022). Postdevelopmental knockout of Orai1 improves muscle pathology in a mouse model of Duchenne muscular dystrophy. J. Gen. Physiol.154, e202213081. 10.1085/jgp.202213081
115
Gardner K. L. Kearney J. A. Edwards J. D. Rafael-Fortney J. A. (2006). Restoration of all dystrophin protein interactions by functional domains in trans does not rescue dystrophy. Gene Ther.13, 744–751. 10.1038/sj.gt.3302686
116
Gaudelli N. M. Komor A. C. Rees H. A. Packer M. S. Badran A. H. Bryson D. I. et al (2017). Programmable base editing of A • T to G • C in genomic DNA without DNA cleavage. Nat. Publ. Group551, 464–471. 10.1038/nature24644
117
Gawlik K. I. Oliveira B. M. Durbeej M. (2011). Transgenic expression of laminin ␣ 1 chain does not prevent muscle disease in the mdx mouse model for duchenne muscular dystrophy. AJPA178, 1728–1737. 10.1016/j.ajpath.2010.12.030
118
Gehlert S. Jacko D. (2019). The role of the immune system in response to muscle damage. Dtsch. Z Sportmed70, 242–249. 10.5960/dzsm.2019.390
119
Giordano C. Mojumdar K. Liang F. Lemaire C. Li T. Richardson J. et al (2015). Toll-like receptor 4 ablation in mdx mice reveals innate immunity as a therapeutic target in Duchenne muscular dystrophy. Hum. Mol. Genet.24, 2147–2162. 10.1093/hmg/ddu735
120
Glancy B. Hartnell L. M. Malide D. Yu Z. X. Combs C. A. Connelly P. S. et al (2015). Mitochondrial reticulum for cellular energy distribution in muscle. Nature523, 617–620. 10.1038/nature14614
121
Godin R. Daussin F. Matecki S. Li T. Petrof B. J. Burelle Y. (2012). Peroxisome proliferator-activated receptor gamma coactivator1-gene alpha transfer restores mitochondrial biomass and improves mitochondrial calcium handling in post-necrotic mdx mouse skeletal muscle. J. Physiol.590, 5487–5502. 10.1113/jphysiol.2012.240390
122
Goldmann T. Overlack N. Möller F. Belakhov V. van Wyk M. Baasov T. et al (2012). A comparative evaluation of NB30, NB54 and PTC124 in translational read-through efficacy for treatment of an USH1C nonsense mutation. EMBO Mol. Med.4, 1186–1199. 10.1002/emmm.201201438
123
Gordon A. M. Homsher E. Regnier M. (2000). Regulation of contraction in striated muscle. Physiol. Rev.80, 853–924. 10.1152/physrev.2000.80.2.853
124
Goyenvalle A. Seto J. T. Davies K. E. Chamberlain J. (2011). Therapeutic approaches to muscular dystrophy. Hum. Mol. Genet.20, R69–R78. 10.1093/hmg/ddr105
125
Goyenvalle A. Wright J. Babbs A. Wilkins V. Garcia L. Davies K. E. (2012). Engineering multiple U7snRNA constructs to induce single and multiexon-skipping for Duchenne muscular dystrophy. Mol. Ther.20, 1212–1221. 10.1038/mt.2012.26
126
Gregorevic P. Allen J. M. Minami E. Blankinship M. J. Haraguchi M. Inn E. et al (2006). rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat. Med.12, 787–789. 10.1038/nm1439
127
Gregorevic P. Blankinship M. J. Allen J. M. Crawford R. W. Meuse L. Miller D. G. et al (2004). Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat. Med.10, 828–834. 10.1038/nm1085
128
Grobet L. Pirottin D. Poncelet D. Royo L. J. Christians E. Desmecht D. et al (2003). Modulating skeletal muscle mass by postnatal, muscle-specific inactivation of the myostatin gene. Muscle-Specific Inact. Myostatin Gene238, 227–238. 10.1002/gene.10188
129
Grounds M. D. Terrill J. R. Al-Mshhdani B. A. Duong M. N. Radley-Crabb H. G. Arthur P. G. (2020). Biomarkers for Duchenne muscular dystrophy: Myonecrosis, inflammation and oxidative stress. DMM Dis. Models Mech.13, dmm043638–12. 10.1242/DMM.043638
130
Guiraud S. Aartsma-Rus A. Vieira N. M. Davies K. E. Van Ommen G. J. B. Kunkel L. M. (2015). The pathogenesis and therapy of muscular dystrophies. Annu. Rev. Genomics Hum. Genet.16, 281–308. 10.1146/annurev-genom-090314-025003
131
Guo C. Willem M. Werner A. Raivich G. Emerson M. Neyses L. et al (2006). Absence of alpha 7 integrin in dystrophin-deficient mice causes a myopathy similar to Duchenne muscular dystrophy. Hum. Mol. Genet.15, 989–998. 10.1093/hmg/ddl018
132
Gurpur P. B. Liu J. Burkin D. J. Kaufman S. J. (2009). Valproic acid activates the PI3K/Akt/mTOR pathway in muscle and ameliorates pathology in a mouse model of Duchenne muscular dystrophy. Dystrophy174, 999–1008. 10.2353/ajpath.2009.080537
133
Gushchina L. V. Frair E. C. Rohan N. Bradley A. J. Simmons T. R. Chavan H. D. et al (2021). Lack of toxicity in nonhuman primates receiving clinically relevant doses of an AAV9.U7snRNA vector designed to induce DMD exon 2 skipping. Hum. Gene Ther.32, 882–894. 10.1089/hum.2020.286
134
Hahn D. Kumar R. A. Ryan T. E. Ferreira L. F. (2019). Mitochondrial respiration and H2O2 emission in saponin-permeabilized murine diaphragm fibers: Optimization of fiber separation and comparison to limb muscle. Am. J. Physiol. Cell Physiol.317, C665–C673. 10.1152/ajpcell.00184.2019
135
Hakim C. H. Wasala N. B. Pan X. Kodippili K. Yue Y. Zhang K. et al (2017). A five-repeat micro-dystrophin gene ameliorated dystrophic phenotype in the severe DBA/2J-mdx model of duchenne muscular dystrophy. Mol. Ther. Methods Clin. Dev.6, 216–230. 10.1016/j.omtm.2017.06.006
136
Hannig J. Yu J. Beckett M. Weichselbaum R. Lee R. C. (1999). Poloxamine 1107 sealing of radiopermeabilized erythrocyte membranes. Int. J. Radiat. Biol.75, 379–385. 10.1080/095530099140555
137
Hanson B. Wood M. J. A. Roberts T. C. (2021). Molecular correction of Duchenne muscular dystrophy by splice modulation and gene editing. RNA Biol.18, 1048–1062. 10.1080/15476286.2021.1874161
138
Happi Mbakam C. Lamothe G. Tremblay G. Tremblay J. P. (2022a). CRISPR-Cas9 gene therapy for duchenne muscular dystrophy. Neurotherapeutics19, 931–941. 10.1007/s13311-022-01197-9
139
Happi Mbakam C. Rousseau J. Tremblay G. Yameogo P. Tremblay J. P. (2022b). Prime editing permits the introduction of specific mutations in the gene responsible for duchenne muscular dystrophy. Int. J. Mol. Sci.23, 6160. 10.3390/ijms23116160
140
Harper S. Q. Hauser M. A. DelloRusso C. Duan D. Crawford R. W. Phelps S. F. et al (2002). Modular flexibility of dystrophin: Implications for gene therapy of duchenne muscular dystrophy. Nat. Med.8, 253–261. 10.1038/nm0302-253
141
Heemskerk H. A. de Winter C. L. de Kimpe S. J. van Kuik-Romeijn P. Heuvelmans N. Platenburg G. J. et al (2009). In vivo comparison of 2’-O-methyl phosphorothioate and morpholino antisense oligonucleotides for Duchenne muscular dystrophy exon skipping. J. Gene Med.11, 257–266. 10.1002/jgm.1288
142
Heemskerk H. de Winter C. van Kuik P. Heuvelmans N. Sabatelli P. Rimessi P. et al (2010). Preclinical PK and PD studies on 2’-O-methyl-phosphorothioate RNA antisense oligonucleotides in the mdx mouse model. Mol. Ther.18, 1210–1217. 10.1038/mt.2010.72
143
Heier C. R. Damsker J. M. Yu Q. Dillingham B. C. Huynh T. Van der Meulen J. H. et al (2013). VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol. Med.5, 1569–1585. 10.1002/emmm.201302621
144
Heier C. R. Yu Q. Fiorillo A. A. Tully C. B. Tucker A. Mazala D. A. et al (2019). Vamorolone targets dual nuclear receptors to treat inflammation and dystrophic cardiomyopathy. Life Sci. Alliance2, e201800186. 10.26508/LSA.201800186
145
Heller K. N. Montgomery C. L. Janssen P. M. Clark K. R. Mendell J. R. Rodino-Klapac L. R. (2013). AAV-mediated overexpression of human α7 integrin leads to histological and functional improvement in dystrophic mice. Mol. Ther.21, 520–525. 10.1038/mt.2012.281
146
Hendriksen J. G. M. Vles J. S. H. (2006). Are males with duchenne muscular dystrophy at risk for reading disabilities?Pediatr. Neurol.34, 296–300. 10.1016/j.pediatrneurol.2005.08.029
147
Hendriksen J. G. M. Vles J. S. H. (2008). Neuropsychiatric disorders in males with duchenne muscular dystrophy: Frequency rate of attention-deficit hyperactivity disorder (ADHD), autism spectrum disorder, and obsessive-compulsive disorder. J. Child. Neurol.23, 477–481. 10.1177/0883073807309775
148
Henriques-Pons A. Yu Q. Rayavarapu S. Cohen T. v. Ampong B. Cha H. J. et al (2014). Role of toll-like receptors in the pathogenesis of dystrophin-deficient skeletal and heart muscle. Hum. Mol. Genet.23, 2604–2617. 10.1093/hmg/ddt656
149
Hodges B. L. Hayashi Y. K. Nonaka I. Wang W. Arahata K. Kaufman S. J. (1997). Altered expression of the alpha7beta1 integrin in human and murine muscular dystrophies. J. Cell Sci.2881, 2873–2881. 10.1242/jcs.110.22.2873
150
Hoffman E. P. Bronson A. Levin A. A. Takeda S. Yokota T. Baudy A. R. et al (2011). Restoring dystrophin expression in duchenne muscular dystrophy muscle progress in exon skipping and stop codon read through. Am. J. Pathology179, 12–22. 10.1016/j.ajpath.2011.03.050
151
Hoffman E. P. Brown R. H. Kunkel L. M. (1987). Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell51, 919–928. 10.1016/0092-8674(87)90579-4
152
Hogrel J.-Y. Zagnoli F. Canal A. Fraysse B. Bouchard J.-P. Skuk D. et al (2013). Assessment of a symptomatic Duchenne muscular dystrophy carrier 20years after myoblast transplantation from her asymptomatic identical twin sister. Neuromuscul. Disord.23, 575–579. 10.1016/j.nmd.2013.04.007
153
Hopf F. W. Turner P. R. Denetclaw W. F. Reddy P. Steinhardt R. A. (1996). A critical evaluation of resting intracellular free calcium regulation in dystrophic mdx muscle. Am. J. Physiol. - Cell Physiol.271, C1325–C1339. 10.1152/ajpcell.1996.271.4.c1325
154
Houang E. M. Haman K. J. Filareto A. Perlingeiro R. C. Bates F. S. Lowe D. A. et al (2015). Membrane-stabilizing copolymers confer marked protection to dystrophic skeletal muscle in vivo. Mol. Ther. Methods Clin. Dev.2, 15042. 10.1038/mtm.2015.42
155
Houang E. M. Haman K. J. Kim M. Zhang W. Lowe D. A. Sham Y. Y. et al (2017). Chemical end group modified diblock copolymers elucidate anchor and chain mechanism of membrane stabilization. Mol. Pharm.14, 2333–2339. 10.1021/acs.molpharmaceut.7b00197
156
Houang E. M. Sham Y. Y. Bates F. S. Metzger J. M. (2018). Muscle membrane integrity in duchenne muscular dystrophy: Recent advances in copolymer-based muscle membrane stabilizers. Skelet. Muscle8, 31. 10.1186/s13395-018-0177-7
157
Huang T. P. Newby G. A. Liu D. R. (2021). Precision genome editing using cytosine and adenine base editors in mammalian cells. Nat. Protoc.16, 1089–1128. 10.1038/s41596-020-00450-9
158
Hughes M. C. Ramos S. V. Turnbull P. C. Edgett B. A. Huber J. S. Polidovitch N. et al (2020). Impairments in left ventricular mitochondrial bioenergetics precede overt cardiac dysfunction and remodelling in Duchenne muscular dystrophy. J. Physiol.598, 1377–1392. 10.1113/JP277306
159
Hughes M. C. Ramos S. V. Turnbull P. C. Rebalka I. A. Cao A. Monaco C. M. F. et al (2019). Early myopathy in Duchenne muscular dystrophy is associated with elevated mitochondrial H2 O2 emission during impaired oxidative phosphorylation. J. Cachexia Sarcopenia Muscle10, 643–661. 10.1002/jcsm.12405
160
Hunter R. L. Luo A. Z. Zhang R. Kozar R. A. Moore F. A. (2010). Poloxamer 188 inhibition of ischemia/reperfusion injury: Evidence for a novel anti-adhesive mechanism. Ann. Clin. Lab. Sci.40, 115–125.
161
Hurst S. Hoek J. Sheu S. S. (2017). Mitochondrial Ca2+ and regulation of the permeability transition pore. J. Bioenerg. Biomembr.49, 27–47. 10.1007/s10863-016-9672-x
162
Hynes R. (1992). Integrins: Versatility, modulation, and signaling in cell adhesion. Cell69 (1), 11–25. 10.1016/0092-8674(92)90115-s
163
Ishikawa-Sakurai M. Yoshida M. Imamura M. Davies K. E. Ozawa E. (2004). ZZ domain is essentially required for the physiological binding of dystrophin and utrophin to β-dystroglycan. Hum. Mol. Genet.13, 693–702. 10.1093/hmg/ddh087
164
Iyombe-Engembe J.-P. Ouellet D. L. Barbeau X. Rousseau J. Chapdelaine P. Lagüe P. et al (2016). Efficient restoration of the dystrophin gene reading frame and protein structure in DMD myoblasts using the CinDel method. Mol. Therapy-Nucleic Acids5, e283. 10.1038/mtna.2015.58
165
Jearawiriyapaisarn N. Moulton H. M. Buckley B. Roberts J. Sazani P. Fucharoen S. et al (2008). Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice. Mol. Ther.16, 1624–1629. 10.1038/mt.2008.120
166
Jearawiriyapaisarn N. Moulton H. M. Sazani P. Kole R. Willis M. S. (2010). Long-term improvement in mdx cardiomyopathy after therapy with peptide-conjugated morpholino oligomers. Cardiovasc Res.85, 444–453. 10.1093/cvr/cvp335
167
Jinek M. Chylinski K. Fonfara I. Hauer M. Doudna J. A. Charpentier E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science337, 816–821. 10.1126/science.1225829
168
Jørgensen L. H. Larochelle N. Orlopp K. Dunant P. Dudley R. W. R. Stucka R. et al (2009). Efficient and fast functional screening of microdystrophin constructs in vivo and in vitro for therapy of duchenne muscular dystrophy. Hum. Gene Ther.20, 641–650. 10.1089/hum.2008.162
169
Jung C. Martins A. S. Niggli E. Shirokova N. (2008). Dystrophic cardiomyopathy: Amplification of cellular damage by Ca2+ signalling and reactive oxygen species-generating pathways. Cardiovasc Res.77, 766–773. 10.1093/cvr/cvm089
170
Jung D. Yang B. Meyer J. Chamberlain J. S. Campbell K. P. (1995). Identification and characterization of the dystrophin anchoring site on beta-dystroglycan. J. Biol. Chem.270, 27305–27310. 10.1074/jbc.270.45.27305
171
Jungbluth H. Treves S. Zorzato F. Sarkozy A. Ochala J. Sewry C. et al (2018). Congenital myopathies: Disorders of excitation-contraction coupling and muscle contraction. Nat. Rev. Neurol.14, 151–167. 10.1038/nrneurol.2017.191
172
Kamdar F. Garry D. J. (2016). Dystrophin-deficient cardiomyopathy. J. Am. Coll. Cardiol.67, 2533–2546. 10.1016/j.jacc.2016.02.081
173
Kang C. Badr M. A. Kyrychenko V. Eskelinen E. L. Shirokova N. (2018). Deficit in PINK1/PARKIN-mediated mitochondrial autophagy at late stages of dystrophic cardiomyopathy. Cardiovasc Res.114, 90–102. 10.1093/cvr/cvx201
174
Kawecka K. Theodoulides M. Hasoglu Y. Jarmin S. Kymalainen H. Le-Heron A. et al (2015). Adeno-associated virus (AAV) mediated dystrophin gene transfer studies and exon skipping strategies for duchenne muscular dystrophy (DMD). Curr. Gene Ther.15, 395–415. 10.2174/1566523215666150710123830
175
Kerem E. Konstan M. W. De Boeck K. Accurso F. J. Sermet-Gaudelus I. Wilschanski M. et al (2014). Ataluren for the treatment of nonsense-mutation cystic fibrosis: A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir. Med.2, 539–547. 10.1016/S2213-2600(14)70100-6
176
Kessler P. D. Podsakoff G. M. Chen X. McQuiston S. A. Colosi P. C. Matelis L. A. et al (1996). Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. Proc. Natl. Acad. Sci. U. S. A.93, 14082–14087. 10.1073/pnas.93.24.14082
177
Khairallah R. J. Shi G. Sbrana F. Prosser B. L. Borroto C. Mazaitis M. J. et al (2012). Microtubules underlie dysfunction in duchenne muscular dystrophy. Sci. Signal5, ra56. 10.1126/scisignal.2002829
178
Kharraz Y. Guerra J. Pessina P. Serrano A. L. Munoz-Canoves P. (2014). Understanding the process of fibrosis in Duchenne muscular dystrophy. Biomed. Res. Int.2014, 965631. 10.1155/2014/965631
179
Kim J. H. Kwak H. B. Thompson L. V. Lawler J. M. (2013). Contribution of oxidative stress to pathology in diaphragm and limb muscles with Duchenne muscular dystrophy. J. Muscle Res. Cell Motil.34, 1–13. 10.1007/s10974-012-9330-9
180
Kim M. Haman K. J. Houang E. M. Zhang W. Yannopoulos D. Metzger J. M. et al (2017). PEO-PPO diblock copolymers protect myoblasts from hypo-osmotic stress in vitro dependent on copolymer size, composition, and architecture. Biomacromolecules18, 2090–2101. 10.1021/acs.biomac.7b00419
181
Kim M. J. Bible K. L. Regnier M. Adams M. E. Froehner S. C. Whitehead N. P. (2019). Simvastatin provides long-term improvement of left ventricular function and prevents cardiac fibrosis in muscular dystrophy. Physiol. Rep.7, e14018. 10.14814/phy2.14018
182
Kim S. Kim D. Cho S. W. Kim J. Kim J. S. (2014). Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res.24, 1012–1019. 10.1101/gr.171322.113
183
Kim T. ‐W. Wu K. Black I. B. (1995). Deficiency of brain synaptic dystrophin in human duchenne muscular dystrophy. Ann. Neurol.38, 446–449. 10.1002/ana.410380315
184
Klingler W. Jurkat-Rott K. Lehmann-Horn F. Schleip R. (2012). The role of fibrosis in Duchenne muscular dystrophy. Acta Myol.31, 184–195.
185
Knott G. J. Doudna J. A. (2018). CRISPR-Cas guides the future of genetic engineering. Science361, 866–869. 10.1126/science.aat5011
186
Koenig M. Kunkel L. M. (1990). Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. J. Biol. Chem.265, 4560–4566. 10.1016/s0021-9258(19)39599-7
187
Koenig M. Monaco A. P. Kunkel L. M. (1988). The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell53, 219–228. 10.1016/0092-8674(88)90383-2
188
Komaki H. Takeshima Y. Matsumura T. Ozasa S. Funato M. Takeshita E. et al (2020). Viltolarsen in Japanese duchenne muscular dystrophy patients: A phase 1/2 study. Ann. Clin. Transl. Neurol.7, 2393–2408. 10.1002/acn3.51235
189
Komor A. C. Kim Y. B. Packer M. S. Zuris J. A. Liu D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature533, 420–424. 10.1038/nature17946
190
Koo T. Malerba A. Athanasopoulos T. Trollet C. Boldrin L. Ferry A. et al (2011). Delivery of AAV2/9-microdystrophin genes incorporating helix 1 of the coiled-coil motif in the C-terminal domain of dystrophin improves muscle pathology and restores the level of α1-syntrophin and α-dystrobrevin in skeletal muscles of mdx mice. Hum. Gene Ther.22, 1379–1388. 10.1089/hum.2011.020
191
Kotterman M. A. Schaffer D. V. (2014). Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet.15, 445–451. 10.1038/nrg3742
192
Krag T. O. B. Bogdanovich S. Jensen C. J. Fischer M. D. Hansen-schwartz J. Javazon E. H. et al (2004). Heregulin ameliorates the dystrophic phenotype in mdx mice. Proc. Natl. Acad. Sci. U. S. A.101, 13856–13860. 10.1073/pnas.0405972101
193
Kuzmin D. A. Shutova M. V. Johnston N. R. Smith O. P. Johnstone E. C. Kukushkin Y. S. et al (2021). The clinical landscape for AAV gene therapies. Nat. Rev. Drug Discov.20, 173–174. 10.1038/d41573-021-00017-7
194
Kyrychenko V. Kyrychenko S. Tiburcy M. Shelton J. M. Long C. Schneider J. W. et al (2017). Functional correction of dystrophin actin binding domain mutations by genome editing. JCI Insight2, e95918. 10.1172/jci.insight.95918
195
Kyrychenko V. Poláková E. Janíček R. Shirokova N. (2015). Mitochondrial dysfunctions during progression of dystrophic cardiomyopathy. Cell Calcium58, 186–195. 10.1016/j.ceca.2015.04.006
196
Lacourpaille L. Gross R. Hug F. Guevel A. Pereon Y. Magot A. et al (2017). Effects of duchenne muscular dystrophy on muscle stiffness and response to electrically-induced muscle contraction: A 12-month follow-up. Neuromuscul. Disord.27, 214–220. 10.1016/j.nmd.2017.01.001
197
Lacourpaille L. Hug F. Guevel A. Pereon Y. Magot A. Hogrel J. Y. et al (2015). Non-invasive assessment of muscle stiffness in patients with Duchenne muscular dystrophy. Muscle Nerve51, 284–286. 10.1002/mus.24445
198
Lai Y. Thomas G. D. Yue Y. Yang H. T. Li D. Long C. et al (2009). Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Invest.119, 624–635. 10.1172/JCI36612
199
Lau C.-H. Suh Y. (2017). In vivo genome editing in animals using AAV-CRISPR system: Applications to translational research of human disease. F1000Res6, 2153. 10.12688/f1000research.11243.1
200
Le Guiner C. Servais L. Montus M. Larcher T. Fraysse B. Moullec S. et al (2017). Long-term microdystrophin gene therapy is effective in a canine model of Duchenne muscular dystrophy. Nat. Commun.8, 16105–16115. 10.1038/ncomms16105
201
Lee K. Conboy M. Park H. M. Jiang F. Kim H. J. Dewitt M. A. et al (2017). Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng.1, 889–901. 10.1038/s41551-017-0137-2
202
Lee R. C. Hannig J. Matthews K. L. Myerov A. Chen C. T. (1999). Pharmaceutical therapies for sealing of permeabilized cell membranes in electrical injuries. Ann. N. Y. Acad. Sci.888, 266–273. 10.1111/j.1749-6632.1999.tb07961.x
203
Lee R. C. River L. P. Pan F. S. Ji L. Wollmann R. L. (1992). Surfactant-induced sealing of electropermeabilized skeletal muscle membranes in vivo. Proc. Natl. Acad. Sci. U. S. A.89, 4524–4528. 10.1073/pnas.89.10.4524
204
Lee S. J. (2004). Regulation of muscle mass by myostatin. Annu. Rev. Cell Dev. Biol.20, 61–86. 10.1146/annurev.cellbio.20.012103.135836
205
Lee T. H. Eun L. Y. Choi J. Y. Kwon H. E. Lee Y. M. Kim H. D. et al (2014). Myocardial atrophy in children with mitochondrial disease and Duchenne muscular dystrophy. Korean J. Pediatr.57, 232–239. 10.3345/kjp.2014.57.5.232
206
Levi O. Genin O. Angelini C. Halevy O. Pines M. (2015). Inhibition of muscle fibrosis results in increases in both utrophin levels and the number of revertant myofibers in Duchenne muscular dystrophy. Oncotarget6, 23249–23260. 10.18632/oncotarget.4021
207
Li H. L. Fujimoto N. Sasakawa N. Shirai S. Ohkame T. Sakuma T. et al (2015). Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep.4, 143–154. 10.1016/j.stemcr.2014.10.013
208
Li J. Cao J. Li M. Yu Y. Yang Y. Xiao X. et al (2011). Collagen triple helix repeat containing-1 inhibits transforming growth factor-b1-induced collagen type I expression in keloid. Br. J. Dermatol164, 1030–1036. 10.1111/j.1365-2133.2011.10215.x
209
Li Z. B. Kollias H. D. Wagner K. R. (2008). Myostatin directly regulates skeletal muscle fibrosis. J. Biol. Chem.283, 19371–19378. 10.1074/jbc.M802585200
210
Lieber R. L. Ward S. R. (2013). Cellular mechanisms of tissue fibrosis. 4. Structural and functional consequences of skeletal muscle fibrosis. Am. J. Physiol. Cell Physiol.305, C241–C252. 10.1152/ajpcell.00173.2013
211
Long C. Amoasii L. Mireault A. A. McAnally J. R. Li H. Sanchez-Ortiz E. et al (2016). Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science351, 400–403. 10.1126/science.aad5725
212
Long C. Li H. Tiburcy M. Rodriguez-Caycedo C. Kyrychenko V. Zhou H. et al (2018). Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci. Adv.4, eaap9004. 10.1126/sciadv.aap9004
213
Long C. McAnally J. R. Shelton J. M. Mireault A. A. Bassel-Duby R. Olson E. N. (2014). Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science345, 1184–1188. 10.1126/science.1254445
214
Lostal W. Kodippili K. Yue Y. Duan D. (2014). Full-length dystrophin reconstitution with adeno-associated viral vectors. Hum. Gene Ther.25, 552–562. 10.1089/hum.2013.210
215
Love D. R. Hillt D. F. Dicksont G. Spurr N. K. Byth B. C. Marsden R. F. et al (1989). An autosomal transcript in skeletal muscle with homology to dystrophin. Nature339, 55–58. 10.1038/339055a0
216
Lozanoska-Ochser B. Benedetti A. Rizzo G. Marrocco V. di Maggio R. Fiore P. et al (2018). Targeting early PKCθ-dependent T-cell infiltration of dystrophic muscle reduces disease severity in a mouse model of muscular dystrophy. J. Pathol.244, 323–333. 10.1002/path.5016
217
Lu Q. L. Mann C. J. Lou F. Bou-Gharios G. Morris G. E. Xue S. A. et al (2003). Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat. Med.9, 1009–1014. 10.1038/nm897
218
Ma N. Chen D. Lee J. H. Kuri P. Hernandez E. B. Kocan J. et al (2022). Piezo1 regulates the regenerative capacity of skeletal muscles via orchestration of stem cell morphological states. Sci. Adv.8, eabn0485. 10.1126/sciadv.abn0485
219
Maffioletti S. M. Noviello M. English K. Tedesco F. S. (2014). Stem cell transplantation for muscular dystrophy: The challenge of immune response. Biomed. Res. Int.2014, 964010–964012. 10.1155/2014/964010
220
Maggio I. Liu J. Janssen J. M. Chen X. Goncalves M. A. (2016a). Adenoviral vectors encoding CRISPR/Cas9 multiplexes rescue dystrophin synthesis in unselected populations of DMD muscle cells. Sci. Rep.6, 37051. 10.1038/srep37051
221
Maggio I. Stefanucci L. Janssen J. M. Liu J. Chen X. Mouly V. et al (2016b). Selection-free gene repair after adenoviral vector transduction of designer nucleases: Rescue of dystrophin synthesis in DMD muscle cell populations. Nucleic Acids Res.44, 1449–1470. 10.1093/nar/gkv1540
222
Maino E. Wojtal D. Evagelou S. L. Farheen A. Wong T. W. Y. Lindsay K. et al (2021). Targeted genome editing in vivo corrects a Dmd duplication restoring wild-type dystrophin expression. EMBO Mol. Med.13, e13228. 10.15252/emmm.202013228
223
Makarova K. S. Wolf Y. I. Iranzo J. Shmakov S. A. Alkhnbashi O. S. Brouns S. J. J. et al (2020). Evolutionary classification of CRISPR-cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol.18, 67–83. 10.1038/s41579-019-0299-x
224
Malerba A. Boldrin L. Dickson G. (2011). Long-term systemic administration of unconjugated morpholino oligomers for therapeutic expression of dystrophin by exon skipping in skeletal muscle: Implications for cardiac muscle integrity. Nucleic Acid. Ther.21, 293–298. 10.1089/nat.2011.0306
225
Mali P. Yang L. Esvelt K. M. Aach J. Guell M. DiCarlo J. E. et al (2013). RNA-guided human genome engineering via Cas9. Science339, 823–826. 10.1126/science.1232033
226
Malik V. Rodino-Klapac L. R. Viollet L. Wall C. King W. Al-Dahhak R. et al (2010). Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann. Neurol.67, 771–780. 10.1002/ana.22024
227
Mamchaoui K. (2018). Myoblots: Dystrophin quantification by in-cell Western assay for a streamlined development of duchenne muscular dystrophy (DMD) treatments. Neuropathol. Appl. Neurobiol.44, 463–473. 10.1111/nan.12448
228
Mann C. J. Honeyman K. Cheng A. J. Ly T. Lloyd F. Fletcher S. et al (2001). Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc. Natl. Acad. Sci. U. S. A.98, 42–47. 10.1073/pnas.011408598
229
Mareedu S. Million E. D. Duan D. Babu G. J. (2021). Abnormal calcium handling in duchenne muscular dystrophy: Mechanisms and potential therapies. Front. Physiol.12, 647010–647019. 10.3389/fphys.2021.647010
230
Markham B. E. Kernodle S. Nemzek J. Wilkinson J. E. Sigler R. (2015). Chronic dosing with membrane sealant poloxamer 188 NF improves respiratory dysfunction in dystrophic mdx and mdx/utrophin-/- mice. PLoS One10, e0134832. 10.1371/journal.pone.0134832
231
Mata López S. Balog-Alvarez C. Vitha S. Bettis A. K. Canessa E. H. Kornegay J. N. et al (2020). Challenges associated with homologous directed repair using CRISPR-Cas9 and TALEN to edit the DMD genetic mutation in canine Duchenne muscular dystrophy. PLoS One15, e0228072. 10.1371/journal.pone.0228072
232
Matre P. R. Mu X. Wu J. Danila D. Hall M. A. Kolonin M. G. et al (2019). CRISPR/Cas9-Based dystrophin restoration reveals a novel role for dystrophin in bioenergetics and stress resistance of muscle progenitors. Stem Cells37, 1615–1628. 10.1002/stem.3094
233
Mauro A. (1961). Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol.9, 493–495. 10.1083/jcb.9.2.493
234
Mayer U. Saher G. Fassler R. Bornemann A. Echtermeyer F. Mark H. V. D. et al (1997). Absence of integrin alpha 7 causes a novel form of muscular dystrophy. Nat. Genet.17 (3), 318–323. 10.1038/ng1197-318
235
Mázala D. A. G. Novak J. S. Hogarth M. W. Nearing M. Adusumalli P. Tully C. B. et al (2020). TGF-β–driven muscle degeneration and failed regeneration underlie disease onset in a DMD mouse model. JCI Insight5, e135703. 10.1172/jci.insight.135703
236
McCourt J. L. Rhett K. K. Jaeger M. A. Belanto J. J. Talsness D. M. Ervasti J. M. (2015). In vitro stability of therapeutically relevant, internally truncated dystrophins. Skelet. Muscle5, 13. 10.1186/s13395-015-0040-z
237
McDonald C. M. Henricson E. K. Abresch R. T. Duong T. Joyce N. C. Hu F. et al (2018). Long-term effects of glucocorticoids on function, quality of life, and survival in patients with duchenne muscular dystrophy: A prospective cohort study. Lancet391, 451–461. 10.1016/S0140-6736(17)32160-8
238
McDonald C. M. Marbán E. Hendrix S. Hogan N. Ruckdeschel Smith R. Eagle M. et al (2022). Repeated intravenous cardiosphere-derived cell therapy in late-stage duchenne muscular dystrophy (HOPE-2): A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet399, 1049–1058. 10.1016/S0140-6736(22)00012-5
239
McDonald C. M. Sajeev G. Yao Z. McDonnell E. Elfring G. Souza M. et al (2020). Deflazacort vs prednisone treatment for duchenne muscular dystrophy: A meta-analysis of disease progression rates in recent multicenter clinical trials. Muscle Nerve61, 26–35. 10.1002/mus.26736
240
McDonald D. G. M. Kinali M. Gallagher A. C. Mercuri E. Muntoni F. Frcpch H. R. et al (2002). Fracture prevalence in Duchenne muscular dystrophy. Dev. Med. Child. Neurol.44, 695–698. 10.1017/s0012162201002778
241
McElroy S. P. Nomura T. Torrie L. S. Warbrick E. Gartner U. Wood G. et al (2013). A lack of premature termination codon read-through efficacy of PTC124 (ataluren) in a diverse array of reporter assays. PLoS Biol.11, e1001593. 10.1371/journal.pbio.1001593
242
Mcmahon C. D. Popovic L. Oldham J. M. Jeanplong F. Smith H. K. Kambadur R. et al (2022). Myostatin-deficient mice lose more skeletal muscle mass than wild-type controls during hindlimb suspension, 82–87.
243
McNally E. M. Pytel P. (2007). Muscle diseases: The muscular dystrophies. Annu. Rev. Pathol.2, 87–109. 10.1146/annurev.pathol.2.010506.091936
244
McNally E. M. Wyatt E. J. (2017). Mutation-based therapy for duchenne muscular dystrophy: Antisense treatment arrives in the clinic. Circulation136, 979–981. 10.1161/CIRCULATIONAHA.117.028382
245
McNally E. (2012). Novel targets and approaches to treating skeletal muscle disease. First Edit. Elsevier Inc. 10.1016/B978-0-12-381510-1.00081-8
246
Mcpherron A. C. Lawlert A. M. Lee S. (1997). Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature38711, 83–90. 10.1038/387083a0
247
Mead A. F. Petrov M. Malik A. S. Mitchell M. A. Childers M. K. Bogan J. R. et al (2014). Diaphragm remodeling and compensatory respiratory mechanics in a canine model of Duchenne muscular dystrophy. J. Appl. Physiol.116, 807–815. 10.1152/japplphysiol.00833.2013
248
Mendell J. R. Al-zaidy S. A. Rodino-klapac L. R. Goodspeed K. Gray S. J. Kay C. N. et al (2021). Current clinical applications of in vivo gene therapy with AAVs. Mol. Ther.29, 464–488. 10.1016/j.ymthe.2020.12.007
249
Mendell J. R. Al-Zaidy S. Shell R. Arnold W. D. Rodino-Klapac L. R. Prior T. W. et al (2017). Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med.377, 1713–1722. 10.1056/nejmoa1706198
250
Mendell J. R. Campbell K. Rodino-Klapac L. Sahenk Z. Shilling C. Lewis S. et al (2010). Dystrophin immunity in duchenne’s muscular dystrophy. N. Engl. J. Med.363, 1429–1437. 10.1056/nejmoa1000228
251
Mendell J. R. Goemans N. Lowes L. P. Alfano L. N. Berry K. Shao J. et al (2016). Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. 10.1002/ana.24555
252
Mendell J. R. Sahenk Z. Lehman K. Nease C. Lowes L. P. Miller N. F. et al (2020). Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with duchenne muscular dystrophy: A nonrandomized controlled trial. JAMA Neurol.77, 1122–1131. 10.1001/jamaneurol.2020.1484
253
Mendell J. R. Shilling C. Leslie N. D. Flanigan K. M. Al-Dahhak R. Gastier-Foster J. et al (2012). Evidence-based path to newborn screening for duchenne muscular dystrophy. Ann. Neurol.71, 304–313. 10.1002/ana.23528
254
Meng X. M. Nikolic-Paterson D. J. Lan H. Y. (2016). TGF-Β: The master regulator of fibrosis. Nat. Rev. Nephrol.12, 325–338. 10.1038/nrneph.2016.48
255
Merchant F. A. Holmes W. H. Capelli-Schellpfeffer M. Lee R. C. Toner M. (1998). Poloxamer 188 enhances functional recovery of lethally heat-shocked fibroblasts. J. Surg. Res.74, 131–140. 10.1006/jsre.1997.5252
256
Mercuri E. Bönnemann C. G. Muntoni F. (2019). Muscular dystrophies. Lancet394, 2025–2038. 10.1016/S0140-6736(19)32910-1
257
Mercuri E. Muntoni F. (2013). Muscular dystrophies. Lancet381, 845–860. 10.1016/S0140-6736(12)61897-2
258
Meyer G. A. Lieber R. L. (2011). Elucidation of extracellular matrix mechanics from muscle fibers and fiber bundles. J. Biomech.44, 771–773. 10.1016/j.jbiomech.2010.10.044
259
Meyers T. A. Townsend D. W. (2019). Cardiac pathophysiology and the future of cardiac therapies in duchenne muscular dystrophy. Int. J. Mol. Sci.20, 4098. 10.3390/ijms20174098
260
Michel R. N. Chakkalakal J. V. Miura P. Be G. Jasmin B. J. (2008). Modulation of utrophin A mRNA stability in fast versus slow muscles via an AU-rich element and calcineurin signaling. Nucleic Acids Res.36, 826–838. 10.1093/nar/gkm1107
261
Miller J. B. Zhang S. Kos P. Xiong H. Zhou K. Perelman S. S. et al (2017). Non-viral CRISPR/Cas gene editing in vitro and in vivo enabled by synthetic nanoparticle Co-delivery of Cas9 mRNA and sgRNA. Angew. Chem. Int. Ed. Engl.56, 1059–1063. 10.1002/anie.201610209
262
Min Y. L. Bassel-Duby R. Olson E. N. (2019a). CRISPR correction of duchenne muscular dystrophy. Annu. Rev. Med.70, 239–255. 10.1146/annurev-med-081117-010451
263
Min Y. L. Li H. Rodriguez-Caycedo C. Mireault A. A. Huang J. Shelton J. M. et al (2019b). CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci. Adv.5, eaav4324. 10.1126/sciadv.aav4324
264
Mitrpant C. Fletcher S. Iversen P. L. Wilton S. D. (2009). By‐passing the nonsense mutation in the 4CV mouse model of muscular dystrophy by induced exon skipping. J. Gene Med. A cross‐disciplinary J. Res. Sci. gene Transf. its Clin. Appl.11, 46–56. 10.1002/jgm.1265
265
Moe K. T. Yin N. O. Naylynn T. M. Khairunnisa K. Wutyi M. A. Gu Y. et al (2011). Nox2 and Nox4 mediate tumour necrosis factor-alpha-induced ventricular remodelling in mice. J. Cell Mol. Med.15, 2601–2613. 10.1111/j.1582-4934.2011.01261.x
266
Mojumdar K. Liang F. Giordano C. Lemaire C. Danialou G. Okazaki T. et al (2014). Inflammatory monocytes promote progression of Duchenne muscular dystrophy and can be therapeutically targeted via CCR2. EMBO Mol. Med.6, 1476–1492. 10.15252/emmm.201403967
267
Mokri B. Engel A. G. (1975). Duchenne dystrophy: Electron microscopic findings pointing to a basic or early abnormality in the plasma membrane of the muscle fiber. Neurology25, 1111–1120. 10.1212/wnl.25.12.1111
268
Montanaro F. Lindenbaum M. Carbonetto S. (1999). alpha-Dystroglycan is a laminin receptor involved in extracellular matrix assembly on myotubes and muscle cell viability. J. Cell Biol.145, 1325–1340. 10.1083/jcb.145.6.1325
269
Moore T. M. Lin A. J. Strumwasser A. R. Cory K. Whitney K. Ho T. et al (2020). Mitochondrial dysfunction is an early consequence of partial or complete dystrophin loss in mdx mice. Front. Physiol.11, 690. 10.3389/fphys.2020.00690
270
Moorwood C. Lozynska O. Suri N. Napper A. D. Diamond S. L. Khurana T. S. (2011). Drug discovery for Duchenne muscular dystrophy via utrophin promoter activation screening. Utrophin Promot. Act. Screen.6, 261699–e26211. 10.1371/journal.pone.0026169 via
271
Moretti A. Fonteyne L. Giesert F. Hoppmann P. Meier A. B. Bozoglu T. et al (2020). Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat. Med.26, 207–214. 10.1038/s41591-019-0738-2
272
Morgan J. Muntoni F. (2021). Changes in myonuclear number during postnatal growth–implications for AAV gene therapy for muscular dystrophy. J. Neuromuscul. Dis.1–8, S317–S324. 10.3233/JND-210683
273
Moss F. P. Leblond C. P. (1971). Satellite cells as the source of nuclei in muscles of growing rats. Anat. Rec.170, 421–435. 10.1002/ar.1091700405
274
Mulivor A. Cannell M. Davies M. Sako D. Liu J. Bresnahan D. et al (2014). “ACE-083, a ligand trap for members of the TGFβ superfamily, increases muscle mass locally in a mouse model of Duchenne muscular dystrophy,” in Presented at action Duchenne int. Conf., 12th, london, nov, 7–8.
275
Mullard A. (2021a). N E W S and A N A LY S I S News in BRieF FDA outlines steps for individualized drug development. Nat. Rev. Drug Discov.20 (2), 91. 10.1038/d41573-021-00009-7
276
Mullard A. (2021b). N E W S and A N A LY S I S News in BRieF FDA outlines steps for individualized drug development. Nat. Rev. Drug Discov.20, 6939926.
277
Muntoni F. Tejura B. Spinty S. Roper H. Hughes I. Layton G. et al (2019). A phase 1b trial to assess the pharmacokinetics of Ezutromid in pediatric duchenne muscular dystrophy patients on a balanced diet. Clin. Pharmacol. Drug Dev.8, 922–933. 10.1002/cpdd.642
278
Nallamilli B. R. R. Chaubey A. Valencia C. A. Stansberry L. Behlmann A. M. Ma Z. et al (2021). A single NGS-based assay covering the entire genomic sequence of the DMD gene facilitates diagnostic and newborn screening confirmatory testing. Hum. Mutat.42, 626–638. 10.1002/humu.24191
279
Nelson C. E. Hakim C. H. Ousterout D. G. Thakore P. I. Moreb E. A. Castellanos Rivera R. M. et al (2016). In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science351, 403–407. 10.1126/science.aad5143
280
Nelson C. E. Robinson-Hamm J. N. Gersbach C. A. (2017). Genome engineering: A new approach to gene therapy for neuromuscular disorders. Nat. Rev. Neurol.13, 647–661. 10.1038/nrneurol.2017.126
281
Nelson C. E. Wu Y. Gemberling M. P. Oliver M. L. Waller M. A. Bohning J. D. et al (2019). Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med.25, 427–432. 10.1038/s41591-019-0344-3
282
Nelson D. M. Lindsay A. Judge L. M. Duan D. Chamberlain J. S. Lowe D. A. et al (2018). Variable rescue of microtubule and physiological phenotypes in mdx muscle expressing different miniaturized dystrophins. Hum. Mol. Genet.27, 2090–2100. 10.1093/hmg/ddy113
283
Nguyen Q. Yokota T. (2019). Antisense oligonucleotides for the treatment of cardiomyopathy in duchenne muscular dystrophy. Am. J. Transl. Res.11, 1202–1218.
284
Niks E. H. Aartsma-Rus A. (2017). Exon skipping: A first in class strategy for duchenne muscular dystrophy. Expert Opin. Biol. Ther.17, 225–236. 10.1080/14712598.2017.1271872
285
Nishi M. Yasue A. Nishimatu S. Nohno T. Yamaoka T. Itakura M. et al (2002). A missense mutant myostatin causes hyperplasia without hypertrophy in the mouse muscle. hypertrophy mouse muscle293, 247–251. 10.1016/S0006-291X(02)00209-7
286
Nishida K. Arazoe T. Yachie N. Banno S. Kakimoto M. Tabata M. et al (2016). Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science353, aaf8729. 10.1126/science.aaf8729
287
Novak J. S. Hogarth M. W. Boehler J. F. Nearing M. Vila M. C. Heredia R. et al (2017). Myoblasts and macrophages are required for therapeutic morpholino antisense oligonucleotide delivery to dystrophic muscle. Nat. Commun.8, 941. 10.1038/s41467-017-00924-7
288
Odom G. L. Gregorevic P. Chamberlain J. S. (2007). Viral-mediated gene therapy for the muscular dystrophies: Successes, limitations and recent advances. Biochim. Biophys. Acta1772, 243–262. 10.1016/j.bbadis.2006.09.007
289
Olson E. N. (2021). Toward the correction of muscular dystrophy by gene editing. Proc. Natl. Acad. Sci. U. S. A.118, e2004840117. 10.1073/pnas.2004840117
290
Onopiuk M. Brutkowski W. Wierzbicka K. Wojciechowska S. Szczepanowska J. Fronk J. et al (2009). Mutation in dystrophin-encoding gene affects energy metabolism in mouse myoblasts. Biochem. Biophys. Res. Commun.386, 463–466. 10.1016/j.bbrc.2009.06.053
291
Ousterout D. G. Kabadi A. M. Thakore P. I. Majoros W. H. Reddy T. E. Gersbach C. A. (2015). Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun.6, 6244. 10.1038/ncomms7244
292
Pant M. Sopariwala D. H. Bal N. C. Lowe J. Delfin D. A. Rafael-Fortney J. et al (2015). Metabolic dysfunction and altered mitochondrial dynamics in the utrophin-dystrophin deficient mouse model of duchenne muscular dystrophy. PLoS One10, e0123875. 10.1371/journal.pone.0123875
293
Papa S. Skulachev V. P. (1997). Reactive oxygen species, mitochondria, apoptosis and aging. Mol. Cell Biochem.174, 305–319. 10.1023/A:1006873518427
294
Passaquin A. C. Renard M. Kay L. Challet C. Mokhtarian A. Wallimann T. et al (2002). Creatine supplementation reduces skeletal muscle degeneration and enhances mitochondrial function in mdx mice. Neuromuscul. Disord.12, 174–182. 10.1016/s0960-8966(01)00273-5
295
Percival J. M. Siegel M. P. Knowels G. Marcinek D. J. (2013). Defects in mitochondrial localization and ATP synthesis in the mdx mouse model of Duchenne muscular dystrophy are not alleviated by PDE5 inhibition. Hum. Mol. Genet.22, 153–167. 10.1093/hmg/dds415
296
Perez-Pinera P. Kocak D. D. Vockley C. M. Adler A. F. Kabadi A. M. Polstein L. R. et al (2013). RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods10, 973–976. 10.1038/Nmeth.2600
297
Pertille A. De Carvalho C. L. T. Matsumura C. Y. Neto H. S. Marques M. J. (2010). Calcium-binding proteins in skeletal muscles of the mdx mice: Potential role in the pathogenesis of Duchenne muscular dystrophy. Int. J. Exp. Pathol.91, 63–71. 10.1111/j.1365-2613.2009.00688.x
298
Pescatori M. Broccolini A. Minetti C. Bertini E. Bruno C. D’amico A. et al (2007). Gene expression profiling in the early phases of DMD: A constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J.21, 1210–1226. 10.1096/fj.06-7285com
299
Pestronk A. Parhad I. Drachman D. Price D. L. (1982). Membrane myopathy: Morphological similarities to duchenne muscular dystrophy. Muscle Nerve5, 209–214. 10.1002/mus.880050306
300
Petrillo S. Pelosi L. Piemonte F. Travaglini L. Forcina L. Catteruccia M. et al (2017). Oxidative stress in duchenne muscular dystrophy: Focus on the NRF2 redox pathway. Hum. Mol. Genet.26, 2781–2790. 10.1093/hmg/ddx173
301
Petrof B. J. Shrager J. B. Stedman H. H. Kelly A. M. Sweeney H. L. (1993). Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. U. S. A.90, 3710–3714. 10.1073/pnas.90.8.3710
302
Pfizer (2014). Pfizer initiates phase 2 study of PF-06252616 in Duchenne muscular dystrophy. Press Release. Dec. 17 Available at: http://www.pfizer.com/news/press-release/press-release-detail/pfizer_initiates_phase_.2_study_of_pf_06252616_in_duchenne_muscular_dystrophy.
303
Phelps S. F. Hauser M. A. Cole N. M. Rafael J. A. Hinkle R. T. Faulkner J. A. et al (1995). Expression of full-length and truncated dystrophin mini-genes in transgenic mdx mice. Hum. Mol. Genet.4, 1251–1258. 10.1093/hmg/4.8.1251
304
Pickar-Oliver A. Gough V. Bohning J. D. Liu S. Robinson-Hamm J. N. Daniels H. et al (2021). Full-length dystrophin restoration via targeted exon integration by AAV-CRISPR in a humanized mouse model of Duchenne muscular dystrophy. Mol. Ther.29, 3243–3257. 10.1016/j.ymthe.2021.09.003
305
Pinniger G. J. Terrill J. R. Assan E. B. Grounds M. D. Arthur P. G. (2017). Pre-clinical evaluation of N-acetylcysteine reveals side effects in the mdx mouse model of Duchenne muscular dystrophy. J. Physiol.595, 7093–7107. 10.1113/JP274229
306
Politano L. Nigro G. Nigro V. Pilus G. Papparella S. Paciello O. et al (2003). Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol.22, 15–21.
307
Politano L. (2021). Read-through approach for stop mutations in Duchenne muscular dystrophy. An update. update12, 43–50. 10.36185/2532-1900-041
308
Pons F. Nicholson L. V. Robert A. Voit T. Leger J. J. (1993). Dystrophin and dystrophin-related protein (utrophin) distribution in normal and dystrophin-deficient skeletal muscles. Neuromuscul. Disord.3, 507–514. 10.1016/0960-8966(93)90106-t
309
Powers S. K. Ji L. L. Kavazis A. N. Jackson M. J. (2011). Reactive oxygen species: Impact on skeletal muscle. Compr. Physiol.1, 941–969. 10.1002/cphy.c100054
310
Prayle A. Smyth A. R. (2010). Aminoglycoside use in cystic fibrosis: Therapeutic strategies and toxicity. Curr. Opin. Pulm. Med.16, 604–610. 10.1097/MCP.0b013e32833eebfd
311
Qi L. S. Larson M. H. Gilbert L. A. Doudna J. A. Weissman J. S. Arkin A. P. et al (2013). Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell152, 1173–1183. 10.1016/j.cell.2013.02.022
312
Rafael J. A. Cox G. A. Corrado K. Jung D. Campbell K. P. Chamberlain J. S. (1996). Forced expression of dystrophin deletion constructs reveals structure-function correlations. J. Cell Biol.134, 93–102. 10.1083/jcb.134.1.93
313
Ramos J. N. Hollinger K. Bengtsson N. E. Allen J. M. Hauschka S. D. Chamberlain J. S. (2019). Development of novel micro-dystrophins with enhanced functionality. Mol. Ther.27, 623–635. 10.1016/j.ymthe.2019.01.002
314
Ran F. A. Hsu P. D. Lin C. Y. Gootenberg J. S. Konermann S. Trevino A. E. et al (2013). Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell154, 1380–1389. 10.1016/j.cell.2013.08.021
315
Reid M. B. (2001). Invited review: Redox modulation of skeletal muscle contraction: What we know and what we don’t. J. Appl. Physiol.90, 724–731. 10.1152/jappl.2001.90.2.724
316
Reid M. B. Shoji T. Moody M. R. Entman M. L. (1992). Reactive oxygen in skeletal muscle. II. Extracellular release of free radicals. J. Appl. Physiol.73, 1805–1809. 10.1152/jappl.1992.73.5.1805
317
Reisz-porszasz S. Bhasin S. Artaza J. N. Shen R. Sinha-hikim I. Hogue A. et al (2022). Lower skeletal muscle mass in male transgenic mice with muscle-specific overexpression of myostatin, 876–888.
318
Ricotti V. Spinty S. Roper H. Hughes I. Tejura B. Robinson N. et al (2016). Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-arylbenzoxazole utrophin modulator, following single- and multiple- dose administration to pediatric patients with duchenne muscular dystrophy. PLoS One.11, 1–16. 10.1371/journal.pone.0152840
319
Rimessi P. Sabatelli P. Fabris M. Braghetta P. Bassi E. Spitali P. et al (2009). Cationic PMMA nanoparticles bind and deliver antisense oligoribonucleotides allowing restoration of dystrophin expression in the mdx mouse. Mol. Ther.17, 820–827. 10.1038/mt.2009.8
320
Roberts R. G. Coffey A. J. Bobrow M. Bentley D. R. (1993). Exon structure of the human dystrophin gene. Genomics16, 536–538. 10.1006/geno.1993.1225
321
Rogers G. L. Martino A. T. Aslanidi G. V. Jayandharan G. R. Srivastava A. Herzog R. W. (2011). Innate immune responses to AAV vectors. Front. Microbiol.2, 194. 10.3389/fmicb.2011.00194
322
Rong Z. Fu X. Wang M. Xu Y. (2012). A scalable approach to prevent teratoma formation of human embryonic stem cells. J. Biol. Chem.287, 32338–32345. 10.1074/jbc.M112.383810
323
Rooney J. E. Gurpur P. B. Burkin D. J. (2009a). Laminin-111 protein therapy prevents muscle disease in the mdx mouse model for Duchenne muscular dystrophy, 106.
324
Rooney J. E. Gurpur P. B. Yablonka-reuveni Z. Burkin D. J. (2009b). Laminin-111 restores regenerative capacity in a mouse model for alpha7 integrin congenital myopathy. Am. J. Pathol.174, 256–264. 10.2353/ajpath.2009.080522
325
Rooney J. E. Welser J. V. Dechert M. A. Flintoff-dye N. L. Kaufman S. J. Burkin D. J. (2006). Severe muscular dystrophy in mice that lack dystrophin and 7 integrin. 10.1242/jcs.02952
326
Rosenberg A. S. Puig M. Nagaraju K. Hoffman E. P. Villalta S. A. Rao V. A. et al (2015). Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med.7, 299rv4. 10.1126/scitranslmed.aaa7322
327
Rouger K. Larcher T. Dubreil L. Deschamps J.-Y. le Guiner C. Jouvion G. et al (2011). Systemic delivery of allogenic muscle stem cells induces long-term muscle repair and clinical efficacy in duchenne muscular dystrophy dogs. Am. J. Pathol.179, 2501–2518. 10.1016/j.ajpath.2011.07.022
328
Rybakova I. N. Amann K. J. Ervasti J. M. (1996). A new model for the interaction of dystrophin with F-actin. J. Cell Biol.135, 661–672. 10.1083/jcb.135.3.661
329
Ryu S. M. Koo T. Kim K. Lim K. Baek G. Kim S. T. et al (2018). Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol.36, 536–539. 10.1038/nbt.4148
330
Saclier M. ben Larbi S. My Ly H. Moulin E. Mounier R. Chazaud B. et al (2021). Interplay between myofibers and pro-inflammatory macrophages controls muscle damage in mdx mice. J. Cell Sci.134, jcs258429. 10.1242/jcs.258429
331
Saleh A. F. Arzumanov A. A. Gait M. J. (2012). “Overview of alternative oligonucleotide chemistries for exon skipping,” in Exon skipping: Methods and protocols. Editor Aartsma-RusA. (Totowa, NJ: Humana Press), 365–378. 10.1007/978-1-61779-767-5_23
332
Salva M. Z. Himeda C. L. Tai P. W. Nishiuchi E. Gregorevic P. Allen J. M. et al (2007). Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol. Ther.15, 320–329. 10.1038/sj.mt.6300027
333
Schneider A. E. Aartsma-Rus A. (2021). Developments in reading frame restoring therapy approaches for Duchenne muscular dystrophy. Expert Opin. Biol. Ther.21, 343–359. 10.1080/14712598.2021.1832462
334
Schnepp B. C. Clark K. R. Klemanski D. L. Pacak C. A. Johnson P. R. (2003). Genetic fate of recombinant adeno-associated virus vector genomes in muscle. J. Virol.77, 3495–3504. 10.1128/jvi.77.6.3495-3504.2003
335
Schuh R. A. Jackson K. C. Khairallah R. J. Ward C. W. Spangenburg E. E. (2012). Measuring mitochondrial respiration in intact single muscle fibers. Am. J. Physiol. Regul. Integr. Comp. Physiol.302, R712–R719. 10.1152/ajpregu.00229.2011
336
Sciorati C. Rigamonti E. Manfredi A. A. Rovere-Querini P. (2016). Cell death, clearance and immunity in the skeletal muscle. Cell Death Differ.23, 927–937. 10.1038/cdd.2015.171
337
Sharma G. Sharma A. R. Bhattacharya M. Lee S. S. Chakraborty C. (2021). CRISPR-Cas9: A preclinical and clinical perspective for the treatment of human diseases. Mol. Ther.29, 571–586. 10.1016/j.ymthe.2020.09.028
338
Sheikh O. Yokota T. (2021). Restoring protein expression in neuromuscular conditions: A review assessing the current state of exon skipping/inclusion and gene therapies for duchenne muscular dystrophy and spinal muscular atrophy. BioDrugs35, 389–399. 10.1007/s40259-021-00486-7
339
Sinadinos A. Young C. N. J. Al-Khalidi R. Teti A. Kalinski P. Mohamad S. et al (2015). P2RX7 purinoceptor: A therapeutic target for ameliorating the symptoms of duchenne muscular dystrophy. PLoS Med.12, e1001888. 10.1371/journal.pmed.1001888
340
Skuk D. Goulet M. Roy B. Chapdelaine P. Bouchard J.-P. Roy R. et al (2006). Dystrophin expression in muscles of duchenne muscular dystrophy patients after high-density injections of normal myogenic cells. J. Neuropathol. Exp. Neurol.65, 371–386. 10.1097/01.jnen.0000218443.45782.81
341
Skuk D. Roy B. Goulet M. Chapdelaine P. Bouchard J.-P. Roy R. et al (2004). Dystrophin expression in myofibers of duchenne muscular dystrophy patients following intramuscular injections of normal myogenic cells. Mol. Ther.9, 475–482. 10.1016/j.ymthe.2003.11.023
342
Smythe G. M. Hodgetts S. I. Grounds M. D. (2000). Immunobiology and the future of myoblast transfer therapy. Mol. Ther.1, 304–313. 10.1006/mthe.2000.0049
343
Soblechero-Martín P. López-Martínez A. de la Puente-Ovejero L. Vallejo-Illarramendi A. Arechavala-Gomeza V. (2021). Utrophin modulator drugs as potential therapies for Duchenne and Becker muscular dystrophies. Neuropathol. Appl. Neurobiol.47, 711–723. 10.1111/nan.12735
344
Sonnemann K. J. Heun-johnson H. Turner A. J. Baltgalvis K. A. Lowe D. A. Ervasti J. M. (2009). Functional substitution by TAT-utrophin in dystrophin- deficient mice. PLoS Med.6, e1000083. 10.1371/journal.pmed.1000083
345
Sorichter S. Mair J. Koller A. Pelsers M. M. Puschendorf B. Glatz J. F. (1998). Early assessment of exercise induced skeletal muscle injury using plasma fatty acid binding protein. Br. J. Sports Med.32, 121–124. 10.1136/bjsm.32.2.121
346
Sorichter S. Puschendorf B. Mair J. (1999). Skeletal muscle injury induced by eccentric muscle action: Muscle proteins as markers of muscle fiber injury. Exerc Immunol. Rev.5, 5–21.
347
Spencer M. J. Montecino-Rodriguez E. Dorshkind K. Tidball J. G. (2001). Helper (CD4+) and cytotoxic (CD8+) T cells promote the pathology of dystrophin-deficient muscle. Clin. Immunol.98, 235–243. 10.1006/clim.2000.4966
348
Spencer M. J. Walsh C. M. Dorshkind K. A. Rodriguez E. M. Tidball J. G. (1997). Myonuclear apoptosis in dystrophic mdx muscle occurs by perforin-mediated cytotoxicity. J. Clin. Investigation99, 2745–2751. 10.1172/JCI119464
349
Spinazzola J. M. Kunkel L. M. (2016). Pharmacological therapeutics targeting the secondary defects and downstream pathology of Duchenne muscular dystrophy. Expert Opin. Orphan Drugs4, 1179–1194. 10.1080/21678707.2016.1240613
350
Spurney C. F. (2011a). Cardiomyopathy of duchenne muscular dystrophy: Current understanding and future directions. Muscle Nerve44, 8–19. 10.1002/mus.22097
351
Spurney C. F. Guerron A. D. Yu Q. Sali A. van der Meulen J. H. Hoffman E. P. et al (2011b). Membrane sealant Poloxamer P188 protects against isoproterenol induced cardiomyopathy in dystrophin deficient mice. BMC Cardiovasc Disord.11, 20. 10.1186/1471-2261-11-20
352
Stedman H. H. Sweeney H. L. Shrager J. B. Maguire H. C. Panettieri R. A. Petrof B. et al (1991). The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature352, 536–539. 10.1038/352536a0
353
Stephenson A. A. Flanigan K. M. (2021). Gene editing and modulation for Duchenne muscular dystrophy. Prog. Mol. Biol. Transl. Sci.182, 225–255. 10.1016/bs.pmbts.2021.01.029
354
Straub V. Bittner R. E. Leger J. J. Voit T. (1992). Direct visualization of the dystrophin network on skeletal muscle fiber membrane. J. Cell Biol.119, 1183–1191. 10.1083/jcb.119.5.1183
355
Straub V. Rafael J. A. Chamberlain J. S. Campbell K. P. (1997). Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. Available at: http://www.jcb.org.
356
Suzuki K. Tsunekawa Y. Hernandez-Benitez R. Wu J. Zhu J. Kim E. J. et al (2016). In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature540, 144–149. 10.1038/nature20565
357
Szabo P. L. Ebner J. Koenig X. Hamza O. Watzinger S. Trojanek S. et al (2021). Cardiovascular phenotype of the Dmdmdx rat - a suitable animal model for Duchenne muscular dystrophy. Dis. Model Mech.14, dmm047704. 10.1242/dmm.047704
358
Tabebordbar M. Lagerborg K. A. Stanton A. King E. M. Ye S. Tellez L. et al (2021). Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell184, 4919–4938 e22. 10.1016/j.cell.2021.08.028
359
Tabebordbar M. Zhu K. Cheng J. K. W. Chew W. L. Widrick J. J. Yan W. X. et al (2016). In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science351, 407–411. 10.1126/science.aad5177
360
Taglia A. Petillo R. D’Ambrosio P. Picillo E. Torella A. Orsini C. et al (2015). Clinical features of patients with dystrophinopathy sharing the 45-55 exon deletion of DMD gene. Acta Myol.34, 9–13.
361
Takeda S. Clemens P. R. Hoffman E. P. (2021). Exon-skipping in duchenne muscular dystrophy. J. Neuromuscul. Dis.8, S343–S358. 10.3233/JND-210682
362
Taylor M. Jefferies J. Byrne B. Lima J. Ambale-Venkatesh B. Ostovaneh M. R. et al (2019). Cardiac and skeletal muscle effects in the randomized HOPE-Duchenne trial. Neurology92, e866–e878. 10.1212/WNL.0000000000006950
363
Terrill J. R. Duong M. N. Turner R. Le Guiner C. Boyatzis A. Kettle A. J. et al (2016). Levels of inflammation and oxidative stress, and a role for taurine in dystropathology of the Golden Retriever Muscular Dystrophy dog model for Duchenne Muscular Dystrophy. Redox Biol.9, 276–286. 10.1016/j.redox.2016.08.016
364
Terrill J. R. Radley-Crabb H. G. Grounds M. D. Arthur P. G. (2012). N-Acetylcysteine treatment of dystrophic mdx mice results in protein thiol modifications and inhibition of exercise induced myofibre necrosis. Neuromuscul. Disord.22, 427–434. 10.1016/j.nmd.2011.11.007
365
Terrill J. R. Radley-Crabb H. G. Iwasaki T. Lemckert F. A. Arthur P. G. Grounds M. D. (2013). Oxidative stress and pathology in muscular dystrophies: Focus on protein thiol oxidation and dysferlinopathies. FEBS J.280, 4149–4164. 10.1111/febs.12142
366
Tidball J. G. Villalta S. A. (2010). Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiology-Regulatory, Integr. Comp. Physiology298, R1173–R1187. 10.1152/ajpregu.00735.2009
367
Tidball J. G. Welc S. S. Wehling‐Henricks M. (2018). “Immunobiology of inherited muscular dystrophies,” in Comprehensive physiology (Wiley), 1313–1356. 10.1002/cphy.c170052
368
Tinsley J. Deconinck N. Fisher R. Kahn D. Phelps S. Gillis J. M. et al (1998). Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat. Med.4, 1441–1444. 10.1038/4033
369
Tobin J. F. Celeste A. J. (2005). Myostatin, a negative regulator of muscle mass: Implications for muscle degenerative diseases. Curr. Opin. Pharmacol.5, 328–332. 10.1016/j.coph.2005.01.011
370
Townsend D. Blankinship M. J. Allen J. M. Gregorevic P. Chamberlain J. S. Metzger J. M. (2007). Systemic administration of micro-dystrophin restores cardiac geometry and prevents dobutamine-induced cardiac pump failure. Mol. Ther.15, 1086–1092. 10.1038/sj.mt.6300144
371
Townsend D. Turner I. Yasuda S. Martindale J. Davis J. Shillingford M. et al (2010). Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J. Clin. Investigation120, 1140–1150. 10.1172/JCI41329
372
Townsend D. W. Yasuda S. McNally E. Metzger J. M. (2011). Distinct pathophysiological mechanisms of cardiomyopathy in hearts lacking dystrophin or the sarcoglycan complex. FASEB J.25, 3106–3114. 10.1096/fj.10-178913
373
Townsend D. Yasuda S. Chamberlain J. Metzger J. M. (2009). Cardiac consequences to skeletal muscle-centric therapeutics for Duchenne muscular dystrophy. Trends Cardiovasc Med.19, 50–55. 10.1016/j.tcm.2009.04.006
374
Townsend D. Yasuda S. Li S. Chamberlain J. S. Metzger J. M. (2008). Emergent dilated cardiomyopathy caused by targeted repair of dystrophic skeletal muscle. Mol. Ther.16, 832–835. 10.1038/mt.2008.52
375
Tsuda T. (2018). Clinical manifestations and overall management strategies for duchenne muscular dystrophy. Methods Mol. Biol.1687, 19–28. 10.1007/978-1-4939-7374-3_2
376
Tulangekar A. Sztal T. E. (2021). Inflammation in duchenne muscular dystrophy-exploring the role of neutrophils in muscle damage and regeneration. Biomedicines9, 1366. 10.3390/biomedicines9101366
377
Turner P. R. Westwood T. Regen C. M. Steinhardt R. A. (1988). Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature335, 735–738. 10.1038/335735a0
378
Tyler K. L. (2003). Origins and early descriptions of Duchenne muscular dystrophy. Muscle Nerve28, 402–422. 10.1002/mus.10435
379
Van Deutekom J. C. T. Bremmer-Bout M. Janson A. A. M. Ginjaar I. B. Baas F. Den Dunnen J. T. et al (2001). Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum. Mol. Genet.10, 1547–1554. 10.1093/hmg/10.15.1547
380
Verma A. (2018). Recent advances in antisense oligonucleotide therapy in genetic neuromuscular diseases. Ann. Indian Acad. Neurol.21, 3–8. 10.4103/aian.AIAN_298_17
381
Vidal B. Serrano A. L. Tjwa M. Suelves M. Ardite E. de Mori R. et al (2008). Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev.22, 1747–1752. 10.1101/gad.465908
382
Villalta S. A. Rosenberg A. S. Bluestone J. A. (2015). The immune system in Duchenne muscular dystrophy: Friend or foe. Rare Dis.3, e1010966. 10.1080/21675511.2015.1010966
383
Voisin V. Se C. Yu H. Gillet B. J M. I. De S. et al (2005). L -arginine improves dystrophic phenotype in mdx mice. Neurobiol. Dis.20, 123–130. 10.1016/j.nbd.2005.02.010
384
Vulin A. Leturcq F. Kaplan J. (2004). Splicingexonskippingcorrextion-Mice306, 4.
385
Wagner K. R. Fleckenstein J. L. Amato A. A. Barohn R. J. Bushby K. Escolar D. M. et al (2008). A phase I/II trial of MYO-029 in adult subjects with muscular dystrophy, 561–571. 10.1002/ana.21338
386
Wagner K. R. Hamed S. Hadley D. W. Gropman A. L. Burstein A. H. Escolar D. M. et al (2001). Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann. Neurol.49, 706–711. 10.1002/ana.1023
387
Wallace G. Q. Lapidos K. A. Kenik J. S. McNally E. M. (2008). Long-term survival of transplanted stem cells in immunocompetent mice with muscular dystrophy. Am. J. Pathology173, 792–802. 10.2353/ajpath.2008.080259
388
Walsh F. S. Celeste A. J. (2005). Myostatin: A modulator of skeletal-muscle stem cells, 1513–1517.
389
Wang B. Li J. Xiao X. (2000). Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc. Natl. Acad. Sci. U. S. A.97, 13714–13719. 10.1073/pnas.240335297
390
Wang D. Zhong L. Nahid M. A. Gao G. (2014). The potential of adeno-associated viral vectors for gene delivery to muscle tissue. Expert Opin. Drug Deliv.11, 345–364. 10.1517/17425247.2014.871258
391
Wang J. Z. Wu P. Shi Z. M. Xu Y. L. Liu Z. J. (2017). The AAV-mediated and RNA-guided CRISPR/Cas9 system for gene therapy of DMD and BMD. Brain Dev.39, 547–556. 10.1016/j.braindev.2017.03.024
392
Wang Z. Storb R. Halbert C. L. Banks G. B. Butts T. M. Finn E. E. et al (2012). Successful regional delivery and long-term expression of a dystrophin gene in canine muscular dystrophy: A preclinical model for human therapies. Mol. Ther.20, 1501–1507. 10.1038/mt.2012.111
393
Wang Z. Zhu T. Qiao C. Zhou L. Wang B. Zhang J. et al (2005). Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat. Biotechnol.23, 321–328. 10.1038/nbt1073
394
Wasala L. P. Watkins T. Wasala N. Burke M. Yue Y. Lai Y. et al (2022). The implication of hinge 1 and hinge 4 in micro-dystrophin gene therapy for Duchenne muscular dystrophy. Hum. Gene Ther.34, 459–470. 10.1089/hum.2022.180
395
Watchko J. O’Day T. Wang B. Zhou L. Tang Y. Li J. et al (2002). Adeno-associated virus vector-mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Hum. Gene Ther.13, 1451–1460. 10.1089/10430340260185085
396
Wehling M. Spencer M. J. Tidball J. G. (2001). A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell Biol.155, 123–131. 10.1083/jcb.200105110
397
Wei T. Cheng Q. Min Y. L. Olson E. N. Siegwart D. J. (2020). Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing. Nat. Commun.11, 3232. 10.1038/s41467-020-17029-3
398
Weir A. P. Burton E. A. Harrod G. Davies K. E. (2002). A- and B-utrophin have different expression patterns and are differentially up-regulated in mdx muscle. J. Biol. Chem.277, 45285–45290. 10.1074/jbc.M205177200
399
Weir A. P. Morgan J. E. Davies K. E. (2004). A-utrophin up-regulation in mdx skeletal muscle is independent of regeneration. Neuromuscul. Disord.14, 19–23. 10.1016/j.nmd.2003.09.004
400
Welch E. M. Barton E. R. Zhuo J. Tomizawa Y. Friesen W. J. Trifillis P. et al (2007). PTC124 targets genetic disorders caused by nonsense mutations. Nature447, 87–91. 10.1038/nature05756
401
Whitehead N. P. Kim M. J. Bible K. L. Adams M. E. Froehner S. C. (2015). A new therapeutic effect of simvastatin revealed by functional improvement in muscular dystrophy. Proc. Natl. Acad. Sci. U. S. A.112, 12864–12869. 10.1073/pnas.1509536112
402
Whitehead N. P. Pham C. Gervasio O. L. Allen D. G. (2008). N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J. Physiol.586, 2003–2014. 10.1113/jphysiol.2007.148338
403
Whitehead N. P. Yeung E. W. Froehner S. C. Allen D. G. (2010). Skeletal muscle NADPH oxidase is increased and triggers stretch-induced damage in the mdx mouse. PLoS One5, e15354. 10.1371/journal.pone.0015354
404
Williams I. A. Allen D. G. (2007). The role of reactive oxygen species in the hearts of dystrophin-deficient mdx mice. Am. J. Physiol. Heart Circ. Physiol.293, H1969–H1977. 10.1152/ajpheart.00489.2007
405
Wojtal D. Kemaladewi D. U. Malam Z. Abdullah S. Wong T. W. Hyatt E. et al (2016). Spell checking nature: Versatility of CRISPR/Cas9 for developing treatments for inherited disorders. Am. J. Hum. Genet.98, 90–101. 10.1016/j.ajhg.2015.11.012
406
Wu B. Li Y. Morcos P. A. Doran T. J. Lu P. Lu Q. L. (2009). Octa-guanidine morpholino restores dystrophin expression in cardiac and skeletal muscles and ameliorates pathology in dystrophic mdx mice. Mol. Ther.17, 864–871. 10.1038/mt.2009.38
407
Wu B. Lu P. Benrashid E. Malik S. Ashar J. Doran T. J. et al (2010). Dose-dependent restoration of dystrophin expression in cardiac muscle of dystrophic mice by systemically delivered morpholino. Gene Ther.17, 132–140. 10.1038/gt.2009.120
408
Wu B. Moulton H. M. Iversen P. L. Jiang J. Li J. Li J. et al (2008). Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc. Natl. Acad. Sci. U. S. A.105, 14814–14819. 10.1073/pnas.0805676105
409
Xiao X. A. Li J. A. Samulski R. J. (1996). Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J. Virol.70, 8098–8108. 10.1128/Jvi.70.11.8098-8108.1996
410
Xu L. Park K. H. Zhao L. Xu J. El Refaey M. Gao Y. et al (2016). CRISPR-Mediated genome editing restores dystrophin expression and function in mdx mice. Mol. Ther.24, 564–569. 10.1038/mt.2015.192
411
Yang J. Ratovitski T. Brady J. P. Solomon M. B. Wells K. D. Wall R. J. (2001). Expression of myostatin pro domain results in muscular transgenic mice, 361.
412
Yang Y. Wang L. Bell P. McMenamin D. He Z. White J. et al (2016). A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol.34, 334–338. 10.1038/nbt.3469
413
Yasuda S. Townsend D. Michele D. E. Favre E. G. Day S. M. Metzger J. M. (2005). Dystrophic heart failure blocked by membrane sealant poloxamer. Nature436, 1025–1029. 10.1038/nature03844
414
Yin H. Saleh A. F. Betts C. Camelliti P. Seow Y. Ashraf S. et al (2011). Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol. Ther.19, 1295–1303. 10.1038/mt.2011.79
415
Yokota T. Lu Q. L. Partridge T. Kobayashi M. Nakamura A. Takeda S. et al (2009). Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann. Neurol.65, 667–676. 10.1002/ana.21627
416
Young C. S. Hicks M. R. Ermolova N. V. Nakano H. Jan M. Younesi S. et al (2016). A single CRISPR-cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell18, 533–540. 10.1016/j.stem.2016.01.021
417
Young C. S. Mokhonova E. Quinonez M. Pyle A. D. Spencer M. J. (2017). Creation of a novel humanized dystrophic mouse model of duchenne muscular dystrophy and application of a CRISPR/Cas9 gene editing therapy. J. Neuromuscul. Dis.4, 139–145. 10.3233/JND-170218
418
Yu H. K. Liu X. Pan M. Chen J. W. Liu C. Wu Y. et al (2022). Performance of passive muscle stiffness in diagnosis and assessment of disease progression in duchenne muscular dystrophy. Ultrasound Med. Biol.48, 414–421. 10.1016/j.ultrasmedbio.2021.09.003
419
Yuan J. Ma Y. Huang T. Chen Y. Peng Y. Li B. et al (2018). Genetic modulation of RNA splicing with a CRISPR-guided cytidine deaminase. Mol. Cell72, 380–394. 10.1016/j.molcel.2018.09.002
420
Yuasa K. Miyagoe Y. Yamamoto K. Nabeshima Y. Dickson G. Takeda S. (1998). Effective restoration of dystrophin-associated proteins in vivo by adenovirus-mediated transfer of truncated dystrophin cDNAs. FEBS Lett.425, 329–336. 10.1016/s0014-5793(98)00251-8
421
Yue Y. Ghosh A. Long C. Bostick B. Smith B. F. Kornegay J. N. et al (2008). A single intravenous injection of adeno-associated virus serotype-9 leads to whole body skeletal muscle transduction in dogs. Mol. Ther.16, 1944–1952. 10.1038/mt.2008.207
422
Zhang Y. Long C. Li H. McAnally J. R. Baskin K. K. Shelton J. M. et al (2017). CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv.3, e1602814. 10.1126/sciadv.1602814
423
Zhang Y. Nishiyama T. Olson E. N. Bassel-Duby R. (2021). CRISPR/Cas correction of muscular dystrophies. Exp. Cell Res.408, 112844. 10.1016/j.yexcr.2021.112844
424
Zhao Q. D. Viswanadhapalli S. Williams P. Shi Q. Tan C. Yi X. et al (2015). NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFκB signaling pathways. Circulation131, 643–655. 10.1161/CIRCULATIONAHA.114.011079
425
Zhou L. Porter J. D. Cheng G. Gong B. Hatala D. A. Merriam A. P. et al (2006). Temporal and spatial mRNA expression patterns of TGF-beta1, 2, 3 and TbetaRI, II, III in skeletal muscles of mdx mice. Neuromuscul. Disord.16, 32–38. 10.1016/j.nmd.2005.09.009
426
Zhu P. Wu F. Mosenson J. Zhang H. He T. C. Wu W. S. (2017). CRISPR/Cas9-Mediated genome editing corrects dystrophin mutation in skeletal muscle stem cells in a mouse model of muscle dystrophy. Mol. Ther. Nucleic Acids7, 31–41. 10.1016/j.omtn.2017.02.007
427
Zhu X. Hadhazy M. Wehling M. Tidball J. G. (2000). Dominant negative myostatin produces hypertrophy without hyperplasia in muscle. FEBS Lett.474, 71–75. 10.1016/s0014-5793(00)01570-2
428
Zincarelli C. Soltys S. Rengo G. Rabinowitz J. E. (2008). Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther.16, 1073–1080. 10.1038/mt.2008.76
429
Zuris J. A. Thompson D. B. Shu Y. Guilinger J. P. Bessen J. L. Hu J. H. et al (2015). Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol.33, 73–80. 10.1038/nbt.3081
430
Wagner K. R. Kuntz N. L. Koenig E. East L. Upadhyay S. Shieh P. B. (2021). Safety, tolerability, and pharmacokinetics of casimersen in patients with Duchenne muscular dystrophy amenable to exon dose-titration trial. Muscle Nerve.64, 285–292. 10.1002/mus.27347
Summary
Keywords
muscle disease, therapeutic strategies, skeletal muscle, dystrophin, Duchenne muscular dystrophy, pathophysiology
Citation
Bez Batti Angulski A, Hosny N, Cohen H, Martin AA, Hahn D, Bauer J and Metzger JM (2023) Duchenne muscular dystrophy: disease mechanism and therapeutic strategies. Front. Physiol. 14:1183101. doi: 10.3389/fphys.2023.1183101
Received
09 March 2023
Accepted
24 May 2023
Published
26 June 2023
Volume
14 - 2023
Edited by
Masatoshi Suzuki, University of Wisconsin-Madison, United States
Reviewed by
Mai ElMallah, Duke University, United States
Ornella Cappellari, University of Bari Aldo Moro, Italy
Updates
Copyright
© 2023 Bez Batti Angulski, Hosny, Cohen, Martin, Hahn, Bauer and Metzger.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph M. Metzger, metzgerj@umn.edu
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.