 Di Zhan1,2
Di Zhan1,2 Christopher Y. Park1,2*
Christopher Y. Park1,2*- 1Department of Pathology, New York University Grossman School of Medicine, New York, NY, United States
- 2Perlmutter Cancer Center, New York University Grossman School of Medicine, New York, NY, United States
The myelodysplastic syndromes (MDS) represent a group of clonal disorders characterized by ineffective hematopoiesis, resulting in peripheral cytopenias and frequent transformation to acute myeloid leukemia (AML). We and others have demonstrated that MDS arises in, and is propagated by malignant stem cells (MDS-SCs), that arise due to the sequential acquisition of genetic and epigenetic alterations in normal hematopoietic stem cells (HSCs). This review focuses on recent advancements in the cellular and molecular characterization of MDS-SCs, as well as their role in mediating MDS clinical outcomes. In addition to discussing the cell surface proteins aberrantly upregulated on MDS-SCs that have allowed the identification and prospective isolation of MDS-SCs, we will discuss the recurrent cytogenetic abnormalities and genetic mutations present in MDS-SCs and their roles in initiating disease, including recent studies demonstrating patterns of clonal evolution and disease progression from pre-malignant HSCs to MDS-SCs. We also will discuss the pathways that have been described as drivers or promoters of disease, including hyperactivated innate immune signaling, and how the identification of these alterations in MDS-SC have led to investigations of novel therapeutic strategies to treat MDS. It is important to note that despite our increasing understanding of the pathogenesis of MDS, the molecular mechanisms that drive responses to therapy remain poorly understood, especially the mechanisms that underlie and distinguish hematologic improvement from reductions in blast burden. Ultimately, such distinctions will be required in order to determine the shared and/or unique molecular mechanisms that drive ineffective hematopoiesis, MDS-SC maintenance, and leukemic transformation.
Introduction
The myelodysplastic syndromes (MDS) are a group of clonal myeloid disorder characterized by ineffective hematopoiesis, disordered hematopoiesis evidenced by morphologic dysplasia, and frequent progression to acute myeloid leukemia (AML). Chemotherapy, stem cell transplantation, hypomethylating agents (HMAs) including decitabine (DAC) and azacytidine (AZA), and immunomodulatory drugs such as lenalidomide are used to treat MDS patients (Platzbecker, 2019). Although these treatment strategies can induce clinical remissions accompanied by improvement in hematologic parameters, all patients will eventually become refractory, and relapse occurs in all MDS patients in the absence of curative therapy, which is currently limited to allogeneic transplantation. It has been proposed that MDS is a disease of stem cells and that MDS-SCs persist during therapy and expanded at the time of relapse. The evolution of stem cell clones from the pre-malignant to malignant state is recognized to play a critical role in MDS pathogenesis and disease progression. This review will summarize the alterations in surface proteins and signaling pathways in MDS-SCs, as well as clonal evolution patterns in MDS-SCs and emerging therapies that target them.
Hematopoietic Stem and Progenitor Cell Alterations in MDS
MDS can be classified into different clinical risk categories based on hematopoietic features, age, and cytogenetic/genetic information at diagnosis. The International Prognostic Scoring System (IPSS) divides MDS into low-risk (Low and Intermediate-1 [Int-1]) and high-risk (Int-2 and High) disease, with high-risk MDS associated with higher blast counts, increased incidence of leukemic transformation, and poor clinical outcome (Greenberg et al., 2012; Greenberg et al., 1997). Studies of low-risk MDS showed no increase in the frequency of immunophenotypically defined HSCs (Lin-CD34+CD38-CD90+CD45RA-) (Majeti et al., 2007; Pang et al., 2013), although multiple studies observed loss of granulocyte-macrophage progenitors (GMPs, Lin−CD34+CD38+CD123+CD45RA+) and relative expansion of common myeloid progenitors (CMPs, Lin−CD34+CD38+CD123+CD45RA−) in the bone marrow (BM) of low-risk MDS patients. However, in high-risk MDS [i.e., refractory anemia with excess blasts (RAEB)], GMP frequency was increased compared to healthy individuals (Pang et al., 2013; Will et al., 2012). These studies underscore that MDS with lower and higher blast counts likely represent distinct biologic entities characterized by unique alterations in HSPC number and frequency (Figure 1). Intriguingly, Will et al., showed that phenotypically primitive long-term HSCs (LIN-CD34+CD38-CD90+) in MDS are expanded in higher-risk cases, suggesting that alterations in HSC function are required prior to the accumulation of blasts (Figure 1; Will et al., 2012). Reduction of megakaryocyte-erythroid progenitors (MEPs, Lin−CD34+CD38+CD123-CD45RA−) can be observed on both high-risk and low-risk MDS, with a relatively greater reduction in low-risk MDS (Will et al., 2012; Pang et al., 2013), suggesting a differentiation block in the transition from CMPs to MEPs.
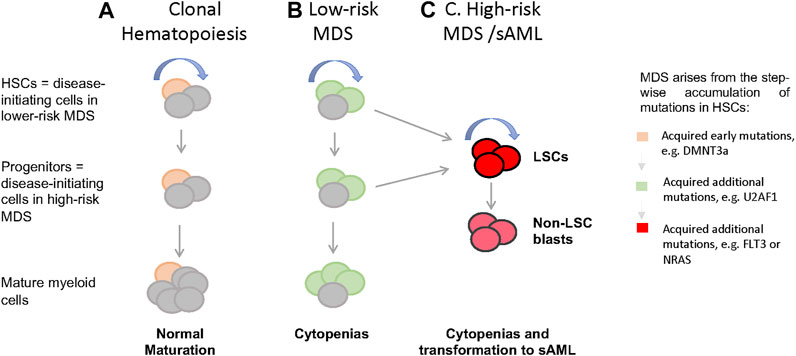
FIGURE 1. Cellular and genetic hierarchies and disease progression in MDS. (A) In healthy individuals, hemotopoiesis is charcterzied as a hierarchy in which self-renewing HSCs give rise to non-self-renewal multipotent and commited progintors and eventually mature cells. When aged HSCs acquire somatic mutations and give rise clones with enhanced self-renewal (yellow), thsi is detectable in the peripheral blood as clonal hematopoiesis (CH), which often characterized by mutations in genes such as DMNT3a, TET, and ASXL1. (B) when HSCs harboring CH associated mutations acquire additional mutations in genes such as U2AF1, SRF2, and SF3B1, this leads to the formation of MSD stem cells, MDS-SCs (green). In low-risk MDS, disease stem cells immunophenotypically resemble normal HSCs, while in high-risk disease with excess blasts, resmbles committed myeloid progenitors. (C) It is presumed that acquisitions of lesions such as FLT3 and NRAS mutations in HSCs or committed proginitors drive the formation of leukemia stem cells (LSCs) and non-self-renewing blasts that accumalate in high-risk MDS and sAML. LSCs immunophenotypically resemble committed progenitors. (curved blue arrow = self-renewal).
MDS is a Hematopoietic Stem Cell Disease
The cancer stem cell hypothesis holds that cancers are initiated and propagated by a rare stem cell population that has the unique ability to self-renew and replenish non-self-renewing malignant cells. Multiple studies have shown that leukemia stem cells (LSCs) in AML immunophenotypically resemble committed progenitor cells such as lymphoid-primed multi-potent progenitors (LMPPs) (Lin-CD34+CD38-CD90-CD45RA+) or GMPs (CD34+CD38+CD123+/loCD110-CD45RA+) (Krivtsov et al., 2006; Goardon et al., 2011; Shlush et al., 2017; van Galen et al., 2019). MDS, in particular lower-risk MDS cases without excess blasts, has been shown to originate in neoplastic HSCs. Earlier studies showed that MDS reconstituting activity is exclusively derived from the CD34+CD38-CD90+ HSC population (Nilsson et al., 2002), demonstrating that MDS initiating cells have a similar phenotype as normal HSCs. Tehranchi et al. evaluated hematopoietic stem and progenitor cell (HSPC) populations in del (5q) MDS patients treated with lenalidomide in remission and relapse (Tehranchi et al., 2010) and identified rare and phenotypically distinct del (5q) MDS-SCs (CD34+CD38−/low CD90+) that are resistant to lenalidomide treatment (Tehranchi et al., 2010). The first xenograft studies demonstrating that HSCs are functional disease-initiating cells in MDS were performed by Pang et al., when they transplanted purified HSCs (LIN-CD34+CD38-CD45RA-CD90+) from MDS samples into immunodeficient mice (Pang et al., 2013). Subsequently Woll et al. demonstrated that HSCs (LIN- CD34+CD38-CD45RA-CD90+), but not CMP, GMP, or MEP, are able to generate long-term cell cultures in vitro and maintain long-term engraftment in immunodeficient mice. Taken together, these studies demonstrate that MDS is a disease that is maintained and propagated by HSCs. In more recent studies exploring the role of the hematopoietic niche in MDS, studies have shown that HSPCs from low-risk MDS patients can reprogram healthy mesenchymal stromal cells (MSCs), and that the reprogrammed MSCs promote MDS HSC engraftment in xenografts (Medyouf et al., 2014). In addition, MDS-SCs from cases with and without excess blasts have been shown to preferentially migrate and engraft into human mesenchymal cell-seeded scaffolds in immunodeficient mice, suggesting that mesenchymal cells interact with MDS-SCs and provide a niche that supports MDS-SCs self-renewal (Mian et al., 2021).
Dysregulated Signaling Pathways in MDS HSCs
Positive regulators of inflammation and innate immunity have been shown to play an important role in promoting MDS pathogenesis. Activation of the p38 MAPK, tumor necrosis factor alpha (TNF-a), and transforming growth factor beta (TGF-b) pathways can promote the expansion of normal HSPC populations while increasing apoptosis in more mature populations (Allampallam et al., 2002; Verma et al., 2002; Saberwal et al., 2003; Navas et al., 2008). HSCs also can be directly activated by pathogen recognition receptor like TLRs or proinflammatory cytokines. Studies have shown that TLRs 1, 2, and 6 are overexpressed in MDS HSPCs (Wei et al., 2013). TLR1 and TLR6 bind to TLR to activate nuclear factor kB (NF-kB), p38 MAPK, and interleukin-1 receptor-associated kinase 1 (IRAK1) signaling pathways in MDS HSPCs (Wei et al., 2013). Overstimulation of these pathways leads to MDS HSPC expansion and a block in erythroid differentiation (Wei et al., 2013). Recent studies also showed IRAK1 is significantly overexpressed in MDS CD34+ cells compared to cord blood CD34+ cells and that IRAK1 phosphorylation leads to TRAF6 and downstream NF-kB activation in MDS HSPCs in both low- and high-risk disease (Rhyasen et al., 2013). In addition, transgenic TRAF6 overexpression in mouse HSPCs leads to an MDS-like phenotype in vivo (Fang et al., 2017; Muto et al., 2020). A recent study showed that the long isoform of interleukin-1 receptor-associated kinase 4 (IRAK4) was the dominant alternatively spliced isoform and highly expressed in CD34+ HSPCs from MDS and AML patients harboring U2AF1 mutations, and inhibition of the long isoform of IRAK4 abrogated disease growth in AML xenografts. These studies indicate that the long isoform of IRAK4 plays a critical role in the activation of chronic innate immune signaling in MDS and AML and thus is a candidate therapeutic target (Smith et al., 2019).
Several other innate immune signaling pathways have been implicated in MDS. The innate immune cytokine IL-8 and its receptor CXCR2 were found to be concomitantly overexpressed in high-risk MDS-SCs and AML LSCs when compared with healthy counterparts, suggesting that an IL-8/CXCR2 autocrine loop promotes MDS-SC and AML LSC function (Schinke et al., 2015). CXCR2 inhibition or downregulation decreased oncogenic MAPK signaling and lead to cell cycle arrest in MDS and AML CD34+/CD38-cells (Schinke et al., 2015). IL1RAP also has been shown to be upregulated in MDS-SCs and AML LSCs and associated with poor clinical outcomes in high-risk MDS and AML (Barreyro et al., 2012). Increased expression of danger-associated molecular pattern (DAMP) molecules S100A8/S100A9 in MDS patients perpetuates noninfectious inflammatory responses that result in the accumulation of Lin- HLA-DR- CD33+ myeloid-derived suppressor cells (MDSCs) in the BM (Chen et al., 2013; Cluzeau et al., 2017; Cheng et al., 2019). Furthermore, S100A9 binds to CD33, which leads to the expansion of MDSCs and suppression of erythroid and myeloid progenitors in a mouse model of MDS (Chen et al., 2013). S100A9 can induce the upregulation of PD-1/PD-L1 expression in MDSCs from S100A9 transgenic mice, and PD-1/PD-L1 blockade restores effective hematopoiesis and improves colony-forming capacity in BM mononuclear cells (MNCs) from MDS patients (Cheng et al., 2019). Collectively, these studies demonstrate the critical role of innate inflammatory pathways in promoting the survival and expansion of MDS-SCs.
One dysregulated non-inflammatory pathway thought to promote MDS HSCs self-renewal is the immunoglobulin-like and endothelial growth factor-like domains 1 (Tie2) pathway. Angiopoietin 1 (ANGPT1) promotes vascular development and angiogenesis by binding to the receptor tyrosine kinase TIE2, which supports normal HSC quiescence and self-renewal (Arai et al., 2004). Recent studies demonstrated that ANGPT-1 is overexpressed in MDS HSPCs and that high ANGPT1 expression is associated with worse clinical outcomes in high-risk MDS and AML patients (Cheng et al., 2011). In addition, Tie2 knockdown and inhibition suppresses leukemic proliferation and enhances hematopoietic differentiation of patient MDS HSPCs (Bachegowda et al., 2016). Another pathway overactivated in MDS HSPCs is the HIF1a signaling pathway. HIF1a protein expression is significantly upregulated in BM MNCs in a subset of MDS patient specimens and high HIF1a expression is associated with worse outcomes in MDS (Tong et al., 2012). Hayashi et al. showed that HIF1a overactivation leads to a MDS-like phenotype in a mouse transgenic model and that HIF1a activation is essential for HSPC expansion in a MLL-PTD mutant mouse model that presents with MDS-associated features (Hayashi et al., 2018).
Defective Ribosome Biogenesis in MDS HSPCs
Defective ribosome biogenesis has been implicated in MDS pathogenesis, as loss of RPS14 expression contributes to del (5q) MDS (Ebert et al., 2008). Similarly, diseases of hematopoiesis resulting in decreased ribosomal protein expression or ribosome assembly – so-called “ribosomopathies” – such as Diamond Blackfan anemia show reductions in mature red cell output, similar to MDS (Ebert et al., 2005; Flygare et al., 2005). Indeed, genetic mouse models deficient in other ribosomal proteins including RPS14, RPS19, and RPS6 exhibited reductions in red cell production (Barlow et al., 2010; Jaako et al., 2011; McGowan et al., 2011). These studies from haploinsufficient genetic mouse models of ribosomal proteins also showed the red cell hypoplasia was dependent on activation of P53 (Barlow et al., 2010; Jaako et al., 2011; McGowan et al., 2011). Taken together, these studies demonstrate that defective ribosome biogenesis likely contributes to the cytopenias observed in low-risk MDS.
Although genetic loss of ribosomal proteins can directly contribute to decreased ribosome formation and MDS-like phenotypes, reduced ribosomal protein gene transcripts have been shown to be common in low-risk MDS. Reduced RPS14 transcripts were described in CD34+ HSPCs from MDS patients without del (5q), defining a subgroup of patients with prolonged survival (Czibere et al., 2009). Other studies showed increased ribosomal protein transcript levels in in CD34+ HSPCs from MDS patients (Sridhar et al., 2009). However, when highly purified MDS HSCs were assessed, global reductions in 21 ribosomal genes were identified, even in patients lacking del (5q) (McGowan et al., 2011), suggesting that ribosomal proteins - and thereby global translation – is reduced in MDS HSCs. We speculate that given the more recent findings that HSCs depend on highly regulated (and low) rates of translation (Signer et al., 2014), decreases in global translation and reduced proteotoxic stress may provide a survival benefit to mutant ribosomal protein HSCs in a cell-intrinsic manner, resulting in their selective advantage.
Genetic Alterations and Clonal Evolution in MDS HSCs
Cytogenetic Abnormalities in MDS HSPCs
Studies utilizing fluorescence in-situ hybridization (FISH) techniques demonstrated the presence of cytogenetic abnormalities in sorted HSPCs from MDS patients (Nilsson et al., 2007; Tehranchi et al., 2010). Studies of del (5q) MDS patients at the time of diagnosis found that the vast majority of CD34+/CD38+ and CD34+/CD38- HSPCs harbor del (5q). This lesion persisted in CD34+/CD38-/CD90+ MDS HSCs despite continuous lenalidomide treatment, consistent with MDS HSCs being relative resistant to therapy and the source of disease maintenance/re-emergence following therapy (Nilsson et al., 2007; Tehranchi et al., 2010). Other studies confirmed that nearly all MDS HSCs harbor cytogenetic alterations such as deletion of chromosome 7 (Will et al., 2012; Pang et al., 2013). Alterations such as trisomy 8, loss of chromosome 7, or deletion of the long arm of chromosome 20 (20q-) also are enriched in MDS HSCs or myeloid progenitors compared to total BM cells or other mature populations such as B-, T- and NK-cells (Miura et al., 2000; Nilsson et al., 2002). Cytogenetically abnormal HSCs also persist through AZA treatment despite achievement of morphologic remission (Will et al., 2012). Despite the fact that many of these cytogenetic abnormalities are considered diagnostic of MDS, these events may not always be “founder” events. For example, Mossner et al. demonstrated that del (5q) is a potential “founder” event in only a minority cases (21.4%) and that cytogenetic lesions including monosomy 7, trisomy 8, and del (5q) can be acquired as late events in MDS (Mossner et al., 2016).
Driver Mutations in MDS HSPCs
A number of large studies have evaluated the presence of somatic mutations in MDS using whole genome- or targeted exome sequencing. Collectively, these studies have demonstrated that MDS shares many driver mutations with other myeloid neoplasms including AML. Genome-wide sequencing analysis of a large cohort of MDS patients showed a mean number of nonsynonymous mutations of approximately 11 per patient (or 0.34/Mb) for all MDS samples with a lower mutation rate in low-risk MDS (0.19/Mb) (Kim et al., 2017). Using targeted sequencing and mass spectrometry–based genotyping, a study of 439 MDS patient BM aspirates demonstrated that 51% of patients had at least one point mutation present at a mutation allele frequency (MAF) of 10% and above, and that mutations in TP53, EZH2, ETV6, RUNX1, and ASXL1 are predictors of poor overall survival in patients, independent of established risk factors such as age, sex and IPSS risk group (Bejar et al., 2011). Papaemmanuil et al. performed targeted sequencing of 111 genes in 738 patients with MDS or closely related neoplasms [e.g., chronic myelomonocytic leukemia (CMML) and mixed MDS-myeloproliferative neoplasms (MPN)] and found that 78% of cases harbored one or more recurrent driver mutations at a MAF ≥10% (Papaemmanuil et al., 2013). Among many categories of somatic mutations, they found that mutations involving RNA spliceosome components including SF3B1, SRSF2, U2AF1 and ZRSR2 occurred most frequently in MDS and were associated with distinct clinical features (Papaemmanuil et al., 2013). More recent studies assessing exome mutations in a cohort of 2,250 MDS patients identified somatic mutations in 10 genes enriched in high-risk MDS, including GATA2, NRAS, KRAS, IDH2, TP53, RUNX1, STAG2, ASXL1, ZRSR2, and TET2, while SF3B1 mutations were almost exclusively found in lower-risk MDS (Makishima et al., 2017). For additional discussion of the mutational spectrum in MDS, we refer the reader to excellent reviews on this subject (Sperling et al., 2017; Ogawa, 2019).
These genetic studies revealed the prevalence and clinical significance of recurrent driver mutations in MDS. Multiple groups have shown that many of the hematologic features of MDS can be partial recapitulated in single-gene genetic mouse models, such as SRSF2, U2AF, SF3B1, and ASXL1 knock-in or knock-out mouse models (Abdel-Wahab et al., 2013; Kim et al., 2015; Shirai et al., 2015; Mupo et al., 2017). However, many of these mouse models require transplantation to elicit physiologically relevant cytopenias, or to generate cytopenias quickly, raising questions about the importance of the microenvironment as well as whether or not any single gene mutation model can truly model MDS.
MDS HSCs and Clonal Evolution
Delineating the hierarchical organization of clones in MDS HSCs is critical to understand mechanisms of disease progression and responses to drug therapy in MDS. Clonal structures and mutation hierarchy are inferred using MAF data, assuming mutations with the highest MAFs are clonal and likely occur early, while mutations with lower MAFs are subclonal and likely occur late in disease pathogenesis. Among different categories of oncogenic mutations, alterations in genes involving RNA splicing components including SF3B1, ZRSF2, SRSF2, and U2AF1 are predicted to represent earlier events that dictate disease evolution and are associated with distinct clinical phenotypes (Papaemmanuil et al., 2013). Another sequencing study found mutations in genes related to DNA methylation and splicing machinery occur earlier during disease evolution while mutations related to signaling pathways expand significantly during progression to secondary AML (sAML) (Kim et al., 2017). Using targeting sequencing of patient and xenografted cells, Mossner et al. evaluated clonal heterogeneity and reconstructed mutational trajectories in BM samples from 54 MDS and CMML patients, including 22 patients covering a cumulative observation time of 75 years (Mossner et al., 2016). They found that mutations in epigenetic modifiers including TET2 and ASXL1, and RNA splicing factors including SF3B1 and SRSF2, are the predominant founder events in MDS, while genes involved in signaling cascades including JAK2 and CBL, transcription factors including RUNX1 and ETV6, and cytogenetic lesions including monosomy 7, trisomy 8, and del (5q), were almost exclusively acquired as late events (Mossner et al., 2016). Other studies showed similar findings (Papaemmanuil et al., 2013; Kim et al., 2017; Makishima et al., 2017). While these studies helped identify the order of mutation acquisition during MDS pathogenesis, they did not resolve which mutation or set of mutations is minimally required for the development of MDS. It is worth noting that mutations in ASXL1, TET2, and DNMT3A, but not RNA spliceosome components, are common in hematopoietic cells from healthy elderly individuals exhibiting clonal hematopoiesis (Genovese et al., 2014; Jaiswal et al., 2014; Xie et al., 2014). It is thus thought that alterations in splicing are required for full manifestation of MDS clinical phenotypes.
Newer sequencing approaches now allow concomitant evaluation of genetic alterations and transcriptomes at the single cell level. Chen et al. performed deep targeted sequencing combined with single-cell sequencing on phenotypically defined malignant stem cells (MDS-SC, AML-SC), premalignant stem cells (pre-MDS-SC, pre-AML-SC), and blast populations in 7 patients with MDS who later progressed to sAML (Chen et al., 2019). They found significantly higher subclonal diversity at the MDS-SC level than in blasts in patients with MDS and sAML. Furthermore, they observed that sAML often developed from a rare subclone contained within the (pre-)MDS-SC pool, and not through further evolution of MDS blasts, indicating a parallel, rather than linear, clonal evolution pattern during MDS progression to sAML (Chen et al., 2019). Thus, this study provided novel insights into MDS disease that were previously unrecognized using bulk-cell sequencing approaches. Unfortunately, these studies did not evaluate genetic evolution or diversity in MDS patients with progressive cytopenias or during treatment.
Therapeutic Targeting of MDS HSCs
Since MDS-SCs are critical for the initiation and propagation of MDS and are the presumed cell that undergoes additional genetic/epigenetic changes to mediate disease progression and relapse, they must be eradicated in order to cure the disease. Not surprisingly, much of the effort to identify therapeutic targets in MDS have focused on high-risk MDS/AML, with relatively few studies directly assessing effects on MDS HSCs in lower-risk disease. This distinction is important to keep in mind when interpreting studies and the potential utility of novel therapeutic strategies in lower-risk MDS. Ideally, therapies in MDS would allow targeting of malignant HSCs without affecting normal HSCs or hematopoiesis. A number of targets have been identified as potential therapeutic targets in MDS.
Multiple studies demonstrated that current therapies including lenalidomide and HMAs treatment are insufficient to eradicate MDS-SCs. Lenalidomide can induce clinical and cytogenetic remissions in MDS patients with del (5q). However, all patients will eventually relapse. A study examined HSPCs from BM specimens obtained from seven MDS patients with del (5q) treated with lenalidomide showed persistence of HSC harboring del (5q), even in patients who achieved cytogenetic remissions and hematologic responses (Tehranchi et al., 2010). AZA treatment leads to increased overall survival in high-risk MDS and was recently been approved for maintenance therapy in AML (Kantarjian et al., 2006; Fenaux et al., 2009; Dombret et al., 2015; Jabbour et al., 2017; Wei et al., 2020). However, responses are difficult to predict, and treatment failure invariably occurs due to the inability of AZA to eliminate MDS-SCs. This was formally demonstrated in a study evaluating LMPP-like and GMP-like populations from 79 patients with high-risk MDS and AML treated with AZA and found that malignant stem cells were never completely eradicated and that expansion of the aberrant pool of HSPCs preceded clinical relapse (Craddock et al., 2013). A similar study of HSCs in lower-risk MDS has not been performed, but presumably MDS HSCs are relatively resistant to therapy, similar to that shown for lenalidomide.
Recently, the combination of AZA plus venetoclax, a BCL2 inhibitor, has been shown to be superior to AZA alone in several large clinical trials, inducing longer overall survival and higher incidence of remission in elderly AML and high-risk MDS patients (DiNardo et al., 2018; Ball et al., 2020; DiNardo et al., 2020; Pollyea et al., 2021). Analyzing LSCs from patients undergoing treatment with venetoclax plus AZA, Pollyea et al. showed that combining venetoclax and AZA eradicates LSC, at least in part, by disruption of the tricarboxylic acid (TCA) cycle and inhibition of electron transport chain complex II (Pollyea et al., 2018).
MDS-SC Antigens and Novel Therapeutics
The ability to distinguish MDS-SCs from normal or preleukemic HSPCs in MDS patients would not only allow more rigorous investigations of MDS-SCs, but also identify potential MDS-SC therapeutic targets. Recent studies have identified surface antigens that are upregulated on MDS-SCs. In lower-risk MDS, the number of cell surface markers shown to be upregulated on HSCs is limited. We demonstrated that CD99/MIC2 is frequently overexpressed in MDS HSCs as well as AML LSC compared to their normal hematopoietic counterparts (Corces-Zimmerman et al., 2014; Chung et al., 2017). In addition, monoclonal antibodies (mAbs) targeting CD99 can induce cell death of MDS HSPCs and AML in vitro, and exhibit antileukemic activity in AML xenografts, without significantly depleting normal HSCs. CD99 may promote MDS-SC and AML LSC self-renewal ability by activating downstream pathways like SRC family kinases (Corces-Zimmerman et al., 2014; Chung et al., 2017). Although the precise functions and mechanisms of action of CD99 in MDS and AML remain to be determined, these studies demonstrate that CD99 is an attractive candidate for targeting stem cells in lower-risk MDS as well as AML.
Surface Markers in High-Risk MDS/AML
Several surface antigens have been proposed as MDS-therapeutic targets, primarily in the context of high-risk MDS/AML.
IL1RAP (IL1R3) is overexpressed on HSPCs in high-risk MDS, but not in low-risk MDS. IL1RAP expression is positively correlated with overall survival in MDS and AML (Barreyro et al., 2012), and antibodies targeting IL1RAP showed therapeutic efficacy in xenograft models of AML (Askmyr et al., 2013; Ågerstam et al., 2015). Mechanistic studies have shown that IL1RAP function is not restricted to the IL-1 receptor pathway, but also mediates signaling and pro-proliferative effects through FLT3 and c-KIT signaling in AML LSCs (Mitchell et al., 2018).
High levels of CD123 (IL3-R alpha chain) expression in immature HSPCs (CD34+/CD38−) has been reported in high-risk, but not lower-risk MDS (Xie et al., 2010; Li et al., 2014). CD123 is also used to distinguish AML LSCs from normal HSCs and thus represent a desirable therapeutic target in AML (Jordan et al., 2000; Jin et al., 2009). CD123+ MDS-SCs also appear to exhibit a unique metabolic signature enriched in oxidative phosphorylation that is associated with stem cell self-renewal and survival in high-risk MDS (Stevens et al., 2018). SL-401, a diphtheria toxin interleukin-3 fusion protein, has been shown to induce cytotoxicity in CD123+ blasts from high-risk MDS and AML patients, and a phase 1/2 clinical trial of SL-401 as single agent therapy showed a predictable and manageable safety profile in patients with myeloid malignancies including high-risk MDS, CMML and blastic plasmacytoid dendritic cell neoplasm (BPDCN) (Alkharabsheh and Frankel, 2019). Another CD123 conjugate, IMGN632, is being evaluated as a single agent therapy as well as in combination with venetoclax and AZA in relapsed/refractory AML (Table 1). CD123-specific chimeric antigen receptor (CAR)-T cells have been developed and have been shown to eliminate LSCs in AML xenografts (Mardiros et al., 2013). MB-102, a CAR T cell therapy, is reported to induce complete responses at low doses in AML and BPDCN without dose-limiting toxicities in a phase I clinical trial (Table 1) (Budde et al., 2017). Although IMGN632 and MB-102 showed promising results in AML and BPDCN, the efficacy of IMGN632 and MB-102 in high-risk MDS needs to be evaluated in future clinical trials.
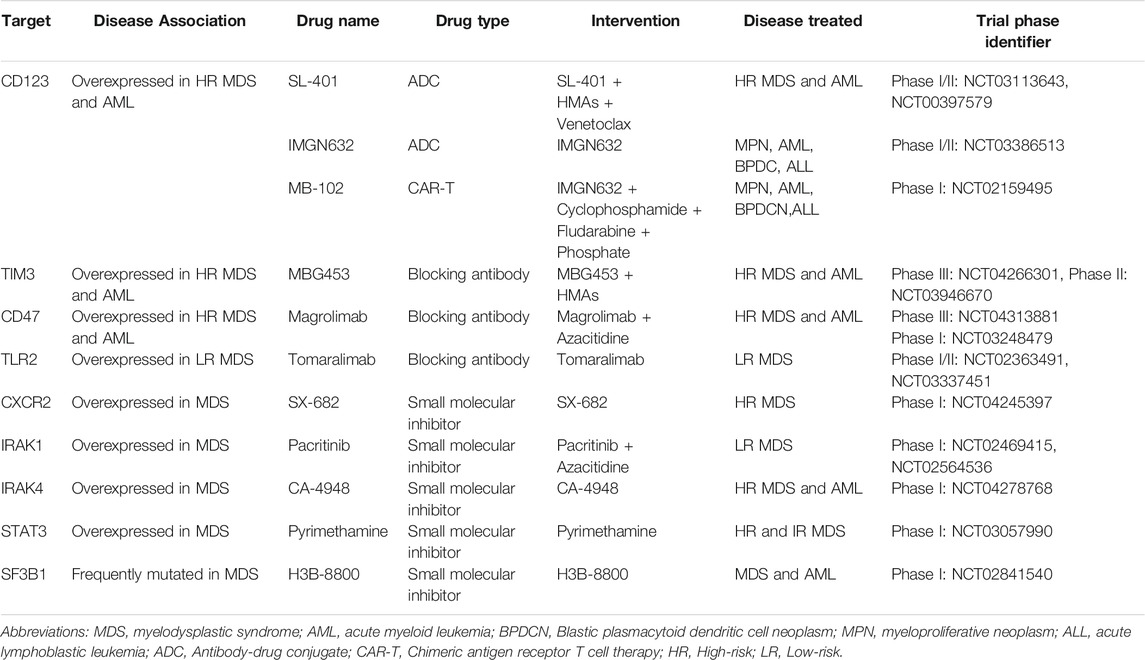
TABLE 1. Therapies targeting MDS-SCs in clinical trials.
CD47 is an antiphagocytic marker that is overexpressed in many human cancers, allowing them to evade immunosurveillance by providing a “don’t eat me” signal (Pang et al., 2013). CD47 is significantly upregulated on committed myeloid progenitors in MDS, which we proposed protects MDS HSPCs from phagocytosis (Pang et al., 2013; Jiang et al., 2013). Thus, CD47 may serve as an important biomarker portending the transition from lower-risk MDS to high-risk MDS (Pang et al., 2013). Magrolimab (previously named 5F9) is a first-in-class antibody that blocks CD47, induces tumor phagocytosis, and eliminates LSCs in AML models (Sallman et al., 2019). AZA synergizes with magrolimab by inducing "eat me" signals to enhance phagocytosis (Sallman et al., 2019). A phase 1b clinical trial of magrolimab plus AZA in high-risk MDS/AML patients demonstrated that this combination shows an excellent safety profile with high response rates in both diseases (Sallman et al., 2019). Registrational clinical trial studies in expansion patient cohorts with high-risk MDS are ongoing (Table 1). Whether or not CD47-directed therapies will show efficacy in the context of low-risk MDS remains uncertain since CD47 upregulation on HSPCs was shown to occur primarily in cases with excess blasts (Pang et al., 2013).
T-cell immunoglobulin mucin-3 (TIM3) is overexpressed on LSCs in AML compared to normal HSPCs. TIM-3 and its ligand, galectin-9 (Gal-9), form an autocrine loop critical for AML LSC self-renewal (Kikushige et al., 2015). In MDS, TIM-3 and Gal-9 have been reported to be overexpressed in high-risk MDS and associated with worse clinical outcomes (Asayama et al., 2017). MBG453, a high-affinity humanized anti-TIM-3 IgG4 antibody, blocks TIM-3 function and is currently being evaluated in clinical trials of patients with high-risk MDS and AML (Table 1) (Borate et al., 2019; Zeidan et al., 2019).
As discussed above, many studies have demonstrated activation of inflammatory and innate immunity pathways in MDS-SCs, and targeting these pathways has shown to be beneficial in preclinical models. Innate immune sensors such as TLR2 are consistently overexpressed in MDS HSPCs from low-risk MDS patients, and high expression of TLR2 is correlated with disease progression. Tomaralimab (OPN-305), an antagonistic IgG4 mAb targeting TLR2, induces differentiation of erythroid cultures (Garcia-Manero et al., 2018). A phase 1 clinical trial of Tomaralimab in low risk/Int-1 MDS patients who previously failed HMA treatment showed good efficacy and safety (Garcia-Manero et al., 2018; Reilly et al., 2013). CXCR2 is an innate immune sensor that closely interacts with IL8 in mediating the activation of innate immunity pathways in malignant stem cells in MDS and AML (Schinke et al., 2015). Several small-molecule antagonists of CXCR2 are currently in development (Table 1) (Ghobrial et al., 2019; Bockorny et al., 2020). Given that IRAK1 and IRAK4 mediate chronic inflammatory signals in MDS, autoimmune disease, and other diseases such as MPNs and diffuse large B-cell lymphoma, there is great interest in IRAKs as therapeutic targets, and several IRAK inhibitors are currently under investigation in early clinical trials (Table 1) (Wiese et al., 2020; Rosenthal et al., 2019). Indeed, a combination of IRAK1 and BCL2 inhibitors has been shown to effectively eliminate MDS clones in xenografts (Rhyasen et al., 2013). Pacritinib is a small molecule that binds to the IRAK1 kinase domain with high-affinity and blocks IRAK1 phosphorylation (Hosseini et al., 2018). Pacritinib reduced AML cell growth in vitro and leukemia burden in AML xenografts (Jeon et al., 2020). IRAK4 inhibitors also are also being investigated in preclinical models (Wiese et al., 2020). Efforts to develop small-molecule inhibitors for other kinases and regulators including PAK1 and STAT3 are also in progress (Table 1) (Semenova and Chernoff, 2017; Yang et al., 2019).
Recent studies have demonstrated that MDS and AML cells bearing mutations in RNA spliceosome components including SF3B1, SRSF2 and U2AF1 are preferentially sensitive to spliceosome inhibition (Fei et al., 2016; Lee et al., 2016; Obeng et al., 2016; Shirai et al., 2017). These findings are presumed to be due to the fact that these cells only possess one functionally intact spliceosome gene allele, and therefore cannot tolerate further reductions in spliceosome function. Small molecule inhibitors including sudemycin, E7107 and H3B-8800 have been developed to target spliceosomal components, and although they are not selective for mutant spliceosome components, have been shown to reduce disease burden in preclinical AML and MDS mouse models (Lee et al., 2016; Shirai et al., 2017; Seiler et al., 2018). E7107, a derivative of the natural product pladienolide B, was investigated in a phase 1 clinical trial in advanced solid tumors, but the clinical trial was discontinued prematurely due to vision loss, which was reported as a major adverse event (Hong et al., 2014). H3B-8800, an orally available small-molecule inhibitor targeting SF3B1, selectively eradicates SF3B1-mutant leukemia cells in xenografts (Seiler et al., 2018) and is currently been investigated in a phase 1/2 clinical trial in AML and MDS (Table 1).
Conclusion
Our understanding of the role of disease-initiating stem cells in MDS has improved tremendously in the past decade. The identification of recurrent somatic mutations in MDS has enabled numerous investigations of specific genes in hematopoiesis, while the evaluation of highly purified MDS-SCs, both in bulk and at the single cell level, has revealed insights into the genetic composition, order of mutational acquisition, mechanisms of disease progression, and mechanisms determining responses to therapy. Improved xenograft models have allowed investigators to interrogate MDS-SC function using patient cells rather than rely solely on mouse or cell line models. Collectively, these studies have demonstrated that dysregulated gene regulation, increased inflammatory signaling, alterations in RNA splicing, and alterations in ribosome assembly/translation are major drivers of MDS HSC function. They also identified cell surface markers including CD123, CD47, TIM3, and CD99 as potential therapeutic targets on MDS-SCs. While much work is still required to develop therapies that can deplete MDS-SCs and induce durable remissions or even cures, there is much reason to be hopeful that such strategies will emerge in the near future. Given the importance of understanding MDS-SC responses to these therapies, we advocate that studies of MDS-SC responses to therapy be performed in conjunction with clinical trials in order to provide the molecular insights necessary to design the next generation of combinatorial therapies. We also encourage investigators to design future studies to distinguish between molecular mechanisms required for SC function in high-risk vs. low-risk MDS, as well as between the types of clinical responses observed (e.g., hematologic improvement vs. blast reductions), in order to better understand the molecular bases of these distinct disease processes.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
CP was supported by NIH Research Project Grant Program R01 CA249054 and R01 CA245502. CP was also supported by a Scholar award from The Leukemia and Lymphoma Society.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdel-Wahab, O., Gao, J., Adli, M., Dey, A., Trimarchi, T., Chung, Y. R., et al. (2013). Deletion of Asxl1 Results in Myelodysplasia and Severe Developmental Defects In Vivo. J. Exp. Med. 210 (12), 2641–2659. doi:10.1084/jem.20131141
Ågerstam, H., Karlsson, C., Hansen, N., Sandén, C., Askmyr, M., von Palffy, S., et al. (2015). Antibodies Targeting Human IL1RAP (IL1R3) Show Therapeutic Effects in Xenograft Models of Acute Myeloid Leukemia. Proc. Natl. Acad. Sci. USA 112 (34), 10786–10791. doi:10.1073/pnas.1422749112
Alkharabsheh, O., and Frankel, A. E. (2019). Clinical Activity and Tolerability of SL-401 (Tagraxofusp): Recombinant Diphtheria Toxin and Interleukin-3 in Hematologic Malignancies. Biomedicines 7 (1). doi:10.3390/biomedicines7010006
Allampallam, K., Shetty, V., Mundle, S., Dutt, D., Kravitz, H., Reddy, P. L., et al. (2002). Biological Significance of Proliferation, Apoptosis, Cytokines, and Monocyte/macrophage Cells in Bone Marrow Biopsies of 145 Patients with Myelodysplastic Syndrome. Int. J. Hematol. 75 (3), 289–297. doi:10.1007/bf02982044
Arai, F., Hirao, A., Ohmura, M., Sato, H., Matsuoka, S., Takubo, K., et al. (2004). Tie2/angiopoietin-1 Signaling Regulates Hematopoietic Stem Cell Quiescence in the Bone Marrow Niche. Cell 118 (2), 149–161. doi:10.1016/j.cell.2004.07.004
Asayama, T., Tamura, H., Ishibashi, M., Kuribayashi-Hamada, Y., Onodera-Kondo, A., Okuyama, N., et al. (2017). Functional Expression of Tim-3 on Blasts and Clinical Impact of its Ligand Galectin-9 in Myelodysplastic Syndromes. Oncotarget 8 (51), 88904–88917. doi:10.18632/oncotarget.21492
Askmyr, M., Ågerstam, H., Hansen, N., Gordon, S., Arvanitakis, A., Rissler, M., et al. (2013). Selective Killing of Candidate AML Stem Cells by Antibody Targeting of IL1RAP. Blood 121 (18), 3709–3713. doi:10.1182/blood-2012-09-458935
Bachegowda, L., Morrone, K., Winski, S. L., Mantzaris, I., Bartenstein, M., Ramachandra, N., et al. (2016). Pexmetinib: A Novel Dual Inhibitor of Tie2 and P38 MAPK with Efficacy in Preclinical Models of Myelodysplastic Syndromes and Acute Myeloid Leukemia. Cancer Res. 76 (16), 4841–4849. doi:10.1158/0008-5472.can-15-3062
Ball, B. J., Famulare, C. A., Stein, E. M., Tallman, M. S., Derkach, A., Roshal, M., et al. (2020). Venetoclax and Hypomethylating Agents (HMAs) Induce High Response Rates in MDS, Including Patients after HMA Therapy Failure. Blood Adv. 4 (13), 2866–2870. doi:10.1182/bloodadvances.2020001482
Barlow, J. L., Drynan, L. F., Hewett, D. R., Holmes, L. R., Lorenzo-Abalde, S., Lane, A. L., et al. (2010). A P53-dependent Mechanism Underlies Macrocytic Anemia in a Mouse Model of Human 5q- Syndrome. Nat. Med. 16 (1), 59–66. doi:10.1038/nm.2063
Barreyro, L., Will, B., Bartholdy, B., Zhou, L., Todorova, T. I., Stanley, R. F., et al. (2012). Overexpression of IL-1 Receptor Accessory Protein in Stem and Progenitor Cells and Outcome Correlation in AML and MDS. Blood 120 (6), 1290–1298. doi:10.1182/blood-2012-01-404699
Bejar, R., Stevenson, K., Abdel-Wahab, O., Galili, N., Nilsson, B., Garcia-Manero, G., et al. (2011). Clinical Effect of point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 364 (26), 2496–2506. doi:10.1056/nejmoa1013343
Bockorny, B., Semenisty, V., Macarulla, T., Borazanci, E., Wolpin, B. M., Stemmer, S. M., et al. (2020). BL-8040, a CXCR4 Antagonist, in Combination with Pembrolizumab and Chemotherapy for Pancreatic Cancer: the COMBAT Trial. Nat. Med. 26 (6), 878–885. doi:10.1038/s41591-020-0880-x
Borate, U., Esteve, J., Porkka, K., Knapper, S., Vey, N., Scholl, S., et al. (2019). Phase Ib Study of the Anti-TIM-3 Antibody MBG453 in Combination with Decitabine in Patients with High-Risk Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML). Blood 134 (Suppl. ment_1), 570. doi:10.1182/blood-2019-128178
Budde, L., Song, J. Y., Kim, Y., Blanchard, S., Wagner, J., Stein, A. S., et al. (2017). Remissions of Acute Myeloid Leukemia and Blastic Plasmacytoid Dendritic Cell Neoplasm Following Treatment with CD123-specific CAR T Cells: A First-In-Human Clinical Trial. Blood 130 (Suppl. l_1), 811. doi:10.1182/blood.v130.suppl_1.811.811
Chen, J., Kao, Y.-R., Sun, D., Todorova, T. I., Reynolds, D., Narayanagari, S.-R., et al. (2019). Myelodysplastic Syndrome Progression to Acute Myeloid Leukemia at the Stem Cell Level. Nat. Med. 25 (1), 103–110. doi:10.1038/s41591-018-0267-4
Chen, X., Eksioglu, E. A., Zhou, J., Zhang, L., Djeu, J., Fortenbery, N., et al. (2013). Induction of Myelodysplasia by Myeloid-Derived Suppressor Cells. J. Clin. Invest. 123 (11), 4595–4611. doi:10.1172/jci67580
Cheng, C.-L., Hou, H.-A., Jhuang, J.-Y., Lin, C.-W., Chen, C.-Y., Tang, J.-L., et al. (2011). High Bone Marrow Angiopoietin-1 Expression Is an Independent Poor Prognostic Factor for Survival in Patients with Myelodysplastic Syndromes. Br. J. Cancer 105 (7), 975–982. doi:10.1038/bjc.2011.340
Cheng, P., Eksioglu, E. A., Chen, X., Kandell, W., Le Trinh, T., Cen, L., et al. (2019). S100A9-induced Overexpression of PD-1/pd-L1 Contributes to Ineffective Hematopoiesis in Myelodysplastic Syndromes. Leukemia 33 (8), 2034–2046. doi:10.1038/s41375-019-0397-9
Chung, S. S., Eng, W. S., Hu, W., Khalaj, M., Garrett-Bakelman, F. E., Tavakkoli, M., et al. (2017). CD99 Is a Therapeutic Target on Disease Stem Cells in Myeloid Malignancies. Sci. Transl Med. 9 (374). doi:10.1126/scitranslmed.aaj2025
Cluzeau, T., McGraw, K. L., Irvine, B., Masala, E., Ades, L., Basiorka, A. A., et al. (2017). Pro-inflammatory Proteins S100A9 and Tumor Necrosis Factor-α Suppress Erythropoietin Elaboration in Myelodysplastic Syndromes. Haematologica 102 (12), 2015–2020. doi:10.3324/haematol.2016.158857
Corces-Zimmerman, M. R., Hong, W.-J., Weissman, I. L., Medeiros, B. C., and Majeti, R. (2014). Preleukemic Mutations in Human Acute Myeloid Leukemia Affect Epigenetic Regulators and Persist in Remission. Proc. Natl. Acad. Sci. 111 (7), 2548–2553. doi:10.1073/pnas.1324297111
Craddock, C., Quek, L., Goardon, N., Freeman, S., Siddique, S., Raghavan, M., et al. (2013). Azacitidine Fails to Eradicate Leukemic Stem/progenitor Cell Populations in Patients with Acute Myeloid Leukemia and Myelodysplasia. Leukemia 27 (5), 1028–1036. doi:10.1038/leu.2012.312
Czibere, A., Bruns, I., Junge, B., Singh, R., Kobbe, G., Haas, R., et al. (2009). Low RPS14 Expression Is Common in Myelodysplastic Syndromes without 5q- Aberration and Defines a Subgroup of Patients with Prolonged Survival. Haematologica 94 (10), 1453–1455. doi:10.3324/haematol.2009.008508
DiNardo, C. D., Jonas, B. A., Pullarkat, V., Thirman, M. J., Garcia, J. S., Wei, A. H., et al. (2020). Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 383 (7), 617–629. doi:10.1056/nejmoa2012971
DiNardo, C. D., Pratz, K. W., Letai, A., Jonas, B. A., Wei, A. H., Thirman, M., et al. (2018). Safety and Preliminary Efficacy of Venetoclax with Decitabine or Azacitidine in Elderly Patients with Previously Untreated Acute Myeloid Leukaemia: a Non-randomised, Open-Label, Phase 1b Study. Lancet Oncol. 19 (2), 216–228. doi:10.1016/s1470-2045(18)30010-x
Dombret, H., Seymour, J. F., Butrym, A., Wierzbowska, A., Selleslag, D., Jang, J. H., et al. (2015). International Phase 3 Study of Azacitidine vs Conventional Care Regimens in Older Patients with Newly Diagnosed AML with >30% Blasts. Blood 126 (3), 291–299. doi:10.1182/blood-2015-01-621664
Ebert, B. L., Lee, M. M., Pretz, J. L., Subramanian, A., Mak, R., Golub, T. R., et al. (2005). An RNA Interference Model of RPS19 Deficiency in Diamond-Blackfan Anemia Recapitulates Defective Hematopoiesis and rescue by Dexamethasone: Identification of Dexamethasone-Responsive Genes by Microarray. Blood 105 (12), 4620–4626. doi:10.1182/blood-2004-08-3313
Ebert, B. L., Pretz, J., Bosco, J., Chang, C. Y., Tamayo, P., Galili, N., et al. (2008). Identification of RPS14 as a 5q- Syndrome Gene by RNA Interference Screen. Nature 451 (7176), 335–339. doi:10.1038/nature06494
Fang, J., Bolanos, L. C., Choi, K., Liu, X., Christie, S., Akunuru, S., et al. (2017). Ubiquitination of hnRNPA1 by TRAF6 Links Chronic Innate Immune Signaling with Myelodysplasia. Nat. Immunol. 18 (2), 236–245. doi:10.1038/ni.3654
Fei, D. L., Motowski, H., Chatrikhi, R., Prasad, S., Yu, J., Gao, S., et al. (2016). Wild-Type U2AF1 Antagonizes the Splicing Program Characteristic of U2AF1-Mutant Tumors and Is Required for Cell Survival. Plos Genet. 12 (10), e1006384. doi:10.1371/journal.pgen.1006384
Fenaux, P., Mufti, G. J., Hellstrom-Lindberg, E., Santini, V., Finelli, C., Giagounidis, A., et al. (2009). Efficacy of Azacitidine Compared with that of Conventional Care Regimens in the Treatment of Higher-Risk Myelodysplastic Syndromes: a Randomised, Open-Label, Phase III Study. Lancet Oncol. 10 (3), 223–232. doi:10.1016/s1470-2045(09)70003-8
Flygare, J., Kiefer, T., Miyake, K., Utsugisawa, T., Hamaguchi, I., DaCosta, L., et al. (2005). Deficiency of Ribosomal Protein S19 in CD34+ Cells Generated by siRNA Blocks Erythroid Development and Mimics Defects Seen in Diamond-Blackfan Anemia. Blood 105 (12), 4627–4634. doi:10.1182/blood-2004-08-3115
Garcia-Manero, G., Jabbour, E. J., Konopleva, M. Y., Daver, N. G., Borthakur, G., DiNardo, C. D., et al. (2018). A Clinical Study of Tomaralimab (OPN-305), a Toll-like Receptor 2 (TLR-2) Antibody, in Heavily Pre-treated Transfusion Dependent Patients with Lower Risk Myelodysplastic Syndromes (MDS) that Have Received and Failed on Prior Hypomethylating Agent (HMA) Therapy. Blood 132 (Suppl. 1), 798. doi:10.1182/blood-2018-99-119805
Genovese, G., Kähler, A. K., Handsaker, R. E., Lindberg, J., Rose, S. A., Bakhoum, S. F., et al. (2014). Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 371 (26), 2477–2487. doi:10.1056/nejmoa1409405
Ghobrial, I. M., Liu, C. J., Zavidij, O., Azab, A. K., Baz, R., Laubach, J. P., et al. (2019). Phase I/II Trial of the CXCR4 Inhibitor Plerixafor in Combination with Bortezomib as a Chemosensitization Strategy in Relapsed/refractory Multiple Myeloma. Am. J. Hematol. 94 (11), 1244–1253. doi:10.1002/ajh.25627
Goardon, N., Marchi, E., Atzberger, A., Quek, L., Schuh, A., Soneji, S., et al. (2011). Coexistence of LMPP-like and GMP-like Leukemia Stem Cells in Acute Myeloid Leukemia. Cancer Cell 19 (1), 138–152. doi:10.1016/j.ccr.2010.12.012
Greenberg, P., Cox, C., LeBeau, M. M., Fenaux, P., Morel, P., Sanz, G., et al. (1997). International Scoring System for Evaluating Prognosis in Myelodysplastic Syndromes. Blood 89 (6), 2079–2088. doi:10.1182/blood.v89.6.2079
Greenberg, P. L., Tuechler, H., Schanz, J., Sanz, G., Garcia-Manero, G., Solé, F., et al. (2012). Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 120 (12), 2454–2465. doi:10.1182/blood-2012-03-420489
Hayashi, Y., Zhang, Y., Yokota, A., Yan, X., Liu, J., Choi, K., et al. (2018). Pathobiological Pseudohypoxia as a Putative Mechanism Underlying Myelodysplastic Syndromes. Cancer Discov. 8 (11), 1438–1457. doi:10.1158/2159-8290.cd-17-1203
Hong, D. S., Kurzrock, R., Naing, A., Wheler, J. J., Falchook, G. S., Schiffman, J. S., et al. (2014). A Phase I, Open-Label, Single-Arm, Dose-Escalation Study of E7107, a Precursor Messenger Ribonucleic Acid (pre-mRNA) Splicesome Inhibitor Administered Intravenously on Days 1 and 8 Every 21 Days to Patients with Solid Tumors. Invest. New Drugs 32 (3), 436–444. doi:10.1007/s10637-013-0046-5
Hosseini, M. M., Kurtz, S. E., Abdelhamed, S., Mahmood, S., Davare, M. A., Kaempf, A., et al. (2018). Inhibition of Interleukin-1 Receptor-Associated Kinase-1 Is a Therapeutic Strategy for Acute Myeloid Leukemia Subtypes. Leukemia 32 (11), 2374–2387. doi:10.1038/s41375-018-0112-2
Jaako, P., Flygare, J., Olsson, K., Quere, R., Ehinger, M., Henson, A., et al. (2011). Mice with Ribosomal Protein S19 Deficiency Develop Bone Marrow Failure and Symptoms like Patients with Diamond-Blackfan Anemia. Blood 118 (23), 6087–6096. doi:10.1182/blood-2011-08-371963
Jabbour, E., Short, N. J., Montalban-Bravo, G., Huang, X., Bueso-Ramos, C., Qiao, W., et al. (2017). Randomized Phase 2 Study of Low-Dose Decitabine vs Low-Dose Azacitidine in Lower-Risk MDS and MDS/MPN. Blood 130 (13), 1514–1522. doi:10.1182/blood-2017-06-788497
Jaiswal, S., Fontanillas, P., Flannick, J., Manning, A., Grauman, P. V., Mar, B. G., et al. (2014). Age-related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 371 (26), 2488–2498. doi:10.1056/nejmoa1408617
Jeon, J. Y., Zhao, Q., Buelow, D. R., Phelps, M., Walker, A. R., Mims, A. S., et al. (2020). Preclinical Activity and a Pilot Phase I Study of Pacritinib, an Oral JAK2/FLT3 Inhibitor, and Chemotherapy in FLT3-ITD-Positive AML. Invest. New Drugs 38 (2), 340–349. doi:10.1007/s10637-019-00786-4
Jiang, H., Fu, R., Wang, H., Li, L., Liu, H., and Shao, Z. (2013). CD47 Is Expressed Abnormally on Hematopoietic Cells in Myelodysplastic Syndrome. Leuk. Res. 37 (8), 907–910. doi:10.1016/j.leukres.2013.04.008
Jin, L., Lee, E. M., Ramshaw, H. S., Busfield, S. J., Peoppl, A. G., Wilkinson, L., et al. (2009). Monoclonal Antibody-Mediated Targeting of CD123, IL-3 Receptor α Chain, Eliminates Human Acute Myeloid Leukemic Stem Cells. Cell Stem Cell 5 (1), 31–42. doi:10.1016/j.stem.2009.04.018
Jordan, C., Upchurch, D., Szilvassy, S., Guzman, M., Howard, D., Pettigrew, A., et al. (2000). The Interleukin-3 Receptor Alpha Chain Is a Unique Marker for Human Acute Myelogenous Leukemia Stem Cells. Leukemia 14 (10), 1777–1784. doi:10.1038/sj.leu.2401903
Kantarjian, H., Issa, J.-P. J., Rosenfeld, C. S., Bennett, J. M., Albitar, M., DiPersio, J., et al. (2006). Decitabine Improves Patient Outcomes in Myelodysplastic Syndromes. Cancer 106 (8), 1794–1803. doi:10.1002/cncr.21792
Kikushige, Y., Miyamoto, T., Yuda, J., Jabbarzadeh-Tabrizi, S., Shima, T., Takayanagi, S.-i., et al. (2015). A TIM-3/Gal-9 Autocrine Stimulatory Loop Drives Self-Renewal of Human Myeloid Leukemia Stem Cells and Leukemic Progression. Cell Stem Cell 17 (3), 341–352. doi:10.1016/j.stem.2015.07.011
Kim, E., Ilagan, J. O., Liang, Y., Daubner, G. M., Lee, S. C.-W., Ramakrishnan, A., et al. (2015). SRSF2 Mutations Contribute to Myelodysplasia by Mutant-specific Effects on Exon Recognition. Cancer Cell 27 (5), 617–630. doi:10.1016/j.ccell.2015.04.006
Kim, T., Tyndel, M. S., Kim, H. J., Ahn, J.-S., Choi, S. H., Park, H. J., et al. (2017). The Clonal Origins of Leukemic Progression of Myelodysplasia. Leukemia 31 (9), 1928–1935. doi:10.1038/leu.2017.17
Krivtsov, A. V., Twomey, D., Feng, Z., Stubbs, M. C., Wang, Y., Faber, J., et al. (2006). Transformation from Committed Progenitor to Leukaemia Stem Cell Initiated by MLL-AF9. Nature 442 (7104), 818–822. doi:10.1038/nature04980
Lee, S. C.-W., Dvinge, H., Kim, E., Cho, H., Micol, J.-B., Chung, Y. R., et al. (2016). Modulation of Splicing Catalysis for Therapeutic Targeting of Leukemia with Mutations in Genes Encoding Spliceosomal Proteins. Nat. Med. 22 (6), 672–678. doi:10.1038/nm.4097
Li, L. J., Tao, J. L., Fu, R., Wang, H. Q., Jiang, H. J., Yue, L. Z., et al. (2014). Increased CD34+CD38−CD123+ Cells in Myelodysplastic Syndrome Displaying Malignant Features Similar to Those in AML. Int. J. Hematol. 100 (1), 60–69. doi:10.1007/s12185-014-1590-2
Majeti, R., Park, C. Y., and Weissman, I. L. (2007). Identification of a Hierarchy of Multipotent Hematopoietic Progenitors in Human Cord Blood. Cell Stem Cell 1 (6), 635–645. doi:10.1016/j.stem.2007.10.001
Makishima, H., Yoshizato, T., Yoshida, K., Sekeres, M. A., Radivoyevitch, T., Suzuki, H., et al. (2017). Dynamics of Clonal Evolution in Myelodysplastic Syndromes. Nat. Genet. 49 (2), 204–212. doi:10.1038/ng.3742
Mardiros, A., Dos Santos, C., McDonald, T., Brown, C. E., Wang, X., Budde, L. E., et al. (2013). T Cells Expressing CD123-specific Chimeric Antigen Receptors Exhibit Specific Cytolytic Effector Functions and Antitumor Effects against Human Acute Myeloid Leukemia. Blood 122 (18), 3138–3148. doi:10.1182/blood-2012-12-474056
McGowan, K. A., Pang, W. W., Bhardwaj, R., Perez, M. G., Pluvinage, J. V., Glader, B. E., et al. (2011). Reduced Ribosomal Protein Gene Dosage and P53 Activation in Low-Risk Myelodysplastic Syndrome. Blood 118 (13), 3622–3633. doi:10.1182/blood-2010-11-318584
Medyouf, H., Mossner, M., Jann, J.-C., Nolte, F., Raffel, S., Herrmann, C., et al. (2014). Myelodysplastic Cells in Patients Reprogram Mesenchymal Stromal Cells to Establish a Transplantable Stem Cell Niche Disease Unit. Cell Stem Cell 14 (6), 824–837. doi:10.1016/j.stem.2014.02.014
Mian, S. A., Abarrategi, A., Kong, K. L., Rouault-Pierre, K., Wood, H., Oedekoven, C. A., et al. (2021). Ectopic Humanized Mesenchymal Niche in Mice Enables Robust Engraftment of Myelodysplastic Stem Cells. Blood Cancer Discov. 2 (2), 135–145. doi:10.1158/2643-3230.bcd-20-0161
Mitchell, K., Barreyro, L., Todorova, T. I., Taylor, S. J., Antony-Debré, I., Narayanagari, S.-R., et al. (2018). IL1RAP Potentiates Multiple Oncogenic Signaling Pathways in AML. J. Exp. Med. 215 (6), 1709–1727. doi:10.1084/jem.20180147
Miura, I., Takahashi, N., Kobayashi, Y., Saito, K., and Miura, A. B. (2000). Molecular Cytogenetics of Stem Cells: Target Cells of Chromosome Aberrations as Revealed by the Application of Fluorescence In Situ Hybridization to Fluorescence-Activated Cell Sorting. Int. J. Hematol. 72 (3), 310–317.
Mossner, M., Jann, J.-C., Wittig, J., Nolte, F., Fey, S., Nowak, V., et al. (2016). Mutational Hierarchies in Myelodysplastic Syndromes Dynamically Adapt and Evolve upon Therapy Response and Failure. Blood 128 (9), 1246–1259. doi:10.1182/blood-2015-11-679167
Mupo, A., Seiler, M., Sathiaseelan, V., Pance, A., Yang, Y., Agrawal, A. A., et al. (2017). Hemopoietic-specific Sf3b1-K700e Knock-In Mice Display the Splicing Defect Seen in Human MDS but Develop Anemia without Ring Sideroblasts. Leukemia 31 (3), 720–727. doi:10.1038/leu.2016.251
Muto, T., Walker, C. S., Choi, K., Hueneman, K., Smith, M. A., Gul, Z., et al. (2020). Adaptive Response to Inflammation Contributes to Sustained Myelopoiesis and Confers a Competitive Advantage in Myelodysplastic Syndrome HSCs. Nat. Immunol. 21 (5), 535–545. doi:10.1038/s41590-020-0663-z
Navas, T., Zhou, L., Estes, M., Haghnazari, E., Nguyen, A. N., Mo, Y., et al. (2008). Inhibition of P38α MAPK Disrupts the Pathological Loop of Proinflammatory Factor Production in the Myelodysplastic Syndrome Bone Marrow Microenvironment. Leuk. Lymphoma 49 (10), 1963–1975. doi:10.1080/10428190802322919
Nilsson, L., Edén, P., Olsson, E., Månsson, R., Åstrand-Grundström, I., Strömbeck, B., et al. (2007). The Molecular Signature of MDS Stem Cells Supports a Stem-Cell Origin of 5q− Myelodysplastic Syndromes. Blood 110 (8), 3005–3014. doi:10.1182/blood-2007-03-079368
Nilsson, L., Åstrand-Grundström, I., Anderson, K., Arvidsson, I., Hokland, P., Bryder, D., et al. (2002). Involvement and Functional Impairment of the CD34+CD38−Thy-1+ Hematopoietic Stem Cell Pool in Myelodysplastic Syndromes with Trisomy 8. Blood 100 (1), 259–267. doi:10.1182/blood-2001-12-0188
Obeng, E. A., Chappell, R. J., Seiler, M., Chen, M. C., Campagna, D. R., Schmidt, P. J., et al. (2016). Physiologic Expression of Sf3b1 K700E Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 30 (3), 404–417. doi:10.1016/j.ccell.2016.08.006
Pang, W. W., Pluvinage, J. V., Price, E. A., Sridhar, K., Arber, D. A., Greenberg, P. L., et al. (2013). Hematopoietic Stem Cell and Progenitor Cell Mechanisms in Myelodysplastic Syndromes. Proc. Natl. Acad. Sci. 110 (8), 3011–3016. doi:10.1073/pnas.1222861110
Papaemmanuil, E., Gerstung, M., Malcovati, L., Tauro, S., Gundem, G., Van Loo, P., et al. (2013). Clinical and Biological Implications of Driver Mutations in Myelodysplastic Syndromes. Blood 122 (22), 3616–3699. doi:10.1182/blood-2013-08-518886
Platzbecker, U. (2019). Treatment of MDS. Blood 133 (10), 1096–1107. doi:10.1182/blood-2018-10-844696
Pollyea, D. A., Pratz, K., Letai, A., Jonas, B. A., Wei, A. H., Pullarkat, V., et al. (2021). Venetoclax with Azacitidine or Decitabine in Patients with Newly Diagnosed Acute Myeloid Leukemia: Long Term Follow‐up from a Phase 1b Study. Am. J. Hematol. 96 (2), 208–217. doi:10.1002/ajh.26039
Pollyea, D. A., Stevens, B. M., Jones, C. L., Winters, A., Pei, S., Minhajuddin, M., et al. (2018). Venetoclax with Azacitidine Disrupts Energy Metabolism and Targets Leukemia Stem Cells in Patients with Acute Myeloid Leukemia. Nat. Med. 24 (12), 1859–1866. doi:10.1038/s41591-018-0233-1
Reilly, M., Miller, R. M., Thomson, M. H., Patris, V., Ryle, P., McLoughlin, L., et al. (2013). Randomized, Double-Blind, Placebo-Controlled, Dose-Escalating Phase I, Healthy Subjects Study of Intravenous OPN-305, a Humanized Anti-TLR2 Antibody. Clin. Pharmacol. Ther. 94 (5), 593–600. doi:10.1038/clpt.2013.150
Rhyasen, G. W., Bolanos, L., Fang, J., Jerez, A., Wunderlich, M., Rigolino, C., et al. (2013). Targeting IRAK1 as a Therapeutic Approach for Myelodysplastic Syndrome. Cancer Cell 24 (1), 90–104. doi:10.1016/j.ccr.2013.05.006
Rosenthal, A. C., Tun, H. W., Younes, A., Nowakowski, G. S., Lunning, M. A., Patel, K., et al. (2019). Phase 1 Study of CA-4948, a Novel Inhibitor of Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) in Patients (Pts) with R/r Non-hodgkin Lymphoma. J. Clin. Oncol. 37 (15_Suppl. l), e19055. doi:10.1200/jco.2019.37.15_suppl.e19055
Saberwal, G., Broderick, E., Janssen, I., Shetty, V., Alvi, S., Lisak, L., et al. (2003). Involvement of Cyclin D1 and E2F1 in Intramedullary Apoptosis in Myelodysplastic Syndromes. J. Hematotherapy Stem Cel Res. 12 (4), 443–450. doi:10.1089/152581603322286079
Sallman, D. A., Asch, A. S., Al Malki, M. M., Lee, D. J., Donnellan, W. B., Marcucci, G., et al. (2019). The First-In-Class Anti-CD47 Antibody Magrolimab (5F9) in Combination with Azacitidine Is Effective in MDS and AML Patients: Ongoing Phase 1b Results. Blood 134 (Suppl. ment_1), 569. doi:10.1182/blood-2019-126271
Schinke, C., Giricz, O., Li, W., Shastri, A., Gordon, S., Barreyro, L., et al. (2015). IL8-CXCR2 Pathway Inhibition as a Therapeutic Strategy against MDS and AML Stem Cells. Blood 125 (20), 3144–3152. doi:10.1182/blood-2015-01-621631
Seiler, M., Yoshimi, A., Darman, R., Chan, B., Keaney, G., Thomas, M., et al. (2018). H3B-8800, an Orally Available Small-Molecule Splicing Modulator, Induces Lethality in Spliceosome-Mutant Cancers. Nat. Med. 24 (4), 497–504. doi:10.1038/nm.4493
Semenova, G., and Chernoff, J. (2017). Targeting PAK1. Biochem. Soc. Trans. 45 (1), 79–88. doi:10.1042/bst20160134
Shirai, C. L., Ley, J. N., White, B. S., Kim, S., Tibbitts, J., Shao, J., et al. (2015). Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell 27 (5), 631–643. doi:10.1016/j.ccell.2015.04.008
Shirai, C. L., White, B. S., Tripathi, M., Tapia, R., Ley, J. N., Ndonwi, M., et al. (2017). Mutant U2AF1-Expressing Cells Are Sensitive to Pharmacological Modulation of the Spliceosome. Nat. Commun. 8, 14060. doi:10.1038/ncomms14060
Shlush, L. I., Mitchell, A., Heisler, L., Abelson, S., Ng, S. W. K., Trotman-Grant, A., et al. (2017). Tracing the Origins of Relapse in Acute Myeloid Leukaemia to Stem Cells. Nature 547 (7661), 104–108. doi:10.1038/nature22993
Signer, R. A. J., Magee, J. A., Salic, A., and Morrison, S. J. (2014). Haematopoietic Stem Cells Require a Highly Regulated Protein Synthesis Rate. Nature 509 (7498), 49–54. doi:10.1038/nature13035
Smith, M. A., Choudhary, G. S., Pellagatti, A., Choi, K., Bolanos, L. C., Bhagat, T. D., et al. (2019). U2AF1 Mutations Induce Oncogenic IRAK4 Isoforms and Activate Innate Immune Pathways in Myeloid Malignancies. Nat. Cel Biol 21 (5), 640–650. doi:10.1038/s41556-019-0314-5
Sperling, A. S., Gibson, C. J., and Ebert, B. L. (2017). The Genetics of Myelodysplastic Syndrome: from Clonal Haematopoiesis to Secondary Leukaemia. Nat. Rev. Cancer 17 (1), 5–19. doi:10.1038/nrc.2016.112
Sridhar, K., Ross, D. T., Tibshirani, R., Butte, A. J., and Greenberg, P. L. (2009). Relationship of Differential Gene Expression Profiles in CD34+ Myelodysplastic Syndrome Marrow Cells to Disease Subtype and Progression. Blood 114 (23), 4847–4858. doi:10.1182/blood-2009-08-236422
Stevens, B. M., Khan, N., D'Alessandro, A., Nemkov, T., Winters, A., Jones, C. L., et al. (2018). Characterization and Targeting of Malignant Stem Cells in Patients with Advanced Myelodysplastic Syndromes. Nat. Commun. 9 (1), 3694. doi:10.1038/s41467-018-05984-x
Tehranchi, R., Woll, P. S., Anderson, K., Buza-Vidas, N., Mizukami, T., Mead, A. J., et al. (2010). Persistent malignant stem cells in del(5q) myelodysplasia in remission. N. Engl. J. Med. 363 (11), 1025–1037. doi:10.1056/nejmoa0912228
Tong, H., Hu, C., Zhuang, Z., Wang, L., and Jin, J. (2012). Hypoxia-inducible Factor-1α Expression Indicates Poor Prognosis in Myelodysplastic Syndromes. Leuk. Lymphoma 53 (12), 2412–2418. doi:10.3109/10428194.2012.696637
van Galen, P., Hovestadt, V., Wadsworth II, M. H., Hughes, T. K., Griffin, G. K., Battaglia, S., et al. (2019). Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 176 (6), 1265–1281. doi:10.1016/j.cell.2019.01.031
Verma, A., Deb, D. K., Sassano, A., Uddin, S., Varga, J., Wickrema, A., et al. (2002). Activation of the P38 Mitogen-Activated Protein Kinase Mediates the Suppressive Effects of Type I Interferons and Transforming Growth Factor-β on Normal Hematopoiesis. J. Biol. Chem. 277 (10), 7726–7735. doi:10.1074/jbc.m106640200
Wei, A. H., Döhner, H., Pocock, C., Montesinos, P., Afanasyev, B., Dombret, H., et al. (2020). Oral Azacitidine Maintenance Therapy for Acute Myeloid Leukemia in First Remission. N. Engl. J. Med. 383 (26), 2526–2537. doi:10.1056/nejmoa2004444
Wei, Y., Dimicoli, S., Bueso-Ramos, C., Chen, R., Yang, H., Neuberg, D., et al. (2013). Toll-like Receptor Alterations in Myelodysplastic Syndrome. Leukemia 27 (9), 1832–1840. doi:10.1038/leu.2013.180
Wiese, M. D., Manning-Bennett, A. T., and Abuhelwa, A. Y. (2020). Investigational IRAK-4 Inhibitors for the Treatment of Rheumatoid Arthritis. Expert Opin. Investig. Drugs 29 (5), 475–482. doi:10.1080/13543784.2020.1752660
Will, B., Zhou, L., Vogler, T. O., Ben-Neriah, S., Schinke, C., Tamari, R., et al. (2012). Stem and Progenitor Cells in Myelodysplastic Syndromes Show Aberrant Stage-specific Expansion and Harbor Genetic and Epigenetic Alterations. Blood 120 (10), 2076–2086. doi:10.1182/blood-2011-12-399683
Xie, M., Lu, C., Wang, J., McLellan, M. D., Johnson, K. J., Wendl, M. C., et al. (2014). Age-related Mutations Associated with Clonal Hematopoietic Expansion and Malignancies. Nat. Med. 20 (12), 1472–1478. doi:10.1038/nm.3733
Xie, W., Wang, X., Du, W., Liu, W., Qin, X., and Huang, S. (2010). Detection of Molecular Targets on the Surface of CD34+CD38− Bone Marrow Cells in Myelodysplastic Syndromes. Cytometry 77A (9), 840–848. doi:10.1002/cyto.a.20929
Yang, L., Lin, S., Xu, L., Lin, J., Zhao, C., and Huang, X. (2019). Novel Activators and Small-Molecule Inhibitors of STAT3 in Cancer. Cytokine Growth Factor. Rev. 49, 10–22. doi:10.1016/j.cytogfr.2019.10.005
Zeidan, A. M., Miyazaki, Y., Platzbecker, U., Malek, K., Niolat, J., Kiertsman, F., et al. (2019). A Randomized, Double-Blind, Placebo-Controlled, Phase II Study of MBG453 Added to Hypomethylating Agents (HMAs) in Patients (Pts) with Intermediate, High, or Very High Risk Myelodysplastic Syndrome (MDS): Stimulus-MDS1. Blood 134 (Suppl. ment_1), 4259. doi:10.1182/blood-2019-127041
Keywords: myelodysplastic syndromes, hematopoietic stem cells, novel therapeutics, clonal hematopoiesis, acute myeloid leukemia
Citation: Zhan D and Park CY (2021) Stem Cells in the Myelodysplastic Syndromes. Front. Aging 2:719010. doi: 10.3389/fragi.2021.719010
Received: 01 June 2021; Accepted: 02 July 2021;
Published: 16 July 2021.
Edited by:
Sergei Vatolin, Case Western Reserve University, United StatesReviewed by:
Daniel Starczynowski, Cincinnati Children’s Hospital Medical Center, United StatesValeria Visconte, Cleveland Clinic, United States
Copyright © 2021 Zhan and Park. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher Y. Park, Y2hyaXN0b3BoZXIucGFya0BueXVsYW5nb25lLm9yZw==