 Marti Cadena Sandoval1,2†
Marti Cadena Sandoval1,2† Alexander Martin Heberle1,2†
Alexander Martin Heberle1,2† Ulrike Rehbein1
Ulrike Rehbein1 Cecilia Barile1
Cecilia Barile1 José Miguel Ramos Pittol1
José Miguel Ramos Pittol1 Kathrin Thedieck1,2,3*
Kathrin Thedieck1,2,3*- 1Institute of Biochemistry and Center for Molecular Biosciences Innsbruck, University of Innsbruck, Innsbruck, Austria
- 2Laboratory of Pediatrics, Section Systems Medicine of Metabolism and Signaling, University of Groningen, University Medical Center Groningen, Groningen, Netherlands
- 3Department for Neuroscience, School of Medicine and Health Sciences, Carl von Ossietzky University Oldenburg, Oldenburg, Germany
The mechanistic target of rapamycin complex 1 (mTORC1) kinase is a master regulator of metabolism and aging. A complex signaling network converges on mTORC1 and integrates growth factor, nutrient and stress signals. Aging is a dynamic process characterized by declining cellular survival, renewal, and fertility. Stressors elicited by aging hallmarks such as mitochondrial malfunction, loss of proteostasis, genomic instability and telomere shortening impinge on mTORC1 thereby contributing to age-related processes. Stress granules (SGs) constitute a cytoplasmic non-membranous compartment formed by RNA-protein aggregates, which control RNA metabolism, signaling, and survival under stress. Increasing evidence reveals complex crosstalk between the mTORC1 network and SGs. In this review, we cover stressors elicited by aging hallmarks that impinge on mTORC1 and SGs. We discuss their interplay, and we highlight possible links in the context of aging and age-related diseases.
Introduction
The mechanistic target of rapamycin (MTOR) is a serine/threonine protein kinase conserved across all eukaryotes (Tatebe and Shiozaki, 2017). MTOR constitutes a central hub that integrates metabolic signals and adapts cellular processes to extrinsic and intrinsic changes and stressors in health, disease, and aging (Papadopoli et al., 2019; Liu and Sabatini, 2020).
MTOR resides in two complexes, MTOR complex 1 and 2 (mTORC1 and mTORC2), each of which regulates distinct functions in metabolic control throughout the life course [reviewed by Liu and Sabatini (2020) and Papadopoli et al. (2019)]. The two complexes exhibit different sensitivities to the macrolide rapamycin that gave MTOR its name (Heitman et al., 1991; Brown et al., 1994; Sabatini et al., 1994). Rapamycin directly binds and inhibits mTORC1 (Yang et al., 2013; Aylett et al., 2016) whereas long-term rapamycin exposure indirectly inhibits also mTORC2 (Sarbassov et al., 2006). From yeast to mammals, rapamycin extends lifespan, highlighting the fundamental role of MTOR as a regulator of longevity and aging (Weichhart, 2018; Papadopoli et al., 2019). Aging is a dynamic process whereby physiological functions needed for survival, renewal, and fertility deteriorate over time. Its pace differs among species and individuals due to differences in molecular networks and in stochastic damage of cellular components (Khan et al., 2017). Mitochondrial malfunction, loss of proteostasis, genomic instability and telomere shortening, dysregulated nutrient sensing, and altered cell communication are considered as hallmarks of aging (Lopez-Otin et al., 2013) and are directly regulated by MTOR [reviewed in detail by Papadopoli et al. (2019)]. Conversely, MTOR also responds to their dysregulation, thus contributing to age-related processes upstream and downstream of aging hallmarks. In this review, we cover stressors elicited by aging hallmarks that impinge on the signaling network converging on mTORC1. We highlight the complex crosstalk of mTORC1 with the formation of stress granules (SGs), a stress-dependent non-membranous cellular compartment, and we discuss the impact of their interplay in aging and age-related diseases.
The mTORC1 Network Under Nutrient Sufficiency and Stress
mTORC1 Activation by Growth Factors
mTORC1 responds to a plethora of environmental cues, including growth factors [e.g. insulin or insulin like growth factor 1 (IGF-1)], nutrients (e.g. amino acids) and stressors (Figure 1) (Heberle et al., 2015; Ben-Sahra and Manning, 2017; Gonzalez and Hall, 2017; Liu and Sabatini, 2020). Upon insulin or IGF-1 binding, the insulin receptor (INSR) or insulin like growth factor 1 receptor (IGF1R) auto-phosphorylate their cytoplasmic domains (Vigneri et al., 2016; Razquin Navas and Thedieck, 2017), allowing the recruitment of several insulin receptor substrate (IRS) protein isoforms (Sun et al., 1991; Razquin Navas and Thedieck, 2017). The INSR phosphorylates the IRS at tyrosine residues, which in turn act as scaffolds for other proteins, including phosphatidylinositol 3-kinases (PIK3C, also known as PI3Ks) (Hadari et al., 1992). Class I PI3Ks convert phosphatidylinositol-4,5-bisphosphate (PI4,5P2) to phosphatidylinositol-3,4,5-trisphosphate (PI3,4,5P3), serving as an anchor site for proteins at the plasma membrane (Czech, 2000; Dibble and Cantley, 2015; Bilanges et al., 2019; Hoxhaj and Manning, 2020) (Figure 1). The pleckstrin homology (PH) domains of PDPK1 (phosphoinositide-dependent kinase-1) and AKT1 (AKT serine/threonine kinase 1) both bind to PI3,4,5P3 (Anderson et al., 1998; Vanhaesebroeck et al., 2012; Dibble and Cantley, 2015). PDPK1 phosphorylates and activates AKT1 (Alessi et al., 1996; Alessi et al., 1997). In turn, AKT1 inhibits the mTORC1 suppressors AKT1 substrate 1 (AKT1S1, also known as PRAS40) (Kovacina et al., 2003; Haar et al., 2007; Nascimento et al., 2010) and the tuberous sclerosis (TSC) protein complex (Inoki et al., 2002). The TSC complex contains the TSC complex subunit 1 (TSC1, also known as hamartin), TSC2 (also known as tuberin), and TBC1 domain family member 7 (TBC1D7) (Dibble et al., 2012; Ramlaul et al., 2021; Yang et al., 2021). When AKT1 is inactive, the TSC complex inhibits mTORC1 at the lysosomes, constituting its central signaling platform (Carroll and Dunlop, 2017). TSC2 harbors a GTPase activating protein (GAP) function towards the small GTPase RHEB (Ras homolog, mTORC1 binding) (Inoki et al., 2003; Tee et al., 2003b; Zhang et al., 2003) (Figure 1). When AKT1 is activated by insulin, it phosphorylates TSC2 (Inoki et al., 2002; Menon et al., 2014), and the lysosomal localization of the TSC complex is reduced (Menon et al., 2014). Also other growth factor responsive pathways including MAPK (mitogen-activated protein kinase) (Tee et al., 2003a; Ma et al., 2005) and WNT (Wnt family member) (Inoki et al., 2006) converge on TSC2, leading to its phosphorylation and inactivation. GTP-bound RHEB binds and activates mTORC1 at the lysosomal surface (Castro et al., 2003; Garami et al., 2003; Inoki et al., 2003; Tee et al., 2003b; Zhang et al., 2003; Long et al., 2005). mTORC1 restricts its own activity via several negative feedback loops (Figure 1) mediated by the mTORC1 substrates RPS6KB1 (ribosomal protein S6 kinase B1, also known as S6K1) that phosphorylates and inhibits the IRS (Um et al., 2004; Tzatsos and Kandror, 2006), and GRB10 (growth factor receptor-bound protein 10) causing the inhibition of the INSR (Hsu et al., 2011; Yu et al., 2011).
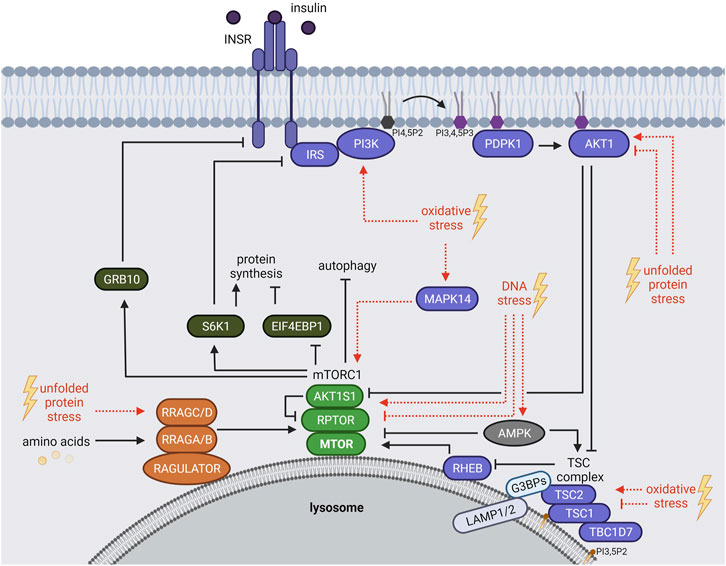
FIGURE 1. The mTORC1 signaling network. mTORC1 is regulated by growth factors (insulin), amino acids and stressors. Insulin binds and activates the INSR, which recruits IRS and PI3Ks. PI3Ks convert PI4,5P2 to PI3,4,5P3 that serves as an anchor for PDPK1 and AKT1 at the plasma membrane. PDPK1 activates AKT1, which in turn inhibits AKT1S1 and the TSC complex. Hence, TSC-mediated RHEB inhibition is repressed resulting in mTORC1 activation at the lysosome. Amino acids regulate mTORC1 recruitment to the lysosomal surface mainly through the RRAG GTPases. mTORC1 phosphorylates many substrates including EIF4BP1, RPS6KB1 (S6K1) and GRB10. S6K1 and GRB10 mediate negative feedback loops towards the IRS and the INSR, respectively. Age-related stressors (depicted in red) activate or inhibit the mTORC1 network via different mechanisms. See main text for details and abbreviations.
mTORC1 Response to Amino Acids
Amino acids mediate the lysosomal recruitment of mTORC1 (Kim et al., 2008; Sancak et al., 2008; Sancak et al., 2010). This finding explains the long-known hierarchy of insulin and amino acid signals to mTORC1 whereby the insulin-RHEB axis can only activate mTORC1 in the presence of amino acids (Hara et al., 1998), when mTORC1 resides at the lysosomes (Sancak et al., 2010; Carroll and Dunlop, 2017). The machineries recruiting mTORC1 to lysosomes are complex and converge mainly on the four Ras related GTP binding GTPases (RRAG A, B, C, D) (Figure 1) [reviewed by Gonzalez and Hall (2017), Wolfson and Sabatini (2017), Kim and Guan (2019), Carroll (2020)]. In their active form, GTP-bound RRAGA or RRAGB and GDP-bound RRAGC or RRAGD assemble as heterodimers (A or B with C or D). They bridge mTORC1 to the RAGULATOR complex [LAMTOR, formed by the late endosomal/lysosomal adaptor MAPK and MTOR activator proteins 1-5, (LAMTOR 1-5)] which resides at the lysosomal surface (Teis et al., 2002; Sancak et al., 2010; Bar-Peled et al., 2012; de Araujo et al., 2017). Specific sensors transduce distinct amino acid signals that activate the RRAGs (Figure 1). For example, 1) leucine is sensed by SESN2 (sestrin 2) (Chantranupong et al., 2014; Parmigiani et al., 2014; Peng et al., 2014; Saxton et al., 2016b; Wolfson et al., 2016) and enhances acetylation of the mTORC1 scaffold protein RPTOR (regulatory associated protein of MTOR complex 1, also known as raptor) and mTORC1-RRAGs binding (Son et al., 2019). 2) Arginine is sensed by CASTOR1 (cytosolic arginine sensor for mTORC1 subunit 1) (Chantranupong et al., 2014; Saxton et al., 2016a) and SLC38A9 (solute carrier family 38 member 9) (Jung et al., 2015; Rebsamen et al., 2015; Wang et al., 2015), 3) S-adenosylmethionine (SAM) by BMT2 (base methyltransferase of 25S rRNA 2 homolog, also known as SAMTOR) (Gu et al., 2017; Kitada et al., 2020), and 4) glutamine and leucine activate mTORC1 via α-ketoglutarate, produced by glutaminolysis, in a RRAG-dependent manner (Duran et al., 2012). Amino acids also activate mTORC1 via RRAG-independent routes (Melick and Jewell, 2020). For instance, glutamine and asparagine signal to lysosomal ATP6V0D1 (ATPase H+ transporting V0 subunit d1) and ARF1 (ADP ribosylation factor 1), thereby promoting mTORC1 activation through a yet unknown mechanism (Stracka et al., 2014; Jewell et al., 2015; Bernfeld et al., 2018; Melick and Jewell, 2020; Meng et al., 2020; Takahara et al., 2020).
Not only mTORC1 but also the TSC protein complex shuttles between the cytoplasm and the lysosomal surface. Recent evidence has shed light on the underlying molecular mechanisms: 1) TSC2 is tethered by the G3BP stress granule assembly factors 1 and 2 (G3BP1 and G3BP2, G3BPs) to the cytoplasmic portion of the lysosomal associated proteins 1 and 2 (LAMP1/2) (Prentzell et al., 2021) (Figure 1). Also the RHEB and RRAG GTPases are required for lysosomal TSC2 recruitment (Demetriades et al., 2014; Menon et al., 2014; Carroll et al., 2016; Yang et al., 2020; Prentzell et al., 2021). 2) TSC1 binds lysosomal phosphatidylinositol-3,5-bisphosphate (PI3,5P2) via its N-terminal domain (Fitzian et al., 2021). The interplay of TSC2 and TSC1 tethering mechanisms in the lysosomal recruitment of the TSC complex remains to be determined. While the TSC complex is widely recognized as a transducer of insulin signals to RHEB and mTORC1 (Inoki et al., 2002; Hoxhaj and Manning, 2020), TSC complex association with the RRAG GTPases suggests also a responsiveness to amino acids (Demetriades et al., 2014; Menon et al., 2014; Carroll et al., 2016). Furthermore, several stressors including hypoxia, osmotic, pH and glycolytic stress enhance lysosomal recruitment of the TSC complex (Plescher et al., 2015; Demetriades et al., 2016). Hence, the control of the lysosomal localization of the TSC complex emerges as a central regulatory event that balances mTORC1 activity in response to growth factors, amino acids, and stresses.
mTORC1 and Age-Related Stressors
Next to growth factors and amino acids, mammalian mTORC1 responds to a variety of stressors, including oxidative, DNA and unfolded protein stress (Heberle et al., 2015; Su and Dai, 2017; Ma et al., 2018). These stressors are connected to hallmarks of aging: 1) oxidative stress, promoted by the accumulation of reactive oxygen species (ROS), arises from dysfunctional oxidative phosphorylation in mitochondria (Desler et al., 2011), oxidative protein folding in the endoplasmic reticulum (ER) (Marciniak et al., 2004; Margittai and Sitia, 2011; Yoboue et al., 2018), and peroxisome metabolism (Titorenko and Terlecky, 2011). ROS accumulation results in oxidative damage of biomolecules including proteins and DNA (Liguori et al., 2018). 2) DNA stress is attributed to DNA damage, genome instability and telomere attrition (Maynard et al., 2015). DNA stress arises intrinsically from insufficient repair of replication errors or spontaneous hydrolytic reactions and telomere shortening during DNA replication (Maynard et al., 2015; Yousefzadeh et al., 2021), and upon damage by extrinsic agents, including electromagnetic radiation and chemical agents (Yousefzadeh et al., 2021). 3) ROS and DNA stress both promote proteasomal stress (loss of proteostasis) (Malhotra and Kaufman, 2007; Gonzalez-Quiroz et al., 2020), arising from imbalanced protein synthesis, folding, and turnover (declining autophagy and proteasome function) and resulting in an accumulation of unfolded proteins in the cytoplasm and/or in the ER (unfolded protein stress). The complex molecular mechanisms via which these stressors impinge on the mTORC1 network have been reviewed in detail by Heberle et al. (2015), Su and Dai (2017) and Ma et al. (2018).
Being mostly perceived as inhibitory, also activating stress inputs to the mTORC1 network have been reported that contribute to the delicate balance of mTORC1 activity under stress. In brief, oxidative stress inhibits mTORC1 by TSC complex-mediated RHEB-repression (Alexander et al., 2010; Zhang et al., 2013; Demetriades et al., 2016) and by inhibiting the lysosomal localization of mTORC1 (Yuan et al., 2015). ROS-mediated activation of mTORC1 also involves the TSC complex, as TSC1 and TSC2 are directly oxidized and inhibited by ROS (Yoshida et al., 2011). Furthermore, oxidative stress by mitochondrial ROS or arsenite activates mTORC1 via RAS (RAS proto-oncogene, GTPase) dependent activation of the PI3K-AKT1 pathway (Kim et al., 2018; Heberle et al., 2019). Arsenite also induces mTORC1 via the stress sensitive MAPK14 (mitogen-activated protein kinase 14, also known as p38) (Wang and Proud, 1997; Heberle et al., 2019) that directly phosphorylates the mTORC1 scaffold protein RPTOR (Wu et al., 2011) (Figure 1). UV-induced ROS activate mTORC1 via PI3K but independent of AKT1 via an unknown mechanism (Huang et al., 2002).
Unfolded protein stress inhibits mTORC1 via AKT1-repression (Qin et al., 2010; Kato et al., 2012; Li et al., 2018), for example through the negative AKT1 regulator TRIB3 (tribbles pseudokinase 3) (Ohoka et al., 2005). Prolonged unfolded protein stress triggered by the ER-stress inducers thapsigargin or tunicamycin inhibits AKT1 and phosphorylation of TSC2 at threonine 1462 and results in mTORC1 inactivation. In contrast, short-term unfolded protein stress mildly enhances AKT1 activity and phosphorylation of TSC2 (Di Nardo et al., 2009). Hence, activating and inhibitory cues converge on the TSC complex, depending on the stress duration and level. Interestingly, the RRAG GTPases may also contribute to unfolded protein stress sensing by mTORC1, as the ER stress inducer tunicamycin promotes RRAGC expression (Guha et al., 2017).
DNA stress is sensed via DNA damage response sensor proteins including PARP (poly ADP-ribose polymerase), ATM (ataxia telangiectasia mutated), DNA-PK (DNA protein kinase) and ATR (ataxia telangiectasia and Rad3 related) (Ma et al., 2018). PARP (Munoz-Gamez et al., 2009; Rodriguez-Vargas et al., 2012) and ATM (Alexander et al., 2010; Zhang et al., 2013; Ma et al., 2018) inhibit mTORC1 upon prolonged DNA stress by activating AMPK (AMP-activated protein kinase) which phosphorylates and activates TSC2, upstream of mTORC1 (Figure 1). Upon short term DNA stress (4 h etoposide), ATM/ATR activate mTORC1 by upregulating the level of MTOR, possibly by stabilizing the protein (Selvarajah et al., 2015). In contrast, prolonged DNA stress (24 h etopisode) results in mTORC1 inactivation and decreased MTOR protein levels (Selvarajah et al., 2015).
When active, mTORC1 enhances virtually all anabolic processes including protein synthesis, and inhibits catabolism, most notably autophagy [comprehensively reviewed by Ben-Sahra and Manning (2017), Rabanal-Ruiz and Korolchuk (2018), Tee (2018), Kim and Guan (2019), Liu and Sabatini (2020)]. Upon stress, mTORC1 suppression limits biosynthesis to essential processes needed for survival (Heberle et al., 2015), and enhances the degradation of cellular macromolecules and organelles by autophagy (Dossou and Basu, 2019), mitigating their damage and supplying the cell with intermediary metabolites as building blocks (Wong et al., 2020). Why does stress also elicit activating inputs to mTORC1? A certain level of tightly controlled mTORC1 activity may sustain processes required for stress survival (Thedieck et al., 2013; Heberle et al., 2015). This may concern the synthesis of stress response proteins (Chou et al., 2012; Hsieh et al., 2012; Thedieck et al., 2013) as well as the formation of SGs (Fournier et al., 2013; Mazan-Mamczarz et al., 2015; Sfakianos et al., 2018; Heberle et al., 2019), a stress-induced cytoplasmic compartment promoting survival (Reineke and Neilson, 2019). Hence, balanced mTORC1 activity might be required for SG-mediated cell survival and stress-recovery.
SGs and mTORC1 Signaling
Control of SG Formation by mTORC1
SGs are cytoplasmic non-membranous assemblies of proteins and mRNAs whose interaction involves liquid-liquid phase separation (LLPS) (Ivanov and Anderson, 2019). A rapidly growing field investigates the molecular mechanisms underlying SG formation (Van Treeck and Parker, 2018; Alberti et al., 2019; Mathieu et al., 2020; Peran and Mittag, 2020; Hofmann et al., 2021; Wiedner and Giudice, 2021). SG formation has been linked with different physiological consequences that are context-dependent and are currently under debate. Depending on the stress and its duration, SGs are rapidly turned over or they persist over long periods of time (Aulas et al., 2017; Markmiller et al., 2018). SGs buffer cellular stress by minimizing energy consumption [reviewed by Mahboubi and Stochaj (2017)] and by anti-apoptotic mechanisms (Arimoto et al., 2008; Takahashi et al., 2013; Thedieck et al., 2013). Such protective functions have been assigned to short-lived SGs (Reineke and Neilson, 2019). However, SGs might also exert pro-apoptotic effects (Fujimura et al., 2012; Aulas et al., 2018; Reineke and Neilson, 2019; Amen and Kaganovich, 2020) and they contribute to the formation of pathogenic protein aggregates (Aulas et al., 2018; Jeon and Lee, 2021). Chronic SG assembly has been linked with age-related disorders including neurotoxicity and cancer cell survival (Reineke and Neilson, 2019; Advani and Ivanov, 2020; Alberti and Hyman, 2021).
SGs form in a highly dynamic process within minutes upon stress exposure (Cao et al., 2020; Peran and Mittag, 2020). Via LLPS proteins and nucleic acids condense into liquid-like droplets surrounded by a liquid uncondensed environment (Alberti et al., 2019; Hofmann et al., 2021). LLPS involves the RNA content as well as proteins with LLPS-promoting domains such as RNA-binding domains (RBDs) (Alberti and Hyman, 2021; Hofmann et al., 2021) and intrinsically disordered regions (IDRs) (Alberti et al., 2019). The list of proteins that promote LLPS upon stress is rapidly growing (Youn et al., 2019). Early on, bona fide SG markers were defined based on the granular pattern that they acquire upon different stressors (Kedersha and Anderson, 2007). Some of these proteins were later shown to be required for SG assembly and are thus considered as core SG components (Kedersha et al., 2013). The core SG proteins include G3BP1/2 (Tourriere et al., 2003; Matsuki et al., 2013), TIA1 (TIA1 cytotoxic granule associated RNA binding protein) (Anderson and Kedersha, 2002; Gilks et al., 2004), and FMR1 (FMRP translational regulator 1) (Mazroui et al., 2002; Didiot et al., 2009). SG assembly is influenced by covalent modifications of RNAs and proteins that alter their physicochemical properties, such as surface charge, hydrophobicity, and binding strength between proteins and RNAs (Wiedner and Giudice, 2021). Post-translational modifications (PTMs) directly affecting SG formation include SUMOylation, methylation and phosphorylation (Kedersha et al., 2013; Mahboubi and Stochaj, 2017; Snead and Gladfelter, 2019; Cao et al., 2020). These PTMs are mediated by cellular signaling networks which thus directly impinge on SG assembly (Kedersha et al., 2013; Mahboubi and Stochaj, 2017; Reineke and Neilson, 2019).
SG formation is intimately linked with translation inhibition. When translation is inhibited, polysomes run off their mRNAs and “naked” mRNAs assemble with SG nucleating proteins to undergo LLPS (Kedersha et al., 2013; Hofmann et al., 2021). Under non-stress conditions, cap-dependent translation is initiated by the assembly of the EIF4F complex (EIF4E, EIF4G, EIF4B and EIF4A) at the 5′ 7-methylguanosine cap (5′ cap) of mRNAs (Sonenberg and Hinnebusch, 2009). One key regulatory event of EIF4F complex assembly is the phosphorylation of the eukaryotic translation initiation factor 4E-binding protein 1 (EIF4EBP1, also known as 4E-BP1) (Thoreen, 2017; Roux and Topisirovic, 2018; Tahmasebi et al., 2018). EIF4EBP1 competes with EIF4G for EIF4E binding, and prevents EIF4F complex assembly (Sonenberg and Hinnebusch, 2009). EIF4EBP1 phosphorylation by mTORC1 prevents EIF4EBP1-EIF4E binding and promotes EIF4F complex formation (Roux and Topisirovic, 2018) and recruitment of the 43S pre-initiation complex, consisting of the small ribosomal subunit (40S) bound to the eukaryotic translation initiation factor-2 complex (EIF2), GTP and Met-tRNAiMet (Sonenberg and Hinnebusch, 2009). This complex is required for ribosome assembly and translation initiation (Merrick and Pavitt, 2018). EIF2 is a heterotrimeric complex consisting of alpha (EIF2S1), beta (EIF2S2), and gamma (EIF2S3) subunits (Merrick and Pavitt, 2018). EIF2S1 phosphorylation at serine 51 inhibits EIF2 (Sonenberg and Hinnebusch, 2009). Four kinases (HRI or EIF2AK1, PKR or EIF2AK2, PERK or EIF2AK3, GCN2 or EIF2AK4) phosphorylate EIF2S1-S51 in response to different stress situations (Donnelly et al., 2013) (Figure 2). This is considered as one of the main regulatory events for translation inhibition and SG initiation (Kedersha et al., 1999; Kedersha et al., 2002; Hofmann et al., 2021). However, SG formation can also be EIF2-independent, e.g. upon translation inhibition at the level of EIF4F complex assembly or activity (Hofmann et al., 2021).
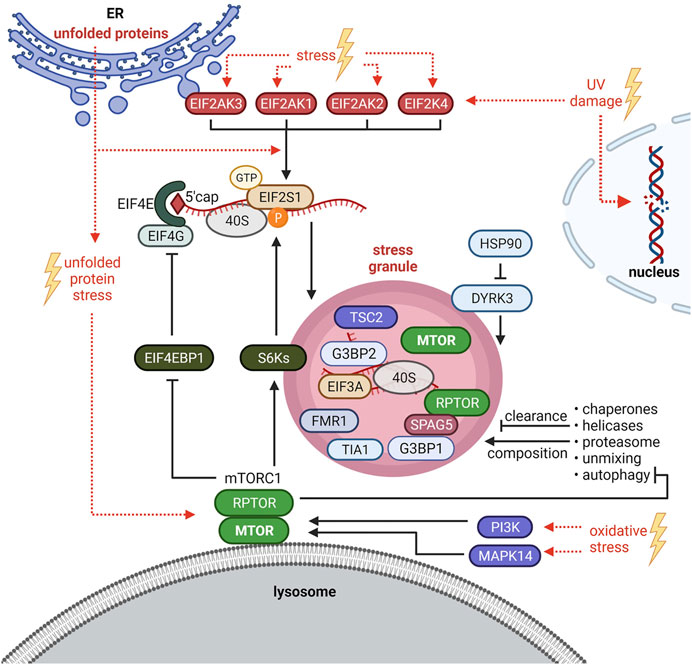
FIGURE 2. Crosstalk between stress granules (SGs) and mTORC1 signaling. SG formation is triggered by EIF2S1 phosphorylation at serine 51 or at the level of the EIF4F complex. HRI/EIF2AK1, PKR/EIF2AK2, PERK/EIF2AK3 and GCN2/EIF2AK4 phosphorylate EIF2S1-S51 in response to different stresses. Under oxidative stress, also the mTORC1-S6Ks axis enhances phosphorylation of EIF2S1-S51. Upon Bortezomib (unfolded protein stress), SGs are induced by mTORC1-driven phosphorylation of EIF4EBP1 to sustain EIF4F complex assembly. PI3K and MAPK14 activate mTORC1 to promote SG formation. Autophagy is inhibited by mTORC1 and mediates SG clearance and composition. SGs inhibit mTORC1 by sequestration of MTOR and RPTOR and via the HSP90-DYRK3 axis. SGs also recruit TSC2 and S6K1 and 2 (S6Ks). See main text for details and abbreviations.
mTORC1 inhibition has been proposed to initiate SG formation in mammalian cells (Fujimura et al., 2012; Hofmann et al., 2012; Panas et al., 2016) as it prevents the phosphorylation of EIF4EBP1 and thus the assembly of the EIF4F complex. In line with this idea, EIF4EBP1 shifts in size or is dephosphorylated, and increases its binding to the mRNA 5′ cap upon several stressors (H2O2, cold shock, selenite, nitric oxide) (Emara et al., 2012; Fujimura et al., 2012; Hofmann et al., 2012; Aulas et al., 2018). Based on observations that EIF4EBP1 or EIF4E inhibition by knockdown impairs SG formation upon selenite (Fujimura et al., 2012) or H2O2 (Emara et al., 2012) stress, respectively, EIF4EBP1-cap association has been proposed to enhance SG formation (Emara et al., 2012; Fujimura et al., 2012; Panas et al., 2016). Thus, unphosphorylated active EIF4EBP1 may promote SG assembly. However, none of the studies tested by inhibitors or knockdowns whether this process depends on mTORC1 (Emara et al., 2012; Fujimura et al., 2012; Hofmann et al., 2012). EIF4EBP1 is targeted by several kinases (Qin et al., 2016) and phosphatases (Kolupaeva, 2019) other than mTORC1. It remains thus open whether EIF4EBP1-mediated SG formation relates to inactive mTORC1, or if it is mediated by inactivation of another kinase or by a phosphatase. Sfakianos et al. (2018) showed that without stress, neither RPTOR knockdown nor rapamycin induced SGs. Hence, mTORC1 inhibition by itself is not sufficient to initiate SG formation. In contrast, mTORC1 inhibition has been shown by several studies to reduce SG formation upon heat shock, arsenite, and the proteasome inhibitor Bortezomib (Fournier et al., 2013; Mazan-Mamczarz et al., 2015; Sfakianos et al., 2018; Heberle et al., 2019). Conversely, TSC2 deficiency, known to hyperactivate mTORC1, increases the number of SGs formed upon arsenite or heat stress (Kosmas et al., 2021). As discussed above, mTORC1 activity under stress is enhanced–at least in part–by PI3Ks and MAPK14 (Heberle et al., 2019). Those kinases promote SG formation (Brown et al., 2011; Heberle et al., 2019), further supporting that a stress-activated signaling network converging on mTORC1 promotes SG formation.
All molecular mechanisms known so far to mediate mTORC1-driven SG formation impinge on the translation machinery. Upon arsenite, the kinases S6K1 and 2 (S6Ks) downstream of mTORC1 promote EIF2S1-S51 phosphorylation in mammalian cells (Sfakianos et al., 2018) (Figure 2), and this mechanism is conserved upon heat stress in the nematode Caenorhabditis elegans. Mammalian S6Ks enhanced SGs only under moderate arsenite stress. Higher concentrations abolished the S6Ks’ impact on SG formation, although it still depended on mTORC1 (Sfakianos et al., 2018). This might be explained by findings of Fournier et al. (2013) who showed that mTORC1-driven phosphorylation of EIF4EBP1 preserves EIF4E-EIF4G interaction, consequently enhancing SG formation upon high concentrations of arsenite as well as Bortezomib (Figure 2). Thus, mTORC1 enhances SG assembly via phosphorylation of S6Ks and EIF4EBPs, both events that are known to enhance translation (Roux and Topisirovic, 2018; Liu and Sabatini, 2020). This indicates that next to translation arrest (Van Treeck and Parker, 2018; Reineke and Neilson, 2019) activating signals to the translation machinery also contribute to SG formation.
Whether and which SGs form independently of mTORC1 remains to be investigated. To the best of our knowledge this has been so far claimed twice, for UV (Ying and Khaperskyy, 2020) and heat stress (Brown et al., 2011), based on the finding that MTOR inhibitors partially inhibit SG formation, but a certain fraction of cells with SGs remains. This observation is in agreement with several studies under different stresses (Fournier et al., 2013; Mazan-Mamczarz et al., 2015; Sfakianos et al., 2018; Heberle et al., 2019). The properties of SGs that are refractory to mTORC1 inhibition are therefore an intriguing topic for future studies. Where does mTORC1 control SG formation? mTORC1’s best described site of activity are the lysosomes (Rabanal-Ruiz and Korolchuk, 2018; Carroll, 2020). Recent evidence shows that SGs physically associate with lysosomes (Liao et al., 2019). Thus, lysosomal mTORC1 may enhance SG assembly. It is interesting to note that the core SG proteins G3BP1 and 2 have non-granule functions as mTORC1 suppressors at lysosomes (Prentzell et al., 2021). It remains open whether the G3BP pools at lysosomes and SGs are separate, or whether G3BPs shuttle between these two compartments. Lysosomes have also been linked to SGs in the context of autophagy: as autophagy degrades aggregated proteins (Yang and Klionsky, 2020) it is straightforward to assume that autophagy contributes to SG clearance. Indeed this has been reported in mammals (Ryu et al., 2014; Marrone et al., 2018; Silva et al., 2019), Saccharomyces cerevisiae (Buchan et al., 2013) and Caenorhabditis elegans (Zhang et al., 2018). mTORC1 is a key suppressor of autophagy (Rabanal-Ruiz et al., 2017). Thus, mTORC1 may enhance SG assembly, at least in part, by inhibiting autophagy. Furthermore, autophagy not only controls SG turnover (Buchan et al., 2013; Lee, 2015) but also their composition (Seguin et al., 2014; Advani and Ivanov, 2020) (Figure 2). This raises the possibility that mTORC1 affects SG composition by inhibiting autophagy.
On a broader level, SG clearance and composition is affected not only by autophagy but also by chaperones, RNA helicases, the proteasomal machinery and unmixing of LLPS condensates (Alberti et al., 2017; Alberti and Hyman, 2021). The cooperation of these processes in SG dynamics is currently investigated by a highly active and growing field of research. The interplay of mTOR with the proteasome is also a matter of active scientific debate (Adegoke et al., 2019). It will be intriguing to link these fields and unravel mTOR’s role in SG turnover.
SGs Inhibit mTORC1
Not only does mTORC1 regulate SG formation and clearance. Conversely, SGs also inhibit mTORC1 by several mechanisms in yeast as well as in mammalian cells (Takahara and Maeda, 2012; Thedieck et al., 2013; Wippich et al., 2013; Mediani et al., 2021). In mammalian cells, SG recruitment of RPTOR is mediated by SPAG5 (sperm associated antigen 5, also known as astrin) and leads to the disassembly and inhibition of mTORC1 (Thedieck et al., 2013) (Figure 2). MTOR localizes to SGs too (Wippich et al., 2013), but the molecule mediating this recruitment is unknown (Figure 2). Likewise, the Saccharomyces cerevisiae RPTOR orthologue KOG1 and TOR1 localize to SGs (Takahara and Maeda, 2012). Also the TSC subunit TSC2 (Kosmas et al., 2021) and the mTORC1 substrates S6K1 and 2 (Sfakianos et al., 2018) have been recently reported at SGs (Figure 2), which might impinge on mTORC1 activity as well. It will be interesting to delineate the coordination of the recruitment and disassembly of the TSC and mTORC1 complexes and their substrates at SGs. SGs also regulate mTORC1 via the kinase DYRK3 (dual specificity tyrosine phosphorylation regulated kinase 3) (Wippich et al., 2013; Mediani et al., 2021). DYRK3 binds HSP90 (heat shock protein 90 family) an essential chaperone which regulates the folding and stability of many clients including stress response factors important to resolve a variety of proteotoxic stresses [reviewed in detail by Schopf et al. (2017), Calderwood (2018), Moran Luengo et al. (2019), Lang et al. (2021)]. Under non-stress conditions, HSP90 keeps DYRK3 in an active confirmation. DYRK3 phosphorylates the mTORC1 inhibitor AKT1S1 at threonine 246, thus de-repressing mTORC1 (Wippich et al., 2013). Upon stress or HSP90 inhibition (Wippich et al., 2013; Mediani et al., 2021), inactive DYRK3 is recruited to SGs via its N-terminal IDR, resulting in AKT1S1 activation and mTORC1 inhibition. SG-localized DYRK3 also stabilizes SGs, thereby enhancing their inhibitory effect on mTORC1. As mTORC1 is inhibited by SGs, mTORC1-driven SG formation may constitute a negative feedback mechanism that restricts mTORC1 activation by stress, contributing to the fine-tuning of cellular anabolism and catabolism that maintains cellular homeostasis under stress.
Discussion
Linkage of MTOR and SGs in Aging
Stresses linked to hallmarks of aging (see mTORC1 and Age-Related Stressors) impinge on mTORC1 activity and SG formation (see SGs and mTORC1 Signaling). Conversely, enhanced mTORC1 signaling (Papadopoli et al., 2019) and SG formation (Cao et al., 2020) have been linked to age-related processes, and MTOR and SG levels often correlate with the severity of age-related diseases (Papadopoli et al., 2019; Liu and Sabatini, 2020; Chrienova et al., 2021). However, the crosstalk between mTORC1 and SG formation in the context of aging progression is poorly explored.
Aging is characterized by increased numbers of senescent cells that have been assigned to stress stimuli which result in irreversible cell cycle arrest (Lopez-Otin et al., 2013; Papadopoli et al., 2019). Senescent cells are impaired in their proliferative capacity, but they maintain an active metabolism (Papadopoli et al., 2019) and exhibit reduced apoptosis (Wanner et al., 2020) despite stress-induced damage (Song et al., 2020). Although SG formation is mainly recognized to counteract senescence (Omer et al., 2018; Cao et al., 2020), senescent cells can form SGs (Lian and Gallouzi, 2009). Interestingly, senescent cells exhibit mTORC1 uncoupling from its suppressors, resulting in mTORC1 hyperactivity (Carroll et al., 2017). Hence, chronic mTORC1 activity might sensitize senescent cells to SG formation. In line with this, senescent cells present a higher number of SGs upon acute stress (Lian and Gallouzi, 2009) and show slowed SG disassembly after stress recovery (Gallouzi, 2009; Lian and Gallouzi, 2009). SGs enhance survival by sequestering pro-apoptotic proteins (Arimoto et al., 2008). mTORC1-driven SG formation may also exert a negative feedback on mTORC1 that restricts its excessive activity, known to result in apoptosis (Appenzeller-Herzog and Hall, 2012; Thedieck et al., 2013). Hence, SG formation might contribute to the survival and increased presence of senescent cells in aging tissues by sequestering pro-apoptotic factors and by dampening mTORC1 activity.
Neurodegeneration and cancer are age-related diseases associated with senescence (Baker and Petersen, 2018; Sikora et al., 2021). Both aberrant mTORC1 activity (Chrienova et al., 2021; Querfurth and Lee, 2021) and chronic SG formation (Wolozin and Ivanov, 2019; Asadi et al., 2021) link with neurodegenerative diseases including ALS (amyotrophic lateral sclerosis), FTD (frontotemporal dementia), AD (Alzheimer’s disease) or PD (Parkinson’s disease). However, their crosstalk in neurodegenerative diseases is largely unknown. It is conceivable that hyperactive mTORC1 drives chronic SG formation, and thereby promotes the progression of neurodegeneration. In cancer, MTOR is widely recognized as a key driver and drug target (Mossmann et al., 2018; Liu and Sabatini, 2020), whereas the importance of SG proteins for tumorigenesis and treatment response is only beginning to emerge (Anderson et al., 2015; Gao et al., 2019). Many SG proteins are dysregulated in cancer (Adjibade et al., 2015; Somasekharan et al., 2015; Vilas-Boas Fde et al., 2016; Sim et al., 2019) and their altered expression has been linked with drug response and disease outcome (Gao et al., 2019). The role of mTORC1-SG crosstalk in neurodegeneration and cancer therefore deserves in depth investigation regarding its role in therapy response and to develop new therapy concepts.
Conclusion
The mTORC1 cascade and SGs are key mediators of cell growth and survival that are closely intertwined in a network whose complexity we are only beginning to understand. Current research on MTOR signaling and on SGs is largely confined to separate fields. Comparatively few studies tackle their interplay and are often correlative in nature. Toward a comprehensive understanding, the challenge of investigating mechanistic inhibitory and activating links between the SG and MTOR networks will be key to identify causal relationships between them. These mechanisms may provide leads for treatments that account for specific metabolic alterations and stresses in age-related conditions such as cellular senescence, cancer, and neurodegeneration.
Author Contributions
AH, MCS, and KT wrote the manuscript, supported by CB, UR, and JRP. AH and MCS have contributed equally to this work.
Funding
KT acknowledges support from the German Tuberous Sclerosis Foundation; Stichting TSC Fonds; the German Research Foundation (TH 1358/3-1); and the PoLiMeR Innovative Training Network (Marie Skłodowska-Curie grant agreement 812616) and the MESI-STRAT project (grant agreement 754688), which received funding from the European Union Horizon 2020 Research and Innovation Program. MCS acknowledges the Graduate School of Medical Sciences of the University of Groningen.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Figures were created with BioRender.com. The proteins were named according to HGNC (HUGO Gene Nomenclature Committee) standards.
References
Adegoke, O. A. J., Beatty, B. E., Kimball, S. R., and Wing, S. S. (2019). Interactions of the Super Complexes: When mTORC1 Meets the Proteasome. Int. J. Biochem. Cell. Biol. 117, 105638. doi:10.1016/j.biocel.2019.105638
Adjibade, P., St-Sauveur, V. G., Quevillon Huberdeau, M., Fournier, M.-J., Savard, A., Coudert, L., et al. (2015). Sorafenib, a Multikinase Inhibitor, Induces Formation of Stress Granules in Hepatocarcinoma Cells. Oncotarget. 6 (41), 43927–43943. doi:10.18632/oncotarget.5980
Advani, V. M., and Ivanov, P. (2020). Stress Granule Subtypes: an Emerging Link to Neurodegeneration. Cell. Mol. Life Sci. 77 (23), 4827–4845. doi:10.1007/s00018-020-03565-0
Alberti, S., Gladfelter, A., and Mittag, T. (2019). Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell. 176 (3), 419–434. doi:10.1016/j.cell.2018.12.035
Alberti, S., and Hyman, A. A. (2021). Biomolecular Condensates at the Nexus of Cellular Stress, Protein Aggregation Disease and Ageing. Nat. Rev. Mol. Cell. Biol. 22 (3), 196–213. doi:10.1038/s41580-020-00326-6
Alberti, S., Mateju, D., Mediani, L., and Carra, S. (2017). Granulostasis: Protein Quality Control of RNP Granules. Front. Mol. Neurosci. 10, 84. doi:10.3389/fnmol.2017.00084
Alessi, D. R., Andjelkovic, M., Caudwell, B., Cron, P., Morrice, N., Cohen, P., et al. (1996). Mechanism of Activation of Protein Kinase B by Insulin and IGF-1. EMBO J. 15 (23), 6541–6551. doi:10.1002/j.1460-2075.1996.tb01045.x
Alessi, D. R., James, S. R., Downes, C. P., Holmes, A. B., Gaffney, P. R. J., Reese, C. B., et al. (1997). Characterization of a 3-Phosphoinositide-Dependent Protein Kinase Which Phosphorylates and Activates Protein Kinase Bα. Curr. Biol. 7 (4), 261–269. doi:10.1016/s0960-9822(06)00122-9
Alexander, A., Cai, S.-L., Kim, J., Nanez, A., Sahin, M., MacLean, K. H., et al. (2010). ATM Signals to TSC2 in the Cytoplasm to Regulate mTORC1 in Response to ROS. Proc. Natl. Acad. Sci. 107 (9), 4153–4158. doi:10.1073/pnas.0913860107
Amen, T., and Kaganovich, D. (2020). Fasnall Induces Atypically Transient Stress Granules Independently of FASN Inhibition. iScience. 23 (10), 101550. doi:10.1016/j.isci.2020.101550
Anderson, K. E., Coadwell, J., Stephens, L. R., and Hawkins, P. T. (1998). Translocation of PDK-1 to the Plasma Membrane Is Important in Allowing PDK-1 to Activate Protein Kinase B. Curr. Biol. 8 (12), 684–691. doi:10.1016/s0960-9822(98)70274-x
Anderson, P., and Kedersha, N. (2002). Stressful Initiations. J. Cell. Sci. 115 (Pt 16), 3227–3234. doi:10.1242/jcs.115.16.3227
Anderson, P., Kedersha, N., and Ivanov, P. (2015). Stress Granules, P-Bodies and Cancer. Biochim. Biophys. Acta (Bba) - Gene Regul. Mech. 1849 (7), 861–870. doi:10.1016/j.bbagrm.2014.11.009
Appenzeller-Herzog, C., and Hall, M. N. (2012). Bidirectional Crosstalk Between Endoplasmic Reticulum Stress and mTOR Signaling. Trends Cell. Biol. 22 (5), 274–282. doi:10.1016/j.tcb.2012.02.006
Arimoto, K., Fukuda, H., Imajoh-Ohmi, S., Saito, H., and Takekawa, M. (2008). Formation of Stress Granules Inhibits Apoptosis by Suppressing Stress-Responsive MAPK Pathways. Nat. Cell. Biol. 10 (11), 1324–1332. doi:10.1038/ncb1791
Asadi, M. R., Sadat Moslehian, M., Sabaie, H., Jalaiei, A., Ghafouri-Fard, S., Taheri, M., et al. (2021). Stress Granules and Neurodegenerative Disorders: A Scoping Review. Front. Aging Neurosci. 13, 650740. doi:10.3389/fnagi.2021.650740
Aulas, A., Fay, M. M., Lyons, S. M., Achorn, C. A., Kedersha, N., Anderson, P., et al. (2017). Stress-Specific Differences in Assembly and Composition of Stress Granules and Related Foci. J. Cell. Sci. 130 (5), 927–937. doi:10.1242/jcs.199240
Aulas, A., Lyons, S. M., Fay, M. M., Anderson, P., and Ivanov, P. (2018). Nitric Oxide Triggers the Assembly of "Type II" Stress Granules Linked to Decreased Cell Viability. Cell Death Dis. 9 (11), 1129. doi:10.1038/s41419-018-1173-x
Aylett, C. H. S., Sauer, E., Imseng, S., Boehringer, D., Hall, M. N., Ban, N., et al. (2016). Architecture of Human mTOR Complex 1. Science. 351 (6268), 48–52. doi:10.1126/science.aaa3870
Baker, D. J., and Petersen, R. C. (2018). Cellular Senescence in Brain Aging and Neurodegenerative Diseases: Evidence and Perspectives. J. Clin. Invest. 128 (4), 1208–1216. doi:10.1172/JCI95145
Bar-Peled, L., Schweitzer, L. D., Zoncu, R., and Sabatini, D. M. (2012). Ragulator Is a GEF for the Rag GTPases That Signal Amino Acid Levels to mTORC1. Cell. 150 (6), 1196–1208. doi:10.1016/j.cell.2012.07.032
Ben-Sahra, I., and Manning, B. D. (2017). mTORC1 Signaling and the Metabolic Control of Cell Growth. Curr. Opin. Cell. Biol. 45, 72–82. doi:10.1016/j.ceb.2017.02.012
Bernfeld, E., Menon, D., Vaghela, V., Zerin, I., Faruque, P., Frias, M. A., et al. (2018). Phospholipase D-Dependent mTOR Complex 1 (mTORC1) Activation by Glutamine. J. Biol. Chem. 293 (42), 16390–16401. doi:10.1074/jbc.RA118.004972
Bilanges, B., Posor, Y., and Vanhaesebroeck, B. (2019). PI3K Isoforms in Cell Signalling and Vesicle Trafficking. Nat. Rev. Mol. Cell. Biol. 20 (9), 515–534. doi:10.1038/s41580-019-0129-z
Brown, E. J., Albers, M. W., Bum Shin, T., Ichikawa, K., Keith, C. T., Lane, W. S., et al. (1994). A Mammalian Protein Targeted by G1-Arresting Rapamycin-Receptor Complex. Nature. 369 (6483), 756–758. doi:10.1038/369756a0
Brown, J. A. L., Roberts, T. L., Richards, R., Woods, R., Birrell, G., Lim, Y. C., et al. (2011). A Novel Role for hSMG-1 in Stress Granule Formation. Mol. Cell Biol. 31 (22), 4417–4429. doi:10.1128/MCB.05987-11
Buchan, J. R., Kolaitis, R.-M., Taylor, J. P., and Parker, R. (2013). Eukaryotic Stress Granules Are Cleared by Autophagy and Cdc48/VCP Function. Cell. 153 (7), 1461–1474. doi:10.1016/j.cell.2013.05.037
Calderwood, S. K. (2018). Heat Shock Proteins and Cancer: Intracellular Chaperones or Extracellular Signalling Ligands? Phil. Trans. R. Soc. B. 373, 20160524. doi:10.1098/rstb.2016.0524
Cao, X., Jin, X., and Liu, B. (2020). The Involvement of Stress Granules in Aging and Aging‐Associated Diseases. Aging Cell. 19 (4), e13136. doi:10.1111/acel.13136
Carroll, B., and Dunlop, E. A. (2017). The Lysosome: a Crucial Hub for AMPK and mTORC1 Signalling. Biochem. J. 474 (9), 1453–1466. doi:10.1042/BCJ20160780
Carroll, B., Maetzel, D., Maddocks, O. D., Otten, G., Ratcliff, M., Smith, G. R., et al. (2016). Control of TSC2-Rheb Signaling Axis by Arginine Regulates mTORC1 Activity. Elife. 5, e11058. doi:10.7554/eLife.11058
Carroll, B., Nelson, G., Rabanal-Ruiz, Y., Kucheryavenko, O., Dunhill-Turner, N. A., Chesterman, C. C., et al. (2017). Persistent mTORC1 Signaling in Cell Senescence Results From Defects in Amino Acid and Growth Factor Sensing. J. Cell. Biol. 216 (7), 1949–1957. doi:10.1083/jcb.201610113
Carroll, B. (2020). Spatial Regulation of mTORC1 Signalling: Beyond the Rag GTPases. Semin. Cell Developmental Biol. 107, 103–111. doi:10.1016/j.semcdb.2020.02.007
Castro, A. F., Rebhun, J. F., Clark, G. J., and Quilliam, L. A. (2003). Rheb Binds Tuberous Sclerosis Complex 2 (TSC2) and Promotes S6 Kinase Activation in a Rapamycin- and Farnesylation-Dependent Manner. J. Biol. Chem. 278 (35), 32493–32496. doi:10.1074/jbc.C300226200
Chantranupong, L., Wolfson, R. L., Orozco, J. M., Saxton, R. A., Scaria, S. M., Bar-Peled, L., et al. (2014). The Sestrins Interact With GATOR2 to Negatively Regulate the Amino-Acid-Sensing Pathway Upstream of mTORC1. Cell. Rep. 9 (1), 1–8. doi:10.1016/j.celrep.2014.09.014
Chou, S.-D., Prince, T., Gong, J., and Calderwood, S. K. (2012). mTOR Is Essential for the Proteotoxic Stress Response, HSF1 Activation and Heat Shock Protein Synthesis. PLoS One. 7 (6), e39679. doi:10.1371/journal.pone.0039679
Chrienova, Z., Nepovimova, E., and Kuca, K. (2021). The Role of mTOR in Age-Related Diseases. J. Enzyme Inhib. Med. Chem. 36 (1), 1679–1693. doi:10.1080/14756366.2021.1955873
de Araujo, M. E. G., Naschberger, A., Fürnrohr, B. G., Stasyk, T., Dunzendorfer-Matt, T., Lechner, S., et al. (2017). Crystal Structure of the Human Lysosomal mTORC1 Scaffold Complex and its Impact on Signaling. Science. 358 (6361), 377–381. doi:10.1126/science.aao1583
Demetriades, C., Doumpas, N., and Teleman, A. A. (2014). Regulation of TORC1 in Response to Amino Acid Starvation via Lysosomal Recruitment of TSC2. Cell. 156 (4), 786–799. doi:10.1016/j.cell.2014.01.024
Demetriades, C., Plescher, M., and Teleman, A. A. (2016). Lysosomal Recruitment of TSC2 Is a Universal Response to Cellular Stress. Nat. Commun. 7, 10662. doi:10.1038/ncomms10662
Desler, C., Marcker, M. L., Singh, K. K., and Rasmussen, L. J. (2011). The Importance of Mitochondrial DNA in Aging and Cancer. J. Aging Res. 2011, 1–9. doi:10.4061/2011/407536
Di Nardo, A., Kramvis, I., Cho, N., Sadowski, A., Meikle, L., Kwiatkowski, D. J., et al. (2009). Tuberous Sclerosis Complex Activity Is Required to Control Neuronal Stress Responses in an mTOR-Dependent Manner. J. Neurosci. 29 (18), 5926–5937. doi:10.1523/JNEUROSCI.0778-09.2009
Dibble, C. C., and Cantley, L. C. (2015). Regulation of mTORC1 by PI3K Signaling. Trends Cell. Biol. 25 (9), 545–555. doi:10.1016/j.tcb.2015.06.002
Dibble, C. C., Elis, W., Menon, S., Qin, W., Klekota, J., Asara, J. M., et al. (2012). TBC1D7 Is a Third Subunit of the TSC1-TSC2 Complex Upstream of mTORC1. Mol. Cell. 47 (4), 535–546. doi:10.1016/j.molcel.2012.06.009
Didiot, M.-C., Subramanian, M., Flatter, E., Mandel, J.-L., and Moine, H. (2009). Cells Lacking the Fragile X Mental Retardation Protein (FMRP) Have Normal RISC Activity but Exhibit Altered Stress Granule Assembly. MBoC. 20 (1), 428–437. doi:10.1091/mbc.E08-07-0737
Donnelly, N., Gorman, A. M., Gupta, S., and Samali, A. (2013). The eIF2α Kinases: Their Structures and Functions. Cell. Mol. Life Sci. 70 (19), 3493–3511. doi:10.1007/s00018-012-1252-6
Dossou, A. S., and Basu, A. (2019). The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers. 11 (10), 1422. doi:10.3390/cancers11101422
Durán, R. V., Oppliger, W., Robitaille, A. M., Heiserich, L., Skendaj, R., Gottlieb, E., et al. (2012). Glutaminolysis Activates Rag-mTORC1 Signaling. Mol. Cell. 47 (3), 349–358. doi:10.1016/j.molcel.2012.05.043
Emara, M. M., Fujimura, K., Sciaranghella, D., Ivanova, V., Ivanov, P., and Anderson, P. (2012). Hydrogen Peroxide Induces Stress Granule Formation Independent of eIF2α Phosphorylation. Biochem. Biophysical Res. Commun. 423 (4), 763–769. doi:10.1016/j.bbrc.2012.06.033
Fitzian, K., Brückner, A., Brohée, L., Zech, R., Antoni, C., Kiontke, S., et al. (2021). TSC1 Binding to Lysosomal PIPs Is Required for TSC Complex Translocation and mTORC1 Regulation. Mol. Cell. 81 (13), 2705–2721. doi:10.1016/j.molcel.2021.04.019
Fournier, M.-J., Coudert, L., Mellaoui, S., Adjibade, P., Gareau, C., Côté, M.-F., et al. (2013). Inactivation of the mTORC1-Eukaryotic Translation Initiation Factor 4E Pathway Alters Stress Granule Formation. Mol. Cell. Biol. 33 (11), 2285–2301. doi:10.1128/MCB.01517-12
Fujimura, K., Sasaki, A. T., and Anderson, P. (2012). Selenite Targets eIF4E-Binding Protein-1 to Inhibit Translation Initiation and Induce the Assembly of Non-Canonical Stress Granules. Nucleic Acids Res. 40 (16), 8099–8110. doi:10.1093/nar/gks566
Gallouzi, I.-E. (2009). Could Stress Granules Be Involved in Age-Related Diseases? Aging. 1 (9), 753–757. doi:10.18632/aging.100090
Gao, X., Jiang, L., Gong, Y., Chen, X., Ying, M., Zhu, H., et al. (2019). Stress Granule: A Promising Target for Cancer Treatment. Br. J. Pharmacol. 176 (23), 4421–4433. doi:10.1111/bph.14790
Garami, A., Zwartkruis, F. J. T., Nobukuni, T., Joaquin, M., Roccio, M., Stocker, H., et al. (2003). Insulin Activation of Rheb, a Mediator of mTOR/S6K/4E-BP Signaling, Is Inhibited by TSC1 and 2. Mol. Cell. 11 (6), 1457–1466. doi:10.1016/s1097-2765(03)00220-x
Gilks, N., Kedersha, N., Ayodele, M., Shen, L., Stoecklin, G., Dember, L. M., et al. (2004). Stress Granule Assembly Is Mediated by Prion-Like Aggregation of TIA-1. MBoC. 15 (12), 5383–5398. doi:10.1091/mbc.e04-08-0715
González, A., and Hall, M. N. (2017). Nutrient Sensing and TOR Signaling in Yeast and Mammals. EMBO J. 36 (4), 397–408. doi:10.15252/embj.201696010
González-Quiroz, M., Blondel, A., Sagredo, A., Hetz, C., Chevet, E., and Pedeux, R. (2020). When Endoplasmic Reticulum Proteostasis Meets the DNA Damage Response. Trends Cell Biol. 30 (11), 881–891. doi:10.1016/j.tcb.2020.09.002
Gu, X., Orozco, J. M., Saxton, R. A., Condon, K. J., Liu, G. Y., Krawczyk, P. A., et al. (2017). SAMTOR Is an S-Adenosylmethionine Sensor for the mTORC1 Pathway. Science. 358 (6364), 813–818. doi:10.1126/science.aao3265
Guha, P., Kaptan, E., Gade, P., Kalvakolanu, D. V., and Ahmed, H. (2017). Tunicamycin Induced Endoplasmic Reticulum Stress Promotes Apoptosis of Prostate Cancer Cells by Activating mTORC1. Oncotarget. 8 (40), 68191–68207. doi:10.18632/oncotarget.19277
Haar, E. V., Lee, S.-i., Bandhakavi, S., Griffin, T. J., and Kim, D.-H. (2007). Insulin Signalling to mTOR Mediated by the Akt/PKB Substrate PRAS40. Nat. Cell. Biol. 9 (3), 316–323. doi:10.1038/ncb1547
Hadari, Y. R., Tzahar, E., Nadiv, O., Rothenberg, P., Roberts, C. T., LeRoith, D., et al. (1992). Insulin and Insulinomimetic Agents Induce Activation of Phosphatidylinositol 3'-Kinase Upon its Association With Pp185 (IRS-1) in Intact Rat Livers. J. Biol. Chem. 267 (25), 17483–17486. doi:10.1016/s0021-9258(19)37065-6
Hara, K., Yonezawa, K., Weng, Q.-P., Kozlowski, M. T., Belham, C., and Avruch, J. (1998). Amino Acid Sufficiency and mTOR Regulate P70 S6 Kinase and eIF-4E BP1 through a Common Effector Mechanism. J. Biol. Chem. 273 (23), 14484–14494. doi:10.1074/jbc.273.23.14484
Heberle, A. M., Prentzell, M. T., van Eunen, K., Bakker, B. M., Grellscheid, S. N., and Thedieck, K. (2015). Molecular Mechanisms of mTOR Regulation by Stress. Mol. Cell Oncol. 2 (2), e970489. doi:10.4161/23723548.2014.970489
Heberle, A. M., Razquin Navas, P., Langelaar-Makkinje, M., Kasack, K., Sadik, A., Faessler, E., et al. (2019). The PI3K and MAPK/p38 Pathways Control Stress Granule Assembly in a Hierarchical Manner. Life Sci. Alliance. 2 (2), e201800257. doi:10.26508/lsa.201800257
Heitman, J., Movva, N., and Hall, M. (1991). Targets for Cell Cycle Arrest by the Immunosuppressant Rapamycin in Yeast. Science. 253 (5022), 905–909. doi:10.1126/science.1715094
Hofmann, S., Cherkasova, V., Bankhead, P., Bukau, B., and Stoecklin, G. (2012). Translation Suppression Promotes Stress Granule Formation and Cell Survival in Response to Cold Shock. MBoC. 23 (19), 3786–3800. doi:10.1091/mbc.E12-04-0296
Hofmann, S., Kedersha, N., Anderson, P., and Ivanov, P. (2021). Molecular Mechanisms of Stress Granule Assembly and Disassembly. Biochim. Biophys. Acta (Bba) - Mol. Cell. Res. 1868 (1), 118876. doi:10.1016/j.bbamcr.2020.118876
Hoxhaj, G., and Manning, B. D. (2020). The PI3K-AKT Network at the Interface of Oncogenic Signalling and Cancer Metabolism. Nat. Rev. Cancer. 20 (2), 74–88. doi:10.1038/s41568-019-0216-7
Hsieh, A. C., Liu, Y., Edlind, M. P., Ingolia, N. T., Janes, M. R., Sher, A., et al. (2012). The Translational Landscape of mTOR Signalling Steers Cancer Initiation and Metastasis. Nature. 485 (7396), 55–61. doi:10.1038/nature10912
Hsu, P. P., Kang, S. A., Rameseder, J., Zhang, Y., Ottina, K. A., Lim, D., et al. (2011). The mTOR-Regulated Phosphoproteome Reveals a Mechanism of mTORC1-Mediated Inhibition of Growth Factor Signaling. Science. 332 (6035), 1317–1322. doi:10.1126/science.1199498
Huang, C., Li, J., Ke, Q., Leonard, S. S., Jiang, B. H., Zhong, X. S., et al. (2002). Ultraviolet-Induced Phosphorylation of p70(S6K) at Thr(389) and Thr(421)/Ser(424) Involves Hydrogen Peroxide and Mammalian Target of Rapamycin but Not Akt and Atypical Protein Kinase C. Cancer Res. 62 (20), 5689–5697.
Inoki, K., Li, Y., Xu, T., and Guan, K. L. (2003). Rheb GTPase Is a Direct Target of TSC2 GAP Activity and Regulates mTOR Signaling. Genes Development. 17 (15), 1829–1834. doi:10.1101/gad.1110003
Inoki, K., Li, Y., Zhu, T., Wu, J., and Guan, K.-L. (2002). TSC2 Is Phosphorylated and Inhibited by Akt and Suppresses mTOR Signalling. Nat. Cell. Biol. 4 (9), 648–657. doi:10.1038/ncb839
Inoki, K., Ouyang, H., Zhu, T., Lindvall, C., Wang, Y., Zhang, X., et al. (2006). TSC2 Integrates Wnt and Energy Signals via a Coordinated Phosphorylation by AMPK and GSK3 to Regulate Cell Growth. Cell. 126 (5), 955–968. doi:10.1016/j.cell.2006.06.055
Ivanov, P., and Anderson, P. (2019). Stress Granules and Processing Bodies in Translational Control. Cold Spring Harb Perspect. Biol. 11 (5), a032813. doi:10.1101/cshperspect.a032813
Jeon, P., and Lee, J. A. (2021). Dr. Jekyll and Mr. Hyde? Physiology and Pathology of Neuronal Stress Granules. Front. Cell. Dev. Biol. 9, 609698. doi:10.3389/fcell.2021.609698
Jewell, J. L., Kim, Y. C., Russell, R. C., Yu, F.-X., Park, H. W., Plouffe, S. W., et al. (2015). Differential Regulation of mTORC1 by Leucine and Glutamine. Science. 347 (6218), 194–198. doi:10.1126/science.1259472
Jung, J., Genau, H. M., and Behrends, C. (2015). Amino Acid-dependent mTORC1 Regulation by the Lysosomal Membrane Protein SLC38A9. Mol. Cell. Biol. 35 (14), 2479–2494. doi:10.1128/MCB.00125-15
Kato, H., Nakajima, S., Saito, Y., Takahashi, S., Katoh, R., and Kitamura, M. (2012). mTORC1 Serves ER Stress-Triggered Apoptosis via Selective Activation of the IRE1-JNK Pathway. Cell Death Differ. 19 (2), 310–320. doi:10.1038/cdd.2011.98
Kedersha, N., and Anderson, P. (2007). Mammalian Stress Granules and Processing Bodies. Methods Enzymol. 431, 61–81. doi:10.1016/S0076-6879(07)31005-7
Kedersha, N., Chen, S., Gilks, N., Li, W., Miller, I. J., Stahl, J., et al. (2002). Evidence That Ternary Complex (eIF2-GTP-tRNAiMet)-Deficient Preinitiation Complexes Are Core Constituents of Mammalian Stress Granules. MBoC. 13 (1), 195–210. doi:10.1091/mbc.01-05-0221
Kedersha, N., Ivanov, P., and Anderson, P. (2013). Stress Granules and Cell Signaling: More Than Just a Passing Phase? Trends Biochem. Sci. 38 (10), 494–506. doi:10.1016/j.tibs.2013.07.004
Kedersha, N. L., Gupta, M., Li, W., Miller, I., and Anderson, P. (1999). RNA-Binding Proteins Tia-1 and Tiar Link the Phosphorylation of Eif-2α to the Assembly of Mammalian Stress Granules. J. Cell. Biol. 147 (7), 1431–1442. doi:10.1083/jcb.147.7.1431
Khan, S. S., Singer, B. D., and Vaughan, D. E. (2017). Molecular and Physiological Manifestations and Measurement of Aging in Humans. Aging Cell. 16 (4), 624–633. doi:10.1111/acel.12601
Kim, E., Goraksha-Hicks, P., Li, L., Neufeld, T. P., and Guan, K.-L. (2008). Regulation of TORC1 by Rag GTPases in Nutrient Response. Nat. Cell. Biol. 10 (8), 935–945. doi:10.1038/ncb1753
Kim, J.-H., Choi, T. G., Park, S., Yun, H. R., Nguyen, N. N. Y., Jo, Y. H., et al. (2018). Mitochondrial ROS-Derived PTEN Oxidation Activates PI3K Pathway for mTOR-Induced Myogenic Autophagy. Cell. Death Differ. 25 (11), 1921–1937. doi:10.1038/s41418-018-0165-9
Kim, J., and Guan, K.-L. (2019). mTOR as a central Hub of Nutrient Signalling and Cell Growth. Nat. Cell. Biol. 21 (1), 63–71. doi:10.1038/s41556-018-0205-1
Kitada, M., Xu, J., Ogura, Y., Monno, I., and Koya, D. (2020). Mechanism of Activation of Mechanistic Target of Rapamycin Complex 1 by Methionine. Front. Cell. Dev. Biol. 8, 715. doi:10.3389/fcell.2020.00715
Kolupaeva, V. (2019). Serine-threonine Protein Phosphatases: Lost in Translation. Biochim. Biophys. Acta (Bba) - Mol. Cell. Res. 1866 (1), 83–89. doi:10.1016/j.bbamcr.2018.08.006
Kosmas, K., Filippakis, H., Khabibullin, D., Turkiewicz, M., Lam, H. C., Yu, J., et al. (2021). TSC2 Interacts With HDLBP/Vigilin and Regulates Stress Granule Formation. Mol. Cancer Res. 19 (8), 1389–1397. doi:10.1158/1541-7786.mcr-20-1046
Kovacina, K. S., Park, G. Y., Bae, S. S., Guzzetta, A. W., Schaefer, E., Birnbaum, M. J., et al. (2003). Identification of a Proline-Rich Akt Substrate as a 14-3-3 Binding Partner. J. Biol. Chem. 278 (12), 10189–10194. doi:10.1074/jbc.M210837200
Lang, B. J., Guerrero, M. E., Prince, T. L., Okusha, Y., Bonorino, C., and Calderwood, S. K. (2021). The Functions and Regulation of Heat Shock Proteins; Key Orchestrators of Proteostasis and the Heat Shock Response. Arch. Toxicol. 95 (6), 1943–1970. doi:10.1007/s00204-021-03070-8
Lee, J.-A. (2015). Autophagy Manages Disease-Associated Stress Granules. Oncotarget. 6 (31), 30421–30422. doi:10.18632/oncotarget.5902
Li, L. J., Chai, Y., Guo, X. J., Chu, S. L., and Zhang, L. S. (2018). Effects of Endoplasmic Reticulum Stress on Autophagy and Apoptosis of Human Leukemia Cells via Inhibition of the PI3K/AKT/mTOR Signaling Pathway. Mol. Med. Rep. 17 (6), 7886–7892. doi:10.3892/mmr.2018.8840
Lian, X. J., and Gallouzi, I.-E. (2009). Oxidative Stress Increases the Number of Stress Granules in Senescent Cells and Triggers a Rapid Decrease in P21 Translation. J. Biol. Chem. 284 (13), 8877–8887. doi:10.1074/jbc.M806372200
Liao, Y.-C., Fernandopulle, M. S., Wang, G., Choi, H., Hao, L., Drerup, C. M., et al. (2019). RNA Granules Hitchhike on Lysosomes for Long-Distance Transport, Using Annexin A11 as a Molecular Tether. Cell. 179 (1), 147–164. doi:10.1016/j.cell.2019.08.050
Liguori, I., Russo, G., Curcio, F., Bulli, G., Aran, L., Della-Morte, D., et al. (2018). Oxidative Stress, Aging, and Diseases. Cia. 13, 757–772. doi:10.2147/CIA.S158513
Liu, G. Y., and Sabatini, D. M. (2020). mTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell. Biol. 21 (4), 183–203. doi:10.1038/s41580-019-0199-y
Long, X., Lin, Y., Ortiz-Vega, S., Yonezawa, K., and Avruch, J. (2005). Rheb Binds and Regulates the mTOR Kinase. Curr. Biol. 15 (8), 702–713. doi:10.1016/j.cub.2005.02.053
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., and Kroemer, G. (2013). The Hallmarks of Aging. Cell. 153 (6), 1194–1217. doi:10.1016/j.cell.2013.05.039
Ma, L., Chen, Z., Erdjument-Bromage, H., Tempst, P., and Pandolfi, P. P. (2005). Phosphorylation and Functional Inactivation of TSC2 by Erk. Cell. 121 (2), 179–193. doi:10.1016/j.cell.2005.02.031
Ma, Y., Vassetzky, Y., and Dokudovskaya, S. (2018). mTORC1 Pathway in DNA Damage Response. Biochim. Biophys. Acta (Bba) - Mol. Cell. Res. 1865 (9), 1293–1311. doi:10.1016/j.bbamcr.2018.06.011
Mahboubi, H., and Stochaj, U. (2017). Cytoplasmic Stress Granules: Dynamic Modulators of Cell Signaling and Disease. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1863 (4), 884–895. doi:10.1016/j.bbadis.2016.12.022
Malhotra, J. D., and Kaufman, R. J. (2007). Endoplasmic Reticulum Stress and Oxidative Stress: a Vicious Cycle or a Double-Edged Sword? Antioxid. Redox Signaling. 9 (12), 2277–2294. doi:10.1089/ars.2007.1782
Marciniak, S. J., Yun, C. Y., Oyadomari, S., Novoa, I., Zhang, Y., Jungreis, R., et al. (2004). CHOP Induces Death by Promoting Protein Synthesis and Oxidation in the Stressed Endoplasmic Reticulum. Genes Development. 18 (24), 3066–3077. doi:10.1101/gad.1250704
Margittai, É., and Sitia, R. (2011). Oxidative Protein Folding in the Secretory Pathway and Redox Signaling Across Compartments and Cells. Traffic. 12 (1), 1–8. doi:10.1111/j.1600-0854.2010.01108.x
Markmiller, S., Soltanieh, S., Server, K. L., Mak, R., Jin, W., Fang, M. Y., et al. (2018). Context-Dependent and Disease-Specific Diversity in Protein Interactions Within Stress Granules. Cell. 172 (3), 590–604. e513. doi:10.1016/j.cell.2017.12.032
Marrone, L., Poser, I., Casci, I., Japtok, J., Reinhardt, P., Janosch, A., et al. (2018). Isogenic FUS-eGFP iPSC Reporter Lines Enable Quantification of FUS Stress Granule Pathology that Is Rescued by Drugs Inducing Autophagy. Stem Cell. Rep. 10 (2), 375–389. doi:10.1016/j.stemcr.2017.12.018
Mathieu, C., Pappu, R. V., and Taylor, J. P. (2020). Beyond Aggregation: Pathological Phase Transitions in Neurodegenerative Disease. Science. 370 (6512), 56–60. doi:10.1126/science.abb8032
Matsuki, H., Takahashi, M., Higuchi, M., Makokha, G. N., Oie, M., and Fujii, M. (2013). Both G3BP1 and G3BP2 Contribute to Stress Granule Formation. Genes Cells. 18 (2), 135–146. doi:10.1111/gtc.12023
Maynard, S., Fang, E. F., Scheibye-Knudsen, M., Croteau, D. L., and Bohr, V. A. (2015). DNA Damage, DNA Repair, Aging, and Neurodegeneration. Cold Spring Harb Perspect. Med. 5 (10), a025130. doi:10.1101/cshperspect.a025130
Mazan-Mamczarz, K., Peroutka, R. J., Steinhardt, J. J., Gidoni, M., Zhang, Y., Lehrmann, E., et al. (2015). Distinct Inhibitory Effects on mTOR Signaling by Ethanol and INK128 in Diffuse Large B-Cell Lymphoma. Cell Commun Signal. 13, 15. doi:10.1186/s12964-015-0091-0
Mazroui, R., Huot, M. E., Tremblay, S., Filion, C., Labelle, Y., and Khandjian, E. W. (2002). Trapping of Messenger RNA by Fragile X Mental Retardation Protein into Cytoplasmic Granules Induces Translation Repression. Hum. Mol. Genet. 11 (24), 3007–3017. doi:10.1093/hmg/11.24.3007
Mediani, L., Antoniani, F., Galli, V., Vinet, J., Carrà, A. D., Bigi, I., et al. (2021). Hsp90‐Mediated Regulation of DYRK3 Couples Stress Granule Disassembly and Growth via mTORC1 Signaling. EMBO Rep. 22 (5), e51740. doi:10.15252/embr.202051740
Melick, C. H., and Jewell, J. L. (2020). Regulation of mTORC1 by Upstream Stimuli. Genes. 11 (9), 989. doi:10.3390/genes11090989
Meng, D., Yang, Q., Wang, H., Melick, C. H., Navlani, R., Frank, A. R., et al. (2020). Glutamine and Asparagine Activate mTORC1 Independently of Rag GTPases. J. Biol. Chem. 295 (10), 2890–2899. doi:10.1074/jbc.AC119.011578
Menon, S., Dibble, C. C., Talbott, G., Hoxhaj, G., Valvezan, A. J., Takahashi, H., et al. (2014). Spatial Control of the TSC Complex Integrates Insulin and Nutrient Regulation of mTORC1 at the Lysosome. Cell. 156 (4), 771–785. doi:10.1016/j.cell.2013.11.049
Merrick, W. C., and Pavitt, G. D. (2018). Protein Synthesis Initiation in Eukaryotic Cells. Cold Spring Harb Perspect. Biol. 10 (12), a033092. doi:10.1101/cshperspect.a033092
Morán Luengo, T., Mayer, M. P., and Rüdiger, S. G. D. (2019). The Hsp70-Hsp90 Chaperone Cascade in Protein Folding. Trends Cell. Biol. 29 (2), 164–177. doi:10.1016/j.tcb.2018.10.004
Mossmann, D., Park, S., and Hall, M. N. (2018). mTOR Signalling and Cellular Metabolism Are Mutual Determinants in Cancer. Nat. Rev. Cancer. 18 (12), 744–757. doi:10.1038/s41568-018-0074-8
Muñoz-Gámez, J. A., Rodríguez-Vargas, J. M., Quiles-Pérez, R., Aguilar-Quesada, R., Martín-Oliva, D., de Murcia, G., et al. (2009). PARP-1 Is Involved in Autophagy Induced by DNA Damage. Autophagy. 5 (1), 61–74. doi:10.4161/auto.5.1.7272
Nascimento, E. B. M., Snel, M., Guigas, B., van der Zon, G. C. M., Kriek, J., Maassen, J. A., et al. (2010). Phosphorylation of PRAS40 on Thr246 by PKB/AKT Facilitates Efficient Phosphorylation of Ser183 by mTORC1. Cell Signal. 22 (6), 961–967. doi:10.1016/j.cellsig.2010.02.002
Ohoka, N., Yoshii, S., Hattori, T., Onozaki, K., and Hayashi, H. (2005). TRB3, a Novel ER Stress-Inducible Gene, Is Induced via ATF4-CHOP Pathway and Is Involved in Cell Death. EMBO J. 24 (6), 1243–1255. doi:10.1038/sj.emboj.7600596
Omer, A., Patel, D., Lian, X. J., Sadek, J., Di Marco, S., Pause, A., et al. (2018). Stress Granules Counteract Senescence by Sequestration of PAI‐1. EMBO Rep. 19 (5), e44722. doi:10.15252/embr.201744722
Panas, M. D., Ivanov, P., and Anderson, P. (2016). Mechanistic Insights into Mammalian Stress Granule Dynamics. J. Cell. Biol. 215 (3), 313–323. doi:10.1083/jcb.201609081
Papadopoli, D., Boulay, K., Kazak, L., Pollak, M., Mallette, F., Topisirovic, I., et al. (2019). mTOR as a central Regulator of Lifespan and Aging. F1000Res. 8, 998. doi:10.12688/f1000research.17196.1
Parmigiani, A., Nourbakhsh, A., Ding, B., Wang, W., Kim, Y. C., Akopiants, K., et al. (2014). Sestrins Inhibit mTORC1 Kinase Activation Through the GATOR Complex. Cell. Rep. 9 (4), 1281–1291. doi:10.1016/j.celrep.2014.10.019
Peng, M., Yin, N., and Li, M. O. (2014). Sestrins Function as Guanine Nucleotide Dissociation Inhibitors for Rag GTPases to Control mTORC1 Signaling. Cell. 159 (1), 122–133. doi:10.1016/j.cell.2014.08.038
Peran, I., and Mittag, T. (2020). Molecular Structure in Biomolecular Condensates. Curr. Opin. Struct. Biol. 60, 17–26. doi:10.1016/j.sbi.2019.09.007
Plescher, M., Teleman, A. A., and Demetriades, C. (2015). TSC2 Mediates Hyperosmotic Stress-Induced Inactivation of mTORC1. Sci. Rep. 5, 13828. doi:10.1038/srep13828
Prentzell, M. T., Rehbein, U., Cadena Sandoval, M., De Meulemeester, A.-S., Baumeister, R., Brohée, L., et al. (2021). G3BPs Tether the TSC Complex to Lysosomes and Suppress mTORC1 Signaling. Cell. 184 (3), 655–674. doi:10.1016/j.cell.2020.12.024
Qin, L., Wang, Z., Tao, L., and Wang, Y. (2010). ER Stress Negatively Regulates AKT/TSC/mTOR Pathway to Enhance Autophagy. Autophagy. 6 (2), 239–247. doi:10.4161/auto.6.2.11062
Qin, X., Jiang, B., and Zhang, Y. (2016). 4E-BP1, a Multifactor Regulated Multifunctional Protein. Cell Cycle. 15 (6), 781–786. doi:10.1080/15384101.2016.1151581
Querfurth, H., and Lee, H.-K. (2021). Mammalian/Mechanistic Target of Rapamycin (mTOR) Complexes in Neurodegeneration. Mol. Neurodegeneration. 16 (1), 44. doi:10.1186/s13024-021-00428-5
Rabanal-Ruiz, Y., and Korolchuk, V. (2018). mTORC1 and Nutrient Homeostasis: The Central Role of the Lysosome. Ijms. 19 (3), 818. doi:10.3390/ijms19030818
Rabanal-Ruiz, Y., Otten, E. G., and Korolchuk, V. I. (2017). mTORC1 as the Main Gateway to Autophagy. Essays Biochem. 61 (6), 565–584. doi:10.1042/EBC20170027
Ramlaul, K., Fu, W., Li, H., de Martin Garrido, N., He, L., Trivedi, M., et al. (2021). Architecture of the Tuberous Sclerosis Protein Complex. J. Mol. Biol. 433 (2), 166743. doi:10.1016/j.jmb.2020.166743
Razquin Navas, P., and Thedieck, K. (2017). Differential Control of Ageing and Lifespan by Isoforms and Splice Variants Across the mTOR Network. Essays Biochem. 61 (3), 349–368. doi:10.1042/EBC20160086
Rebsamen, M., Pochini, L., Stasyk, T., de Araújo, M. E. G., Galluccio, M., Kandasamy, R. K., et al. (2015). SLC38A9 Is a Component of the Lysosomal Amino Acid Sensing Machinery That Controls mTORC1. Nature. 519 (7544), 477–481. doi:10.1038/nature14107
Reineke, L. C., and Neilson, J. R. (2019). Differences Between Acute and Chronic Stress Granules, and How These Differences May Impact Function in Human Disease. Biochem. Pharmacol. 162, 123–131. doi:10.1016/j.bcp.2018.10.009
Rodríguez-Vargas, J. M., Ruiz-Magaña, M. J., Ruiz-Ruiz, C., Majuelos-Melguizo, J., Peralta-Leal, A., Rodríguez, M. I., et al. (2012). ROS-Induced DNA Damage and PARP-1 Are Required for Optimal Induction of Starvation-Induced Autophagy. Cell Res. 22 (7), 1181–1198. doi:10.1038/cr.2012.70
Roux, P. P., and Topisirovic, I. (2018). Signaling Pathways Involved in the Regulation of mRNA Translation. Mol. Cell. Biol. 38 (12), e00070. doi:10.1128/MCB.00070-18
Ryu, H.-H., Jun, M.-H., Min, K.-J., Jang, D.-J., Lee, Y.-S., Kim, H. K., et al. (2014). Autophagy Regulates Amyotrophic Lateral Sclerosis-Linked Fused in Sarcoma-Positive Stress Granules in Neurons. Neurobiol. Aging. 35 (12), 2822–2831. doi:10.1016/j.neurobiolaging.2014.07.026
Sabatini, D. M., Erdjument-Bromage, H., Lui, M., Tempst, P., and Snyder, S. H. (1994). RAFT1: a Mammalian Protein That Binds to FKBP12 in a Rapamycin-Dependent Fashion and Is Homologous to Yeast TORs. Cell. 78 (1), 35–43. doi:10.1016/0092-8674(94)90570-3
Sancak, Y., Bar-Peled, L., Zoncu, R., Markhard, A. L., Nada, S., and Sabatini, D. M. (2010). Ragulator-Rag Complex Targets mTORC1 to the Lysosomal Surface and Is Necessary for its Activation by Amino Acids. Cell. 141 (2), 290–303. doi:10.1016/j.cell.2010.02.024
Sancak, Y., Peterson, T. R., Shaul, Y. D., Lindquist, R. A., Thoreen, C. C., Bar-Peled, L., et al. (2008). The Rag GTPases Bind Raptor and Mediate Amino Acid Signaling to mTORC1. Science. 320 (5882), 1496–1501. doi:10.1126/science.1157535
Sarbassov, D. D., Ali, S. M., Sengupta, S., Sheen, J.-H., Hsu, P. P., Bagley, A. F., et al. (2006). Prolonged Rapamycin Treatment Inhibits mTORC2 Assembly and Akt/PKB. Mol. Cell. 22 (2), 159–168. doi:10.1016/j.molcel.2006.03.029
Saxton, R. A., Chantranupong, L., Knockenhauer, K. E., Schwartz, T. U., and Sabatini, D. M. (2016a). Mechanism of Arginine Sensing by CASTOR1 Upstream of mTORC1. Nature. 536 (7615), 229–233. doi:10.1038/nature19079
Saxton, R. A., Knockenhauer, K. E., Wolfson, R. L., Chantranupong, L., Pacold, M. E., Wang, T., et al. (2016b). Structural Basis for Leucine Sensing by the Sestrin2-mTORC1 Pathway. Science. 351 (6268), 53–58. doi:10.1126/science.aad2087
Schopf, F. H., Biebl, M. M., and Buchner, J. (2017). The HSP90 Chaperone Machinery. Nat. Rev. Mol. Cell. Biol. 18 (6), 345–360. doi:10.1038/nrm.2017.20
Seguin, S. J., Morelli, F. F., Vinet, J., Amore, D., De Biasi, S., Poletti, A., et al. (2014). Inhibition of Autophagy, Lysosome and VCP Function Impairs Stress Granule Assembly. Cel Death Differ. 21 (12), 1838–1851. doi:10.1038/cdd.2014.103
Selvarajah, J., Elia, A., Carroll, V. A., and Moumen, A. (2015). DNA Damage-Induced S and G2/M Cell Cycle Arrest Requires mTORC2-dependent Regulation of Chk1. Oncotarget. 6 (1), 427–440. doi:10.18632/oncotarget.2813
Sfakianos, A. P., Mellor, L. E., Pang, Y. F., Kritsiligkou, P., Needs, H., Abou-Hamdan, H., et al. (2018). The mTOR-S6 Kinase Pathway Promotes Stress Granule Assembly. Cell. Death Differ. 25 (10), 1766–1780. doi:10.1038/s41418-018-0076-9
Sikora, E., Bielak-Zmijewska, A., Dudkowska, M., Krzystyniak, A., Mosieniak, G., Wesierska, M., et al. (2021). Cellular Senescence in Brain Aging. Front. Aging Neurosci. 13, 646924. doi:10.3389/fnagi.2021.646924
Silva, J. M., Rodrigues, S., Sampaio-Marques, B., Gomes, P., Neves-Carvalho, A., Dioli, C., et al. (2019). Dysregulation of Autophagy and Stress Granule-Related Proteins in Stress-Driven Tau Pathology. Cel Death Differ. 26 (8), 1411–1427. doi:10.1038/s41418-018-0217-1
Sim, E., Irollo, E., and Grabocka, E. (2019). Evaluating Stress Granules in Pancreatic Cancer In Vitro and In Vivo. Methods Mol. Biol. 1882, 183–195. doi:10.1007/978-1-4939-8879-2_17
Snead, W. T., and Gladfelter, A. S. (2019). The Control Centers of Biomolecular Phase Separation: How Membrane Surfaces, PTMs, and Active Processes Regulate Condensation. Mol. Cell. 76 (2), 295–305. doi:10.1016/j.molcel.2019.09.016
Somasekharan, S. P., El-Naggar, A., Leprivier, G., Cheng, H., Hajee, S., Grunewald, T. G. P., et al. (2015). YB-1 Regulates Stress Granule Formation and Tumor Progression by Translationally Activating G3BP1. J. Cell Biol. 208 (7), 913–929. doi:10.1083/jcb.201411047
Son, S. M., Park, S. J., Lee, H., Siddiqi, F., Lee, J. E., Menzies, F. M., et al. (2019). Leucine Signals to mTORC1 via Its Metabolite Acetyl-Coenzyme A. Cell Metab. 29 (1), 192–201. doi:10.1016/j.cmet.2018.08.013
Sonenberg, N., and Hinnebusch, A. G. (2009). Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell. 136 (4), 731–745. doi:10.1016/j.cell.2009.01.042
Song, S., Lam, E. W.-F., Tchkonia, T., Kirkland, J. L., and Sun, Y. (2020). Senescent Cells: Emerging Targets for Human Aging and Age-Related Diseases. Trends Biochem. Sci. 45 (7), 578–592. doi:10.1016/j.tibs.2020.03.008
Stracka, D., Jozefczuk, S., Rudroff, F., Sauer, U., and Hall, M. N. (2014). Nitrogen Source Activates TOR (Target of Rapamycin) Complex 1 via Glutamine and Independently of Gtr/Rag Proteins. J. Biol. Chem. 289 (36), 25010–25020. doi:10.1074/jbc.M114.574335
Su, K.-H., and Dai, C. (2017). mTORC1 Senses Stresses: Coupling Stress to Proteostasis. Bioessays. 39 (5), 1600268. doi:10.1002/bies.201600268
Sun, X. J., Rothenberg, P., Kahn, C. R., Backer, J. M., Araki, E., Wilden, P. A., et al. (1991). Structure of the Insulin Receptor Substrate IRS-1 Defines a Unique Signal Transduction Protein. Nature. 352 (6330), 73–77. doi:10.1038/352073a0
Tahmasebi, S., Khoutorsky, A., Mathews, M. B., and Sonenberg, N. (2018). Translation Deregulation in Human Disease. Nat. Rev. Mol. Cell. Biol. 19 (12), 791–807. doi:10.1038/s41580-018-0034-x
Takahara, T., Amemiya, Y., Sugiyama, R., Maki, M., and Shibata, H. (2020). Amino Acid-dependent Control of mTORC1 Signaling: a Variety of Regulatory Modes. J. Biomed. Sci. 27 (1), 87. doi:10.1186/s12929-020-00679-2
Takahara, T., and Maeda, T. (2012). Transient Sequestration of TORC1 Into Stress Granules During Heat Stress. Mol. Cell. 47 (2), 242–252. doi:10.1016/j.molcel.2012.05.019
Takahashi, M., Higuchi, M., Matsuki, H., Yoshita, M., Ohsawa, T., Oie, M., et al. (2013). Stress Granules Inhibit Apoptosis by Reducing Reactive Oxygen Species Production. Mol. Cel Biol. 33 (4), 815–829. doi:10.1128/MCB.00763-12
Tatebe, H., and Shiozaki, K. (2017). Evolutionary Conservation of the Components in the TOR Signaling Pathways. Biomolecules. 7 (4), 77. doi:10.3390/biom7040077
Tee, A. R., Anjum, R., and Blenis, J. (2003a). Inactivation of the Tuberous Sclerosis Complex-1 and -2 Gene Products Occurs by Phosphoinositide 3-kinase/Akt-Dependent and -Independent Phosphorylation of Tuberin. J. Biol. Chem. 278 (39), 37288–37296. doi:10.1074/jbc.M303257200
Tee, A. R., Manning, B. D., Roux, P. P., Cantley, L. C., and Blenis, J. (2003b). Tuberous Sclerosis Complex Gene Products, Tuberin and Hamartin, Control mTOR Signaling by Acting as a GTPase-Activating Protein Complex toward Rheb. Curr. Biol. 13 (15), 1259–1268. doi:10.1016/s0960-9822(03)00506-2
Tee, A. (2018). The Target of Rapamycin and Mechanisms of Cell Growth. Ijms. 19 (3), 880. doi:10.3390/ijms19030880
Teis, D., Wunderlich, W., and Huber, L. A. (2002). Localization of the MP1-MAPK Scaffold Complex to Endosomes Is Mediated by P14 and Required for Signal Transduction. Developmental Cell. 3 (6), 803–814. doi:10.1016/s1534-5807(02)00364-7
Thedieck, K., Holzwarth, B., Prentzell, M. T., Boehlke, C., Kläsener, K., Ruf, S., et al. (2013). Inhibition of mTORC1 by Astrin and Stress Granules Prevents Apoptosis in Cancer Cells. Cell. 154 (4), 859–874. doi:10.1016/j.cell.2013.07.031
Thoreen, C. C. (2017). The Molecular Basis of mTORC1-Regulated Translation. Biochem. Soc. Trans. 45 (1), 213–221. doi:10.1042/BST20160072
Titorenko, V. I., and Terlecky, S. R. (2011). Peroxisome Metabolism and Cellular Aging. Traffic. 12 (3), 252–259. doi:10.1111/j.1600-0854.2010.01144.x
Tourriere, H., Chebli, K., Zekri, L., Courselaud, B., Blanchard, J. M., Bertrand, E., et al. (2003). The RasGAP-Associated Endoribonuclease G3BP Assembles Stress Granules. J. Cell. Biol. 160 (6), 823–831. doi:10.1083/jcb.200212128
Tzatsos, A., and Kandror, K. V. (2006). Nutrients Suppress Phosphatidylinositol 3-Kinase/Akt Signaling via Raptor-dependent mTOR-Mediated Insulin Receptor Substrate 1 Phosphorylation. Mol. Cell. Biol. 26 (1), 63–76. doi:10.1128/MCB.26.1.63-76.2006
Um, S. H., Frigerio, F., Watanabe, M., Picard, F., Joaquin, M., Sticker, M., et al. (2004). Absence of S6K1 Protects against Age- and Diet-Induced Obesity While Enhancing Insulin Sensitivity. Nature. 431 (7005), 200–205. doi:10.1038/nature02866
Van Treeck, B., and Parker, R. (2018). Emerging Roles for Intermolecular RNA-RNA Interactions in RNP Assemblies. Cell. 174 (4), 791–802. doi:10.1016/j.cell.2018.07.023
Vanhaesebroeck, B., Stephens, L., and Hawkins, P. (2012). PI3K Signalling: the Path to Discovery and Understanding. Nat. Rev. Mol. Cel Biol. 13 (3), 195–203. doi:10.1038/nrm3290
Vigneri, R., Goldfine, I. D., and Frittitta, L. (2016). Insulin, Insulin Receptors, and Cancer. J. Endocrinol. Invest. 39 (12), 1365–1376. doi:10.1007/s40618-016-0508-7
Vilas-Boas, F. d. A. S., da Silva, A. M., de Sousa, L. P., Lima, K. M., Vago, J. P., Bittencourt, L. F. F., et al. (2016). Impairment of Stress Granule Assembly via Inhibition of the eIF2alpha Phosphorylation Sensitizes Glioma Cells to Chemotherapeutic Agents. J. Neurooncol. 127 (2), 253–260. doi:10.1007/s11060-015-2043-3
Wang, S., Tsun, Z.-Y., Wolfson, R. L., Shen, K., Wyant, G. A., Plovanich, M. E., et al. (2015). Lysosomal Amino Acid Transporter SLC38A9 Signals Arginine Sufficiency to mTORC1. Science. 347 (6218), 188–194. doi:10.1126/science.1257132
Wang, X., and Proud, C. G. (1997). p70 S6 Kinase Is Activated by Sodium Arsenite in Adult Rat Cardiomyocytes: Roles for Phosphatidylinositol 3-kinase and P38 MAP Kinase. Biochem. Biophysical Res. Commun. 238 (1), 207–212. doi:10.1006/bbrc.1997.7273
Wanner, E., Thoppil, H., and Riabowol, K. (2020). Senescence and Apoptosis: Architects of Mammalian Development. Front. Cel Dev. Biol. 8, 620089. doi:10.3389/fcell.2020.620089
Weichhart, T. (2018). mTOR as Regulator of Lifespan, Aging, and Cellular Senescence: A Mini-Review. Gerontology. 64 (2), 127–134. doi:10.1159/000484629
Wiedner, H. J., and Giudice, J. (2021). It's Not Just a Phase: Function and Characteristics of RNA-Binding Proteins in Phase Separation. Nat. Struct. Mol. Biol. 28 (6), 465–473. doi:10.1038/s41594-021-00601-w
Wippich, F., Bodenmiller, B., Trajkovska, M. G., Wanka, S., Aebersold, R., and Pelkmans, L. (2013). Dual Specificity Kinase DYRK3 Couples Stress Granule Condensation/Dissolution to mTORC1 Signaling. Cell. 152 (4), 791–805. doi:10.1016/j.cell.2013.01.033
Wolfson, R. L., Chantranupong, L., Saxton, R. A., Shen, K., Scaria, S. M., Cantor, J. R., et al. (2016). Sestrin2 Is a Leucine Sensor for the mTORC1 Pathway. Science. 351 (6268), 43–48. doi:10.1126/science.aab2674
Wolfson, R. L., and Sabatini, D. M. (2017). The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cel Metab. 26 (2), 301–309. doi:10.1016/j.cmet.2017.07.001
Wolozin, B., and Ivanov, P. (2019). Stress Granules and Neurodegeneration. Nat. Rev. Neurosci. 20 (11), 649–666. doi:10.1038/s41583-019-0222-5
Wong, S. Q., Kumar, A. V., Mills, J., and Lapierre, L. R. (2020). Autophagy in Aging and Longevity. Hum. Genet. 139 (3), 277–290. doi:10.1007/s00439-019-02031-7
Wu, X.-N., Wang, X.-K., Wu, S.-Q., Lu, J., Zheng, M., Wang, Y.-H., et al. (2011). Phosphorylation of Raptor by P38β Participates in Arsenite-Induced Mammalian Target of Rapamycin Complex 1 (mTORC1) Activation. J. Biol. Chem. 286 (36), 31501–31511. doi:10.1074/jbc.M111.233122
Yang, H., Rudge, D. G., Koos, J. D., Vaidialingam, B., Yang, H. J., and Pavletich, N. P. (2013). mTOR Kinase Structure, Mechanism and Regulation. Nature. 497 (7448), 217–223. doi:10.1038/nature12122
Yang, H., Yu, Z., Chen, X., Li, J., Li, N., Cheng, J., et al. (2021). Structural Insights into TSC Complex Assembly and GAP Activity on Rheb. Nat. Commun. 12 (1), 339. doi:10.1038/s41467-020-20522-4
Yang, S., Zhang, Y., Ting, C.-Y., Bettedi, L., Kim, K., Ghaniam, E., et al. (2020). The Rag GTPase Regulates the Dynamic Behavior of TSC Downstream of Both Amino Acid and Growth Factor Restriction. Developmental Cell. 55 (3), 272–288. doi:10.1016/j.devcel.2020.08.006
Yang, Y., and Klionsky, D. J. (2020). Autophagy and Disease: Unanswered Questions. Cel Death Differ. 27 (3), 858–871. doi:10.1038/s41418-019-0480-9
Ying, S., and Khaperskyy, D. A. (2020). UV Damage Induces G3BP1-Dependent Stress Granule Formation That Is Not Driven by Translation Arrest via mTOR Inhibition. J. Cell. Sci. 133 (20). doi:10.1242/jcs.248310
Yoboue, E. D., Sitia, R., and Simmen, T. (2018). Redox Crosstalk at Endoplasmic Reticulum (ER) Membrane Contact Sites (MCS) Uses Toxic Waste to Deliver Messages. Cell. Death Dis. 9 (3), 331. doi:10.1038/s41419-017-0033-4
Yoshida, S., Hong, S., Suzuki, T., Nada, S., Mannan, A. M., Wang, J., et al. (2011). Redox Regulates Mammalian Target of Rapamycin Complex 1 (mTORC1) Activity by Modulating the TSC1/TSC2-Rheb GTPase Pathway. J. Biol. Chem. 286 (37), 32651–32660. doi:10.1074/jbc.M111.238014
Youn, J.-Y., Dyakov, B. J. A., Zhang, J., Knight, J. D. R., Vernon, R. M., Forman-Kay, J. D., et al. (2019). Properties of Stress Granule and P-Body Proteomes. Mol. Cell. 76 (2), 286–294. doi:10.1016/j.molcel.2019.09.014
Yousefzadeh, M., Henpita, C., Vyas, R., Soto-Palma, C., Robbins, P., and Niedernhofer, L. (2021). DNA Damage-How and Why We Age? Elife. 10, e62852. doi:10.7554/eLife.62852
Yu, Y., Yoon, S.-O., Poulogiannis, G., Yang, Q., Ma, X. M., Villen, J., et al. (2011). Phosphoproteomic Analysis Identifies Grb10 as an mTORC1 Substrate that Negatively Regulates Insulin Signaling. Science. 332 (6035), 1322–1326. doi:10.1126/science.1199484
Yuan, H.-X., Wang, Z., Yu, F.-X., Li, F., Russell, R. C., Jewell, J. L., et al. (2015). NLK Phosphorylates Raptor to Mediate Stress-Induced mTORC1 Inhibition. Genes Dev. 29 (22), 2362–2376. doi:10.1101/gad.265116.115
Zhang, G., Wang, Z., Du, Z., and Zhang, H. (2018). mTOR Regulates Phase Separation of PGL Granules to Modulate Their Autophagic Degradation. Cell. 174 (6), 1492–1506. doi:10.1016/j.cell.2018.08.006
Zhang, J., Kim, J., Alexander, A., Cai, S., Tripathi, D. N., Dere, R., et al. (2013). A Tuberous Sclerosis Complex Signalling Node at the Peroxisome Regulates mTORC1 and Autophagy in Response to ROS. Nat. Cell. Biol. 15 (10), 1186–1196. doi:10.1038/ncb2822
Keywords: MTOR, aging hallmarks, stress, insulin, amino acids, cellular signaling, stress granules (SGs), autophagy
Citation: Cadena Sandoval M, Heberle AM, Rehbein U, Barile C, Ramos Pittol JM and Thedieck K (2021) mTORC1 Crosstalk With Stress Granules in Aging and Age-Related Diseases. Front. Aging 2:761333. doi: 10.3389/fragi.2021.761333
Received: 19 August 2021; Accepted: 28 September 2021;
Published: 13 October 2021.
Edited by:
Dudley Lamming, University of Wisconsin-Madison, United StatesReviewed by:
Alessandro Bitto, University of Washington, United StatesEstela Jacinto, The State University of New Jersey, United States
Copyright © 2021 Cadena Sandoval, Heberle, Rehbein, Barile, Ramos Pittol and Thedieck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathrin Thedieck, a2F0aHJpbi50aGVkaWVja0B1aWJrLmFjLmF0, a2F0aHJpbi50aGVkaWVja0B1bmktb2xkZW5idXJnLmRl, ay50aGVkaWVja0B1bWNnLm5s
†These authors have contributed equally to this work