 August Yue Huang
August Yue Huang Eunjung Alice Lee
Eunjung Alice Lee- Division of Genetics and Genomics, Manton Center for Orphan Diseases, Boston Children's Hospital, Boston, MA, United States, Department of Pediatrics, Harvard Medical School, Boston, MA, United States
Somatic mutations are DNA variants that occur after the fertilization of zygotes and accumulate during the developmental and aging processes in the human lifespan. Somatic mutations have long been known to cause cancer, and more recently have been implicated in a variety of non-cancer diseases. The patterns of somatic mutations, or mutational signatures, also shed light on the underlying mechanisms of the mutational process. Advances in next-generation sequencing over the decades have enabled genome-wide profiling of DNA variants in a high-throughput manner; however, unlike germline mutations, somatic mutations are carried only by a subset of the cell population. Thus, sensitive bioinformatic methods are required to distinguish mutant alleles from sequencing and base calling errors in bulk tissue samples. An alternative way to study somatic mutations, especially those present in an extremely small number of cells or even in a single cell, is to sequence single-cell genomes after whole-genome amplification (WGA); however, it is critical and technically challenging to exclude numerous technical artifacts arising during error-prone and uneven genome amplification in current WGA methods. To address these challenges, multiple bioinformatic tools have been developed. In this review, we summarize the latest progress in methods for identification of somatic mutations and the challenges that remain to be addressed in the future.
Introduction
The human body consists of more than 1013 cells developed from a single fertilized zygote and experiences about 1016 cell divisions throughout its lifespan (Sender et al., 2016). Previously, all the cells from a single individual were thought to carry an identical genome, but this has been proven wrong due to the widespread occurrence of somatic mutations even in healthy individuals (Evrony et al., 2012; Lupski, 2013; Huang et al., 2014). Somatic mutations occur postzygotically as a result of errors in DNA replication and exposure to exogenous and endogenous mutagenic factors (Vijg and Dong, 2020). Once fixed in the genome, somatic mutations can be inherited from parental cells to daughter cells through cell division; when somatic mutations occasionally affect lines of germ cells, the mutations may be transmitted to offspring (Ye et al., 2018). The scale of somatic mutation varies from single-nucleotide variant and short indel to structural variation and chromosomal anomaly, and the somatic single-nucleotide variant (sSNV) is the most common mutation type in the human genome (De, 2011).
Somatic mutations have increasingly been implicated in various diseases. Somatic mutations in oncogenes and tumor-suppressor genes are the major cause of cancer (Watson et al., 2013). Accumulation of somatic mutations in cancer driver genes has also been reported in precancerous and apparently normal samples of blood and epithelial tissues, and is associated with increased cancer risks (Kakiuchi and Ogawa, 2021). In addition to cancer, somatic mutations have been found to play a critical role in an increasing list of non-cancer overgrowth diseases, such as Proteus syndrome (Lindhurst et al., 2011), arteriovenous malformation (Couto et al., 2017), and brain malformation (Jamuar et al., 2014). As a previously overlooked genetic factor, somatic mutation has been implicated in more and more non-Mendelian, complex diseases including autism (Dou et al., 2017; Lim et al., 2017), schizophrenia (Fullard et al., 2019), and congenital heart disease (Hsieh et al., 2020). Using single-cell sequencing, an increased genome-wide burden of somatic mutation in neurons was found to be associated with aging and neurodegenerative conditions (Lodato et al., 2018).
Different mutational processes generate distinct profiles of mutational genomic contexts, termed “mutational signatures,” and the landscape of somatic mutations observed in tissue samples or single cells often reflects the combined impact of multiple mutational processes (Helleday et al., 2014). The large collection of somatic mutations from cancer samples has enabled the decomposition of mutational profiles from different cancer types into mutational signatures. By using non-negative matrix factorization (Lee and Seung, 1999), Alexandrov et al. analyzed the tri-nucleotide sSNV profiles across 30 cancer types and successfully identified 27 mutational signatures (Alexandrov et al., 2013). The catalogue of mutational signatures has then been extended by incorporating more cancer data and other mutation types including short indels and double-nucleotide variants (Alexandrov et al., 2020). A similar analysis strategy has also been widely applied to somatic mutations identified from healthy human tissues or cells (Lodato et al., 2018; Martincorena et al., 2018) as well as from cultured cell lines (Kucab et al., 2019).
Theoretically, sequencing reads from reference and mutant alleles of a given mutation should follow a binomial sampling process, where the expected number of mutant reads is positively correlated with total depth and mutant allele fraction. The mutant allele fraction is one of the key variables for somatic mutation detection, which is largely determined by the timing of the occurrence of the mutation and the selective pressure acting on the cell carrying the mutation (Figure 1). Somatic mutations occurring during embryogenesis or subjected to clonal expansion can achieve high allele fractions (>1%) in the cell population so that such mutations can be detected when sequencing bulk samples at high depth (Huang et al., 2018). However, next-generation sequencing (NGS) is not perfect: the error-prone processes of base-calling and alignment can produce ubiquitous technical artifacts that resemble true somatic mutations (Ma et al., 2019). Random variation and systemic bias in sequencing cause the deviation of allele fractions of heterozygous germline mutations from the expected 50%, which can also lead to false calls of somatic mutation. More recently, single-cell sequencing has been developed as a powerful strategy to enable identification of somatic mutations that are carried by a very small number of cells or that are even restricted to a single cell (Baslan and Hicks, 2017). Due to the low DNA content in every single cell, various methods have been applied to amplify genomic DNA before sequencing (Gundry et al., 2012; Chen et al., 2017; Gonzalez-Pena et al., 2021), but they also introduce numerous amplification errors and severe coverage unevenness that need to be addressed for somatic mutation calling.
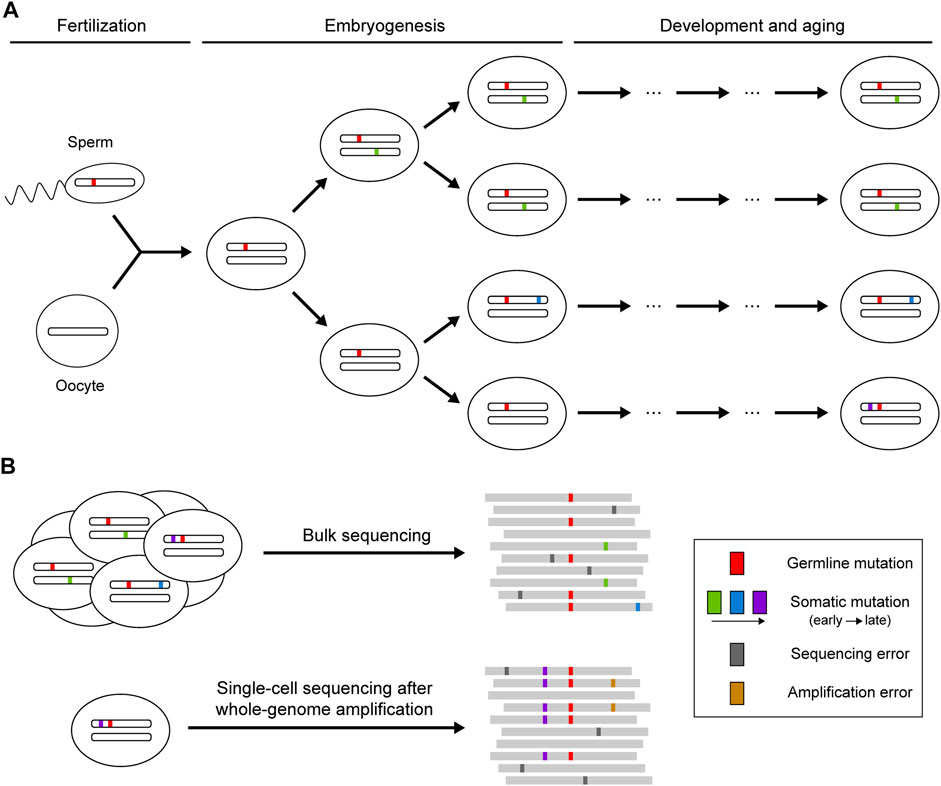
FIGURE 1. Occurrence of somatic mutations and their identification in next-generation sequencing data. (A) Somatic mutations that occur postzygotically after fertilization. Mutations arising during embryogenesis or under clonal expansion (green and blue) are shared in a fraction of the cell population, whereas mutations accumulating during the aging process (purple) may only be present in a single cell. (B) Identification of somatic mutations using bulk or single-cell sequencing. Bulk sequencing is suitable for detecting somatic mutations shared across multiple cells, though mutations with low allele fractions are difficult to distinguish from sequencing errors. Private somatic mutations can be detected with single-cell sequencing, but the whole-genome amplification before sequencing may introduce additional artifacts resulting from amplification errors.
Calling Somatic Mutations From Bulk DNA Sequencing Data
Early attempts on somatic mutation calling were made in cancer studies, where the sequencing data from a tumor sample were typically compared to a matched normal control sample obtained from the same donor. Strelka (Saunders et al., 2012) and VarScan2 (Koboldt et al., 2012) compared mutant allele fractions between tumor and normal samples to test whether any given site showed a significantly higher fraction in the tumor sample. JointSNVMix (Roth et al., 2012) further considered the base-quality information and deployed a Bayesian model to jointly analyze tumor and normal samples, in which germline mutations could be ruled out if they were predicted to be present in both samples. Moreover, MuTect (Cibulskis et al., 2013) generated a probabilistic model to calculate the likelihood of the presence of a mutant allele that could not be explained by base-calling error or sample contamination, and then utilized a panel of normal samples to reduce false positives and filter out germline mutations. In addition to these statistical models, these somatic mutation callers also incorporated a series of error filters to further remove technical artifacts based on aberrant read alignment patterns, such as strand bias or poor mapping scores.
Although clonal expansion events led by driver mutations are not rare in healthy tissues, they usually involve relatively small clones, making it hard to attain high allele fractions in bulk tissue sequencing (Martincorena and Campbell, 2015). Moreover, the lack of matched control samples in non-cancer studies poses further challenges to somatic mutation identification in healthy individuals. MosaicHunter (Huang et al., 2014) addressed these difficulties by introducing a mosaic genotype into the Bayesian model to identify sSNVs without the need for control samples; it also designed more stringent empirical filters to achieve high precision when the signal-to-noise ratio is lower in non-cancer tissues. For whole-exome sequencing data, the additional exome enrichment steps in library preparation could result in over-dispersed distribution of mutant allele fractions when compared to binomial expectation (Huang et al., 2017); MosaicHunter and EM-mosaic (Hsieh et al., 2020) introduced beta-binomial models to capture the over-dispersion estimated from each whole-exome sample. MosaicForecast (Dou et al., 2020) leveraged machine-learning methods to incorporate multiple classifiers to distinguish somatic mutations from false positives, and demonstrated a better balance of sensitivity and specificity than previous methods where error filters had been empirically defined.
Targeted ultra-deep sequencing has been widely used as a cost-efficient strategy to increase sequencing depth and thus improve sensitivity in detecting somatic mutations, especially for screening mutations in cancer-related genes (Martincorena et al., 2015; Keogh et al., 2018). However, conventional somatic mutation callers designed for whole-genome or whole-exome sequencing usually cannot produce high-confidence calls of somatic candidates with lower allele fractions (<1%), because a large number of technical artifacts can reach allele fractions of 0.1–1% in ultra-deep sequencing data (Fox et al., 2014). To address this, RareVar (Hao et al., 2017) built a position-specific error model considering genomic contexts including mutation type and GC content, enabling identification of sSNVs with a 0.5% allele fraction. RePlow (Kim et al., 2019) utilized technical replicates of the same sequenced sample to estimate the background error rate during library preparation and sequencing, which greatly reduced false positives in ultra-deep sequencing data.
Calling Somatic Mutations From Single-Cell DNA Sequencing Data
Somatic mutation in single-cell data has emerged as a powerful endogenous marker to comprehend underlying mutational mechanisms across different cell types (Brazhnik et al., 2020), and to reconstruct developmental lineage during embryogenesis (Bizzotto et al., 2021). Theoretically, somatic and germline heterozygous mutations should appear similarly at the single-cell level, both following a binomial distribution for allele fraction with an expected probability of 0.5; therefore, a bulk sample from the same individual is usually necessary to facilitate distinguishing the two types of mutations. Current whole-genome amplification (WGA) methods in single-cell sequencing can result in widespread amplification errors arising during multiple rounds of PCR, highly variable read coverage across the genome, and severe allelic dropout events when one allele of a genomic locus completely failed to be captured and amplified (Gawad et al., 2016).
Early pioneering works have demonstrated success in applying bulk-sequencing-based methods to sSNV calling in single cells (Wang et al., 2014; Lodato et al., 2015), despite potentially high false positive rates with the lack of refined modeling of single-cell-sequencing-specific features. Monovar (Zafar et al., 2016) derived the conventional binomial model by considering global allelic dropout and amplification error rates for every single cell estimated by using heterozygous germline mutations. SCcaller (Dong et al., 2017) further applied a kernel smoothing method which enabled the estimation of local allelic dropout across different genomic loci, and achieved better performance. To eliminate false positives arising during amplification, LiRA (Bohrson et al., 2019) and Conbase (Hard et al., 2019) utilized the read phasing information between somatic mutation candidates and adjacent germline heterozygous mutations, where only true mutations but not artifacts would be completely linked to one of the two alleles of a germline heterozygous mutation. Moreover, SCAN-SNV (Luquette et al., 2019) estimated genome-wide allelic imbalance using germline heterozygous mutations and then checked whether a somatic candidate had a similar level of allelic fraction to local expectation.
Single cells may share some somatic mutations if those mutations occurred in their common ancestral cell (Woodworth et al., 2017). Compared to mutations that are present in only a single cell, shared mutations can be more reliably called and distinguished from random amplification errors if somatic mutation callers can jointly consider sequencing data from multiple single cells or bulk cell populations. Monovar and Conbase applied a similar set intersection strategy, in which somatic mutations from every single cell were called independently and then only mutations recurrently called in multiple cells were considered as true clonal events, although Conbase showed a much lower false positive rate due to its usage of read phasing information. With the consideration of single-cell-specific allelic dropout and amplification error rates, single-cell MosaicHunter (Huang et al., 2020) incorporated the genotype probability of single-cell and bulk sequencing data into a single Bayesian graphical model where bulk data was generated either from the actual bulk cell population or an in silico mixture of multiple single cells, and outperformed other tools on calling clonal mutations.
Calling Somatic Mutations From Non-DNA Sequencing Data
Somatic mutations can also be called from other types of sequencing data beyond DNA sequencing data. RNA-MuTect identified exonic somatic mutations from bulk RNA-seq data by comparing mutation calls against DNA sequencing of a matched control sample (Yizhak et al., 2019). Somatic mutation candidates from RNA-seq data need to be distinguished from RNA editing sites and germline mutations with allelic expression bias There are successful attempts on calling somatic mutations from single-cell RNA-seq (Vu et al., 2019) and ATAC-seq (Bizzotto et al., 2021) data, but these analyses were limited to re-capture mutations that had been identified by other DNA-based methods. Mitochondrial DNA is known to have a higher mutation rate than the nuclear counterpart, likely due to the abundant mutagenic oxidative radicals and lack of DNA repair machinery (Schon et al., 2012). A recent study demonstrated the possibility of calling mitochondrial somatic mutations in single-cell RNA-seq and ATAC-seq data and using the mutations as lineage markers (Ludwig et al., 2019).
Conclusion and Future Perspectives
Many bioinformatic methods have been developed to study somatic mutation in healthy and diseased human samples using bulk or single-cell sequencing (Table 1). In bulk-sequencing-based methods, the detectable allele fraction of somatic mutation is largely restricted by the intrinsic base-calling error rate of ∼0.01–0.1% in current sequencing technologies. Molecular barcoding has been suggested as a promising solution since it generates a consensus sequence from multiple sequencing reads derived from the same DNA fragment and dramatically reduces the base-calling error rate (Hiatt et al., 2013; Hoang et al., 2016; Abascal et al., 2021); however, the requirement of high sequencing depth and efficient tools for consensus sequence calling currently prevents its broad application. On the other hand, alternative experimental methods have recently emerged to bypass the WGA step in single-cell DNA sequencing, including cell culture of isolated single cells into clones (Bae et al., 2018) or organoids (Behjati et al., 2014; Nanki et al., 2020), micro-dissection of monoclonal cells from tissue sections (Martincorena et al., 2015; Li et al., 2020), and even direct sequencing without pre-amplification (Zahn et al., 2017).
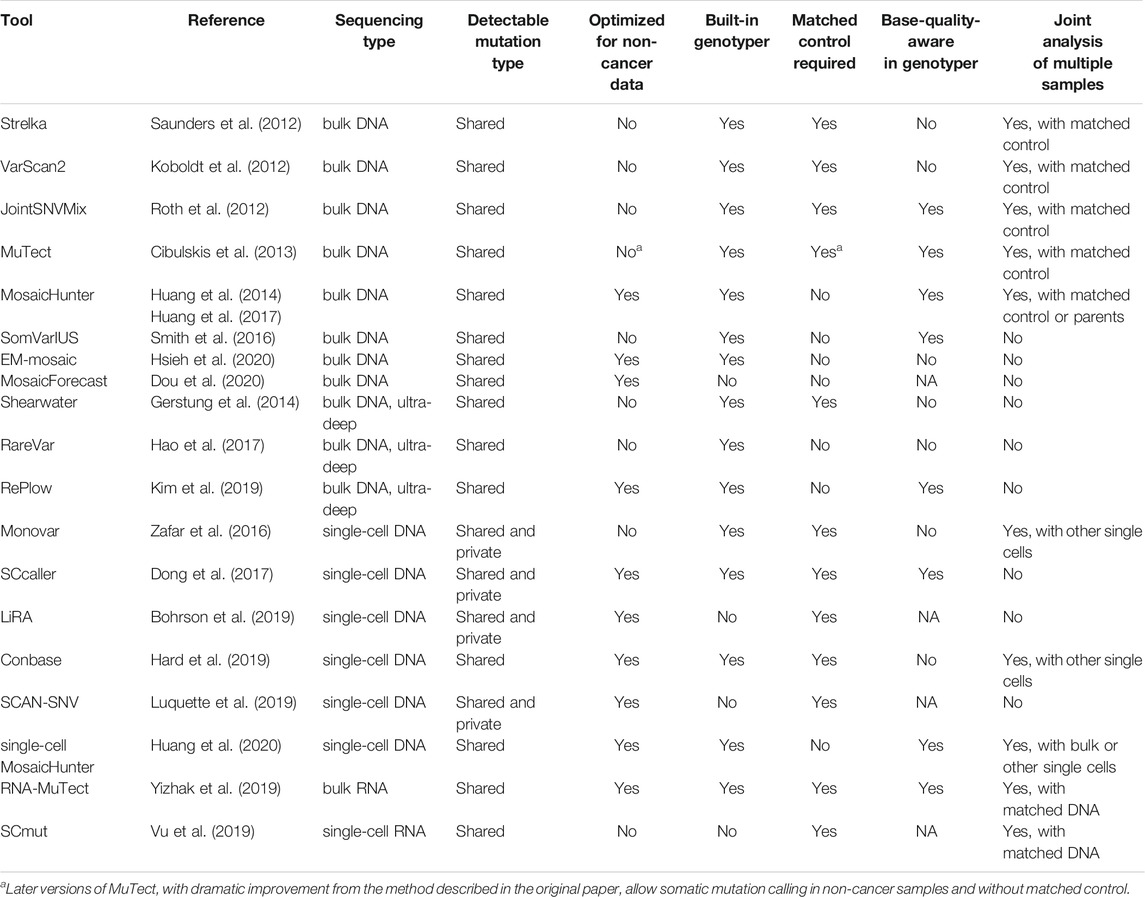
TABLE 1. A selected list of tools for somatic mutation calling.
In the past decade, genomic studies have benefited from the development of single-molecule sequencing technologies that can directly read nucleotide sequences from DNA or RNA molecules and deliver much longer reads than previously available NGS technologies (Logsdon et al., 2020). Long sequencing reads unlock the possibility of exploring repetitive genomic regions that are generally inaccessible with short-read sequencing and characterizing large and complex genetic variants involving copy number variation or structural variation (Ameur et al., 2019). New bioinformatic tools specialized for long-read sequencing have emerged for read alignment (Li, 2018) and variant calling (Sedlazeck et al., 2018) that have been successfully implemented in cancer studies (Nattestad et al., 2018; Aganezov et al., 2020). However, the relatively high cost of single-molecule sequencing limits its broad application to genome-wide detection of somatic mutations with low allele fractions since such detection requires high sequencing depth. Rapid advances in sequencing technologies and bioinformatic methods will allow more comprehensive identification and deeper understanding of somatic mutations in healthy and diseased human genomes in the future.
Author Contributions
AYH wrote the original draft of the manuscript. EAL reviewed and edited the manuscript.
Funding
The work was supported by the NIH R01 (R01AG070921) and DP2 (DP2AG072437) Grants to EAL.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abascal, F., Harvey, L. M. R., Mitchell, E., Lawson, A. R. J., Lensing, S. V., Ellis, P., et al. (2021). Somatic Mutation Landscapes at Single-Molecule Resolution. Nature 593, 405–410. doi:10.1038/s41586-021-03477-4
Aganezov, S., Goodwin, S., Sherman, R. M., Sedlazeck, F. J., Arun, G., Bhatia, S., et al. (2020). Comprehensive Analysis of Structural Variants in Breast Cancer Genomes Using Single-Molecule Sequencing. Genome Res. 30, 1258–1273. doi:10.1101/gr.260497.119
Alexandrov, L. B., Kim, J., Kim, J., Haradhvala, N. J., Huang, M. N., Tian Ng, A. W., et al. (2020). The Repertoire of Mutational Signatures in Human Cancer. Nature 578, 94–101. doi:10.1038/s41586-020-1943-3
Alexandrov, L. B., Nik-Zainal, S., Nik-Zainal, S., Wedge, D. C., Aparicio, S. A. J. R., Behjati, S., et al. (2013). Signatures of Mutational Processes in Human Cancer. Nature 500, 415–421. doi:10.1038/nature12477
Ameur, A., Kloosterman, W. P., and Hestand, M. S. (2019). Single-Molecule Sequencing: Towards Clinical Applications. Trends Biotechnol. 37, 72–85. doi:10.1016/j.tibtech.2018.07.013
Bae, T., Tomasini, L., Mariani, J., Zhou, B., Roychowdhury, T., Franjic, D., et al. (2018). Different Mutational Rates and Mechanisms in Human Cells at Pregastrulation and Neurogenesis. Science 359, 550–555. doi:10.1126/science.aan8690
Baslan, T., and Hicks, J. (2017). Unravelling Biology and Shifting Paradigms in Cancer with Single-Cell Sequencing. Nat. Rev. Cancer 17, 557–569. doi:10.1038/nrc.2017.58
Behjati, S., Huch, M., Van Boxtel, R., Karthaus, W., Wedge, D. C., Tamuri, A. U., et al. (2014). Genome Sequencing of Normal Cells Reveals Developmental Lineages and Mutational Processes. Nature 513, 422–425. doi:10.1038/nature13448
Bizzotto, S., Dou, Y., Ganz, J., Doan, R. N., Kwon, M., Bohrson, C. L., et al. (2021). Landmarks of Human Embryonic Development Inscribed in Somatic Mutations. Science 371, 1249–1253. doi:10.1126/science.abe1544
Bohrson, C. L., Barton, A. R., Lodato, M. A., Rodin, R. E., Luquette, L. J., Viswanadham, V. V., et al. (2019). Linked-read Analysis Identifies Mutations in Single-Cell DNA-Sequencing Data. Nat. Genet. 51, 749–754. doi:10.1038/s41588-019-0366-2
Brazhnik, K., Sun, S., Alani, O., Kinkhabwala, M., Wolkoff, A. W., Maslov, A. Y., et al. (2020). An Erratum for the Research Article: "Single-Cell Analysis Reveals Different Age-Related Somatic Mutation Profiles Between Stem and Differentiated Cells in Human Liver" by K. Brazhnik, S. Sun, O. Alani, M. Kinkhabwala, A. W. Wolkoff, A. Y. Maslov, X. Dong, and J. Vijg. Sci. Adv. 6, eaax2659. doi:10.1126/sciadv.abe8055
Chen, C., Xing, D., Tan, L., Li, H., Zhou, G., Huang, L., et al. (2017). Single-cell Whole-Genome Analyses by Linear Amplification via Transposon Insertion (LIANTI). Science 356, 189–194. doi:10.1126/science.aak9787
Cibulskis, K., Lawrence, M. S., Carter, S. L., Sivachenko, A., Jaffe, D., Sougnez, C., et al. (2013). Sensitive Detection of Somatic Point Mutations in Impure and Heterogeneous Cancer Samples. Nat. Biotechnol. 31, 213–219. doi:10.1038/nbt.2514
Couto, J. A., Huang, A. Y., Konczyk, D. J., Goss, J. A., Fishman, S. J., Mulliken, J. B., et al. (2017). Somatic MAP2K1 Mutations Are Associated with Extracranial Arteriovenous Malformation. Am. J. Hum. Genet. 100, 546–554. doi:10.1016/j.ajhg.2017.01.018
De, S. (2011). Somatic Mosaicism in Healthy Human Tissues. Trends Genet. 27, 217–223. doi:10.1016/j.tig.2011.03.002
Dong, X., Zhang, L., Milholland, B., Lee, M., Maslov, A. Y., Wang, T., et al. (2017). Accurate Identification of Single-Nucleotide Variants in Whole-Genome-Amplified Single Cells. Nat. Methods 14, 491–493. doi:10.1038/nmeth.4227
Dou, Y., Kwon, M., Rodin, R. E., Cortés-Ciriano, I., Doan, R., Luquette, L. J., et al. (2020). Accurate Detection of Mosaic Variants in Sequencing Data Without Matched Controls. Nat. Biotechnol. 38, 314–319. doi:10.1038/s41587-019-0368-8
Dou, Y., Yang, X., Li, Z., Wang, S., Zhang, Z., Ye, A. Y., et al. (2017). Postzygotic Single‐nucleotide Mosaicisms Contribute to the Etiology of Autism Spectrum Disorder and Autistic Traits and the Origin of Mutations. Hum. Mutat. 38, 1002–1013. doi:10.1002/humu.23255
Evrony, G. D., Cai, X., Lee, E., Hills, L. B., Elhosary, P. C., Lehmann, H. S., et al. (2012). Single-neuron Sequencing Analysis of L1 Retrotransposition and Somatic Mutation in the Human Brain. Cell 151, 483–496. doi:10.1016/j.cell.2012.09.035
Fox, E. J., Reid-Bayliss, K. S., Emond, M. J., and Loeb, L. A. (2014). Accuracy of Next Generation Sequencing Platforms. Next Gener Seq. Appl. 1, 1000106. doi:10.4172/jngsa.1000106
Fullard, J. F., Charney, A. W., Voloudakis, G., Uzilov, A. V., Haroutunian, V., and Roussos, P. (2019). Assessment of Somatic Single-Nucleotide Variation in Brain Tissue of Cases with Schizophrenia. Transl Psychiatry 9, 21. doi:10.1038/s41398-018-0342-0
Gawad, C., Koh, W., and Quake, S. R. (2016). Single-cell Genome Sequencing: Current State of the Science. Nat. Rev. Genet. 17, 175–188. doi:10.1038/nrg.2015.16
Gerstung, M., Papaemmanuil, E., and Campbell, P. J. (2014). Subclonal Variant Calling with Multiple Samples and Prior Knowledge. Bioinformatics 30, 1198–1204.
Gonzalez-Pena, V., Natarajan, S., Xia, Y., Klein, D., Carter, R., Pang, Y., et al. (2021). Accurate Genomic Variant Detection in Single Cells with Primary Template-Directed Amplification. Proc. Natl. Acad. Sci. U S A. 118, e2024176118. doi:10.1073/pnas.2024176118
Gundry, M., Li, W., Maqbool, S. B., and Vijg, J. (2012). Direct, Genome-wide Assessment of DNA Mutations in Single Cells. Nucleic Acids Res. 40, 2032–2040. doi:10.1093/nar/gkr949
Hao, Y., Xuei, X., Li, L., Nakshatri, H., Edenberg, H. J., and Liu, Y. (2017). RareVar: A Framework for Detecting Low-Frequency Single-Nucleotide Variants. J. Comput. Biol. 24, 637–646. doi:10.1089/cmb.2017.0057
Hård, J., Al Hakim, E., Kindblom, M., Björklund, Å. K., Sennblad, B., Demirci, I., et al. (2019). Conbase: A Software for Unsupervised Discovery of Clonal Somatic Mutations in Single Cells Through Read Phasing. Genome Biol. 20, 68. doi:10.1186/s13059-019-1673-8
Helleday, T., Eshtad, S., and Nik-Zainal, S. (2014). Mechanisms Underlying Mutational Signatures in Human Cancers. Nat. Rev. Genet. 15, 585–598. doi:10.1038/nrg3729
Hiatt, J. B., Pritchard, C. C., Salipante, S. J., O'roak, B. J., and Shendure, J. (2013). Single Molecule Molecular Inversion Probes for Targeted, High-Accuracy Detection of Low-Frequency Variation. Genome Res. 23, 843–854. doi:10.1101/gr.147686.112
Hoang, M. L., Kinde, I., Tomasetti, C., Mcmahon, K. W., Rosenquist, T. A., Grollman, A. P., et al. (2016). Genome-wide Quantification of Rare Somatic Mutations in Normal Human Tissues Using Massively Parallel Sequencing. Proc. Natl. Acad. Sci. USA 113, 9846–9851. doi:10.1073/pnas.1607794113
Hsieh, A., Morton, S. U., Willcox, J. A. L., Gorham, J. M., Tai, A. C., Qi, H., et al. (2020). EM-mosaic Detects Mosaic Point Mutations that Contribute to Congenital Heart Disease. Genome Med. 12, 42. doi:10.1186/s13073-020-00738-1
Huang, A. Y., Li, P., Rodin, R. E., Kim, S. N., Dou, Y., Kenny, C. J., et al. (2020). Parallel RNA and DNA Analysis After Deep Sequencing (PRDD-Seq) Reveals Cell Type-specific Lineage Patterns in Human Brain. Proc. Natl. Acad. Sci. USA 117, 13886–13895. doi:10.1073/pnas.2006163117
Huang, A. Y., Xu, X., Ye, A. Y., Wu, Q., Yan, L., Zhao, B., et al. (2014). Postzygotic Single-Nucleotide Mosaicisms in Whole-Genome Sequences of Clinically Unremarkable Individuals. Cell Res 24, 1311–1327. doi:10.1038/cr.2014.131
Huang, A. Y., Yang, X., Wang, S., Zheng, X., Wu, Q., Ye, A. Y., et al. (2018). Distinctive Types of Postzygotic Single-Nucleotide Mosaicisms in Healthy Individuals Revealed by Genome-wide Profiling of Multiple Organs. Plos Genet. 14, e1007395. doi:10.1371/journal.pgen.1007395
Huang, A. Y., Zhang, Z., Ye, A. Y., Dou, Y., Yan, L., Yang, X., et al. (2017). MosaicHunter: Accurate Detection of Postzygotic Single-Nucleotide Mosaicism Through Next-Generation Sequencing of Unpaired, Trio, and Paired Samples. Nucleic Acids Res. 45, e76. doi:10.1093/nar/gkx024
Jamuar, S. S., Lam, A.-T. N., Kircher, M., D’Gama, A. M., Wang, J., Barry, B. J., et al. (2014). Somatic Mutations in Cerebral Cortical Malformations. N. Engl. J. Med. 371, 733–743. doi:10.1056/nejmoa1314432
Kakiuchi, N., and Ogawa, S. (2021). Clonal Expansion in Non-cancer Tissues. Nat. Rev. Cancer 21, 239–256. doi:10.1038/s41568-021-00335-3
Keogh, M. J., Wei, W., Aryaman, J., Walker, L., Van Den Ameele, J., Coxhead, J., et al. (2018). High Prevalence of Focal and Multi-Focal Somatic Genetic Variants in the Human Brain. Nat. Commun. 9, 4257. doi:10.1038/s41467-018-06331-w
Kim, J., Kim, D., Lim, J. S., Maeng, J. H., Son, H., Kang, H.-C., et al. (2019). The Use of Technical Replication for Detection of Low-Level Somatic Mutations in Next-Generation Sequencing. Nat. Commun. 10, 1047. doi:10.1038/s41467-019-09026-y
Koboldt, D. C., Zhang, Q., Larson, D. E., Shen, D., Mclellan, M. D., Lin, L., et al. (2012). VarScan 2: Somatic Mutation and Copy Number Alteration Discovery in Cancer by Exome Sequencing. Genome Res. 22, 568–576. doi:10.1101/gr.129684.111
Kucab, J. E., Zou, X., Morganella, S., Joel, M., Nanda, A. S., Nagy, E., et al. (2019). A Compendium of Mutational Signatures of Environmental Agents. Cell 177, 821–836. doi:10.1016/j.cell.2019.03.001
Lee, D. D., and Seung, H. S. (1999). Learning the Parts of Objects by Non-negative Matrix Factorization. Nature 401, 788–791. doi:10.1038/44565
Li, H. (2018). Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 34, 3094–3100. doi:10.1093/bioinformatics/bty191
Li, R., Du, Y., Chen, Z., Xu, D., Lin, T., Jin, S., et al. (2020). Macroscopic Somatic Clonal Expansion in Morphologically Normal Human Urothelium. Science 370, 82–89. doi:10.1126/science.aba7300
Lim, E. T., Uddin, M., Uddin, M., De Rubeis, S., Chan, Y., Kamumbu, A. S., et al. (2017). Rates, Distribution and Implications of Postzygotic Mosaic Mutations in Autism Spectrum Disorder. Nat. Neurosci. 20, 1217–1224. doi:10.1038/nn.4598
Lindhurst, M. J., Sapp, J. C., Teer, J. K., Johnston, J. J., Finn, E. M., Peters, K., et al. (2011). A Mosaic Activating Mutation in AKT1 Associated with the Proteus Syndrome. N. Engl. J. Med. 365, 611–619. doi:10.1056/nejmoa1104017
Lodato, M. A., Rodin, R. E., Bohrson, C. L., Coulter, M. E., Barton, A. R., Kwon, M., et al. (2018). Aging and Neurodegeneration Are Associated with Increased Mutations in Single Human Neurons. Science 359, 555–559. doi:10.1126/science.aao4426
Lodato, M. A., Woodworth, M. B., Lee, S., Evrony, G. D., Mehta, B. K., Karger, A., et al. (2015). Somatic Mutation in Single Human Neurons Tracks Developmental and Transcriptional History. Science 350, 94–98. doi:10.1126/science.aab1785
Logsdon, G. A., Vollger, M. R., and Eichler, E. E. (2020). Long-read Human Genome Sequencing and its Applications. Nat. Rev. Genet. 21, 597–614. doi:10.1038/s41576-020-0236-x
Ludwig, L. S., Lareau, C. A., Ulirsch, J. C., Christian, E., Muus, C., Li, L. H., et al. (2019). Lineage Tracing in Humans Enabled by Mitochondrial Mutations and Single-Cell Genomics. Cell 176, 1325–1339. doi:10.1016/j.cell.2019.01.022
Lupski, J. R. (2013). Genome Mosaicism-One Human, Multiple Genomes. Science 341, 358–359. doi:10.1126/science.1239503
Luquette, L. J., Bohrson, C. L., Sherman, M. A., and Park, P. J. (2019). Identification of Somatic Mutations in Single Cell DNA-Seq Using a Spatial Model of Allelic Imbalance. Nat. Commun. 10, 3908. doi:10.1038/s41467-019-11857-8
Ma, X., Shao, Y., Tian, L., Flasch, D. A., Mulder, H. L., Edmonson, M. N., et al. (2019). Analysis of Error Profiles in Deep Next-Generation Sequencing Data. Genome Biol. 20, 50. doi:10.1186/s13059-019-1659-6
Martincorena, I., and Campbell, P. J. (2015). Somatic Mutation in Cancer and Normal Cells. Science 349, 1483–1489. doi:10.1126/science.aab4082
Martincorena, I., Fowler, J. C., Wabik, A., Lawson, A. R. J., Abascal, F., Hall, M. W. J., et al. (2018). Somatic Mutant Clones Colonize the Human Esophagus with Age. Science 362, 911–917. doi:10.1126/science.aau3879
Martincorena, I., Roshan, A., Gerstung, M., Ellis, P., Van Loo, P., Mclaren, S., et al. (2015). High Burden and Pervasive Positive Selection of Somatic Mutations in Normal Human Skin. Science 348, 880–886. doi:10.1126/science.aaa6806
Nanki, K., Fujii, M., Shimokawa, M., Matano, M., Nishikori, S., Date, S., et al. (2020). Somatic Inflammatory Gene Mutations in Human Ulcerative Colitis Epithelium. Nature 577, 254–259. doi:10.1038/s41586-019-1844-5
Nattestad, M., Goodwin, S., Ng, K., Baslan, T., Sedlazeck, F. J., Rescheneder, P., et al. (2018). Complex Rearrangements and Oncogene Amplifications Revealed by Long-Read DNA and RNA Sequencing of a Breast Cancer Cell Line. Genome Res. 28, 1126–1135. doi:10.1101/gr.231100.117
Roth, A., Ding, J., Morin, R., Crisan, A., Ha, G., Giuliany, R., et al. (2012). JointSNVMix: A Probabilistic Model for Accurate Detection of Somatic Mutations in Normal/tumour Paired Next-Generation Sequencing Data. Bioinformatics 28, 907–913. doi:10.1093/bioinformatics/bts053
Saunders, C. T., Wong, W. S. W., Swamy, S., Becq, J., Murray, L. J., and Cheetham, R. K. (2012). Strelka: Accurate Somatic Small-Variant Calling from Sequenced Tumor-normal Sample Pairs. Bioinformatics 28, 1811–1817. doi:10.1093/bioinformatics/bts271
Schon, E. A., Dimauro, S., and Hirano, M. (2012). Human Mitochondrial DNA: Roles of Inherited and Somatic Mutations. Nat. Rev. Genet. 13, 878–890. doi:10.1038/nrg3275
Sedlazeck, F. J., Rescheneder, P., Smolka, M., Fang, H., Nattestad, M., Von Haeseler, A., et al. (2018). Accurate Detection of Complex Structural Variations Using Single-Molecule Sequencing. Nat. Methods 15, 461–468. doi:10.1038/s41592-018-0001-7
Sender, R., Fuchs, S., and Milo, R. (2016). Revised Estimates for the Number of Human and Bacteria Cells in the Body. Plos Biol. 14, e1002533. doi:10.1371/journal.pbio.1002533
Smith, K. S., Yadav, V. K., Pei, S., Pollyea, D. A., Jordan, C. T., and De, S. (2016). SomVarIUS: Somatic Variant Identification from Unpaired Tissue Samples. Bioinformatics 32, 808–813.
Vijg, J., and Dong, X. (2020). Pathogenic Mechanisms of Somatic Mutation and Genome Mosaicism in Aging. Cell 182, 12–23. doi:10.1016/j.cell.2020.06.024
Vu, T. N., Nguyen, H.-N., Calza, S., Kalari, K. R., Wang, L., and Pawitan, Y. (2019). Cell-level Somatic Mutation Detection from Single-Cell RNA Sequencing. Bioinformatics 35, 4679–4687. doi:10.1093/bioinformatics/btz288
Wang, Y., Waters, J., Leung, M. L., Unruh, A., Roh, W., Shi, X., et al. (2014). Clonal Evolution in Breast Cancer Revealed by Single Nucleus Genome Sequencing. Nature 512, 155–160. doi:10.1038/nature13600
Watson, I. R., Takahashi, K., Futreal, P. A., and Chin, L. (2013). Emerging Patterns of Somatic Mutations in Cancer. Nat. Rev. Genet. 14, 703–718. doi:10.1038/nrg3539
Woodworth, M. B., Girskis, K. M., and Walsh, C. A. (2017). Building a Lineage from Single Cells: Genetic Techniques for Cell Lineage Tracking. Nat. Rev. Genet. 18, 230–244. doi:10.1038/nrg.2016.159
Ye, A. Y., Dou, Y., Yang, X., Wang, S., Huang, A. Y., and Wei, L. (2018). A Model for Postzygotic Mosaicisms Quantifies the Allele Fraction Drift, Mutation Rate, and Contribution to De Novo Mutations. Genome Res. 28, 943–951. doi:10.1101/gr.230003.117
Yizhak, K., Aguet, F., Kim, J., Hess, J. M., Kübler, K., Grimsby, J., et al. (2019). RNA Sequence Analysis Reveals Macroscopic Somatic Clonal Expansion Across Normal Tissues. Science 364, eaaw0726. doi:10.1126/science.aaw0726
Zafar, H., Wang, Y., Nakhleh, L., Navin, N., and Chen, K. (2016). Monovar: Single-Nucleotide Variant Detection in Single Cells. Nat. Methods 13, 505–507. doi:10.1038/nmeth.3835
Keywords: somatic mutation, bulk sequencing, single-cell sequencing, bioinformatic tool, single-nucleotide variant
Citation: Huang AY and Lee EA (2022) Identification of Somatic Mutations From Bulk and Single-Cell Sequencing Data. Front. Aging 2:800380. doi: 10.3389/fragi.2021.800380
Received: 23 October 2021; Accepted: 08 December 2021;
Published: 03 January 2022.
Edited by:
Michael Lodato, University of Massachusetts Medical School, United StatesReviewed by:
Prescott Deininger, Tulane University, United StatesCopyright © 2022 Huang and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eunjung Alice Lee, ZWFsaWNlLmxlZUBjaGlsZHJlbnMuaGFydmFyZC5lZHU=