 Francesco Donati
Francesco Donati Livia Concetti1
Livia Concetti1 Francesco Botrè
Francesco Botrè- 1Laboratorio Antidoping, Federazione Medico Sportiva Italiana, Rome, Italy
- 2Beijing Anti-Doping Laboratory, Beijing Sport University, Beijing, China
- 3REDs—Research and Expertise in Anti-Doping Sciences, ISSUL—Institute of Sport Sciences, University of Lausanne, Lausanne, Switzerland
Background: In doping control, the presence of the exon5 c.577del variant in the human erythropoietin gene may be a confounding factor in the interpretation of the results from the analytical method currently in force for the detection of human recombinant erythropoietin, based on immunoelectrophoresis on SDS-PAGE and/or SAR-PAGE. This variant, determining the transcription of a higher molecular weight protein, can erroneously suggest the presence of recombinant erythropoietin in a biological sample, causing the possibility of a false positive result. Although the variant was now identified only in East Asian populations and with a very low frequency, it can threaten the reliability of current anti-doping tests.
Methods: We have implemented a genetic test to identify the presence of this variant in the biological samples that are presently collected for anti-doping analysis (whole blood, urine, and dried blood spots). The test is based on the Sanger sequencing of the human erythropoietin gene exon 5, where the c.577del variant falls.
Results and Discussion: The method has a specificity of 100% and allows identification of the possible presence of the variant starting from 100 pg of genomic DNA extracted from each biological sample. The efficacy of the test has been confirmed by the analysis of real samples from subjects showing and not showing the exon c.577del variant.
1 Introduction
The abuse of recombinant human erythropoietin (rEPO) by athletes causes a fraudulent improvement of oxygen delivery to the tissues, and the consequent improvement in sports performance, especially in endurance disciplines. For this reason, rEPO is included in the list of prohibited substances and doping methods, annually updated by the World Anti-Doping Agency (WADA) (World Anti-doping Agency, 2023). At present, the detection of the abuse of rEPO in doping control is performed, in urine and plasma, by immunoelectrophoresis, using Western Blot originally preceded by polyacrylamide gel isoelectric focusing (IEF-PAGE), and more recently by either N-lauroysarcosinate SAR-polyacrylamide gel (SAR- PAGE), or sodium dodecyl sulfate, gel electrophoresis (SDS-PAGE) (Jelkmann and Lundby, 2011; Reichel, 2014). Hence, the presence of rEPO in a sample can be detected due to the difference in the molecular mass compared to the endogenous erythropoietin isoform (Pascual et al., 2004).
In 2021, Zhou et al. (2022) reported the occurrence of unusual cases in the application of the rEPO routine identification method. The anomaly concerns the presence of a non-specific electrophoretic band in the specific identification region of the rEPO during the SAR-PAGE step analysis applied to serum and plasma samples. However, the unspecific band does not show a similar high intensity in the corresponding urine samples. Subsequent investigations made it possible to understand that the presence of this uncommon band is due to a rare genetic variant present in the sequence of the human EPO gene. In the human EPO gene sequence, the variant c.577del (rs369859204) is a single nucleotide deletion (SND) at position 577 of the cDNA in exon 5. The deletion causes the loss of the nearby stop codon (at position 580) during gene transcription and the change in the wild-type protein’s last amino acid (AA) with the final formation of a longer AA sequence than the normally transcribed sequence.
Moreover, the new AA sequence does not bring any additional N-glycosylation sites concerning the typical sequence; hence the wild-type EPO protein (WT-EPO) has the same N-glycosylation pattern as the variant protein (VAR-EPO). At present, variant c.577del has been detected only in the populations of Asian ethnicity, being present only in the East Asian population, where it shows a frequency between 0.7% and 0.8%. Heterozygous individuals for the c.577 variant, therefore, encode for two EPO proteins of different molecular mass (the wild-type protein and a variant protein that is 3.3 kDa heavier than the normal one). Consequently, athletes who naturally carry this genetic variant may be suspected of taking rEPO, with the risk of the laboratory reporting “false positive” results. Given the above, the WADA technical document TDEPO2022 suggests screening for the presence of the c.577del variant whenever a suspected double band is detected in blood or a “smear” of rEPO detected in urine by the SAR-PAGE analysis, with the final aim to avoid the possibility of a false positive case (World Anti-Doping Agency, 2022).
We are here presenting the implementation of a gDNA Sanger sequencing-based test to identify the c.577del variant on samples that are routinely analyzed for the purposes and the aims of doping control. Sanger sequencing is still a gold standard for recognizing mutations in the genome (whether they are insertions or deletions) and is commonly applied for forensic investigations, human genetics, genotyping, and molecular-biology assay development (Heather and Chain, 2016). Despite being an older-generation sequencing method, it is still widely used in many laboratories as one of the most reliable DNA sequencing approaches. A further advantage with respect to other procedures (e.g., CRISPS-CA9), recently presented for indirectly identifying c.577del variant (Yi et al., 2023), is the possibility of detecting also any additional potential polymorphisms that may occur in different regions of the sequence.
2 Materials and methods
Whole blood and urines used to implement the method were aliquots obtained from routine anti-doping tests, therefore anonymized, belonging to athletes that had consented to the use of the samples for research uses and were aliquoted after all tests had given negative results. Whole blood and urine were processed as such, while DBS samples were prepared by spotting 20 µL of whole blood on QIAcard FTA DMPK-C (Qiagen) to simulate an actual, routine Dried Blood Spot (DBS) sampling carried out for anti-doping purposes. Whole blood samples belonging to individuals carrying a heterozygous genotype for the c.577del mutation were provided by the Beijing Anti-Doping Laboratory of Beijing (China). The workflow for human EPO exon 5 sequencing consists of the following steps: 1) genomic DNA extraction and quantification; 2) dsDNA amplification; 3) dsDNA amplicon purity check and quantitation; 4) ssDNA Sanger sequencing reaction; 5) capillary Electrophoresis Run; 6) data analysis and basecalling.
Genomic DNA (gDNA) was extracted from whole blood (20 µL), urine (1 mL), and DBS (20 µL of spotted blood) using the prepFiler forensic DNA extraction kit (Thermo Fisher) with a manual method following manufacturer specification. gDNA was quantified by real-time PCR 7500 Fast (Thermo Fisher) using the Quantifiler DNA Kit (Thermo Fisher), which allows an absolute quantification through a calibration curve prepared with a DNA standard of known concentration. Primers Hs00289435 (FWD = GGGAGAAGGGTCTTGCTAAGGA, 22bp and REV = AGTTTAAAGCTGCTCTCTGAATGCT, 25bp)) and Hs00637921 (FWD = CCACTCCGAACAATCACTGCT, 21bp and REV = TGAGATGTCATTGCTGGCACT, 21bp) for dsDNA amplification and final Sanger sequencing reaction were purchased from Thermo Fisher Scientific. We selected these two primers on their ability to 1) identify the sequence of exon 5 where the c.577 deletion falls and 2) ability to generate two amplicons of different molecular weights. dsDNA amplification was performed with the AmpliTaq Gold master Mix (Thermo Fisher) and conducted on a 9,700 GeneAmp Thermocycler (Thermo Fisher) according to the following condition: 95°C 10 min (denaturation) followed by 95° 30 s, 63°C 30 s and 72°C 60 s for 25–40 cycles (according to gDNA input available), 72°C 7 min (final stage). ExoSap-IT kit (Thermo Fisher) was used to purify the dsDNA amplicons before moving on the next stages. Verification of the occurred amplification and of the purity of the amplified dsDNA was performed by SDS-PAGE chip electrophoresis on an Agilent 2,100 Bionalyzer using the DNA Chip Kit (Agilent). Estimating amplicon concentration was necessary to assess the optimal input DNA quantity to proceed to the following steps. ssDNA Sanger sequencing reaction was performed using the Big Dye Terminator V1.1 Cycle Sequencing Kit (Thermo Fisher). The reaction mixture was prepared according to manufacturer instructions and the same primers used for dsDNA PCR amplification were employed separately after diluting 1 pmol/μL. Sanger sequencing reaction was conducted on the 9,700 GeneAmp Thermocycler (Thermo Fisher) according to the following condition: 96°C for 1 min followed by 25 cycles (96°C 10 s, 50°C 5 s, 60°C 4 min) and 60°C 4 min before proceedings to the next step. The purification of the DNA sequencing reaction was performed using the BigDye XTerminator purification kit (Thermo Fisher) following manufacturer instructions. Single-strand sequencing was performed on the Thermo Fisher SeqStudio 4-capillaries Genetic Analyzer by loading 10 µL of the purified Sanger product and running the MediumSeq Module. Data analysis and basecalling was made using the Thermo Fisher Sequencing Analysis Software v.7.
3 Results and discussion
Different amplification cycles (25–40) were tested to select the optimal operating conditions in relation to the initial amount of available gDNA, with the aim to obtain a suitable amount of double-stranded amplicon sufficient to proceed with Sanger sequencing. This was necessary because, especially from urine samples, the quantity of gDNA that can be extracted is generally low and not of the best quality. By progressively increasing the amplification cycles, enough double-strand amplicon is obtainable to carry out the Sanger sequencing under optimal conditions. We have found that 35 amplification cycles are ideal for obtaining a suitable amount of amplicon with at least 5–10 ng of product. Additional amplification cycles may be necessary in the case of problematic cases, e.g., those due to significantly degraded urine samples that are poor in cellular material: we have confirmed that by increasing the amplification cycles up to 40, it is still possible to obtain the desired quantity of amplicon to proceed with the sequencing. Nonetheless, it is not advisable to perform more than 40 amplification cycles due to the appearance of some artifacts which significantly decrease the quality of the amplicon.
The quality control of the double-strand amplicon is essential before starting the sequencing reaction. In this method, the estimation of the purity and concentration of the amplicon was performed by Chip Electrophoresis, which allows for verifying whether: 1) the ds amplicon produced has the expected length based on the primers used, and 2) the amplicon is pure and free from contamination (see Figure 1). The two primers we selected to develop the method can generate amplicons of suitable molecular weight to perform the sequencing. This is particularly relevant since dsPCR-produced amplicons that are too small (<100bp) or too large (>1000bp) are not suitable for performing the Sanger reaction on capillary electrophoresis.

FIGURE 1. DNA chip electrophoresis for the quality control assessment of the ds amplicons. The concentration of the double-stranded amplicon produced is determined by comparison with a ladder of known concentration. The presence of a single peak of the expected size evidences the purity of the amplicon.
Primer Hs00289435 produces a “Long” double-strand amplicon of 501 bp which, after sequencing, returns the exon 5 sequence in its entirety. On the contrary, Primer Hs00637921 produces a “Short” double-strand amplicon of approximatively 268bp. This does not produce a complete exon 5 sequence as the primer already binds within the exon 5. However, the portion of the exon containing the c.577 position is anyway included in the sequenced segment. The purity of the double-stranded amplicon is essential for obtaining a good quality Sanger sequencing. Indeed, samples that were sequenced without purifying the double-strand amplicons resulted in poor quality sequences in which the base call assignment was problematic or even incorrect. The presence of some contaminants, such as proteins or other organic substances, can be inferred by analyzing the product on a spectrophotometer by measuring the A260/A280 ratio. A ratio higher than 1.6 is sufficient to assess the good quality of the extract and the absence of proteins or other organic contaminants. In cases where the presence of traces of contaminants is found, a sufficient dilution of the sample prevents the contaminants themselves from interfering with the sequence reaction. However, in cases where the contamination is excessive, it is necessary to repeat the extraction of the DNA sample. In the same way, the purification step performed downstream of the Sanger reaction has also proved crucial for obtaining a correct final result.
For the Sanger sequencing reaction, 5–10 ng of the ds amplicon generated by the Hs00637921 primers and 5–20 ng of the ds amplicon generated by the Hs00289435 primers were established to be optimal. Sequencing data analysis were assessed by using the commonly parameters that the automated data analysis software assigns as quality indexes of the obtained sequences. More in detail, a Quality Value (QV) is assigned to each base identified in the sequence and is related to the probability of error in calling the base (QV = −10 x log “error probability”). This depends generally on the shape of the peak and on its signal-to-noise ratio hence allowing to evaluate the accuracy of the base assigned by the software. As an example, a QV = 10 has 1 to 10 probability of a wrong base call and an accuracy of a base call of 90%; a QV = 50 has 1 to 100,000 probability of a wrong base call and an accuracy of a base call of 99.999%. Sample Score (SS) sometimes also called as Quality Score represents the average value of all the QV of the peaks to which a base assigned in the sequence corresponds. This is a measure of the quality of the whole sequence obtained. S/N is the ratio of the Intensity Value (expressed in Relative Fluorescence Units, RFU) of the detected base peak and the RFU of the background noise under the area of the same peak. RFU intensity signal is an indicator of the robustness of the peak obtained or, in the case of average signal intensity, of the robustness of the reaction.
For assessing the actual presence of the c.577del variant in a sample, we have established a series of quali-quantitative interpretation criteria. Preliminary to any data analysis and interpretation, a visual inspection of the entire sequence must always be performed to identify critical sequencing areas and to assess whether those critical areas can represent a risk in the correct interpretation of the result.
As shown in Figure 2, wild-type individuals show a linear sequence without nucleotide bases overlapping. The position in which the c.577 variant falls is identifiable by the configuration of the sequence in its neighborhood: a short sequence composed of four guanine precedes the position in which the A-deletion (A) can occur: GGGGAC(A)GATGACCA (NCBI, 2023). Upstream of this, two other sequences made of four guanine aid in the identification: a short GGGGAGGC placed 23 bases prior the c.577 position and even further upstream a short GGGGAAAGC starting 44 bases prior the c.577 position. Downstream is a short ACCAGGT sequence helps again in the identification.
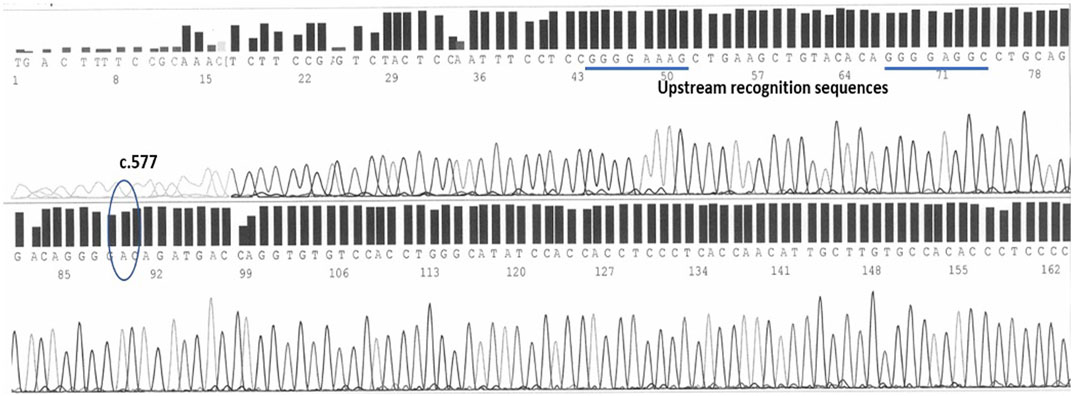
FIGURE 2. The c.577 position on human epo exon 5 sequenced by forward primers. Two upstream sequences, both characterized by 4 guanines, help in the identification.
On the complementary strand, sequenced by the reverse primers, at the .577 position is the nucleotide Timine (complementary to the Adenine of the sense strand) recognizable within the following sequence: TCATC(T)GTCCCC. 15 bases upstream the .577 position, a short ACACACC sequence helps in the recognition of the spot (Figure 3).
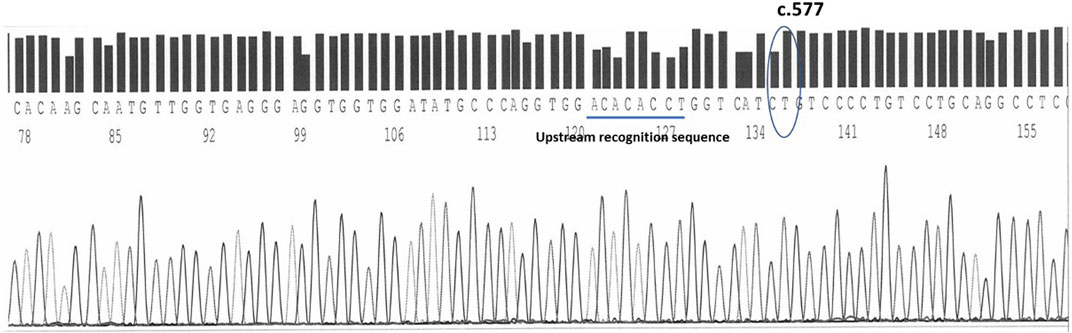
FIGURE 3. The timine at the c.577 position on human epo exon 5 sequenced by reverse primers. An upstream sequence ACACACCT helps in the identification.
Carriers of the variant in the heterozygous state have 50% of their chromosomes carrying the wild type chromosomes and the other 50% with the mutated chromosomes carriers of the c.577del. The presence of the variant is thus identified in the electropherogram by the appearance, starting from the mutation site, of a mixed sequence in which the tracings of the normal sequence and the mutated one overlap each other (Figure 4).
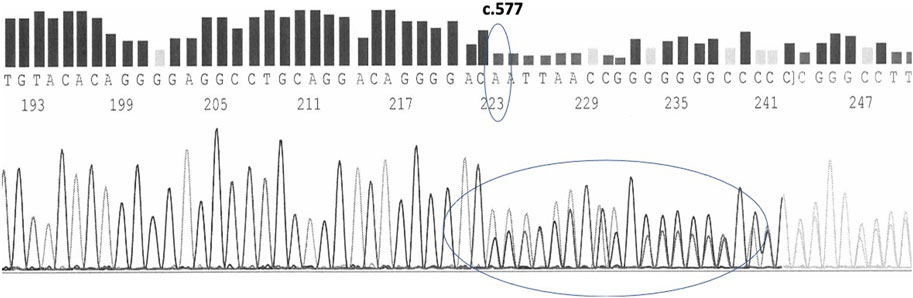
FIGURE 4. c.577del variant as sequenced by forward primers in a heterozygous carrier.
Individuals carrying the variant in the homozygous state will show a linear sequence without nucleotides overlapping, but shifted by one position due to the total deletion of Adenine/Timine in position .577. Figure 5 summarizes the sequence of Epo gene exon 5 in the absence of the c.577del variant and with its presence in both the homozygous and heterozygous state.
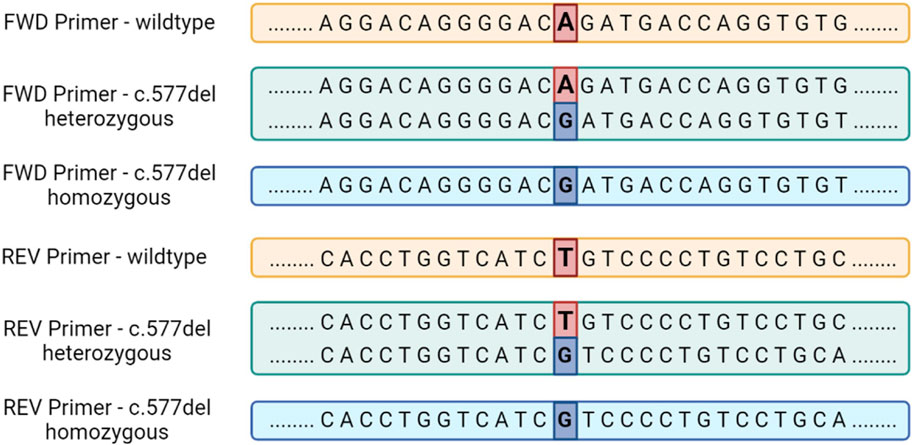
FIGURE 5. The sequence of the human Epo gene exon 5 in the presence or in the absence of the c.577del variant for sequencing performed with both forward and reverse primers.
Interpretation criteria can be summarized as follows.
1) c.577del must be determined by at least two different primers. Bi-directional sequencing is not mandatory. Even though the analysis is usually performed using the same primer (both forward and reverse), the interpretation can be valid even using two different primers, be they both forward or reverse. This is because the bi-directional analysis could not always be possible due to the presence of mutated sequences, which prevent one of the primers from aligning correctly and giving the expected sequencing result.
2) A negative (wild-type) and a positive control (c.577del from a heterozygous or from a homozygous carrier) must always be included in the same experiment and have to return the expected result.
3) The neighborhood of the sequence (at least 10 nucleotides upstream and 10 downstream where the c.577 position falls) must be sequenced entirely without any ambiguity in basecalling.
4) A minimum QV of 20 is necessary to ensure the reliable identification of a nucleotide. This criterion applies perfectly for wild-type sequences or for sequences homozygous for the c.577del variant. For sequences with c.577del in the heterozygous state, the QV often has a low value due to the software interpretive problems on the traits of mixed sequences. In this case we have obtained positive identification of the variant when the height of the peak in position c.577del is at least 200 RFU with a S/N > 3.
5) A minimum Sample Score of is 20 is necessary, but global Samples Scores lower than 20 can be acceptable whenever the part of the sequence that contains the variant and its around has Sample Score greater than 20 and the presence of the variant is clearly visible.
Table 1 shows the results of 8 gDNA samples extracted from urine, whole blood and DBS samples for the two different forward and reverse primers. A whole blood sample carrying the heterozygous c.577del variant was used as positive control. None of the samples analyzed reported the presence of the variant. This is in line with the fact that this variant, already rare, is present only in samples of Asian origin.
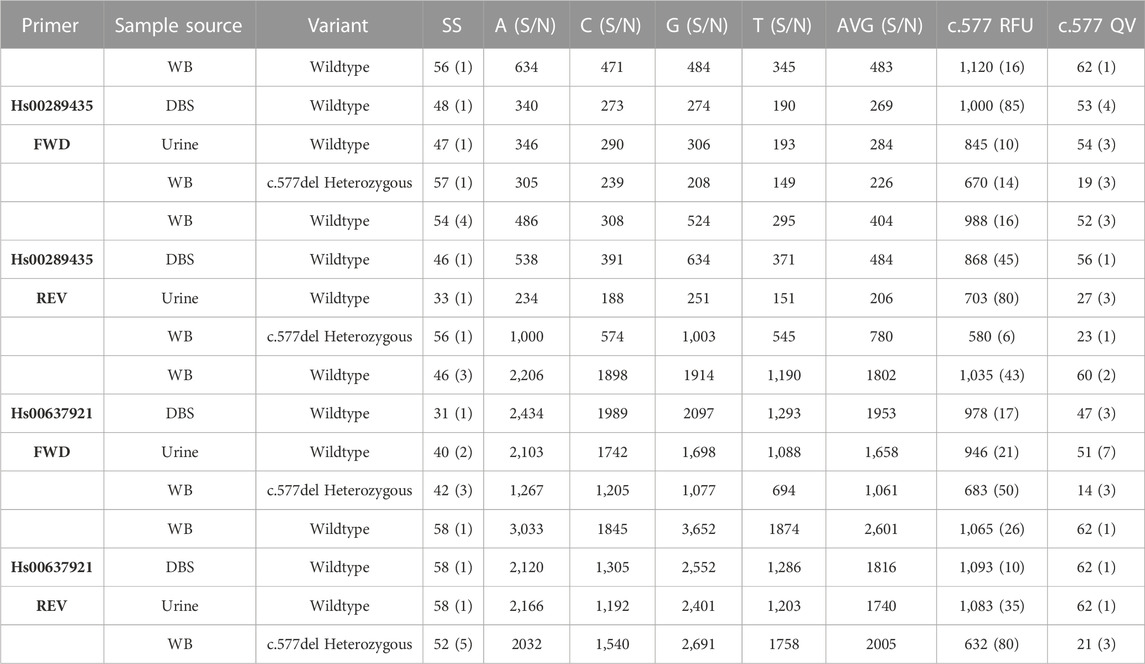
TABLE 1. Results of 8 urine, whole blood (WB), and Dried Blood Spots (DBS) samples all sequenced starting from 1 ng of gDNA amplified with 35 cycles. In parentheses the standard deviation of the score values. 10 ng of ds amplicon were used in each sample for Sanger reaction.
Both primers returned a sequenced fragment of the expected size and, after the trimming of the primers themselves, clearly allowed the sequencing of the exon 5 c.577 position and its around. Despite the fact that it is generally easier to obtain good quality sequences from short amplicons, no appreciable differences are detected comparing the results of the sequences made by the two different primers. Heterozygous positive controls were correctly assessed by both primers. Results obtained from whole blood samples provided the most accurate and precise results after sequencing. Whole blood is the biological sample that provides the best qualitative and quantitative DNA. gDNA sequencing obtained from whole blood samples is much more reproducible regarding the number of bases detected and quality values. Sequences obtained from DBS provided similar quality and interpretability to those obtained from whole blood. In this case, the stability of the dried spots over time should be studied, which has not been taken into consideration for the purposes of this work. In some cases, a lower qualitative result found for gDNA extracted from urine samples is most likely due to the lower quality due to some degradation of gDNA. It is important to note that also in some cases of low QV index, the identification of the variant was anyway carried out without any doubt. No wild-type sample gave a different result than expected. Thus the specificity of the method is therefore 100%. The sensitivity, understood as the minimum amount of gDNA needed to produce an amplicon in quantity suitable for sequencing, was determined to be 100 pg. This amount of gDNA can also be obtained from poor DNA matrices, as in the case of urine samples.
From a practical point of view, the entire process requires 2 working days. Day 1 is dedicated to extraction, quantification, and purification of DNA; while amplification and electrophoretic run are performed on Day 2. The above refers to a totally manual procedure, that can be significantly shortened by automation. The costs depend mostly on the primers and the sequencing kits; clearly, costs per sample drastically decrease if multiple samples have to be assayed. We believe this method can be easily implemented by the WADA-accredited antidoping laboratories for routine application whenever a sample tests positive by the SAR-Page reference method for human recombinant.
4 Conclusion
The presence of c.577del variant (rs369859204) in the human erythropoietin gene may be a confounding factor in the interpretation of the results of the analytical procedure for the detection of human recombinant erythropoietin in doping control. This variant, determining the transcription of a higher molecular weight protein, can erroneously suggest the presence of recombinant erythropoietin in a biological sample, causing the possibility of a false positive result. Although the variant is present at a very low frequency (0.5%–1%) and has been identified only in East Asian populations, it can represent a threat to the accuracy with which anti-doping tests are usually carried out. Consequently, developing a method for identifying the variant mentioned above in a biological sample falls within the aims and objectives of anti-doping purposes. In our laboratory, we have implemented a procedure to perform genetic screening to recognize the rs369859204 deletion site based on Sanger sequencing. The test has 100% specificity and can identify the presence or absence of the rs369859204 variant starting from 100 pg of genomic DNA extracted from biological samples of blood, urine, and dried blood spots, which are typically used in daily anti-doping tests. The use of primers capable of producing double-stranded amplicons of different molecular weight allows to reduce significantly any sequencing problems that may arise due to random mutations in the genome of individuals. The identification of the presence of the genetic variant is specific by respecting the specially developed interpretation criteria. Finally, the possibility of performing this genetic test within the same laboratory where the anti-doping analyzes are performed allows the chain of custody of the samples to be kept intact without having to send the sample to an external facility.
Data availability statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Ethics statement
The studies involving human samples were performed in compliance with the World Anti-Doping Agency ethical code for the accredited laboratories. All samples considered in the present study were from athletes who explicitly expressed their written consent for the samples to be used for research purposes, after further anonymization.
Author contributions
FD preliminarily designed the original research plan, processed the results and revised the paper; LC contributed to the final research plan, performed the experimental part of the study, collected and processed the results and drafted the paper; XdT discussed the results and contributed to writing the paper; XZ isolated the samples with exon c.577del variant and reviewed the data and the final version of the manuscript; LZ supervised the study, discussed the results, and revised the final version of the manuscript; FB supervised the study, discussed the results, revised and formatted the paper and submitted it. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the research funds of the Laboratorio Anti-doping FMSI. Open access funding by University of Lausanne.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Heather, J. M., and Chain, B., The sequence of sequencers: The history of sequencing DNA. Genomics (2016) 107(1):1–8. doi:10.1016/j.ygeno.2015.11.003
Jelkmann, W., and Lundby, C., Blood doping and its detection. Blood (2011) 118(9):2395–2404. doi:10.1182/blood-2011-02-303271
NCBI. NCBI human genome database (2023). Available at: https://www.ncbi.nlm.nih.gov/nuccore/1243022497?report=graph (accessed May 17, 2023).
Pascual, J. A., Belalcazar, V., de Bolos, C., Gutiérrez, R., Llop, E., and Segura, J., Recombinant erythropoietin and analogues: A challenge for doping control. Ther. Drug Monit. (2004) 26(2):175–179. doi:10.1097/00007691-200404000-00016
Reichel, C., Detection of peptidic erythropoiesis-stimulating agents in sport. Br. J. Sports Med. (2014) 48(10):842–847. doi:10.1136/bjsports-2014-093555
World Anti-doping Agency. Harmonization of analysis and reporting of erythropoietin (EPO) and other EPO-receptor agonists (ERAs) by polyacrylamide gel electrophoretic (PAGE) analytical methods (2022). Available at: https://www.wada.ama.org/sites/default/files/2022-01/td2022epo_v1.0_final_eng_0.pdf.
World Anti-doping Agency. World anti-doping agency (2023). Available at: https://www.wada-ama.org/en/prohibited-list.
Yi, J. Y., Kim, M., Jeon, M., Min, H., Kim, B-G., Son, J., et al. Simple visualization method for the c.577del of erythropoietin variant: CRISPR/dCas9-based single nucleotide polymorphism diagnosis. Drug Test. Anal. (2023) 13:871–875. doi:10.1002/dta.2980
Keywords: erythropoietin gene, genetic variant, Sanger sequencing, sport doping, antidoping analysis
Citation: Donati F, Concetti L, de la Torre X, Zhou X, Zhang L and Botrè F (2023) Detection of exon 5 c.577del variant of human erythropoietin gene in whole blood, dried blood spots and urine samples for doping control. Front. Anal. Sci. 3:1202074. doi: 10.3389/frans.2023.1202074
Received: 07 April 2023; Accepted: 26 May 2023;
Published: 04 July 2023.
Edited by:
Zhanjun Yang, Yangzhou University, ChinaCopyright © 2023 Donati, Concetti, de la Torre, Zhou, Zhang and Botrè. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Donati, Zi5kb25hdGlAbGFiYW50aWRvcGluZy5pdA==; Francesco Botrè, Zi5ib3RyZUBsYWJhbnRpZG9waW5nLml0