 Gian Luca Forni
Gian Luca Forni Giuliano Grazzini
Giuliano Grazzini Jeanne Boudreaux3
Jeanne Boudreaux3 Vanessa Agostini
Vanessa Agostini Laurel Omert
Laurel Omert- 1Center for Congenital Anemias, Galliera Hospital, Genoa, Italy
- 2Consultant, Hemanext, Inc., Lexington, MA, United States
- 3Aflac Cancer and Blood Disorders Center, Children’s Healthcare of Atlanta, Atlanta, GA, United States
- 4Transfusion Medicine Department, IRCCS Ospedale Policlinico San Martino, Genoa, Italy
- 5Hemanext, Inc., Lexington, MA, United States
Beta thalassemia (β-thalassemia) is part of a group of inherited hemoglobinopathies caused by a mutation in the beta globin gene, leading to minimal functional hemoglobin and resulting in damaged red blood cells and anemia. β-Thalassemia is most common in the Mediterranean region, South-East Asia, the Indian subcontinent, and the Middle East. Many of these regions include low- and middle-income countries where there are significant unmet needs in the adequate care and management of thalassemia. Patients with transfusion-dependent β-thalassemia, the most severe form of the disease, require regular blood transfusions. Chronic transfusions are often accompanied by iron chelation therapy to manage ferritin levels. Complications caused by transfusions and iron overload are only partially addressed by current treatment strategies, which negatively affect the quality of life of patients with transfusion-dependent β-thalassemia. Until curative modalities become available for all patients worldwide, methods of optimizing supportive treatments are needed to reduce the symptoms of ineffective erythropoiesis; minimize transfusion-related reactions and side effects; reduce rates of alloimmunization and transfusion-transmitted infections; and to reduce the psychosocial burden on both patients and their caregivers. This review aims to provide an overview and comparison of the ways transfusion-dependent β-thalassemia is identified and treated in different geographic regions, to assess unmet needs specific to these regions, and to discuss how therapies currently in development may improve care.
1 Introduction
Thalassemia is a group of inherited hemoglobinopathies in which an autosomal recessive mutation in a globin gene produces too little functional hemoglobin (Hb), resulting in anemia. The most common types are α-thalassemia and β-thalassemia (Mediterranean/Cooley’s anemia) (1). Prevalence is highest in people of Mediterranean, Middle Eastern, Indian, South-East Asian, and African origin (tropical and subtropical areas) (Figure 1) (3, 4).
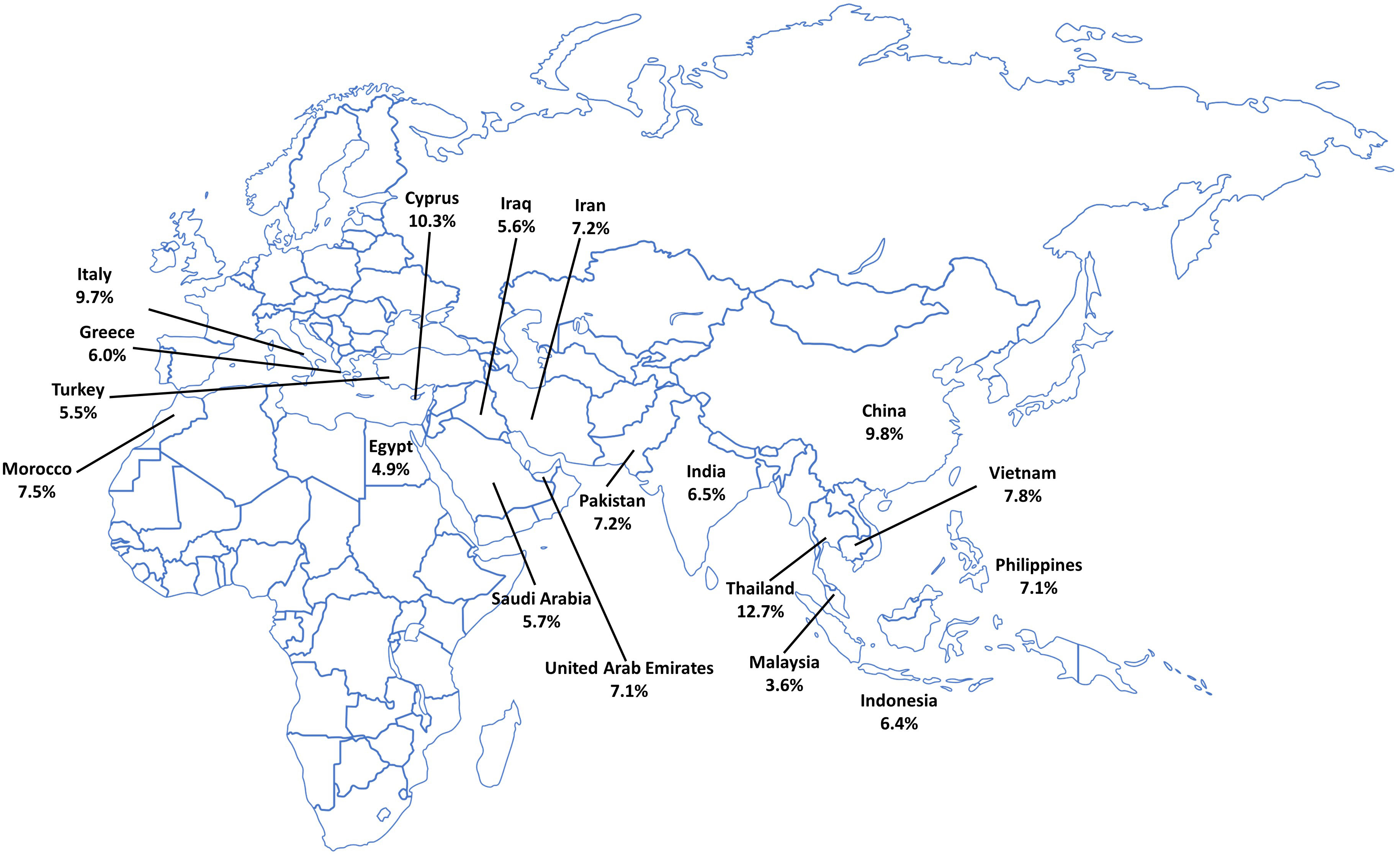
Figure 1 Carrier rate of β-thalassemia in endemic countries. Data taken from the Global Burden of Disease Collaborative Network (2).
The deficit in one type of globin (α- or β-subunit) is generally accompanied by normal levels of the other, resulting in an imbalance of globin chain types, with a relative excess of the non-affected globin (1). Therefore, in β-thalassemia, the deficiency in the β-globin subunit leads to an excess of α-globin. The α-globin aggregates to form inclusions that damage red blood cells (RBCs), causing hemolysis in the bone marrow and/or spleen. As a consequence, patients with β-thalassemia experience anemia and resulting weakness, fatigue, shortness of breath, headaches or dizziness, hepatosplenomegaly as a result of ineffective erythropoiesis, growth retardation, and heart problems (5). The severity of the condition is related to the extent of the imbalance in α-globin and non-α-globin synthesis (1, 5).
Diagnosis of thalassemia is made based on clinical presentation and on hematologic and molecular/genetic analyses (6). Many countries have preconception screening and prevention programs in place to identify carriers of thalassemia (7). The most severe types of thalassemia are transfusion-dependent thalassemia (TDT), in which patients have a low level of functional Hb and require regular RBC transfusions. Transfusion-dependent β-thalassemia (TDβT) is the most common form of TDT; previous studies have shown that, worldwide, over 25,500 infants born annually have β-thalassemia that will become transfusion dependent (8, 9). TDβT is a multifaceted disorder associated with a wide variety of disease manifestations and management strategies. The aim of the present review is to provide an overview and comparison of treatment strategies for TDβT in different geographic regions, to identify unmet needs specific to these regions, and to discuss how therapies currently in development may improve care.
2 β-Thalassemia epidemiology and screening programs
The World Health Organization (WHO) reports that, globally, 40,000 infants annually are born with thalassemia, and the majority of these have β-thalassemia (8). Most children with thalassemia are born in low- and middle-income countries (LMICs), predominantly in South-East Asia (Figure 1) (8). Migration from regions where β-thalassemia is endemic has resulted in approximately 1.5% of the global population becoming carriers of β-thalassemia genes (4), and countries where β-thalassemia is less common, such as the United States, have seen an increase in prevalence. In the last 50 years, prevalence of β-thalassemia in the United States has risen by 7.5% (10), partly due to the movement of peoples from Asian and Middle Eastern countries by immigration and adoption (11). Furthermore, according to the Cooley’s Anemia Foundation, approximately 12% of US patients with β-thalassemia were adopted from other countries (12).
National programs to prevent β-thalassemia via carrier screening, counseling, and prenatal diagnosis in at-risk populations in endemic countries have existed since the 1970s (13, 14). Since then, several high-income countries (e.g., Italy) have initiated prevention programs that not only include population screening and prenatal diagnosis for thalassemia, but also include public education to remove any stigma associated with the detection of thalassemia genes (13). This has resulted in a reduction in the number of children born with thalassemia (14). In countries where thalassemia is prevalent, premarital or antenatal screening programs are largely voluntary initiatives offering information to at-risk populations. Therefore, to ensure widespread acceptance and to have maximum impact, they must take into account local needs and customs (7).
β-Thalassemia is considered endemic in Italy; the country has had policies in place to reduce the incidence of hemoglobinopathies, including thalassemia, since the 1970s, with free carrier screening and genetic diagnostics widely available (15, 16). These preventative measures have led to a reduction in the number of children born with thalassemia (17), particularly in Sicily, which has seen an 85% decrease in the incidence of β-thalassemia over the last 30 years (17). In addition to reducing incidence, research into improving the overall standard of care in Italy has allowed patients with thalassemia to live long into adulthood (Figure 2). An Italian study followed patients with β-thalassemia over 50 years and found that those in younger birth cohorts demonstrated a better rate of survival, owing to an improvement in the standard of care over the years; however, despite these advances, the median age of death in patients with β-thalassemia was still 23.2 years (19).
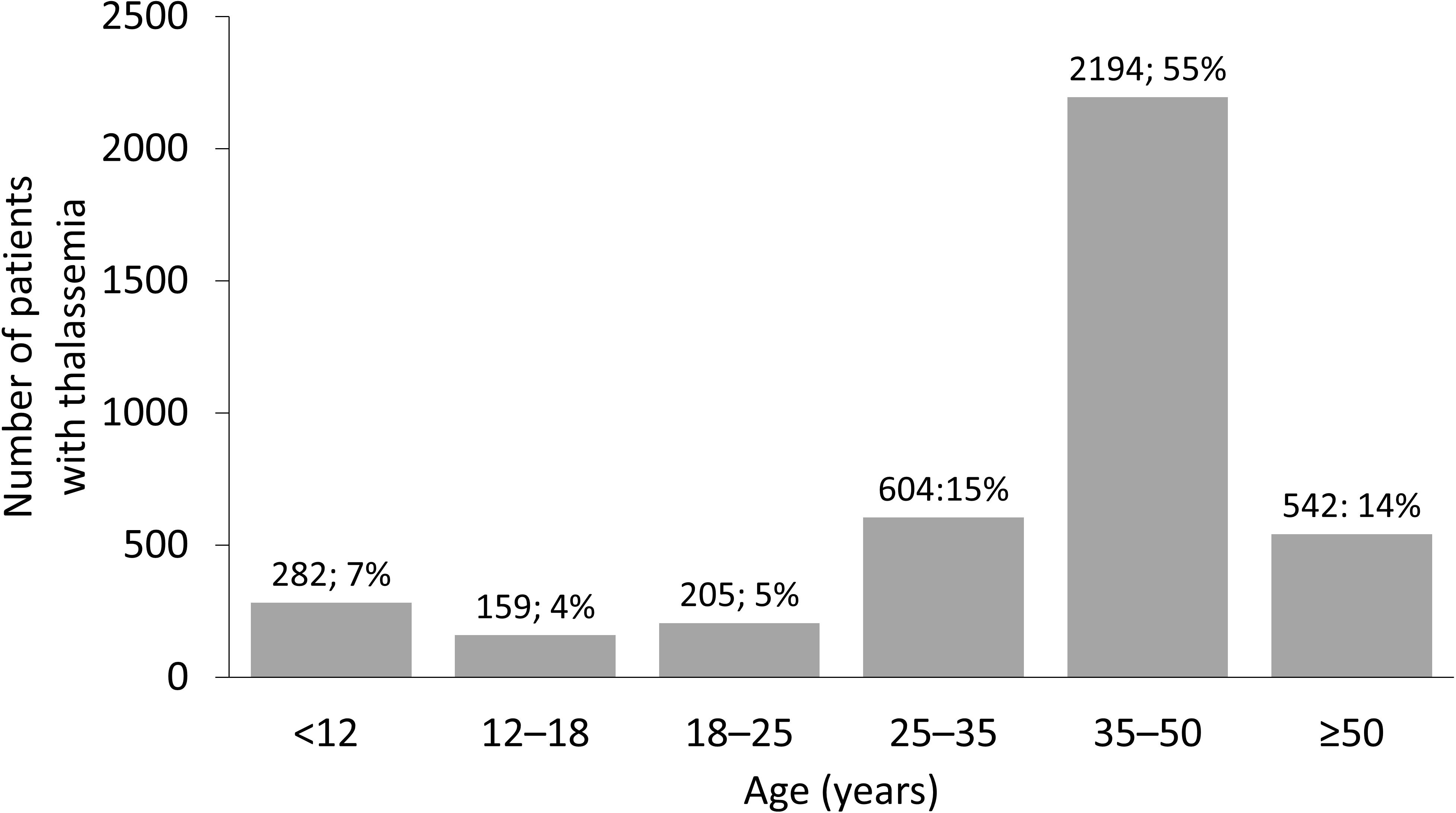
Figure 2 Age distribution of thalassemia in Italy. Distribution of age of patients with thalassemia syndromes at 36 treatment centers in Italy. Reproduced with permission from Longo et al, 2021 (18).
In the United States, although the prevalence of β-thalassemia has increased in recent years, it is not a core condition listed in the United States Recommended Uniform Screening Panel (RUSP) (20). Despite this, methodologies used to screen for sickle cell disease (a RUSP core condition) detect a diminished ratio of Hb A to fetal Hb, which is suggestive of β-thalassemia. As such, appropriate care can be received if abnormal Hb levels are detected, to help minimize the impact of the disease (20). In countries where β-thalassemia is prevalent, national thalassemia prevention and control programs have also been successfully implemented (Table 1). In Thailand, an upper middle-income country, widespread public education, genetic counseling, and preconception and prenatal screening and diagnosis have been effective in reducing the incidence of thalassemia (13). Evidence suggests that despite no change in the prevalence of thalassemia genes among the population, new cases of the disease have been prevented by screening programs (41). In Turkey, the Haemoglobinopathy Prevention Program has led to the creation of diagnostic centers for public education on β-thalassemia. Premarital screening tests were performed in 30% of all couples in the first year of the program, increasing to 81% in 2008, resulting in a 90% reduction in new thalassemia births in Turkey (42). In Brazil, the β-thalassemia trait is present in around 1.5% of the Caucasian population, with suspected thalassemia major in 0.8% of the total population (43); although the prevalence is lower than in Mediterranean or South-East Asian countries, this is still of concern in a country with over 214 million people (44). The Brazilian National Neonatal Screening Program provides access to the majority of the country through separate State Neonatal Screening Programs (43). The availability of screening for thalassemia and other hemoglobinopathies is important in a country with a diverse genetic background related to previous colonization and migration from the Mediterranean region and Africa (43).
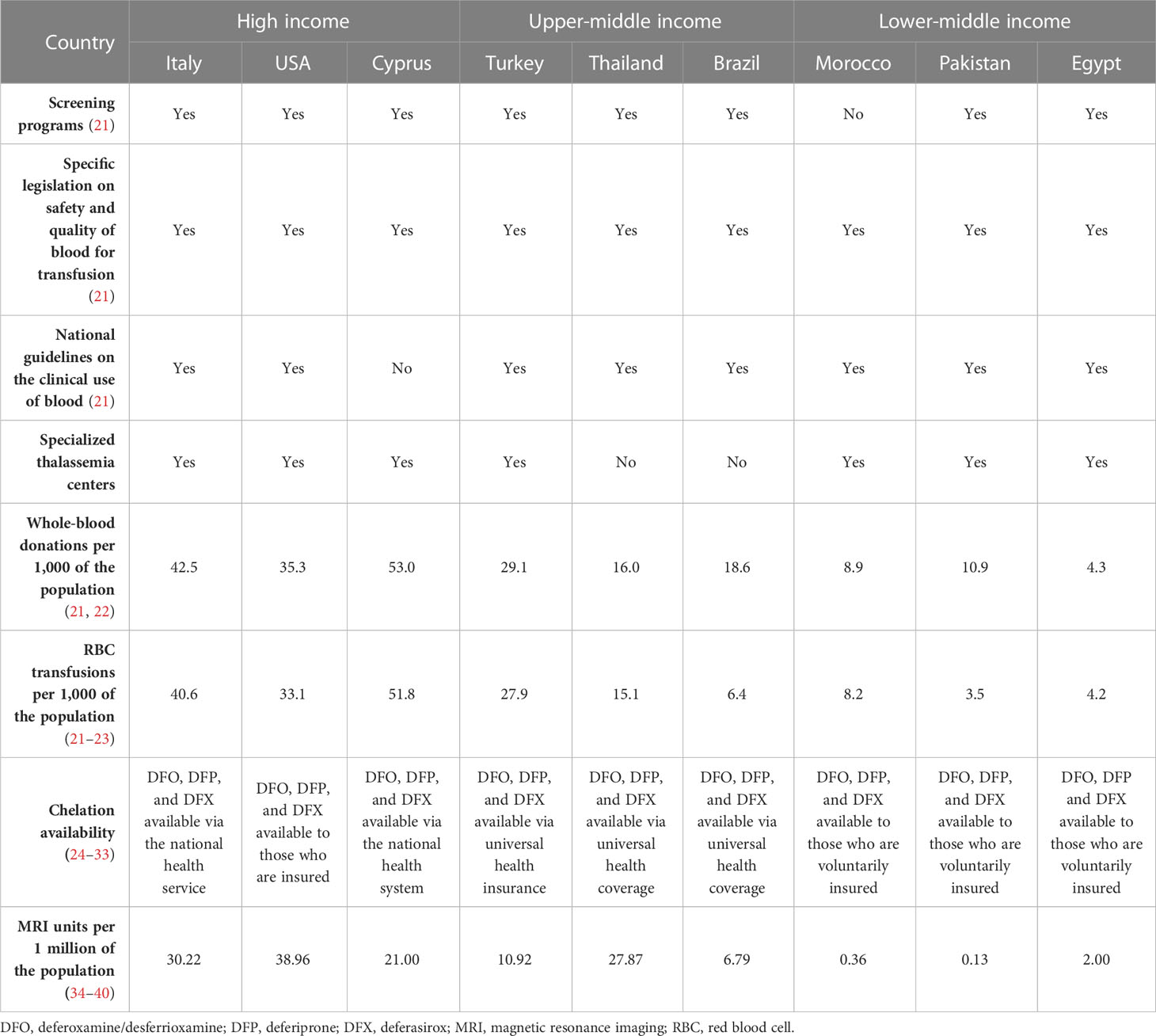
Table 1 Availability of management strategies for transfusion-dependent β-thalassemia in countries where thalassemia is prevalent.
In lower-income countries, awareness of β-thalassemia plays a role in the epidemiology of the disease. In Pakistan, the burden of β-thalassemia is high and life expectancy of patients with thalassemia is around 10–12 years owing to a lack of healthcare resources and the limited availability of safe donor RBCs (45). The Punjab Thalassemia Prevention Project offers cascade screening for relatives of children with β-thalassemia (45). A study of northern Moroccans with hemoglobinopathies found that 50% of couples were consanguineous compared with 30% in the general population. In the hemoglobinopathy population studied, a significant decrease of 14% was found in the prevalence of consanguineous marriages compared with the previous generation. This may be attributed to awareness campaigns informing the population of the risks of consanguineous marriage (46).
The migration of people from regions where β-thalassemia is prevalent has an impact on the healthcare systems of the host country. Overlooking new residents will likely change the epidemiology of β-thalassemia and it is important that these people are included in relevant screening programs (2, 12). Despite the relative success of screening programs worldwide, not offering the services to migrants negatively affects the epidemiology of the disease. Following an influx of refugees at the beginning of the Syrian civil war, over one-quarter of a million babies were born to Syrian couples living in Turkey between 2011 and 2017 (47). Difficulties in communication and understanding of medical terminology led to these refugees being overlooked by screening programs (48). Owing to the large volume of migrant workers coming to Thailand, it has been recommended that the Thai government extend prevention and control programs to include migrant workers living in the country, many of whom come from other countries where β-thalassemia is endemic (49).
3 Management of transfusion-dependent β-thalassemia
3.1 Treatment guidelines
The aim of β-thalassemia treatments is primarily to suppress the ineffective erythropoiesis that causes symptoms of anemia, with conventional treatments focused on RBC transfusion and iron chelation (6). In Europe, few countries provide dedicated thalassemia services, and details of measures for patient monitoring and treatment are, overall, very scarce (50). Italy has, arguably, one of the best standards of care for thalassemia as a result of the high prevalence and availability of resources. A survey of 114 Italian treatment centers reported that complete blood count, ferritin level, echocardiography, and T2*-weighted magnetic resonance imaging (MRI) were commonly performed in the preceding 12 months. Approximately half of patients reviewed were reported to have received ≥ 3 units of RBCs per month, with deferasirox chelation therapy (Table 2) administered in two-thirds of patients (54).
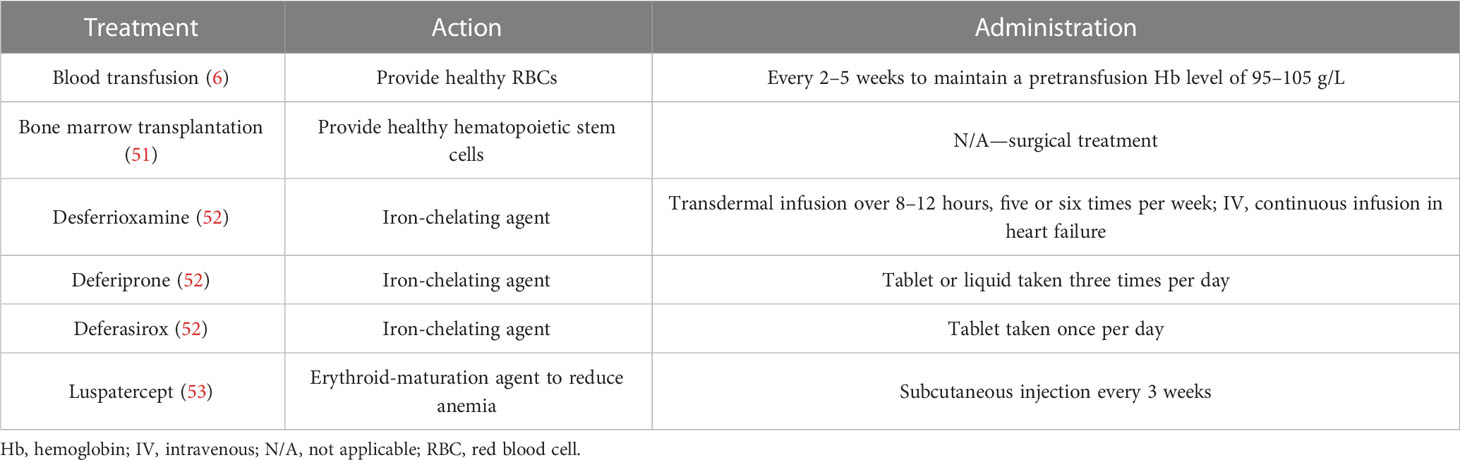
Table 2 Key available treatments for transfusion-dependent β-thalassemia.
Many countries have their own national guidelines with recommendations for the care of patients with thalassemia (Table 1). For example, in South America, Brazil has comprehensive guidelines for the management of TDβT (55), and in South-East Asia, Thailand has recommendations for RBC transfusions in patients with TDT (56). In recent years, six major TDT management guidelines have been published (the Thalassemia International Federation, and national guidelines for Australia, Canada, Italy, the United Kingdom, and the United States of America). These guidelines are aligned on most aspects of management, although there are some differences in iron overload monitoring and when to administer chelation therapy (57). Iron loading can be monitored with MRI; however, access differs depending on the income level of a country (Table 1) (34). Italian guidelines specifically recommend liver biopsy with iron measurement by atomic absorption spectroscopy as the gold standard, with MRI as an accepted alternative. Italian patients over the age of 5 years with β-thalassemia and without a transfusion history must also have serum ferritin levels monitored 1–2 months apart to determine a baseline level before iron chelation can be initiated (24). In children who are frequently transfused, Italian guidelines recommend that iron chelation can be initiated when they have received > 10 units of blood or if their serum ferritin level is ≥ 1,000 ng/mL (24), which is also recommended by the 2021 Thalassemia International Federation guidelines (6).
3.2 Blood transfusions
Blood transfusions are the primary treatment to suppress ineffective erythropoiesis and reduce anemia (Table 2), with guidelines recommending that a clinical assessment of the need for regular transfusions should quickly follow a confirmed genetic diagnosis of thalassemia. International guidelines for the management of TDT state that to initiate transfusion therapy, patients must satisfy the following criteria (6). There must be a confirmed diagnosis of thalassemia, a Hb level of < 70 g/L on two occasions more than 2 weeks apart (excluding other causes), and/or the following clinical criteria irrespective of Hb level: significant symptoms of anemia, poor growth/failure to thrive, complications from excessive intramedullary hematopoiesis, and clinically significant extramedullary hematopoiesis.
For patients with TDT, lifelong regular blood transfusions are recommended every 2–5 weeks to maintain a pretransfusion Hb level of 95–105 g/L (6). This transfusion regimen promotes normal growth in children and allows for a normal level of physical activity (9). Maintaining the guideline-recommended Hb level may require shorter intervals between transfusions, but an appropriate interval between treatments must consider other factors, such as a patient’s school or work schedule, and fit to their lifestyle to prevent a negative effect on quality of life (QoL) (6). However, maintaining an adequate supply of blood in countries where β-thalassemia is endemic is difficult, as many of these are LMICs that lack the volunteers, the resources, and the national organization of high-income countries. This can be seen in the overall volume of whole blood donated, where lower-income countries may have as much as 10-fold fewer donations per 1,000 of the population compared with higher-income countries (Egypt, 4.3 vs. Italy, 42.5; Table 1). Globally, only 12% of children born with TDβT received a RBC transfusion, many of whom live in lower-income countries (8).
3.2.1 Transfusion-related complications
In a study of Italian patients with thalassemia, the most frequent transfusion-related clinical complication was iron-related heart disease, reported in 30% of patients, with many other common complications being endocrine in nature (18). As a result of suboptimal monitoring and management, symptoms of comorbidities, such as endocrine disorders, may increase with cumulative iron exposure and can manifest in early adulthood in patients who are exposed to chronic transfusions without adequate chelation (58). Reducing the intervals between transfusions may be necessary to maintain guideline-recommended Hb levels, but can cause patient discomfort and inconvenience. Increasing the exposure to allogeneic blood with short intervals between transfusions leads to the accumulation of excess iron in the heart, liver, and other organs; cardiac iron overload can have serious consequences, with a serum ferritin level > 1,000 ng/mL reported as an independent risk factor for death (19). In addition to the excess iron from blood transfusions, patients with thalassemia have inappropriate increased absorption of iron from their diet as a result of hepcidin suppression (59–61). Furthermore, there is the risk of alloimmunization with RBC transfusions through the development of anti-RBC antibodies (62) and allergic, febrile, or delayed hemolytic transfusion reactions (63), as well as the risk of transmission of blood-borne infections.
3.3 Management of transfusion-related complications
Chelation therapy (deferoxamine [desferrioxamine, Desferal], deferiprone, deferasirox) is used to remove the iron build-up resulting from blood transfusions and iron absorption through the gastrointestinal tract (6). It must be taken regularly to be effective (6), but factors impacting adherence include route of administration, side effects, and forgetfulness. Adherence to a medication that must be taken every day, sometimes multiple times per day, is very difficult for many patients to maintain (25, 64). Although chelation therapy may be available globally (Table 1), the cost is prohibitive in some countries. Worldwide, less than half of children born with TDβT receive adequate iron chelation therapy (8). According to a 2008 WHO report, of the 100,000 patients living with regular transfusions, around 3,000 die annually in their teens or early adulthood from uncontrolled iron overload (8).
Alloimmunization is not uncommon, with 25% of thalassemia patients in one study having positive alloantibodies (65) and 18% of patients in another study (66). It depends on several factors, including the homogeneity of the donor–recipient population (67). Blood antigen matching is performed to reduce alloimmunization (62) and Rh genotyping for the purpose of identifying genetic variants is possible, although cost prohibitive in many countries (68). A systematic review of 41 studies determined that 78% of antibodies identified in patients with TDT were anti-Rh and anti-Kell antibodies; therefore, matching these antigens may reduce the risk of alloimmunization by approximately 80% (62). In a Moroccan study over 9 years, the prevalence of RBC alloimmunization was 9%, with a univariate analysis indicating that a transfusion interval < 3 weeks was a significant risk factor (71% vs. 36% for intervals > 3 weeks; p = 0.01). As previously discussed, this may be due to increased exposure to allogeneic blood compared with longer intervals between transfusions (67).
3.3.1 Patient management strategies
Management of patients with β-thalassemia in specialized treatment centers has a positive impact on life expectancy (69); however, they are often only accessible to patients with TDβT who reside in high-income countries. Although many lower-income countries where thalassemia is endemic do possess specialized treatment centers to provide safe blood and proper care to patients (Table 1), they are often only present in the major cities, and in some cases, there may only be a single specialized center serving the whole country. In Morocco, most patients with thalassemia reside in rural areas (46), and do not have the regular access to care that they require. Conversely, in Italy, there are several specialized centers across the country (70). Patients treated at specialized centers have longer life expectancies (69), as they are treated by a multidisciplinary team and provided with the best possible standard of care (Figure 3); however, longer life expectancy has associated complications, as older people are more likely to develop comorbidities that must be treated in tandem with their thalassemia (70). In many parts of Asia, prognosis remains poor, as there is limited treatment availability and treatments are performed by non-specialized local hospitals; supportive treatments such as RBC transfusion are often the only available treatment (14). In many Asian countries, treatment is only available to those who can afford it, particularly in the case of iron chelation (14). Although blood transfusion may be widely available in these countries, access to chelators to manage iron overload are not (14), leading to associated complications and, ultimately, death.
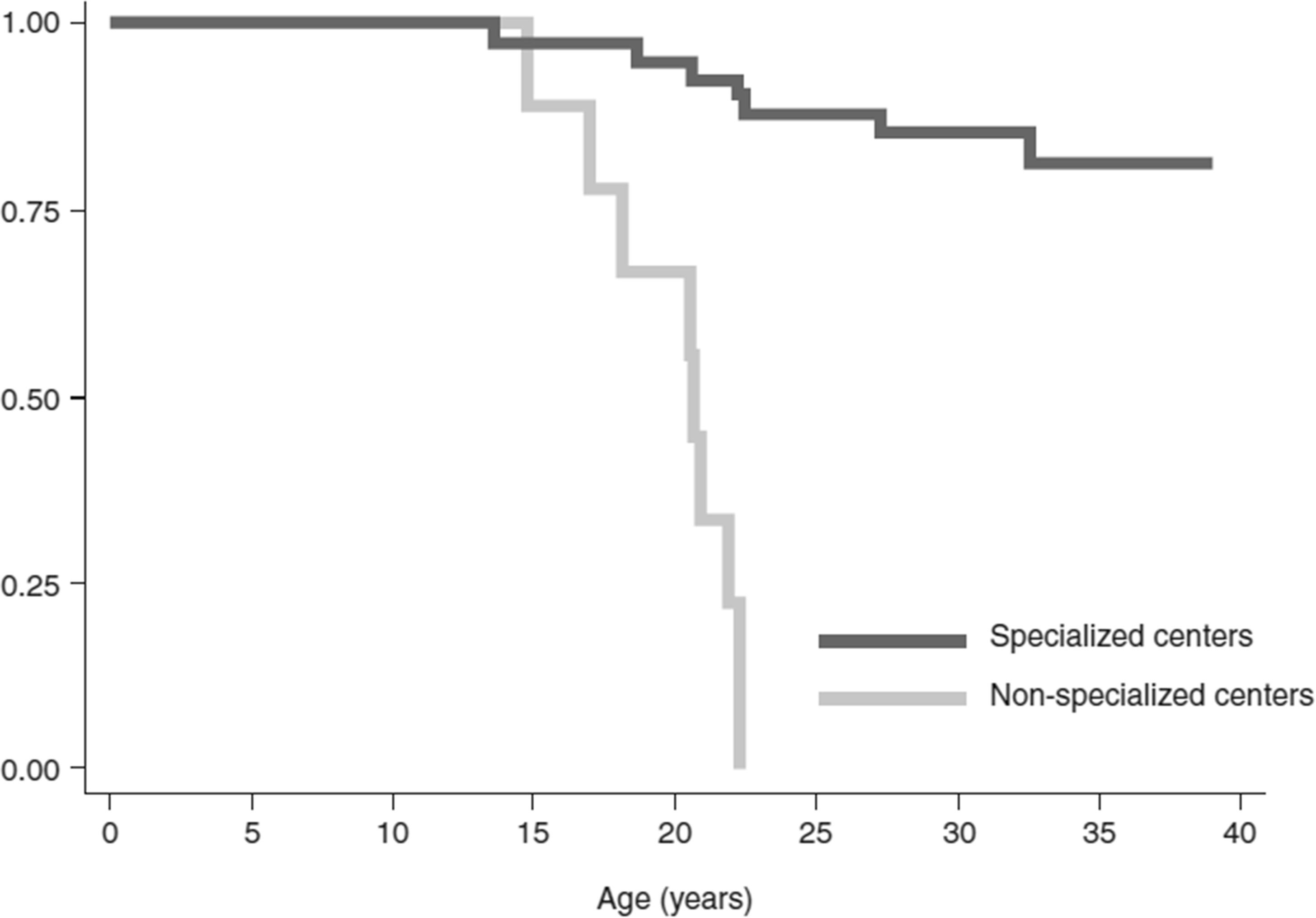
Figure 3 Survival rates of patients with thalassemia major treated in specialized centers for hemoglobinopathies in Italy. Kaplan–Meier overall survival curves of patients referred to specialized centers compared with patients referred to non-specialized centers. Log-rank p-value < 0.0001; hazard ratio of specialized compared with non-specialized centers, adjusted for sex (Cox model): 18.1, 95% confidence interval 4.7 to 69.0; p < 0.001. Reproduced with permission from Forni et al, 2009 (69).
Luspatercept is a treatment for anemia in adults with β-thalassemia who require regular blood transfusions (Table 2). In the Phase III BELIEVE trial, luspatercept reduced the transfusion burden of patients with TDβT compared with placebo. Luspatercept has received approval for use in TDβT in both the US and Europe (71, 72); however, this treatment may not yet be covered by many national healthcare systems or private insurers. Another treatment, hydroxyurea, is an orally administered ribonucleotide reductase inhibitor approved to reduce the need for blood transfusions in sickle cell disease. This has the potential to reduce the cost of transfusion; however, there are inconsistent results in patients with TDβT and those receiving hydroxyurea should be closely monitored for response (9). In addition, potential therapies in the pipeline, such as pyruvate kinase activators etavopivat and mitapivat, may decrease transfusion needs in patients with β-thalassemia (73, 74).
3.3.2 Safety of blood transfusions
Chronic blood transfusion comes with many risks, and many patients in lower-income countries are unable to access safe blood (21). International guidelines recommend that blood should be obtained from “carefully selected voluntary, non-remunerated donors” to prevent transfusion-transmitted infections (6). Two-thirds of countries have specific legislation on the safety and quality of transfused blood, but this only includes 39% of low-income countries (21). Lower-income countries often have decentralized blood banking systems that rely heavily on non-governmental organizations, family replacement donations, and charities. This is often due to a lack of centralized systems, specialized centers, and voluntary donations, and improper screening (75, 76). The WHO recommends that all blood donations should be screened for infectious agents prior to use, and screening for the human immunodeficiency virus (HIV), hepatitis B (HBV), hepatitis C (HCV), and syphilis should be mandatory. Further testing for locally relevant infectious diseases, such as malaria or West Nile virus, may also be necessary (6). Nearly all of the blood donated in high-income countries and upper-middle-income countries is screened (99.8% and 99.9%, respectively), but screening rates are only 83% and 76% in LMICs and low-income countries, respectively. This has led to a higher prevalence of transfusion-transmitted infections (TTIs) in lower-income than in higher-income countries (HIV: 0.7% vs. 0.002%; HBV: 2.8% vs. 0.02%; HCV: 1.0% vs. 0.007%; syphilis: 0.9% vs. 0.02%) (21). Furthermore, LMICs often have difficulty in sourcing and storing sufficient volumes of blood, and these shortages may be addressed via unbanked directed blood transfusion, where donors undergo rapid TTI tests before transfusing blood to the recipient. There is concern that the use of rapid testing may lead to increased infection rates (77); only 34% of blood laboratories in low-income countries undergo external quality assessment, compared with over 80% in high-income countries. Chronic transfusions required during the management of TDβT increase the risk of acquiring TTIs; in one study in Pakistan, older thalassemia patients had a significantly higher risk of HCV infection than thalassemia patients aged ≤ 10 years (22% vs. 8%; p = 0.005) (75). This difference can, however, be attributed to better screening in younger people and the fact that HCV is also sexually transmitted (75). Countries where thalassemia is prevalent have some of the highest rates of TTIs, such as HCV. The rates of thalassemia patients with HCV in Iran and Bangladesh are approximately 15% (78), and an Indian study found a significant HCV viral load in 16% of chronically transfused patients with thalassemia (79). In a study of almost 4,000 patients with thalassemia in Pakistan, HCV was present in the serum of 26% of patients assessed (75). The introduction of serologic testing and the development of direct-acting antiviral agents has led to HCV being all but eradicated in higher-income countries, such as Italy (80). Although the risk of acquiring TTIs, including HIV, HBV, HCV, and syphilis, is higher in LMICs than higher-income countries, it is decreasing globally and may reduce further as new technologies, such as pathogen inactivation systems for RBCs, become widely available (81).
4 Unmet needs in transfusion-dependent β-thalassemia
As outlined, there are multiple facets to disease presentation in thalassemia, and most are only partially addressed by current treatment strategies. Despite improvements in treatment, disease- and transfusion-associated complications are still a challenge (82). The burden of regular transfusions in TDT and the associated side effects, particularly iron overload, is central to this (83), with a number of thalassemia-related complications, and subsequent healthcare resource utilization, positively correlated with increases in the number of units of RBCs transfused (84). A study in the United States of America estimated that patients with TDβT required an average of 17 RBC transfusions annually with an annual healthcare cost of US$130,000/year compared with US$5,000/year in non-thalassemia controls; these costs were attributed to RBC transfusion and chelation therapy (85). The incidence of comorbidities in patients with TDβT is higher than people without the disease and may be largely attributable to iron overload (85). With respect to cancer incidence, the liver is a common site of tumor development in patients with thalassemia, possibly as a result of the stress caused by iron overload and infection with HBV/HCV, although overall cancer risk is not increased as a result of the presence of the hemoglobinopathy (86). An Italian study assessing COVID-19 vaccinations reported that the immune response of patients with TDT resembled that of healthy elderly subjects, indicating that the immune system may prematurely age in these patients (87). Poor physical health-related quality of life (HRQoL) in patients with β-thalassemia was positively associated with somatic comorbidities and depression score in an Iranian study. Variables associated with poor mental HRQoL were anxiety and depression scores (88). In another Iranian study, all aspects of QoL were affected in adults with TDβT, with QoL correlating to laboratory findings. The authors concluded that managing patients’ laboratory indices may improve QoL and regular screening of QoL may allow for more efficient management of disease complications (89). This impairment in QoL is not exclusive to LMICs, as patients with TDβT in Italy reported significantly lower general health and vitality as well as total psychological well-being than the general population (90). In Sri Lanka, children with TDβT had lower mean HRQoL scores than healthy children, and splenectomy, short stature, under nutrition, and longer hospital stays were associated with lower QoL scores (91). In Malaysia, HRQoL in children with TDT has improved since 2005, but psychosocial health was still lacking, with school functioning being the lowest dimension of this subscale (92). In children with TDT in the United Arab Emirates, increasing age was associated with worse QoL scores, possibly due to older children suffering more from iron overload and having missed out on more school (93). Despite a high burden on QoL in TDT, there is some evidence indicating that this may be higher in patients who do not require regular transfusion (94), that is, non-transfusion-dependent thalassemia. A recent study found that anxiety/depression and perceived barriers were significant negative predictors of QoL in these patients, whereas a health-promoting lifestyle was a positive predictor. Where budgets are limited, the authors suggested that factors more strongly affecting QoL should be prioritized (95). In addition, there are other burdens that may negatively impact the QoL of the patient. First, there are the financial costs for the treatment itself (85, 96), but there are also the costs associated with undergoing treatments, such as transport, and lost opportunities for the patient and their family (97). Finally, the loss in productivity at work as well as the inability to do unpaid work may negatively impact the patient (98).
Current curative measures utilizing stem cell transplants are limited and may be high risk, particularly in LMICs where the standard of care is suboptimal. As such, recent research has focused on improving the collection, preparation, testing, storage, transport, and administration of blood and blood components. The COVID-19 pandemic has led to a severe shortage of donated blood, with donor attendance falling by up to 30% and many hospitals unable to supply blood to the patients who need it most (99, 100), particularly in lower-income countries where supply is already limited (Table 1). In the case of patients with TDβT, there is a need to reduce the requirement for RBC transfusion or to extend the shelf life of RBCs to prevent supply issues and allow patients to receive the care they need. A reduction in the required transfusion frequency and volume may also reduce or delay iron overload and its effects on patient health and QoL.
5 Treatments in development
Many therapies and technologies are currently in development for TDT that attempt to address currently unmet needs (Table 3) (106, 107). There are therapeutic interventions in development that aim to increase Hb levels, with different combinations of chelators aiming to optimize the reduction in iron overload. In addition, there are several therapies that aim to resolve the underlying disease process, such as stem cell transplantation and gene therapy. Genome editing may help to reduce transfusion dependence; alteration of the BCL11A erythroid enhancer has been shown to induce long-term transfusion independence in patients with TDβT (108), while mutation of α-globin enhancers has demonstrated benefits in patients with TDβT via a clinically significant knockdown of α-globin (103). Furthermore, a technology in development in clinical trials is aiming to improve the quality of transfused blood via hypoxic storage of RBCs (109). This may help to reduce the frequency of RBC transfusions and the overall transfusion burden by providing patients with cells of better quality.
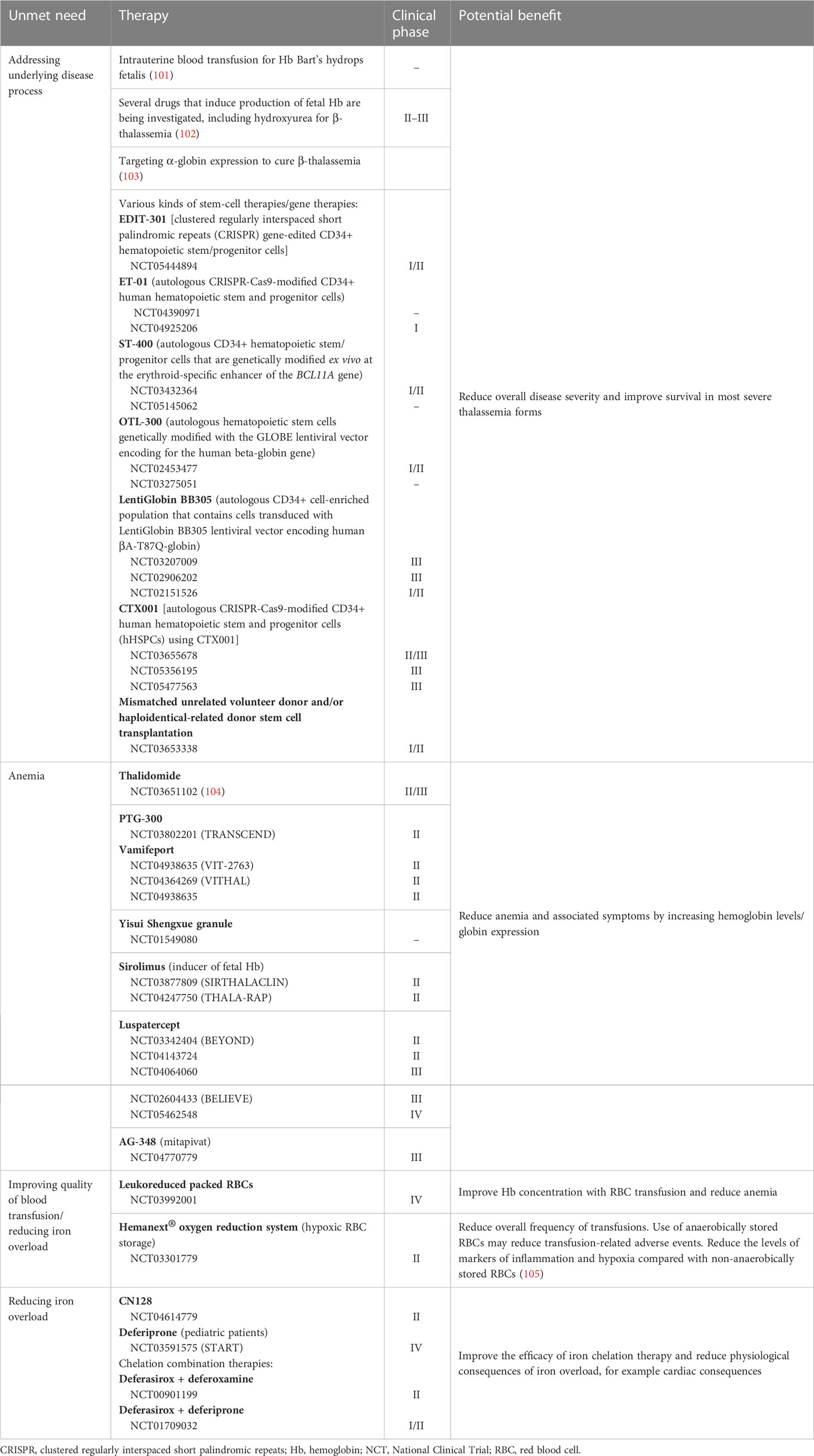
Table 3 Overview of therapies/technologies in clinical development for transfusion-dependent thalassemia.
6 Discussion
TDβT is a severe form of thalassemia that affects many people worldwide, with thousands of infants born annually eventually becoming dependent on transfusions in later life (8, 9). This multifaceted disorder is associated with a large disease burden, and the wide variety of symptoms require individualized management strategies and adequate resources. Patients with thalassemia have a high unmet need as a result of the effects of the disease and also the side effects of the primary management strategy, RBC transfusion. Most patients with thalassemia are in LMICs where the standard of care is suboptimal compared with higher-income countries. As these needs continue to go unmet throughout a patient’s life, both their physical and mental health are negatively affected, impacting both their life expectancy and their QoL as a whole.
Curative treatments are not yet available for every patient worldwide; they may be high risk and are expensive, regardless of availability. Iron chelation therapies and new treatments, such as luspatercept, are not widely available. Therefore, RBC transfusion with iron chelation currently remains the main treatment for most patients. However, the availability of a safe and adequate blood supply is suboptimal in many countries, particularly in those lower-income countries without bloodbanking infrastructure, which rely on the family of the patients and paid volunteers to maintain supply. In addition to the low volumes of available blood in these countries, there is a higher incidence of TTIs than in higher-income countries.
In conclusion, until curative modalities become available for all patients, modalities to optimize blood transfusion are needed to reduce anemia and its impact on physical and mental well-being; minimize transfusion-related reactions, side effects, alloimmunization, and TTIs; and reduce the psychosocial burden on both the patient and their caregivers. In addition, all patients need to have access to adequate chelation to reduce the sequelae associated with iron overload.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of interest
GG reports independent consultancy work for Hemanext, Inc. LO reports employment by and stock in Hemanext, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Galanello R, Origa R. Beta-thalassemia. Orphanet J. Rare Dis. (2010) 5:11. doi: 10.1186/1750-1172-5-11
2. Global Burden of Disease Collaborative Network. Global burden of disease study 2019 (Gbd 2019) results (2019). Available at: https://vizhub.healthdata.org/gbd-results/ (Accessed January 2023).
3. Piel FB, Weatherall DJ. The alpha-thalassemias. N Engl. J. Med. (2014) 371(20):1908–16. doi: 10.1056/NEJMra1404415
4. Colah R, Gorakshakar A, Nadkarni A. Global burden, distribution and prevention of beta-thalassemias and hemoglobin e disorders. Expert Rev. Hematol. (2010) 3(1):103–17. doi: 10.1586/ehm.09.74
5. Thein SL. The molecular basis of beta-thalassemia. Cold Spring Harb. Perspect. Med. (2013) 3(5):a011700. doi: 10.1101/cshperspect.a011700
6. Cappellini MD, Farmakis D, Porter J, Taher. A. 2021 Guidelines for the management of transfusion dependent thalassaemia (Tdt). Thalassaemia Int. Fed. (2021).
7. Kattamis A, Forni GL, Aydinok Y, Viprakasit V. Changing patterns in the epidemiology of beta-thalassemia. Eur. J. Haematol (2020) 105(6):692–703. doi: 10.1111/ejh.13512
8. Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull. World Health Organ (2008) 86(6):480–7. doi: 10.2471/blt.06.036673
9. Shah FT, Sayani F, Trompeter S, Drasar E, Piga A. Challenges of blood transfusions in beta-thalassemia. Blood Rev. (2019) 37:100588. doi: 10.1016/j.blre.2019.100588
10. Sayani FA, Kwiatkowski JL. Increasing prevalence of thalassemia in America: implications for primary care. Ann. Med. (2015) 47(7):592–604. doi: 10.3109/07853890.2015.1091942
11. Lal A, Wong T, Keel S, Pagano M, Chung J, Kamdar A, et al. The transfusion management of beta thalassemia in the united states. Transfusion (2021) 61(10):3027–39. doi: 10.1111/trf.16640
12. Cooley’s Anemia Foundation. Cooley’s anemia foundation. resources on adoption of children with thalassemia (2022). Available at: https://www.thalassemia.org/adoption/ (Accessed January 2023).
13. Cao A, Kan YW. The prevention of thalassemia. Cold Spring Harb. Perspect. Med. (2013) 3(2):a011775. doi: 10.1101/cshperspect.a011775
14. Weatherall DJ. The challenge of haemoglobinopathies in resource-poor countries. Br. J. Haematol (2011) 154(6):736–44. doi: 10.1111/j.1365-2141.2011.08742.x
15. Aguilar Martinez P, Angastiniotis M, Eleftheriou A, Gulbis B, Manu Pereira Mdel M, Petrova-Benedict R, et al. Haemoglobinopathies in Europe: health & migration policy perspectives. Orphanet J. Rare Dis. (2014) 9:97. doi: 10.1186/1750-1172-9-97
16. Monni G, Peddes C, Iuculano A, Ibba RM. From prenatal to preimplantation genetic diagnosis of beta-thalassemia. prevention model in 8748 cases: 40 years of single center experience. J. Clin. Med. (2018) 7(2):35. doi: 10.3390/jcm7020035
17. Giambona A, Damiani G, Vinciguerra M, Jakil C, Cannata M, Cassara F, et al. Incidence of haemoglobinopathies in Sicily: the impact of screening and prenatal diagnosis. Int. J. Clin. Pract. (2015) 69(10):1129–38. doi: 10.1111/ijcp.12628
18. Longo F, Corrieri P, Origa R, Barella S, Sanna PMG, Bitti PP, et al. Changing patterns of thalassaemia in Italy: a webthal perspective. Blood Transfus (2021) 19(3):261–8. doi: 10.2450/2020.0143-20
19. Forni GL, Gianesin B, Musallam KM, Longo F, Rosso R, Lisi R, et al. Overall and complication-free survival in a Large cohort of patients with beta-thalassemia major followed over 50 years. Am. J. Hematol. (2023) 98(3):381–7. doi: 10.1002/ajh.26798
20. Bender MA, Hulihan M, Dorley MC, Aguinaga MDP, Ojodu J, Yusuf C. Newborn screening practices for beta-thalassemia in the united states. Int. J. Neonatal Screen (2021) 7(4):83. doi: 10.3390/ijns7040083
21. World Health Organization. Blood safety and availability (2022). Available at: https://www.who.int/news-room/fact-sheets/detail/blood-safety-and-availability (Accessed January 2023).
22. Jones JM, Sapiano MRP, Mowla S, Bota D, Berger JJ, Basavaraju SV. Has the trend of declining blood transfusions in the united states ended? findings of the 2019 national blood collection and utilization survey. Transfusion (2021) 61 Suppl 2:S1–S10. doi: 10.1111/trf.16449
23. Facco G, Bennardello F, Fiorin F, Galassi C, Monagheddu C, Berti P, et al. A nationwide survey of clinical use of blood in Italy. Blood Transfus (2021) 19(5):384–95. doi: 10.2450/2021.0083-21
24. Angelucci E, Barosi G, Camaschella C, Cappellini MD, Cazzola M, Galanello R, et al. Italian Society of hematology practice guidelines for the management of iron overload in thalassemia major and related disorders. Haematologica (2008) 93(5):741–52. doi: 10.3324/haematol.12413
25. Theppornpitak K, Trakarnsanga B, Lauhasurayotin S, Poparn H, Chiengthong K, Sosothikul D, et al. A study to assess and improve adherence to iron chelation therapy in transfusion-dependent thalassemia patients. Hemoglobin (2021) 45(3):171–4. doi: 10.1080/03630269.2021.1934010
26. AAMDS. Iron chelation (2022). Available at: https://www.aamds.org/treatments/therapies/iron-chelation (Accessed January 2023).
27. Kontoghiorghe CN, Kolnagou A, Kontoghiorghes GJ. Phytochelators intended for clinical use in iron overload, other diseases of iron imbalance and free radical pathology. Molecules (2015) 20(11):20841–72. doi: 10.3390/molecules201119725
28. Uygun V, Kurtoglu E. Iron-chelation therapy with oral chelators in patients with thalassemia major. Hematology (2013) 18(1):50–5. doi: 10.1179/1607845412Y.0000000046
29. Verissimo M, Loggetto SR, Fabron Junior A, Baldanzi GR, Hamerschlak N, Fernandes JL, et al. Brazilian Thalassemia association protocol for iron chelation therapy in patients under regular transfusion. Rev. Bras. hematologia e hemoterapia (2013) 35:428–34. doi: 10.5581/2F1516-8484.20130106
30. Ruger JP, Kress D. Health financing and insurance reform in Morocco. Health Aff (Millwood) (2007) 26(4):1009–16. doi: 10.1377/hlthaff.26.4.1009
31. Hassaine A, Ghanam A, Elouali A, Babakhouya A, Rkain M, NBenajiba N. Iron chelation of thalassemics in the Eastern region of Morocco. Saudi J. Med. Pharm. Sci. (2020) 6(7):507–10. doi: 10.36348/sjmps.2020.v06i07.005
32. Hussain S, Hoodbhoy Z, Ali F, Hasan E, Alvi N, Hussain A, et al. Reduction of cardiac iron overload by optimising iron chelation therapy in transfusion dependent thalassaemia using cardiac T2* mri: a quality improvement project from Pakistan. Arch. Dis. Child (2020) 105(11):1041–8. doi: 10.1136/archdischild-2020-319203
33. Hagag A A, Hamam M A, Taha O A, M Hazaa S. Therapeutic efficacy of different iron chelators in Egyptian children with beta thalassemia with iron overload. Infect. Disorders-Drug Targets (Formerly Curr. Drug Targets-Infectious Disorders) (2015) 15(2):98–105. doi: 10.2174/1871526515666150724111721
34. Ogbole GI, Adeyomoye AO, Badu-Peprah A, Mensah Y, Nzeh DA. Survey of magnetic resonance imaging availability in West Africa. Pan Afr Med. J. (2018) 30:240. doi: 10.11604/pamj.2018.30.240.14000
35. The Global Economy. Italy: Magnetic resonance imaging units (2019). Available at: https://www.theglobaleconomy.com/Italy/magnetic_resonance_imaging_units (Accessed January 2023).
36. Kantaris M, Theodorou M, Angelopoulos G, Kaitelidou D. Aξoνική/Υπoλoγιστική Kαι Mαγνητική To-Moγραφία Στην Kύπρo: Aνάλυση Tης Aγo-Pάς. Nurs. Care Research/Nosileia kai Ereuna (2017) 9(48):112–27.
37. The Global Economy. Turkey: Magnetic resonance imaging units (2019). Available at: https://www.theglobaleconomy.com/Turkey/magnetic_resonance_imaging_units (Accessed January 2023).
38. World Health Organization. Global health observatory medical equipment - data by country (2016). Available at: https://apps.who.int/gho/data/node.main-searo.510 (Accessed January 2023).
39. The Global Economy. Brazil: Magnetic resonance imaging units (2012). Available at: https://www.theglobaleconomy.com/Brazil/magnetic_resonance_imaging_units (Accessed January 2023).
40. Sajjad Z. Neuro-imaging facilities in Pakistan. Journal-Pakistan Med. Assoc. (2003) 53(12):621–2.
41. Chaibunruang A, Sornkayasit K, Chewasateanchai M, Sanugul P, Fucharoen G, Fucharoen S. Prevalence of thalassemia among newborns: a re-visited after 20 years of a prevention and control program in northeast Thailand. Mediterr J. Hematol. Infect. Dis. (2018) 10(1):e2018054. doi: 10.4084/MJHID.2018.054
42. Canatan D. Haemoglobinopathy prevention program in Turkey. Thalassemia Rep. (2011) 1(1):9–11. doi: 10.4081/thal.2011.s2.e4
43. Rosenfeld LG, Bacal NS, Cuder MAM, Silva AGD, Machado IE, Pereira CA, et al. Prevalence of hemoglobinopathies in the Brazilian adult population: national health survey 2014-2015. Rev. Bras. Epidemiol. (2019) 22Suppl 02(Suppl 02):E190007 SUPL 2. doi: 10.1590/1980-549720190007.supl.2
44. World Bank. Population, total - Brazil (2021). Available at: https://data.worldbank.org/indicator/SP.POP.TOTL?locations=BR (Accessed January 2023).
45. Ahmed S, Jafri H, Rashid Y, Ehsan Y, Bashir S, Ahmed M. Cascade screening for beta-thalassemia in Pakistan: development, feasibility and acceptability of a decision support intervention for relatives. Eur. J. Hum. Genet. (2022) 30(1):73–80. doi: 10.1038/s41431-021-00918-6
46. Laghmich A, Alaoui Ismaili FZ, Zian Z, Barakat A, Ghailani Nourouti N, Bennani Mechita M. Hemoglobinopathies in the north of Morocco: consanguinity pilot study. BioMed. Res. Int. (2019) 2019:6857417. doi: 10.1155/2019/6857417
47. Ombudsman Institute. Syrians in Turkey special report (2018). Available at: https://www.Ombudsman.Gov.Tr/Syrians/Special_Report.Pdf (Accessed June 24, 2022).
48. Yazal Erdem A, Demir Yenigurbuz F, Pekpak E, Akinci B, Aktekin E, Bayram C, et al. Refugee children with beta-thalassemia in Turkey: overview of demographic, socioeconomic, and medical characteristics. Pediatr. Blood Cancer (2019) 66(5):e27636. doi: 10.1002/pbc.27636
49. Xu JZ, Foe M, Tanongsaksakul W, Suksangpleng T, Ekwattanakit S, Riolueang S, et al. Identification of optimal thalassemia screening strategies for migrant populations in Thailand using a qualitative approach. BMC Public Health (2021) 21(1):1796. doi: 10.1186/s12889-021-11831-4
50. Conte R, Ruggieri L, Gambino A, Bartoloni F, Baiardi P, Bonifazi D, et al. The Italian multiregional thalassemia registry: centers characteristics, services, and patients’ population. Hematology (2016) 21(7):415–24. doi: 10.1080/10245332.2015.1101971
51. Baronciani D, Angelucci E, Potschger U, Gaziev J, Yesilipek A, Zecca M, et al. Hemopoietic stem cell transplantation in thalassemia: a report from the European society for blood and bone marrow transplantation hemoglobinopathy registry, 2000-2010. Bone Marrow Transplant. (2016) 51(4):536–41. doi: 10.1038/bmt.2015.293
52. Borgna-Pignatti C, Marsella M. Iron chelation in thalassemia major. Clin. Ther. (2015) 37(12):2866–77. doi: 10.1016/j.clinthera.2015.10.001
53. Cappellini MD, Taher AT. The use of luspatercept for thalassemia in adults. Blood Adv. (2021) 5(1):326–33. doi: 10.1182/bloodadvances.2020002725
54. Angelucci E, Burrows N, Losi S, Bartiromo C, Hu XH. Beta-thalassemia (Bt) prevalence and treatment patterns in Italy: a survey of treating physicians. Blood (2016) 128(22):3533. doi: 10.1182/blood.V128.22.3533.3533
55. Langhi D Jr., Ubiali EM, Marques JF Jr., Verissimo MA, Loggetto SR, Silvinato A, et al. Guidelines on beta-thalassemia major - regular blood transfusion therapy: associacao brasileira de hematologia, hemoterapia e terapia celular: project guidelines: associacao medica brasileira - 2016. Rev. Bras. Hematol. Hemoter (2016) 38(4):341–5. doi: 10.1016/j.bjhh.2016.09.003
56. Teawtrakul N. (2020)., in: Thai Thalassemia Guideline 2020. The 36th Annual Meeting of The Royal College of Physicians of Thailand, PEACH Royal Cliff Beach Resort, Pattaya, Chonburi, Thailand, 29th - 31st October 2020.
57. Angastiniotis M, Eleftheriou A, Porter JB. Cross-talk between available guidelines for the management of patients with beta-thalassemia major. Acta Haematol (2013) 130(2):64–73. doi: 10.1159/000345734
58. Taher AT, Cappellini MD. How I manage medical complications of beta-thalassemia in adults. Blood (2018) 132(17):1781–91. doi: 10.1182/blood-2018-06-818187
59. Shah FT, Porter JB, Sadasivam N, Kaya B, Moon JC, Velangi M, et al. Guidelines for the monitoring and management of iron overload in patients with haemoglobinopathies and rare anaemias. Br. J. Haematol (2022) 196(2):336–50. doi: 10.1111/bjh.17839
60. Taher AT, Saliba AN. Iron overload in thalassemia: different organs at different rates. Hematol. Am. Soc. Hematol. Educ. Program (2017) 2017(1):265–71. doi: 10.1182/asheducation-2017.1.265
61. Gardenghi S, Grady RW, Rivella S. Anemia, ineffective erythropoiesis, and hepcidin: interacting factors in abnormal iron metabolism leading to iron overload in beta-thalassemia. Hematol. Oncol. Clin. North Am. (2010) 24(6):1089–107. doi: 10.1016/j.hoc.2010.08.003
62. Franchini M, Forni GL, Marano G, Cruciani M, Mengoli C, Pinto V, et al. Red blood cell alloimmunisation in transfusion-dependent thalassaemia: a systematic review. Blood Transfus (2019) 17(1):4–15. doi: 10.2450/2019.0229-18
63. Tubman VN, Fung EB, Vogiatzi M, Thompson AA, Rogers ZR, Neufeld EJ, et al. Guidelines for the standard monitoring of patients with thalassemia: report of the thalassemia longitudinal cohort. J. Pediatr. Hematol. Oncol. (2015) 37(3):e162–9. doi: 10.1097/MPH.0000000000000307
64. Reddy PS, Locke M, Badawy SM. A systematic review of adherence to iron chelation therapy among children and adolescents with thalassemia. Ann. Med. (2022) 54(1):326–42. doi: 10.1080/07853890.2022.2028894
65. Davoudi-Kiakalayeh A, Mohammadi R, Pourfathollah AA, Siery Z, Davoudi-Kiakalayeh S. Alloimmunization in thalassemia patients: new insight for healthcare. Int. J. Prev. Med. (2017) 8:101. doi: 10.4103/ijpvm.IJPVM_246_16
66. El-Beshlawy A, Salama AA, El-Masry MR, El Husseiny NM, Abdelhameed AM. A study of red blood cell alloimmunization and autoimmunization among 200 multitransfused Egyptian beta thalassemia patients. Sci. Rep. (2020) 10(1):21079. doi: 10.1038/s41598-020-78333-y
67. El Kababi S, Benajiba M, El Khalfi B, Hachim J, Soukri A. Red blood cell alloimmunizations in beta-thalassemia patients in Casablanca/Morocco: prevalence and risk factors. Transfus Clin. Biol. (2019) 26(4):240–8. doi: 10.1016/j.tracli.2019.06.004
68. Waldis SJ, Uter S, Kavitsky D, Flickinger C, Vege S, Friedman DF, et al. Rh Alloimmunization in chronically transfused patients with thalassemia receiving rhd, c, e, and K matched transfusions. Blood Adv. (2021) 5(3):737–44. doi: 10.1182/bloodadvances.2020003732
69. Forni GL, Puntoni M, Boeri E, Terenzani L, Balocco M. The influence of treatment in specialized centers on survival of patients with thalassemia major. Am. J. Hematol. (2009) 84(5):317–8. doi: 10.1002/ajh.21398
70. Pinto VM, Poggi M, Russo R, Giusti A, Forni GL. Management of the aging beta-thalassemia transfusion-dependent population - the Italian experience. Blood Rev. (2019) 38:100594. doi: 10.1016/j.blre.2019.100594
71. Food and Drug Administration. Fda approves luspatercept-aamt for anemia in patients with beta thalassemia (2019). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-luspatercept-aamt-anemia-patients-beta-thalassemia (Accessed January 2023).
72. European Medicines Agency. Reblozyl (2020). Available at: https://www.ema.europa.eu/en/medicines/human/EPAR/reblozyl (Accessed January 2023).
73. Musallam KM, Taher AT, Cappellini MD. Right in time: mitapivat for the treatment of anemia in alpha- and beta-thalassemia. Cell Rep. Med. (2022) 3(10):100790. doi: 10.1016/j.xcrm.2022.100790
74. Matte A, Federti E, De Franceschi L. Erythrocyte pyruvate kinase activation in red cell disorders. Curr. Opin. Hematol. (2023) 30(3):93–8. doi: 10.1097/MOH.0000000000000758
75. Ehsan H, Wahab A, Anwer F, Iftikhar R, Yousaf MN. Prevalence of transfusion transmissible infections in beta-thalassemia major patients in Pakistan: a systematic review. Cureus (2020) 12(8):e10070. doi: 10.7759/cureus.10070
76. Al Kanaani Z, Mahmud S, Kouyoumjian SP, Abu-Raddad LJ. The epidemiology of hepatitis c virus in Pakistan: systematic review and meta-analyses. R Soc. Open Sci. (2018) 5(4):180257. doi: 10.1098/rsos.180257
77. Jenny HE, Saluja S, Sood R, Raykar N, Kataria R, Tongaonkar R, et al. Access to safe blood in low-income and middle-income countries: lessons from India. BMJ Glob Health (2017) 2(2):e000167. doi: 10.1136/bmjgh-2016-000167
78. Jafroodi M, Davoudi-Kiakalayeh A, Mohtasham-Amiri Z, Pourfathollah AA, Haghbin A. Trend in prevalence of hepatitis c virus infection among beta-thalassemia major patients: 10 years of experience in Iran. Int. J. Prev. Med. (2015) 6:89. doi: 10.4103/2008-7802.164832
79. Bhattacharyya KK, Biswas A, Gupta D, Sadhukhan PC. Experience of hepatitis c virus seroprevalence and its genomic diversity among transfusion-dependent thalassemia patients in a transfusion center. Asian J. Transfus Sci. (2018) 12(2):112–6. doi: 10.4103/ajts.AJTS_73_17
80. Maffei L, Sorrentino F, Caprari P, Taliani G, Massimi S, Risoluti R, et al. Hcv infection in thalassemia syndromes and hemoglobinopathies: new perspectives. Front. Mol. Biosci. (2020) 7:7. doi: 10.3389/fmolb.2020.00007
81. Scott SR, Wu Z. Risks and challenges of hiv infection transmitted Via blood transfusion. Biosafety Health (2019) 1(3):124–8. doi: 10.1016/j.bsheal.2019.12.001
82. Betts M, Flight PA, Paramore LC, Tian L, Milenkovic D, Sheth S. Systematic literature review of the burden of disease and treatment for transfusion-dependent beta-thalassemia. Clin. Ther. (2020) 42(2):322–37 e2. doi: 10.1016/j.clinthera.2019.12.003
83. Paramore C, Levine L, Bagshaw E, Ouyang C, Kudlac A, Larkin M. Patient- and caregiver-reported burden of transfusion-dependent beta-thalassemia measured using a digital application. Patient (2021) 14(2):197–208. doi: 10.1007/s40271-020-00473-0
84. Tang CH, Furnback W, Wang BCM, Tang J, Tang D, Lu MY, et al. Relationship between transfusion burden, healthcare resource utilization, and complications in patients with beta-thalassemia in Taiwan: a real-world analysis. Transfusion (2021) 61(10):2906–17. doi: 10.1111/trf.16636
85. Weiss M, Parisi Jun M, Sheth S. Clinical and economic burden of regularly transfused adult patients with beta-thalassemia in the united states: a retrospective cohort study using payer claims. Am. J. Hematol. (2019) 94(5):E129–E32. doi: 10.1002/ajh.25429
86. Origa R, Gianesin B, Longo F, Di Maggio R, Cassinerio E, Gamberini MR, et al. Incidence of cancer and related deaths in hemoglobinopathies: a follow-up of 4631 patients between 1970 and 2021. Cancer (2023) 129(1):107–17. doi: 10.1002/cncr.34509
87. Carsetti R, Agrati C, Pinto VM, Gianesin B, Gamberini R, Fortini M, et al. Premature aging of the immune system affects the response to sars-Cov-2 mrna vaccine in beta-thalassemia: role of an additional dose. Blood (2022) 140(15):1735–8. doi: 10.1182/blood.2022017594
88. Azarkeivan A, Hajibeigi B, Alavian SM, Lankarani MM, Assari S. Associates of poor physical and mental health-related quality of life in beta thalassemia-Major/Intermedia. J. Res. Med. Sci. (2009) 14(6):349–55.
89. Khodashenas M, Mardi P, Taherzadeh-Ghahfarokhi N, Tavakoli-Far B, Jamee M, Ghodrati N. Quality of life and related paraclinical factors in Iranian patients with transfusion-dependent thalassemia. J. Environ. Public Health (2021) 2021:2849163. doi: 10.1155/2021/2849163
90. Tedone F, Lamendola P, Lopatriello S, Cafiero D, Piovani D, Forni GL. Quality of life and burden of disease in Italian patients with transfusion-dependent beta-thalassemia. J. Clin. Med. (2021) 11(1):15. doi: 10.3390/jcm11010015
91. Mettananda S, Pathiraja H, Peiris R, Bandara D, de Silva U, Mettananda C, et al. Health related quality of life among children with transfusion dependent beta-thalassaemia major and haemoglobin e beta-thalassaemia in Sri Lanka: a case control study. Health Qual Life Outcomes (2019) 17(1):137. doi: 10.1186/s12955-019-1207-9
92. Shafie AA, Chhabra IK, Wong JHY, Mohammed NS, Ibrahim HM, Alias H. Health-related quality of life among children with transfusion-dependent thalassemia: a cross-sectional study in Malaysia. Health Qual Life Outcomes (2020) 18(1):141. doi: 10.1186/s12955-020-01381-5
93. Alshamsi S, Hamidi S, Narci HO. Health-related quality of life and associated factors of children with transfusion-dependent thalassemia in Dubai, united Arab Emirates. Global J. Health Sci. (2021) 13(7):18–31. doi: 10.5539/gjhs.v13n7p18
94. Cappellini MD, Kattamis A, Viprakasit V, Sutcharitchan P, Pariseau J, Laadem A, et al. Quality of life in patients with beta-thalassemia: a prospective study of transfusion-dependent and non-Transfusion-Dependent patients in Greece, Italy, Lebanon, and Thailand. Am. J. Hematol. (2019) 94(10):E261–E4. doi: 10.1002/ajh.25584
95. Maheri M, Rohban A, Sadeghi R, Joveini H. Predictors of quality of life in transfusion-dependent thalassemia patients based on the precede model: a structural equation modeling approach. J. Epidemiol. Glob Health (2020) 10(2):157–63. doi: 10.2991/jegh.k.191001.001
96. Alshamsi S, Hamidi S, Narci HO. Healthcare resource utilization and direct costs of transfusion-dependent thalassemia patients in Dubai, united Arab Emirates: a retrospective cost-of-Illness study. BMC Health Serv. Res. (2022) 22(1):304. doi: 10.1186/s12913-022-07663-6
97. Esmaeilzadeh F, Azarkeivan A, Emamgholipour S, Akbari Sari A, Yaseri M, Ahmadi B, et al. Economic burden of thalassemia major in Iran, 2015. J. Res. Health Sci. (2016) 16(3):111–5.
98. Alshamsi S, Hamidi S, Ozgen Narci H. Productivity loss and associated costs among patients with transfusion-dependent thalassemia in Dubai, united Arab Emirates. Clinicoecon Outcomes Res. (2021) 13:853–62. doi: 10.2147/CEOR.S334724
99. Stanworth SJ, New HV, Apelseth TO, Brunskill S, Cardigan R, Doree C, et al. Effects of the covid-19 pandemic on supply and use of blood for transfusion. Lancet Haematol (2020) 7(10):e756–e64. doi: 10.1016/S2352-3026(20)30186-1
100. Pagano MB, Hess JR, Tsang HC, Staley E, Gernsheimer T, Sen N, et al. Prepare to adapt: blood supply and transfusion support during the first 2 weeks of the 2019 novel coronavirus (Covid-19) pandemic affecting Washington state. Transfusion (2020) 60(5):908–11. doi: 10.1111/trf.15789
101. Zhang HJ, Amid A, Janzen LA, Segbefia CI, Chen S, Athale U, et al. Outcomes of haemoglobin bart’s hydrops fetalis following intrauterine transfusion in Ontario, Canada. Arch. Dis. Child Fetal Neonatal Ed (2021) 106(1):51–6. doi: 10.1136/archdischild-2019-317626
102. Yasara N, Premawardhena A, Mettananda S. A comprehensive review of hydroxyurea for beta-haemoglobinopathies: the role revisited during covid-19 pandemic. Orphanet J. Rare Dis. (2021) 16(1):114. doi: 10.1186/s13023-021-01757-w
103. Mettananda S. Genetic and epigenetic therapies for beta-thalassaemia by altering the expression of alpha-globin gene. Front. Genome Ed (2021) 3:752278. doi: 10.3389/fgeed.2021.752278
104. Yang K, Wu Y, Zhou Y, Long B, Lu Q, Zhou T, et al. Thalidomide for patients with beta-thalassemia: a multicenter experience. Mediterr J. Hematol. Infect. Dis. (2020) 12(1):e2020021. doi: 10.4084/MJHID.2020.021
105. Williams AT, Jani VP, Nemkov T, Lucas A, Yoshida T, Dunham A, et al. Transfusion of anaerobically or conventionally stored blood after hemorrhagic shock. Shock (2020) 53(3):352–62. doi: 10.1097/SHK.0000000000001386
106. Musallam KM, Bou-Fakhredin R, Cappellini MD, Taher AT. 2021 Update on clinical trials in beta-thalassemia. Am. J. Hematol. (2021) 96(11):1518–31. doi: 10.1002/ajh.26316
107. Cappellini MD, Motta I. New therapeutic targets in transfusion-dependent and -independent thalassemia. Hematol. Am. Soc. Hematol. Educ. Program (2017) 2017(1):278–83. doi: 10.1182/asheducation-2017.1.278
108. Fu B, Liao J, Chen S, Li W, Wang Q, Hu J, et al. Crispr-Cas9-Mediated gene editing of the Bcl11a enhancer for pediatric Beta(0)/Beta(0) transfusion-dependent beta-thalassemia. Nat. Med. (2022) 28(8):1573–80. doi: 10.1038/s41591-022-01906-z
Keywords: anemia, hemoglobinopathies, transfusion-dependent beta thalassemia, unmet need, epidemiology, blood transfusion
Citation: Forni GL, Grazzini G, Boudreaux J, Agostini V and Omert L (2023) Global burden and unmet needs in the treatment of transfusion-dependent β-thalassemia. Front. Hematol. 2:1187681. doi: 10.3389/frhem.2023.1187681
Received: 16 March 2023; Accepted: 23 May 2023;
Published: 20 June 2023.
Edited by:
Annarita Miccio, INSERM U1163 Institut Imagine, FranceReviewed by:
Sachith Mettananda, University of Kelaniya, Sri LankaZahra Pakbaz, University of California, Irvine, United States
Copyright © 2023 Forni, Grazzini, Boudreaux, Agostini and Omert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laurel Omert, TGF1cmVsLk9tZXJ0QGhlbWFuZXh0LmNvbQ==