 Christian Kjellander1,2*
Christian Kjellander1,2* Emma Hernlund3†Moa Ivergård3†
Emma Hernlund3†Moa Ivergård3† Axel Svedbom3†Therese Dibbern4†Anna Stenling4Fredrik Sjöö4†Simona Vertuani4
Axel Svedbom3†Therese Dibbern4†Anna Stenling4Fredrik Sjöö4†Simona Vertuani4 Andreas Glenthøj5
Andreas Glenthøj5 Honar Cherif6
Honar Cherif6- 1Department of Internal Medicine, Capio St Göran Hospital, Stockholm, Sweden
- 2Department of Laboratory Medicine, Karolinska Institutet, Stockholm, Sweden
- 3ICON plc, Stockholm, Sweden
- 4Novartis Sverige AB, Stockholm, Sweden
- 5Center for Hemoglobinopathies, Department of Hematology, Rigshospitalet, Copenhagen, Denmark
- 6Department of Hematology, Uppsala University Hospital, Uppsala, Sweden
Introduction: Sickle cell disease (SCD) describes a group of inherited disorders of hemoglobin. Globally, SCD occurs in approximately 300,000-400,000 births annually and is most prevalent in malaria-endemic countries. However, migration has impacted the epidemiology of SCD but data on the matter are scarce. The objective of this study was to describe the epidemiology, treatment uptake, and economic burden of SCD in Sweden, a country with substantial immigration over the last decades.
Methods: This nationwide retrospective observational registry cohort study identified patients with SCD from 2001 to 2018 and followed them from 2006 to 2018. Using data from high-quality population-based Swedish registers, we estimated prevalence, treatment uptake, and SCD-related health care resource use, sick leave and disability pension.
Results: Between 2006 and 2018 the number of patients with SCD increased from 504 to 670; inpatient hospital stays and outpatient visits increased by 200% and 300%, respectively. Patients with pain crises had approximately twice the number of inpatient episodes and outpatient visit per year, and had higher productivity losses compared to patients without crises.
Conclusion: In an era of emerging treatments for SCD, we have, to the best of our knowledge, for the first time comprehensively described epidemiological and economic aspects of SCD in a country where the disease is still rare and not well recognized by the healthcare system.
Introduction
Sickle cell disease (SCD) describes a group of autosomal recessive blood disorders caused by structurally abnormal variants of adult hemoglobin (HbA). The disease arises from a genetic mutation in the β-globin chain gene (HBB) that is critical for the proper structure of HbA (1). The abnormal hemoglobin responsible for SCD, hemoglobin-S (HbS), causes red blood cells to become rigid and adopt a sickle-like shape upon deoxygenation (2). The irregular erythrocyte morphology leads to hemolytic anemia, episodes of vascular occlusion, acute pain and progressive organ damage (3). These vaso-occlusive crises are the hallmark of SCD and are the most common reason for hospitalization.
Globally, SCD occurs in approximately 300,000-400,000 births annually and is most prevalent in malaria-endemic parts of the world, primarily sub-Saharan Africa and the Middle East (4); also countries such as Brazil and India have comparatively high frequencies of SCD (5). The prevalence estimates of SCD are heterogeneous and changing due to migration and variations in diagnostic methods and disease management (6). In some high-prevalence countries screening program have improved life expectancy (7) and premarital screening even reduce the at risk marriages (8). Identification of all patients with SCD is challenging in countries such as Sweden, Norway, and Finland that lack systematic screening at birth or at immigration, including those from regions with high SCD prevalence (7); presumably for historical reasons or uncertain cost-effectiveness. This makes it difficult to accurately describe the epidemiology of SCD. In Sweden, the number of SCD patients was estimated at 584 patients during 1987 to 2010, all SCD subtypes and SCD carriers counted (Hb-SS disease, Hb-SC disease, Hb-SD disease, Hb-SE disease, Hb-S thalassemia disease, other sickle cell disease, or Hb-S trait) (9). Immigration from countries with high prevalence of hemoglobinopathies during the last decade has increased substantially in Sweden (10) and accordingly SCD prevalence may have changed dramatically. To the best of our knowledge, there are no recent studies conducted in the Nordics or in Sweden specifically reporting the epidemiology, treatments, and costs of SCD. To address this research gap, we conducted this retrospective population-based nationwide register study.
Material and methods
Study design and patients
This was a nationwide retrospective observational cohort study of patients with SCD in Sweden using secondary data from Swedish national registers. The study population was identified from the Swedish National Patient Register (NPR). Patients were included in the cohort if they had any inpatient stay or outpatient visit to specialized care with an ICD-10 diagnosis code for SCD (ICD-10 codes D57.0 “Sickle cell disease with crisis” [Hb-SS disease with crisis], D57.1 “Sickle cell disease without crisis” [Hb-SS disease without crisis], D57.2 “Double heterozygous disease” [Hb-SC, Hb-SD, Hb-SE, or Hb-S thalassemia disease] or D57.8 “Other sickle cell disease” [Hb-SO disease or other sickle cell disease]) from 2001 to 2018. The follow-up period was up to 13 years, 2006-2018; patients that were not alive or had emigrated from Sweden before the start of the follow-up period were excluded. Patients were classified into four mutually exclusive groups based on the following criteria: i) Any record of a D57.0 diagnosis classified the patient as SCD with crisis, ii) No record of D57.0 and any record of a D57.1 diagnosis classified the patient as SCD without crisis; iii) No record of a D57.0 or D57.1 diagnosis, and any record of a diagnosis classified the patient as compound heterozygous, and iv) No record of a D57.0, D57.1 or D57.2 diagnosis, but a D57.8 diagnosis classified the patient as having other SCD. Diagnoses were considered as primary and accompanying diagnoses.
Data sources
Data were retrieved from five sources: the NPR, the Prescribed Drugs Register (PDR), the Causes of Death Register (CDR), Micro-Data for the Analysis of Social Insurance (MiDAS) and Statistics Sweden. These registers have a well-documented reliability and degree of coverage and are frequently used for population-based health care research (11–15).
The NPR contains information on all hospital care episodes together with information about the patient, and information about the visit/stay. Costs were inflated to 2019 value using the health-care component of the Swedish consumer price index. The PDR holds data on filled prescriptions, including date of filling, substance name and anatomical therapeutic class (ATC) codes. The CDR contains information on all deaths in Sweden, including date of death. MiDAS contains data on disability pension and sick leaves longer than 14 days together with an ICD-10 diagnosis code for the underlying cause of the productivity loss. All costs are presented in 2019 Swedish krona (SEK). 1SEK ≈ 0.11 USD.
Outcomes
Individuals were assessed for 1-year prevalence for the follow-up period 2006-2018. Hospital related resource use, as recorded in NPR, was deemed SCD-related if it was registered as having an ICD-10 code of D57, either as main or accompanying diagnosis during the follow-up. Crisis was solely defined by the ICD-10 code for Sickle cell disease with crisis (D57.0).
Costs for hospital resource use were estimated through remuneration amounts based on diagnosis related groups (DRG). Presence of comorbidities commonly associated with SCD were assessed by inpatient stays or outpatients visits with relevant ICD-10 codes, regardless of co-registration of a SCD diagnosis (D57). This was done for ICD-10 diagnosis chapters due to the lack of availability of more granular ICD-10 diagnoses for this study. The selected comorbidities were: cerebrovascular disease: chapter I60-I69; disorders of the choroid and retina: H30-H36; influenza and pneumonia: J09-J18; acute upper respiratory tract infections: J00:J06; pulmonary heart disease and diseases of pulmonary circulation: I26-I28; other forms of heart disease: I30-I52; renal failure: N17-N19; other diseases of blood and blood-forming organs: D70-D77; other osteopathies: M86-M90; other dorsopathies: M50-M54; and symptoms and signs involving the digestive system and abdomen: R10-R19.
Treatment uptake considered any record of a relevant prescription by calendar year. Relevant prescriptions were based on ATC codes; hydroxyurea: L01XX05; iron chelators: V03AC; opioids: N02A; paracetamol: N02BE01; anticoagulants: B01; antibiotics: J01, A07AA, R02AB; antidepressants: N06A, neuroleptics: N05, and antiepileptics: N03A. At the time of the study, neither voxelotor, nor crizanlizumab or L-glutamine were available in Sweden.
This study assumed that sick leave or disability pension episodes recorded in MiDAS were SCD-related when they were recorded with an SCD diagnosis or a diagnosis for stroke, sequelae from stroke, pneumonia or pulmonary embolism, which are likely related to SCD (ICD-10 codes I63, I69, I26 and J12-J18). Sick leaves and days with disability pension were assessed in the working-age patient population (i.e. patients aged 18-65 years in each calendar year). In the calculation of costs due to sick leaves, only days of sick leave in working patients were included (i.e. excluding sick leave days in students and patients on parental leaves etc.), and the extent of sick leave (i.e. 25%, 50%, 75% or 100%) was also taken into account. Days of sick leave or disability pension were costed with the mean annual salary in Sweden, plus employer fees and social security contributions paid by the employer. The average monthly salary for the working population in Sweden was 35,300 SEK [Statistics Sweden, 2019 (16)] and employer’s social security contribution was 31.42% of the total salary.
The study was approved by the Swedish Ethical Review Authority and all registry holders. Informed consent is not required for administrative database studies in Sweden.
Analysis
An individual was considered prevalent from birth or immigration to death or emigration and was included in the analysis if living in Sweden during any point of the calendar year during the follow-up period (2006-2018), i.e. individuals were considered prevalent before the first identification in the NPR.
Treatment uptake was assessed as the proportion of prevalent individuals with SCD in one calendar year filling any prescription with a relevant ATC code within that calendar year. Resource use with any D57 as the main or secondary diagnosis was assessed as total numbers of outpatient visits and inpatient stays per calendar year during the follow-up period 2006-2018, as well as mean number per patient year (all prevalent patients included) in respective calendar year.
Results
Prevalence
The SCD cohort included 776 patients during the follow-up period. Of these, 282 were classified as SCD with crisis, 304 were classified as SCD without crisis, 124 were classified as compound heterozygous and 66 as having other SCD. A total of 100 of these patients were born in Sweden during the follow-up period, i.e. years 2006 to 2018. Among these individuals, the mean time from birth to identification in the NPR was 2.6 years.
The one-year prevalence by calendar year increased from 504 in 2006 to 670 in 2018 (Table 1), corresponding to an increase in number of patients by 33%. The patients classified as having SCD with crisis showed the largest increase in prevalence, from 139 in 2006 to 260 in 2018. The proportion of prevalent individuals that were born in Sweden decreased over the years, from approximately 55% in the beginning of the follow-up period to 45% in the end of the study period. After Swedish born, the most common region of birth for the prevalent individuals was Africa, increasing from approximately 24% to 31% over the follow-period. The mean age of the prevalent SCD population decreased over the study period.
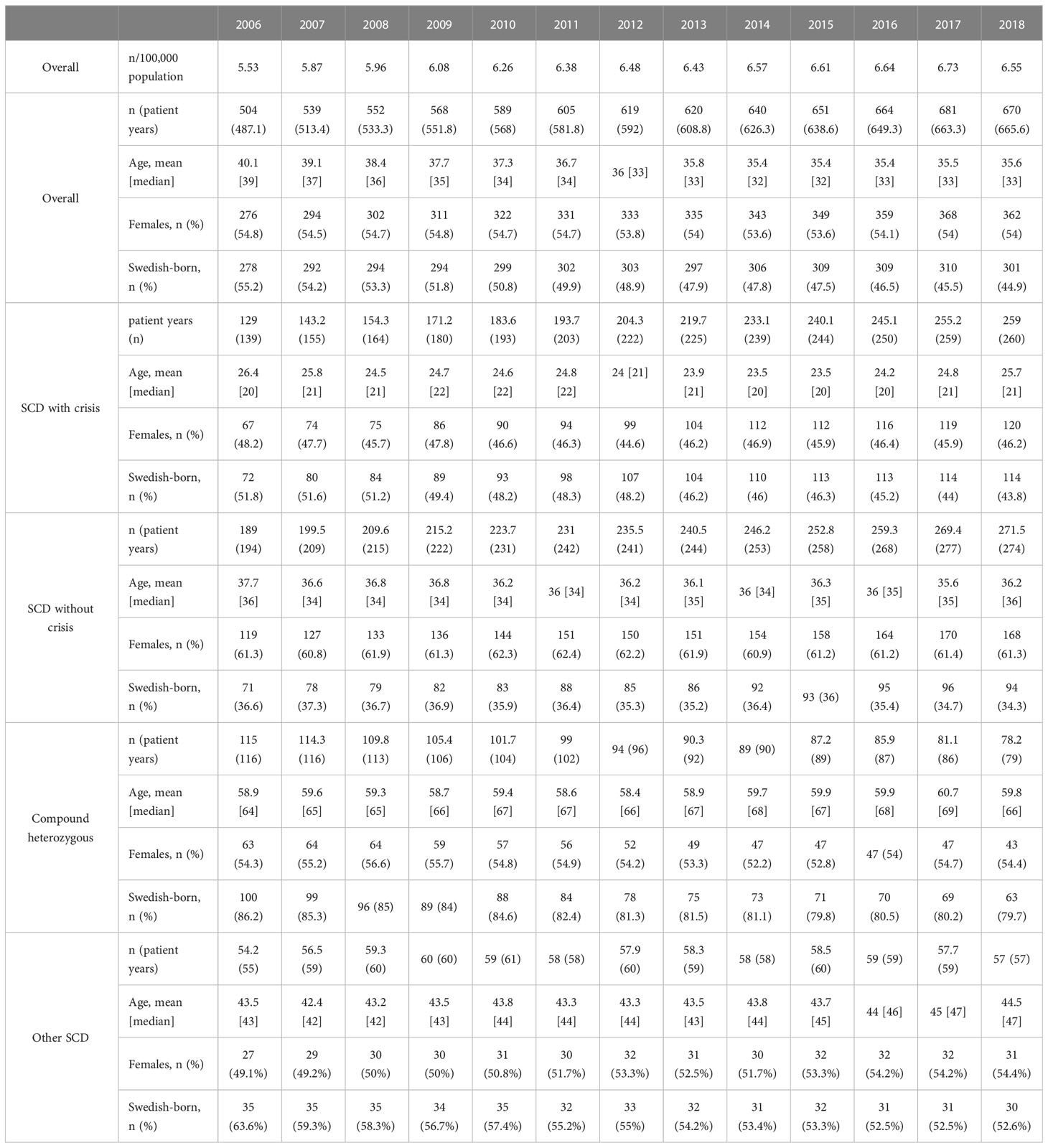
Table 1 One-year prevalence and patient characteristics of patients with SCD diagnoses by calendar year.
Treatment uptake
The prescribed treatments with the highest increase in uptake over the study period were hydroxyurea, vitamins and paracetamol. Hydroxyurea was primarily used by patients classified as SCD with crisis, and similarly so in pediatric (0-17 years) and adult patients (18 years and above) (Figure 1). Antibiotics were on average prescribed to more than 60% of pediatric patients classified as SCD with crisis in each year.
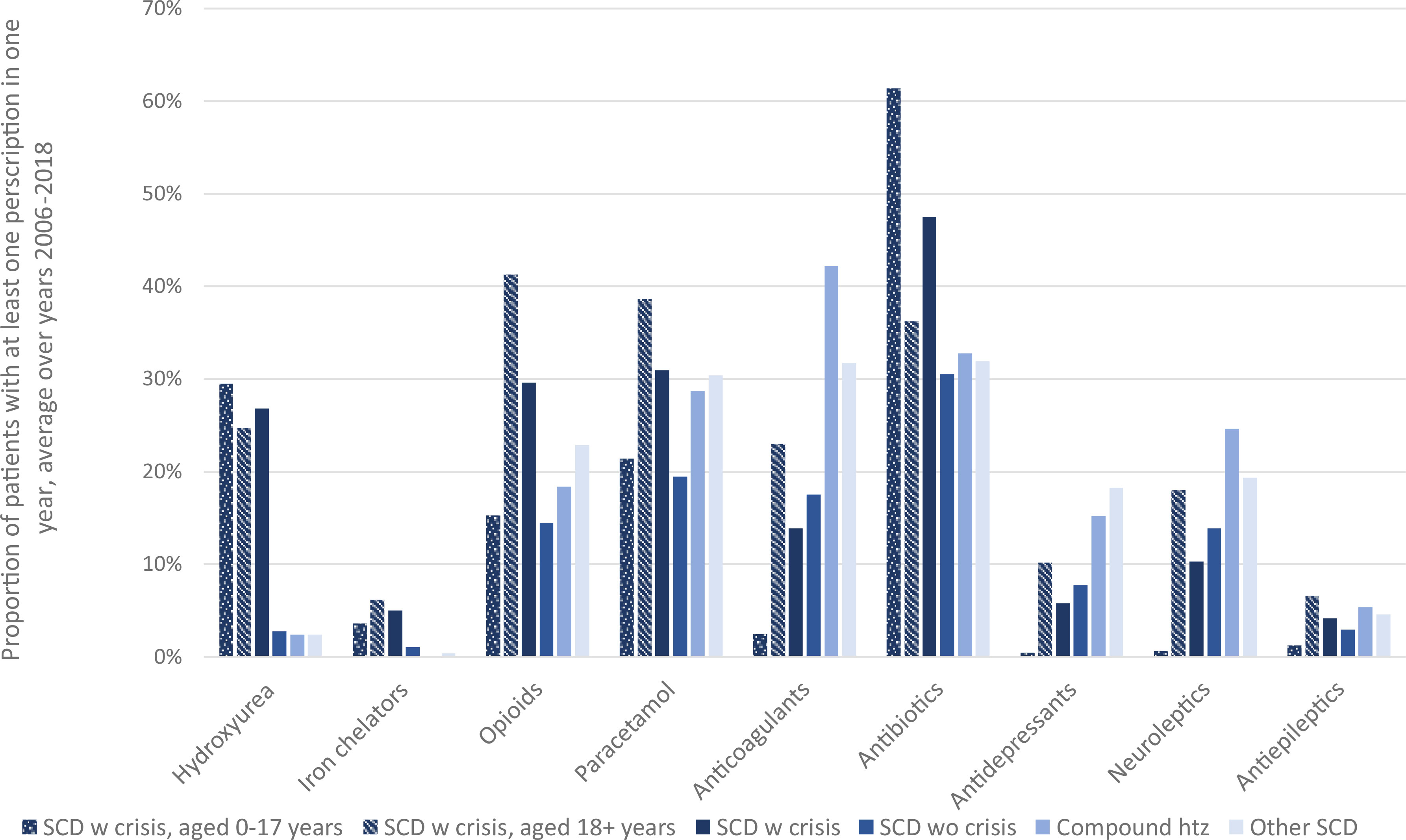
Figure 1 Uptake of prescribed treatment. Proportion of patients with at least one filled prescription of selected treatments.
Hospital related resource use
The number of SCD-related inpatient stays increased from 98 in 2006 to a plateau of 340-365 from 2012 to 2017 (Figure 2). In 2018, a slight decrease was observed, without an obvious explanation. The total number of inpatient admissions amounted to 3,600 over the 13-year study period, and were observed in 7,679 patient years, and the mean number of inpatient stays per patient year ranged between approximately 0.2 and 0.6 per calendar year over the follow-up period. The vast majority of SCD-inpatient stays occurred in patients with SCD with crisis; 3,293 out of 3,600 inpatient stays were recorded in these patients. The mean number of inpatient stays per patient year in patients with SCD with crisis ranged from 0.7 to 1.6 per calendar year over the study period.
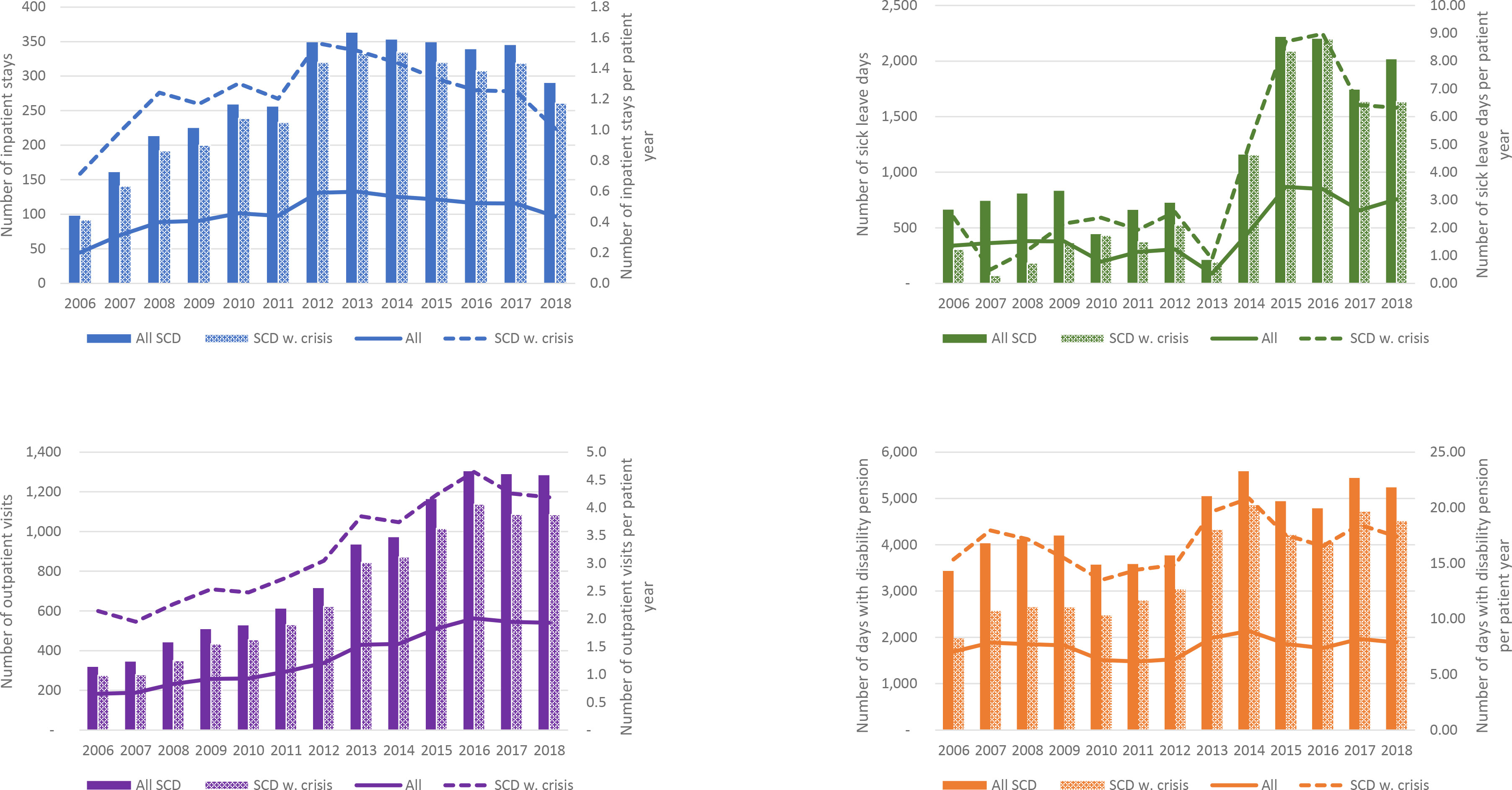
Figure 2 Hospital resource use and sick leave/disability pension days per calendar year. The bars and the lines represent total numbers and numbers per patient year, respectively. For days with sick leaves and disability pension, analyses only include patients in aged 18 to 65 years.
Yearly numbers of outpatient visits with a SCD diagnosis also increased substantially over the follow-up period, from 318 outpatient visits in 2006 to a plateau of approximately 1,300 in years 2016-2018. Due to the increase in SCD prevalence during this time, the mean number of outpatient visits per patient years did not increase with the same magnitude; however, an increase from 0.7 outpatient visits per patient year in 2006 to 2.0 outpatient visits per patient year in 2016 was observed. As for inpatient stays, the vast majority of outpatient visits were recorded in patients with SCD with crisis. The mean number of outpatient visits per patient year in patients with SCD with crisis was estimated at approximately four visits for each calendar year between 2015 and 2018. The mean number of specialist outpatient visits and inpatient days by patient group and calendar year are presented in the Supplementary Appendix. Notably, patients with compound heterozygous disease and “other sickle cell disease” had practically no health care resource use related to SCD.
Sick leaves and days with disability pension
Lower numbers of sick leave days were observed in the first part of the follow-up period (years 2006-2013), followed by an increase to approximately triple the number of days per calendar year (years 2015-2018) (Figure 3). For the years with highest numbers of sick leave days, the majority of sick leave days were registered in patients with SCD with crisis. Due to the relatively low number of patients in the analysis, the observed numbers can change substantially between years if a few patients are on long-term sick leaves.
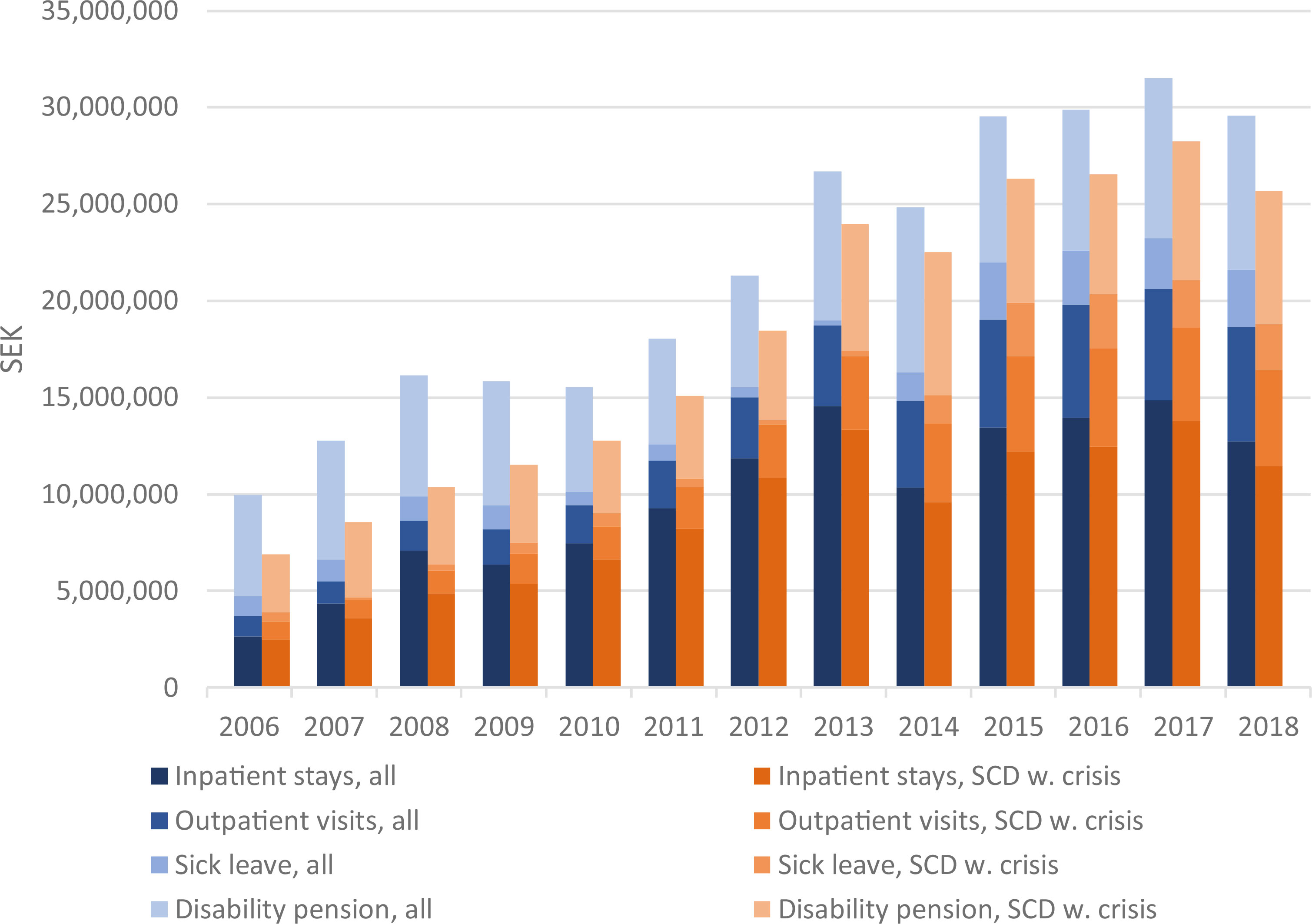
Figure 3 Health care costs and costs due to sick leave and disability pension per calendar year for all patients and those with crises. For days with sick leaves and disability pension, analyses only include patients in age 18-65 years.
Days with disability pension per year were more stable over the follow-up period. The mean number of days with disability pension per patient year ranged between 15 and 23 days per calendar year. Over the follow-up period, 57,800 days of disability pension were recorded, on average approximately 4,400 days per year.
Costs
The costs of hospital resource use and days with sick leaves and disability pension were estimated for the follow-up period. The largest contribution to costs were due to inpatient stays, followed by days with disability pension, outpatient visits and lastly sick leave days. Yearly costs increased about three-fold over the follow-up period, from a total of 10,000,000 SEK for year 2006 to around 30,000,000 in years 2014-2018 (Figure 3). Mean total costs per patient year increased from approximately 20,500 SEK per patient year in 2006 to 44,500 SEK per patient year in 2018. For patients classified as SCD with crisis, the corresponding costs per patient year were 53,500 SEK in 2006 and 99,000 SEK in 2018.
SCD comorbidities
The one-year prevalence of any inpatient stay and/or outpatient visit recorded with a relevant diagnosis for patients classified as SCD with crisis are shown in Table 2. In this patient population, influenza, pneumonia, and acute upper respiratory tracts infections were frequent reasons for hospitalization.
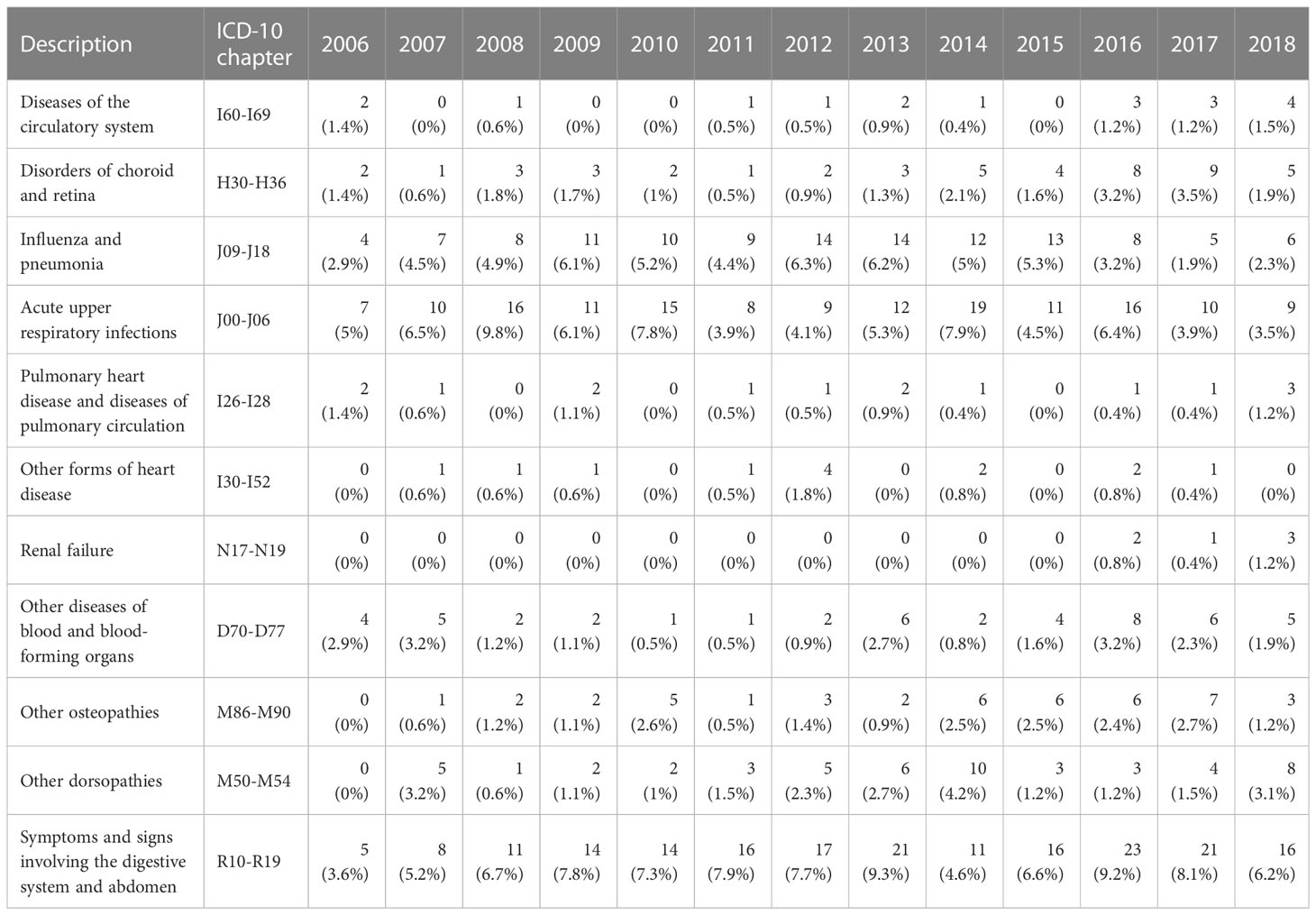
Table 2 Presence of selected comorbidities by calendar year, % of 1-year prevalent patients with any diagnosis in patients classified as SCD with crisis.
Discussion
This study describes the prevalence and characteristics of SCD patients in Sweden where the disease is still uncommon and – to the best of our knowledge – has not been well described previously. Although the prevalence is lower compared to many other countries it has increased in recent years, likely due to previous and/or current migration patterns. We show that there is substantial health care resource use due to SCD, especially in patients classified as SCD with crisis. Another important finding is the atypical characteristics of patients with double heterozygous disease: These patients were old (median age 66 years) with very low levels of SCD related health care contacts, indicating that these patient has a mild phenotype not typically seen at reference centers or are misclassified sickle cell trait carriers. This potential misclassification points to the need to raise awareness of the disease, even among specialists, and the need for diagnostic guidelines.
Guddati et al (17) analyzed the economic burden of sickle cell anemia among adults in the USA. Similar to our findings, they reported a significant increase in the number of hospitalizations (25% within 15 years) and hospital costs (150% increase).
The burden of SCD highlights the importance of identifying those who suffer from the disease as early as possible. Early identification facilitates early intervention which improves quality of life and reduce the risk of complications and future costs. To these ends, it may be beneficial to implement a screening program for individuals at high risk of SCD in Sweden. For many years, hydroxyurea has been the only available disease modifying agent for SCD. It is inexpensive, available and often well tolerated (18). During the last few years, new medications with different modes of action have passed pivotal clinical trials and are now in clinical use for SCD patients. These agents include the essential amino acid L-Glutamine (19), the P-selectin blocker crizanlizumab (20) and the HbS polymerization inhibitor voxelotor (21). Common for these agents is that the annual cost of treatment is at a minimum 50 times higher than that of hydroxyurea. The increasing prevalence combined with the expected increase in medication costs need to be accounted for by the health care system. Moreover, these costs are to be weighed against the improvement in morbidity and decrease in related costs for the society.This study has a number of limitations. Firstly, the NPR is an administrative register and not designed to give details on the patient’s medical history including disease-related morbidities or disease type (Hb-SS, Hb-SC, Hb-SD, Hb-SE, Hb-SO, or Hb-S thalassemia disease). This limitation is highlighted by the potential misclassification of patients carrying sickle cell trait as having compound heterozygous disease. It could be addressed in an updated study by linking the NPR to electronic medical records or biobanks. Such a study could also stratify the analysis by sickle cell disease types. However, that is not possible given the ethics approval of the study at hand. In this context it may be noted that the prevalence presented in this study (504 to 670 depending on year) is in line with data from Thalassaemia International Federation which estimates that approximately 580 individuals in Sweden have SCD, supporting the veracity of the estimates in this study. In addition, hospital resource use that is caused by SCD but SCD was not included in the diagnoses codes are not accounted for. In terms of sick leave and disability pension, it should be noted that episodes lasting fewer than 14 days are generally not captured and that some of the episodes – e.g., those due to pneumonia, stroke, or pulmonary embolism – may not be related to SCD. Another limitation related to data availability is that procedure codes that could be used to identify interventions such as blood transfusions, are not fully registered in the NPR, limiting our analysis of treatment uptake. It is a possible area of future research as blood transfusions are an important component of SCD care and better data on the matter could inform improvements. An important bias to consider in the study is survivorship bias. Patients need to be alive to enter the study, likely resulting in a relatively old and relatively healthy population. This effect is also reflected in the observed decrease in mean age over time: Patients who were prevalent at the start of start of the study are likely to be older compared to incident patients who enter the study population later, especially those who were born in Sweden (on average 2.6 years old at diagnosis). The age of newly diagnosed in a non-screening setting is comparable with what was found by Vichinsky et al. (22). As the study progresses, the older patients included at study start leave the population due to death or emigration, while younger patients who are newly diagnosed join the population.
Even though the study is associated with some limitations, the Swedish registers can provide meaningful information regarding patients with SCD. The registers used cover all individuals registered in Sweden and personal identification numbers unique to each person are used throughout the registers, enabling data linkage. Thus, even though some medical information can be missed, it is likely that all symptomatic SCD patients are included in this study, unless they are misdiagnosed.
In conclusion, we found increasing prevalence and costs of SCD in Sweden, factors that need to be accounted for when planning and structuring the care for patients with SCD. These findings are important from an international perspective: In an era of substantial migration and emerging treatments for SCD, this is – to the best of our knowledge – the first study that has comprehensively described epidemiological, disease-related and economic aspects of SCD in a country where the disease is still rare and not well recognized by the health care system.
Data availability statement
The datasets generated during and/or analyzed during the current study are not publicly available due to Swedish laws on individual patient level data. However, the same data can be extracted from the registry holders given approval from the Swedish ethical review authority and the registry holders.
Ethics statement
The studies involving human participants were reviewed and approved by Swedish Ethical Review Authority. Written informed consent from the participants’ legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
Author contributions
All authors conceived and designed the analysis. EH, MI, TD, and ASv acquired the data from the registry holders. SV, TD, ASt, and FS acquired funding, EH and MI performed the analyses. EH, MI, CK, HC, and AG interpreted the analyses. EH, ASv, and CK wrote the paper. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by Novartis.
Conflict of interest
At the time of the study, authors EH, MI, and ASv were employed by the company ICON plc; authors TD, ASt, FS, and SV were employed by the company Novartis Sweden AB; AG reports consultancy Agios, Bristol Myers Squibb, Novo Nordisk and Novartis and research funding Sanofi, Saniona, and Alexion.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that this study received funding from Novartis. The funder had the following involvement in the study: design of the analyses.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/frhem.2023.1205941/full#supplementary-material
References
1. Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bul World Health Organ (2008) 86(6):480–7. doi: 10.2471/BLT.06.036673
2. Sundd P, Gladwin MT, Novelli EM. Pathophysiology of sickle cell disease. Annu Rev pathology: Mech Dis (2019) 14:263–92. doi: 10.1146/annurev-pathmechdis-012418-012838
3. Pecker LH, Lanzkron S. Sickle cell disease. Ann Internal Med (2021) 174(1):ITC1–ITC16. doi: 10.7326/AITC202101190
4. Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, et al. Sickle cell disease. Nat Rev Dis Primers (2018) 4:18010. doi: 10.1038/nrdp.2018.10
5. Lervolino LG, Baldin PE, Picado SM, Calil KB, Viel AA, Campos LA. Prevalence of sickle cell disease and sickle cell trait in national neonatal screening studies. Rev Bras Hematol Hemoter (2011) 33(1):49–54. doi: 10.5581/1516-8484.20110015
6. Roberts I, de Montalembert M. Sickle cell disease as a paradigm of immigration hematology: new challenges for hematologists in Europe. Haematologica (2007) 92(7):865–71. doi: 10.3324/haematol.11474
7. Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med (2017) 376(16):1561–73. doi: 10.1056/NEJMra1510865
8. Memish ZA, Saeedi MY. Six-year outcome of the national premarital screening and genetic counseling program for sickle cell disease and β-thalassemia in Saudi Arabia. Ann Saudi Med (2011) 31(3):229–35. doi: 10.4103/0256-4947.81527
9. Hemminki K, Li X, Forsti A, Sundquist J, Sundquist K. Thalassemia and sickle cell anemia in Swedish immigrants: genetic diseases have become global. SAGE Open Med (2015) 3:2050312115613097. doi: 10.1177/2050312115613097
10. Karlsdottìr A, Rispling L, Norlén G, Randall L, Gassen NS, Heleniak T, et al. State of the Nordic region 2018: immigration and integration edition: Nordic council of ministers (Copenhagen, Denmark: Nordregio). (2018).
11. Ludvigsson JF, Andersson E, Ekbom A, Feychting M, Kim JL, Reuterwall C, et al. External review and validation of the Swedish national inpatient register. BMC Public Health (2011) 11:450. doi: 10.1186/1471-2458-11-450
12. Wettermark B, Hammar N, Fored CM, Leimanis A, Otterblad Olausson P, Bergman U, et al. The new Swedish prescribed drug register–opportunities for pharmacoepidemiological research and experience from the first six months. Pharmacoepidemiol Drug Saf (2007) 16(7):726–35. doi: 10.1002/pds.1294
13. Brooke HL, Talbäck M, Hörnblad J, Johansson LA, Ludvigsson JF, Druid H, et al. The Swedish cause of death register. Eur J Epidemiol (2017) 32(9):765–73. doi: 10.1007/s10654-017-0316-1
14. Leijon O, Josephson M, Österlund N. How common is change of primary diagnosis during an episode of sickness benefit? a register study of medical sickness certificates issued 2010–2012 in Sweden. Scandinavian J Public Health (2015) 43(1):44–51. doi: 10.1177/1403494814560843
15. Ludvigsson JF, Almqvist C, Bonamy AK, Ljung R, Michaëlsson K, Neovius M, et al. Registers of the Swedish total population and their use in medical research. Eur J Epidemiol (2016) 31(2):125–36. doi: 10.1007/s10654-016-0117-y
16. SCB. Genomsnittlig månadslön och lönspridning efter sektor, yrke (SSYK 2012), kön, tabellinnehåll och år (2019). Available at: https://www.scb.se/hitta-statistik/sverige-i-siffror/utbildning-jobb-och-pengar/medelloner-i-sverige/ (Accessed 31 August 2020).
17. Guddati AK, Kota V. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med (2018) 379(19):1879. doi: 10.1056/NEJMoa1715971
18. McGann PT, Ware RE. Hydroxyurea therapy for sickle cell anemia. Expert Opin Drug Saf (2015) 14(11):1749–58. doi: 10.1517/14740338.2015.1088827
19. Niihara Y, Miller ST, Kanter J, Lanzkron S, Smith WR, Hsu LL, et al. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med (2018) 379(3):226–35. doi: 10.1056/NEJMoa1715971
20. Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med (2017) 376(5):429–39. doi: 10.1056/NEJMoa1611770
21. Vichinsky E, Hoppe CC, Ataga KI, Ware RE, Nduba V, El-Beshlawy A, et al. A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med (2019) 381(6):509–19. doi: 10.1056/NEJMoa1903212
Keywords: epidemiology, health economics, costs, sick leave, sickle cell disease
Citation: Kjellander C, Hernlund E, Ivergård M, Svedbom A, Dibbern T, Stenling A, Sjöö F, Vertuani S, Glenthøj A and Cherif H (2023) Economic burden of sickle cell disease in Sweden: a population-based national register study with 13 years follow up. Front. Hematol. 2:1205941. doi: 10.3389/frhem.2023.1205941
Received: 14 April 2023; Accepted: 31 May 2023;
Published: 29 June 2023.
Edited by:
Samit Ghosh, University of Pittsburgh, United StatesReviewed by:
Zahra Pakbaz, University of California, Irvine, United StatesImo J. Akpan, Columbia University, United States
Copyright © 2023 Kjellander, Hernlund, Ivergård, Svedbom, Dibbern, Stenling, Sjöö, Vertuani, Glenthøj and Cherif. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Kjellander, Q2hyaXN0aWFuLktqZWxsYW5kZXJAY2FwaW9zdGdvcmFuLnNl
†Present addresses: Emma Hernlund, Drottningholm, Sweden
Moa Ivergård, Rönninge, Sweden
Axel Svedbom, Karolinska Universitetssjukhuset, Stockholm, Sweden
Therese Dibbern, Nordic Market Access, Stockholm, Sweden
Fredrik Sjöö, CSL Behring, Danderyd, Sweden