1
Department of Pharmacology, The School of Pharmacy, London, UK
2
Institute of Life Science, School of Medicine, Swansea University, Swansea, UK
3
Laboratoire de Neurobiologie, CNRS, Ecole Normale Supérieure, Paris, France
4
Institute of Medical Genetics, School of Medicine, Cardiff University, Wales, UK
Defects in mammalian glycinergic neurotransmission result in a complex motor disorder characterized by neonatal hypertonia and an exaggerated startle reflex, known as hyperekplexia (OMIM 149400). This affects newborn children and is characterized by noise or touch-induced seizures that result in muscle stiffness and breath-holding episodes. Although rare, this disorder can have serious consequences, including brain damage and/or sudden infant death. The primary cause of hyperekplexia is missense and non-sense mutations in the glycine receptor (GlyR) α1 subunit gene (GLRA1) on chromosome 5q33.1, although we have also discovered rare mutations in the genes encoding the GlyR β subunit (GLRB) and the GlyR clustering proteins gephyrin (GPNH) and collybistin (ARHGEF9). Recent studies of the Na+/Cl–-dependent glycine transporters GlyT1 and GlyT2 using mouse knockout models and human genetics have revealed that mutations in GlyT2 are a second major cause of hyperekplexia, while the phenotype of the GlyT1 knockout mouse resembles a devastating neurological disorder known as glycine encephalopathy (OMIM 605899). These findings highlight the importance of these transporters in regulating the levels of synaptic glycine.
We have employed an alternative approach to the study of inhibitory synapses, integrating data from mouse models and proteomic studies to identify biologically plausible candidate genes for genetic analysis in human neurological disorders. This approach has resulted in the identification of mutations in the genes for the GlyR α1 and β subunits (Rees et al., 1994
, 2001
, 2002
), gephyrin (Rees et al., 2003
) and the RhoGEF collybistin (Harvey et al., 2004
), all postsynaptic proteins found at inhibitory synapses (Figure 1
). However, the relative dearth of remaining well-characterized postsynaptic targets, taken together with new phenotypic data from glycine transporter (GlyT) knockout models (Gomeza et al., 2003a
,b
) have resulted in a shift of focus towards presynaptic and glial mechanisms of disease.
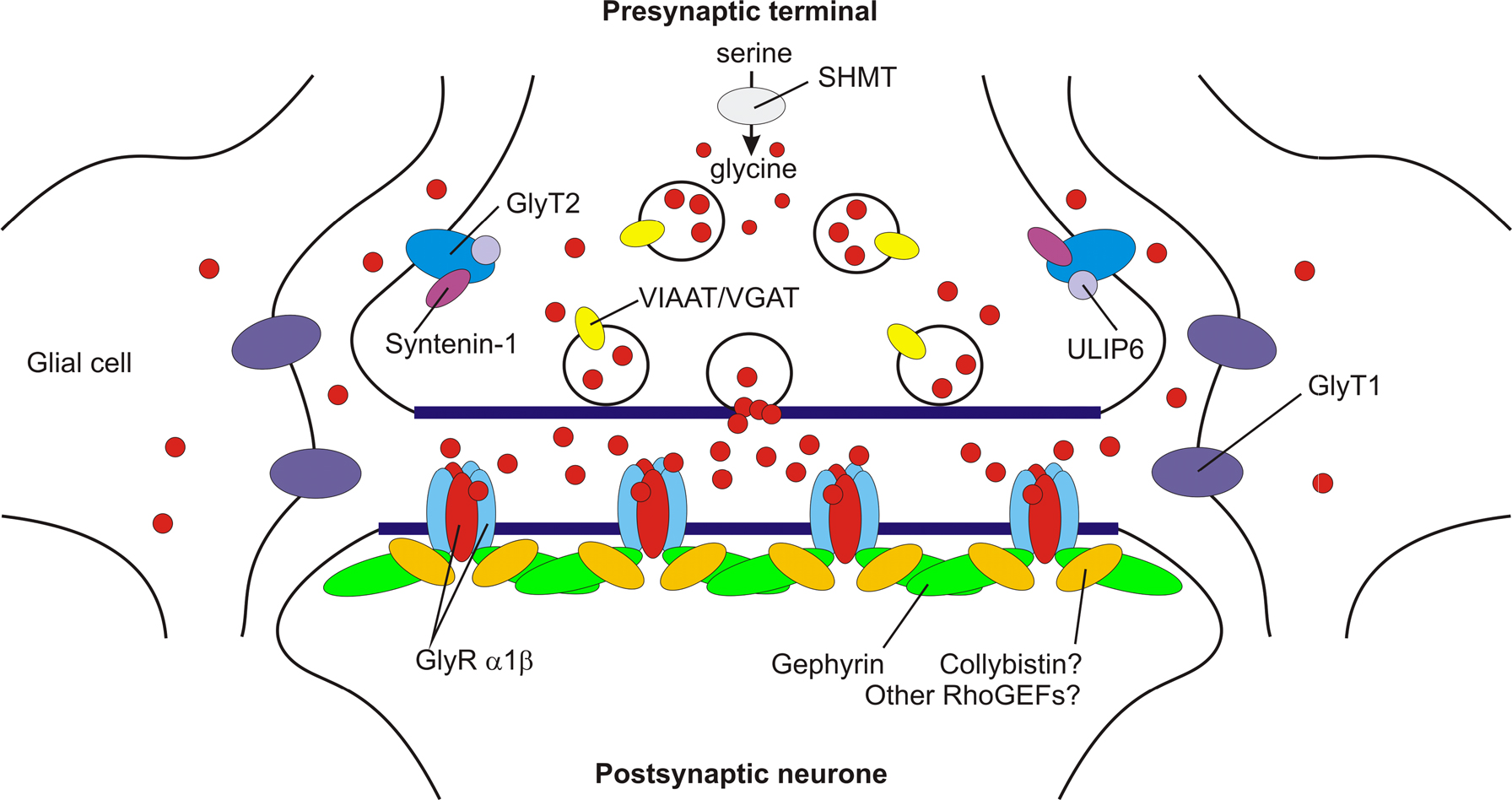
Figure 1. A model showing key proteins at mature glycinergic synapses. Postsynaptic α1β subunit GlyRs are clustered by gephyrin. Although the RhoGEF collybistin is responsible for the translocation of gephyrin to GABAergic synapses (Harvey et al., 2004
; Papadopoulos et al., 2007
), collybistin may be dispensable for GlyR clustering, since collybistin knockout mice do not exhibit developmental onset of exaggerated acoustic or tactile startle responses (Papadopoulos et al., 2007
). This suggests that additional RhoGEFs involved in the synaptic localisation of gephyrin and inhibitory receptors remain to be identified. The glial glycine transporter GlyT1 removes glycine from the synaptic cleft, thereby terminating neurotransmission. By contrast, GlyT2 (which binds ULIP6 and syntenin 1) is responsible for glycine re-uptake into the nerve terminal cytosol. This in turn provides glycine for the vesicular transporter VIAAT to refill synaptic vesicles. Glycine is also synthesised by a de novo pathway involving serine hydroxymethyl transferase (SHMT). Loss of glial GlyT1 uptake may cause raised CSF and serum glycine, leading to glycine encephalopathy.
Mutations in the GlyT2 Gene (SLC6A5) Define a Presynaptic Component of Hyperekplexia
Mouse models of GlyR dysfunction, such as spastic, spasmodic and oscillator have traditionally served as excellent leads for genetic analysis of the corresponding genes in human neurological illness, in particular hyperekplexia (Buckwalter et al., 1994
; Kingsmore et al., 1994
; Kling et al., 1997
; Mulhardt et al., 1994
; Ryan et al., 1994
; Saul et al., 1994
). For this reason, it was of interest that Gomeza et al. (2003b)
reported that the phenotype of knockout mice for the presynaptic glycine transporter GlyT2 also resembled human hyperekplexia. GlyT2 knockout mice gain weight slowly and die prematurely at the end of the second postnatal week, displaying a complex neurological phenotype characterized by spasticity, rigid muscle tone, strong spontaneous tremor and a severely impaired righting reflex (Gomeza et al., 2003b
). GlyTs are members of the neurotransmitter transporter superfamily (Betz et al., 2006
; Eulenburg et al., 2005
; Supplisson and Roux, 2002
), integral membrane proteins that utilize Na+ and Cl– electrochemical gradients to recapture neurotransmitters at central synapses. This superfamily includes four subfamilies of transporters for biogenic amines (norepinephrine, dopamine, serotonin), GABA and osmolytes (betaine, taurine, creatine), amino acids (glycine, proline) as well as several ‘orphan’ transporters (Broer, 2006
). GlyTs have dual functions at both inhibitory and excitatory synapses, resulting from the differential localisation of two distinct transporters, GlyT1 and GlyT2. GlyT1 is expressed in astroglial cells, exhibits a 2 Na+/1 Cl–/1 glycine stoichiometry and bi-directional glycine transport (Jursky and Nelson, 1995,
1996
; Roux and Supplisson, 2000
). These properties are appropriate for the control of extracellular glycine concentrations in the submicromolar range for modulation of excitatory N-methyl-D-aspartate (NMDA) selective glutamate receptors (Gabernet et al., 2005
; Martina et al., 2005
; Singer et al., 2007
; Tsai et al., 2004
; Yee et al., 2006
), where glycine acts as a co-agonist, and also for terminating neurotransmission at inhibitory glycinergic synapses (Gomeza et al., 2003a
). By contrast, GlyT2 is found in glycinergic axons, exhibits a 3 Na+/1 Cl–/1 glycine stoichiometry and does not display reverse uptake (Roux and Supplisson, 2000
). These peculiar properties reflect an essential role for GlyT2 in maintaining a high presynaptic pool of neurotransmitter at glycinergic synapses (Figure 1
). To assess whether mutations in the human GlyT2 gene could cause hyperekplexia, we scanned all 16 coding exons of the SLC6A5 gene (11p15.1) by dHPLC analysis in an international cohort of 83 sporadic and familial hyperekplexia patients devoid of mutations in GLRA1, GLRB, GPHN and ARHGEF9. Direct sequencing of aberrant dHPLC profiles revealed a mosaic of missense and nonsense mutations in SLC6A5 (Figure 2
) and additional regions of high-frequency SNPs (Rees et al., 2006
). In the majority of cases, GlyT2 mutations were inherited as compound heterozygotes indicating that SLC6A5 is predominantly associated with recessive hyperekplexia, although one dominant mutation was identified. Subcellular localisation studies using EGFP-tagged wild-type and mutant GlyT2 proteins, [3H]-glycine uptake assays and voltage-clamp electrophysiological analysis in Xenopus oocytes revealed that GlyT2 mutations result in defective subcellular localisation and/or decreased glycine uptake, with selected mutations affecting glycine and Na+ binding to the transporter (Rees et al., 2006
). Insights into the precise molecular mechanisms underlying GlyT2 missense mutations were provided using the high resolution structure of the bacterial leucine transporter (LeuT) (Yamashita et al., 2005
). Alignments of GlyT2 and LeuT revealed that mutations that disrupt glycine or Na+ binding to GlyT2 were predicted to form part of equivalent leucine or Na+ binding sites on LeuT (Figure 2
). These results demonstrated that SLC6A5 was a major gene for hyperekplexia and defined the first neurological disorder linked to mutations in a Na+/Cl–-dependent transporter for a classical fast neurotransmitter. It is also of interest to note that GlyT2 mutations are not lethal in humans: although patients harbouring mutations in SLC6A5 presented with hypertonia, an exaggerated startle response to tactile or acoustic stimuli and life-threatening neonatal apnoea episodes, in most cases these symptoms resolved in the first year of life following intensive postnatal and infant care. This may reflect a stronger contribution of GABAergic vs. glycinergic signalling in humans in postnatal development.
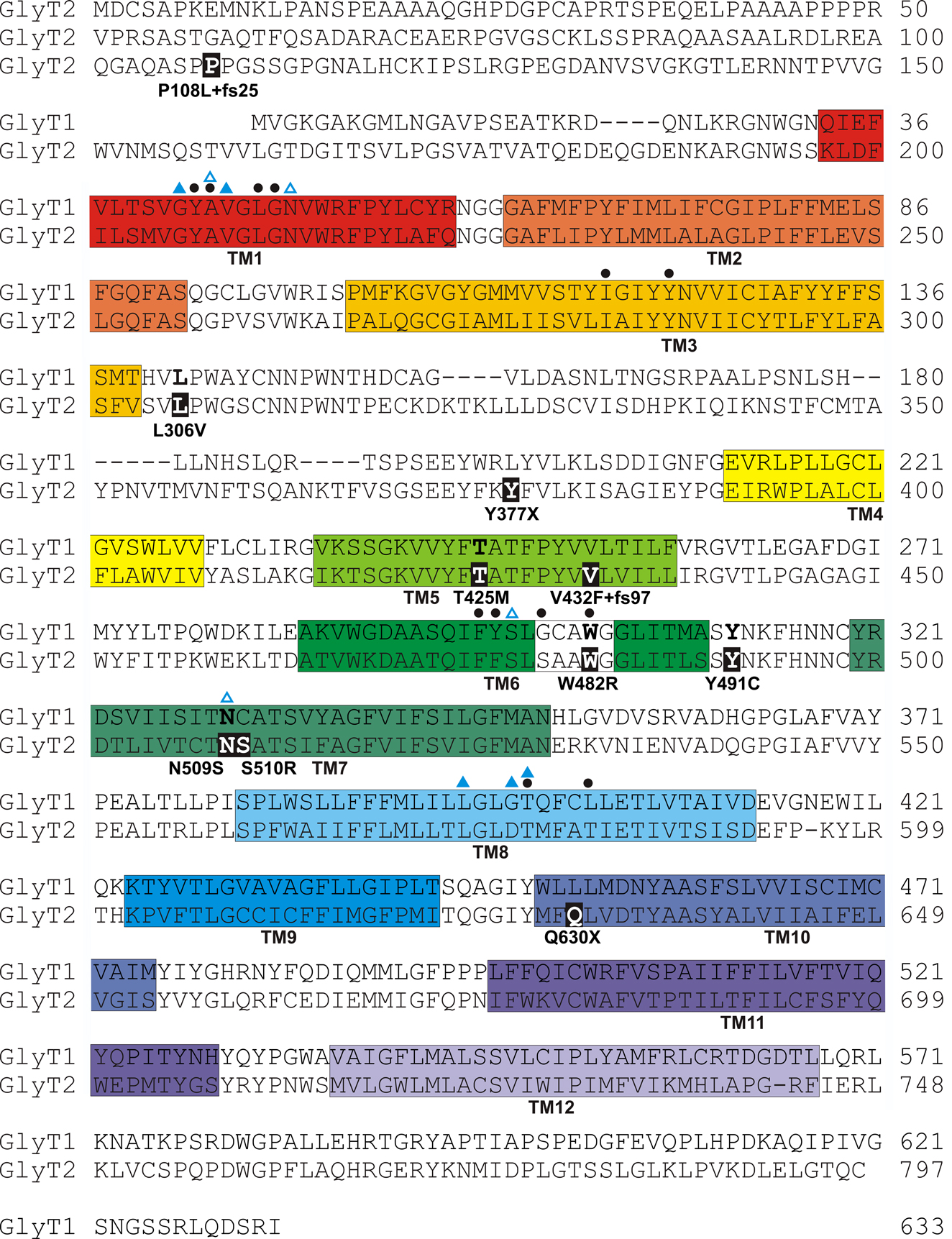
Figure 2. Amino acid sequence of human GlyT1 and GlyT2 indicating the revised positions of putative transmembrane (TM) domains (coloured boxes) based on the structure of the bacterial leucine transporter (LeuT) (Yamashita et al., 2005
). Mutations identified in human GlyT2 in hyperekplexia (Rees et al., 2006
) are indicated by black boxes. Blue triangles above the sequence indicate residues in hGlyT1 and hGlyT2 that are likely to coordinate Na+ ions based on sequence alignments with the bacterial LeuT (Yamashita et al., 2005
). However, it is noteworthy that GlyT2 binds three Na+ ions, while LeuT and GlyT1 bind two, suggesting that other residues involved in Na+ co-ordination remain to be identified in GlyT2. Filled black circles above the sequences indicate residues predicted to be involved in glycine binding. Note that mutations W482R and N509S alter putative glycine and Na+ binding residues, respectively.
GlyT2 Interacting Proteins and VIAAT as Candidate Genes for Further Genetic Analysis in Hyperekplexia
Despite significant advances in the genetics of hyperekplexia, our cohort still contains ~60 single, unrelated, sporadic cases of hyperekplexia which are gene-negative for mutations in GLRA1, GLRB, ARHGEF9, GPHN and SLC6A5. This suggests that further hyperekplexia genes remain undiscovered, and underlines the genetic heterogeneity associated with this disorder. Further presynaptic candidates for possible genetic analysis include the vesicular inhibitory amino acid transporter (VIAAT) and the GlyT2 accessory proteins syntenin-1 and ULIP6. ULIP6 is encoded by DPYSL5 (2p23.3) and is a brain-specific phosphoprotein of the Ulip/collapsin response mediator protein family. ULIP6 interacts with amino acids 135–184 of the GlyT2 N-terminal intracellular domain in a phosphorylation-dependent manner (Horiuchi et al., 2005
). The association of ULIP6 with GlyT2 is relatively specific, since other members of this gene family (ULIP1-5) do not interact with GlyT2. Since ULIP6 has been implicated in GlyT2 endocytosis and recycling (Eulenburg et al., 2005
; Horiuchi et al., 2005
), it is possible that mutations in ULIP6 could cause hyperekplexia by altering steady-state levels of presynaptic GlyT2. By contrast, the PDZ containing protein syntenin-1, encoded by the gene SDCBP (8q12.1) is thought to regulate the trafficking or presynaptic localisation of GlyT2 in glycinergic neurones (Ohno et al., 2004
). Although syntenin-1 and GlyT2 show extensive co-localisation in brainstem sections, syntenin-1 has numerous binding partners (Hirbec et al., 2002
, 2005
), including AMPA, kainate and metabotropic glutamate receptors, syndecans, neurexins and ephrin B, so any mutations in this protein might be expected to have diverse effects. Another plausible hyperekplexia candidate is VIIAT (Chaudry et al., 1998
; McIntire et al., 1997
; Sagné et al., 1997
),
encoded by the gene SLC32A1 on human chromosome 20q11.23. VIAAT is a transporter present in both GABAergic and glycinergic neurones (Chaudry et al., 1998
), and is responsible for the loading of presynaptic vesicles with glycine and GABA. Although a recent knockout mouse for VIAAT has shown a severe phenotype (embryonic lethality, omphalocele and cleft palate) (Wojcik et al., 2006
), it remains possible that missense mutations in VIAAT could lead to the loss of either GABA or glycine loading into synaptic vesicles. VIAAT was first identified (McIntire et al., 1997
) as a mammalian homologue of the ‘uncoordinated’ C. elegans mutant unc-47, which was known to be defective in a presynaptic component of GABA release. Three mutations in unc-47 were characterized by McIntire et al. (1997)
: (1) a G to A transition affecting a splice acceptor site, (2) a 238-bp deletion that removes part of the coding region and (3) a G to A transition causing the substitution G462R. Despite the fact that C. elegans does not appear to use glycine as a neurotransmitter (Schuske et al., 2004
), using a sophisticated cellular assay, Aubrey et al. (2007)
were able to demonstrate that UNC-47 is able to transport both GABA and glycine into vesicles. However, mutation G462R abolishes GABA, but not glycine uptake. Since residue G462 is conserved at the equivalent position (G500) in rodent and human VIAAT sequences, mutations in SLC32A1 could compromise GABA uptake into synaptic vesicles while leaving glycine uptake intact. Since similar mutations could affect glycine uptake, leaving GABA transport intact, missense mutations in SLC32A1 could potentially cause hyperekplexia.
Disruption of the Mouse GlyT1 Gene: A Mouse Model of Glycine Encephalopathy
Knockout mouse models of GlyT1 dysfunction (Gomeza et al., 2003a)
have suggested the intriguing possibility that mutations in the corresponding gene in humans (SLC6A9) could cause glycine encephalopathy (OMIM 605899), also known as non-ketotic hyperglycinemia (Applegarth and Toone, 2006
). Homozygous GlyT1 knockout mice exhibit severe motor deficits accompanied by lethargy, hypotonia and hyporesponsivity, and die within 6–14 hours after birth as a result of respiratory failure, although wasting and dehydration caused by an inability to suckle may also play a role (Gomeza et al., 2003a
; Tsai et al., 2004
). In a series of elegant experiments, Gomeza et al. (2003a)
demonstrated that this phenotype was not due to increased NMDA receptor activity, but rather that glial glycine clearance at glycinergic synapses was compromised in GlyT1 knockouts. GlyT1 deficient mice displayed increased chloride conductance at brainstem synapses, consistent with a tonic activation of GlyRs by elevated glycine concentrations. Furthermore, spontaneous inhibitory postsynaptic currents had longer decay time constants than those in wild-type littermates. Potentiation of inhibitory glycinergic transmission is also observed in the zebrafish mutant shocked, where a missense mutation causing a G81D substitution in TM2 disrupts GlyT1 function, causing motor deficits such as reduced spontaneous coiling of the trunk, diminished escape responses when touched, and an absence of swimming (Cui et al., 2005
). These observations suggest that GlyT1 plays a fundamental role in terminating glycinergic transmission by removing glycine from the synaptic cleft (Cui et al., 2005
; Gomeza et al., 2003a
). Potentiation of glycinergic neurotransmission is likely to be responsible for the early postnatal death of GlyT1 knockout mice, since recordings of neuronal activity in the brain stem circuitry responsible for generating the respiratory rhythm disclosed a slowed and irregular breathing pattern, which could be normalized by the GlyR antagonist strychnine (Gomeza et al., 2003a
). Antagonism of exaggerated glycinergic neurotransmission by strychnine also restored spontaneous coiling and swimming behaviours in zebrafish shocked mutants (Cui et al., 2005
).
Gomeza et al. (2003a)
also speculated that the overall reduction of motosensory functions in GlyT1 knockout mice was similar to symptoms of glycine encephalopathy in humans, a disease associated with disruption of the mitochondrial glycine cleavage system, which degrades excess glycine (Applegarth and Toone, 2006
; Sakata et al., 2001
). In typical cases with early neonatal onset, convulsive seizures, coma or respiratory distress develop within a few days after birth. One third of patients die within the first year of life, many in the neonatal period, often of respiratory impairment which can be treated with appropriate support. Those who survive can live for many years, but suffer varying degrees of mental retardation, having an average developmental age of between 3–6 months, as well as intractable seizures (Applegarth and Toone, 2006
; Conter et al., 2006
; Sakata et al., 2001
). Confirmation of diagnosis requires: (1) detection of raised cerebrospinal fluid (CSF) and plasma glycine concentrations and exclusion of organic acid disease by urinary organic acid assay; (2) a biochemical assay revealing a disrupted glycine cleavage enzyme complex in liver and (3) identification of mutations in one of the four genes of the mitochondrial glycine cleavage enzyme complex, encoding the P-protein (a pyridoxyl-dependent glycine decarboxylase, GLDC), the H-protein (a lipoic acid-containing hydrogen carrier protein), the T protein (a tetrahydrofolate-dependent aminomethyltransferase) and the L protein (lipoamide dehydrogenase). Mutations in the P-, H- and T-proteins have been identified in glycine encephalopathy (Applegarth and Toone, 2006
; Conter et al., 2006
; Sakata et al., 2001
). More than 80% of patients have a defect in the P-protein gene (GLDC, 9p24.1), while 15% of patients have defects in the T-protein gene (AMT, 3p21.31). Defects in the H-protein gene (GCSH, 16q23.2) are extremely rare. However, there are also reports of individuals with glycine encephalopathy who have normal GCS activity in liver and brain, but defective CNS glycine transport in postmortem tissue (Applegarth and Toone, 2006
; Mayor et al., 1985
). We consider that mutations in the human GlyT1 gene (SLC6A9; 1p34.1) could underlie glycine encephalopathy in these cases, since the loss of glial GlyT1 uptake would be expected to raise CSF glycine, while loss of GlyT1 from peripheral sites, including the liver (Jursky and Nelson, 1996
) (Figure 3
) could also lead to raised serum glycine. The activity of the isolated glycine cleavage enzyme complex would not be affected, since this complex is assayed in a cell-free lysate isolated from liver biopsy, and would therefore be independent of GlyT1 function. However, it has not yet been demonstrated that GlyT1 knockout mice actually have the main features expected of a mouse model of glycine encephalopathy, i.e., raised CSF and serum glycine in the presence of normal mitochondrial glycine cleavage enzyme complex activity (Applegarth and Toone, 2006
). Such measurements might allow researchers to establish a biochemical ‘profile’ of a human patient with mutations in the GlyT1 gene.
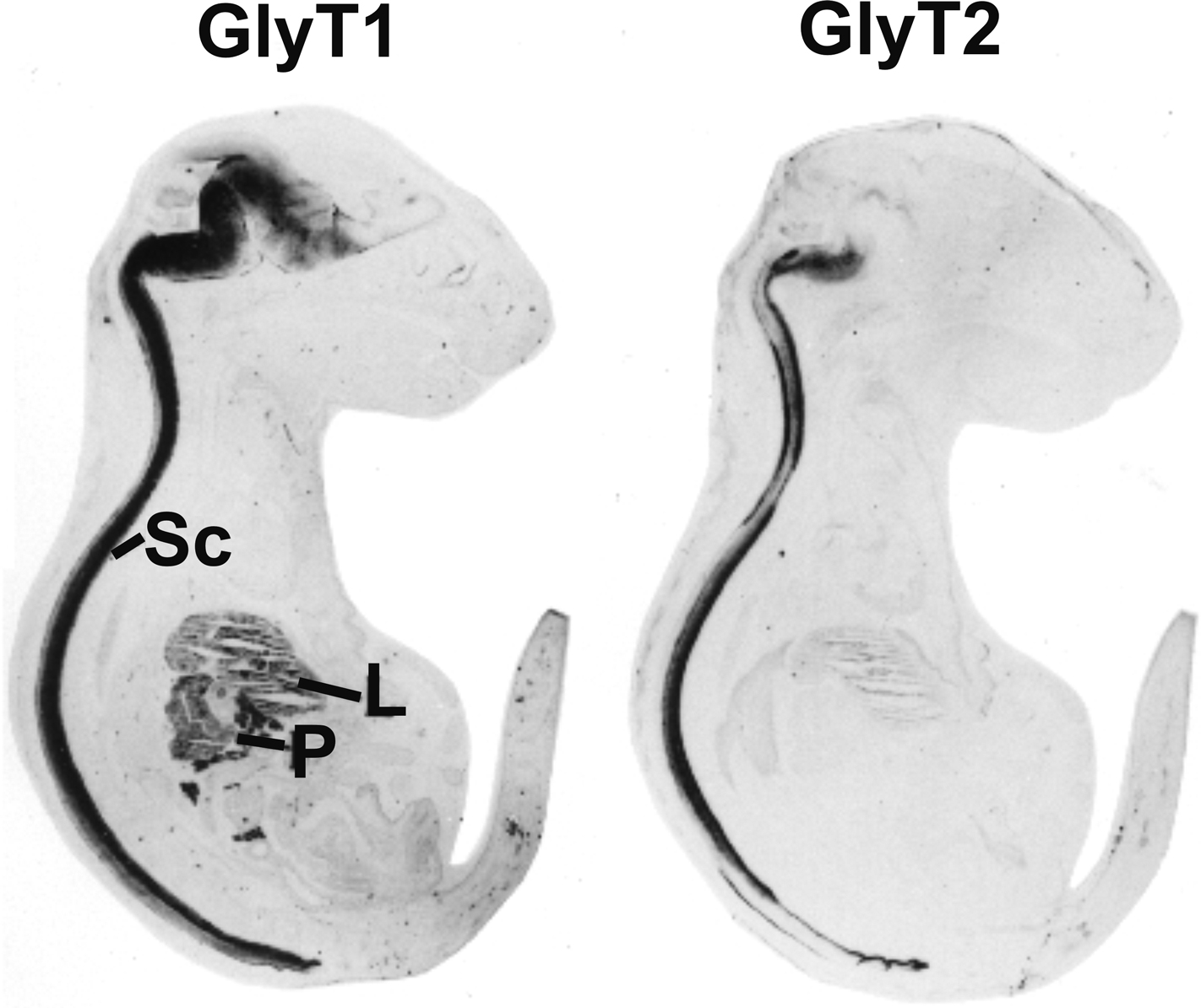
Figure 3. Distribution of GlyT1 and GlyT2 in sections from mouse E18 embryos. Note that GlyT2 immunoreactivity is mostly confined to spinal cord (Sc) and brainstem, while GlyT1 is also expressed in some higher brain regions and peripheral tissues, especially liver (L) and pancreas (P). Reproduced with permission from: F. Jursky and N. Nelson: Developmental expression of the glycine transporters GlyT1 and GlyT2 in mouse brain. Journal of Neurochemistry 67(1), 336–344 (1996), Wiley-Blackwell, Oxford, UK.
Modulation of Excitatory Glutamate Receptor Function by GlyT1
Knockout mice for GlyT1 have also underlined a key role for GlyTs in modulating excitatory NMDAR activity (Gabernet et al., 2005
; Martina et al., 2005
; Singer et al., 2007
; Tsai et al., 2004
; Yee et al., 2006
). Historically, there have been reservations about the in vivo role of the NR1 glycine co agonist site of NMDARs (Johnson and Ascher, 1987
) because the average concentration of glycine in CSF was expected to be in low micromolar range, while the affinity of glycine for the glycine modulatory site of the NMDAR ranges from 0.1 to 3 μM depending on the type of NR2 subunit present. Under these conditions, the NMDAR co-agonist site would always be fully saturated and therefore of no physiological or pharmacological relevance. Moreover, other studies have suggested that glial-derived D-serine, rather than glycine, is the endogenous ligand for the NMDAR co-agonist site (Mothet et al., 2000
; Panatier et al., 2006
). However, the location of the glycine transporter GlyT1 on astroglia (Zafra et al., 1995
) in close proximity to synaptic NMDARs could cause a localized reduction in glycine levels in the sub micromolar range (Berger et al., 1998
; Supplisson and Bergman, 1997
). More recently, GlyT1 has been localized to pre- and postsynaptic aspects of glutamatergic synapses, particularly in the neocortex and hippocampus (Cubelos et al., 2005a
) and have been shown to interact (Cubelos et al., 2005b
) with PDZ domains I and II of PSD-95, a scaffolding protein that also binds to NMDARs. Interestingly, the GlyT1-PSD95 interaction stabilises the transporter at the plasma membrane and produces a significant increase in the Vmax of glycine uptake (Cubelos et al., 2005b
). Studies of GlyT1 modulation of NMDAR function are slightly complicated by the fact that homozygous GlyT1 knockout mice die soon after birth. However, experiments using heterozygous GlyT1 mice (GlyT1+/–) or tissue-specific GlyT1 knockouts have been more illuminating. Unlike homozygous GlyT1 knockout mice, heterozygous GlyT1 mice are phenotypically normal, but have a demonstrable loss of [3H]glycine uptake in brain homogenates compared to wild-type mice (Gomeza et al., 2003a
; Tsai et al., 2004
). Heterozygous GlyT1 mice exhibit an enhanced NMDA/AMPA response ratio, suggesting that increased glycine levels lead to major functional changes at glutamatergic synapses (Gabernet et al., 2005
; Martina et al., 2005
; Tsai et al., 2004
). In addition, GlyT1+/– mice show resistance to pharmacological disruption of prepulse inhibition (PPI) by amphetamine (Tsai et al., 2004
). PPI is a neurological phenomenon in which a weaker prestimulus (prepulse) inhibits the reaction of an organism to a subsequent strong startling stimulus (pulse). Deficits of PPI manifest in the inability to filter out the unnecessary information and have been linked to abnormalities of sensorimotor gating such as those observed in schizophrenia. Lastly, GlyT1+/– mice show improved spatial memory retention (Tsai et al., 2004
), while selective disruption of GlyT1 in forebrain neurones results in enhanced associative and object recognition memory (Singer et al., 2007
; Yee et al., 2006
). Taken together, these studies suggest that reduced expression of GlyT1 enhances memory retention, and protects against disruptions of sensory gating, suggesting that GlyT1 may represent a target for novel cognition-enhancing and antipsychotic drugs.
The study of existing and newly characterized GlyT2 hyperekplexia mutations is expected to reveal important residues and domains of interest that will influence structure/function studies on other members of the Na+/Cl–-dependent transporter superfamily. Screening of the genes encoding ULIP-6, syntenin-1 and VIAAT in individuals with hyperekplexia may reveal additional presynaptic disease genes, and provide a solution for those families awaiting a definitive genetic diagnosis. GlyT1 knockout mice will also be instrumental in establishing a ‘patient profile’ for possible human glycine encephalopathy cases involving mutations in the GlyT1 gene. Screening of individuals with glycine encephalopathy that do not have obvious glycine cleavage system deficiencies will reveal whether GlyT1 has a role in this devastating postnatal neurological disorder.
The authors declare that this research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Research into the genetic causes of hyperekplexia and glycine encephalopathy in the author’s laboratories is supported by a grant from the Medical Research Council to RJH, KH and MIR (G0601585).
Harvey, K., Duguid, I. C., Alldred, M. J., Beatty, S. E., Ward, H., Keep, N. H., Lingenfelter, S. E., Pearce, B. R., Lundgren, J., Owen, M. J., Smart, T. G., Luscher, B., Rees, M. I., and Harvey, R. J. (2004). The GDP-GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J. Neurosci. 24, 5816–5826.
Horiuchi, M., Loebrich, S., Brandstaetter, J. H., Kneussel, M., and Betz, H. (2005). Cellular localization and subcellular distribution of Unc-33-like protein 6, a brain-specific protein of the collapsin response mediator protein family that interacts with the neuronal glycine transporter 2. J. Neurochem. 94, 307–315.
Papadopoulos, T., Korte, M., Eulenburg, V., Kubota, H., Retiounskaia, M., Harvey, R. J., Harvey, K., O’Sullivan, G. A., Laube, B., Hülsmann, S., Geiger, J. R., and Betz, H. (2007). Impaired GABAergic transmission and altered hippocampal synaptic plasticity in collybistin-deficient mice. EMBO J. 26, 3888–3899.
Rees, M. I., Harvey, K., Pearce, B. R., Chung, S. K., Duguid, I. C., Thomas, P., Beatty, S., Graham, G. E., Armstrong, L., Shiang, R., Abbott, K. J., Zuberi, S. M., Stephenson, J. B., Owen, M. J., Tijssen, M. A., van den Maagdenberg, A. M., Smart, T. G., Supplisson, S., and Harvey, R. J. (2006). Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of human startle disease. Nat. Genet. 38, 801–806.
Rees, M. I., Harvey, K., Ward, H., White, J. H., Evans, L., Duguid, I. C., Hsu, C. C., Coleman, S. L., Miller, J., Baer, K., Waldvogel, H. J., Gibbon, F., Smart, T. G., Owen, M. J., Harvey, R. J., and Snell, R. G. (2003). Isoform heterogeneity of the human gephyrin gene (GPHN), binding domains to the glycine receptor, and mutation analysis in hyperekplexia. J. Biol. Chem. 278, 24688–24696.
Rees, M. I., Lewis, T. M., Vafa, B., Ferrie, C., Corry, P., Muntoni, F., Jungbluth, H., Stephenson, J. B., Kerr, M., Snell, R. G., Schofield, P. R., and Owen, M. J. (2001). Compound heterozygosity and nonsense mutations in the α1-subunit of the inhibitory glycine receptor in hyperekplexia. Hum. Genet. 109, 267–270.
Yee, B. K., Balic, E., Singer, P., Schwerdel, C., Grampp, T., Gabernet, L., Knuesel, I., Benke, D., Feldon, J., Möhler, H., and Boison, D. (2006). Disruption of glycine transporter 1 restricted to forebrain neurons is associated with a procognitive and antipsychotic phenotypic profile. J. Neurosci. 26, 3169–3181.