RNA Editing and Hyperexcitability Disorders Helmholtz Group, Max Delbrück Center for Molecular Medicine, Berlin, Germany
Information transfer in the brain requires a homeostatic control of neuronal excitability. Therefore, a functional balance between excitatory and inhibitory systems is established during development. This review contains recent information about the molecular mechanisms orchestrating the establishment and maintenance of this excitation-inhibition (E-I) balance, and it reviews examples of deregulation of inhibitory and excitatory systems at a molecular, network and disease level of investigation.
By keeping the neuronal firing rate within a narrow range, homeostatic plasticity provides a dynamic basis for adequate information transfer in the brain (Turrigiano and Nelson, 2000
). To this end, a functional balance between excitatory and inhibitory synapses (E-I balance; Figure 1
A) is established during development and maintained throughout life (Turrigiano and Nelson, 2004
). Excitatory synaptic transmission is driven mainly by glutamatergic synapses whereas inhibitory synaptic transmission involves GABAergic and glycinergic signaling. In addition to such synaptic working mode of neurons, tonic ion conductances play an important role in controlling overall neuronal excitability (Mody and Pearce, 2004
). Together, these systems provide the spatial and temporal framework for physiological information transfer in the brain. Because deregulation of the E-I balance is frequently associated with a number of nervous system disorders, such as epilepsy, mental retardation and autism, this topic gains influence in both fundamental and clinical research.
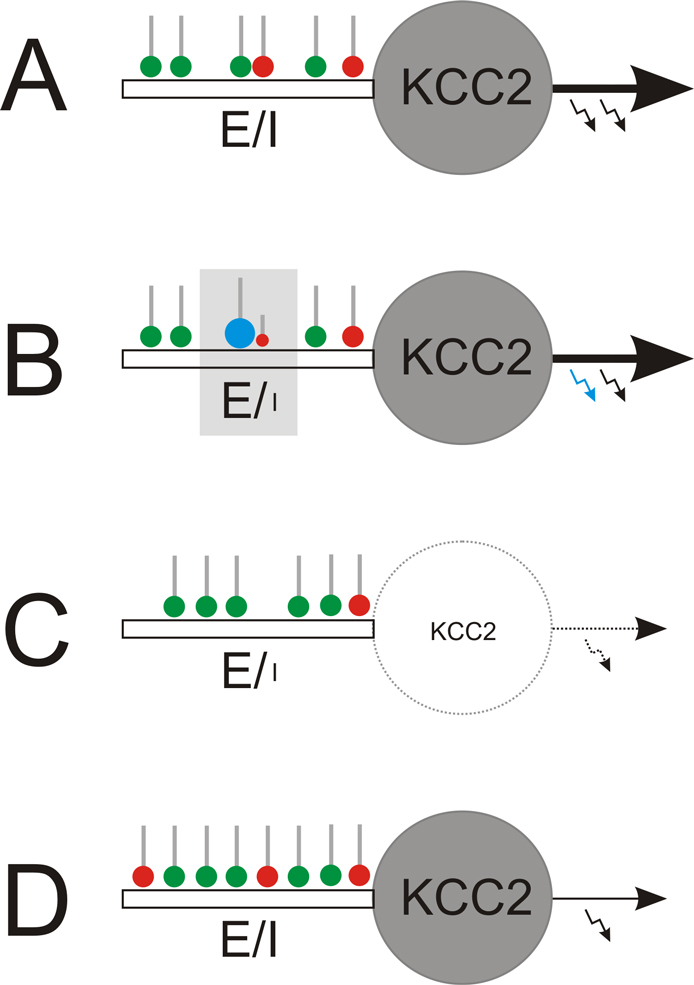
Figure 1. Local and global regulation of the E-I balance. (A) Starting point: homeostatically established E-I ratio of synaptic inputs to mature neurons with KCC2. (B) Local regulation: The E-I ratio of synaptic input can locally be enhanced to facilitate long-term potentiation of glutamatergic synapses (green). This strengthens glutamatergic inputs (blue) that temporally coincide with the neuronal output (blue arrow). Among others, endocannobinoids were identified as retrograde messengers involved in functional suppression of GABAergic synapses (red) in the neighborhood of potentiated glutamatergic terminals. (C, D) Global regulation. (C) Early in development or in the epileptic brain, where KCC2 expression is low, weakening or suppression of neuronal firing (silencing) provokes up-scaling of the glutamatergic system and down-scaling of the GABAergic system and, finally, elimination of silenced neurons through excitotoxic mechanisms. (D) In contrast, in the mature nervous system, neuron silencing provokes concomitant up-scaling of both, GABAergic and glutamatergic systems, which homeostatically compensates for reduced neuronal output while keeping the E-I balance.
.
Information transfer in the brain requires a constancy of the internal environment, originally specified by the American physiologist Walter Bradford Cannon (1932). Today, it is known that neuronal activity controls itself in order to avoid runaway excitation or quiescence (Turrigiano and Nelson, 2004
). For this purpose, a neuron has to establish mechanisms that not only provide neurons with a basic configuration of synaptic inputs, but also allow approximation of a homeostatic setpoint value of the E-I ratio of synaptic input in response to alterations in neuronal activity. Individual synapses within such a basic configuration can then be weighted differently according to their potential to coincide with the neuronal output. Because setpoint approximation requires intracellular signaling and engages enzymatic processes it is expected to always lag behind sudden changes in neuronal activity, which therefore may generate vulnerable periods.
Plasticity at the Level of Individual Synapses
Apart from alterations in presynaptic transmitter release, which has already been reviewed before (for review see Nicoll and Schmitz, 2005
), synaptic plasticity directly involves lateral mobility of surface neurotransmitter receptors. That neurotransmitter receptors diffuse laterally in the plane of the neuronal plasma membrane was recently exemplified with glycine receptors (Meier et al., 2001
). This study contributed to the concept of reversible postsynaptic neurotransmitter receptor anchoring that forms the basis of a dynamic equilibrium between highly mobile, non-synaptic and less mobile, postsynaptic receptors. This mechanism contributes to the determination of the ratio between non-synaptic and postsynaptic receptors at a given time. Several publications then showed that other neurotransmitter receptors, such as GABAA receptors (Bogdanov et al., 2006
) and ionotropic glutamate receptors (Borgdorff and Choquet, 2002
), are also mobile within the neuronal plasma membrane, providing a basis for dynamic activity-dependent regulation of postsynaptic receptor numbers. In fact, recent data show that the number of postsynaptic ionotropic glutamate receptors is regulated by synaptic usage. The elegant experiments conducted by Ehlers et al.
(2007)
disclosed that enrichment of postsynaptic domains with AMPA-type glutamate receptors is facilitated by activation of glutamatergic fibers. As a result of glutamate receptor activation, postsynaptic Ca2+ transients slow down receptor diffusion, according to the degree of synaptic usage (Borgdorff and Choquet, 2002
). This self-organizing system ensures that a sufficient number of receptors reside at postsynaptic domains, capable of appropriately answering presynaptic glutamate release. As receptor diffusion influences postsynaptic receptor numbers on a time scale of minutes, postsynaptic responsiveness may change very rapidly. Thereby, the temporal coincidence of synapse usage with adjacent depolarizing currents or with the respective neuronal output operates as a setpoint device for postsynaptic receptor numbers (Bi and Poo, 1998
), and if sustained, ultimately assigns different weights to different synapses by long-term potentiation or depression (Malinow and Malenka, 2002
).
A number of other regulatory mechanisms were identified to contribute to differential weighting of individual synapses. For example, endocannabinoids were shown to locally increase the E-I ratio in response to repetitive activation of glutamatergic fibers, a phenomenon originally termed depolarization-induced suppression of inhibition (Freund et al., 2003
). Thereby, increased E-I ratios are generated locally (Figure 1
B) by inducing long term depression at neighboring GABAergic synapses via retrograde endocannabinoid signaling (Chevaleyre and Castillo, 2004
). This mechanism facilitates induction of long-term potentiation of glutamatergic synapses adjacent to the depressed GABAergic input.
Altogether, synaptic plasticity mechanisms ensure local finetuning of individual synapses for weighted information transfer from neuron to neuron and stimulus-appropriate information processing in the brain.
Plasticity at the Neuronal Level – Delayed Setpoint Approximation
In 1994 already, Ger Ramakers noticed that homeostatic regulation increases the E-I ratio of synaptic input in response to chronic neuron silencing (Ramakers et al., 1994
). In fact, a large number of studies employing pharmacological modulation of neuronal activity showed that homeostatic mechanisms adjust the neuronal firing rate in response to altered neuronal activity. For example, prolonged pharmacological inactivation of ionotropic glutamate receptors or tetrodotoxin (TTX)-sensitive sodium channels evokes compensatory, homeostatic, mechanisms orchestrating an up-scaling of the glutamatergic system, which prevents neuronal quiescence as long as neurons are silenced (Ramakers et al., 1990
).
Per definition, homeostatic regulation is bidirectional. In this respect, it is conceivable that pharmacological blockade of GABAA receptors, which increases the neuronal output, engages homeostatic mechanisms in order to rectify the experimentally induced deviation from the setpoint of homeostasis. In fact, the neuron ends up with a down-scaled firing rate that prevents runaway excitation as long as GABAA receptors are blocked (Corner and Ramakers, 1992
).
These experimentally determined homeostatic rules are in step with actual practice where intrinsic factors temporarily interfere with homeostasis. For example, progesterone potentiates GABAergic transmission during the menstrual cycle, which temporarily deviates the neuronal output from the setpoint of homeostasis and therefore elicits homeostatic counter-regulation, i.e., down-scaling of inhibitory systems. A sudden fall of progesterone levels at the end of the menstrual cycle then increases seizure susceptibility in individuals with pre-existing epileptic foci (Herzog, 1999
; Klein and Herzog, 1998
; Smith, 2001
). Likewise, chronic ethanol consumption provokes homeostatic down-scaling of the ethanol-potentiated GABAergic system, and sudden ethanol withdrawal from these individuals lowers their seizure threshold (Cagetti et al., 2004
; Kokka et al., 1993
). This indicates that the ethanol-driven homeostatically down-scaled GABAergic system alone no longer is capable of adequately balancing neuronal excitation. As setpoint approximation lags behind sudden alterations in neuronal activity vulnerable periods inevitably follow because neurons may become hyperexcitable and generate rebound activity.
A New Hotspot for Homeostatic Regulation at the Neuronal Level
By scrutinizing hippocampal glycine receptors we identified a novel potential mechanism that may contribute to homeostatic setpoint approximation in response to hyperexcitability. So far, hippocampal glycine receptors were enigmatic: On the one hand they do not contribute to hippocampal synaptic transmission (Bedet et al., 2006
; Meier and Grantyn, 2004
; Song et al., 2006
; Zhang et al., 2008
) and on the other hand they may not even contribute to tonic hippocampal inhibition because their affinity for glycine (Legendre, 2001
) is far below hippocampal ambient glycine (Sherwin, 1999
).
Yet, these receptors are expressed and synthesized in hippocampal neurons. However, a reconcilement of this discrepancy recently emerged from the discovery of high affinity glycine receptor (haGlyR) variants (Meier et al., 2005
). It was shown that haGlyRs are generated by post-transcriptional, activity-dependent C-to-U RNA editing. Because their affinity for glycine approximates hippocampal ambient glycine, these gain-of-function receptors are very well adapted to translate the ambient glycine and taurine concentrations in the hippocampus into tonic inhibition of neuron firing, which may play a homeostatic role in cases of hyperexcitability. Further studies are necessary to determine a specific role for haGlyRs in hyperexcitability cases.
According to the developmental state of neurons homeostatic regulation may have a completely different impact on the neuronal network. In the developing brain GABAergic transmission is still excitatory due to reduced expression of the potassium chloride co-transporter 2 (KCC2) (Rivera et al., 1999
). In that condition chronic neuron silencing is detrimental because the E-I balance is going to be established, and this requires activity-dependent functional adjustment of developing glutamatergic and GABAergic synapses. If prohibited through neuron silencing, GABAergic synapses are down-scaled according to the proposed default elimination pathway for “un-synchronizable” GABAergic synapses (Tao and Poo, 2005
), and neurons that appear not to fit into the developing network will ultimately be eliminated (Figure 1
C; Eichler et al., unpublished). This rule may also apply to the epileptic brain, where neuronal hypoactivity occurs (Sloviter et al., 2006
) and which was found to adopt an earlier developmental status in terms of KCC2 down-regulation and associated impairment of Cl– extrusion (Hartmann et al., 2001
; Rivera et al., 2002
; Wake et al., 2007
). By contrast, in the adult neuronal tissue, neuron silencing provokes concomitant up-regulation of both glutamatergic and GABAergic synapses (Figure 1
D), which maintains the E-I ratio of synaptic input (Burrone et al., 2002
). Although exaggerated BDNF-signaling might be involved (Rivera et al., 2004
; Singh et al., 2006
), the identity of the molecular machinery responsible for such differential homeostatic regulation still has to be disclosed. In conclusion, the capacity of neurons to dynamically adjust the E-I ratio of synaptic input in response to alterations in neuronal activity certainly provides a homeostatic basis for information transfer in the adult brain. However, earlier in development as well as in the epileptic brain, alterations in neuronal activity are differently interpreted by the homeostatic machinery because, at that time, a physiologically meaningful neuronal integration into the developing network has to be set up.
On the way to mature synapses, appropriate expression and targeting of synapse-scaffolding molecules is required for the establishment, regulation and maintenance of the E-I balance. In the following, some molecular key players involved in these processes will be discussed.
Cell Adhesion Molecules Neuroligin-1 and -2
At a molecular level, excitatory and inhibitory synapses can be dissected according to their postsynaptic enrichment of neuroligin-1 and -2 (Chih et al., 2005
; Craig and Kang, 2007
). In excitatory glutamatergic synapses neuroligin-1 provides a physical link between pre- and postsynaptic membranes and thereby ensures transmitter-specific apposition of pre- and postsynaptic protein scaffolds (Fu et al., 2003
). Correspondingly, neuroligin-2 links pre- and postsynaptic compartments at inhibitory GABAergic synapses (Varoqueaux et al., 2004
). As the expression of neuroligins and their presynaptic counterpart neurexin suffices to induce the respective molecular scaffolds in fibroblast cells (Graf et al., 2004
; Scheiffele et al., 2000
) alterations in the expression and/or targeting of these cell adhesion molecules are expected to significantly affect the E-I balance. As preformed scaffold protein packages are postsynaptic hotspots for the formation of excitatory synapses (Gerrow et al., 2006
), it certainly is not surprising that overexpression of neuroligin-1 and the scaffolding protein PSD-95 enhances the E-I ratio of synaptic input (Prange et al., 2004
).
Scaffolding Proteins
Postsynaptic domains of excitatory synapses contain a multitude of different proteins, and there is substantial evidence that highlights the fundamental role of appropriate expression and targeting of scaffolding proteins during excitatory synapse formation and maintenance (El-Husseini et al., 2000
). Actually, neuronal overexpression of PSD-95 alone is sufficient to enhance the ratio between glutamatergic and GABAergic synapses (Prange et al., 2004
). Likewise, knockdown of PSD-95 results in partial loss of excitatory synapses, which decreases the E-I ratio of synaptic input (Prange et al., 2004
). As PSD-95 couples NMDA-type glutamate receptors to complex signaling pathways that control bidirectional synaptic plasticity these findings have direct implications for learning and memory (Lau and Zukin, 2007
; Migaud et al., 1998
).
It is tempting to speculate that other scaffolding proteins exert similar effects on the E-I ratio of synaptic input. Indeed, acute knockdown of SAP-97 diminishes excitatory synapse transmission and postsynaptic glutamate receptor clustering (Nakagawa et al., 2004
). As these scaffolding proteins provide protein–protein interaction platforms at excitatory synapses other constituents of these postsynaptic domains are expected to follow their rules. For example, targeting of guanylate kinase domain-associated protein (GKAP) and AMPA-type glutamate receptors are dependent on the availability of PSD-95 (El-Husseini et al., 2000
; Gerrow et al., 2006
).
As the total number of synapses remains constant, a shift of the E-I balance apparently always occurs at the expense of one type of synapse (Lardi-Studler et al., 2007
; Prange et al., 2004
), suggesting that glutamatergic and GABAergic synapses are co-regulated. Therefore, it is reasonable to assume that alterations in the expression and/or targeting of gephyrin, a scaffolding molecule of glycinergic and GABAergic postsynaptic domains, influence the E-I ratio of synaptic input. In line with this assumption, overexpression of gephyrin was found to decrease the E-I ratio (Lardi-Studler et al., 2007
). Also, post-transcriptional mechanisms were identified that could be relevant for the control of the E-I ratio of synaptic input: The recently identified C5-gephyrin splice variant (Meier et al., 2000
) was shown not only to be capable of controlling glycine receptor surface stability, but also to exert dominant negative effects on postsynaptic gephyrin (Bedet et al., 2006
). Whether the expression of C5-gephyrins thereby affects the E-I ratio remains to be determined. In conclusion, appropriate expression and targeting of scaffolding proteins contribute to the control of the E-I ratio of synaptic input.
Deregulation of the E-I ratio of synaptic input has been associated with a variety of human nervous system disorders. In the following, potential coherences between deregulated E-I ratios and the phenomenology of nervous system disorders will be discussed.
Increased E-I Ratios of Synaptic Input
In epilepsy patients a multitude of genes can be afflicted with genomic loss- or gain-of-function mutations, which generates a persistent deviation of the neuronal output from the setpoint of homeostasis. For example, epilepsy-associated mutations were found in GABA(A) receptor (GABRG2), sodium (SCN1, SCN2), potassium (KCNA, KCNQ) and calcium channel (CACNA1A) genes (Kullmann, 2002
; Scheffer and Berkovic, 2003
; Sloviter, 1987
). Although it is known that increased E-I ratios of synaptic input accompany the phenomenology of epilepsy, it remains to be elucidated whether this is causative for the disease or whether it occurs secondarily to pathological patterns of enhanced synchronous neuronal network activity (Kofke et al., 1997
; Stief et al., 2007
).
Decreased E-I Ratios of Synaptic Input
Decreased E-I ratios of synaptic input are associated with nervous system disorders, and recent findings underscore the relevance of functional cell adhesion systems in nervous system physiology. For example, loss-of-function mutations in genes encoding neuroligin (NLGN-3 and NLGN-4) and β-neurexin (NRXN1) were implicated in autism and mental retardation (Feng et al., 2006
; Jamain et al., 2003
; Laumonnier et al., 2004
). Interestingly, a loss-of-function mutation in the scaffolding protein and neuroligin binding partner SHANK3 could also be related to autism (Durand et al., 2007
). Likewise, interference with PSD-95 protein synthesis through deletion of the fragile X mental retardation protein (FMRP) gene causes mental retardation (Todd et al., 2003
). Finally, decreased E-I ratios of synaptic input were associated with Rett Syndrome, a pervasive neurodevelopmental disorder associated with mental retardation and autistic behavior. In this case, however, decreased E-I ratios result from loss-of-function mutations of the MECP2 gene, which encodes a methyl CpG-binding protein potentially involved in global repression of transcription (Dani et al., 2005
). Altogether, it seems as if one could reduce these diverse genetic origins of mental retardation and autism to a common denominator, which could be the neuronal failure to establish an adequate connectivity with glutamatergic neurons.
The AMPA-to-NMDA Receptor Ratio
At glutamatergic synapses, the ratio between AMPA- and NMDA-type glutamate receptors determines the postsynaptic responsiveness to presynaptic glutamate release: Decreased postsynaptic AMPA-to-NMDA receptor ratios reduce the threshold for LTP-induction and synaptic plasticity, but may generate silent synapses if AMPA-receptors disappear completely. Likewise, increased postsynaptic AMPA-to-NMDA receptor ratios ensure basal synaptic transmission at the expense of synaptic plasticity. For example, increased AMPA-to-NMDA receptor ratios were found in patients afflicted with Alzheimer’s disease (Snyder et al., 2005
) and schizophrenia (Mohn et al., 1999
). Therefore, subtle changes in the neurotransmitter receptor composition of postsynaptic densities may directly contribute to pathophysiological mechanisms involved in nervous system disorders, without necessarily affecting the morphologically determined E-I ratio of synaptic input.
Critical Role for Neurotransmitter Transporters
As afore mentioned, purely morphological criteria are not sufficient to draw conclusions at a functional level. In that respect, it is important to note that the level of ambient neurotransmitters critically depends on re-uptake mechanisms, which includes both a steady clearance of the extracellular space as well as removal of presynaptically released neurotransmitters. For example, impairment of glycine re-uptake by glial cells (GlyT-1, steady clearance) and by neurons (GlyT-2, presynaptic clearance) generates deficits in respiration (Gomeza et al., 2003a
) and provokes hyperexcitability (Gomeza et al., 2003b
; Rees et al., 2006
), respectively. Moreover, glycine as a co-agonist at NMDA-type glutamate receptors critically influences NMDA-receptor responsiveness. Therefore, disruption of the glial glycine transporter GlyT-1 in the forebrain clearly compensates for NMDA-receptor hypofunctionality in a disease model of schizophrenia (Yee et al., 2006
). In addition, loss-of-function mutations in the GlyT-1 gene (SLC6A9) were uncovered to cause glycine encephalopathy by enhancing tonic glycine receptor activation, as a result of deficits in the glial glycine uptake system (Applegarth and Toone, 2001
). Likewise, as reviewed previously (Dalby, 2003
; Schousboe, 2003
), GABA and glutamate transporters are essential for physiological neuronal information transfer.
The brain works appropriately only if all molecular constituents (among others: receptors, neurotransmitters, synapses, neurons) are properly assembled and installed, which actually establishes a functional syncytium of the brain. However, there is an overwhelming number of laboratories and studies investigating items of this functional syncytium separately. Although a significant and increasing number of studies point to multiple interrelations between homeostatic glutamatergic and GABAergic mechanisms still little is known about the molecular identity of the heterosynaptic plasticity machinery and their reference to nervous system disorders. Therefore, future experimental approaches should give consideration to such interrelation between excitatory and inhibitory systems in the brain, which may open avenues in the treatment of nervous system disorders afflicted with a deregulated E-I balance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by German science foundation DFG (ME2075/ 3-1 to JCM) and Helmholtz association (VH-NG-246 to JCM).
Laumonnier, F., Bonnet-Brilhault, F., Gomot, M., Blanc, R., David, A., Moizard, M. P., Raynaud, M., Ronce, N., Lemonnier, E., Calvas, P., Laudier, B., Chelly, J., Fryns, J. P., Ropers, H. H., Hamel, B. C., Andres, C., Barthelemy, C., Moraine, C., and Briault, S. (2004). X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 74, 552–557.
Migaud, M., Charlesworth, P., Dempster, M., Webster, L. C., Watabe, A. M., Makhinson, M., He, Y., Ramsay, M. F., Morris, R. G., Morrison, J. H., O’Dell, T. J., and Grant, S. G. (1998). Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396, 433–439.
Rees, M. I., Harvey, K., Pearce, B. R., Chung, S. K., Duguid, I. C., Thomas, P., Beatty, S., Graham, G. E., Armstrong, L., Shiang, R., Abbott, K. J., Zuberi, S. M., Stephenson, J. B., Owen, M. J., Tijssen, M. A., van den Maagdenberg, A. M., Smart, T. G., Supplisson, S., and Harvey, R. J. (2006). Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of human startle disease. Nat. Genet. 38, 801–806.
Rivera, C., Li, H., Thomas-Crusells, J., Lahtinen, H., Viitanen, T., Nanobashvili, A., Kokaia, Z., Airaksinen, M. S., Voipio, J., Kaila, K., and Saarma, M. (2002). BDNF-induced TrkB activation down-regulates the K+-Cl– cotransporter KCC2 and impairs neuronal Cl– extrusion. J. Cell Biol. 159, 747–752.
Singh, B., Henneberger, C., Betances, D., Arevalo, M. A., Rodriguez-Tebar, A., Meier, J. C., and Grantyn, R. (2006). Altered balance of glutamatergic/GABAergic synaptic input and associated changes in dendrite morphology after BDNF expression in BDNF-deficient hippocampal neurons. J. Neurosci. 26, 7189–7200.
Yee, B. K., Balic, E., Singer, P., Schwerdel, C., Grampp, T., Gabernet, L., Knuesel, I., Benke, D., Feldon, J., Mohler, H., and Boison, D. (2006). Disruption of glycine transporter 1 restricted to forebrain neurons is associated with a procognitive and antipsychotic phenotypic profile. J. Neurosci. 26, 3169–3181.