Institute of Psychiatry, King’s College London, London, UK
GSK3β and Cdk5 are the two kinases in the center of research on Alzheimer’s disease (AD), involved in the pathological symptoms of AD, Aβ plaque formation, tau hyperphosphorylation and neurodegeneration. So far, both kinases have mostly been examined in isolation, leading to a schism of the research field into defenders of the GSK3β-versus the Cdk5 hypotheses of AD. However, in this debate the fact that activities of GSK3β and Cdk5 can influence each other deserves more attention. Recent evidence from p25 transgenic mice suggests that there is a
dynamic crosstalk: during aging or prolonged overactivation of Cdk5, GSK3β activity may alter in favor of AD pathogenesis. In this review we summarize the connections between GSK3β and Cdk5 and discuss implications for AD hypotheses.
Alzheimer’s disease (AD) is the most prevalent form of dementia, currently affecting more than 24 million people worldwide (Ferri et al., 2005
). The predominant symptoms include memory impairments, later followed by a decline of intellectual skills and loss of control over body functions (Mayeux, 2003
). Pathologically, AD is characterized by three hallmarks: (1) Neurofibrillary tangles (NFT), consisting of filamentous aggregates of hyperphosphorylated tau (Lovestone and Reynolds, 1997
). (2) Senile plaques, which are mostly derived from β-amyloid or “Aβ” (Selkoe, 1994
); Aβ1–42 is an aberrant cleavage product from amyloid precursor protein (APP) and can aggregate to complexes with toxic properties (Hardy and Higgins, 1992
). (3) Brain atrophy, which is caused by loss of both synapses and whole neurons (Gómez-Isla et al., 1997
).
As putative mediators of these symptoms, two proline-directed serine/threonine kinases became the focus of molecular AD research: Glycogen synthase kinase 3β (GSK3β) and cyclin-dependent kinase 5 (Cdk5), whose activities appear to be increased in the AD brain (Lee et al., 1999
; Pei et al., 1997
, 1998
; Yamaguchi et al., 1996
).
GSK3β is starring in a variety of physiological processes such as regulation of cellular morphology, neuronal outgrowth and motility and synaptic plasticity (Peineau et al., 2008
). GSK3β integrates a variety of intracellular and extracellular pathways, including wnt signalling, the insulin pathway, G-protein coupled receptors and others – the effect is a change in GSK3β activity by phosphorylation, protein binding and/or cleavage (Jope and Johnson, 2004
).
Cdk5 is named after its structural similarity to members of the serine/threonine cyclin-dependent kinase family. Cdk5 reaches a peak kinase activity in neurons due to restricted expression of its activators p35 and p39 (Hellmich et al., 1992
). Cleavage of p35 to p25 by calpain leads to overactivation of Cdk5 (Patrick et al., 1999
). Cdk5 is a main player in processes of neural development, synaptic signalling, learning and memory (Angelo et al., 2006
). Under physiological conditions, p25 occurs in only minor quantities. Formation of p25 may play a role in learning processes (Angelo et al., 2003
; Fischer et al., 2005
). It has been suggested that p25 levels and therefore Cdk5 activity are increased in AD brain (Patrick et al., 1999
), but studies by other groups did not confirm this finding or even reported a down regulation of p25 in AD (Takashima et al., 2001
; Yoo and Lubec, 2001
).
Like GSK3β, Cdk5 integrates various signalling pathways, induced by NMDA receptor activity, Rac and growth factors (Dhavan and Tsai, 2001
; Nikolic, 2002
). Several risk factors of AD such as inflammation and oxidative stress have been shown to lead to increased Cdk5 activity (Muyllaert et al., 2008
; Strocchi et al., 2003
).
Both GSK3β and Cdk5 can regulate synaptic plasticity via regulation of long-term potentiation (LTP), synaptic vesicle release and modification of the NMDA Receptor (Angelo et al., 2006
; Giese, 2009
).
Besides sharing both up- and downstream pathways, Cdk5 and GSK3β are linked more directly in neurons. In this review we will summarize the mechanisms of crosstalk between GSK3β and Cdk5 and discuss its implications for tau hyperphosphorylation, a main pathological feature of AD.
The first studies addressing crosstalk between Cdk5 and GSK3β were performed by Morfini et al. (2004)
using cultured neurons: Their investigations to determine a regulatory pathway for kinesin-driven motility in axons revealed that Cdk5 is neccessary for kinesin-driven motility within the axon but cannot directly phosphorylate kinesin. Inhibition of Cdk5 by olomoucine enhanced phosphorylation of kinesin, suggesting that the effect was mediated by negative regulation of other kinases. Therefore activities of the other kinases Erk, CK1 and GSK3β were tested after oloumucine treatment. Altered phosphorylation of CREB phosphopeptide, a substrate of GSK3β, suggested that GSK3β may be the mediator of Cdk5 activity in kinesin-mediated axonal transport.
Activity of GSK3β is regulated by phosphorylation; phosphorylation of Tyr216 can enhance enzyme activity, and conversely phosphorylation of Ser9 is inhibitory. Inhibition of Cdk5 resulted in dephosphorylation of the inhibitory Ser9 site on GSK3β and an increase of activating phosphorylation at Tyr216. Cdk5 does not phosphorylate GSK3β directly on these sites and Morfini et al. (2004)
investigated whether the altered inhibitory phosphorylation at Ser9 may be mediated by a phosphatase. They found that GSK3β and phosphatase 1 (PP1) co-immunoprecipitate and that Cdk5 can indirectly inhibit GSK3β activity via inhibition of PP1 (Figure 1
). GSK3β is activated upon PP1-dependent dephosphorylation and is therefore inhibited by Cdk5. In support of this idea, immunostaining revealed that Cdk5, PP1 and GSK3β co-localize at growth cones. Additionally, incubation of cortical neurons with the non-specific phosphatase inhibitor okadaic acid increased phosphorylation of GSK3β-Ser9. Later evidence indicated that PP1 is a substrate of Cdk5 (Li et al., 2007
). Additionally, regulation of PP1 by Cdk5 may occur via phosphorylation of PP1 inhibitor 1 (Nguyen et al., 2007
). Furthermore, Morfini et al. (2004)
showed for the first time that Cdk5 can influence tau phosphorylation indirectly via regulation of GSK3β. However, these studies were performed on cell culture and the results had to be confirmed by in vivo experiments.
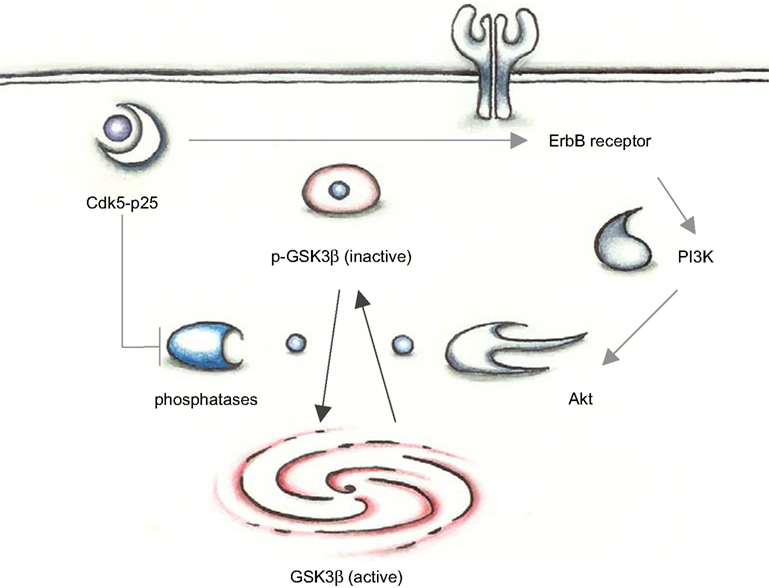
Figure 1. In young mutant mice with low p25 expression, GSK3β is inhibited by phosphorylation at Ser 9. Cdk5-p25 may influence the balance between p-GSK3 and an activatory dephosphorylation via regulation of phosphatases and the PI3K/Akt pathway.
Tau is a microtubule binding protein, which occurs in the healthy brain and functions in axonal transport, assembly and stabilization of microtubules (Buée et al., 2000
). Compared to healthy controls, the AD brain contains four to eight times more tau, which is hyperphosphorylated (Khatoon et al., 1994
). More than 40 phosphorylation sites have been detected on tau (Hanger et al., 1998
) and several kinases and phosphatases have been suggested as deregulators of tau in AD (Buée et al., 2000
; Iqbal and Grundke-Iqbal, 2008
) – amongst these, Cdk5 and GSK3β are thought to be the major tau kinases in vivo (Flaherty et al., 2000
). The fact that the number of NFTs, but not senile plaques correlates highly with the severity of dementia in AD (Dickson et al., 1988
) has stimulated a great amount of research into the misregulation of tau. GSK3β and Cdk5 were proposed as tau kinases after isolating them in a complex with neurofibrillary tangles from AD brain (Yamaguchi et al., 1996
). This association between GSK3β, Cdk5 and tau has been confirmed in post-mortem tissue (Pei et al., 1998
, 1999
), as well as in cell and mouse models (Flaherty et al., 2000
; Li et al., 2006
; Noble et al., 2003
; Pei et al., 1997
; Yamaguchi et al., 1996
). However, it is currently debated which kinase is most strongly associated with tangle formation.
Conditional GSK3β transgenic mice demonstrate tau hyperphosphorylation and neurodegeneration (Lucas et al., 2001
), indicating that GSK3β activity is sufficient to hyperphosphorylate tau. Despite the accumulating evidence for GSK3β as major tau kinase, Cdk5 overactivation may also play a role in tau phosphorylation. Tau is a substrate of Cdk5 and is regulated by phosphorylation under physiological conditions, e.g. during G-protein mediated growth cone collapse (Nakayama et al., 1999
). Furthermore, numerous studies have shown that Cdk5 can phosphorylate tau at sites which are hyperphosphorylated in AD (Imahori and Uchida, 1997
; Lew and Wang, 1995
; Sengupta et al., 1997
; Tang and Wang, 1996
).
A negative correlation between activities of the tau kinases Cdk5 and GSK3β, as observed by Morfini et al. (2004)
, may provide a new perspective on the interplay between tau and its kinases. Studies with p25 transgenic mice, expressing low levels of p25 predominantly in the hippocampus (Angelo et al., 2003
) that is affected in the early stages of AD (Braak and Braak, 1991
), showed that there is crosstalk between Cdk5 and GSK-3β in vivo (Plattner et al., 2006
). This was confirmed with another p25 mouse line (Wen et al., 2008
). In young age the p25-induced increase in Cdk5 activity inhibits GSK-3β activity by enhancing the inhibitory phosphorylation at Ser-9 of GSK-3β (Plattner et al., 2006
; Wen et al., 2008
). Importantly, at this age the increased Cdk5 activity does not cause tau hyperphosphorylation. However, in old age tau becomes hyperphosphorylated in these p25 mutants (Plattner et al., 2006
; Wen et al., 2008
). This tau hyperphorylation is a result of an age-dependent loss of the Cdk5 inhibition of GSK-3β. In fact in the older age GSK-3β activity is enhanced in the p25 mutants. Thus, increased GSK-3β activity, and not Cdk5 activity, leads to tau hyperphosphorylation.
Plattner et al. (2006)
inspected whether in young age Cdk5 and GSK3β interact with other signalling molecules in vivo. They found that GSK3β and Cdk5 aggregate in a complex with protein phosphatase 2A (PP2A), but not with PP1 as found by Morfini et al. (2004)
in vitro. Furthermore, blocking phosphatases with okadaic acid resulted in less GSK3β activity in young p25 mice, which supports the evidence that the inhibitory Ser9 site is regulated by phosphatise activity. Thus, the inhibitory crosstalk on GSK-3β activity in young age is linked to altered balance in phosphatase activity. However, it is unclear why the inhibitory crosstalk gets lost with age and why in older age GSK-3β activity is increased in the p25 mutants.
The correlation between the time courses of increasing GSK3β activity and tau hyperphosphorylation suggests that GSK3β is the major tau kinase in these p25 transgenic mice. This idea was confirmed by treatment with lithium, an inhibitor of GSK3β. Mutant mice treated with lithium do not exhibit hyperphosphorylation of tau epitopes. In fact, other mouse models with high p25 expression with tau hyperphosphorylation and tangle formation possessed elevated GSK3β activity too (Noble et al., 2003
) but for other reasons as the activating Tyr-216 phosphorylation is increased in comparison to a control group, which was not found by Plattner et al. (2006)
Moreover, in p35 knockout mice tau is hyperphosphorylated and GSK3β activity increased while activity of Cdk5 is decreased (Hallows et al., 2003
). These mice may reflect the scenario of young p25 mutants, in which Cdk5 activity and GSK3β activity correlate inversely. At the same time this study shows that Cdk5 activity is not a prerequisite for tau hyperphosphorylation.
Cdk5 and GSK3β may be linked by more intricate pathways: It has been shown that phosphorylation of Ser9 after Cdk5 activation can be mediated by ErbB and PI3K/Akt pathway (Wen et al., 2008
): Cdk5 can phosphorylate T871 on ErbB2 and S1120 on Erb3 (Li et al., 2003
) and Erb phosphorylation and GSK3 phosphorylation correlate in p25 transgenic mice (Figure 1
).
The activity of GSK3β was found up-regulated in AD brain (Giese, 2009
), while the role of Cdk5 regulators in AD brain is still controversial. Further studies with post-mortem tissue may elucidate the inverse relationship between the two kinases during disease progression.
Recent evidences have shown that the activities of Cdk5 and GSK3β are linked and the crosstalk depends on ageing. The proposed mechanisms for inhibition of GSK-3β by Cdk5 include phosphatases and ErbB signaling. However, it is unclear why the impact from Cdk5 on GSK3β activity renders from an inhibitory into an activatory state in ageing p25 transgenic mice. One possibility is that ageing neurons may become more sensitive to p25 exposure, in which case research on mouse models of p25 expression should put more emphasis on studying aged mice. On the other hand it is conceivable that GSK3β activity changes during steady exposure to p25, be it short periods of high levels of p25 or prolonged exposure to low p25 levels. As Plattner et al. (2006)
have demonstrated, Cdk5 may exert its effects on tau phosphorylation mostly via GSK3β since inhibition of GSK3β by lithium reversed this effect. Therefore, tau hyperphosphorylation in other p25 models may also result from changes in GSK3β activity (Figure 2
).
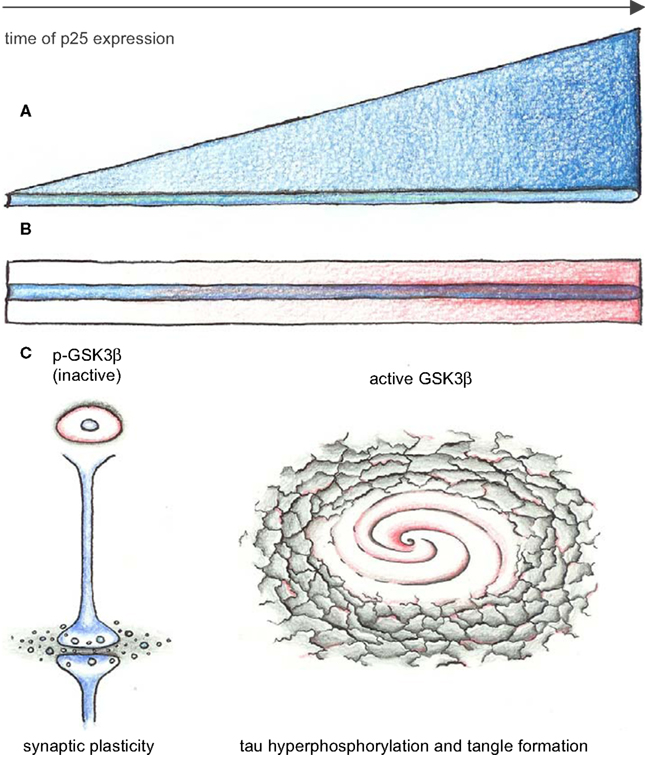
Figure 2. Continued exposure to low p25 levels changes activity of GSK3β. (A,B) Time axes of p25 expression (blue). Two mechanisms for changing effects of Cdk5-p25 on GSK3β are conceivable: (A) Molecular changes accumulate as consequence of continuous exposure to overactive Cdk5. (B) Ageing effects (red) make neurons more vulnerable to effects of Cdk5-p25, resulting in overactive GSK3β. (C) Changes of GSK3β activity from inhibited into an overactive state in p25 mutants may mediate improved L&M and synaptic plasticity in young adult mice while leading to tau hyperphosphorylation and tangle formation in old mice.
Young female mice with constant expression of low p25 or inducible mice with short exposure to high p25 levels (Fischer et al., 2005
; Ris et al., 2005
) exhibit improved learning and memory (L&M). This observation has led to the hypothesis that p25 expression is not a cause of AD but a reaction to stimuli that impair L&M, ultimately turning from beneficial to detrimental as p25 exposure continues (Angelo et al., 2006
). The learning ability during p25 expression in mice from early improvements toward neurodegeneration may be mediated by changing GSK3β activity. Therefore, studies of these mice at various time points should not only include substrates of Cdk5 but also focus on GSK3β pathways.
P25 has a half life of less than 30 min in cultured neurons (Patrick et al., 1999
). If lifelong exposure to p25 insults leads to changes in GSK3β activity, information about Cdk5 activity may be stored within the cell, for instance in phosphorylation of substrates with low turnover rate such as tau or within the genome. Identifying how this information is stored and resetting it in ageing p25 mutants may be useful to reverse insults that have already occurred during a life time.
Mutant mice with low p25 expression model ageing effects as the most important risk factors of AD. Understanding why GSK3β turns from an inhibited into a hyperactive state during ageing of these mice and testing the mutants with drugs that can revert this process seems a promising approach to prevent or treat tau hyperphosphorylation in AD.
We declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
We thank Prof. Chris Miller and Dr. Andrew Thompson for helpful comments on the manuscript. This work has been supported by a Medical Research Council Ph.D. studentship.
Ferri, C. P., Prince, M., Brayne, C., Brodaty, H., Fratiglioni, L., Ganguli, M., Hall, K., Hasegawa, K., Hendrie, H., Huang, Y., Jorm, A., Mathers, C., Menezes, P. R., Rimmer, E., Scazufca, M., and Alzheimer’s Disease International (2005). Global prevalence of dementia: a Delphi consensus study. Lancet 366, 2112–2117.
Noble, W., Olm, V., Takata, K., Casey, E., Mary, O., Meyerson, J., Gaynor, K., LaFrancois, J., Wang, L., Kondo, T., Davies, P., Burns, M., Veeranna, Nixon, R., Dickson, D., Matsuoka, Y., Ahlijanian, M., Lau, L. F., and Duff, K. (2003). Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 38, 555–565.
Wen, Y., Planel, E., Herman, M., Figueroa, H. Y., Wang, L., Liu, L., Lau, L. F., Yu, W. H., and Duff, K. E. (2008). Interplay between cyclin-dependent kinase 5 and glycogen synthase kinase 3 beta mediated by neuregulin signaling leads to differential effects on tau phosphorylation and amyloid precursor protein processing. J. Neurosci. 28, 2624–2632.
Yamaguchi, H., Ishiguro, K., Uchida, T., Takashima, A., Lemere, C. A., and Imahori, K. (1996). Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3 beta and cyclin-dependent kinase 5, a component of TPK II. Acta Neuropathol. 92, 232–241.