Laboratory for Neuronal Circuit Dynamics, Neuronal Circuit Dynamics, Brain Science Insitute, RIKEN, Japan
Over the last decade, optical neuroimaging methods have been enriched by engineered biosensors derived from fluorescent protein (FP) reporters fused to protein detectors that convert physiological signals into changes of intrinsic FP fluorescence. These FP-based indicators are genetically encoded, and hence targetable to specific cell populations within networks of heterologous cell types. Among this class of biosensors, the development of optical probes for membrane potential is both highly desirable and challenging. A suitable FP voltage sensor would indeed be a valuable tool for monitoring the activity of thousands of individual neurons simultaneously in a non-invasive manner. Previous prototypic genetically-encoded FP voltage indicators achieved a proof of principle but also highlighted several difficulties such as poor cell surface targeting and slow kinetics. Recently, we developed a new series of FRET-based Voltage-Sensitive Fluorescent Proteins (VSFPs), referred to as VSFP2s, with efficient targeting to the plasma membrane and high responsiveness to membrane potential signaling in excitable cells. In addition to these FRET-based voltage sensors, we also generated a third series of probes consisting of single FPs with response kinetics suitable for the optical imaging of fast neuronal signals. These newly available genetically-encoded reporters for membrane potential will be instrumental for future experimental approaches directed toward the understanding of neuronal network dynamics and information processing in the brain. Here, we review the development and current status of these novel fluorescent probes.
Significant progress in our understanding of neuronal network dynamics underlying brain function requires the ability to monitor the activity of multiple neurons simultaneously. Optical imaging based on voltage-sensitive dyes offers the spatio-temporal resolution necessary to fulfill this requirement (reviewed in Baker et al., 2005
; Grinvald and Hildesheim, 2004
; Grinvald et al., 1988
; Knöpfel et al., 2006
). These organic dyes have been successfully used during the past 20 years to report changes in membrane potential from single or large numbers of neurons in a variety of preparations, including mammalian brain tissue (Ferezou et al., 2007
; Grinvald and Hildesheim, 2004
). However, conventional voltage-sensitive dyes are generally not suitable for the labeling of specific cell populations. In the absence of targeting to a defined cell population, the optical signal is often drowned out either by background fluorescence from inactive cells or by signals from cells that are not the focus of interest. Moreover, unspecific staining of brain tissue prevents the unequivocal attribution of the fluorescence output signal to a defined cell population. To overcome these limitations of classical voltage-sensitive dyes, it is greatly desirable to find a way to target specific cell populations.
With the molecular cloning of green fluorescent protein (GFP) from Aequorea victoria (Chalfie et al., 1994
) and subsequent generation of new and improved spectral variants derived from various sea organisms (reviewed in Shaner et al., 2007
; Verkhusha and Lukyanov, 2004
), the construction of genetically-encoded sensors for visualization of cellular dynamics became conceivable. A now broadly used approach to fluorescent biosensor engineering involves the molecular fusion of a GFP-based reporter protein to a second protein that undergoes conformational transitions in response to a physiological signal such as fluctuations in calcium or membrane potential (recently reviewed in Knöpfel et al., 2006
; Qiu et al., 2008
; Van Engelenburg and Palmer, 2008
). Since protein-based sensors are encoded in DNA, they can be expressed under the control of cell specific promoters and introduced in vivo using gene transfer techniques. In a transgenic animal, a genetically-encoded voltage sensor could be expressed in practically any cell type and would have the advantage of staining only the cell population determined by the promoter used to drive the expression.
During recent years, several designs of genetically-encoded optical probes for membrane potential have been explored. FlaSh, the first prototype, was obtained by inserting GFP within the C-terminal tail of the voltage-gated Shaker potassium channel (Siegel and Isacoff, 1997
). Concomitantly, our laboratory explored a FRET (Fluorescence Resonance Energy Transfer) design principle based on the voltage-dependent conformational change associated with the voltage-sensing domain of the Kv2.1 potassium channel, resulting in a voltage sensor we named VSFP1 (Sakai et al., 2001
). Finally, the third prototype, SPARC, was generated by introducing GFP into a reversibly nonconducting form of the rat μI skeletal muscle sodium channel (Ataka and Pieribone, 2002
). Although these first generation fluorescent protein voltage sensors were shown to optically report changes in membrane potential, their application in mammalian systems is severely hindered by their poor targeting to the plasma membrane in transfected cells (Baker et al., 2007
). Indeed, confocal microscopy analysis revealed a prominent intracellular expression for Flare (a Kv1.4 FlaSh variant), VSFP1 and SPARC with little, if any, fluorescence associated with the cell surface in both HEK293 cells and hippocampal neurons. Unfortunately, neither the mutagenesis of potential ER retention sites nor the introduction of ER export motifs has resulted in a significant improvement of the low plasma membrane expression displayed by the first generation FP voltage-sensitive probes (Baker et al., 2008
). Despite this setback, the functional concept underlying VSFP1 (Sakai et al., 2001
) has proven to be the most successful for the following generation of VSFPs.
Recently, a self-contained voltage sensing domain (VSD) was isolated from the non-ion channel protein Ciona intestinalis voltage sensor-containing phosphatase (Ci-VSP) (Murata et al., 2005
). Interestingly, a single VSD was shown to be functional in Ci-VSP (Kohout et al., 2008
) while four VSD-containing subunits are required for the gating of the Kv potassium channel pore region (Bezanilla, 2000
). Furthermore, the VSD of Ci-VSP operates as a sensor by itself since robust sensing currents were shown in the absence of the enzyme region (Murata et al., 2005
). In contrast, sensing or gating currents of voltage-gated ion channels have so far been elusive if the voltage sensor is separated from the pore region (Okamura et al., 2009
).
We thus reasoned that the limited cell surface targeting of first generation voltage-sensitive proteins could be resolved by using the structurally much simpler Ci-VSP scaffold and tested this hypothesis by exchanging the VSD of VSFP1 with that of Ci-VSP (Dimitrov et al., 2007
; He et al., 2007
). The resulting series of voltage sensors were termed VSFP2s (Dimitrov et al., 2007
; Lundby et al., 2008
; Mutoh et al., 2009
). The initial series included the variant VSFP2.1 in which a positive arginine was mutated to a neutral glutamine at position 217 (R217Q) within the charged S4 membrane segment of the VSD. As a result of this charge neutralization, the voltage-activity curve of the native Ci-VSP showed a leftward shift to the physiological range of membrane potential fluctuations in electrically active mammalian cells (Dimitrov et al., 2007
). As expected from the self-contained properties of Ci-VSP, VSFP2s displayed a predominant targeting to the plasma membrane in transfected PC12 cells. Figure 1
A illustrates the expression and clear membrane localization of VSFP2.1, which represents a major advance when compared to the poor cell surface trafficking exhibited by the first generation FP voltage sensors (Baker et al., 2007
).
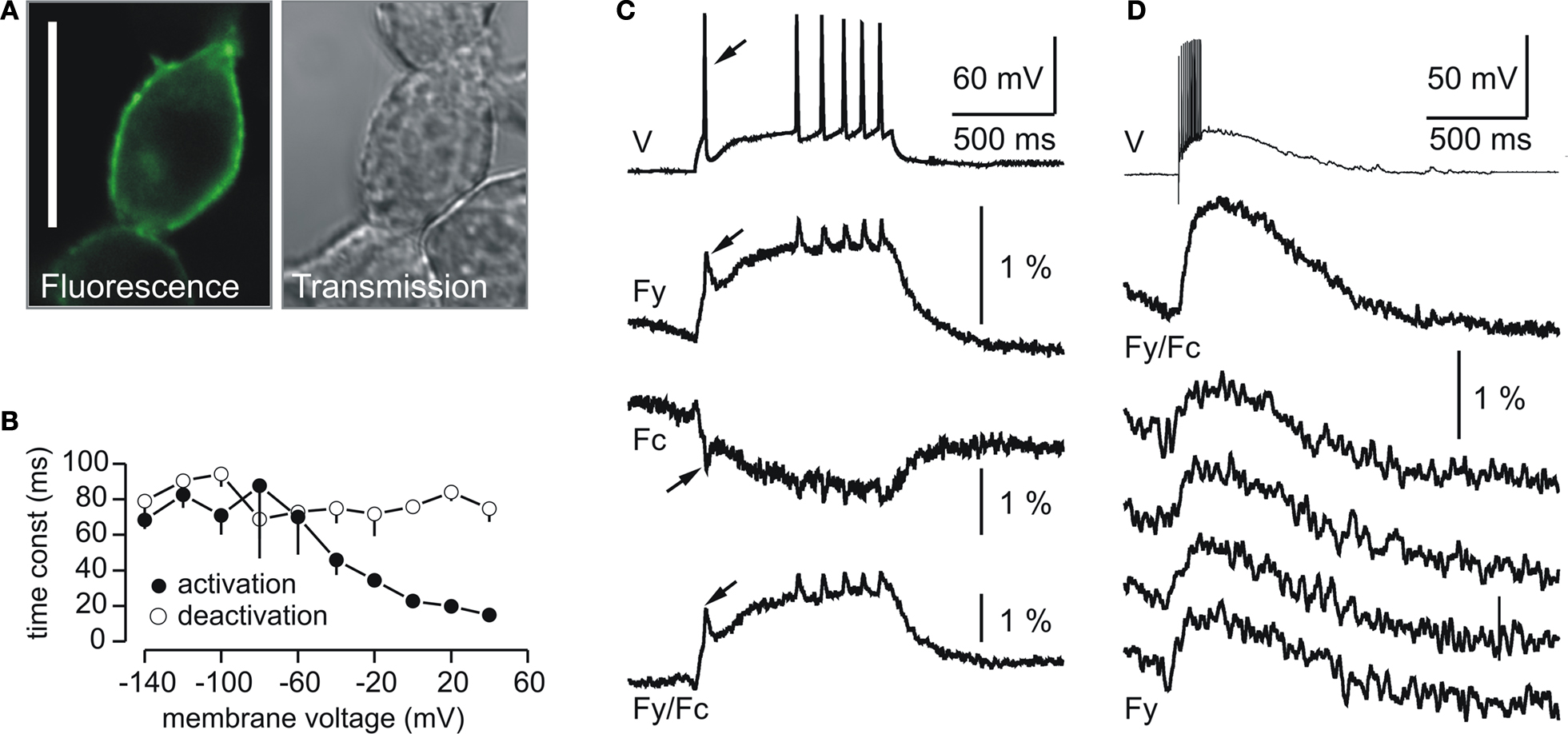
Figure 1. Plasma membrane expression and fluorescence response properties of VSFP2.1. (A) Confocal fluorescence and transmission images of PC12 cells transfected with VSFP2.1. Note the targeting of VSFP2.1 to the plasma membrane. Scale bar is 25 μm. (B) Voltage dependence of apparent activation and deactivation time constants of VSFP2.1 fluorescence signals in PC12 cells at 35°C. (C,D) VSFP2.1 fluorescence responses to physiological neuronal membrane signals. VSFP2.1-expressing PC12 cells were voltage-clamped with membrane voltage traces recorded from olfactory mitral cells that were stimulated to generate a series of action potentials by direct current injection (C) or by electrical stimulation of the olfactory nerve (D). Traces in (C) and upper trace in (D) are the average of 50 and 90 sweeps, respectively. The lower four traces in (D) are single sweeps. Traces show membrane potential (V), yellow fluorescence (Fy), cyan fluorescence (Fc) and the ratio of yellow and cyan fluorescence (Fy/Fc). Fluorescence signals were digitally low pass filtered (0.2 kHz) and were not corrected for photobleaching. Recordings were done at 35°C (from Dimitrov et al., 2007
).
VSFP2.1 responded to depolarizing voltage pulses by a decrease in cyan fluorescence and a concomitant raise in yellow florescence, resulting from increased energy transfer from the cyan to the yellow-emitting FP chromophore following the translocation of the S4 transmembrane segment (Dimitrov et al., 2007
). Moreover, VSFP2.1 showed relatively fast kinetics with an apparent on-time constant of ∼15 ms upon a depolarization from a holding potential of −70 to +40 mV as shown in Figure 1
B. To investigate whether VSFP2.1 could be a candidate for optical measurements of neuronal activity, PC12 cells were voltage-clamped with membrane voltage transients recorded from olfactory mitral cells that were stimulated either by direct current injection to generate a series of action potentials (Figure 1
C) or synaptically to induce a burst of fast action potentials (Figure 1
D). Fluorescence traces revealed that VSFP2.1 could clearly resolve individual action potentials as well as the underlying membrane depolarization (Figure 1
C). However, VSFP2.1 mainly reported the slow components of these voltage transients, as expected from its response kinetics (Dimitrov et al., 2007
). Indeed, the optical readout of the fast action potentials was significantly reduced relative to the slower component of the membrane potential change. This phenomenon was also clearly noticeable when using the membrane voltage transients from a mitral cell succeeding a single shock electrical stimulation of the olfactory nerve (Figure 1
D). Most importantly, it should be noted that the responses shown in Figure 1
D could be resolved in single sweeps. Optimizing the length of the amino acid linkers connecting either the donor chromophore to the VSD or the FRET donor/acceptor pair in VSFP2.1 resulted in VSFP2.3 with both improved fluorescence response kinetics and FRET efficiency (D. Dimitrov et al., unpublished; Akemann et al., 2009
; Lundby et al., 2008
; Mutoh et al., 2009
; Villalba-Galea et al., 2008
, 2009
).
Spectral variants of VSFP2.1
Cyan- and yellow-emitting variants of GFP from A. Victoria are most often used as a fluorescent reporter component for FRET-based sensors. However, the photophysical properties of this FP pair are less than ideal for FRET imaging since both chromophores have broad emission/absorption spectra with relatively small Stokes shift (Chapman et al., 2005
) and considerable FRET donor emission within the acceptor emission band. Furthermore, red-shifted emitting variants would ultimately yield a higher signal-to-noise ratio if tissue autofluorescence is an issue. Indeed, red fluorescence should provide better spectral separation from the intrinsic green autofluorescence of brain tissue given that the fluorescence of flavins, vitamins and NADPH is considerably lower in the red region of the spectrum than in the blue-green region. Additionally, long-wavelength light is usually associated with reduced phototoxicity and deeper penetration into biological tissue. To this aim, we generated a red-shifted VSFP2.1 variant comprising a pair of yellow and far-red emitting FPs that we termed VSFP2.4 (Mutoh et al., 2009
). The absorption spectrum of the acceptor, mKate2, shows considerable overlap with the emission spectrum of the donor, Citrine, with a calculated Förster distance of 5.82 nm (Mutoh et al., 2009
). Furthermore, both spectra are well enough separated to allow independent excitation of the chromophores, limiting spectral bleed through from the donor emission into the acceptor channel. In parallel, Tsutsui et al. (2008)
reported another VSFP2.1 spectral variant based on FPs isolated from corals (mUKG and mKOκ) referred to as Mermaid. The conceptual design of the original series of VSFP2s and its color variants is illustrated in Figure 2
A.
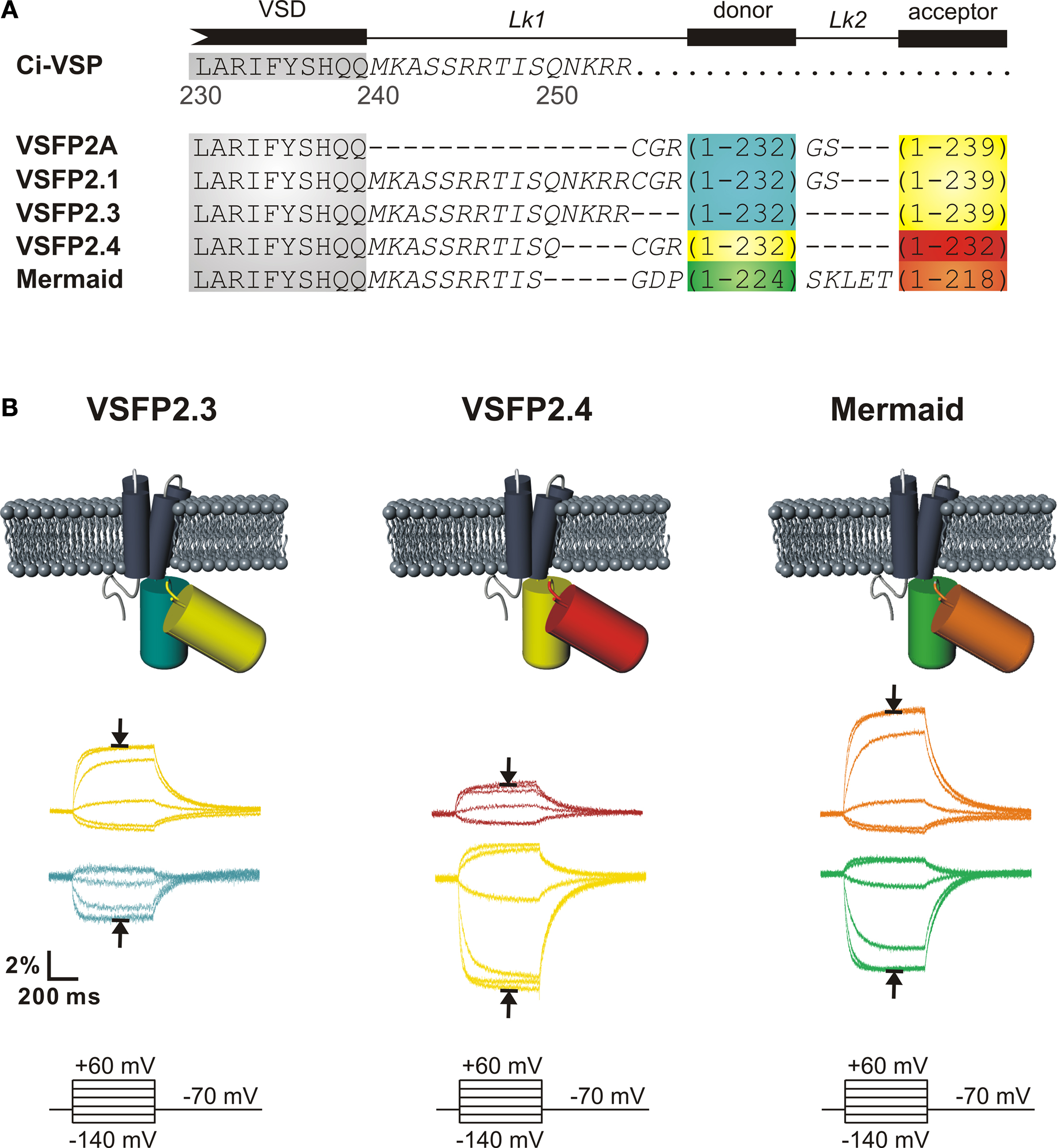
Figure 2. Second generation voltage-sensitive fluorescent proteins. (A) Alignment of the amino acid sequences of VSFP2.1 variants. The C-terminal residues and downstream segment (240–254) of the VSD of Ci-VSP are shown in gray and italic text, respectively. FPs are mCerulean (blue), Citrine/mCitrine (yellow), mKate2 (red), mUKG (green) and mKOκ (orange). (B) Acceptor (upper color traces) and donor (lower color traces) signals of PC12 cells in response to a family of 500 ms voltage steps from a holding potential of −70 mV to test potentials of −140 to +60 mV recorded at 35°C. Traces corresponding to a depolarization to +60 mV are indicated by arrows (from Mutoh et al., 2009
).
Quantitative comparison of these three most advanced FRET-based voltage probes (VSFP2.3, VSFP2.4 and Mermaid) revealed relatively similar steady state spectrally-resolved maximal change in fluorescence (ΔR/R) upon a depolarization from −100 to +40 mV (13.3 ± 3.4, 12.4 ± 1.0 and 12.9 ± 4.8% for VSFP2.3, VSFP2.4 and Mermaid, respectively) (Mutoh et al., 2009
). Acceptor and donor fluorescence signals in response to voltage steps from a holding potential of −70 mV to test potentials of −140 to +60 mV are shown in Figure 2
B. Likewise, VSFP2.1 spectral variants displayed comparable voltage dependencies (V1/2 = −54.2 mV, V1/2 = −54.2 mV and V1/2 = −43.6 mV for VSFP2.3, VSFP2.4 and Mermaid, respectively). Upon depolarization from a holding potential of −70 mV, all three sensors exhibited fluorescence signals that could be fitted with two main time constants (Table 1
; Akemann et al., 2009
; Lundby et al., 2008
; Mutoh et al., 2009
; Tsutsui et al., 2008
; Villalba-Galea et al., 2009
) that likely correspond to the conformational transition states of Ci-VSP (Villalba-Galea et al., 2008
). The values for these on-time response components were very similar except that the fast on-time constant contributed to a larger fraction of the total signal in VSFP2.4 when compared to Mermaid (40 ± 4 and 23.5 ± 5% at +60 mV, respectively) (Mutoh et al., 2009
). The off-time kinetics did not differ among the VSFP2.1 variants under this assay protocol. The fluorescence response properties of the VSFP2.1 variants detailed above are summarized in Table 1
.

To validate the expression pattern of the latest FRET-based voltage-sensitive probes in neurons, we transfected primary hippocampal neurons after 6 days of culture with VSFP2.3, VSFP2.4 and Mermaid and evaluated them by confocal fluorescence imaging 1 week later. As shown in Figure 3
, VSFP2.3, VSFP2.4 and Mermaid fluorescence was distributed over the cell body, dendrites and axons of a variety of neurons including pyramidal cells.
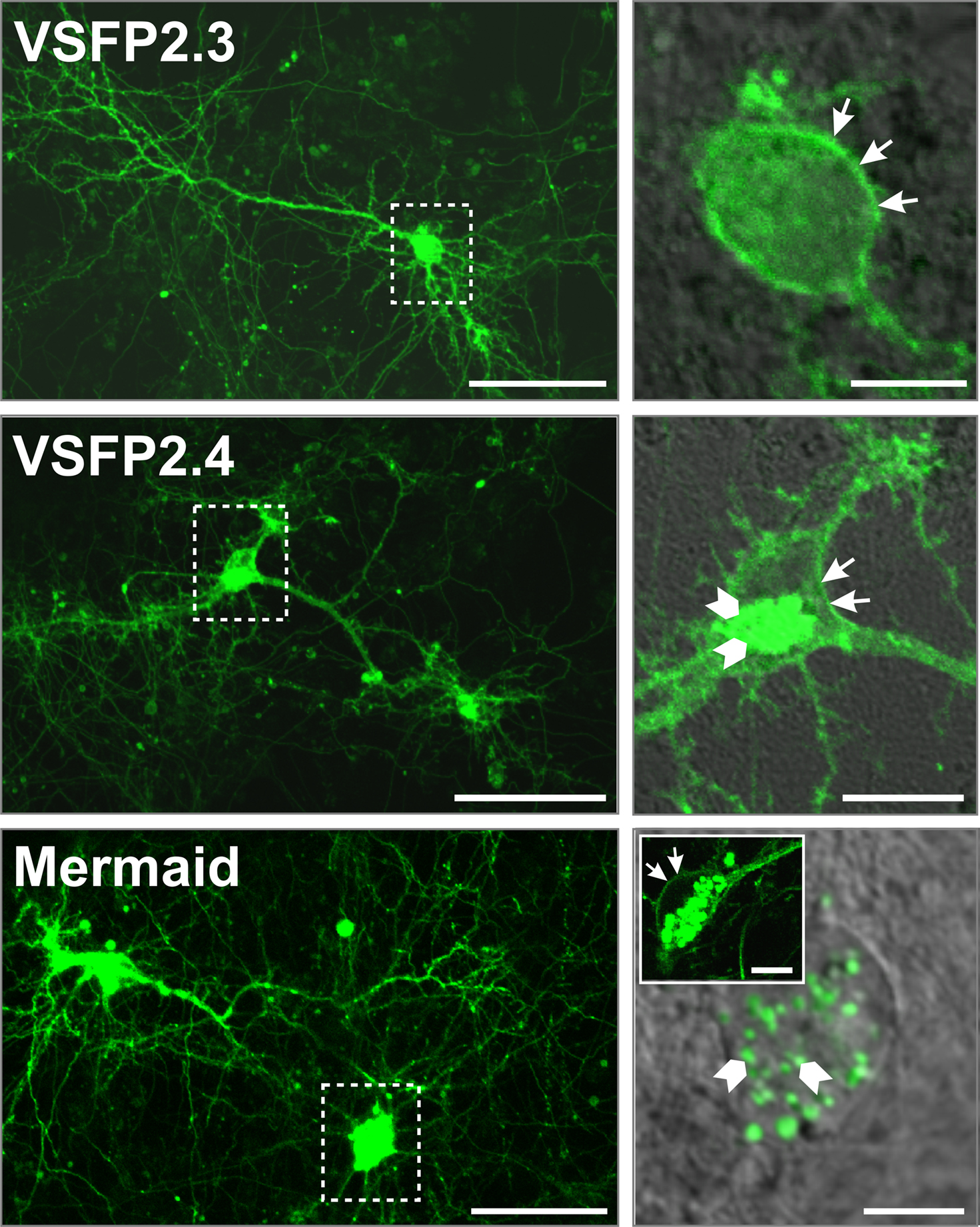
Figure 3. Expression pattern of VSFP2.1 variants in transfected hippocampal neurons. Primary hippocampal cultures derived from mouse E18 embryos were transfected with either VSFP2.3, VSFP2.4 or Mermaid 6 days after plating and imaged by confocal fluorescence microscopy 1 week later. Fluorescence was allowed to saturate locally to optimize the visualization of neuronal processes. Boxed areas in the left panel are shown magnified on the right panel. Arrows indicate cell surface expression while arrowheads show intracellular expression. Note the targeting of VSFP2.3 and VSFP2.4 to the plasma membrane and Mermaid-associated intracellular aggregates in magnified views. The insert in the lower right image shows an example of a cell with clear expression of Mermaid at the cell surface. Scale bars are 50 and 10 μm for left and right panels, respectively.
In particular, VSFP2.3 showed efficient targeting to the plasma membrane as indicated by arrows in magnified views. Likewise, VSFP2.4 fluorescence was mainly found at the cell surface while some fluorescence was also observed intracellularly within a juxtanuclear trans-Golgi network-like structure (arrowheads) which is likely involved in endosome trafficking. Mermaid was also in part targeted to the plasma membrane but the extent of membrane-associated fluorescence was largely overwhelmed by much stronger fluorescence derived from structures reminiscent of intracellular vesicles as indicated by arrowheads in Figure 3
. These punctuate structures have previously been reported for fluorescent proteins isolated from reef coral anthozoan species which are known for their high tendency to form aggregates (Hirrlinger et al., 2005
; Katayama et al., 2008
). Indeed, bright fluorescent clusters were observed in cell somata and processes of a series of transgenic mouse lines expressing various reef coral FPs within early postnatal weeks, which were shown to increase substantially with the age of the animal (Hirrlinger et al., 2005
). In contrast, fluorescent aggregates were not detected in mice expressing A. victoria GFP variants even at older ages (Hirrlinger et al., 2005
; Nolte et al., 2001
). Since the establishment of transgenic mice expressing VSFP2s requires long-term expression of the reporter proteins, VSFP2.3 and VSFP2.4 would likely be better candidates due to their reduced intracellular accumulation and aggregation (Figure 3
). Accordingly, with the relatively small signal amplitude of these second generation FP voltage sensors, optimal responses are largely dependent on proper trafficking to the plasma membrane since intracellular expression principally contributes to background fluorescence, decreasing the signal-to-noise ratio (RS/N) significantly.
In order to investigate the relationship between VSFP activation kinetics and VSFP-mediated optical report of neuronal activity, we represented VSFP2.3 and VSFP2.4 by an eight state Markov process kinetic model reflecting their experimental response properties (Figures 4
A1,B1; see Akemann et al., 2009
).
Inclusion of these kinetic models into a realistic conductance-based computational version of a rat somato-sensory layer 5 pyramidal neuron given by Mainen and Sejnowski (1996)
enabled us to predict in silico the possible VSFP fluorescence readouts that would be obtained from neuronal activity (Akemann et al., 2009
). The simulations indicated that the second generation VSFPs can provide an activation mechanism sufficiently fast to track burst firing of action potentials albeit with significant attenuation as shown in Figure 4
, wherein individual action potentials were resolved as narrow peaks superimposed on a slowly rising (VSFP2.3; Figure 4
A2) or declining (VSFP2.4; Figure 4
B2) curve representing the activation state of both voltage reporters. Additionally, simulation of VSFP2.3 and VSFP2.4 optical readouts in response to a subthreshold or suprathreshold activation of a distal synaptic conductance (causing an excitatory postsynaptic potential, EPSP) showed that both FP voltage sensors are able to readily resolve synaptic potentials in individual L5 pyramidal neurons (Figures 4
A3,B3). However, as the fluorescence response properties of VSFP2s to membrane potential changes consist of a slow and fast kinetic response with time constants differing by more than an order of amplitude (Lundby et al., 2008
; Mutoh et al., 2009
; Villalba-Galea et al., 2009
), the optical resolution of any membrane voltage transient rising faster than the slow kinetic component will critically depend on the relative contribution of the slow versus fast component of the VSFP activation response.
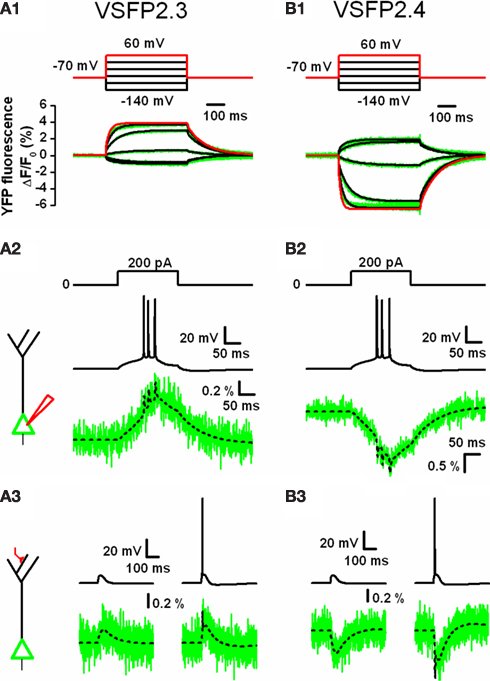
Figure 4. Computer simulations of VSFP2.3 and VSFP2.4 optical response signals in cortical layer 5 (L5) pyramidal neurons. (A1) Family of YFP fluorescence (ΔF/F0) responses (lower panel; green traces) to 500 ms voltage steps (upper panel) from a holding membrane voltage of −70 mV to test potentials of −140 to +60 mV recorded from voltage-clamped PC12 cells expressing VSFP2.3. Overlaid to the experimental traces are simulated traces obtained from a VSFP2.3 model wherein the kinetics are represented as an eight state Markov process as given in (Akemann et al., 2009
). The simulated response to +60 mV is highlighted in red. (A2) Predicted VSFP2.3 fluorescence signal (ΔF/F0; lower panel) in response to an action potential burst (middle panel) in the cell body of a simulated L5 pyramidal neuron evoked by constant current injection (upper panel). The schematic to the left depicts the neuron with a point current source attached to the cell body (red electrode). The electrical response was simulated using a conductance-based model of a reconstructed rat L5 neuron given by Mainen and Sejnowski (1996)
. VSFP2.3 was homogenously inserted within the membrane (all 177 compartments) at a density of 800 units/μm2. The fluorescence signal (lower panel) represents the response of VSFP2.3 in the somatic membrane with (green) or without (dashed black) photon quantum noise calculated assuming a sampling rate of 2 kHz and excitation light level that bleaches GFP within 10 s of illumination. (A3) VSFP2.3 fluorescence signal (ΔF/F0; lower panel) as predicted by the simulation in response to a subthreshold (left column) or suprathreshold (right column) activation of a distal synaptic conductance (schematically depicted to the left with the activated synapse in red). The voltage signal in the cell body (upper row left: EPSP; upper row right: EPSP plus evoked action potential) together with the associated fluorescence signals (lower row), with (green) or without (dashed black) photon quantum noise are shown. (B1–B3) Same as in (A1–A3), but using a model of VSFP2.4 instead of VSFP2.3. VSFP2.4 was simulated as an eight state Markov chain model analogously to VSFP2.3. For methodological details see Akemann et al. (2009)
.
To address whether the relatively slow fluorescence response kinetics of the second generation FP voltage probes is due to intrinsically slow operations of Ci-VSP, we measured fluorescence signals along with sensing currents (i.e. currents resulting from the displacement of charges within the VSD) in VSFP2.3-expressing PC12 cells and found that the voltage dependency of the fluorescence read-outs closely resembles the activation curve of the sensing currents, indicating that the fluorescence signal effectively reports the voltage-dependent conformational change of the VSD (Lundby et al., 2008
). However, the sensing charge movement was found to be two orders of magnitude faster than the dominant slow component of the fluorescence response (∼1 versus ∼100 ms), suggesting a relatively weak coupling between the VSD and the VSFP2 class reporter proteins (Lundby et al., 2008
).
Already during the analysis of our initial series of Ci-VSP-based VSFP2s, we noted that the ratio between the CFP and YFP signal components increases when the length of the linker connecting the VSD to the donor chromophore is shortened (Dimitrov et al., 2007
). Furthermore, a short linker version of VSFP2.1 (VSFP2A R217Q) clearly exhibited a fast on-time component in the CFP response which was not observed in the YFP channel (CFP: 7 ± 2 and 180 ± 15 ms, YFP: 171 ± 15 ms; Figure 5
A), suggesting the presence of a FRET-independent response component (Lundby et al., 2008
). To test this hypothesis, we measured the response characteristics of VSFP2A(R217Q) before and after photobleaching of the acceptor chromophore (YFP) as illustrated in Figures 5
A,B, respectively. Single cell spectrofluorometry confirmed the disappearance of the 530-nm peak in the emission spectrum and revealed an increase in CFP emission at 470 nm due to donor dequenching (Figure 5
B) (Lundby et al., 2008
). Most importantly, a significant signal remained in the cyan channel after acceptor photobleaching (Figure 5
B, lower panel), indicating that the fast CFP response component does not require the presence of a FRET acceptor. To investigate whether the removal of YFP would favor the fast intrinsic CFP response further, we generated a novel Ci-VSP-based voltage sensor comprising a single cyan fluorescent reporter protein that we termed VSFP3.1. Interestingly, VSFP3.1 responded to voltage steps with a very fast initial transient (activation time constant of 1.3 ± 0.1 ms at +70 mV; Figure 5
C), indicating that truncation of the acceptor chromophore significantly improved the coupling of the VSD to the fluorescent reporter protein. Indeed, the fluorescence output of VSFP3.1 showed a dominant fast response component matching closely the sensing currents of Ci-VSP (Lundby et al., 2008
; Villalba-Galea et al., 2009
).
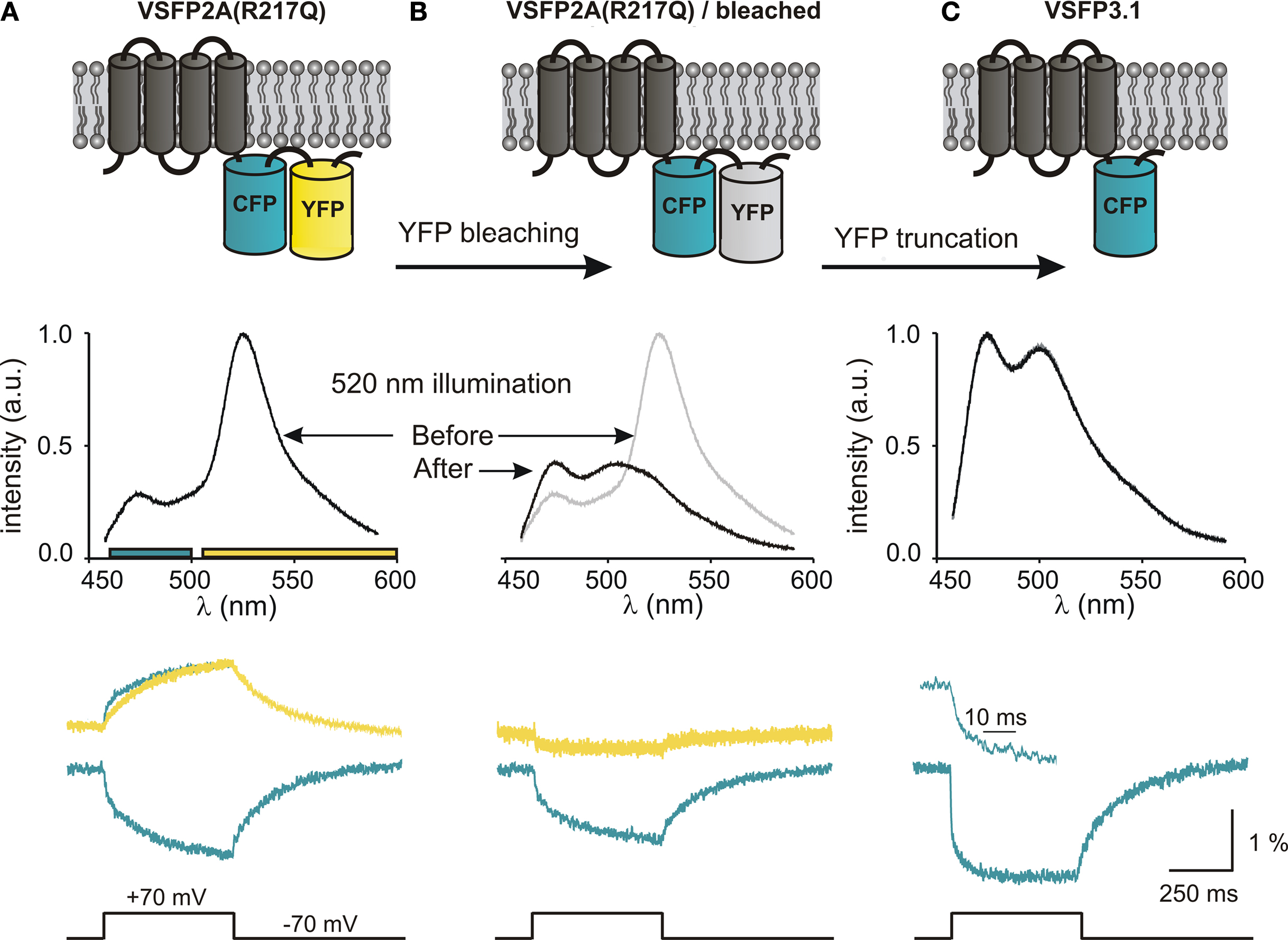
Figure 5. Development of third generation voltage-sensitive fluorescent proteins. The membrane topology of VSFP2A(R217Q) (A), VSFP2A(R217Q) after acceptor photobleaching (B) and VSFP3.1 (C) is illustrated in top panels. Emission spectra recorded for each constructs using 440-nm excitation light are shown below. The lower panel shows the depolarization-induced fluorescence signals recorded in the yellow and cyan channels. For VSFP2A(R217Q), a scaled mirror image of the cyan signal is shown aligned with the yellow signal; note the fast CFP component. For VSFP3.1, the top trace represents the onset of the fluorescence signal at a magnified time scale (from Lundby et al., 2008
).
As shown in Figure 6
, VSFP3.1 was efficiently targeted to the plasma membrane of transfected hippocampal neurons with a pattern similar to that of VSFP2.3 and VSFP2.4 (Figure 3
). We anticipate that VSFP3.1 and its color variants will facilitate improved optical measurements of fast neuronal signals.
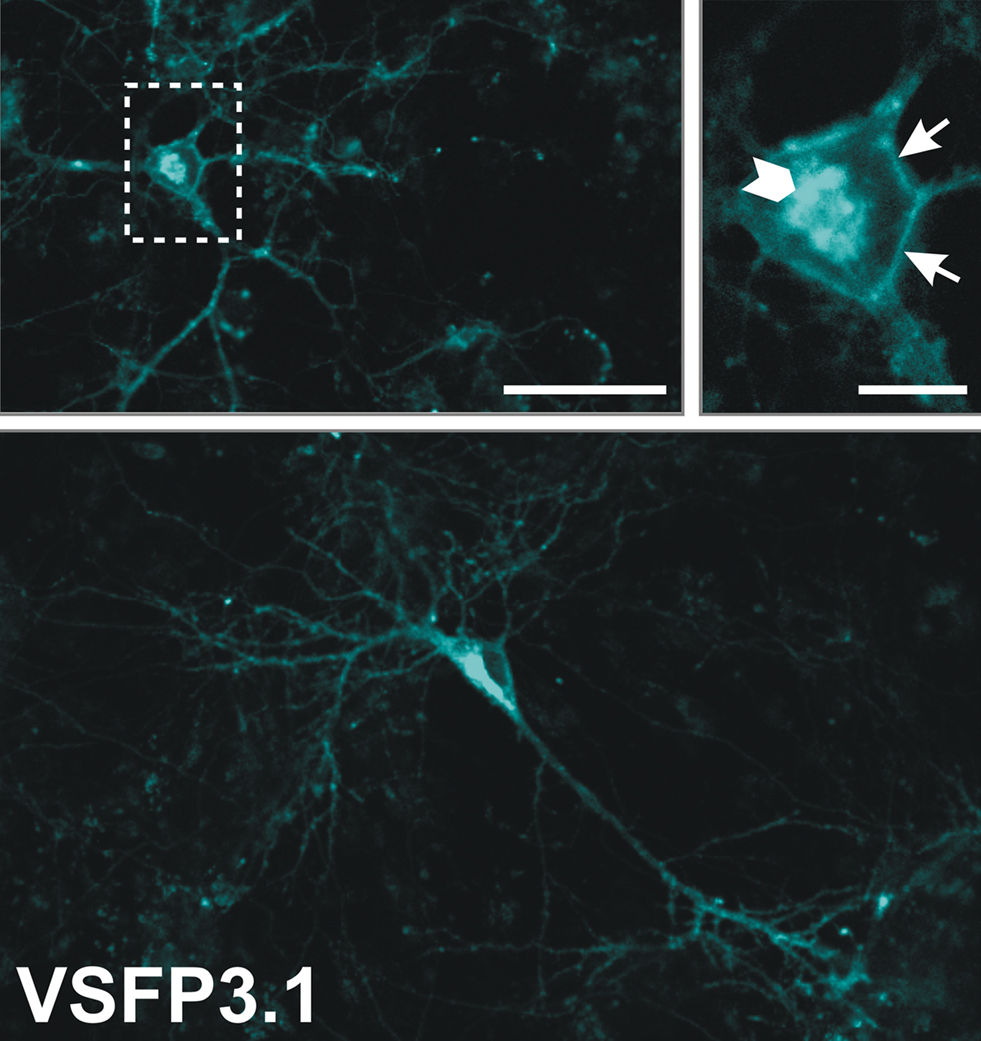
Figure 6. Expression pattern of VSFP3.1 in cultured mouse hippocampal neurons. Primary hippocampal cultures were transfected with VSFP3.1 6 days after plating and confocal images were taken 6 days later. Overviews and magnified views of VSFP3.1 expression in transfected hippocampal neurons are shown. The boxed region in the left panel is shown magnified on the right panel. Arrows indicate plasma membrane expression whereas the arrowhead shows intracellular expression. Scale bar is 50 μm (upper left and lower panel) and 10 μm (upper right).
In this review, we have focused on the most advanced genetically-encoded voltage sensors reported thus far based on a fusion between the voltage-sensing domain of Ci-VSP and a FP reporter module comprising either a fluorescent protein FRET pair or a single FP.
Despite comparable amplitude and kinetics of the voltage-dependent fluorescence response of the VSFP2.1 spectral variants described in this review, each of these fluorescent probes has particular advantages when considering potential applications. For instance, VSFP2.3 is a suitable sensor for instrumentation with standard optical components since it is based on the most commonly used FRET pair. On the other hand, the spectral properties of VSFP2.4 are strongly preferable for either in vivo imaging due to the elimination of green/yellow autofluorescence or deep tissue imaging using two-photon excitation fluorescence microscopy. Finally, Mermaid is, in principle, a good candidate for ratiometric measurements due to the good dynamic range of the donor and acceptor fluorescence responses. However, the tendency of the FPs used in Mermaid to form bright fluorescent aggregates may severely limit the usefulness of this variant.
To our knowledge, VSFP3.1 is the fastest FP voltage sensor reported to date, exhibiting an activation time constant matching that of fast neuronal signals, which makes this single-FP voltage sensor a promising candidate for the generation of transgenic animals. However, VSFP2-type sensors have practical advantages over single color variants of the VSFP3 class. Indeed, FRET-based voltage sensors provide signals at two different colors with opposite polarity. This feature enables ratiometric measurements and thereby, at least theoretically, absolute calibration of membrane voltage. Furthermore, ratiometric measurements are less sensitive to movement artifacts. Versions like VSFP2.3 that exhibit a large baseline FRET efficacy may also be used monochromatically by exciting CFP and recording YFP fluorescence, in which case the larger separation of excitation and emission wavelengths can have practical advantages over single color variants of the VSFP3 class.
Current work aims at characterizing the performance of these voltage-sensitive fluorescent protein probes in differentiated neurons, which will undeniably constitute an important step towards the realization of an optical sensor for neuronal circuit activity. Furthermore, expression of voltage-sensitive fluorescent proteins under cell-specific promoters will allow the labeling of defined neuronal populations within assemblies composed of diverse cell types (Diez-Garcia et al., 2005
, 2007
; Metzger et al., 2002
; Qiu et al., 2008
).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Thomas Launey for essential help with preparing neuronal primary cultures and all members of the Knöpfel laboratory for discussions and support. The work of this laboratory is supported by grants from RIKEN BSI (TK), the RIKEN BSI director’s fund (TK), a NIH grant NS057631 (under a sub award granted by Yale University to TK), the JSPS (Japanese Society for the Promotion of Science)-CIHR (Canadian Institutes of Health Research) postdoctoral fellowship program (AP), a grant-in-aid for JSPS fellows (AP, TK) and a grant-in-aid for young scientists from MEXT (Ministry of Education, Culture, Sports and Technology) (HM, TK).
Metzger, F., Repunte-Canonigo, V., Matsushita, S., Akemann, W., Diez-Garcia, J., Ho, C. S., Iwasato, T., Grandes, P., Itohara, S., Joho, R. H., and Knöpfel, T. (2002). Transgenic mice expressing a pH and Cl-sensing yellow-fluorescent protein under the control of a potassium channel promoter. Eur. J. Neurosci. 15, 40–50.