Physiology and Pharmacology Department, University of Bristol, Bristol, UK
Ion channels are the determinants of excitability; therefore, manipulation of their levels and properties provides an opportunity for the investigator to modulate neuronal and circuit function. There are a number of ways to suppress electrical activity in Drosophila neurons, for instance, over-expression of potassium channels (i.e. Shaker Kv1, Shaw Kv3, Kir2.1 and DORK) that are open at resting membrane potential. This will result in increased potassium efflux and membrane hyperpolarisation setting resting membrane potential below the threshold required to fire action potentials. Alternatively over-expression of other channels, pumps or co-transporters that result in a hyperpolarised membrane potential will also prevent firing. Lastly, neurons can be inactivated by, disrupting or reducing the level of functional voltage-gated sodium (Nav1 paralytic) or calcium (Cav2 cacophony) channels that mediate the depolarisation phase of action potentials. Similarly, strategies involving the opposite channel manipulation should allow net depolarisation and hyperexcitation in a given neuron. These changes in ion channel expression can be brought about by the versatile transgenic (i.e. Gal4/UAS based) systems available in Drosophila allowing fine temporal and spatial control of (channel) transgene expression. These systems are making it possible to electrically inactivate (or hyperexcite) any neuron or neural circuit in the fly brain, and much like an exquisite lesion experiment, potentially elucidate whatever interesting behaviour or phenotype each network mediates. These techniques are now being used in Drosophila to reprogram electrical activity of well-defined circuits and bring about robust and easily quantifiable changes in behaviour, allowing different models and hypotheses to be rapidly tested.
Drosophila with its 200,000 neuron brain displays a range of different behaviours such as learning, courtship, circadian rhythms, sleep, aggression and response to addictive drugs. Each of these behaviours has been mapped to well defined neural circuits with particular genes known to influence or control different aspects of each behaviour as determined by a powerful combination of genetic screens, promoter-based transgenic manipulations and studies of gene product expression (Baker et al., 2001
; McGuire et al., 2004
; Holmes et al., 2007
). In Drosophila, like any other animal, information flows through the nervous system via patterned changes in membrane depolarisation along a neuron, interspersed with synaptic transmission between neurons. Ion channels are the electrical components of the circuit controlling membrane depolarisation and synaptic currents. Information is processed and stored in a network via changes in synaptic strength and connectivity (synaptic plasticity) as well as changes in excitability in the rest of the neuron connecting input and output synapses (intrinsic plasticity). Drosophila has a similar range of interacting intrinsic and synaptic plasticity mechanisms, some pre- and some post-synaptic. These have mostly been studied at glutamatergic or cholinergic synapses with conserved roles for CaMKII, PKA, CREB, potassium (K+) channels and NMDA receptors in mechanisms of plasticity and learning between Drosophila and humans (Littleton and Ganetzky, 2000
; Giese et al., 2001
; Haghighi et al., 2003
; Rohrbough et al., 2003
; Gasque et al., 2006
; Wu et al., 2007
; Schmid et al., 2008
; Turrigiano, 2008
).
Approximately 350 genes encode ion channels in human (ca. 1–2% of the coding genome), with over 60 of these genes causing disease (channelopathies) when mutated (Ashcroft, 2006
; Cannon, 2006
). Drosophila has around 150 ion channels (most of which have been mutated), again about 1–2% of its coding genome. Drosophila channels are clearly related to the human channels, with flies often having one prototypic member of an ion channel family containing many exons that are alternatively spliced (for instance the voltage-gated Shaker K+ channel), while humans have many separate genes possibly through gene duplication of the prototypic channel gene (i.e. there are eight human Shaker genes called KCNA1-8 encoding channel proteins Kv1.1–Kv1.8). This makes it relatively trivial in flies to make an animal completely lacking a class of ion channels and then determining the functional consequence (Salkoff et al., 1992
; Littleton and Ganetzky, 2000
; Giese et al., 2001
; Wicher et al., 2001
).
Ways to Manipulate Ion Channel Expression to Silence Neurons
Since ion channels are the determinants of excitability, manipulation of their levels and properties allows one to modulate neuronal and circuit function. There are a number of ways to suppress electrical activity in neurons, for instance manipulating the expression of channels that regulate resting membrane potential (RMP) setting it below the threshold required to fire action potentials. This can be achieved in several ways (listed below, Table 1 and sections ‘Ion channel manipulations that have been used to electrically inactivate Drosophila neurons’ and ‘Gal4/UAS promoter system for broad spatial and temporal control of inactivation of neurons’) mainly involving over-expression of K+ channels that are open at RMP, causing increased K+ efflux and therefore membrane hyperpolarisation. Alternatively one can over-express channels or pumps that conduct chloride (Cl−) ions into the cell or over-express a sodium (Na+)/K+ ATPase co-transporter, again to hyperpolarise RMP and prevent action potential firing. Neurons can also be silenced by disrupting or reducing the level of functional voltage-gated Na+ or calcium (Ca++) channels that mediate the depolarisation phase of action potentials. Similarly, a strategy involving the opposite channel manipulation should allow net depolarisation and hyperexcitation in a given neuron (Wicher et al., 2001
; Wulff and Wisden, 2005
).
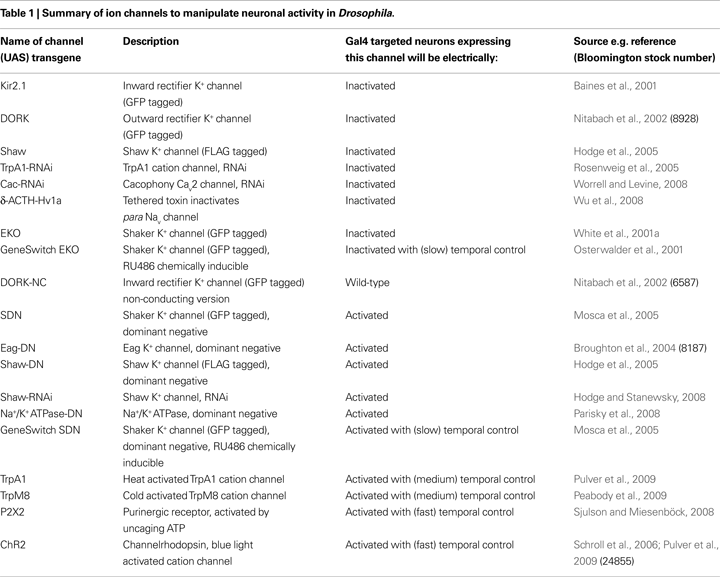
Combining these approaches with the versatile (Gal4/UAS) transgenic systems available in Drosophila allows one temporal and spatial control of (channel) transgene expression (McGuire, et al., 2004
). This now makes it possible to electrically silence (or hyperexcite) any neuron or neural circuit in the fly brain (Peabody et al., 2008
, 2009
; Gordon and Scott, 2009
), and much like an exquisite lesion experiment, potentially elucidate whatever interesting behaviour or phenotype each mediate. Likewise, this approach can determine if the expression, mechanism of action or structure of a molecule, pathway or process is regulated by electrical activity. This special issue on new genetic techniques revolutionising the study of neural circuits, should facilitate the transfer of technology and channels between model systems and help researchers wishing to formulate a theoretical or computational understanding of this research.
Homeostatic Plasticity Mechanisms
A key issue to consider when ion channels are expressed in order to silence neurons or reduce the activity of neurons, is will the resulting change in electrical activity induce homeostatic plasticity mechanisms that compensate and try to return the neuron back to its normal activity state. Put another way, neuronal networks have a tendency not to like to be silenced or, have extreme patterns of hyperexcitability imposed upon them. In fact, they may have evolved a diverse repertoire of homeostatic mechanisms to compensate for large net changes in synaptic and intrinsic excitability in order to preserve important network functions (Marder and Prinz, 2002
; Davis, 2006
; Turrigiano, 2008
).
Ion channel mutants (i.e. nulls or ‘knock outs’), although useful for determining the basic function of a channel, can be limited as tools for the spatial and temporal control of neural activity in a given circuit, as channels tend to be broadly expressed. Furthermore, removing a channel throughout development can induce compensatory mechanisms. An example from mouse is the GABAA subunits that mediate persistent inward Cl− currents important for tonic inhibition of cerebellar granule cells when knocked-out trigger a homeostatic mechanism resulting in up-regulation of leak TASK-1 K+ channels that preserve these particular neuron’s characteristic electrophysiological properties (Brickley et al., 2001
). Even single point mutations in Drosophila channels can result in a slew of transcriptional changes in activity dependent genes (Guan et al., 2005
). These genes presumably form an inherently robust genetic network with loss of any branch (gene) of the network being compensated by a complementary rearrangement of the remaining interacting branches (genes) maintaining functional output from the network (van Swinderen and Greenspan, 2005
).
Another example, this time from lobster, is when Shal (Kv4 or KCND) RNA is injected into neurons there is an increase in the fast transient IA current. However this change is also accompanied by a compensatory up-regulation of a hyperpolarisation activated Ih cation current in order to maintain the neuron’s firing behaviour. Interestingly expression of a non-conducting version of Shal also induced Ih up-regulation, suggesting that changes in channel protein level, as opposed to the accompanying changes in membrane activity, can be the trigger of this activity-independent homeostatic response. Not all changes in channel level are compensated, for instance the reciprocal increase in Ih channel was not compensated and did alter the neuron’s behaviour (MacClean et al., 2003
).
Loss-of-Function Mutations in Neuronal Na+ Or Ca2+ Channels
Removing the main depolarising channels from neurons is one strategy to inactivate Drosophila neurons. However these ion channels tend to have a number of specific roles and serve a range of pleiotropic functions making their presence necessary for viability; this is reflected in these channels having widespread tissue or developmental expression. For instance, Drosophila’s single member of the Cav2 family, the pan-neural expressed cacophony, when null mutated causes embryonic lethality, while partial loss of function alleles cause changes in synaptic transmission, increased convulsions and aberrant courtship and vision (Smith et al., 1998
; Wicher et al., 2001
). Likewise, null alleles of paralytic, the tetrodotoxin-sensitive Drosophila Nav1 are lethal, while partial loss of function or temperature sensitive-alleles (parats at restrictive temperature) have disrupted action potential propagation and paralysis (Suzuki and Wu, 1984
; Wicher et al., 2001
). Some point mutations may however, confer some spatial control of functional inactivation of neurons, for instance, Smellblind mutant alleles are thought to remove the olfactory specific splice form of para by specific exon-skipping events (Reenan et al., 1995
). Similar splicing defects of human Cav and Nav channel genes can cause channelopathies resulting from brain region- or tissue specific-compromised channel function with accompanying pathophysiology such as cerebellar ataxia, familial hemiplegic migraine and cardiac arrhythmias (Ashcroft, 2006
; Cannon, 2006
).
Gain-of-Function Mutations in Neuronal K+ Channels
K+ channels are the most diverse ion channel family, with over 30 such channels in Drosophila, therefore mutations are seldom lethal with the possibility of having adult flies lacking two major classes of K+ channel (Littleton and Ganetzky, 2000
; Vähäsöyrinki et al., 2006
). However K+ channel mutants, like other channel chromosomal mutants, maybe of limited use to suppress neuronal electrical activity, especially as unlike human or worm channel mutants, few are gain-of-function alleles required for electrical inactivation (Ashcroft, 2006
; Cardnell et al., 2006
). Increased repolarising current can also be achieved by mutants that contain a chromosomal rearrangement that result in duplicated copies of a K+ channel translocated onto an additional chromosome (Haugland and Wu, 1990
). Similar changes in channel gene dosage likely occur in Downs (Trisomy 21) syndrome resulting in changes in neural development, long-term potentiation (LTP) and learning in mouse models of Downs (Morice et al., 2008
). However all these approaches are limited in that they do not allow the experimenter both spatial and temporal control of electrical inactivation of just a chosen neural circuit.
The disadvantages of using mutants can be overcome by using selective over-expression strategies. The Gal4/UAS bipartite system has one part that consists of a fly containing a transgenic copy of the Gal4 transcription factor inserted into its genome. The expression of the integrated Gal4 transgene is dependent on whatever endogenous enhancers it is inserted next to (e.g. enhancer-trapping). The second half of the system is a fly containing a transgene of interest downstream of a Gal4 upstream activation sequence (UAS), which by itself: is not expressed. By crossing a fly containing a Gal4 insert with a fly containing a UAS-transgene, the resulting progeny will have both, therefore expressing your gene of interest in a given pattern of cells. Over the last 15 years, UAS-transgenes to over-express most of the genes in the fly genome have been collected (http://flybase.org
) some containing fluorescent tags, some point mutations and some homologues from other species such as human (Brand and Perrimon, 1993
; White et al., 2001b
). Recent inclusions to this arsenal include publicly available UAS-RNAi lines to all 14,000 genes in the fly genome (Dietzl et al., 2007
).
Married to these are collections of 10,000s of Gal4 lines that have been screened for expression in different tissues and times of development using UAS-GFP. Many Drosophila promoter sequences are relatively compact so can be placed before Gal4, allowing the resulting Gal4 insertion to report the expression of the original gene promoter. Gene promoters for neuropeptide, neurotransmitter synthetic enzymes and different channels and receptors have revealed their neural expression pattern and also allowed mis-expression in dopaminergic, cholinergic and GABAergic neurons (Holmes et al., 2007
). However the expression of Gal4 lines can be complex, for instance, there are many mushroom body (MB) Gal4 lines, however these seldom express in MB alone or just in the adult MB learning centre. This is because most genes are expressed in development and often in a number of tissues serving a number of functions (pleiotropic; Hall, 2005
). For instance Rdl, a Drosophila GABAA receptor subunit is expressed in the MB and is important for learning (Liu et al., 2007
), but is also expressed in clock neurons regulating sleep (Parisky et al., 2008
). This is probably not a coincidence as sleep and memory influence one another and might be co-regulated by molecules expressed in over-lapping circuits controlling both behaviours. The combination of well-defined neural circuits, robust behavioural assays and powerful molecular genetics of Drosophila is now revealing the molecular nature of the plasticity mechanisms connecting sleep and memory (Donlea et al., 2009
; Gilestro et al., 2009
). Such circuit-breaking studies are more difficult in the more complex mammalian brain as there are many more cells of each type, a paucity of well-defined promoters, and neuronal groups (for instance GABAergic neurons) are often dispersed between brain structures (Wulff and Wisden, 2005
).
A number of variations of the Gal4 promoter and similar systems are continually being developed in Drosophila (McGuire et al., 2004
) to increase spatial and temporal control of transgene expression. For instance, Gal4 expression has been made to be dependent on the presence of an activator, a progesterone analogue called RU486. The resulting system called GeneSwitch makes flies only express Gal4 and hence transgenes such as Shaker when they are placed on RU486 containing food, thereby potentially bypassing any problems arising from lethality, developmental defects and compensation through long term (developmental) changes in excitability (Osterwalder et al., 2001
). It is not exactly known why over-expressing K+ channels sometimes causes apoptosis, however it maybe due to the steady depletion of K+ from the cell (Nadeau et al., 2000
). Likewise in order to circumvent lethality sometimes resulting from expression of K+ channel transgenes, such as Kir2.1, the TARGET system can be employed (McGuire et al., 2004
). At 18°C Gal80ts inhibits Gal4 activity blocking Kir2.1 expression and lethality. Whereas keeping the flies at 31°C inactivates Gal80ts releasing Gal4 driven Kir2.1. This technique allowed the critical window in development to be probed where electrical inactivation of a specific group of neurons resulted in lethality (Peabody et al., 2008
).
In the following sections I will catalogue in detail these inactivation strategies that have been successfully used with the Gal4/UAS system.
Human (KCNJ2/Kir2.1) Channels
A pioneering study by Baines et al. (2001)
wished to tease apart the relationship between intrinsic excitability and synaptic strength. In order to study the effect of electrical inactivation of fly neurons on synaptic transmission, the authors expressed an N-terminal GFP tagged human inwardly rectifying Kir2.1 channel. This channel had been used previously (Johns et al., 1999
), to genetically suppress excitability of rat neurons in culture using an inducible promoter this time responsive for the Drosophila hormone, ecdysone. Gal4 promoters with different distributions of pre- and post-synaptic expression at defined synapses in the embryonic fly brain were used. Kir2.1 expression in the postsynaptic neuron (a motorneuron) removed evoked but not spontaneous release of neurotransmitter. No compensatory changes were seen. The same approach of developmental cell specific expression of Kir2.1 has also been used to determine the role of neural activity in generating the mouse olfactory sensory map (Yu et al., 2004
). Also hyperpolarising Kir2.1 expression in hippocampal neurons was found to decrease firing rate, but this time in these mammalian neurons a homeostatic mechanism returned firing rate to normal even though Kir2.1 continued to be expressed (Burrone et al., 2002
).
At the Drosophila larval neuromuscular junction (NMJ) synapse (Paradis et al., 2001
), muscle expression of Kir2.1 resulted in an outward (∼10nA) leak current accompanied by 10–15mV hyperpolarised shifts in muscle RMP. Despite this reduction in postsynaptic excitability, synaptic transmission was maintained by a compensatory increase in presynaptic release (quantal content), implicating an activity-dependent retrograde homeostatic mechanism.
Expression of the Kir2.1 transgene in insulin secreting cells of the fly was then used in order to help characterise a Drosophila model of diabetes (Kim and Rulifson, 2004
). Drosophila has homologues of both the sulphonylurea receptor (SUR1 β-cell KATP channel β-subunit), and the KATP channel α-subunit (Kir6.2 or KCNJ11) it confers glucose sensitivity to. Sulphonylurea disrupted glucose stimulated insulin release in Drosophila by a mechanism involving SUR and Kir channels. Expression of the non-ATP sensitive human Kir2.1 in the Drosophila insulin secreting neurons prevented membrane depolarisation and release from these neurons resulting in flies with disrupted control of circulating glucose (Kim and Rulifson, 2004
; Ashcroft, 2006
).
In addition to learning and memory, another function prescribed to the MB and cAMP signalling within these cells is control of sleep. MB electrical inactivation using promoters that express Kir2.1 in these cells led to increased daily sleep while manipulations thought to increase excitability lead to a reduction in sleep (Joiner et al., 2006
). It should be noted expression of Kir2.1 has the most extreme functional consequences of all the K+ channel transgenes, for instance, Kir2.1 expression in as little as 30–40 crustacean cardioactive peptide (CCAP) neurons is sufficient to cause lethality (Luan et al., 2006
). So what are the longer-term consequences of electrical inactivation of different neurons using this approach, expression of Kir2.1 in a defined type of serotonin neuron, resulted in morphological changes in the neuron’s neurites (Roy et al., 2007
).
Lastly, in order to study the post-mating switch in behaviour of female flies (decrease in receptivity to courting males and increased egg laying), the Jan lab used Kir2.1 based techniques. The switch in behaviour is brought about by sex peptide that is transferred with the male’s sperm into the female’s reproductive organ where it acts on fruitless and pickpocket (ppk, a Drosophila Na+ channel) neurons. Expression of Kir2.1 in ppk sensory neurons increased the post-mating change in behaviour, suggesting sex peptide normally inhibits neuronal transmission in these circuits. Clonal analysis using flies expressing GFP-Kir2.1 in ppk neurons downstream of FRT sites; showed that the virgins that displayed the most post-mating responses where the one’s that had the most Kir2.1 expression in the ppk neurons on the uterus and which projected axons towards the flies brain (Yang et al., 2009
).
Drosophila Shaker EKO (KCNA/Kv1) Channels
Possibly the earliest attempt to electrically inactivate neurons using K+ channel over-expression (Gisselmann et al., 1989
), used a heat shock promoter to express either a Shaker cDNA or a dominant negative truncated version of Shaker (W404 to stop in the pore of the channel). Because a functional Shaker channel consists of four pore-forming α-subunits, the presence of one or more pore-truncated subunits would block the remaining pore intact endogenous Shaker subunits removing channel function. Heat-shock of the full-length or truncated Shaker over-expressing flies caused the predicted hypo- or hyper-excitability at the electrophysiological (NMJ recordings) and behavioural (ether-induced shaking) level.
In order to direct the inactivation of defined neurons, the Gal4/UAS was used to express Shaker channels that were genetically modified to have a more hyperpolarised voltage-dependence of activation, no N-terminal fast inactivation and an N-terminal GFP tag. This ‘electrical knockout channel’ or EKO allowed sustained activation in response to depolarisation suppressing electrical activity of the cells in which it was expressed. Pre or post-synaptic expression of EKO caused an increase in the sustained K+ current, RMP hyperpolarisation and reduced firing. These changes were partially reversed by the Shaker channel blocker, 4-AP. No homeostatic changes were reported although the level of endogenous Shaker transient IA current was reduced in muscles over-expressing EKO (White et al., 2001a
,b
).
As for Kir2.1 channels, EKO expression throughout the nervous system or musculature caused some paralysis and impaired locomotion. These effects increased with EKO gene dosage (it is possible to have multiple copies of UAS-EKO in a fly) resulting in increased embryonic lethality. Reduction in neuronal excitability with EKO over-expression caused some aberrant NMJ connectivity and in the eye a dose-dependent decrease in photoreceptor potential, again partially blocked by 4-AP. A307-Gal4 expression of EKO strongly in the ventral and abdominal ganglion of the fly resulted in adult flies with unexpanded wings and abnormally pigmented thorax. This developmental phenotype was suggested to be due to incomplete adult eclosion (insects divide their life into embryonic, larval, pupal and adult stages, completion of the later stage is called eclosion which involves wing expansion) requiring neuronal release of eclosion hormones (White et al., 2001a
,b
). Later studies have elegantly dissected the role of excitability in control of wing expansion by release of the hormone bursicon from CCAP neurons, three copies of EKO was found to be sufficient to block release of bursicon (Luan et al., 2006
).
A similar approach was then used to increase excitability by expression of an N-terminal GFP-tagged Shaker dominant negative (SDN) subunit truncated after the first transmembrane domain (Mosca et al., 2005
). As predicted SDN caused hyperexcitability associated with reduced IA, enhanced synaptic transmission, enhanced synapse size and increased larval locomotion. In adults, hyperexcitable phenotypes included ether-induced shaking. Serendipitously the identical Shaker mutation was picked up in a forward genetic screen for flies with reduced sleep and called minisleep. This is a mutant that sleeps one-third as much as wild-type flies (Cirelli, et al., 2005
). Electrical inactivation of MB neurons with EKO resulted in flies with a modest increase in sleep (Joiner et al., 2006
).
Neuron specific expression of EKO can therefore be used to decrease intrinsic excitability, while expression of SDN can be used to increase excitability in neurons that endogenously express Shaker channels. Postembryonic expression of either transgene in motorneurons caused increased dendritic outgrowth (Duch et al., 2008
). This study also showed that SDN changed the firing patterns of a motorneuron upon current injection, while EKO decreased firing but did not completely silence neurons. Interestingly, none of the transgenic manipulations effected RMP, input resistance or synaptic properties. Increases in flight behaviour were seen in the SDN flies and a decrease in flies expressing EKO in their motorneurons.
Drosophila ORK (KCNK/TASK) Channels
Drosophila open rectifier K+ channels (DORK) exhibit no voltage or time dependence of opening, essentially forming K+ selective holes in the membranes of the cells they are expressed. A C-terminally GFP tagged DORK transgene (DORK) was expressed in flies using the GAL4/UAS system, and an additional mutant non-conducting version of the channel (DORK-NC) was also expressed as a control (Nitabach et al., 2002
). Unlike non-conducting versions of lobster Shal (MacClean et al., 2003
), no homeostatic changes were triggered. However the trend of DORK-NC overexpression as the sole choice of wild-type control for over-expression of different transgenic channels should also be performed with caution, due to the increasing number of non-conducting functions of ion channels being discovered (Kaczmarek, 2006
).
Not surprisingly, pan-neuronal expression of DORK like Kir2.1 caused embryonic lethality. In order to determine the effect of electrical inactivation on a well-defined behaviour: DORK or Kir2.1 were expressed in a subset of clock neurons (∼20 neurons) called the lateral neurons ventral (LNv) using Pigment dispersing factor (pdf-Gal4) promoter (Nitabach et al., 2002
). These form part of the ∼150 neurons that form the fly clock. Circadian rhythms are generated by the molecular clock in each clock neuron which consists of rhythmically expressed clock genes (i.e. timeless and period) that feedback and control their own expression with an ∼24hour period (Hall, 2005
).
It was claimed that DORK or Kir2.1 expression electrically silenced the clock neurons. Chronic silencing of neurons with such channels can be associated with apoptosis (Nadeau et al., 2000
), however Pigment dispersing factor (PDF) staining of the ‘silenced’ LNv was qualitatively shown not to grossly disrupt their viability or morphology. In constant darkness, ‘silencing’ the LNv resulted in a higher proportion of flies showing arrhythmic or weakly rhythmic behaviour with respect to wild-type controls. LNv expression of DORK-NC resulted in some arrhythmic behaviour but otherwise appeared normal. The rhythmic expression of clock proteins using semi-quantification of antibody stain intensity on fixed brains was dampened in neurons expressing DORK or Kir2.1. The interruption of these results was that electrical silencing of the LNv had stopped the free-running circadian clock. Less dramatic effects were reported for the effect of these transgenic manipulations under normal lighting conditions (12 hours of light, 12 hours dark) (Nitabach et al., 2002
). Expression throughout the clock circuit using timeless-Gal4 resulted in adult arrhythmia in constant darkness and disrupted the molecular clock as judged by timeless staining of the larval LNv (Nitabach et al., 2005
). Later studies showed that electrical inactivation of LNv clock neurons had similar circadian molecular and behavioural effects as pdf mutants and that the molecular clock oscillations in remaining clock neurons were not abolished (Wu et al., 2008a
).
It is now thought that synchronisation of the rhythms between clock neurons and the circadian output from the clock that regulates rhythmic behaviour are mediated by, both chemical (predominantly PDF) and electrical signals (Hall, 2005
; Holmes et al., 2007
). Earlier mammalian suprachiamatic nucleus (SCN) recordings (Kuhlman and McMahon, 2004
) and fly large (l)-LNv (Park and Griffith, 2005
; Cao and Nitabach, 2008
; Sheeba et al., 2008a
,b
; Wu et al., 2008a
) recordings revealed clock neurons display circadian changes in their electrical properties including a (∼10mV) depolarised shift in RMP during the day accompanied by an increase in frequency of spontaneous action potentials (Brown and Piggins, 2007
; Holmes et al., 2007
).
In order to extend the DORK behavioural genetic studies, whole-cell recordings were performed on l-LNv neurons expressing DORK this caused a 10mV hyperpolarising shift in LNv RMP and reduced but did not silence evoked action potential firing (Park and Griffith, 2005
). The chromosomal location of a transgene can also effect expression and hence have functional effects, for instance the pdf-Gal4 expression of C1 insertion of DORK results in most flies being arrhythmic while the C2 insertion causes most flies to be weakly arrhythmic with Kir2.1 causing the greatest arrhythmicity (Nitabach et al., 2002
). A later study confirmed pdf-Gal4 expression of DORK-C2 caused a significant hyperpolarisation of LNv RMP; that was greater for DORK-C1 and greatest for Kir2.1. This resulted in over 60% of flies expressing either Kir2.1 or DORK C1 displaying arrhythmia in constant darkness while over-expression of the hyperpolarising DORK-C2 now gave the same level of arrhythmicity as control (∼20%; Wu et al., 2008a
).
Lastly, DORK expression has been used to explore plasticity mechanisms at fly synapses. It is widely thought that neurons can detect their level of activity via a range of calcium sensitive sensors prominent amongst which is CaMKII which autophosphorylates at T287 during LTP and T306 during long term depression (LTD), both events are thought to be important for learning in a range of animals including flies (Marder and Prinz, 2002
; Davis, 2006
; Nelson and Turrigiano, 2008
). Electrical inactivation of postsynaptic cells using DORK caused an increase in synaptic phosphorylation of T306 and concomitant reduction in T287, these changes were regulated by the PDZ-scaffolding molecule, CASK (Lu et al., 2003
; Hodge et al., 2006
). Activated CaMKII is known to directly bind or modulate a number of K+ channels and glutamate receptors regulating neuronal excitability in a range of systems (Griffith et al., 1994
; Park et al., 2001
; Yao and Wu, 2001
; Haghighi et al., 2003
; Sun et al., 2004
; Nelson et al., 2005
; Gasque et al., 2006
), while CASK also interacts and changes the activity of a number of synaptic ion channels and receptors (Hsueh, 2006
).
Drosophila Shaw (KCNC/Kv3) Channels
Drosophila Shaw is a member of the Kv channel family and encodes a slowly activating and non-inactivating K+ current. Shaw is open at normal RMP and causes hyperpolarisation by K+ efflux; the channel is widely expressed in the nervous system and helps regulate RMP in Drosophila central neurons (Salkoff et al., 1992
; Tsunoda and Salkoff, 1995
; Hodge et al., 2005
; Parisky et al., 2008
). Gal4 over-expression of full-length Shaw was detected via a C terminal FLAG tag on the transgenic channel. A Flag tagged dominant negative form of Shaw was also made (Hodge et al., 2005
) with the analogous pore truncation as the heat shock Shaker dominant negative transgene described previously (Gisselmann et al., 1989
). This subunit behaved in a dominant negative fashion causing ∼10mV depolarising shift in RMP and a doubling of spike frequency. This hyperexcitation did not lead to any homeostatic changes in the remaining K+ currents. Like EKO expression, widespread neuronal expression of Shaw resulted in developmental lethality and wing expansion deficits consistent with the transgene causing electrical inactivation of the neurons it is expressed in. This wing expansion phenotype was mapped to the 30–40 CCAP positive neurons. Widespread expression of dominant negative Shaw caused hyperexcitable phenotypes such as ether-induced shaking and wing expansion phenotype. Endogenous Shaw was expressed widely in the nervous system including CCAP neurons (Hodge et al., 2005
).
Because RMP-hyperpolarising DORK over-expression in clock neurons caused behavioural arrhythmicity (Nitabach et al., 2002
, 2005
), we wished to explore which channels might endogenously regulate clock neuron RMP. We started with Shaw as this channel is widely expressed in the Drosophila brain and regulates neuronal RMP (Hodge et al., 2005
). Furthermore mammalian homologues of Shaw, Kv3, are widely expressed in SCN and the magnitude of their current varies between day and night and even in constant darkness. Blocking the currents prevented the daily rhythm in firing of SCN neurons (Itri et al., 2005
). Using a C-terminal antibody, Shaw was found widely expressed in the adult brain including a subset of clock neurons. In normal light conditions expression of membrane hyperpolarising Shaw in all clock neurons (timeless-Gal4) increased locomotor activity at night. Under constant conditions, electrical inactivation of all clock neurons resulted in extreme arrhythmia as was also reported for expression of Kir2.1 or DORK. This had little effect on the molecular clock as measured by per-luciferase oscillations in the dorsal clock neurons. The rhythmic accumulation of PDF in terminals of small LNv neurons was however disrupted by changing the level of Shaw throughout the clock. Hyperpolarising Shaw over-expression caused PDF accumulation in terminals while reduction in functional Shaw depleted the levels of the neuropeptide in terminals. Hyperexciting the clock by tim-Gal4 expression of dominant negative Shaw also caused weaker rhythms (Hodge and Stanewsky, 2008
).
As mentioned over-expression of GABAA mediated inward Cl− currents would inactivate neurons with GABAergic input and has been shown to inhibit Drosophila olfactory associative learning (Liu et al., 2007
). In order to determine the role of GABA mediated regulation of sleep, Shaw was expressed in GABAergic neurons using glutamic acid decarboxylase (GAD)-Gal4, in order to decrease GABA release. This resulted in a reduction in the initiation and maintenance of sleep (Agosto et al., 2008
). Likewise reduction of expression of GABAA receptors using RNAi in just the LNv neurons caused a similar reduction in sleep. Electrical inactivation of LNv using EKO also caused increased initiation and maintenance of sleep. In order to hyperactivate the LNvs in a physiologically relevant manner, Shaw-RNAi or dominant negative Na+/K+-ATPase was over-expressed. Both gave an ∼10mV depolarised shift of RMP accompanied by increased firing of action potential, resulting in a reciprocal decreased initiation and maintenance of sleep. These results together suggest the LNvs are important for light-induced arousal (Parisky et al., 2008
).
Targeted Reduction in Depolarising Currents in Order to Inactivate Drosophila Neurons
As Na+ and Ca2+ channels gene products are large and do not tetramerise to form a function channel, dominant negative strategies targeting these channels are therefore more problematic. RNAi expression has proved affective at reducing Drosophila TrpA1 resulting in flies that do not show normal avoidance of elevated temperatures (Rosenweig et al., 2005
). RNAi has also been used for targeted reduction of Cav2 currents, thereby decreasing evoked and spontaneous neuronal activity by a similar amount as hypomorphic cacophony mutants (Worrell and Levine, 2008
). With genome-wide collections of Gal4 driven RNAi lines now available (Dietzl et al., 2007
), one can expect this approach to be used increasingly to target Drosophila ion channels in inactivate neurons. Another new technology tested using the Drosophila circadian system, is cell autonomous expression of membrane-tethered toxins. Pdf-Gal4 expression of δ-ACTH-Hv1a inhibited the para Nav channel inactivation and induced rhythmic action potential firing. This resulted in PDF accumulating in LNv terminals earlier and flies starting to become active earlier (anticipating) before lights on (Wu et al., 2008b
).
Eag-DN, Narrow Abdomen, Trp-M, Trp-A and NaChBac Transgenic Channels to Change Drosophila Neuron Activity
An alternative Gal4 dominant negative K+ channel strategy used an eag transgene truncated before the first transmembrane segment (eag-Dominant Negative). This was shown to increase excitability (Duch et al., 2008
) and has been used to study courtship in flies (Broughton et al., 2004
).
Another study exploring the relationship between the molecular clock and electrical activity used a range of mutations in Narrow Abdomen, a NALCN/ Na+ non-selective leak channel endogenously expressed in the clock. Mutants are expected to hyperpolarise clock neuron RMP and caused arrhythmia in constant darkness, however as for Shaw expression, this activity manipulation did not disrupt the molecular clock but did change output from the clock via elevated PDF levels in LNv terminals (Lear et al., 2005
).
In order to achieve finer temporal control of electrical activity, the heat-sensitive Drosophila TrpA1 channel has been used acutely increase LNv activity with temperature shifts sufficient to disrupt sleep (Parisky et al., 2008
). Likewise, the cold and menthol sensitive rat TrpM8 channel has been expressed in CCAP neurons imposing a rapid cold-sensitive switch in network activity controlling wing expansion (Peabody et al., 2009
).
Flies have also been made that express a voltage-gated bacterial Na+ channel (NaChBac). Expression in CCAP neurons resulted in the wing expansion phenotype (Luan et al., 2006
). pdf-Gal4 expression of NaChBac was found to cause hyper-excitation of LNvs in terms of causing massive increases in action potential amplitude and duration. However at the same time NaChBac expression was found to cause strong hypoexcitation of LNvs with RMP going from a wild-type value of −41.5 to −103mV with an accompanying drop in firing frequency. These changes in LNv excitability caused by NaChBac expression were not homeostatically compensated (Sheeba et al., 2008a
,b
). In addition, NaChBac LNv terminal PDF levels remain high at night. This resulted in subsets of clock neurons becoming desynchronised causing the generation of complex behavioural rhythms (splitting) (Nitabach et al., 2006
), similar effects were seen with hyperpolarising expression of Shaw in some clock neurons (Hodge and Stanewsky, 2008
). Likewise, l-LNv NacBac expression caused the normal day-night shift in excitability to be reversed, so that the flies were more active at night (Sheeba et al., 2008b
). A mosaic technique allowed single cell manipulation of electrical activity. It was found the more l-LNvs neurons expressing NaChBac the more nocturnal the fly’s behaviour became (Shang et al., 2008
).
A current technology set to revolutionise the study of neural activity in circuits is the use of light-activated channels. Expression of the microbial channelorhodopsin using Gal4 has been used to depolarise and activate Drosophila neurons upon blue light stimulation. Likewise UV-uncaging of ATP has been used to cell autonomously stimulate rat P2X2 receptors expressed in Drosophila neural circuits. Conversely light stimulation of inhibitory channels, such as the Natronomonas pharaonis (NpHR) halorhodopsin chloride pump with yellow light has also been demonstrated (Zhang et al., 2007
; Sjulson and Miesenböck, 2008
). Generation of Gal4 inducible NpHR flies holds great promise for acute electrical inactivation of neural circuits in flies, however as it is a pump, it might be expected to have slower kinetics and different trafficking than ChR2. However, such Gal4 driven approaches would allow acute bi-directional changes in activation of Drosophila neurons that may bypass some of the confounding problems that can be caused by chronic channel activity mentioned earlier in this review.
Therefore these channels have the promise to provide millisecond control of membrane potential and neuronal spike firing (with activated TRP channels allowing longer stimulation; Pulver et al., 2009
) and allow robust remote control of different fly behaviours. For instance, blue-light photoexcitation of ChR2 expressed in acj6 (abnormal chemosensory jump 6 transcription factor) neurons was able to elicit the innate escape response of flies (Zimmermann et al., 2009
). Furthermore, light induced activation of ChR2 expressed in larval dopaminergic neurons when paired with a specific odour could induce aversive memory formation, while activation of octopaminergic/tyraminergic neurons with another odour could induce positively reinforced memories (Schroll et al., 2006
). Likewise, UV uncaging of ATP activated P2X2 receptors expressed in giant fibre system elicited the predicted escape behaviours of jumping, wing beating and flight. While expression in dopamine neurons caused increased locomotion (Lima and Miesenböck, 2005
). Finally, photo-triggering P2X2 expressed in fruitless neurons that form part of the neural circuit that helps generate the male courtship song, caused flies of either sex to perform the courtship song (Clyne and Miesenböck, 2008
).
Generally it should be noted that many of the ion channel manipulations described in this review did not cause any obvious compensatory changes, suggesting that Drosophila neurons and circuits may contain fewer compensatory mechanisms than mammalian ones. However in order to bypass any potential compensatory problems caused by chronic channel expression, pharmacological modulation of ectopically expressed transgenic channel could be added, allowing further temporal control of electrical activity to the investigator (Wulff and Wisden, 2005
; Wulff et al., 2007
). Likewise synthetic photoisomerisable small molecules have now been used to specifically target and inactivate endogenous Shaker channels causing their inactivation and acute changes in neural activity in rodent and leech preparations (Fortin et al., 2008
). In order to get round any unwanted potential leaky expression of UAS channel transgenes independent of Gal4, it maybe possible to use new tools (retrovirus insulators and phiC31 integration) to minimise positional effects of host chromatin and target integration of transgenes to defined sites known not to be leaky. These technologies should help standardise reverse genetics in Drosophila by improving leakiness and allowing robust transgene expression at a defined level (Markstein et al., 2008
).
So what else can we hope for in the future? Certainly, bi directional translation of these new technologies between model organisms and also into new organisms that offer unique experimental features (Baker et al., 2001
; Marder, 2002
). Early examples of this trend include the fly’s phototransduction pathway namely arrestin, rhodopsin G-protein coupled receptor (GPCR) and Gqα collectively termed chARGe being used to allow light triggered excitation of hippocampal neurons (Zemelman et al., 2002
). Likewise, ectopic expression of the Drosophila allatostatin GPCR which couples to a Gi/o to open inwardly rectifying K+ channel (GIRK) can be used to cause a neuronal hyperpolarisation and resulting decrease in spiking on addition of allatostatin and has been used to investigate mouse spinal cord networks in vivo (Gosgnach et al., 2008
; Zhang et al., 2008
).
Also, as an increasing number of CNS disorders are found to involve abnormal neural activity of circuits (Saper et al., 2005
; Ashcroft, 2006
; Cannon, 2006
; Beck and Yaari, 2008
), modelling these diseases in a range of model organisms should in future elucidate the fundamental properties of these disorders at the molecular, electrophysiological and behavioural level (Mackay and Anholt, 2006
; Zhang et al., 2007
; Morice et al., 2008
; Song and Tanouye, 2008
). These model systems together have the potential to allow screening or validation of new candidate genes, drug targets, and therapies using small molecules, biologicals or even gene therapies using transgenic channels to repair or rescue functional output (Herlitze and Landmesser, 2007
).
Researchers championing the different technologies described in this review can now think about reprogramming electrical activity in well-defined circuits and bring about predictable changes in behaviour. This approach can be used in a manner somewhat analogous to dynamic clamp (Prinz et al., 2004
) except it is optically controlled, allowing the researcher to add or remove different currents to a circuit, testing theoretical models or replaying different activity patterns to try and instruct different changes in behaviour.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
I am grateful for the collaborative work performed with Drs Leslie Griffith, Ralf Stanewsky and Cahir O’Kane that allowed my entry into this field. My research has been supported by an EMBO short-term fellowship (337-2006), FP7 MIRG-CT-2007-200632, Royal Society (RG0870156) and BBSRC grant (BB/G008973/1). Thanks to Dr Leslie Griffith, Jack Mellor, Maria Usowicz and Benjamin White for useful comments on earlier versions of this manuscript.
Nitabach, M. N., Wu, Y., Sheeba, V., Lemon, W. C., Strumbos, J., Zelensky, P. K., Benjamin, B. H., and Holmes, T. C. (2006). Electrical hyperexcitation of lateral ventral pacemaker neurons desynchronizes downstream circadian oscillators in the fly circadian circuit and induces multiple behavioural periods. J. Neurosci. 26, 479–489.
Wulff, P., Goetz, T., Leppä, E., Linden, A. M., Renzi, M., Swinny, J. D., Vekovischeva, O. Y., Sieghart, W., Somogyi, P., Korpi, E. R., Farrant, M., and Wisden, W. (2007). From synapse to behaviour: rapid modulation of defined neuronal types with engineered GABAA receptors. Nat. Neurosci. 10, 923–929.