1
INSERM U952, Paris, France
2
CNRS UMR 7224, Paris, France
3
Université Pierre et Marie Curie, Paris, France
4
Fachbereich Biologie, Chemie, Pharmazie, Freie Universität Berlin, Berlin, Germany
5
RNA Editing and Hyperexcitability Disorders, Max Delbrück Center for Molecular Medicine, Berlin-Buch, Germany
6
Developmental Neurobiology, Max Delbrück Center for Molecular Medicine, Berlin-Buch, Germany
Information processing in the brain requires a delicate balance between excitation and inhibition. Glycine receptors (GlyR) are involved in inhibitory mechanisms mainly at a synaptic level, but potential novel roles for these receptors recently emerged due to the discovery of posttranscriptional processing. GLR transcripts are edited through enzymatic modification of a single nucleotide leading to amino acid substitution within the neurotransmitter binding domain. RNA editing produces gain-of-function receptors well suited for generation and maintenance of tonic inhibition of neuronal excitability. As neuronal activity deprivation in early stages of development or in epileptic tissue is detrimental to neurons and because RNA editing of GlyR is up-regulated in temporal lobe epilepsy patients with a severe course of disease a pathophysiological role of these receptors emerges. This review contains a state-of-the-art discussion of (patho)physiological implications of GlyR RNA editing.
Glycine receptors (GlyRs) are pentameric chloride (Cl−) channels. They contribute to inhibition of neuron firing in brain. Expression of GlyR genes (GLRA1-4) produces four different α subunits that are capable of forming functional, homomeric channels (Betz and Laube, 2006
; Lynch, 2009
), while a single gene (GLRB) delivers neurons with GlyR β subunit mRNA. Besides modulation of ligand binding (Grudzinska et al., 2005
) the GlyR β subunit mediates receptor stabilization at postsynaptic sites (Kirsch et al., 1991
; Meyer et al., 1995
; Meier et al., 2000
, 2001
; Meier and Grantyn, 2004
). GlyR genes are located on several chromosomes in humans, with GLRA1 on chromosome 5 (5q32), GLRA2 on chromosome X (Xp22.1-p21.3) and GLRA3 as well as GLRB on chromosome 4 (4q33-q34 and 4q31.3, respectively). GLRA4 is a pseudogene (Simon et al., 2004
) located on the X-chromosome, position Xq22.2. Expression of all other genes occurs in a wide range of brain regions (Lynch, 2009
), and generally it is thought that GlyR α2 expression predominates in the juvenile brain and declines with development (Lynch, 2009
). However, at least in retina (Haverkamp et al., 2004
) and in the hippocampus (Eichler et al., 2008
) GlyR α2 expression persists throughout development.
Through RNA editing the genetically encoded information can be modified. Enzymatic deamination of adenosine and cytidine is mediated by adenosine deaminases acting on RNA (ADAR) and apolipoprotein B mRNA editing complex (APOBEC) or activation induced deaminase (AID), respectively (Anant and Davidson, 2001
; Seeburg and Hartner, 2003
; Honjo et al., 2005
). The resulting inosine (equivalent to guanosine) or uracil may then lead to amino acid substitutions in corresponding proteins, provided that the resulting amino acid triplet codons engage different transfer RNAs. These enzyme machineries thus allow for diversification of the proteome or even correction of the genome code. Although the role of diversification by cytidine-to-uracil (C-to-U) RNA editing in generation of immunoglobulin variability is under debate (Honjo et al., 2005
) the critical impact of RNA editing on correction of genome codes is well established. For example, the permeability of glutamate receptors for calcium ions increases when adenosine-to-inosine (A-to-I) RNA editing is prohibited, resulting in severe epilepsy (Brusa et al., 1995
). Therefore in this case, the A-to-I RNA editing machinery is required for maintenance of physiological brain state through correction of unwanted genome codes (Seeburg and Hartner, 2003
). The number of identified RNA-edited transcripts steadily increases, and for example serotonin receptors, potassium channels, GABA and glycine receptors were recently included in this register (Gurevich et al., 2002
; Bhalla et al., 2004
; Buckingham et al., 2005
; Meier et al., 2005
).
A cDNA clone corresponding to GlyR α3185L was originally isolated from Sprague Dawley rat brain, and molecular analysis revealed the involvement of C-to-U RNA editing in proline-to-leucine substitution at position 185 of the mature GlyR α3 protein (Meier et al., 2005
). Molecular analysis of hippocampi from pharmacoresistant temporal lobe epilepsy (TLE) patients further revealed expression of mRNAs coding for the high affinity GlyR α3185L variant (Eichler et al., 2008
). In addition, GLRA2 transcripts of GlyR α2192L were found in these patients. Sequencing of corresponding exons demonstrated lack of genomic α2192L codons, again supporting the involvement of C-to-U RNA editing in proline-to-leucine substitution in TLE patients (Eichler et al., 2008
). However, the amount of RNA-edited GLR transcripts was not constant between TLE patients, but increased according to the frequency of secondarily generalized tonic-clonic seizures or the degree of hippocampal sclerosis. That hippocampal sclerosis was associated with increased amounts of RNA-edited GLRA2/3 transcripts indicates a pathophysiological role of high affinity GlyRs in human hippocampus, which already was suggested by our data on experimentally induced brain lesion (Meier et al., 2005
). Consequently, in TLE patients without hippocampal sclerosis the amount of RNA-edited GLR transcripts was very low, particularly in case of GlyR α3 (below 1% of GLRA3 transcripts). This renders quantification of RNA-edited transcripts rather difficult (Nakae et al., 2008
). However, our methods for quantification were recently shown to be suitable for detection of RNA-edited messenger fractions below 1% (Eichler et al., 2008
).
Leucine at Positions 192 (α2) and 185 (α1 and α3) is Responsible for High Apparent GlyR Agonist Affinity
GlyRs are members of the ligand-gated ion channel (LGIC) superfamily, which among others comprises A- and C-type GABA receptors (GABAA/CR) and the nicotinic acetylcholine receptor (nAChR). All members share common structural features, particularly in the ligand binding domain (Celie et al., 2005
). In fact, all GlyR subunits and GABAAR β1-3 contain proline at the position corresponding to amino acid 185 of the mature signal-peptide cleaved α3 protein.
However, GABAARs that require a high agonist affinity because of their involvement in tonic inhibition of neuronal excitability don’t have proline at this position. Instead, GABAARs containing α4, α6 or δ subunits are equipped with aliphatic hydrophobic neutral amino acids (such as alanine, leucine or valine) (Meier et al., 2005
), and these receptors are well known to respond to low neuronal ambient GABA with tonic chloride currents (Mody, 2001
; Wisden et al., 2002
; Caraiscos et al., 2004
; Mody and Pearce, 2004
). Therefore, amino acid substitution at this position of the ligand binding domain should increase the apparent GlyR agonist affinity. Indeed, proline-to-leucine substitution by C-to-U editing of GLRA3 transcripts was shown to increase the apparent agonist affinity (Meier et al., 2005
). Similarly, editing of GLRA2 transcripts at the corresponding position (192) produces receptors with increased apparent agonist affinity (Eichler et al., 2008
).
To further emphasize the critical role of position 185/192 in determining apparent receptor agonist affinity dose-response curves were obtained from HEK293 cells expressing GlyR α1185P or α1185L (Figure 1
). The short GlyR α1 splice variant lacking insert SPMLNLFQ (Malosio et al., 1991
) was used throughout. Again, compared with GlyR α1185P, the apparent affinity of α1185L channels was increased several fold [EC50(Glycine) (μM) α1185L: 17.7 ± 1.5 vs. α1185P: 85.3 ± 4.4, EC50(Taurine) (μM) α1185L: 67.0 ± 6. 8 vs. α1185P: 378 ± 33, mean ± SD].
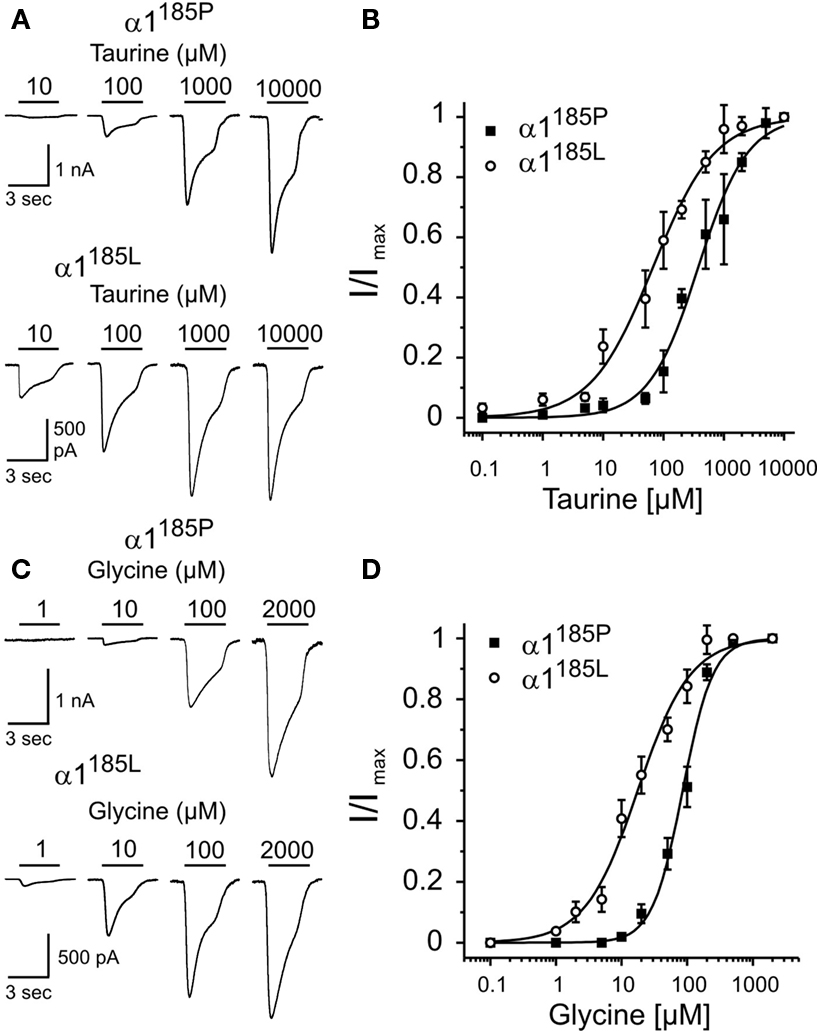
Figure 1. Apparent agonist affinities of GlyR α1185P and α1185L. Dose-response curves were obtained from transfected HEK293 cells. (A,C) Example traces of Cl− currents obtained at indicated agonist concentrations. At −70 mV holding potential, maximal current responses (α1185P, taurine: 3.3 ± 0.3 nA, glycine: 2.5 ± 0.3 nA; α1185L, taurine: 1.7 ± 0.3 nA, glycine: 1.6 ± 0.3 nA) were obtained with 2 mM glycine and 10 mM taurine. Taurine (B) and glycine (D) dose-response curves of HEK293 cells expressing GlyR α1185P or α1185L are shown. Taurine and glycine current amplitudes were normalized to maximal current amplitudes. Each data point (B,D) represents the average current amplitude of 13–21 sampled cells. Hill coefficients: α1185P, taurine: 1.06 ± 0.09, glycine: 1.88 ± 0.17; α1185L, taurine: 0.84 ± 0.07, glycine: 1.08 ± 0.09. Values represent mean ± SD.
It should be noted here that the apparent affinity of GlyRs does not seem to be influenced by RNA splicing because GlyR α1 with insertion (α1ins) (Malosio et al., 1991
) exhibited similar current profiles (not shown). In addition, compared with α3K, the long splice variant of α3-GlyRs (α3L) (Nikolic et al., 1998
) also had increased apparent glycine affinity when RNA-edited (Figure 2
).
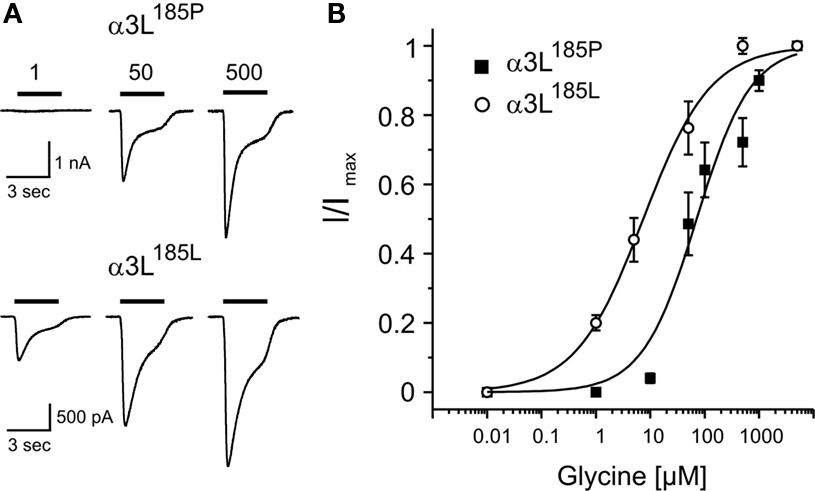
Figure 2. Apparent agonist affinities of GlyR α3L185P and α3L185L. Dose-response curves were obtained from transfected HEK293 cells. (A) Example traces of Cl− currents obtained at indicated agonist concentrations. (B) Glycine dose-response curves of HEK293 cells expressing GlyR α3L185P or α3L185L are shown. Glycine current amplitudes were normalized to maximal current amplitudes. Each data point represents the average current amplitude of 13–21 sampled cells. At −70 mV holding potential, the apparent affinities for glycine (EC50(Glycine) [μM]) were 70.9 ± 16.1 (α3L185P) and 7.4 ± 0.9 (α3L185L). Hill coefficients were 0.87 ± 0.17 (α3L185P) and 0.70 ± 0.06 (α3L185L). Thus, the long splice variant of RNA-edited GlyR α3L185L is a high affinity receptor as well. Values represent mean ± SD.
Altogether, proline-to-leucine substitution at positions 192 (α2) and 185 (α1 and α3) confers high apparent agonist affinities to GlyRs. On the contrary, GlyR apparent agonist affinity is not regulated by RNA splicing, except for GlyR α2 where changes in the apparent affinity were observed upon RNA splicing in the region coding for the N-terminal domain (Miller et al., 2004
; Eichler et al., 2008
).
RNA editing involves a single amino acid substitution in the N-terminal GlyR domain. The main consequence of RNA editing is an increase in the apparent affinities (EC50) of GlyRs for glycine and taurine. RNA editing does not modify GlyR desensitization kinetics (Meier et al., 2005
), which is consistent with previous studies showing that receptor desensitization kinetics are mainly affected by change in amino acid sequences in the TM3-4 loop (Legendre, 2001
; Breitinger and Becker, 2002
; Breitinger et al., 2002
). A single amino acid substitution in the N-terminal GlyR domain at positions 192 (α2) or 185 (α1 and α3) could result in a change in the transduction process between agonist binding and channel gating, but it cannot be excluded at this stage that proline-to-leucine substitution modifies the conformation of the N-terminal domain and hence the access of the ligand to the binding pocket. Changes in EC50 values do therefore not necessarily imply changes in the affinity of the receptor for its agonist. Effectively, in the simplest case, the EC50 depends both on the agonist dissociation constant (Kd) and on the receptor-channel efficacy (E), and EC50 = Kd/(1 + E) [for a more detailed explanation see (Colquhoun, 1998
; Legendre, 2001
)]. Several models have been proposed to describe GlyR activation kinetics, e.g. the “reluctant gating model” (Legendre, 1998
) and the “flip model” (Burzomato et al., 2004
). The flip model was recently extended to nAChR (Lape et al., 2008
), although it has partially been challenged recently (Mukhtasimova et al., 2009
). In any case, these studies demonstrate that receptor channels can have intermediate conformational states between binding steps and opening states, which must be taken into account when interpreting changes in EC50. Accordingly, the best way to determine how GlyR RNA editing influences their apparent glycine affinity will be to compare activation kinetics of regular and RNA-edited GlyRs at the single channel level, as was already done in case of the hyperekplexia amino acid substitution α1K276E (Lewis et al., 1998
), the hyperekplexia amino acid substitution α1Q266H (Moorhouse et al., 1999
) and the spasmodic amino acid substitution α1A52S (Plested et al., 2007
). Nonetheless, the net outcome of proline-to-leucine substitution is a gain-of-function GlyR.
The crystal structure of the acetylcholine binding protein (AChBP) was recently determined at high resolution (Celie et al., 2005
). Because members of the LGIC superfamily have a common ancestor (Celie et al., 2005
) we have used the AChBP structure as a model for the ligand binding domain of GlyR. At least from the point of view of intramolecular location of β-sheets a projection of GlyR sequences on AChBP protein structure can be made (Grenningloh et al., 1987
). According to these thoughts proline 185/192 of GlyR α subunits is located directly at the beginning of β-sheet 9 (Figure 3
, β9), a position that is close to the plasma membrane where these receptors enter the cytosol through transmembrane domain (TM) 1 (Figure 3
, C-terminus). The glycine binding pocket (Figure 3
) is formed by two distant amino acid groups (Legendre, 2001
), namely GY (160 and 161, respectively) and KYT (200, 202 and 204, respectively). These two domains are separated by β-sheets 7 and 10, and prolines 185/192 are part of the β8/β9-flanked connecting F-loop (Figure 3
).
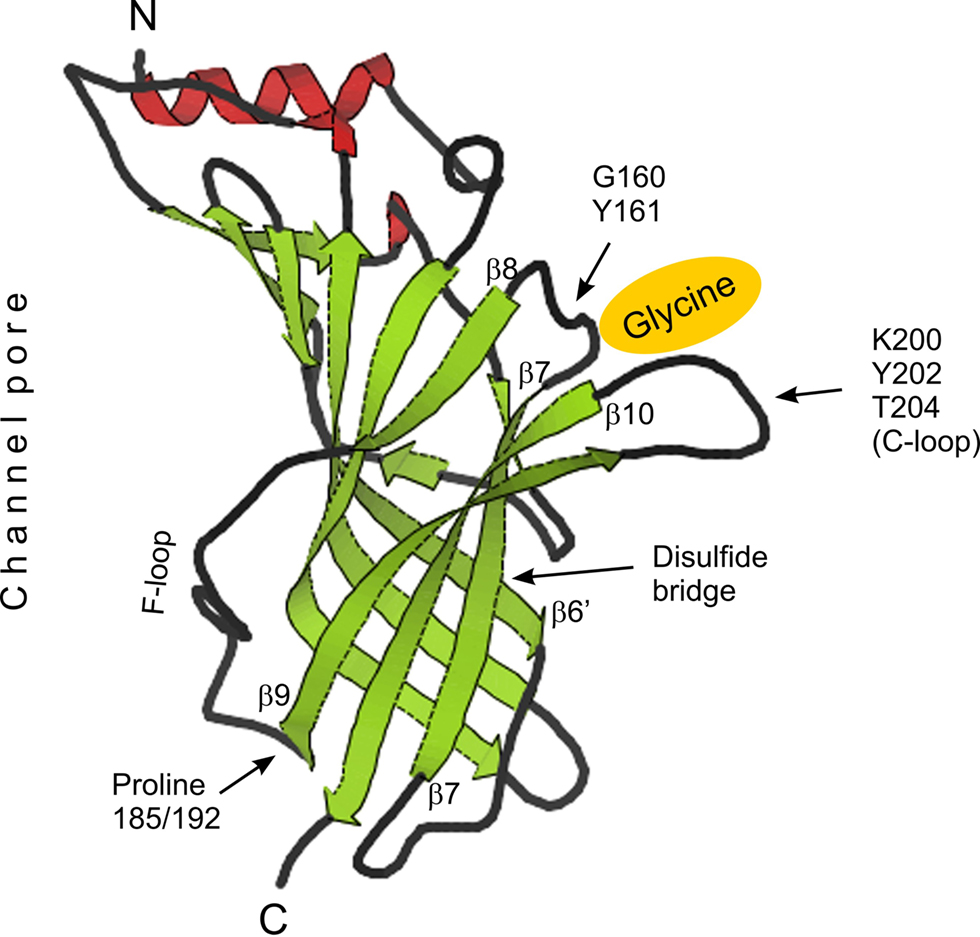
Figure 3. Tertiary structure of AChBP and projection of GlyR α subunit associated structural determinants. Positions of the glycine (yellow) binding pocket, formed by amino acids GY and KYT (C-loop), of the disulfide bridge between β-sheets 6′ and 7 as well as of proline residues at positions 185 (α1 and α3) and 192 (α2) are shown. N- and C-termini and the location of the chloride channel pore are indicated. β-sheets, α-helices and coils are color-coded (green, red and black, respectively). GlyR sequence was projected on the structure view of AChBP downloaded from www.pdb.org
. The position of β-sheets served as reference points.
Further pursuing these considerations we have arguments that support a conformational role of proline-to-leucine substitution. First, leucine residues have bifurcated non-polar side chains that can mediate interaction with membrane lipids, while proline is frequently found in regions where the protein backbone has to make a sharp turn. In fact, increasing local hydrophobicity at position 250 (located between TM1 and TM2 close to the intracellular side of the plasma membrane) was shown to impact on receptor apparent affinity (Breitinger et al., 2001
; Breitinger and Becker, 2002
). In this case however, increased hydrophobicity decreased apparent GlyR affinity and favored rapid desensitization. Second, the disulfide bridge that connects β-sheets 6’ and 7 (Figure 3
) could serve as a hinge (Laube et al., 1993
), potentially transmitting conformational rearrangement by proline-to-leucine substitution to the glycine binding pocket. Third, the glycine binding sequence KYT is located within the C-loop (Figure 3
), which was shown to undergo conformational change upon AChR agonist binding (Unwin et al., 2002
), and fourth, F-loop dynamics are involved in benzodiazepine mediated GABAAR activation (Padgett and Lummis, 2008
). More recently, the F-loop was involved in sensing conformational rearrangements upon ligand docking (Pless and Lynch, 2009
). Therefore, we put forward the idea that structural rearrangement by proline-to-leucine substitution will impact on the accessibility of GlyR agonists to the C-loop binding pocket.
Because proline-to-leucine substitution can cause structural rearrangement and therefore impact on accessibility of GlyR agonists to the C-loop binding pocket we have considered possible GABA responsiveness of RNA-edited GlyRs. Non-edited GlyRs can be activated by GABA at concentrations in the millimolar range (De Saint et al., 2001
), which apparently precluded any physiological role for GABA on GlyRs. However, an elegant study recently demonstrated the opposite. Lu and colleagues (Lu et al., 2008
) show that co-release of GABA and glycine accelerates glycinergic transmission by acting directly on GlyRs. This effect is explained by competition of GABA and glycine with the same binding site. The deactivation phase time constant of a synaptic event reflects the duration of bursts of channel openings (Legendre, 2001
). Since burst duration partly depends on the dissociation rate constant of the agonist, a weak partial agonist will evoke bursts of short duration, which will in turn result in a decrease in the deactivation time constant of the synaptic response. It is tempting to speculate that postsynaptic GlyRs facing GABAergic nerve endings as observed in some CNS area such as hippocampus, and brain stem and spinal cord during development, could be activated by synaptic GABA release (Geiman et al., 2002
; Levi et al., 2004
; Meier and Grantyn, 2004
; Muller et al., 2006
). However, the concentration of GABA, released into the synaptic clef, must be high enough (>1 mM) to activate GlyRs. This limitation can be overcome as GlyRs acquire a higher agonist affinity if RNA-edited (Meier et al., 2005
; Eichler et al., 2008
). As GABA is a week competitive partial agonist on GlyRs it is likely that an increase in the apparent affinity for glycine will also result in an increase in the apparent affinity for GABA. This is the case (Figure 4
).
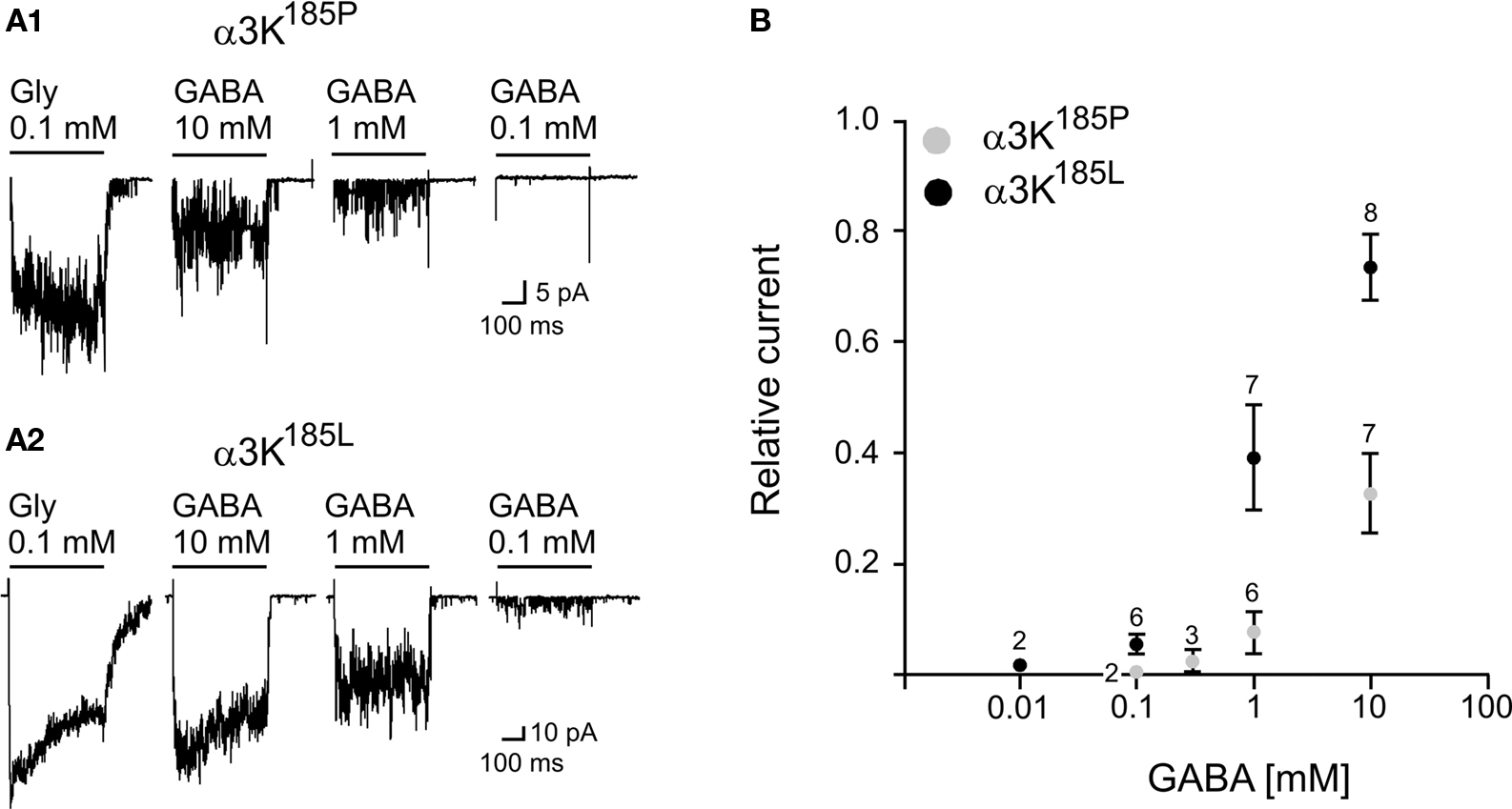
Figure 4. Activation of GlyR α3K185P and GlyR α3K185L by GABA applications. (A1) Example traces of outside-out currents evoked by activation of GlyR α3K185P in response to the application of glycine (0.1 mM) and various concentrations of GABA (Vh = −50 mV; Filter 1 kHz). (A2) Example traces of outside-out currents evoked by activation of GlyR α3K185L in response to the application of glycine (0.1 mM) and various concentrations of GABA (Vh = −50 mV; Filter 1 kHz). (B) Concentration-response curves for GABA-evoked currents on outside-out patches containing GlyR α3K185P (○) or GlyR α3K185L (●). Measurements were performed on averaged traces. The amplitude of GABA-evoked currents was normalized to the peak amplitude of the current evoked by the application of 0.1 mM glycine (relative current). Numbers above data points indicate the number of measurements per point (mean ± SD).
As shown in Figure 4
the threshold for GlyR activation by GABA is >0.3 mM in case of GlyR α3K185P, whereas it is close to 0.1 mM for RNA-edited GlyR α3K185L. Interestingly, application of 10 mM GABA (Figure 4
B) on outside-out patches containing GlyR α3K185L evoked responses with amplitudes representing ∼80% of current amplitudes obtained with saturating glycine concentration (0.1 mM) (Meier et al., 2005
). We did not test the effect of GABA concentrations above 10 mM, but according to the amplitude of GABA-evoked responses relative to current amplitudes obtained with 0.1 mM glycine, the apparent affinity of GlyR α3K185L for GABA (EC50) is in the range of 2−3 mM (Figure 4
B).
At GABAergic synapses the peak concentration of GABA molecules released per vesicle is likely to range from 0.5 to 1 mM (Jones and Westbrook, 1995
). Accordingly, GlyR α3185L could be partially activated if located vis-à-vis GABAergic nerve endings. In fact, we have shown recently that GlyR α3K preferentially associates with GABAergic hippocampal nerve endings (Eichler et al., 2009
). However, it still remains to be shown that RNA-edited α3K behaves similarly.
In addition to these findings, recent analysis of α3-GlyR distribution revealed an unexpected location of the long splice variant of these receptors (α3L) adjacent to glutamatergic nerve endings in hippocampal principle cell layers in adult mice (Eichler et al., 2009
). It therefore is conceivable that activation of GlyRs can evoke local chloride currents at glutamatergic synapses, which will reduce the efficacy of these synapses. However, these GlyRs must be activated at the level of glutamatergic postsynaptic domains. Presence of glycine transporter 1 at hippocampal glutamatergic synapses (Cubelos et al., 2005
) together with the observation of postsynaptic glutamatergic responses with recurrent small strychnine-sensitive components (Müller et al., 2007
) can support this hypothesis, particularly in case of RNA-edited receptors (Meier et al., 2005
; Eichler et al., 2008
). Again, it remains to be shown whether RNA editing impacts on the subcellular distribution of these receptors.
It is now well established that GlyRs are highly mobile entities of the neuronal plasma membrane (Meier et al., 2001
; Dahan et al., 2003
), which implies that receptors have a certain probability to be encountered at non-synaptic locations.
In the hippocampus, functional glycinergic synapses have not been described, and the majority of α3-GlyR immunoreactivity indeed is found at non-synaptic places (Eichler et al., 2009
). Furthermore, it has recently been shown that glycine uptake can regulate hippocampal network activity via GlyR-mediated non-synaptic tonic inhibition (Zhang et al., 2008
), and the source of extracellular glycine was suggested to be glial cells (Zhang et al., 2008
). A continuous activation of non-synaptic receptors requires a sufficient amount of extracellular glycine or taurine. However, in the central nervous system, the concentration of glycine is tightly regulated by glycine transporters (Gomeza et al., 2003
), and the residual glycine concentration in the extracellular space is likely to be close to 0.1 μM in normal conditions (Roux and Supplisson, 2000
).
Extracellular glycine can increase temporarily in case of sustained presynaptic activity (Roux and Supplisson, 2000
; Supplisson and Roux, 2002
), as for example in the epileptic tissue where hyper-synchronous high frequency network activity occurs (Fisher et al., 1992
; Bragin et al., 2007
). Accordingly, this might have important functional consequences, because non-synaptic slightly desensitizing GlyRs could be continuously activated as a result of temporarily altered GlyT activity (e.g. reverse transport) or even due to glycine spillover. Remarkably, the embryonic form of GlyRs, which is composed of five α2 subunits and expressed before synaptogenesis displays functional properties adapted to non-synaptic release of the agonist (Mangin et al., 2003
). Therefore, it can sustain a long lasting activation state when the extracellular glycine concentration increases above its activation threshold (Mangin et al., 2003
).
Irrespective of the subunit, remarkable features of RNA-edited GlyRs are their high apparent affinities for glycine (Meier et al., 2005
; Eichler et al., 2008
). Effectively, these receptors can be activated by very low glycine concentrations. In case of GlyR α3185L, 300 nM glycine already is sufficient for receptor activation, which in turn results in the occurrence of single channel openings. Independently of the chloride equilibrium potential, the activation of several GlyR α3185L can evoke an inhibition of the neuronal activity. Effectively, the opening of several channels can lead to a decrease in the cell input resistance, which will reduce the propagation of synaptic events and of action potentials, resulting in an inhibitory shunting process (Eichler et al., 2008
).
Although GlyRs were reported to have anticonvulsant effects (Kirchner et al., 2003
; Song et al., 2006
) results obtained more recently (Eichler et al., 2008
) point to a pathophysiological role of enhanced GlyR-mediated signaling, particularly in the context of high chloride equilibrium potential as is the case in TLE (Palma et al., 2006
). Under these circumstances, neuron silencing triggers up-regulation of the ratio between glutamatergic and GABAergic nerve terminals and is associated with abnormal dendrite morphology (Lohmann et al., 2005
; Eichler et al., 2008
), both of which phenomena are reminiscent of the TLE histopathology (Loup et al., 2000
; Stief et al., 2007
). Most importantly, silenced hippocampal neurons were prone to die due to glutamate excitotoxicity, but amongst others could be rescued by expression of the potassium chloride cotransporter type 2 (KCC2) (Eichler et al., 2008
). KCC2 expression provides neurons with an efficient chloride extrusion mechanism that decreases their chloride equilibrium potential (Lee et al., 2005
). Although the mechanisms underlying this neuroprotective effect of KCC2 remained obscure our results support the previously postulated developmentally dichotomous effects of single cell activity deprivation on neuron integrity within an otherwise active neuronal network (Tao and Poo, 2005
).
Furthermore considering altered GLRA3 splicing in the hippocampus of TLE patients, favouring association of α3-GlyRs with hippocampal GABAergic synapses, the net outcome of anomalous posttranscriptional GLRA3 processing is predicted to add to the pathophysiology of TLE. In a worst case scenario, provided that the chloride equilibrium potential is high enough (Rivera et al., 2004
; Palma et al., 2006
; Eichler et al., 2008
), up-regulation of RNA-edited α3K-GlyRs (associated preferentially with GABAergic synapses) accompanied by down-regulation of RNA-edited α3L-GlyRs (associated preferentially with glutamatergic synapses) could increase glycinergic signalling at depolarizing GABAergic synapses while reducing inhibitory effects on glutamatergic postsynaptic currents.
In any case, it is necessary to prove the existence of RNA-edited GlyR protein. However, this figured out to be rather difficult. Because of the position of leucine 185/192 at the interior of the channel pore close to the plasma membrane antibody accessibility is predicted to be low. Indeed, a potentially GlyR α3185L-specific antibody obtained by immunization of guinea pigs with peptide EGLTLLQFLLK (185L, underlined and bold) and cross-absorption against peptide EGLTLPQFLLK (185P, underlined and bold) was not able to visualize RNA-edited GlyR α3185L in transfected HEK293 (not shown). Even variation of fixation protocols and reagents did not ameliorate cellular protein detection. Therefore, it will be necessary to isolate a particular single channel conductance specific to RNA-edited GlyRs or any other differentiating factor in order to validate existence of these proteins in neuronal tissue. It is conceivable to screen ligand databases for the presence of agonists or antagonists specific to RNA-edited GlyRs. So far, however, poor availability of reliable chloride indicators applicable to living cells was a rather rate limiting step in identification of novel ligands.
RNA-edited GlyRs may open avenues to medication of excitability disorders (Eichler and Meier, 2008
) because they could provide researchers and clinicians with a battery of new therapeutically relevant drugs, yet these enigmatic receptors still have to be captured at the protein level of investigation. In particular, a possible cell-type specific hippocampal expression has to be determined.
Whole-Cell Patch-Clamp Recordings
Whole-cell patch-clamp recordings of HEK293 cells were performed 2–3 days after Ca2+-phosphate transfection with α1-GlyR constructs. For electrophysiological recordings, the growth medium was replaced with a bath solution containing (in mM): 145 NaCl, 2.5 KCl, 20 HEPES, 10 glucose, 2 CaCl2, 1 MgCl2, adjusted to pH 7.3. All recordings were carried out at room temperature using patch-clamp amplifier EPC-9 (HEKA Elektronik, Lambrecht, Germany). Patch electrodes were fabricated from borosilicate glass capillaries tubing using a P-97 puller (Sutter Instruments, Novato, CA, USA). The pipette solution contained (in mM): 4 NaCl, 130 KCl, 5 EGTA, 10 HEPES, 10 glucose, 0.5 CaCl2, 4 MgCl2, adjusted to pH 7.3. The pipette-to-bath DC resistance of patch electrodes ranged from 2–4 MOhm. Recordings were performed at a holding potential of −70 mV. Glycine or taurine was locally applied for 3 s via a gravity-driven 5-channel superfusion system. An interval of at least 60 s was left between successive agonist concentrations to allow for recovery from receptor desensitization. Different agonist concentrations were applied randomly. Electrophysiological signals were sampled at a rate of 5 kHz, filtered at 1 kHz and analyzed off-line using software TIDA 5.1 (HEKA Electronics, Lambrecht, Germany). To determine the EC50 value for agonists, peak currents obtained with different glycine or taurine concentrations were plotted and fitted to the Hill equation using software Origin 7.0 (OriginLab, Northampton, USA).
Outside-Out Patch-Clamp Recordings
Outside-out patch-clamp recordings of HEK293 cells were performed 2 days after Ca2+-phosphate transfection with α3K185P or α3K185L GlyR constructs. Cells were continuously perfused at room temperature (20–22°C) with bathing solution (2 ml/min) containing (in mM): NaCl 147, KCl 2.4, CaCl2 2, MgCl2 2, HEPES 10, glucose 10 (pH 7.2, osmolarity 320 mOsm). Patch-clamp electrodes (5–10 MΩ) were pulled from thick-wall borosilicate glass (Harvard Apparatus, Kent, UK) using a Brown-Flaming puller (Sutter Instrument Co., Navato, USA). They were fire-polished and filled with (in mM): CsCl 130, MgCl2 4, Na2ATP 4, EGTA 10, HEPES 10 (pH 7.2, osmolarity 290 mOsm). Currents were recorded using an Axopatch 1D amplifier (Axon Instruments, Foster City, USA). Recordings were filtered at 10 kHz using an eight-pole bessel filter (Frequency Devices, Haverhill, USA), sampled at 50 kHz and stored on a PC computer using pClamp software 10.1. (Axon Instruments, Foster City, USA). The membrane potential was held at −50 mV throughout the experiment. Outside-out single-channel currents were evoked using a fast-flow operating system (Legendre, 1998
). Control and drug solution were gravity-fed into two channels of a thin-wall glass theta tube (2-mm outer diameter; Hilgenberg, Malsfeld, Germany) pulled and broken to obtain a 200-μm tip diameter. The outside-out patch was positioned 100 μm away from the glass theta tube, one lumen of the application pipette was connected to reservoirs filled with solutions containing glycine or GABA. The solution exchange was performed by rapidly moving the solution interface across the tip of the patch pipette, using a piezo-electric translator (model P245.30, Physik Instrument, Waldbronn, Germany). Concentration steps of glycine or GABA were applied with an interval of ≥10 s. Exchange time solution (<100 μs) was determined before each set of experiments by monitoring the change in the liquid junction potential evoked by the application of a 10%-diluted control solution to the open tip of the patch pipette.
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
This study was supported by the DFG (ME2075/3-1 to JCM and RA424/5-1 to RJ, and Sonderforschungsbereich SFB-TR3 B5 to Jochen C. Meier) and the Helmholtz Association (VH-NG-246 to Jochen C. Meier), and Institut National de la Santé Et de la Recherche Médicale (INSERM to Pascal Legendre), and Centre de la Recherché Scientifique (CNRS to Pascal Legendre).
Caraiscos, V. B., Elliott, E. M., You, T., Cheng, V. Y., Belelli, D., Newell, J. G., Jackson, M. F., Lambert, J. J., Rosahl, T. W., Wafford, K. A., MacDonald, J. F., and Orser, B. A. (2004). Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by alpha5 subunit-containing gamma-aminobutyric acid type A receptors. Proc. Natl. Acad. Sci. U.S.A 101, 3662–3667.
Celie, P. H., Klaassen, R. V., Rossum-Fikkert, S. E., van Elk, R., van Nierop, P., Smit, A. B., and Sixma, T. K. (2005). Crystal structure of AChBP from Bulinus truncatus reveals the conserved structural scaffold and sites of variation in nicotinic acetylcholine receptors. J. Biol. Chem. 280, 26457–26466.
Eichler, S. A., Kirischuk, S., Jüttner, R., Schäfermeier, P. K., Legendre, P., Lehmann, T. N., Gloveli, T., Grantyn, R., and Meier, J. C. (2008). Glycinergic tonic inhibition of hippocampal neurons with depolarising GABAergic transmission elicits histopathological signs of temporal lobe epilepsy. J. Cell. Mol. Med. 12, 2848–2866.
Nikolic, Z., Laube, B., Weber, R. G., Lichter, P., Kioschis, P., Poustka, A., Mulhardt, C., and Becker, C. M. (1998). The human glycine receptor subunit alpha3. Glra3 gene structure, chromosomal localization, and functional characterization of alternative transcripts. J. Biol. Chem. 273, 19708–19714.
Palma, E., Amici, M., Sobrero, F., Spinelli, G., Di, A. S., Ragozzino, D., Mascia, A., Scoppetta, C., Esposito, V., Miledi, R., and Eusebi, F. (2006). Anomalous levels of Cl− transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc. Natl. Acad. Sci. U.S.A 103, 8465–8468.
Wisden, W., Cope, D., Klausberger, T., Hauer, B., Sinkkonen, S. T., Tretter, V., Lujan, R., Jones, A., Korpi, E. R., Mody, I., Sieghart, W., and Somogyi, P. (2002). Ectopic expression of the GABA(A) receptor alpha6 subunit in hippocampal pyramidal neurons produces extrasynaptic receptors and an increased tonic inhibition. Neuropharmacology 43, 530–549.