Department of Biology, University of Miami, Coral Gables, FL, USA
The zebrafi sh glial glycine transporter 1 (GlyT1) mutant provides an animal model in which homeostatic plasticity at glycinergic synapses restores rhythmic motor behaviors. GlyT1 mutants, initially paralyzed by the build-up of the inhibitory neurotransmitter glycine, stage a gradual recovery that is associated with reductions in the strength of evoked glycinergic responses.Gradual motor recovery suggests sequential compensatory mechanisms that culminate in the down-regulation of the neuronal glycine receptor. However, how motor recovery is initiated and how other forms of plasticity contribute to behavioral recovery are still outstanding questions that we discuss in the context of (1) glycinergic synapses as they function in spinal circuits that produce rhythmic motor behaviors, (2) the proteins involved in regulating glycinergic synaptic strength, (3) current models of glycinergic synaptogenesis, and (4) plasticity mechanisms that modulate the strength of glycinergic synapses. Concluding remarks (5) explore the potential for distinct plasticity mechanisms to act in concert at different spatial and temporal scales to achieve a dynamic stability that results in balanced motor behaviors.
The zebrafish, Danio rerio, provides a vertebrate animal model of inherited human diseases that impact glycinergic synapse function and plasticity (Oda et al., 1998
; Cui et al., 2005
; Hirata et al., 2005
; Downes and Granato, 2006
; Rigo and Legendre, 2006
; Mongeon et al., 2008
). As the predominant inhibitory neurotransmitter in vertebrate brain stem and spinal cord, glycine is critically important for the generation of rhythmic motor behaviors (Moss and Smart, 2001
). Indeed, in humans, rhythmic motor behaviors are disrupted by mutations that either reduce glycinergic signaling in the case of the startle syndrome hyperekplexia (Bakker et al., 2006
; Harvey et al., 2008
), or augment glycinergic signaling in the case of glycine encephalopathy (Applegarth and Toone, 2006
). In patients with glycine encephalopathy, elevated glycine disrupts respiratory circuits so that babies with the disease require a ventilator to breathe (Applegarth and Toone, 2004
). With time however, many affected infants recover the ability to breathe on their own, and a small subset of these infants recover normal neurological functions (Boneh et al., 2008
), suggesting compensatory or homeostatic mechanisms can reduce the severity of the disease. Variable outcomes in human patients with glycine encephalopathy stand in contrast to 100% motor impairment in the mouse, or 100% motor recovery in the zebrafish models of the disease (Gomeza et al., 2003a
; Luna et al., 2004
). Zebrafish, therefore, are ideal models for studying homeostatic mechanisms that can restore motor behaviors. In zebrafish, gradual motor recovery is mirrored by reductions in evoked glycinergic post-synaptic potentials. This gradual reduction in the strength of glycinergic signaling culminates with the down-regulation of glycine receptor RNA and protein (Mongeon et al., 2008
). Still, there are outstanding questions, such as how motor recovery is initiated, and how sequential plasticity mechanisms orchestrate behavioral recovery. We discuss these questions in the context of what is known about synaptogenesis and plasticity at glycinergic synapses.
Patterned motor output is generated in the spinal cord by neuronal circuits made up of excitatory (mostly glutamatergic) and inhibitory (mostly glycinergic) interneurons that synapse both with each other and with motor neurons. The balance between excitatory and inhibitory synapses onto interneurons and motor neurons underlies normal functioning of locomotor circuits that produce rhythmic motor output (Grillner et al., 1995
; Hultborn and Nielsen, 2007
). In mammals, for example, individual motor neurons coordinate between 20,000 to 50,000 synaptic inputs (Gelfan, 1963
; Hochman, 2007
). These pre-synaptic inputs align with post-synaptic receptors to determine the duration and frequency with which post-synaptic neurons fire action potentials. Genetic disruption of glycinergic synapses onto both interneurons and motor neurons results in locomotory dysfunction (Kingsmore et al., 1994
; Mulhardt et al., 1994
; Gomeza et al., 2003a
, b
; Cui et al., 2005
; Hirata et al., 2005
). Such locomotory dysfunction is exhibited by two human genetic diseases, hyperekplexia and glycine encephalopathy. Hyperekplexia is characterized by insufficient glycinergic inhibition leading to an excessive startle response (Harvey et al., 2008
). Glycine encephalopathy, in contrast, is marked by excess glycinergic inhibition leading to hypotonia and subsequent difficulties in breathing (Applegarth and Toone, 2006
).
As a model for genetically inherited diseases of the nervous system, zebrafish have significant advantages that compliment existing mammalian models. In particular, compared to mammals, zebrafish develop externally over a compressed time scale, facilitating both observations and experimental manipulations to understand underlying mechanisms by which genetic mutations manifest themselves in stereotyped behaviors (Granato et al., 1996
; Gahtan and Baier, 2004
; Burgess and Granato, 2007
; Fetcho et al., 2008
; McLean and Fetcho, 2008
). At the interface of behavior and genetics lie neuronal circuits that in zebrafish are accessible to both in vivo electrophysiological and imaging analyses (Ali et al., 2000
; Saint-Amant and Drapeau, 2001
; Ibanez-Tallon et al., 2004
; Wen and Brehm, 2005
; McLean et al., 2008
; Wyart et al., 2009
). Taken together, the ability to visualize development in vivo as well as to study genetic correlates of human disease make zebrafish an ideal and accessible model to address the ontogeny of inherited nervous system deficits.
The accessibility of the zebrafish nervous system makes it feasible to directly record inhibitory glycinergic responses in motor neurons. Specifically, inputs onto primary motor neurons from morphologically and genetically identified glycinergic inhibitory CoLo (Commissural Local) interneurons can be recorded. The ability to record from these interneurons in vivo has revealed their specific contribution to escape behaviors in zebrafish (Liao and Fetcho, 2008
; Mongeon et al., 2008
; Satou et al., 2009
). Escape behavior in zebrafish larvae consists of a strong contra-lateral bend away from the stimulus (Faber et al., 1989
). To ensure that only the side of the fish contra-lateral to the stimulus responds, the glycinergic CoLos inhibit motor neurons ipsi-lateral to the stimulus (Satou et al., 2009
). Therefore, recordings of evoked CoLo post-synaptic potentials in motor neurons can reveal the contribution of particular synaptic proteins such as GlyT1 to glycinergic synapse function (Mongeon et al., 2008
).
In the GlyT1 mutant, CoLo/motor neuron synaptic recordings can help explain the progression of GlyT1 mutant motor behaviors (Mongeon et al., 2008
). As embryos and early larvae, GlyT1 mutants exhibit reduced movements that correspond to augmented glycinergic potentials (Mongeon et al., 2008
). Elevated glycinergic inhibition throughout the nervous system would globally reduce excitability and motor output. This period of reduced excitability transitions to motor recovery at which point glycinergic potentials are dramatically reduced (Mongeon et al., 2008
). This reduction in glycinergic synaptic responses reflects a global increase in nervous system tolerance to glycine that underlies motor recovery (Mongeon et al., 2008
). Although the zebrafish spinal cord is less complex than the mammalian spinal cord both in terms of the gross neuron populations as well as the diversity of functional classes of interneurons (Goulding, 2009
), zebrafish and mammalian spinal cords have proven similar in the physiological basis for mutant phenotypes (Tropepe and Sive, 2003
; Ingham, 2009
).
The proteins of the glycinergic inhibitory synapse work together to:(1) package glycine into pre-synaptic vesicles, (2) concentrate post-synaptic glycine receptors so that receptors are juxtaposed with the pre-synaptic terminal, and (3) remove and recycle unbound glycine from the synaptic cleft to achieve temporal control of glycinergic synaptic transmission (Dresbach et al., 2008
).
Glycinergic Pre-Synapse
Many pre-synaptic proteins localized at glycinergic synapses function to package and/or recycle glycine. Packaging glycine involves concentrating glycine into synaptic vesicles. A central player in this process, the Vesicular Inhibitory Amino Acid Transporter, VIAAT, utilizes a proton concentration gradient to transport glycine (as well as GABA) into synaptic vesicles. Because VIAAT binds glycine with low affinity, glycine must be concentrated in the synaptic terminal to millimolar levels by yet another transporter, the neuronal Glycine Transporter 2, GlyT2, expressed in the plasma membrane of the axon terminal. GlyT2 couples transport of one molecule of glycine to the cotransport of three Na+ ions and one Cl- ion, supporting the uni-directional transport of glycine into the axon terminal (Chen et al., 2004
; Rees et al., 2006
). Pre-synaptic release of glycine into the synaptic cleft then sets into motion several glycine uptake mechanisms that help to determine the time-course of glycinergic signaling. GlyT2 is critical to recycling glycine into the pre-synaptic axon terminal (Gomeza et al., 2003b
).
Glia
Like GlyT2, the glial glycine transporter type 1, GlyT1, maintains glycine levels by effectively removing glycine from the synapse and terminating glycinergic transmission (Eulenburg et al., 2005
). GlyT1 is structurally similar to the 12-membrane spanning GlyT2 but differs in its expression domain and the stoichiometry of its transport mechanism. Like GlyT2, GlyT1 also employs sodium and chloride gradients to transport glycine, but unlike GlyT2, GlyT1 can both import and export glycine thus playing an important role in setting the levels of glycine bathing the nervous system (Supplisson and Roux, 2002
; Eulenburg et al., 2005
). While GlyT1 is predominantly expressed in glial cells, GlyT2 expression is specific to the pre-synaptic terminal (Zafra et al., 1995
; Eulenburg et al., 2005
; Betz et al., 2006
).
Glycinergic Post-Synapse
In the glycinergic post-synapse, gephyrin bridges glycine receptors and the cytoskeleton (Prior et al., 1992
; Charrier et al., 2006
) and serves as a docking site for multiple regulatory proteins (Figure 1
; Fritschy et al. 2008
). Microtubules serve as highways along which glycine receptor/gephyrin complexes are delivered to the plasma membrane by dynein light chain 1 and 2 motors (Maas et al., 2006
), while the sub-synaptic actin cytoskeleton provides a gephyrin-mediated anchor for the glycine receptor and is especially important for synaptic localization (Kirsch and Betz, 1995
; Fritschy et al., 2008
).
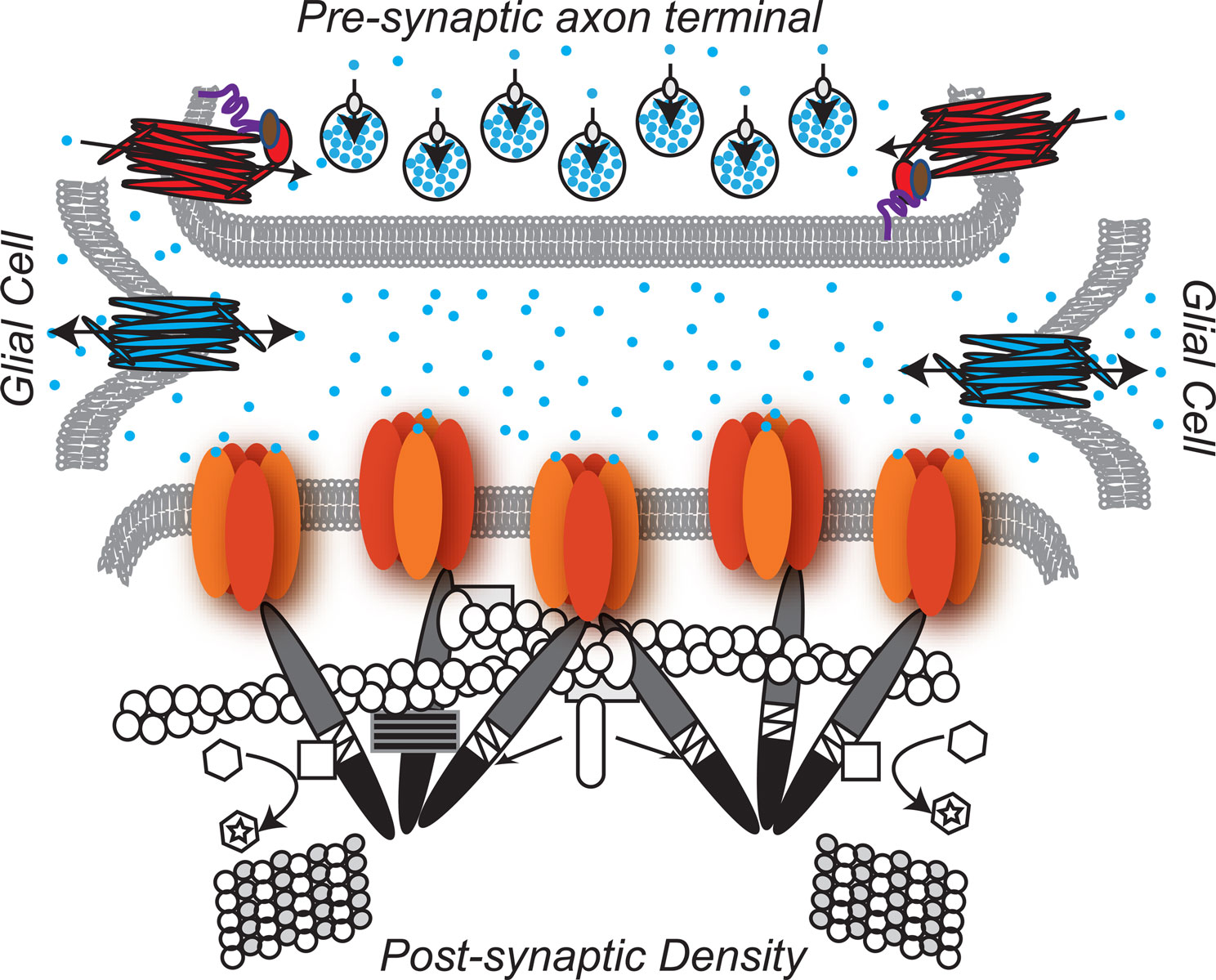
Figure 1. The glycinergic synapse. Pre-Synaptic:  Glycine is the predominant inhibitory neurotransmitter in the spinal cord.
Glycine is the predominant inhibitory neurotransmitter in the spinal cord.  GlyT2-neuronal Glycine Transporter 2 functions in glycine re-uptake from the synaptic cleft (Betz et al., 2006
) and is responsible for neurostransmitter recycling in the pre-synaptic terminal. GlyT2 is localized at the axon terminal through its carboxy-terminal, PDZ, protein-interaction domain
GlyT2-neuronal Glycine Transporter 2 functions in glycine re-uptake from the synaptic cleft (Betz et al., 2006
) and is responsible for neurostransmitter recycling in the pre-synaptic terminal. GlyT2 is localized at the axon terminal through its carboxy-terminal, PDZ, protein-interaction domain  . The GlyT2 PDZ domain anchors the GlyT2 to the PDZ protein syntenin 1
. The GlyT2 PDZ domain anchors the GlyT2 to the PDZ protein syntenin 1  , which in turn binds syntaxin
, which in turn binds syntaxin  , a member of the SNARE complex that governs fusion of vesicles with the plasma membrane (Geerlings et al., 2000
; Geerlings et al., 2001
; Ohno et al., 2004
; Armsen et al., 2007
)
, a member of the SNARE complex that governs fusion of vesicles with the plasma membrane (Geerlings et al., 2000
; Geerlings et al., 2001
; Ohno et al., 2004
; Armsen et al., 2007
)  VIAAT-the Vesicular Inhibitory. Amino Acid Transporter loads glycine into synaptic vesicles
VIAAT-the Vesicular Inhibitory. Amino Acid Transporter loads glycine into synaptic vesicles  (Gasnier, 2004
). Synaptic vesicles accumulate at the active zone of the axon terminal due to the actions of the SNARE complex, a large set of proteins, some of which are expressed on the vesicular membrane, and others of which are expressed on the plasma membrane (Rizo and Rosenmund, 2008
). Glia :
(Gasnier, 2004
). Synaptic vesicles accumulate at the active zone of the axon terminal due to the actions of the SNARE complex, a large set of proteins, some of which are expressed on the vesicular membrane, and others of which are expressed on the plasma membrane (Rizo and Rosenmund, 2008
). Glia : GlyT1-glial Glycine Transpoter 1 regulates the amount of glycine available to bind the glycine receptor and, at some synapses, has been shown to play a key role in terminating glycinergic synaptic transmission (Betz et al., 2006
). Post-synaptic:
GlyT1-glial Glycine Transpoter 1 regulates the amount of glycine available to bind the glycine receptor and, at some synapses, has been shown to play a key role in terminating glycinergic synaptic transmission (Betz et al., 2006
). Post-synaptic:  Enriched in the post-synaptic membrane directly across from the pre-synaptic terminal, glycine receptors are chloride ion channels (Lynch, 2004
). Five subunits, two alpha and three beta, associate to form the functional channel (Grudzinska et al., 2005
). At least four independent genes (Glra1-4) encode alpha subunits
Enriched in the post-synaptic membrane directly across from the pre-synaptic terminal, glycine receptors are chloride ion channels (Lynch, 2004
). Five subunits, two alpha and three beta, associate to form the functional channel (Grudzinska et al., 2005
). At least four independent genes (Glra1-4) encode alpha subunits  in vertebrates. The α subunit genes encode the glycine binding site that when bound to the substrate, glycine, gates the channel. One gene, Glrb, encodes the β subunit
in vertebrates. The α subunit genes encode the glycine binding site that when bound to the substrate, glycine, gates the channel. One gene, Glrb, encodes the β subunit  .β subunits link the glycine receptor to the cytoskeleton through high affinity associations with the post-synaptic scaffold protein gephyrin.
.β subunits link the glycine receptor to the cytoskeleton through high affinity associations with the post-synaptic scaffold protein gephyrin.  Gephyrin (Greek for bridge) links the glycine receptor to the cytoskeleton, forming a submembranous, hexagonal lattice (Bechade et al., 1996
; Fritschy et al., 2008
). The carboxy-terminal E domain
Gephyrin (Greek for bridge) links the glycine receptor to the cytoskeleton, forming a submembranous, hexagonal lattice (Bechade et al., 1996
; Fritschy et al., 2008
). The carboxy-terminal E domain  of gephyrin dimerizes and interacts with the β subunit of the glycine receptor. The E domain also interacts with profilin
of gephyrin dimerizes and interacts with the β subunit of the glycine receptor. The E domain also interacts with profilin  and Mena/VASP
and Mena/VASP  , both proteins involved in actin microfilament polymerization
, both proteins involved in actin microfilament polymerization  . The amino terminal G domain of gephryin
. The amino terminal G domain of gephryin 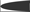 forms trimers. A proline rich C domain
forms trimers. A proline rich C domain 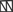 is located between the E and G domains. This C domain is a highly-regulated platform for possible interactions between gephyrin and several other proteins. These proteins include: microtubules
is located between the E and G domains. This C domain is a highly-regulated platform for possible interactions between gephyrin and several other proteins. These proteins include: microtubules 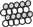 ; GEFs Guanine nucleotide Exchange Factors (collybistins at GABAergic synapses; as yet unknown at glycinergic synapses)
; GEFs Guanine nucleotide Exchange Factors (collybistins at GABAergic synapses; as yet unknown at glycinergic synapses)  that, by activating CDC42
that, by activating CDC42 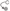 , remodel the actin cytoskeleton; and RAFT1
, remodel the actin cytoskeleton; and RAFT1  that regulates localized protein translation. Finally, Pin-1 Peptidyl-prolyl isomerase 1
that regulates localized protein translation. Finally, Pin-1 Peptidyl-prolyl isomerase 1 alters the configuration of gephyrin by inducing proline isomerization in a phosphorylation-dependent manner (Zita et al., 2007
).
alters the configuration of gephyrin by inducing proline isomerization in a phosphorylation-dependent manner (Zita et al., 2007
).
Glycine is the predominant inhibitory neurotransmitter in the spinal cord. GlyT2-neuronal Glycine Transporter 2 functions in glycine re-uptake from the synaptic cleft (Betz et al., 2006
) and is responsible for neurostransmitter recycling in the pre-synaptic terminal. GlyT2 is localized at the axon terminal through its carboxy-terminal, PDZ, protein-interaction domain . The GlyT2 PDZ domain anchors the GlyT2 to the PDZ protein syntenin 1 , which in turn binds syntaxin , a member of the SNARE complex that governs fusion of vesicles with the plasma membrane (Geerlings et al., 2000
; Geerlings et al., 2001
; Ohno et al., 2004
; Armsen et al., 2007
) VIAAT-the Vesicular Inhibitory. Amino Acid Transporter loads glycine into synaptic vesicles (Gasnier, 2004
). Synaptic vesicles accumulate at the active zone of the axon terminal due to the actions of the SNARE complex, a large set of proteins, some of which are expressed on the vesicular membrane, and others of which are expressed on the plasma membrane (Rizo and Rosenmund, 2008
). Glia : GlyT1-glial Glycine Transpoter 1 regulates the amount of glycine available to bind the glycine receptor and, at some synapses, has been shown to play a key role in terminating glycinergic synaptic transmission (Betz et al., 2006
). Post-synaptic: Enriched in the post-synaptic membrane directly across from the pre-synaptic terminal, glycine receptors are chloride ion channels (Lynch, 2004
). Five subunits, two alpha and three beta, associate to form the functional channel (Grudzinska et al., 2005
). At least four independent genes (Glra1-4) encode alpha subunits in vertebrates. The α subunit genes encode the glycine binding site that when bound to the substrate, glycine, gates the channel. One gene, Glrb, encodes the β subunit .β subunits link the glycine receptor to the cytoskeleton through high affinity associations with the post-synaptic scaffold protein gephyrin. Gephyrin (Greek for bridge) links the glycine receptor to the cytoskeleton, forming a submembranous, hexagonal lattice (Bechade et al., 1996
; Fritschy et al., 2008
). The carboxy-terminal E domain of gephyrin dimerizes and interacts with the β subunit of the glycine receptor. The E domain also interacts with profilin and Mena/VASP , both proteins involved in actin microfilament polymerization . The amino terminal G domain of gephryin forms trimers. A proline rich C domain is located between the E and G domains. This C domain is a highly-regulated platform for possible interactions between gephyrin and several other proteins. These proteins include: microtubules ; GEFs Guanine nucleotide Exchange Factors (collybistins at GABAergic synapses; as yet unknown at glycinergic synapses) that, by activating CDC42 , remodel the actin cytoskeleton; and RAFT1 that regulates localized protein translation. Finally, Pin-1 Peptidyl-prolyl isomerase 1 alters the configuration of gephyrin by inducing proline isomerization in a phosphorylation-dependent manner (Zita et al., 2007
).Diseases of the Glycinergic Synapse
The inherited human diseases glycine encephalopathy and hyperekplexia result from defects in glycinergic signaling. Glycine encephalopathy is caused by tonic activation of glycinergic synapses, and is often a fatal disease as affected infants require artificial ventilation immediately after birth (Applegarth and Toone, 2004
). With ventilation aids, some infants that exhibit glycine encephalopathy recover balanced patterned motor output (Boneh et al., 2008
). Like their human counterparts, phenotypes displayed by GlyT1 mutant mice and zebrafish reflect increased inhibition as a result of inadequate glycine clearance from the synapse (Lopez-Corcuera et al., 2001
; Eulenburg et al., 2005
; Betz et al., 2006
). In contrast, Hyperekplexia, caused by mutations that reduce the strength of glycinergic signaling, is defined by an exaggerated startle response: acoustic or tactile stimuli induce hypertonia and difficulty breathing (Fritschy et al., 2008
; Harvey et al., 2008
). Mutations in either pre-synaptic genes, e.g. GlyT2, or post-synaptic genes, e.g. glycine receptor subunits, are known to cause the disease in humans (Harvey et al., 2008
). Animal models, including GLRA1: spasmodic (Ryan et al., 1994
; Findlay et al., 2003
), GLRB:spastic (Kingsmore et al., 1994
; Mulhardt et al., 1994
), and GlyT2 knockout mice (Gomeza et al., 2003b
) have paved the way to our understanding of the physiological basis of this genetic disease. Unlike the case in humans, the hypertonic phenotype in mice is lethal. Like mouse models, the zebrafish glycine receptor β mutant bandoneon mirrors the hyperekplexia phenotype, but unlike mouse is accessible at earlier developmental stages (Hirata et al., 2005
). There are patients that present with hyperekplexia symptoms but have no mutations in known culprit genes (Rees et al., 2006
; Harvey et al., 2008
; Kalscheuer et al., 2009
). Some potential candidate culprits include the host of other proteins enriched at the glycinergic synapse (Harvey et al., 2008
).
Our understanding of the process of synaptogenesis has been transformed by in vivo studies in zebrafish (Niell et al., 2004
; Meyer and Smith, 2006
). These studies demonstrate that synaptogenesis occurs coincident with neuronal morphogenesis. In vivo synaptogenesis models also confirm the requirement for neuronal activity during synapse establishment (Kirsch and Betz, 1998
). Synaptic glycine receptors exchange with extra-synaptic clusters or individual receptors within or outside the synapse (Meier et al., 2000
). Because differences in glycinergic synaptic strength often rely on receptor density, plasticity mechanisms would depend on the ability of receptors to accumulate at or dissociate away from the synapse (Legendre et al., 2002
).
In the nascent nervous system, synapses form during a time of several fundamental transitions: young neurons extend axons and dendrites to build circuits (Niell et al., 2004
; Meyer and Smith, 2006
); neuronal communication transitions from predominantly gap-junction mediated to predominantly synapse mediated (Saint-Amant and Drapeau, 2001
; Brustein et al., 2003
); and glycine- and GABA-evoked chloride conductances are depolarizing (Reynolds et al., 2008
). Both maturing glycinergic and GABAergic synapses switch from initial excitatory- to inhibitory-signaling due to the expression of the potassium-chloride co-transporter 2, KCC2 (Rivera et al., 1999
). Prior to maturation, both the ligand-gated receptors that underlie synaptic transmission and the voltage-gated ion channels that underlie endogenous firing properties are distinct from their mature counterparts (Ali et al., 2000
). Immature neurons tend to express relatively few channels that underlie qualitatively different, low-frequency, spontaneous forms of electrical activity (Moody and Bosma, 2005
) that trigger neurotransmitter release from growing axons during early steps in synapse formation (Zhang and Poo, 2001
).
Many models of activity-mediated glycinergic synapse formation were derived from neuronal tissue culture (Bechade et al., 1996
; Meier, 2003
). Kneussel and Betz (2000)
proposed the membrane activation model, suggesting that after glycine binds the receptor, post-synaptic depolarization activates calcium channels and triggers the specialization of the post-synaptic density. As the pre-synaptic axon terminal matures, expressing VIAAT, gephyrin clusters become localized to the post-synaptic membrane. Specifically, glycine receptors are clustered when gephyrin anchors itself to the cytoskeleton, and binds the β subunit of glycine receptors (Kneussel and Betz, 2000
; Luscher et al., 2000
). Gephyrin can aggregate independent of synapse formation (Colin et al., 1996
; Dumoulin et al., 2000
), but receptors fail to cluster when gephyrin expression is reduced by antisense oligonucleotides (Kirsch et al., 1993
). Moreover, initial synaptic receptor clustering also requires receptor activation (Levi et al., 1999
): when glycine receptors are blocked with strychnine, the receptors no longer form clusters (Kirsch and Betz, 1998
). Although activation of the receptor appears to be essential for specialization of the post-synaptic density, high levels of glycine in culture media are not sufficient to trigger receptor clusters, suggesting that other factors, such as cell adhesion molecules, associated with the pre-synaptic terminal are essential for glycine receptor clustering (Legendre, 2001
).
Because the process of synaptogenesis establishes the template upon which mature behaviors are formed, how synaptogenesis is impacted by mutations in synaptic genes is likely to inform our understanding of mutant phenotypes. The accessibility of the zebrafish nervous system to both physiological recordings and imaging make this organism especially well-suited to addressing how mutations affect the process of synaptogenesis.
In order to produce appropriate responses to varying stimuli, neuronal synapses must be plastic, having the capacity to change physiological connection strength (Nelson and Turrigiano, 2008
). When subsets of synapses in a neuronal circuit are selectively altered (long-term potentiation or depression), synaptic plasticity alters behavior (Oda et al., 1998
). However, when synapses undergo more global remodeling during development or in response to either environmental or genetic perturbation, synaptic plasticity underlies homeostasis that stabilizes behavior (Levi et al., 2008
; Mongeon et al., 2008
). Thus, behaviors can be either stabilized or changed by different types of neuronal plasticity. Because of the multi-component nature of the synapse, changes in connectivity can take place at multiple levels, including pre-synaptic, glial, and post-synaptic (Gaiarsa et al., 2002
). Here we discuss examples of glycinergic synaptic plasticity in the context of first potentiation and then homeostasis.
That glycinergic synaptic strength can underlie behavioral change has been elegantly demonstrated at glycinergic synapses onto zebrafish Mauthner neurons (Korn et al., 1992
; Charpier et al., 1995
; Oda et al., 1998
). Mauthner neurons are crucial for the speed of the teleost escape behavior (Liu and Fetcho, 1999
). Two, bilateral Mauthner cell bodies in the hindbrain, extend axons that cross the midline before projecting caudally the full length of the spinal cord, exciting contra-lateral motor neurons and causing a rapid bend away from the stimulus (Faber et al., 1989
). In response to sound, for example, the VIII cranial nerve stimulates both the ipsi-lateral Mauthner cell body and a glycinergic interneuron. This glycinergic interneuron synapses onto both bilateral Mauthner cell bodies. The resulting glycinergic inhibition ensures that only the Mauthner neuron ipsi-lateral to the stimulus fires a single action potential because the ipsi-lateral cell receives strong excitation just prior to inhibition. In response to repeated VIII nerve stimulation (sub threshold for inducing an escape response), glycinergic synapses onto both Mauthner cell bodies experience a persistent (more than 5 h) potentiation (Korn et al., 1992
). This long-term potentiation is specific to glycingergic inhibition elicited by the VIII nerve, as recurrent inhibition mediated by a collateral of the Mauthner axon is not similarly potentiated. Most significant, VIII nerve/Mauthner glycinergic synaptic potentiation has a behavioral correlate: After auditory conditioning, fish are less likely to escape in response to stimuli of identical intensity (Oda et al., 1998
). Thus, plasticity at glycinergic synapses has clear implications for altering behavior in response to changing environmental stimuli.
In contrast to long-term potentiation, homeostatic synaptic plasticity stabilizes behavioral output not only as circuits are reconfiguring during development but also in response to environmental and genetic perturbation (Turrigiano and Nelson, 2004
; Davis, 2006
; Mongeon et al., 2008
). Homeostasis at the glycinergic synapses can be mediated by diverse mechanisms. For instance, at GABAergic synapses, pre-synaptic mechanisms involving the expression level of VIAAT have been shown to alter quantal content (the amount of neurotransmitter loaded into the pre-synaptic vesicle) in accordance with the amount of neuronal activity (De Gois et al., 2005
; Erickson et al., 2006
). At glycinergic synapses, two transporter proteins, VIAAT and GlyT2, have been shown to influence quantal content. Depending on the level of VIAAT expression, more or less glycine will be loaded into the synaptic vesicle. Likewise, when GlyT2 expression is reduced, glycinergic synapses lose strength because the amount of cytoplasmic glycine available to the VIAAT transporter becomes limiting (Lopez-Corcuera et al., 2001
; Gomeza et al., 2003b
).
The ability to directly measure in vivo receptor dynamics (membrane area explored per unit time) has identified glycine receptor diffusion dynamics as a post-synaptic mechanism underlying glycinergic synapse homeostasis (Levi et al., 2008
; Renner et al., 2008
). In response to electrical activity and specifically NMDA receptor activation, rapid homeostatic adjustments are triggered in the strength of glycinergic signaling by changing the dynamics of glycine receptor lateral diffusion (Levi et al., 2008
). Receptor transit between synaptic, peri-synaptic, and extra-synaptic domains occurs within minutes, much faster than the rate of receptor turnover: receptor half-life at the plasma membrane is 14 hours (Rasmussen et al., 2002
). Moreover, each domain is associated with a unique mobility with the highest diffusion coefficients associated with extra-synaptic receptors and the lowest diffusion coefficients associated with synaptic receptors (Dahan et al., 2003
). Diffusion of receptors into and out of the post-synapse is regulated by the cytoskeleton (Charrier et al., 2006
). When either actin or microtubules are depolymerized, dwell time at the synapse is decreased and diffusion away from the synapse is increased implicating receptor interactions with the cytoskeleton as crucial to glycine receptor diffusion dynamics.
“The living being is stable. It must be so in order not to be destroyed, dissolved or disintegrated by the colossal forces, often adverse, which surround it. By apparent contradiction it maintains its stability only if it is excitable and capable of modifying itself according to external stimuli and adjusting its response to the stimulation. In a sense it is stable because it is modifiable—the slight instability is the necessary condition for the true stability of the organism.” (Richet, 1900
).
In zebrafish, a glial glycine transporter (GlyT1) mutation triggers a gradual homeostatic plasticity at glycinergic synapses that restores rhythmic motor output (Mongeon et al., 2008
). Enabling rhythmic motor output in high glycine, sequential homeostatic mechanisms function together over a time-course of 2 days to reduce glycinergic post-synaptic potentials (Figure 2
; second panel). Reduced glycinergic potentials are associated with reduced expression of both glycine receptor (Mongeon et al., 2008
) and GlyT2 transcripts (Mongeon et al., 2008
). Both the glycine receptor and GlyT2 have been previously implicated in reducing the strength of glycinergic synapses by their association with the human startle disease hyperekplexia. Although transcriptional mechanisms that reduce the expression of glycine receptor transcripts in mutants are likely to contribute to behavioral recovery, the timing of the reductions in glycine receptor expression occurs after post-synaptic potentials are already significantly reduced, implicating additional mechanisms in the recovery process.
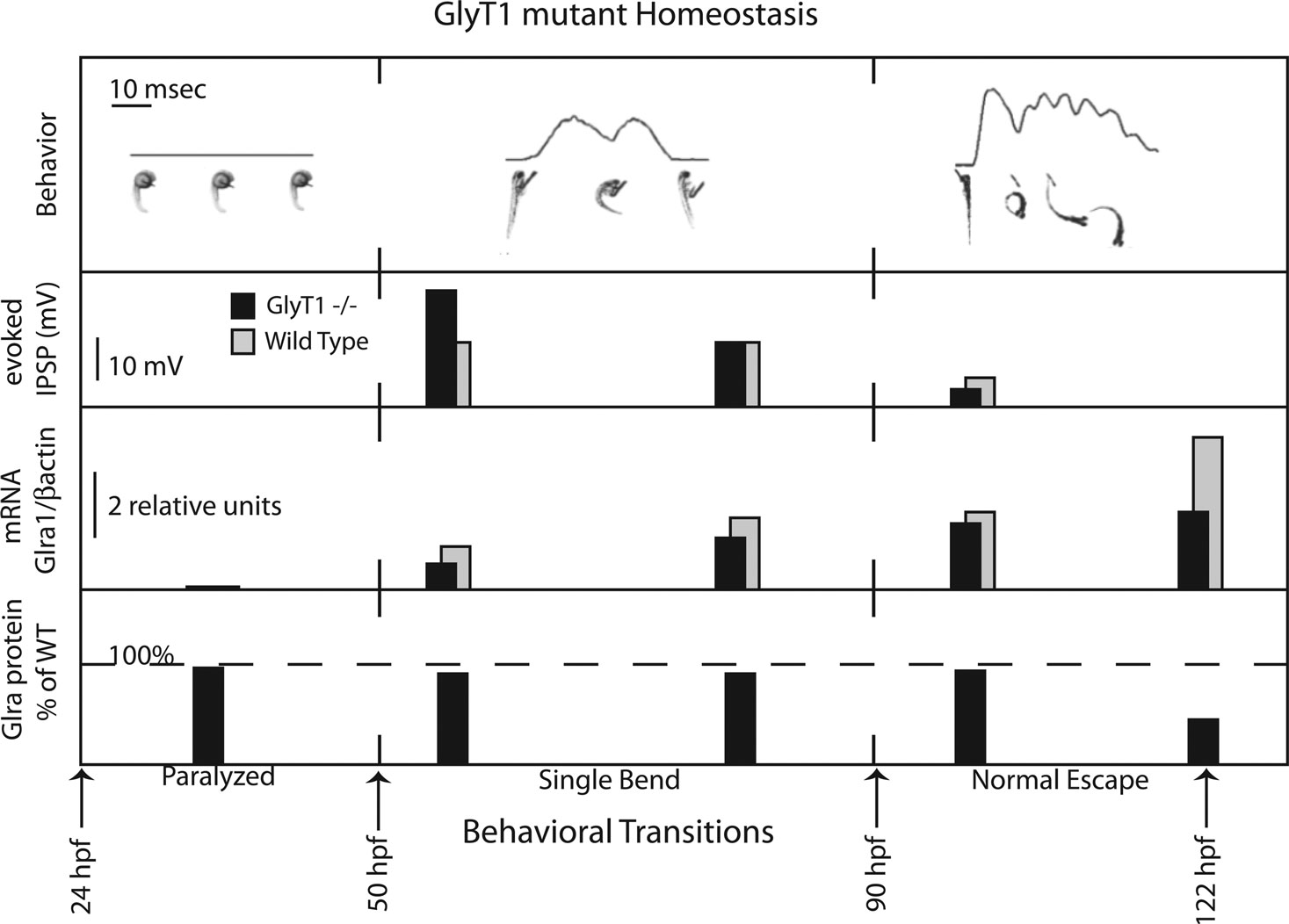
Figure 2. GlyT1 mutant neuronal homeostasis. Top Row: GlyT1 mutants exhibit two behavioral transitions, from paralysis to a single bend at 50-h post fertilization and from single bend to full recovery at 90-h post fertilization. Line graphs are produced by motion detection software that tracks larval pixel displacement over time. Below line graphs till images of the embryos and larvae are placed at corresponding timepoints. Second row: Maximum amplitude in millivolts (mV) of evoked motor neuron glycinergic inhibitory post-synaptic potentials (IPSPs) recorded in GlyT1 mutants versus wild type larvae at three timepoints. Third Row: Relative quantitation of mRNA expression, a1 subunit of the glycine receptor (Glra1) normalized to bactin, using quantitative polymerase chain reaction to compare GlyT1 mutants versus wild type. Fourth Row: Comparison (% of wild type staining) of maximum antibody labeling of motor neuron glycine receptor alpha subunits in GlyT1 mutants.
Additional homeostatic mechanisms in the GlyT1 mutant could involve changes in spinal cord cell fates. The eventual fate of neural precursors is influenced by environmental stimuli that include neurotransmitters like glycine (Nguyen et al., 2001
). In zebrafish, knocking down the α2 glycine receptor subunit reduces the number of interneurons that differentiate (McDearmid et al., 2006
). Consistent with environmental plasticity in cell fate, changes spinal cord neuron excitability in frogs results in homeostatic changes in the ratio of neurons that become excitatory versus inhibitory: when spontaneous electrical activity is elevated by global over-expression of a sodium channel, more inhibitory neurons form at the expense of excitatory neurons; conversely, when spontaneous electrical activity is reduced by global over-expression of a potassium channel, more excitatory neurons form at the expense of inhibitory neurons (Borodinsky et al., 2004
). In the case of the zebrafish glycine transporter 1 mutant, the early paralytic phenotype can be relieved by reducing nervous system glycine or by blocking the glycine receptor with strychnine (Cui et al., 2005
; Mongeon et al., 2008
). Therefore, it is likely that glycine reduces nervous system activity by tonic activation of glycine receptors, making it more difficult for neurons to reach threshold for an action potential. Perhaps the early reductions in glycinergic potentials reflect such changes in numbers of different types of spinal cord neurons.
Such a shift in neuronal cell type would impact synaptic inputs and have the potential to restore excitability in the glycine-inundated nervous system. With this in mind, it is interesting to speculate how changes in cell fate would impact motor behavior. Rhythmic motor behaviors can be mathematically modeled with a segmentally reiterated module of eight neurons: four neurons on the left side of the spinal cord are mirrored by another four neurons on the right side of the spinal cord (Goulding, 2009
). These neurons include (1) the motor neuron that receives excitatory drive from (2) ipsi-lateral glutamatergic and (3) glycinergic interneurons. The fourth neuron type is (4) a glycinergic interneuron that crosses the spinal cord so that when one side of the spinal cord is activated, the other side is inhibited. While many more classes of interneurons are present in the spinal cord (Hale et al., 2001
), the robustness of the simplified mathematical network suggests that if the four basic types of neurons remain, the motor circuit could tolerate changes in cell fate without necessarily losing rhythmicity (Kozlov et al., 2009
).
Other forms of plasticity that could contribute to reduced glycinergic potentials are post-translational modifications of glycine receptor function, including those mechanisms that impact the rate of exchange of glycine receptors from synaptic to extra-synaptic sites (Levi et al., 2008
). Future experiments will test the role of cell fate changes and post-translational regulation of the glycine receptor in GlyT1 mutant synaptic homeostasis. Both establishing the mechanisms that contribute to synaptic homeostasis at glycinergic synapses and determining how these mechanisms are regulated will contribute to our understanding of and hopefully the ability to treat diseases that result from glycinergic synapse dysfunction.
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
We thank Hiromi Hirata and Luis Saint-Amant for improving the manuscript with their careful reviews. We would also like to apologize to those researchers whose work is not cited due to space constraints. James Baker, Qing Yan, Steven Sloan, and Matthew Stark provided critical reading of drafts and helpful commentary. This work was supported by a National Institutes of Health career development award K01NS048200 from the National Institute of Neurological Disease and Stroke to JED.
Granato, M., van Eeden, F. J., Schach, U., Trowe, T., Brand, M., Furutani-Seiki, M., Haffter, P., Hammerschmidt, M., Heisenberg, C. P., Jiang, Y. J., Kane, D. A., Kelsh, R. N., Mullins, M. C., Odenthal, J., and Nusslein-Volhard, C. (1996). Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development 123, 399–413.
Kalscheuer, V. M., Musante, L., Fang, C., Hoffmann, K., Fuchs, C., Carta, E., Deas, E., Venkateswarlu, K., Menzel, C., Ullmann, R., Tommerup, N., Dalpra, L., Tzschach, A., Selicorni, A., Luscher, B., Ropers, H. H., Harvey, K., and Harvey, R. J. (2009). A balanced chromosomal translocation disrupting ARHGEF9 is associated with epilepsy, anxiety, aggression, and mental retardation. Hum. Mutat. 30, 61–68.
Rees, M. I., Harvey, K., Pearce, B. R., Chung, S. K., Duguid, I. C., Thomas, P., Beatty, S., Graham, G. E., Armstrong, L., Shiang, R., Abbott, K. J., Zuberi, S. M., Stephenson, J. B., Owen, M. J., Tijssen, M. A., van den Maagdenberg, A. M., Smart, T. G., Supplisson, S., and Harvey, R. J. (2006). Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of human startle disease. Nat. Genet. 38, 801–806.
Ryan, S. G., Buckwalter, M. S., Lynch, J. W., Handford, C. A., Segura, L., Shiang, R., Wasmuth, J. J., Camper, S. A., Schofield, P., and O’Connell, P. (1994). A missense mutation in the gene encoding the alpha 1 subunit of the inhibitory glycine receptor in the spasmodic mouse. Nat. Genet. 7, 131–135.