Department of Anatomy and Neurobiology, University of California, Irvine, CA, USA
Somatic recordings from CA1 pyramidal cells indicated a persistent upregulation of the h-current (Ih) after experimental febrile seizures. Here, we examined febrile seizure-induced long-term changes in Ih and neuronal excitability in CA1 dendrites. Cell-attached recordings showed that dendritic Ih was significantly upregulated, with a depolarized half-activation potential and increased maximal current. Although enhanced Ih is typically thought to be associated with decreased dendritic excitability, whole-cell dendritic recordings revealed a robust increase in action potential firing after febrile seizures. We turned to computational simulations to understand how the experimentally observed changes in Ih influence dendritic excitability. Unexpectedly, the simulations, performed in three previously published CA1 pyramidal cell models, showed that the experimentally observed increases in Ih resulted in a general enhancement of dendritic excitability, primarily due to the increased Ih-induced depolarization of the resting membrane potential overcoming the excitability-depressing effects of decreased dendritic input resistance. Taken together, these experimental and modeling results reveal that, contrary to the exclusively anti-convulsive role often attributed to increased Ih in epilepsy, the enhanced Ih can co-exist with, and possibly even contribute to, persistent dendritic hyperexcitability following febrile seizures in the developing hippocampus.
Fever-induced (febrile) seizures are the most common form of childhood seizures, occurring in 3–5% of infants (Shinnar and Glauser, 2002
). Because of the potential clinical association of prolonged (>15 min) febrile seizures with persistent temporal lobe epilepsy, it is important to understand how prolonged seizures in the developing brain alter neuronal excitability (Annegers et al., 1987
; Baram et al., 1997
; Chen et al., 2007
; Schuchmann et al., 2006
). In the rat hyperthermia model of experimental febrile seizures (HT) (Baram et al., 1997
; Dube et al., 2000
, 2006
), whole cell somatic patch clamp recordings from CA1 pyramidal neurons demonstrated long-term upregulation of Ih with a depolarized half-activation potential and slower kinetics (Chen et al., 2001
). However, because of the dendritic localization of Ih in pyramidal cells, dendritic recordings are needed to provide direct information about the nature of the febrile seizure-induced plasticity of Ih and its effect on dendritic excitability.
The question of how seizure-related alterations in Ih modulate dendritic excitability is also interesting because persistent alterations in Ih have been reported in other seizure models as well, indicating the general nature of the problem. For example, downregulation of Ih has been characterized as pro-excitatory in several models of epilepsy due to increases in neuronal input resistance, spike-coupling and input summation (Jung et al., 2007
; Kole et al., 2007
; Shah et al., 2004
; Strauss et al., 2004
; Zhang et al., 2006
). In agreement with these results, studies investigating increases in Ih activation have reported decreased neuronal excitability due to decreased input resistance (Fan et al., 2005
; Poolos et al., 2002
; Rosenkranz and Johnston, 2006
).
In order to investigate how febrile seizures change dendritic Ih and dendritic excitability, we used a combination of dendritic cell-attached and whole-cell patch clamp recordings and computational modeling techniques, and asked the following questions: (1) Is Ih persistently upregulated in the dendrites of CA1 pyramidal cells following experimental febrile seizures? (2) Are the pyramidal cell dendrites hypo- or hyperexcitable after febrile seizures? (3) What role may the experimentally observed alterations in Ih play in modulating dendritic excitability after febrile seizures?
The results indicated that febrile seizures result in a persistent upregulation of dendritic Ih as well as a robust increase in the excitability of pyramidal cell dendrites. In addition, computational modeling data suggested that the experimentally observed enhancement in Ih may actually promote dendritic excitability in post-febrile seizure CA1 pyramidal cells.
Hyperthermia-Induced Seizures
The hyperthermia-induced seizure paradigm has been previously described (Baram et al., 1997
; Chen et al., 1999
; Dube et al., 2000
). Briefly, on P10-11, the core temperature of Sprague-Dawley pups (Zivic-Miller, Zelienople, Pennsylvania) was raised using a regulated stream of hot air until hyperthermia (core temperature >39.5°C) was reached and maintained for 30 min. In this model, seizure duration averages 22.8 ± 0.3 min with mean seizure onset temperature threshold at 41.1°C (Chen et al., 1999
; Dube et al., 2000
). Following hyperthermia, rats were monitored on a cool surface for 5 min, then monitored for 1 hour on a heating pad (36.5°C) before return to the home cage. Behavioral seizures in this model have been previously shown to be stereotyped and correlated with rhythmic, epileptiform discharges from the hippocampus and amygdala determined from EEG (Baram et al., 1997
; Dube et al., 2000
).
Slice Preparation
Following hyperthermia-induced seizures at P10-11, experimental (HT) and control (Ctrl) Sprague-Dawley rats (4–5 weeks old) were transcardially perfused through the ascending aorta under deep ketamine (100 mg/kg)/xylazine (8 mg/kg) anesthesia using ice-cold (∼2°C), oxygenated and sucrose-substituted ACSF (in mM: 85 NaCl, 75 sucrose, 2.5 KCl, 25 glucose, 1.25 NaH2PO4, 4 MgCl2, 0.5 CaCl2, 24 NaHCO3, 2.7 Na-pyruvate and 0.8 Na-ascorbate). Transverse hippocampal slices (400 μm) were prepared in sucrose-substituted ACSF and incubated for 30 min at 32°C, then transferred to the ACSF solution subsequently used during recordings (in mM: 126 NaCl, 2.5 KCl, 26 NaHCO3, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 10 glucose, 2.7 Na-pyruvate and 0.8 Na-ascorbate) and incubated for an additional 30 min at room temperature.
Electrophysiology
Slices were visualized with an upright microscope (FN-1; Nikon, Tokyo, Japan) with infrared differential interference contrast (IR-DIC) optics under constant perfusion with oxygenated ACSF (see above) at room temperature (∼21°C). Individual CA1 pyramidal neurons with cell bodies located in the pyramidal layer were selected for recordings after visual evaluation of general cellular health, and additional criteria for determining suitability for recording included stable RMP (when applicable) and low series resistance (see below). The recording distance was measured along the linear path of the apical dendrite after calibrating the IR-DIC video image with a micrometer resolution graticule.
Cell-attached patch-clamp recordings of Ih were obtained in voltage-clamp configuration from CA1 pyramidal cell apical dendrites at 200 μm distance from the soma with seal-resistances >5 GΩ. Patch pipettes (4–6 MΩ) were visually inspected and fire-polished on a microforge (Narishige,Tokyo, Japan) and filled with internal solution containing (in mM): 120 KCl, 20 TEA-Cl, 10 HEPES, 5 4-aminopyridine, 2 CaCl2, 1 MgCl2 and 1 BaCl2 (pH 7.3). P/N leak subtraction was performed online in Clampex 9.2 (Molecular Devices, Union City, CA, USA) using eight sub-sweeps of polarity opposite to command voltages (note that active currents around RMP other than Ih were blocked by components of the internal solution; Magee, 1998
).
Dendritic whole-cell recordings were obtained in current-clamp configuration from CA1 pyramidal cell apical dendrites at 200 μm distance from the soma. Visually inspected patch pipettes (4–6 MΩ) were filled with internal solution containing (in mM): 126 K-gluconate, 4 KCl, 10 HEPES, 4 ATP-Mg, 0.3 GTP-Na, 10 phosphocreatine, and 0.2% biocytin (pH 7.2). Series resistance for whole-cell recordings ranged from 17 to 40 MΩ, with no significant difference between control and HT patches (Ctrl 28.8 ± 1.2 MΩ, HT 27.3 ± 0.7 MΩ, p > 0.25). The dendritic resting membrane potential (RMP) values were determined immediately after break-in. Artificial EPSPs were evoked with a series of 10 EPSC-shaped current injections with peak amplitudes from 0.1 to 1 nA (rise time-constant 0.1 ms, decay time-constant 2 ms; Gasparini et al., 2004
). H-current blockade was achieved by 10-min wash-in of ACSF solution with 100 μM ZD7288 (Tocris, Ellisville, MO, USA).
Recordings were made using a MultiClamp 700B amplifier without compensation for liquid junction potential (Molecular Devices, Union City, CA, USA). Signals were filtered at 4 kHz using a Bessel filter and digitized at 10 kHz with a Digidata 1320A analog–digital interface (Molecular Devices, Union City, CA, USA).
Computer Modeling
The effect of experimentally determined altered Ih characteristics were tested in three published models of CA1 pyramidal neurons downloaded from the SenseLab ModelDB at http://senselab.med.yale.edu/modeldb/ (Crasto et al., 2007
). Each model was based on a reconstructed neuronal morphology and contained several voltage-gated conductances. These models had previously been used to study the effects of Lamotrigine modulation of Ih on dendritic excitability (Poolos et al., 2002
), action potential backpropagation in apical dendrites (Golding et al., 2001
) and synaptic summation (Poirazi et al., 2003
). For control simulations, the original h-conductance representation in each model was replaced by the Boltzmann activation function and time constants determined from our cell-attached recordings (Figures 1
D–F). In all three models, the h-conductance density from the Poolos et al. (2002)
model was used to ensure comparability. For simulations of the post-febrile seizure condition, the experimentally determined Boltzmann activation function and time constants (Figures 1
D–F) were implemented in each model and the conductance density was increased by 68% relative to the control value (Figure 1
C). No other changes in parameters or components of either model were made except for the Poolos et al. (2002)
model, from which a mechanism that was originally used to clamp the membrane potential at a particular value was eliminated. While the Poolos et al. (2002) and Golding et al. (2001)
models are quite similar, the activation curves of the sodium channels in the two models differ, resulting in a substantially more hyperpolarized action potential threshold in the Golding et al. (2001)
model. Because this difference could impact the effect that Ih changes have on cellular excitability, we utilized both models.
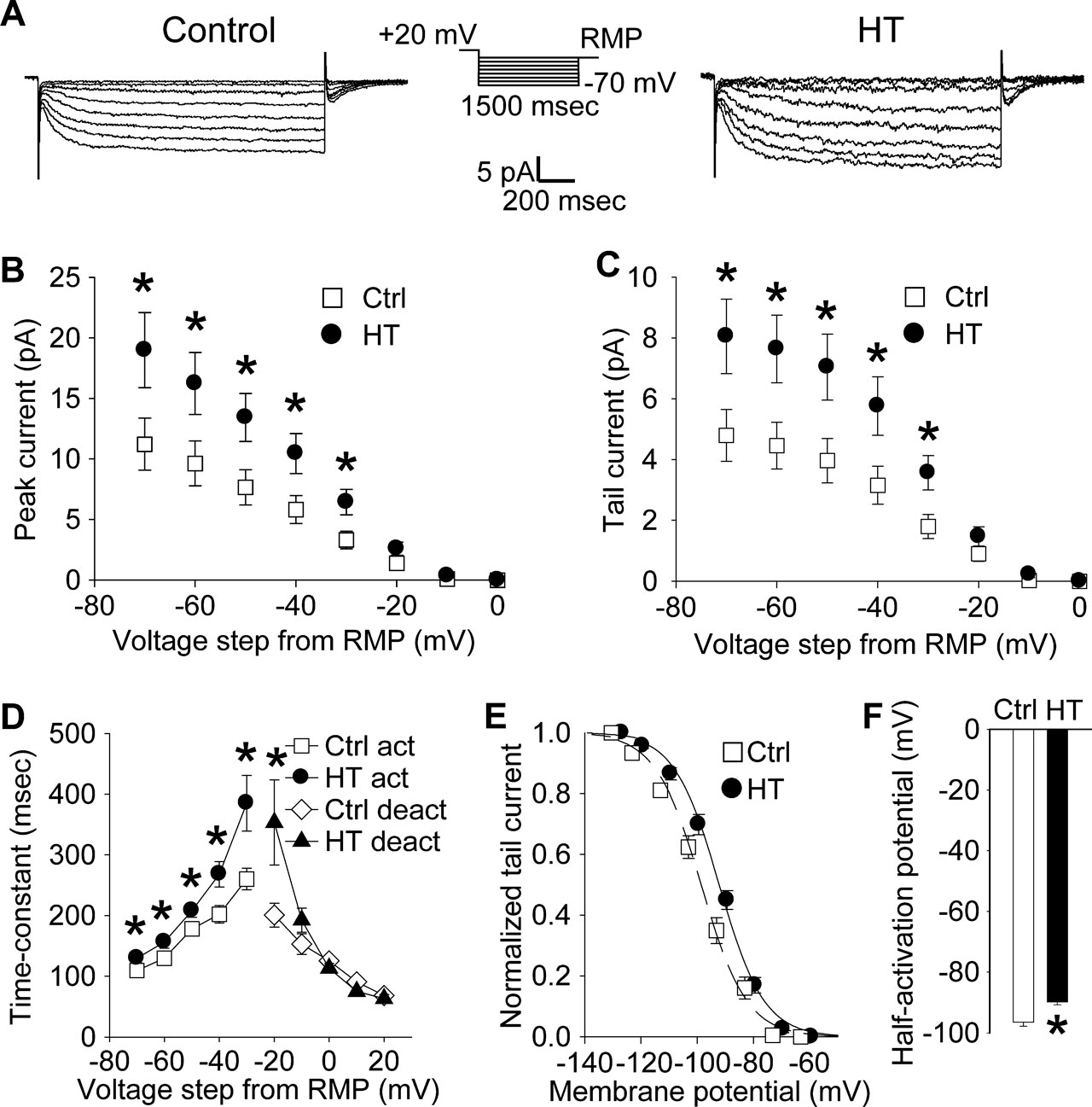
Figure 1. Increased h-current with altered kinetics in CA1 pyramidal apical dendrites following HT. (A) Dendritic cell-attached recordings of Ih at a 200 μm distance from the soma. Hyperpolarizing, 1500 ms long voltage commands from a holding potential 20 mV depolarized from rest produced slowly activating, non-inactivating inward currents characteristic of the h-current in control (left) and HT (right) dendrites. (B) Peak inward currents in control (open squares) and HT (filled circles) dendrites. (C) Tail currents from control (open squares) and HT (filled circles) dendrites. (D) Fitted activation (control, open squares; HT, filled circles) and deactivation (control, open diamonds; HT, filled triangles) time constants for the h-current. (E) Boltzmann activation curves for Ih fitted to normalized tail currents from control (open squares) and HT (filled circles) dendrites. (F) Half activation potential for Ih from control (open bar) and HT (filled bar) dendrites.
EPSP summation was tested by evoking a train of five EPSPs in an apical dendrite (at 200 μm from the soma) of both the control and HT versions of the Poolos et al. (2002) and Poirazi et al. (2003)
model cells. EPSPs were evoked at 50 Hz using a simulated synaptic conductance that was modeled as a double exponential function with rise and decay time constants of 5 and 20 ms, respectively (similar to the values used for simulating synaptic conductances in Poolos et al., 2002
), a maximum conductance of either 1 (small EPSPs in both Poolos et al., 2002
; Poirazi et al., 2003
models) or 7.6/2 nS (large EPSPs in Poolos et al., 2002
; Poirazi et al., 2003
models, respectively), and a reversal potential of 0 mV. Note that qualitatively similar results were obtained with different parameters as well (e.g. 0.5 ms risetime and 5 ms decay at 200 Hz). Recordings were made from the apical dendrite at the stimulation site.
Simulations were performed in the NEURON 5.6 simulation environment (Hines and Carnevale, 1997
) under 64-bit FedoraCore 2.6.12 on a Tyan Thunder 2.0 GHz dual Opteron server.
Analysis
Data were analyzed using Clampfit 9.2 (Molecular Devices, Union City, CA, USA) and SigmaPlot 8.0 (SPSS Inc., Chicago, IL, USA). For directly measured Ih from cell-attached recordings, leak-subtracted currents were low-pass filtered at 500 Hz before analysis. Statistical significance was determined using Student’s t-test. All measures are given as mean ± S.E.M.
Upregulated Dendritic Ih Following Experimental Febrile Seizures
Following prolonged hyperthermia-induced experimental seizures at P10-P11, Ih was characterized using cell-attached recordings from CA1 pyramidal neuron apical dendrites (200 μm from the soma) in 4–5-week-old control and experimental (HT) rats.
Hyperpolarizing voltage steps in the cell-attached configuration from a 20 mV depolarized holding potential (from RMP) elicited robust, slowly activating and non-inactivating inward currents in patches from both control (Figure 1
A, left panel) and HT dendrites (Figure 1
A, right panel). As determined from leak-subtracted currents, the amplitude of the peak inward currents (Figure 1
B) in HT dendrites (n = 13) was significantly increased compared to controls (n = 17) following hyperpolarizing voltage steps of −30 to −70 mV (following −70 mV steps: Ctrl: 11.2 ± 2.2 pA; HT: 18.9 ± 3.1 pA; p < 0.05; 69% increase). Similarly, the amplitude of the tail currents (Figure 1
C) was significantly increased in patches from HT dendrites compared to controls after hyperpolarizing voltage steps of −30 to −70 mV (following −70 mV steps: Ctrl: 4.8 ± 0.8 pA; HT 8.1 ± 1.2 pA; p < 0.05; 68% increase). This increase in the fully activated tail current demonstrates a directly measured 68% up-regulation of h-current density in the apical dendrites of CA1 pyramidal cells following experimental febrile seizures.
The time constants of Ih were determined from single-exponential fits to the leak-subtracted currents from control and HT dendrites. As shown in Figure 1
D, the time constants of Ih activation were significantly increased in HT dendrites compared to controls (at −30 mV steps: Ctrl: 260.3 ± 17.8 ms; HT: 384.9 ± 45.8 ms; p < 0.05; 48% increase). The deactivation time-constant was only significantly increased when stepping back from the −60 mV hyperpolarizing command potential to a −20 mV hyperpolarized holding potential (at −20 mV holding potential: Ctrl: 200.7 ± 19.9 ms; HT: 353.3 ± 69.7 ms; p < 0.05; 76% increase). The slowing of Ih kinetics is similar to previously reported data using whole-cell somatic recordings from CA1 pyramidal neurons following experimental febrile seizures (Chen et al., 2001
).
The Boltzmann activation curve for the measured h-current from control and HT dendrites was fitted using normalized tail-current amplitudes (the mean resting membrane potentials of control and HT dendrites were determined immediately after break-in from the cell-attached configuration; Ctrl: −63.1 ± 0.8 mV, n = 5; HT: −60.1 ± 0.5 mV, n = 5). As illustrated in Figure 1
E, the activation curve for Ih in HT dendrites was shifted towards more depolarized potentials, with the half-activation voltage V1/2 (Figure 1
F) being significantly more depolarized compared to controls (Ctrl: −96.5 ± 1.3 mV; HT: −89.8 ± 0.9 mV; p < 0.001; average shift in V1/2 = 6.7 mV). There was no significant difference in the slope factor k of the Boltzmann function (Ctrl: 8.1 ± 0.3; HT 8.0 ± 0.5; p > 0.8). These results are consistent with previously reported data from whole-cell somatic recordings (Chen et al., 2001
).
CA1 Pyramidal Cell Apical Dendrites are Hyperexcitable Following Experimental Febrile Seizures
Next, in order to establish whether the post-febrile seizure dendrites of CA1 pyramidal cells are hypo- or hyperexcitable, whole-cell current clamp recordings were made from control (Figure 2
A, left) and HT (Figure 2
A, right) dendrites at the same distance from the soma (200 μm) as the cell-attached recordings. As previously reported from somatic recordings (Chen et al., 2001
), the HT dendrites displayed a significantly depolarized RMP compared to controls (Ctrl: −63.1 ± 0.7 mV; HT: −59.5 ± 0.7 mV; p < 0.01; 3.6 mV depolarizing shift). When back-propagating action potentials (APs) were elicited using 1500 ms long depolarizing current injections from the RMP of each dendrite (i.e., when the voltage was allowed to “float”), the number of APs was significantly higher in HT dendrites (n = 7) compared to controls (n = 8) (Figures 2
A,B; at 200 pA: Ctrl: 10.6 ± 1.7 APs; HT: 18.1 ± 3.2 APs: p < 0.05; 71% increase). These data show, for the first time, that experimental febrile seizures induce a long-term increase in dendritic excitability in CA1 dendrites. Furthermore, these results also indicate that, although increased Ih is often thought to be associated with decreased excitability, the post-febrile seizure upregulation of Ih occurs in hyperexcitable CA1 dendrites.
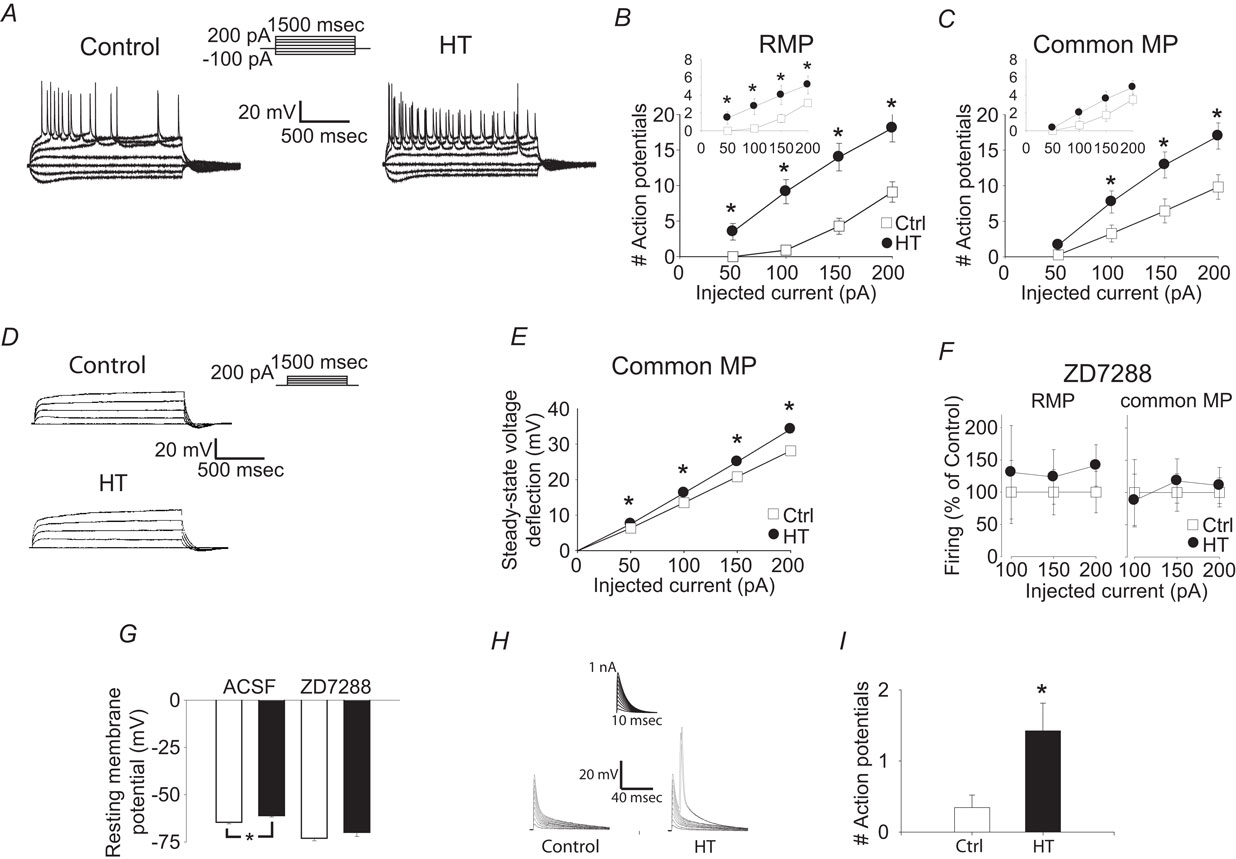
Figure 2. Whole cell recordings from CA1 pyramidal apical dendrites show that h-current alterations are not solely responsible for hyperexcitability. (A) Whole cell, current clamp recordings of control (left) and HT (right) dendrite responses to 1500 ms long current injections from RMP. Recording distance 200 μm from soma. (B) Number of action potentials fired in response to 1500 ms depolarizing current injections in control (open squares) and HT (filled circles) dendrites from the RMP. (C) Number of action potentials fired in response to 1500 ms depolarizing current injections in control (open squares) and HT (filled circles) dendrites from a −60 mV holding potential. Insets: Number of action potentials in the first 300 ms of the depolarizing current injections. Note that the data in (B) and (C) are from the same population of dendrites, before and after compensation of the RMP difference by constant current injection. (D) Whole cell, current clamp recordings of control (top) and HT (bottom) dendrite responses to 1500 ms long current injections (recording distance: 200 μm) in the presence of 1 μM TTX, from a −60 mV holding potential. (E) Steady-state voltage deflection of control (open squares) and HT (filled circles) dendrites in response to 1500 ms current injections from a −60 mV holding potential. (F) Action potentials fired (% of control) in control (open squares) and HT (filled circles) dendrites in the presence of ZD7288, from RMP (left) and from a −70 mV holding potential (right; recording distance: 200 μm). (G) RMP of control (white bars) and HT (black bars) dendrites in the absence or presence of ZD7288. (H) Control and HT dendritic responses to artificial EPSPs generated by current injection from the recording pipette (recording distance: 200 μm) from RMP. (I) Average number of action potentials fired in response to the artificial EPSPs in (H).
Could the upregulated Ih be contributing to hyperexcitability, for example, through depolarization of the dendritic RMP that may overcome the effects of the increased dendritic conductance? In order to address this question, current injections were also performed from a common membrane potential (−60 mV) in the same dendrites as in the previous experiments. When dendritic excitability was tested from −60 mV using long (1500 ms) depolarizing pulses, the dendrites from HT animals were still more excitable compared to the controls (Figure 2
C), indicating that upregulation of Ih cannot be solely responsible for the hyperexcitability observed with these long pulses. When the same dataset was reanalyzed by focusing only on the first 300 ms (after 300 ms, firing is likely not influenced by Ih due to its deactivation time constant of less than 100 ms), the difference in firing was significant at RMP but non-significant at common MP (insets in Figures 2
B,C), suggesting that non-Ih conductances may be especially involved in the generation of hyperexcitability during the later parts of the long current pulses. In order to obtain further insights into the potential involvement of non-Ih current(s) in the febrile seizure-induced persistent hyperexcitability, we performed whole cell voltage clamp experiments in the presence of the Na+-channel blocker tetrodotoxin (TTX). The steady-state voltage deflections following 1500 ms depolarizing current pulses from a common MP (−60 mV) were consistently larger in dendrites from HT animals (n = 9) compared to controls (n = 11) (Figures 2
D,E). Since Ih was fully deactivated by the end of these long depolarizing pulses (see Figure 1
), these data suggest that the dendritic hyperexcitability observed after febrile seizures in CA1 pyramidal cells is at least partially due to long-term alterations in a current (or currents) distinct from Ih (note, however, that the latter conclusion does not exclude some pro excitable role for Ih, see below; note also that an unequivocal experimental assessment of the role of non-Ih conductances in post-febrile seizure dendritic excitability is confounded by the possibility that, when dendritic MPs from control and HT cells are held at the same value by current through the recording pipette, differences in MPs at locations away from the recording electrode could still exist between these two groups, due to the non-isopotential nature of pyramidal dendrites). The Ih blocker ZD7288 (100 μM) decreased both the difference in firing between control and HT cells (Figure 2
F) as well as the difference between the RMPs below statistically significant levels (Figure 2
G; RMPs in ACSF: Ctrl: −64.5 ± 0.9 mV; HT: −61.1 ± 0.7 mV; p < 0.05; in ZD: Ctrl: −73.0 ± 1.4 mV; HT: −69.9 ± 2.2 mV; not significant; n = 9/9), in an apparent contrast to the data in Figures 2
D,E (but see Discussion on potential non-specific effects of ZD7288). Finally, when artificial EPSPs were generated by current injections through the recording pipette, the cells from the HT animals fired significantly more action potentials from RMP compared to controls (Figures 2
H,I; Ctr: n = 27; HT: n = 33), in agreement with the results shown in the inset in Figure 2
B.
Detailed CA1 Pyramidal Cell Models Predict Hyperexcitability due to Altered Ih
The data in Figure 2
highlighted the difficulty in unequivocally determining the role of the upregulated Ih in the post-febrile seizure dendritic hyperexcitability. Therefore, in order to isolate the role of Ih alterations in HT dendritic excitability from other unidentified changes, we turned to computational modeling. To ensure the robustness of our simulation results, the effect of the altered h-current was tested in three previously published models from different research groups. These detailed CA1 pyramidal neuron models differ from each other in their complexity, and they had previously been used to investigate the effects of Lamotrigine modulation of Ih on dendritic excitability (Poolos et al., 2002
), action potential backpropagation in apical dendrites (Golding et al., 2001
) and synaptic summation (Poirazi et al., 2003
). For all simulations, the “HT” model cell was identical to the “control” model cell for the particular model being utilized, except for the inclusion of the experimentally quantified alterations in Ih discussed previously.
The experimentally determined Ih alterations resulted in a depolarized RMP in all three versions of HT model cell (range of RMP shift was 2.9–4.7 mV). As seen in Figures 3Ai, Aii; Bi, Bii; Ci and Cii
, the HT model cells were hyperexcitable compared to control model cells in response to 1500 ms dendritic current injections (at 200 μm distance from the soma), when tested from their respective RMP. In each model, the HT cell fired action potentials in response to smaller current injections than the control (Figures 3Ai, Bi and Ci
), and displayed hyperexcitability (quantified by an increased number of APs) throughout a range of current injection amplitudes (Figures 3Aii, Bii and Cii
). While the Poolos et al. (2002)
HT model cell showed only modest hyperexcitability that was abolished at large current injection amplitudes (Figure 3
Aii), both the Golding et al. (2001) and Poirazi et al. (2003)
HT model cells showed large and persistent increases in excitability (Figures 3
Bii, Cii). These increases in excitability existed despite 14, 24, and 44% decreases in the input resistance of the HT model cells compared to controls in the Poolos et al. (2002)
, Golding et al. (2001)
, and Poirazi et al. (2003)
models, respectively. However, in contrast to the whole cell experiments, when the control and HT cells were held at a common membrane potential, differences between HT and control cells were abolished or even reversed (Figures 3Aiii, Biii, and Ciii
).
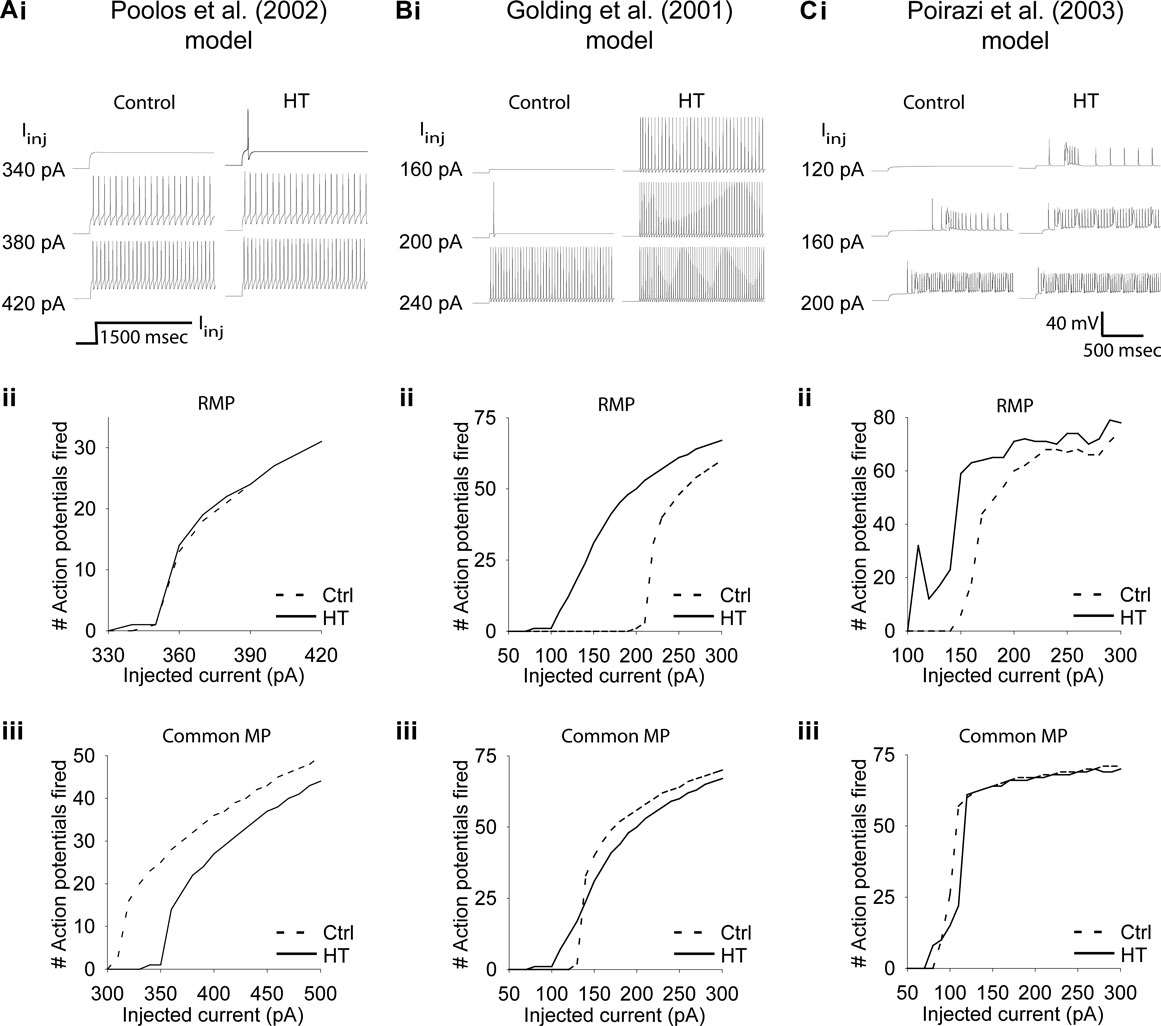
Figure 3. Altered h-current causes hyperexcitability in three detailed CA1 pyramidal neuron models. (Ai, Bi, Ci) Example traces of response to 1500 ms simulated apical dendritic current injections with control (left) and HT (right) h-current parameters. Note that HT models always fire at lower current injection amplitudes than controls. (Aii, Bii, Cii) Number of action potentials fired in response to 1500 ms simulated dendritic current injections in control (dashed line) and HT (solid line) model cells, from their respective RMPs. (Aiii, Biii, Ciii) Number of action potentials fired in response to 1500 ms simulated apical dendritic current injections in control (dashed line) and HT (solid line) model cells, from a common holding potential. Note that due to specific model parameters, the common holding potential varied between models.
To further clarify the effects of Ih on dendritic excitability, we performed modeling experiments to determine the effects of the experimentally observed Ih alterations on EPSP summation in both the Poolos et al. (2002) and Poirazi et al. (2003)
cells. A train of five small EPSPs (see Materials and Methods), evoked from the RMPs of the control and HT model cells showed that the HT cells reached a more depolarized membrane potential than the controls at the end of the train (Figures 4
Figures Ai, Di). However, an overlay of the control and HT responses to the EPSP trains indicates that the EPSPs summated to a lesser degree in the HT model cells (Figures 4
Figures Aii, Dii), in agreement with previous findings regarding the role of Ih in temporal summation (Magee, 1998
; Poolos et al., 2002
). Additionally, trains of large EPSPs evoked action potentials in the HT model cells while no action potentials were fired in controls (Figures 4
Bi, Ei). Again, an overlay shows that there was less EPSP summation in the HT cells (Figures 4
Bii, Eii), but the depolarized RMP overcame this effect and resulted in hyperexcitability. To confirm that the RMP shift was the cause of the increased excitability in the HT model cells, we held both the control and HT cells at the same membrane potential and applied the large trains of EPSPs. In this case, the HT model cells did not fire any action potentials and the control cells were clearly more depolarized than the HT cells at the end of the EPSP trains (Figures 4
C,F).
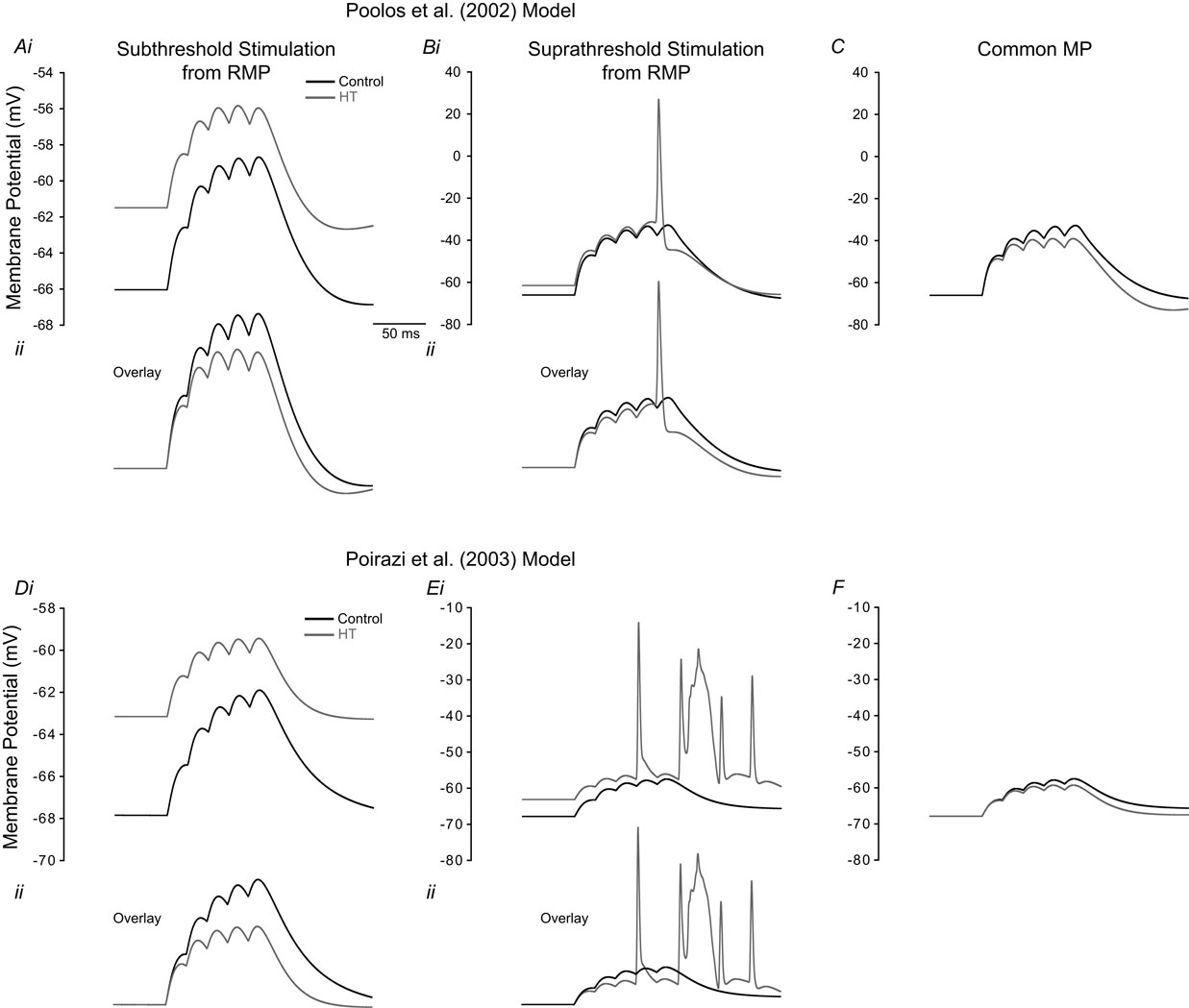
Figure 4. Model HT cell shows reduced EPSP summation but increased firing than control due to depolarized RMP. (A–C) refer to simulations performed using the Poolos et al. (2002)
model, and (D–F) are simulations using the Poirazi et al. (2003)
model. (Ai, Di) Train of five small EPSPs from the RMP in control (black) and HT (grey) model cells. (Aii, Dii) Overlay of the traces in (Ai, Di) showing decreased EPSP summation in the HT model cells. (Bi, Ei) Train of five large EPSPs, resulting in AP firing in the HT model cells (grey), but not in controls (black). (Bii, Eii) Overlay of the traces in (Bi, Ei). (C, F) Train of five large EPSPs in control (black) and HT (grey) model cells, from a common holding potential. Note that compensation of the depolarized RMP in the HT model cells eliminates AP firing.
These modeling results suggest that the reported Ih alterations may contribute to hyperexcitability in CA1 pyramidal cells after febrile seizures. They also support our experimental findings that Ih is unlikely to be the sole cause of hyperexcitability in HT dendrites, as hyperexcitability was invariably abolished upon compensation of the RMP change.
In this study, we report that (1) Three to 4 weeks after experimental febrile seizures, cell-attached dendritic recordings showed that Ih was persistently upregulated in CA1 pyramidal cell dendrites, characterized by depolarized half-activation potential and altered kinetics; (2) In whole cell recordings, dendrites from animals that experienced experimental febrile seizures showed markedly enhanced excitability when tested from the RMP. However, this increased excitability (when tested with long depolarizing pulses) was not eliminated after compensation of the depolarized post-febrile dendritic RMP, indicating that unidentified currents other than Ih may also play a role in long-term, dendritic hyperexcitability after febrile seizures; (3) Simulations implementing the experimentally determined h-current alterations in published CA1 neuron models predicted that the observed Ih changes may contribute to hyperexcitability. Taken together, these experimental and computational data reveal an unexpected complexity of the relationship between Ih plasticity and epilepsy, by demonstrating that increased Ih can exist in hyperexcitable dendrites and may even promote dendritic excitability through its depolarizing effects on the RMP.
Persistently Hyperexcitable Dendrites After Febrile Seizures
The present study was initiated in order to determine whether the Ih changes observed in somatic whole cell patch clamp recordings after experimental febrile seizures (Chen et al., 2001
) are comparable to the Ih alterations measured using direct dendritic cell-attached recordings. The data confirmed that febrile seizures result in a depolarizing shift in Ih activation curves, no change in the slope of the Boltzmann fit (V1/2 shift from somatic recordings, from the 9-week post-febrile data in Chen et al. (2001)
: −91.0–85.1 mV, a significant 5.9 mV shift; from the current dendritic cell-attached data obtained 4–5 weeks after febrile seizures: −96.5–89.8 mV, a significant 6.7 mV shift; slope factor k: from somatic recordings: 8.9–8.8, no change; from current dendritic recording: 8.1–8.0, no change), and a slowing of the activation and deactivation kinetics.
In addition to confirming the previously reported long-term alterations in Ih using more direct electrophysiological techniques, the current study demonstrated, for the first time, that the CA1 pyramidal cell apical dendrites are persistently hyperexcitable following a single episode of prolonged, experimental febrile seizures in infancy. These findings are interesting and important for the ongoing efforts to understand the relationship between early-life prolonged seizures and the development of temporal lobe epilepsy later in life (Annegers et al., 1987
; Baram et al., 1997
; Chen et al., 2007
; Dube et al., 2000
, 2006
;Schuchmann et al., 2006
). Our data also suggest that in addition to Ih, other, currently unidentified conductances may also potentially contribute to the generation of the hyperexcitable responses in pyramidal cell dendrites after febrile seizures. Future research is needed to identify these non-Ih-related, long-term plastic changes occurring in the dendrites following early-life seizures. Nevertheless, it is interesting to point out that upregulated Ih has been observed concurrently with a downregulated TEA-sensitive delayed rectifier current in mossy cells of the dentate gyrus following traumatic brain injury (Howard et al., 2007
), and it is possible that dowregulation of a K+-current or currents (Bernard et al., 2004
) is also involved in the hyperexcitable responses observed after febrile seizures in pyramidal cell dendrites. It is possible that the inability of the Ih blocker ZD7288 to reveal an h-current independent difference in firing between control and HT cells in our experiments is associated with, and thus confounded by, non-specific effects of ZD7288 on non-Ih conductances (Chen, 2004
; Chevaleyre and Castillo, 2002
; Do and Bean, 2003
; Nolan et al., 2004
).
Seizure-Induced Persistent Ih Upregulation and Epilepsy
Studies in animal models of epilepsy have frequently reported down-regulation of Ih and dendritic hyperexcitability (Kole et al., 2007
; Shah et al., 2004
; Strauss et al., 2004
; Zhang et al., 2006
). In an apparent contrast to these prior findings from other epilepsy models, prolonged febrile seizures in the developing hippocampus resulted in an upregulation of Ih (Chen et al., 2001
; present study), which was also accompanied by enhanced dendritic excitability. However, as discussed above, the post-febrile, persistent dendritic hyperexcitability is at least partially due to changes in non-Ih-related conductances, raising the question of precisely what role Ih plays in dendritic hyperexcitability after febrile seizures. In order to study the role of febrile seizure-induced changes in Ih on dendritic excitability in isolation, separate from any known and unknown changes that may occur in the post-febrile tissue, we turned to computational modeling techniques.
The implementation of the experimentally observed febrile seizure-induced long-term modifications in Ih in three distinct CA1 pyramidal cell models consistently revealed that the seizure-induced upregulation in Ih most often resulted in hyperexcitable responses. The actual amount of increased excitability differed greatly between the models, indicating that the effect of the enhanced Ih will be strongly influenced by the electrophysiological parameters of neurons. Additional computational analysis showed that the mechanism underlying the ability of upregulated Ih to enhance excitability was due to its depolarizing effect on the dendritic RMP. Although these computational modeling predictions will need to be further examined and carefully tested in subsequent experimental studies in various epilepsy paradigms, these results highlight the complex relationship between Ih and neuronal excitability, and stress the need for caution in predicting functional effects from the direction of the changes in Ih alone. Indeed, it seems that it is possible to increase dendritic excitability with both a near-complete loss of Ih (as occurs in the entorhinal cortex in the kainic acid model; Shah et al., 2004
) and with a relatively modest increase in Ih (as predicted by the simulation results in the current study). In studies implicating a downregulation of Ih in hyperexcitability (Kole et al., 2007
; Shah et al., 2004
; Strauss et al., 2004
; Zhang et al., 2006
), the increase in input resistance with less active h-conductance outweighed the accompanying hyperpolarization of the RMP. Conversely, if the effect of the depolarized RMP dominates the effect of the decreased input resistance following a modest upregul-ation of Ih, the net effect will be a hyperexcitable cell or dendrite as reported in the present study.
The previous cautionary note is further underlined by the accumulating recent evidence that homeostatic mechanisms often come into play following epileptogenic insults (Echegoyen et al., 2007
; Howard et al., 2007
), which may involve highly specific, concurrently regulated, apparently closely linked changes in multiple ion channels (Gibson et al., 2006
; Prinz et al., 2004
; Schulz et al., 2007
). These considerations suggest that the functional impact of Ih alterations in epilepsy will be model-, and perhaps also cell type-specific, especially when the functional effects of relatively subtle changes are being considered. The complexity of the functional effects (including interactions between Ih, EPSPs, and calcium spikes, Tsay et al., 2007
) is likely governed by correspondingly diverse molecular mechanisms ranging from modulation by second messengers, alterations in channel distribution, and transcriptional channelopathies (Jung et al., 2007
; Poolos et al., 2006
; Richichi et al., 2007
; Shin and Chetkovich, 2007
).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors wish to thank Drs Jeffrey C Magee (Howard Hughes Medical Institute, Janelia Farms), Frederic Pouille and Massimo Scanziani (UCSD) for their generous help and advice concerning dendritic recording techniques, and Ms Rose Zhu for excellent technical assistance. This study was supported by the Epilepsy Foundation/Milken Family Foundation (EFA-36656 to JDJ) and the NIH (NS38580 to IS).
Crasto, C. J., Marenco, L. N., Liu, N., Morse, T. M., Cheung, K. H., Lai, P. C., Bahl, G., Masiar, P., Lam, H. Y., Lim, E., Chen, H., Nadkarni, P., Migliore, M., Miller, P. L., and Shepherd, G. M. (2007). SenseLab: new developments in disseminating neuroscience information. Brief Bioinform. 8, 150–162.
Nolan, M. F., Malleret, G., Dudman, J. T., Buhl, D. L., Santoro, B., Gibbs, E., Vronskaya, S., Buzsaki, G., Siegelbaum, S. A., Kandel, E. R., and Morozov, A. (2004). A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell 119, 719–732.