Laboratory for Neuronal Circuit Dynamics, RIKEN Brain Science Institute, Wako, Japan
The relatively simple and highly modular circuitry of the cerebellum raised expectations decades ago that a realistic computational model of cerebellar circuit operations would be feasible, and prove insightful for unraveling cerebellar information processing. To this end, the biophysical properties of most cerebellar cell types and their synaptic connections have been well characterized and integrated into realistic single cell models. Furthermore, large scale models of cerebellar circuits that extrapolate from single cell properties to circuit dynamics have been constructed. While the development of single cell models have been constrained by microelectrode recordings, guidance and validation as these models are scaled up to study network interactions requires an experimental methodology capable of monitoring cerebellar dynamics at the population level. Here we review the potential of optical imaging techniques to serve this purpose.
The cerebellar cortex, the deep cerebellar nuclei and the inferior olive form the three main hubs of the cerebellar circuit. Each of these components of the olivo-cortico-nuclear circuit involves a relatively modest number of cell types, with synaptic connectivity within and between the hubs being highly parallel and modular. To date, the electrophysiological properties of most cerebellar cell types have been characterized in detail, resulting in data that has allowed the construction of compartmental conductance-based single cell models. Thus, for most cerebellar cell types, a digital representative already exists, that faithfully implements its active membrane properties and the transformation of synaptic inputs into spiking outputs. It is these cellular models which are in turn used as the foundations to computationally rebuild the cerebellar circuit (for review see Medina and Mauk, 2000
). In order for these models to generate realistic data when scaled up to the network level, additional parameters such as functional connectivity and population dynamics must be incorporated, however much of this information is either conflicting or missing.
It should be stressed here that functional connectivity cannot be derived from anatomical information only. One particular example related to cerebellar circuitry where a discrepancy between anatomical and functional connectivity became an issue, involves the synapses between granule cells and Purkinje cells. It is well established anatomically that granule cell axons form synapses with Purkinje cells, via the ascending axons entering the molecular layer, and also following bifurcation as the parallel fibers contact with Purkinje cell dendrites (see Huang et al., 2009 and references cited therein). However, it is still under debate as to, whether granule cells influence the firing of Purkinje cells predominantly via the ascending axon synapses or via the parallel fiber system. With this in mind, optical imaging data combines both structural and functional information and therefore is, at least in principle, well suited in resolving such issues (For further discussion on these issues see: Cohen and Yarom, 1998
; Coutinho et al., 2004
; Vranesic et al., 1994
).
Whilst there remains a significant lack of detailed information related to functional connectivity between major cerebellar neuronal populations, “educated guesswork-based” network models have been constructed, with emerging circuit properties that can replicate experimental observations. However, since these cerebellar circuit models are based on connectivity assumptions, their power of prediction is somewhat limited. This problem can be ameliorated not only by providing better information about functional connectivity but also by validating experimentally the dynamics that emerge from circuit modeling. Here we review the potential of optical imaging methodologies to serve these purposes. This focused review does not cover systems other than the cerebellum, and highlights how optical imaging has been applied in the context discussed above.
Optical imaging most often refers to imaging modalities based on fluorescent probes that transform network activity into optical signals. There are, however, approaches that use “intrinsic” optical signals that correlate with neuronal activity. These methods rely on changes in light absorption and scattering or tissue autofluorescence, negating the need for exogenous application of chemical dyes (Chance et al., 1962
; Coutinho et al., 2004
; Reinert et al., 2004
, 2007
; Rossini et al., 1991
). None of these “intrinsic” optical imaging methods have been extensively used in the cerebellum to obtain information that is relevant for construction of large scale circuit models. An exception is perhaps FAD (flavin adenine dinucleotide) green autofluorescence imaging, which has been used to investigate the metabolic consequences of synaptic excitation and inhibition (Gao et al., 2006
), the functional connectivity in the mossy fiber-granule cell-Purkinje cell pathway and the topology of climbing fiber projections in vitro and in vivo (Coutinho et al., 2004
; Reinert et al., 2004
, 2007
). Imaging based on hemodynamic events used extensively for studying changes in cerebrocortical neuronal activity (Vanzetta et al., 2005
) has yet to be successfully exploited for the cerebellum. There are optical imaging approaches other than those covered in this review, most notably are the exciting advances in application of second harmonic generation microscopy to monitor cerebellar electrical activity (Sacconi et al., 2008
).
Fluorescent calcium indicators can be added to the internal solution of glass electrodes used for intracellular recordings. Filling cells with such calcium sensitive dyes allows for measurements of calcium dynamics at the single cell level with excellent sensitivity and spatio-temporal resolution. There are, however, only a few cases where single cell calcium imaging data were used to fine-tune realistic computational models of cerebellar neurons. Examples include the optically determined localization of calcium channels in Purkinje cells, granule cells and deep cerebellar nuclei (Gall et al., 2005
; Knöpfel et al., 1991
; Muri and Knöpfel, 1994
; Tank et al., 1988
) and the corresponding compartmental models (De Schutter and Bower, 1994a
,b
; Diwakar et al., 2009
; Quadroni and Knöpfel, 1994
). Whilst calcium imaging in single cells has not led to useful constraints for circuit models, calcium imaging of large populations of cerebellar neurons at single cell resolution could potentially provide information that is not readily accessible by microelectrode techniques.
The most widely used calcium imaging approach to investigate activity in intact circuits is based on staining with membrane permeable calcium indicator dyes (that become impermeable and trapped following intracellular cleavage of a lipophilic side group), which also allows for the simultaneous sampling of many single cells (Ikegaya et al., 2004
; Kerr et al., 2005
). In the cerebellum, this AM dye technology when combined with cutting edge optical systems and specific staining protocols (Garaschuk et al., 2006
) has proven successful to permit the monitoring of CF activities of several Purkinje cells in the living, and even, freely moving rodent cerebellum (Engelbrecht et al., 2008
; Sullivan et al., 2005
). It should be noted however, that AM calcium based imaging in vivo has been demonstrated only in the molecular layer with staining of parallel fibers, interneurons and Purkinje cells (e.g. Sullivan et al., 2005
). Functional imaging of deeper circuit elements (like granule cells and Golgi cells) has not yet been clearly demonstrated and is, according to some investigators, hampered by excessive light scattering in the cerebellar granule cell layer (Sullivan et al., 2005
).
Taken together, calcium imaging techniques in the cerebellum in vivo, are still being methodologically refined, but it is anticipated these techniques have a high potential to provide experimental data by which network simulations can be constrained and validated in the near future.
In contrast to calcium imaging, which is limited by the temporal precision of the calcium signals themselves, voltage sensitive dyes bind to the plasma membrane, where their fluorescence is modulated directly by changes in membrane potential, allowing faithful optical reporting of electrical signals with very rapid kinetics, including single action potentials. Indeed, cerebellar voltage sensitive dye imaging was demonstrated over a decade ago, enabling the activity of single cerebellar Purkinje cells to be imaged in cultured neurons (Staub et al., 1994
), see Figure 1
.
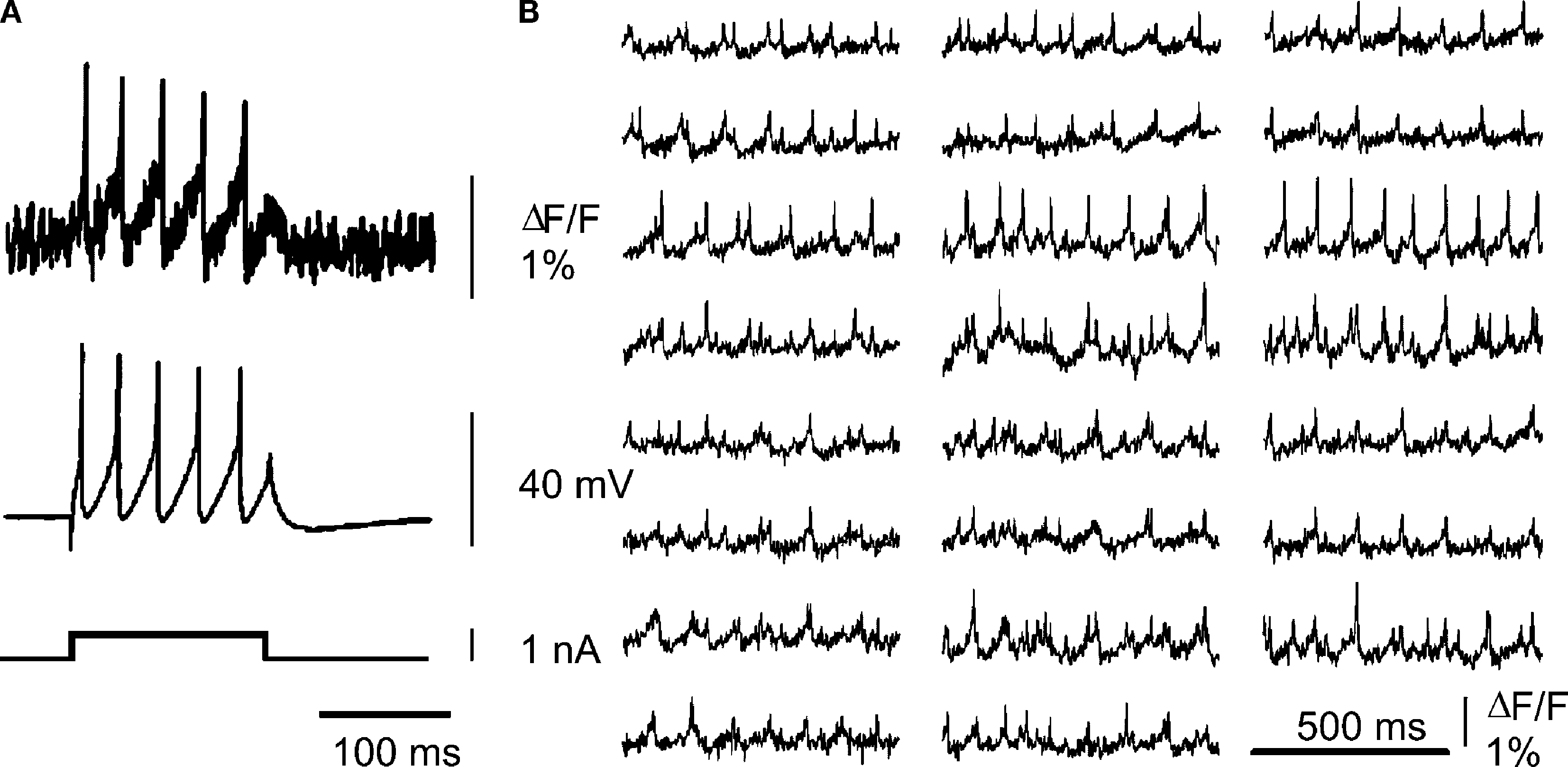
Figure 1. Voltage sensitive dye imaging in cerebellar slice cultures. Cerebellar cultures containing granule cells and Purkinje cells were stained with the voltage sensitive dye di-4-ANEPPS. (A) Optical voltage signal (upper trace) and simultaneous microelectrode recording from a Purkinje cell during injection of a current pulse (lower trace). (B) Simultaneous optical recording of spontaneous spiking activity of several Purkinje cells. For methodological details see Staub et al. (1994)
.
The promise of resolving single action potentials from acute cerebellar slices however, was only achieved when dyes were intracellularly applied via patch pipettes (Zhou et al., 2008
). Of course the rationale behind using imaging over conventional electrophysiology, is that theoretically, optical approaches would permit the electrical activity of large populations of neurons to be monitored, at single cell resolution in intact tissue. However to date this has not yet proven feasible. The reason that success with voltage sensitive dye imaging has been limited, involves their main drawback, in that, they (when exogenously applied) stain tissue indiscriminately (including the membranes of all neurons and glial cells within the tissue). The implications this has for recording of optical activity is that signals of interest from specific morphologically identified neurons are diluted by a large background of stained cells within the tissue, meaning that in reality signals are too small to allow for isolation of specific cellular responses.
Despite the outlined drawbacks, voltage sensitive dyes have proven useful for monitoring network activity, in which the imaged signals reflect summations over many cells. In some cases these compound optical signals have been dissected by pharmacological means, allowing them to be attributed to specific cellular subpopulations. This approach has been used to isolate parallel fiber action potentials, and to characterize their transverse propagation through the molecular layer and their excitation of Purkinje neurons (Cohen and Yarom, 1998
; Vranesic et al., 1994
). Whilst the dream scenario of classical voltage sensitive dyes providing a tool to monitor the activity of many hundreds of neurons (or whole networks) simultaneously at single cell resolution still seems far away, their ability to monitor population responses has already been successfully exploited. This is a particularly pertinent property when one considers computational modeling of neuronal structures such as the cerebellum, since more detailed single-cell electrophysiology recordings are readily available characterizing specific cellular behaviors. The forte of optical voltage imaging is that by varying the spatial scale, activity of whole networks can be recorded, demonstrating the spatial extent over which, and with what temporal precision, areas within a given network can synchronize, with the added advantage that these data can be gathered non-invasively (i.e. without altering intracellular ionic compositions that are associated with patch-clamping).
It is well-established that neuronal networks, including those residing within the cerebellar cortex generate oscillatory activity (Middleton et al., 2008
; Niedermeyer, 2004
; Soteropoulos and Baker, 2006
; Timofeev and Steriade, 1997
), and these have been shown in cerebrocortical regions to be associated with the temporal coding of various features of sensory stimuli (Canolty et al., 2006
; Gray et al., 1989
; Womelsdorf et al., 2007
). However when considering the precise, spatio-temporal patterning and propagation of these rhythms, conventional microelectrode techniques become impractical. To this end, voltage dye imaging has been successfully used to study network dynamics in the inferior olive (IO) in vitro. In this study of Rodolfo Llinas and co-workers, IO local electrical stimulation was used to reset the phase of subthreshold oscillations and entrain a large population of IO neurons. Whilst intracellular recordings demonstrated that individual cells display membrane potential oscillations, optical imaging was used to reveal that at the network level, such stimulation produced oscillations which clustered in to coherent populations comprising hundreds of neurons (Leznik et al., 2002
; Llinas et al., 2002
). Notably these population responses were used to validate and interpret the behavior of computational simulations of the IO network (Llinas et al., 2002
).
More recently, voltage imaging was used to elucidate the spatial expression of cerebellar high frequency oscillations (Middleton et al., 2008
). Nicotinic receptor activation in the cerebellum is associated with generation of high frequency network oscillations, which field potential recordings reveal to be a mixture consisting of classical gamma band oscillations (40 Hz), co-existing with very fast oscillations (VFOs, 80–160 Hz). Optical signals however, accurately localize the changes in membrane potential, demonstrating that gamma frequency rhythms are confined predominantly to the Purkinje cell layer and distal white matter, whilst VFO peak power originates in the granule cell layer and diffusely throughout the white matter (see Figure 2
).
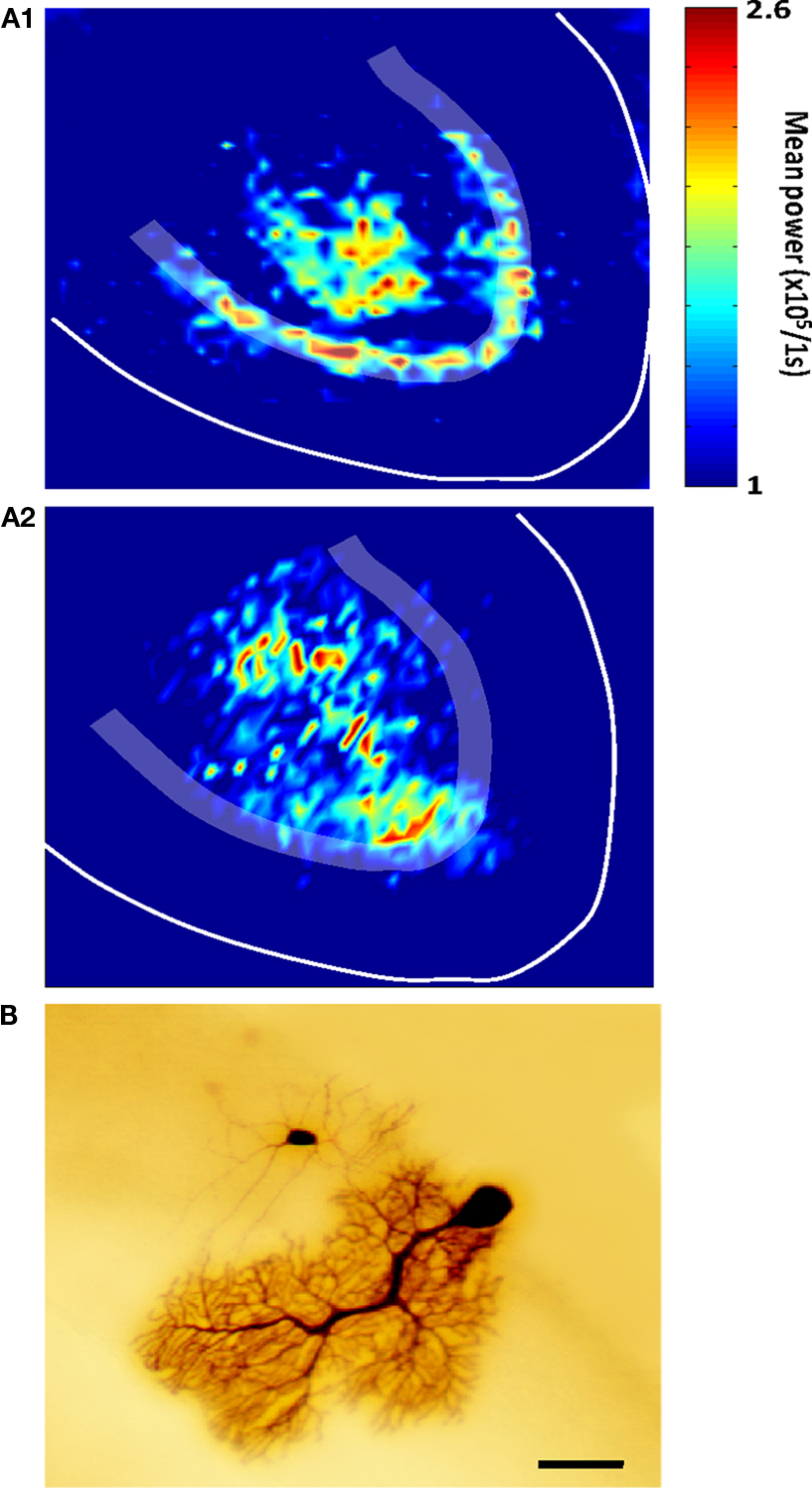
Figure 2. Optical imaging of cerebellar high-frequency oscillations. (A1) Pooled peak power in the gamma band (20–60 Hz) from power spectra of optical data morphed onto an idealized cerebellar slice, with similar data shown for VFOs (80–160 Hz) shown in (A2) (n = 5, 1s epochs). Note that gamma frequency and very fast oscillations are predominant in different spatial locations, an observation confirmed by electrophysiological recordings. (B) An example of heterocellular dye-coupling between a Purkinje cell and an inhibitory interneuron. Note: biocytin was injected only into the Purkinje neuron during extracellular high frequency oscillations and is presumed to diffuse to the other cell via gap junctions. Taken from Middleton et al. (2008)
.
Given the laminar localization of VFOs in the optical signals together with pharmacological data with gap junction blockers, it was hypothesized that gap junctions between Purkinje cell axons may be responsible, or at least contribute to the VFOs. For this reason, a computational model based on these data was constructed to show that a network of Purkinje cells with gap junctions located on their proximal axons was sufficient to reproduce VFOs (Traub et al., 2008
). Furthermore, predictions from the model, namely that the frequency of the VFOs was gated by sodium channel inactivation were subsequently proven experimentally by pharmacologically prolonging the corresponding inactivation time constant (Traub et al., 2008
). Further examples where voltage sensitive dye data has been incorporated into computational models are detailed elsewhere (Sergio Solinas and Egidio D’ Angelo, A realistic large-scale model of the cerebellum granular layer predicts circuit spatio-temporal dynamics, submitted; Jonathan Mapelli, Daniela Gandolfi and Egidio D’ Angelo, High-pass filtering properties of granular to molecular layer transmission revealed by high-resolution voltage-sensitive dye imaging, submitted).
These studies demonstrate that whilst optical imaging with voltage sensitive dyes has not yet been exploited to its full potential, for understanding interactions macroscopically at the network level and in combination with computational approaches, it remains a powerful tool. Of course it remains that for most cases it would be more preferable to simultaneously monitor single cells, whilst also observing the network in which they are embedded. In order to make this feasible in terms of signal to noise and interpretation of the recorded signals, it would be preferable to have specific populations of cells labeled (i.e. only Purkinje cells). To overcome these issues, genetically encoded sensors (of either calcium or membrane potential) are preferable; these are discussed in detail below.
Ideally, computed cerebellar circuit dynamics could be directly matched with experimental data if recording methods existed that would allow recordings of large populations of identified neurons in vivo. Whilst, to date, this hasn’t yet been achieved, imaging using genetically encoded probes could, at least in principle, make this possible. These nascent optogenetic approaches utilize fluorescent protein calcium or voltage sensors, with expression driven by cell type specific promoters, thus reporting activity from genetically defined cell populations (Knöpfel et al., 2006
; Luo et al., 2008
). Moreover, fluorescent protein-based probes can be constructed with multiple colors such that signals from “color tagged” cells or cell populations can be spectroscopically isolated even if single cells are not optically resolved. Figure 3
illustrates genetic color tagging in a line of mice where green fluorescent protein is expressed in GABAergic cells (via the GAD 67 promoter) and yellow fluorescent protein is expressed in granule cells via the Kv3.1 promoter (Metzger et al., 2002
; Tamamaki et al., 2003
).
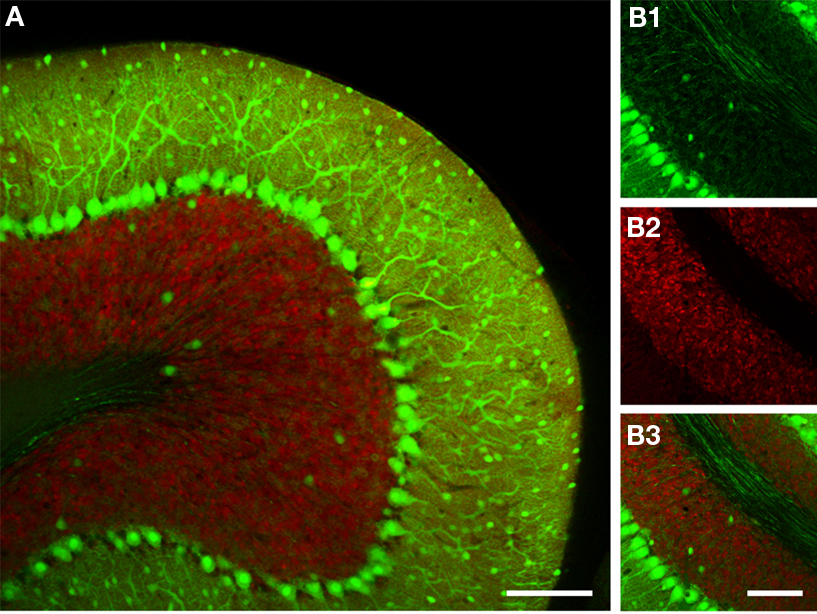
Figure 3. The use of fluorescent protein color variants to genetically define multiple cell populations. (A) A confocal image of a cerebellar slice from a transgenic mouse expressing GFP and EYFP via the GAD 67 and Kv3.1 promoters respectively. Images were acquired with a 32-channel spectral resolution on a Nikon C1si system and deconvoluted with GFP and EYFP spectra. The resulting GFP and EYFP signals are shown in green, and red false colors to enhance contrast. In (B) are shown images with larger magnification of the separate channels demonstrating the very high resolution that can be achieved with genetic approaches (note individual Purkinje cell axons). (B1) shows only GFP fluorescence highlighting the dense axonal network formed by the Purkinje cells and interneurons, whilst in (B2) only EYFP is visible in the numerous small granule cells, finally (B3) shows both channels. Scale bar throughout: 100 μm.
Genetically-encoded calcium probes (GECPs) are proteins with engineered, calcium-sensitive fluorescence properties, as reviewed in (Knöpfel et al., 2006
; Mank and Griesbeck, 2008
). Within the cerebellar cortical network, cell-specific targeting of the calcium probe GCaMP2 to granule cells and Purkinje neurons was achieved by recombinant expression of GCaMP2 under control of the Kv3.1 potassium channel extended promoter (Figure 4
A1) and the L7/pcp2 promoter (Figure 4
B1), respectively, in transgenic mice (Figures 4
A2–4 and 4
B2–4) (Diez-Garcia et al., 2005
). Furthermore, reportedly exclusive targeting of GCaMP2 to Bergmann glia was obtained after acute adenoviral infection of the cerebellar cortex with GCaMP2 under the cytomegalovirus immediate-early CMV promoter (Hoogland et al., 2009
). The GCaMP2 probe permitted in-vivo recording of calcium transients from population activity of granule cells and Purkinje neurons, respectively (Figures 4
A5,6 and 4
B5,6). These results, together with data obtained for members of other families of protein probes in cells of the mouse cerebral cortex and hippocampus (Hasan et al., 2004
; Heim et al., 2007
; Mank et al., 2008
; Wallace et al., 2008
) provide a proof of principle for a cell-specific readout of network activity using GECPs. However, full exploitation of the potential of GECPs, requires further development. For instance, it still has to be shown that a sufficiently complete set of promoters/enhancers can be established to permit comprehensive dissection of the cerebellar circuitry. To expand the set of suitable promoters it appears advantageous to drive Cre recombinase by a cell type specific promoter and use this enzyme to activate the probe (Cre/lox system, for recent review see Bucholtz, 2008
). The potential benefits include that transcription of the probe gene can be under the control of a strong promoter to drive high expression levels, while the protein is produced in only a subset of cells where the activator is present to switch on the probe gene. This allows the use of a weak, but highly specific promoter for the recombinase, with the combination resulting in a strong, highly specific protein expression. Furthermore cross-breeding of already available Cre mouse lines with probe mice, provides an efficient means to generate animals with GECPs targeting different components of the network. In the future virus-mediated gene delivery may eventually substitute for the need of transgenic animals carrying the probe gene while maintaining the power of the Cre/lox system (Atasoy et al., 2008
). At present, conditional expression strategies have not yet been fully employed for optimizing GECP expression in specific neuron populations in experimental animals.
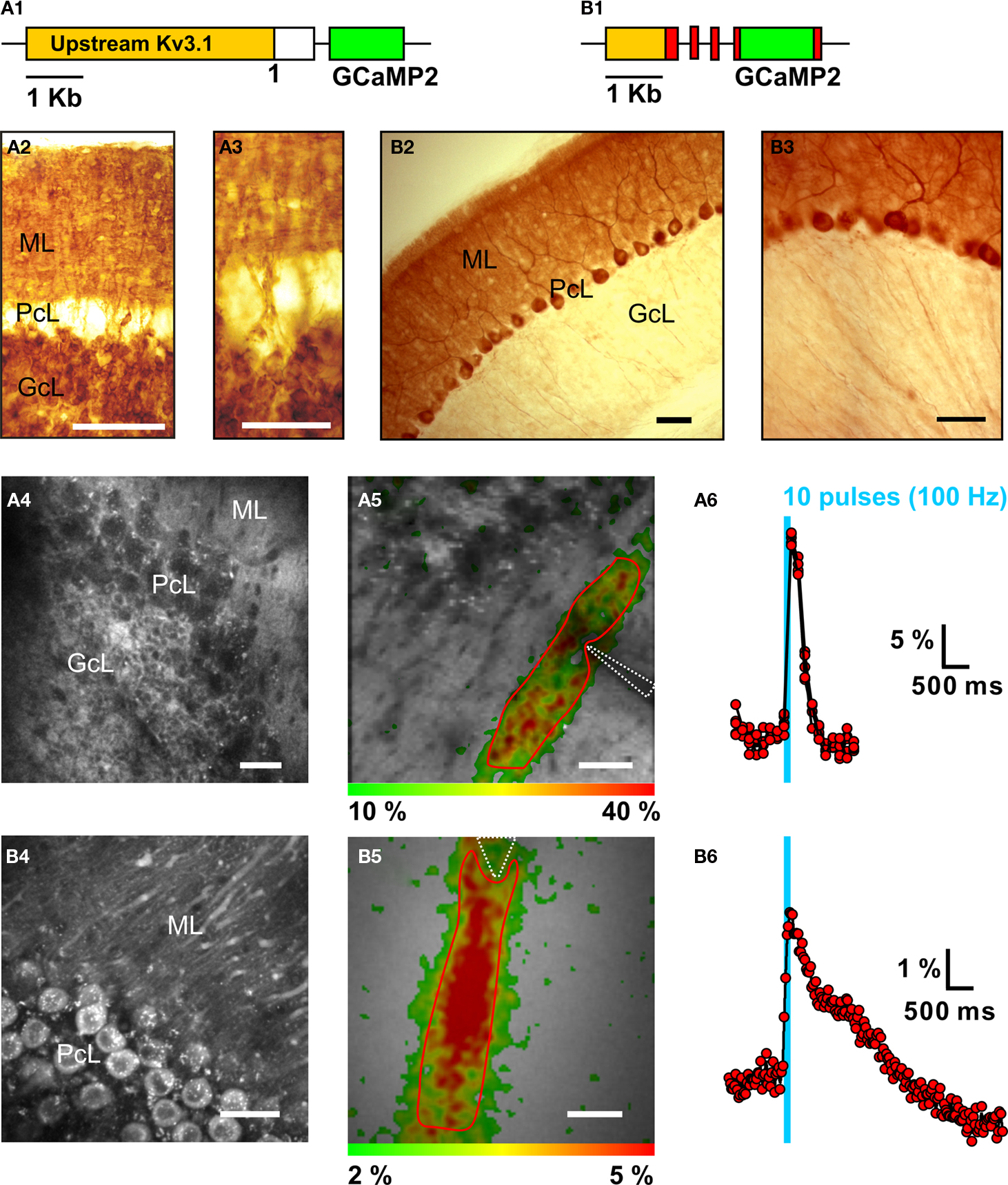
Figure 4. Targeting of the genetically-encoded calcium probe GCaMP2 to granule cells and Purkinje neurons and in-vivo recording of calcium population transients in transgenic mice. (A1) Schematic of the pKv3.1-GCaMP2 gene construct consisting of the extended (∼6 kb) promoter of the Kv3.1 potassium channel (orange) and GCaMP2 (green). (A2) Immunoreactivity against green fluorescent protein (GFP) in a coronal cerebellar section from a pKv3.1-GCaMP2-positive mouse showing the granule cell layer (GcL), Purkinje cell layer (PcL) and molecular layer (ML). Scale: 100 μm. Data from: (Diez-Garcia et al., 2005
) (A3) Same as (A2), but different field of view. Scale: 50 μm. (A4) Two-photon image of a craniotomized area (crus I/II; 190 μm below pia) of the cerebellum in a pKv3.1-GCaMP2-positive mouse in-vivo. Scale: 25 μm. (A5) In-vivo stimulation of a parallel fiber bundle using an external electrode (white dashed lines; 10 pulses; 100 Hz) with the GCaMP2 responses (ΔF/F) coded according the color scale below. Scale: 50 μm. (A6) Time course of the fluorescence response (ΔF/F) extracted from the region of interest (red) in (A5). Duration of the stimulus indicated in blue. Data from (Diez-Garcia et al., 2007
) (B1) Schematic of the pL7-GCaMP2 gene construct consisting of the L7(pcp2) (∼1 kb) promoter (orange) and GCaMP2 (green) inserted into exon 4 of the L7(pcp2) gene. Natural translation initiation sites in L7(pcp2) exons (red) have been silenced. (B2) GFP immunoreactivity in a sagittal cerebellar section from a pL7-GCaMP2-positive mouse. Scale: 50 μm. (B3) Same as (B2) but different field of view. Scale: 50 μm. (B4) Two-photon image of a craniotomized area (crus I/II; 170 μm below pia) of the cerebellum in a pL7-GCaMP2-positive mouse in-vivo. Scale: 50 μm. (B5) In-vivo GCaMP2 response (ΔF/F; coded according to the color scale below) in the molecular layer evoked by a local stimulation electrode (white dashed line; 10 pulse; 100 Hz) as obtained by single-photon fluorescence excitation (490 ± 10 nm). Scale: 50 μm. (B6) Time course of the fluorescence response (ΔF/F) extracted from the region of interest (red) in (B5).
Apart from the need to specifically target individual cell types as a key requirement for complete mapping of the cerebellar circuit, another crucial aspect of currently available GECP-based imaging concerns the ability to resolve the temporal spike code of neurons in the network. The principal limitation of the GECP approach, as for calcium imaging of neuronal activity in general, derives from the intrinsic slowness of neuronal calcium transients (typical decay of spike triggered neuronal calcium events ∼50–150 ms) as compared to action potentials (∼0.4–2 ms). Using GECPs, the time resolution is further degraded because of the slow kinetics of calcium binding to GECPs (typical off binding time constants ∼200–1500 ms) (Hendel et al., 2008
; Hires et al., 2008
).
From the point of view of their kinetic properties, GECPs are best suited to report electrical activity of cells that use sparse temporal codes (infrequent single spikes or action potential bursts at low incidence). Given that sparse activity is the rule rather than the exception in many brain circuits, e.g. (Kerr et al., 2005
, 2007
; Lee et al., 2006
) the temporal filtering property of GECPs, is not expected to prevent recording of in-vivo neuronal activity patterns and, in fact, has the advantage to bestow these recordings with improved signal-to-noise ratios. In spite of the large dynamic range of spike-evoked elevation in intracellular calcium concentration, presently available GECPs do not, however, reliably report action potentials in all preparations (Hendel et al., 2008
; Mank et al., 2008
; Mao et al., 2008
). In the case of GCaMP2, single spike sensitivity seems to be disfavored by a rather high calcium dissociation constant (1.2 μM) and Hill coefficient (2.5) (Hires et al., 2008
; Pologruto et al., 2004
). General problems that might reduce GECP performance in certain applications include: specific interactions of the calcium binding domain with intrinsic cytosolic proteins, GECP sequestration in intracellular compartments, and/or too low expression level. In the case of cameleons (Miyawaki et al., 1997
) reengineering of the calmodulin binding interface yielded substantial improvement in the in-vivo performance of the probe (Palmer et al., 2006
; Wallace et al., 2008
). Furthermore, acute viral transfer of GECPs into neurons can elevate GECP expression to levels that enable reliable detection of single spikes from somatic calcium transients in-vivo (Wallace et al., 2008
). Thus, virus-mediated gene transfer in combination with improved GECPs (Palmer et al., 2006
; Souslova et al., 2007
) is likely to overcome the technical limitations experienced in earlier studies (Diez-Garcia et al., 2007
) and provide sufficient sensitivity to map the in-vivo activity of identified neurons. Using constitutive cell type specific GCaMP2 expression in transgenic mice, we have already succeeded to dissect the pre- and postsynaptic calcium responses evoked by compound activity of parallel fibers in the molecular layer of the cerebellum in-vivo (Figure 4
). The postsynaptic response in Purkinje neurons, representing calcium signals from the Purkinje neuron dendrites, takes the form of a “beam-like” territory (Figure 4
B5) indicating synaptic activation of Purkinje neurons along the direction of the parallel fibers (Figure 4
A5). The ability to unambiguously discriminate and quantify pre- and postsynaptic events based on targeted GCaMP2 expression allowed the mechanism of presynaptic plasticity of the granule cell (Gc) to Purkinje neuron (PN) transmission upon repetitive 4 Hz (120 pulses) stimulation of parallel fibers (Qiu and Knöpfel, 2007
), to be isolated in vitro. This protocol gives rise to a long-lasting potentiation (LTP) of Gc-PN synaptic efficacy with the presynaptic site of modification indicated by an associated increase of calcium influx into parallel fiber synaptic boutons. The observed plasticity required activation of protein kinase A, NMDA receptors and NO synthase in a presumably multi-synaptic pathway involving the activation of NMDA receptors on local interneurons (Qiu and Knöpfel, 2007
). The Gc-PN presynaptic site was also susceptible to long-term synaptic depression (LTD), which was unmasked when presynaptic LTP at this synapse was pharmacologically inhibited (Qiu and Knöpfel, 2009
). Thus, genetically targetable calcium probes provide a new experimental approach to trace synaptic mechanisms in neuronal circuits which, when extended to in-vivo preparations, can help address the relationship between molecular cellular events and behavior.
The kinetic limitations associated with genetically encoded calcium sensors can, in principle, be overcome by genetically encoded voltage probes. This theoretical advantage is important for structures such as the cerebellum where cells typically have very high spike rates, making GECPs impractical for resolving single spikes. There is no doubt that multisite cellular voltage signals are the type of data that can bridge cellular neurophysiology and circuit modeling, but as already outlined above, voltage sensitive dye imaging is technically more challenging than calcium imaging in terms of signal-to-noise ratio. This fact results from the smaller number of dye molecules that can be accommodated on the surface of cells when compared to cytosolic volume and a generally smaller dynamic range of optical voltage signals as compared to optical calcium signals. Attempts to generate fluorescent protein voltage probes started around the time calcium probes were being designed, but progress has been much slower. After failure of a first generation of such probes (Baker et al., 2007
), a second generation proved much more promising. Currently the best available probes follow the original design of Sakai et al., termed VSFP1 (Sakai et al., 2001
), but make use of a different voltage sensor domain (the part of the sensor that is voltage sensitive) (Dimitrov et al., 2007
; Lundby et al., 2008
; Mutoh et al., 2009
). The third generation variants with the largest steady state voltage response are VSFP2.3, VSFP2.4 and Mermaid (Mutoh et al., 2009
; Tsutsui et al., 2008
) having dynamic ranges of around 12% ΔR/R in a side-by-side PC12 cell assay (Mutoh et al., 2009
). Responses to transient electrical events in neurons are smaller, amounting in the best cases to 1–2% ΔR/R for slow potentials and 0.2–1% for action potentials or presumed action potential like voltage signals (Dimitrov et al., 2007
; Tsutsui et al., 2008
). The relatively small amplitudes of the optical signals induced by action potentials is a direct consequence of the intrinsic activation kinetics of VSFP which comprises a fast initial response and a subsequent relatively slow response (Mutoh et al., 2009
). To explore the low-pass filter function imposed by these probes we used a combination of voltage clamped VSFP2.1 expressing PC12 cells together with computer simulations (Figures 5
A1,A2).
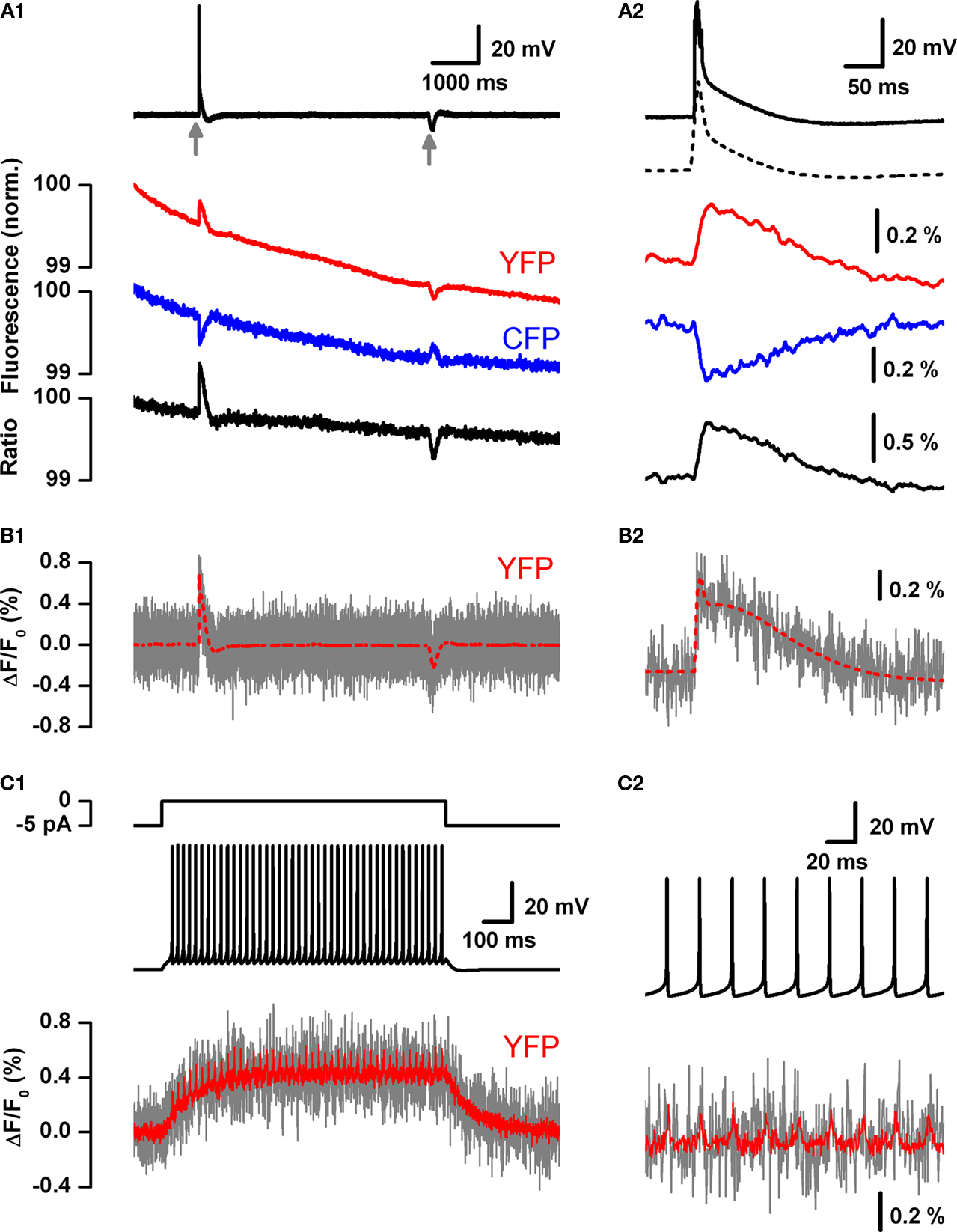
Figure 5. Estimation of the theoretical readout of simple and complex spikes from Purkinje neurons by voltage imaging using present generation VSFPs. (A1) A PC12 cell expressing VSFP2.1 was voltage-clamped with a voltage trace (top black trace) from a cerebellar Purkinje neuron during induction of a climbing fiber response (complex spike; marked by arrow) and during intracellular injection of a small hyperpolarizing current pulse (marked by a second arrow). Traces show membrane potential (top trace), yellow fluorescence (YFP; red trace), cyan fluorescence (CFP; blue trace) and the ratio of yellow and cyan fluorescence (bottom black trace). The voltage trace was recorded with 1 kHz cutoff frequency. Fluorescence signals were digitally low pass filtered (0.1 kHz), averaged over 20 consecutive sweeps and scaled in units of normalized absolute fluorescence. (Mutoh and Knöpfel, unpublished data, Methods as in Dimitrov et al., 2007
). (A2) Same traces as in (A1) at expanded time scale. The black dotted trace is the low pass-filtered complex spike obtained from the upper voltage trace (black continuous line) by digital filtering with 0.1 kHz cutoff. The fluorescence traces are represented in units of the differential fluorescence change (ΔF/F0). (B1) Computer simulation of the fluorescence response of VSFP2.3 in a Purkinje cell body as evoked by the same voltage waveform as in (A1; top panel). The Purkinje soma is represented by a spherical membrane (25 μm diameter) containing VSFP2.3 at a surface density of 800 units per μm2. VSFP2.3 was represented as a kinetic reaction model developed from experimental fluorescence data of this protein (Akemann et al., 2009
). The traces show the simulated yellow (YFP) fluorescence response (ΔF F0) with (gray) and without (red line) photon quantum noise at 2 kHz sampling frequency (and otherwise unlimited bandwidth). (B2) Expanded view of (B1) in the range of the complex spike-evoked response. (C1) Simulated response of VSFP2.3 to Purkinje neuron simple spikes. A spiking model of a Purkinje cell body producing intrinsic action potentials (black trace) at 40 Hz (zero injected current; 37°C) was loaded with VSFP2.3 at 800 units per μm2. The lower trace shows the VSFP2.3 fluorescence response (2 kHz sampling) in single trial (gray) and after averaging 20 trials (without jitter; red). (C2) View of (C1) at expanded time scale. For details regarding the simulations see Akemann et al. (2009)
.
VSFP2.1 when expressed in PC12 cells could clearly monitor (without averaging over sweeps) the slow component of the voltage transient associated with a climbing fiber response, however single action potentials were not resolved. Notably, this experiment was done with the early variant VSFP2.1 and improved signals are expected from newer variants, e.g. VSFP2.3 and VSFP2.4. With the aim to characterize the performance of VSFPs as indicators of Purkinje neuron electrical activity, we incorporated a Markov chain model of VSFP2.3 (where the sensor moves between different states in a voltage-dependent manner) into a previously established single compartment model of a Purkinje neuron (Akemann and Knöpfel, 2006
). Consistent with the experimental results on the time course of VSFP2.1 signals (Figures 5
A1,A2) the simulations predicted similar response shapes reported by VSFP2.3 but with a slightly larger amplitude (Figures 5
B1,B2). In contrast to complex spikes, single simple spikes are predicted to be resolved only after averaging over >20 sweeps (Figures 5
C1,C2). Attempts to generate faster (and possibly more sensitive) VSFP versions resulted in VSFP3s (Lundby et al., 2008
). Although this new generation of VSFPs displays an optical response consisting of a very fast component which dominates over a slower one, the dynamic range of the fast component still needs to be extended to enable routine optical reading of fast electrical activity at the single neuron level in brain tissue (Akemann et al., 2009
).
While this review does not aim to cover methodological aspects, we shall mention a few points of caution related to optical imaging. One of the key arguments for use of optical methods is that neuronal activity can be monitored non-invasively, giving an advantage over conventional microelectrode recording. Whilst this statement is true, illumination of tissue can also be associated with tissue damage. In particular it is established that photo-chemical reactions of certain dyes (in particular conventional voltage sensitive dyes) generate toxic by-products. Modern dyes are now available that have been optimized to minimize these effects. Furthermore voltage sensitive dyes (including fluorescent protein voltage sensors) add displaceable charges to the membrane, acting to increase the membrane capacitance. This “capacitance effect” has been recently investigated in detail (Akemann et al., 2009
). Calcium indicators meanwhile suffer from the fact they directly bind calcium and thus act as calcium buffers, care must therefore be taken to ensure that calcium dynamics (and thus cellular properties) are not adversely altered. The problem becomes even more complicated with protein based calcium indicators which can potentially interact not only with Ca2+ but also other endogenous proteins (Wallace et al., 2008
). One further point to note is the possibility of interference of the signal of interest (from the fluorescent reporter) with intrinsic fluorescence signals. A prominent case is the spectral overlap between green fluorescent protein-based sensors and flavoproteins (demonstrated and discussed in detail in Diez-Garcia et al., 2007
). This issue can be resolved by using fluorescent proteins whose emission has a large spectral separation from the flavoprotein signal (e.g. Mutoh et al., 2009
; Perron et al., 2009
).
The aim of this review is to discuss and highlight the potential of optical imaging as a methodological platform that may serve as a link between cellular neurophysiology and circuit modeling. Clearly, realization of this potential has only begun. We anticipate that genetically encoded probes in combination with modern in vivo imaging techniques will allow gathering of data that can drive and validate large scale circuit models. At present, questions that address the responses of larger but genetically defined cell populations are difficult to answer using traditional electrophysiology. These include questions like the spatio-temporal interaction of different cell types during oscillatory activity, revisiting sensory receptive fields, and the spatial organization of signals related to motor commands. It is the hope that, in the near future, imaging circuit dynamics at the resolution of single genetically defined cells will help answer these questions and also shed light on the neuronal codes used in the cerebellum.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all members of the Knöpfel laboratory for discussions and support. The work of this laboratory is supported by grants from RIKEN BSI (TK), the RIKEN BSI directors fund (TK), NIH grant NS057631 (under a sub award granted by Yale University to TK), and JSPS (Japanese Society for the Promotion of Science).
Metzger, F., Repunte-Canonigo, V., Matsushita, S., Akemann, W., Diez-Garcia, J., Ho, C. S., Iwasato, T., Grandes, P., Itohara, S., Joho, R. H., and Knöpfel, T. (2002). Transgenic mice expressing a pH and Cl- sensing yellow-fluorescent protein under the control of a potassium channel promoter. Eur. J. Neurosci. 15, 40–50.