1
Division of Biology, California Institute of Technology, Pasadena, CA, USA
2
Division of Biology, Howard Hughes Medical Institute, Chevy Chase, MD, USA
Many lines of evidence suggest that the efficacy of synapses on CA1 pyramidal neuron dendrites increases as a function of distance from the cell body. The strength of an individual synapse is also dynamically modulated by activity-dependent synaptic plasticity, which raises the question as to how a neuron can reconcile individual synaptic changes with the maintenance of the proximal-to-distal gradient of synaptic strength along the dendrites. As the density of A-type potassium channels exhibits a similar gradient from proximal (low)-to-distal (high) dendrites, the A-current may play a role in coordinating local synaptic changes with the global synaptic strength gradient. Here we describe a form of homeostatic plasticity elicited by conventional activity blockade (with tetrodotoxin) coupled with a block of the A-type potassium channel. Following A-type potassium channel inhibition for 12 h, recordings from CA1 somata revealed a significantly higher miniature excitatory postsynaptic current (mEPSC) frequency, whereas in dendritic recordings, there was no change in mEPSC frequency. Consistent with mEPSC recordings, we observed a significant increase in AMPA receptor density in stratum pyramidale but not stratum radiatum. Based on these data, we propose that the differential distribution of A-type potassium channels along the apical dendrites may create a proximal-to-distal membrane potential gradient. This gradient may regulate AMPA receptor distribution along the same axis. Taken together, our results indicate that A-type potassium channels play an important role in controlling synaptic strength along the dendrites, which may help to maintain the computational capacity of the neuron.
CA1 pyramidal neurons in the hippocampus receive approximately 30,000 excitatory inputs along their dendrites (Megias et al., 2001
). Due to voltage attenuation by cable filtering, distal synapses must be stronger than proximal synapses to provide the same amplitude of voltage change at the soma (London and Segev, 2001
). Recent studies, indeed, documented that the synapses on distal dendrites of CA1 pyramidal neurons have higher synaptic strength than those on proximal dendrites (Magee and Cook, 2000
; Smith et al., 2003
; Nicholson et al., 2006
). These data suggest that neurons adjust synaptic strength so that synapses at different dendritic locations can have a comparable impact on the soma. A potential mechanism for this adjustment is the regulation of AMPA receptor distribution along the dendrites.
A number of recent studies have emphasized the importance of homeostatic control of the synaptic strength. After a long-term reduction in neural activity with a sodium channel blocker, tetrodotoxin (TTX), synaptic strength becomes larger, whereas after a chronic enhancement of activity (with a GABAA receptor blocker) synaptic strength becomes reduced (Turrigiano et al., 1998
; Burrone et al., 2002
; Stellwagen and Malenka, 2006
). This homeostatic plasticity allows neurons to keep their firing frequency within an appropriate range and neuronal networks to remain stable (Turrigiano, 2008
). This type of scaling, elicited by neuronal activity, is often called global scaling because the synaptic strength is increased multiplicatively or decreased divisively for (presumably) all synapses of a neurons (Rabinowitch and Segev, 2008
).
Synaptic strength can also be controlled locally by spontaneously released synaptic vesicles (i.e., miniature synaptic transmission) (Sutton and Schuman, 2006
; Sutton et al., 2006
). This mechanism does not require action potentials, but instead, each synapse monitors and adjusts synaptic strength according to the level of miniature synaptic transmission. The calcium-mediated signal evoked by miniature synaptic transmission controls dendritic protein synthesis and adjusts synaptic efficacy. This homeostatic scaling occurs locally, allowing for neurons to control individual synapses independently.
Although homeostatic scaling mechanisms can work either globally or locally, it remains unexplored how neurons maintain the distance-dependent synaptic strength along the proximal-distal axis in dendrites in the face of conventional global scaling. Previous studies have provided several clues to potential mechanisms. For example, the distance-dependent distribution of AMPA receptors is abolished in animals lacking either GluR1 or Kv4.2 channel, a molecular component of A-type-mediated potassium current (Andrasfalvy et al., 2003
, 2008
). This implies that both GluR1 and A-type channels may contribute to the distance-dependent (along the somatic dendritic axis) AMPA receptor distribution. Interestingly, similar to AMPA receptors, A-type potassium channels are also differentially distributed, exhibiting higher channel density in distal, compared to proximal, regions of apical CA1 dendrites (Hoffman et al., 1997
). The distribution pattern of A-type channels may be important for the proximal-to-distal gradient of synaptic strength because layer V pyramidal neurons in the neocortex exhibit a uniform A-type channel density along the dendrites (Bekkers, 2000
; Korngreen and Sakmann, 2000
) and do not exhibit the distance-dependent increase in synaptic strength (Williams and Stuart, 2002
). Here, we asked whether A-type channel activity can directly influence the distance-dependent differences in synaptic strength observed in the dendrites of hippocampal neurons.
Hippocampal Slice Preparation
Slices were prepared from 21 to 32-day-old Sprague-Dawley rats (Charles River). In brief, a vibrating microtome (Leica VT1000S) or a tissue chopper (Stoelting) was used to cut transverse hippocampal sections (500 μm thickness) in ice-cold oxygenated artificial cerebrospinal fluid (ACSF) containing (in mM) 119 NaCl, 2.5 KCl, 1.3 MgSO4, 2.5 CaCl, 1.0 NaH2PO4, 26.2 NaHCO3, 11.0 glucose. After a 2-h recovery at room temperature, slices were treated for 12-h at 32°C with pharmacological inhibitors (as indicated) and nutritional supplements (1/10 concentration of GlutaMax, MEM vitamin and MEM amino acids, purchased from Invitrogen). For electric field-application, a pair of Ag/AgCl pellets (WPI) were positioned parallel to one another for current application from the current source (Axon Instruments). Between current injecting electrodes, a pair of Ag/AgCl wires was positioned to measure the voltage difference between the electrodes using a voltmeter. The amplitude of current injection was appropriately adjusted by monitoring the voltmeter. Since we observed a ∼2-mV difference in resting membrane potential along the 300 μm length of dendrites, we estimate the strength of dendritic electric field to be approximately 6–7 mV/mm, which is comparable to the strength of externally applied electric field (10 mV/mm).
Electrophysiology
Whole-cell voltage-clamp recordings from CA1 pyramidal neuron somata or dendrites were made (without visualization) with an Axopatch 200B (Axon Instruments). The internal solution of patch pipettes was as follows: for the miniature excitatory postsynaptic current (mEPSC) recordings (in mM) 115 cesium gluconate, 20 cesium chloride, 10 sodium phosphocreatine, 10 HEPES, 2 MgATP, 0.3 NaGTP (pH 7.3); for the measurement of the resting membrane potential (in mM) 115 potassium gluconate, 20 potassium chloride, 10 sodium phosphocreatine, 10 HEPES, 2 MgATP, 0.3 NaGTP (pH 7.3). After recovery from slicing or a pharmacological treatment, slices were transferred to a submerged recording chamber perfused with ACSF at 24.5–25.5°C or 32–34°C (for resting membrane potential recording). The mEPSC recordings were conducted in the presence of TTX (1 μM) and bicuculline (20 μM). Membrane voltage was clamped at −70 mV (without liquid junction potential correction). After recordings were stabilized, current traces were acquired for 3 min and all detected events were analyzed to calculate the mean amplitude and frequency of mEPSCs. The mEPSCs were detected using a template-matching algorithm from Clampfit 9 (Molecular Devices); the waveform template was made by averaging mEPSCs in untreated slices. In Figure 3
, a different program for mEPSC detection (Mini Analysis, Synaptosoft) was used, which is based on threshold-cutting for event amplitude and area (threshold used for the analysis: amplitude > 5 pA, area > 10 fC). For the resting membrane potential measurement, potentials were corrected for liquid junction potentials, which were approximately −11 mV for TTX alone and −9 mV for TTX + 4AP (10 mM). Recordings were discarded when the series resistance was over 20 MΩ (50 MΩ in dendritic recordings) or either series or membrane resistance changed more than 20% during data acquisition. Data were collected by DigiData 1200 and pClamp 9 (Axon Instruments). All numerical values listed represent mean ± SEM, unless noted otherwise. The error bars in all figures represent SEM. A student’s t-test was performed for all statistical analysis.
Immunohistochemistry
Slices (500 μm thickness) were prepared using the same procedure as for electrophysiology recordings. After the 12-h incubation in pharmacological inhibitors (as indicated), slices were washed once with ice-cold ACSF and quickly fixed in 4% paraformaldehyde in phosphate-buffered saline for at least 2 days. An NMDA receptor antagonist, APV (50 μM), was always applied during the incubation for the immunohistochemistry to avoid the influence of dendritic protein synthesis (Sutton et al., 2006
). Thin (50 μm) sections were cut with a vibrating microtome (Leica VT1000S). The sections were incubated overnight with one of the following antibody dilutions: 1:1000 of anti-Synaptophysin I (Millipore), 1:1000 of anti-Bassoon (Stressgen), 1:1000 of anti-GluR1 (Millipore), 1:100 of GluR2/3 (Millipore), or 1:1000 of mouse anti-MAP2 (Sigma) antibodies. The incubation was carried out at room temperature in Tris-buffered saline containing 0.2% Triton X-100, BSA 2%, NGS 4 %, followed by 4 h of secondary-antibody incubation with 1:1000 of Alexa 488-conjugated anti-rabbit and 1:1000 of Alexa 543-conjugated anti-mouse antibodies (Invitrogen). For the analysis of immunohistochemistry experiments, images were obtained with a Zeiss LSM 510 laser scanning confocal microscope using a Plan-Neofluor 10×/0.3 air objective. Alexa 488 and 546 were visualized by excitation with the 488 line of an argon ion laser and the 543 nm line of a HeNe laser, respectively. The optical section was 20 μm and fluorescent signals were acquired and summed throughout the slice thickness (50 μm). A student’s t-test was performed for statistical analysis.
Cell Culture and Infection
Dissociated postnatal (P1–P2) rat hippocampal neuron cultures, plated at a density of 55,000–70,000 cells/cm2 in poly-D-lysine-coated glass-bottom Petri dishes (Mattek), were prepared as previously described (Aakalu et al., 2001
) and maintained for 18–22 days in vitro at 37°C in growth medium [Neurobasal A supplemented with B27 and Glutamax-1 (Invitrogen)]. For infection with a Sindbis viral vector expressing N-terminal GFP-tagged GluR1 (Shi et al., 2001
) cells were incubated with virus (diluted in conditioned medium) for 17–20 h at 37°C.
Chronic Blockade of A-Type Potassium Channels Enhanced mEPSC Frequency in the Region Near the Soma of CA1 Pyramidal Neurons
The observation that slices prepared from Kv4.2 knockout animals do not exhibit a distance-dependent increase in synaptic strength (Andrasfalvy et al., 2008
), suggests that A-type potassium channel activity may participate in the differential distribution of ion channels along the dendritic axis. To initially test this idea, we asked whether chronic blockade of A-type potassium channels (by the antagonist 4-aminopyridine, 4AP; Thompson, 1982
) can result in changes in synaptic strength. To avoid the influence of seizure-like activities induced by 4AP, we included TTX in our experiments.
Using acute hippocampal slices to preserve the channel or receptor distribution present in vivo, we examined miniature excitatory synaptic transmission by recording from the soma of CA1 pyramidal neurons after treatment with either TTX alone or TTX + 4AP for 12 h. We found that mEPSC frequency was significantly enhanced in 4AP-treated slices, relative to slices treated with TTX alone (TTX: 3.12 ± 0.32 Hz, TTX + 4AP: 6.88 ± 0.41 Hz) (Figure 1
A). Treatment with 4AP did not elicit a significant change in mEPSC amplitude (TTX: 12.24 ± 0.76 pA, TTX + 4AP: 13.24 ± 0.76 pA). The observed differences are mediated by homeostatic synaptic scaling, rather than direct action of 4AP on synaptic transmission, because under our recording conditions, neither an acute application of 4AP nor a short-term (1 h) treatment with 4AP influenced the mEPSC amplitude or frequency (Figure 2
). A similar treatment with a blocker for a different class of potassium channels (tetraethylammonium; TEA) did not influence synaptic scaling (mEPSC amplitude: TTX 12.15 ± 0.61 pA, TTX + TEA 10.96 ± 0.29 pA; mEPSC frequency: TTX 2.49 ± 0.25 Hz, TTX + TEA 2.14 ± 0.16 Hz) (Figure 1
C), thus this scaling is specifically mediated by A-type potassium channels.
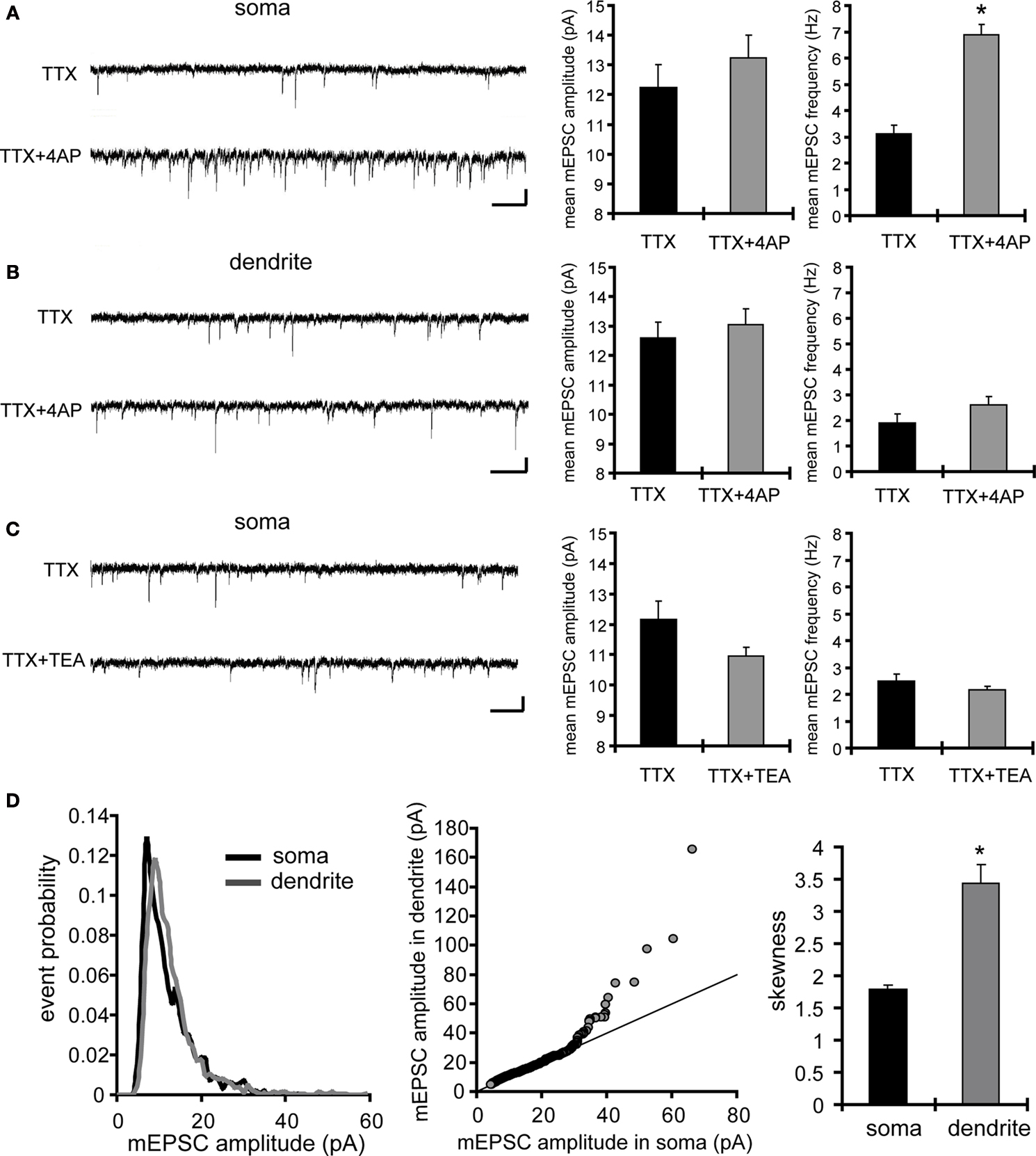
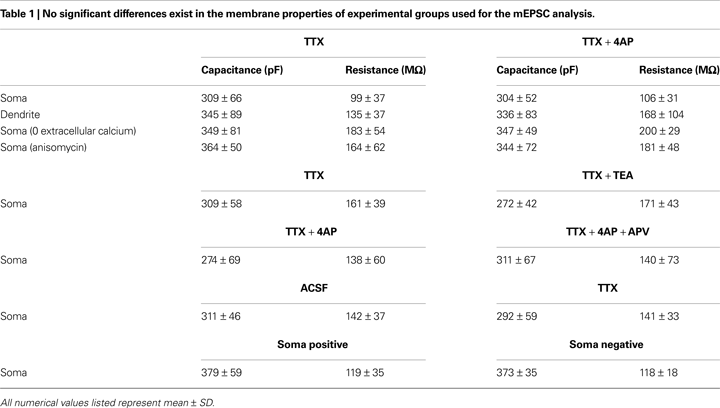
Figure 1. A-type potassium channel blockade enhances the frequency of mEPSCs recorded in the soma, but not the dendrites, of CA1 pyramidal neurons. (A) Representative mEPSCs recorded from the somata of CA1 pyramidal neurons. Hippocampal slices were treated with either TTX (2 μM) alone or TTX + 4AP (5 mM) for 12 h. The slices treated with 4AP showed enhanced mEPSC frequency (TTX: n = 8, TTX + 4AP: n = 9) (scale bar = 10 pA, 500 ms) (*p < 0.05). No apparent difference in membrane properties was observed between experimental groups (Table 1
). (B) Representative mEPSCs recorded from the dendrites of CA1 pyramidal neurons. The recorded sites were in the middle of the stratum radiatum, which is approximately 200–250 μm away from the soma. The slices treated with 4AP did not exhibit a significant enhancement of mEPSC frequency in dendrites (n = 10 for each group) (scale bar = 10 pA, 500 ms). No apparent difference in membrane properties was observed between experimental groups (Table 1
). (C) Representative mEPSCs recorded from the somata of CA1 pyramidal neurons. Hippocampal slices were treated with either TTX alone or TTX + TEA (10 mM) for 12 h. TEA-treated slices did not exhibit enhanced mEPSC frequency (n = 8 for each group) (scale bar = 10 pA, 500 ms) (*p < 0.05). No apparent difference in membrane properties was observed between experimental groups (Table 1
). (D) A comparison of amplitude distribution between somatic and dendritic mEPSCs. The left figure showed the event probability of mEPSC amplitude in somatic and dendritic recordings from slices treated with TTX alone [n = 1,920 mEPSCs chosen randomly from recording data used in (A) and (B)]. In the middle figure, the ranked mEPSC amplitudes in somatic recordings were plotted against ranked mEPSC amplitudes in dendritic recordings (black line: slope of 1). A cluster of dendritic mEPSCs shows larger amplitude compared to somatic mEPSCs, although the distributions are similar for small-sized (e.g. ∼ < 30 pA) mEPSCs. The right figure showed a mean skewness of amplitude distribution for somatic and dendritic mEPSCs recorded from slices treated with TTX alone. The skewness is significantly larger in dendritic mEPSCs, suggesting that the amplitude distribution of dendritic mEPSCs is distorted toward larger amplitude compared to that of somatic mEPSCs.
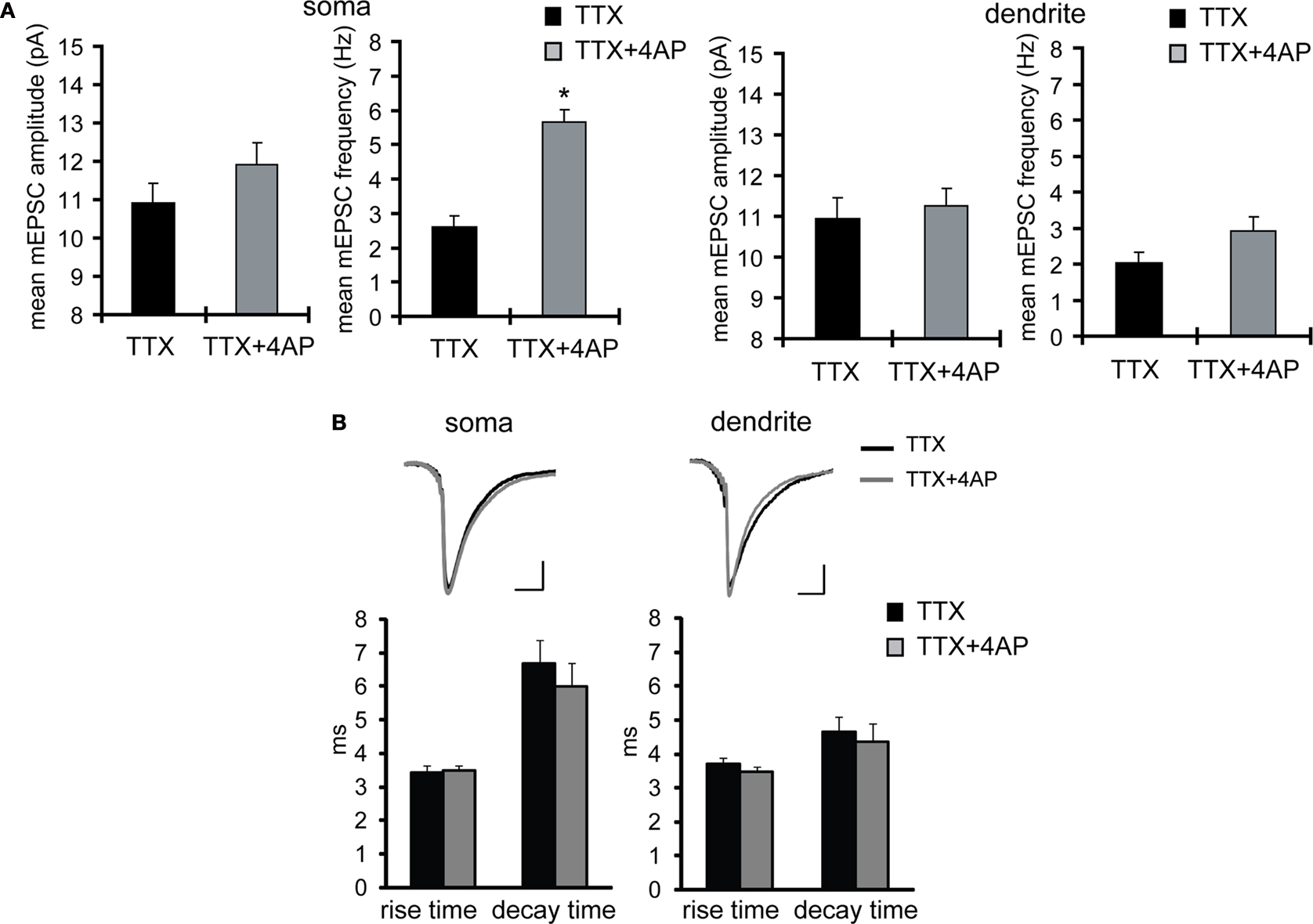
Figure 2. Acute application of 4AP influences neither the amplitude nor the frequency of mEPSCs. (A) 4AP (5 mM) was applied during mEPSC recording from the soma. Data from individual experiments are represented by gray circles and black diamonds represent the mean (n = 6). The results indicate that 4AP does not have an immediate influence on mEPSC amplitude or frequency under our recording conditions. Note that most postsynaptic A-type channels are probably not functional under the voltage-clamp recordings at −70 mV (without liquid junction potential correction) with the cesium-based internal solution we use. (B) mEPSCs were recorded from the soma after 1 h, rather than 12 h, incubation with either TTX alone or TTX + 4AP (n = 5 for each group). A 1-h treatment with 4AP has no significant influence on mEPSC amplitude or frequency.
Next we examined whether A-type channel blockade may differentially influence miniature synaptic transmission in the somatic region vs. the apical dendrites. We recorded from dendrites of CA1 pyramidal neuron by targeting the middle of stratum radiatum (approximately 200 to 250 μm from the stratum pyramidale). Consistent with previous reports (Magee and Cook, 2000
; Smith et al., 2003
; Nicholson et al., 2006
), we observed a cluster of large mEPSC events in dendritic recordings, compared to those in somatic recordings (Figure 1
D). We then examined dendritic mEPSCs in slices treated with TTX alone or TTX + 4AP, and found that 4AP had a significantly smaller influence on mEPSC frequency in dendrites (mEPSC amplitude: TTX 12.60 ± 0.55 pA, TTX + 4AP 13.04 ± 0.54 pA; mEPSC frequency: TTX 1.91 ± 0.34 Hz, TTX + 4AP 2.61 ± 0.33 Hz) (Figure 1
B). Our basic observation of 4AP-mediated scaling was reconfirmed with a different algorithm for mEPSC detection (Figure 3
A), and we did not find any significant difference either in membrane properties (Table 1
) or kinetics of mEPSC waveform (Figure 3
B) after chronic 4AP treatment. Thus, prolonged A-type potassium channel blockade appears to exert a much greater influence on mEPSC frequency in the regions near the soma, suggesting that 4AP disrupted the synaptic balance usually observed along the dendritic axis.
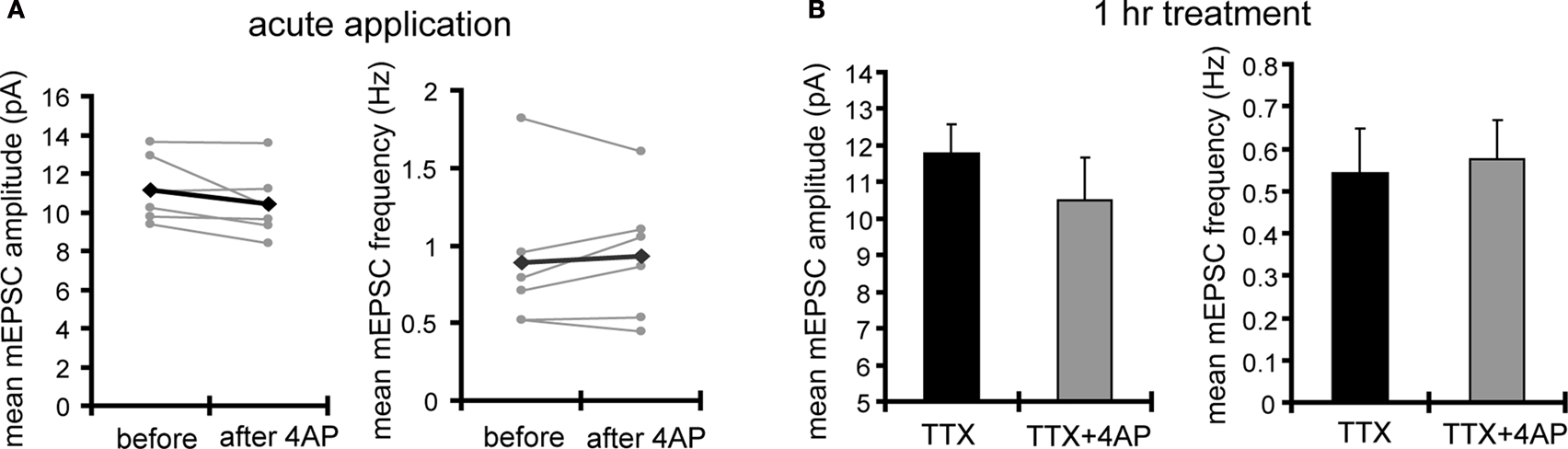
Figure 3. The kinetics of the mEPSC waveform is not significantly different between TTX- and TTX + 4AP-treated slices. (A) The data in Figures 1
A,B were reanalyzed using a different program for mEPSC detection (Mini Analysis, Synaptosoft), which is based on threshold-cutting for event amplitude and area, rather than template-matching as used in Figure 1
(Clampfit 9, Molecular Devices). The same pattern of results was obtained. (B) The averaged mEPSC waveforms detected by the Mini Analysis program were shown (top) (scale bar = 2 pA, 10 ms) (mEPSC waveforms were aligned at the 50% of rise time). The bottom figures show the mean rise (10–90% time point) and decay time (tau) of mEPSCs. No significant difference was observed in the kinetics of the mEPSC waveform between groups.
A-Type Channel Blockade Enhances mEPSC Frequency by a Protein-Synthesis Dependent Mechanism
Miniature synaptic transmission regulates synaptic scaling by controlling dendritic protein synthesis and enhancing the synaptic insertion of GluR2-lacking AMPA receptors, an effect mediated by NMDA receptor activity (Sutton et al., 2006
). Thus, we examined whether a NMDA receptor blocker, APV, influences synaptic scaling by A-type channel blockade. We did not find a significant influence of concurrent APV treatment on 4AP-induced synaptic scaling (mEPSC amplitude: TTX + 4AP 11.39 ± 1.04 pA, TTX + 4AP + APV 12.06 ± 0.73 pA; mEPSC frequency: TTX + 4AP 8.17 ± 1.41 Hz, TTX + 4AP + APV 8.79 ± 1.26 Hz) (Figure 4
A). In addition, a specific blocker for GluR2-lacking AMPA receptors, 1-Naphthylacetyl spermine (Naspm), had no influence on 4AP-treated slices (mEPSC amplitude: before 12.87 ± 0.40 pA, after Naspm 12.68 ± 0.57 pA; mEPSC frequency: before 5.93 ± 0.59 Hz, after Naspm 5.89 ± 0.56 Hz) (Figure 4
B), suggesting a different mechanism is involved in 4AP-induced scaling. Furthermore, even when slices were treated in a nominally zero calcium solution during the 12-h incubation, we still observed the enhancement of mEPSC frequency by A-type channel blockade (mEPSC amplitude: TTX 9.33 ± 0.40 pA, TTX + 4AP 9.76 ± 0.45 pA; mEPSC frequency: TTX 2.25 ± 0.32 Hz, TTX + 4AP 5.06 ± 0.58 Hz) (Figure 4
C), suggesting that this scaling is mediated by a calcium-independent mechanism.
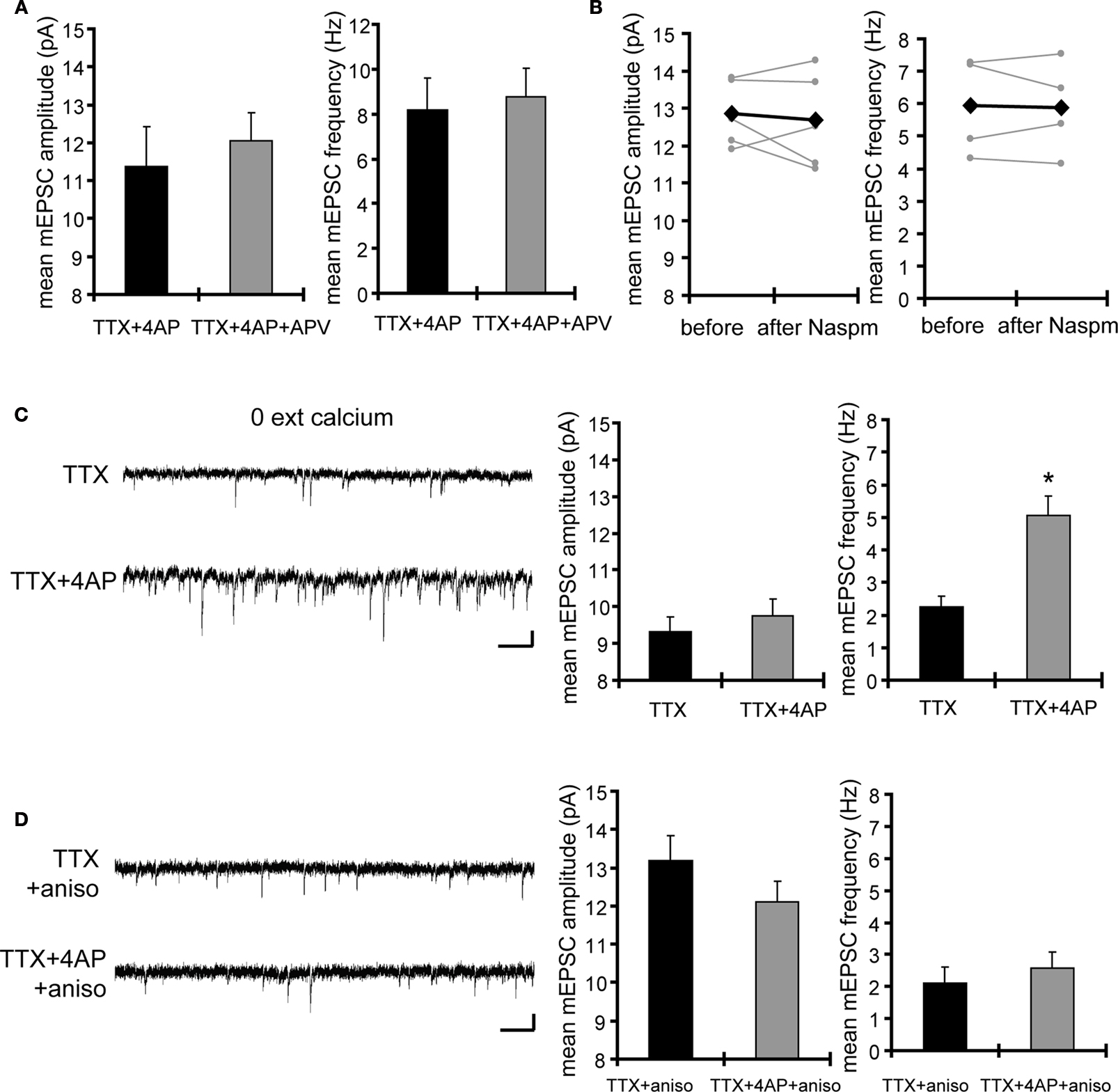
Figure 4. Synaptic scaling induced by A-type channel blockade is protein synthesis dependent, but calcium-signaling independent. (A) Co-treatment with the NMDA receptor antagonist, APV (50 μM), did not influence synaptic scaling by A-type channel blockade (n = 7 for each). No apparent difference in membrane properties was observed between experimental groups (Table 1
). (B) Synaptic scaling induced by A-type channel blockade does not require GluR2-lacking receptors. After 12-h treatment with TTX + 4AP, mEPSCs were recorded and the acute influence of Naspm (10 μM) application was examined. Data from individual experiments are represented by gray circles and black diamonds represent the mean (n = 5 for each). (C) A reduction in extracellular calcium did not influence 4AP-induced synaptic scaling. Slices were treated in ACSF which contained (nominal) zero calcium; the divalent cation concentration was adjusted with magnesium (TTX: n = 5, TTX + 4AP: n = 7) (*p < 0.05). No apparent difference in membrane properties was observed between experimental groups (Table 1
). (D) 4AP-induced synaptic scaling is protein-synthesis dependent. Slices were treated with either TTX alone or TTX + 4AP, together with anisomycin (50 μM) for 12 h (n = 9 for each). No apparent difference in membrane properties was observed between experimental groups (Table 1
).
We then asked whether 4AP-mediated scaling is protein-synthesis dependent. When slices were co-treated with a protein-synthesis inhibitor, anisomycin, we did not observe significant influence of 4AP (mEPSC amplitude: TTX + aniso 13.18 ± 0.66 pA, TTX + 4AP + aniso 12.1 ± 0.55 pA; mEPSC frequency: TTX + aniso 2.09 ± 0.53 Hz, TTX + 4AP + aniso 2.57 ± 0.51 Hz) (Figure 4
D), suggesting that 4AP-mediated scaling is protein-synthesis dependent.
A-Type Channel Blockade Enhanced GluR1 Density in the Stratum Pyramidale, but not in the Stratum Radiatum, of Area CA1
Because synaptic scaling can be detected by electrophysiological recordings from the soma, but is much diminished in dendritic recordings, we examined whether there are any differences in somatic vs. dendritic synaptic protein expression levels after A-type channel blockade. We performed immunohistochemistry on slices after either TTX alone or TTX + 4AP treatment, examining the distribution patterns of synaptic proteins along the somatic-dendritic axis. Although we did not find any 4AP-induced differences in presynaptic protein distribution in area CA1 (signal ratio of s. pyramidale to s. radiatum; synaptophysin: TTX 0.46 ± 0.03, TTX + 4AP 0.43 ± 0.04, bassoon: TTX 0.08 ± 0.02, TTX + 4AP 0.12 ± 0.01) (Figure 5
A), we observed a significant enhancement of GluR1 distribution in the soma, relative to the dendrites, following 4AP treatment (TTX: 1.10 ± 0.09, TTX + 4AP: 1.59 ± 0.13) (Figure 5
A). The selective enhancement of GluR1 density near the soma is consistent with mEPSC recording results, where the enhancement was observed only in regions near the soma (Figure 1
). A similar change in distribution was also observed for other AMPA receptor subunits, GluR2/3 (TTX: 0.83 ± 0.04, TTX + 4AP: 1.31 ± 0.06) (Figure 5
B). Because TTX-treatment alone did not influence either mEPSC or GluR1 distribution compared to untreated slices (Figure 6
), the differential synaptic modulation along the somatic-dendritic axis is mediated by A-type potassium channels.
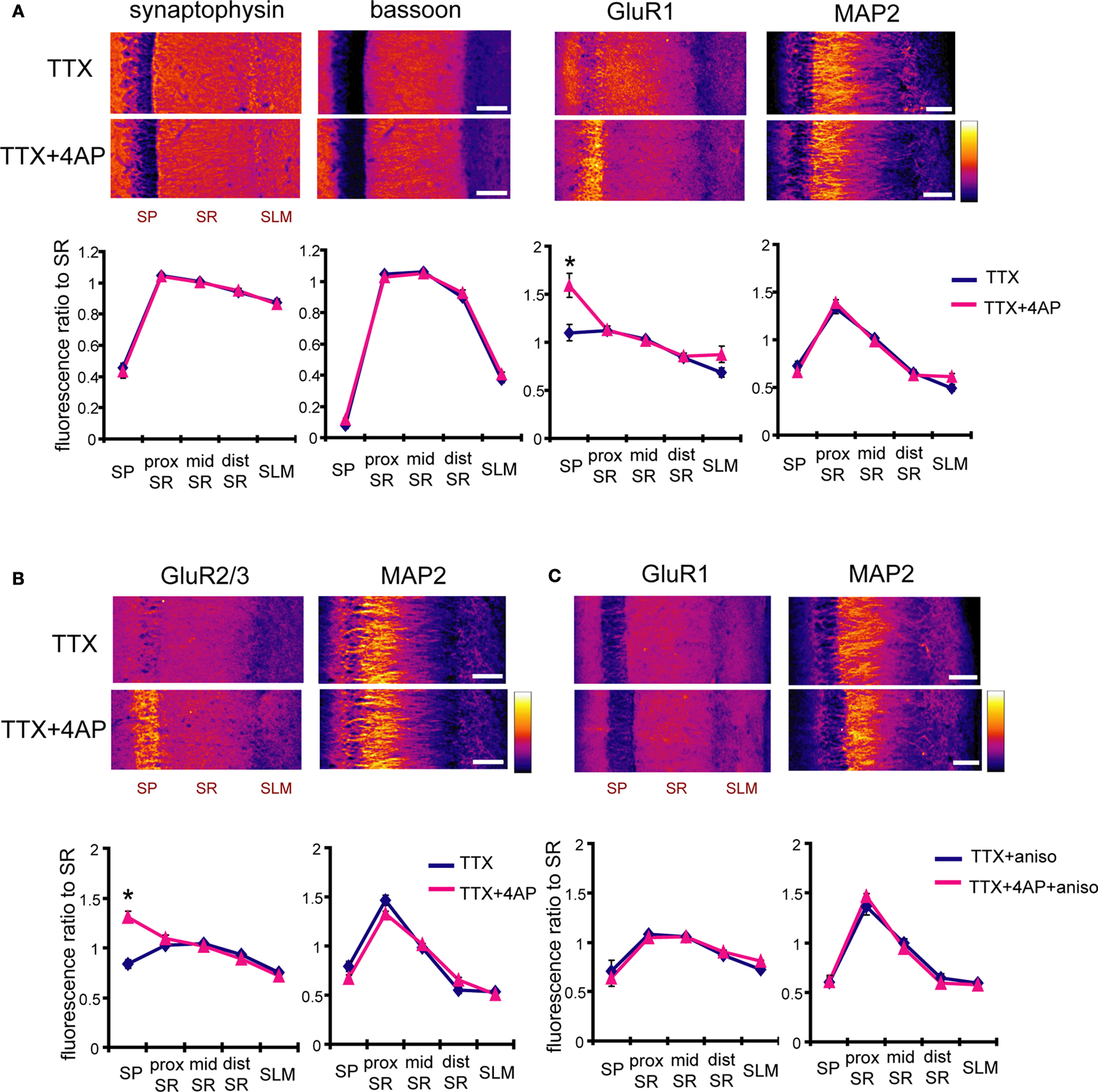
Figure 5. A-type channel blockade enhances the relative density of GluR1 in the stratum pyramidale. (A) Immunohistochemistry of hippocampal slices after 12-h treatment with either TTX (2 μM) alone or TTX + 4AP (5 mM). Hippocampal slices were stained with antibodies raised against a pair of presynaptic proteins (synaptophysin and bassoon), or a pair of postsynaptic proteins (GluR1 and MAP2). The fluorescent signal was measured along the apical dendrites from the stratum pyramidale to the stratum lacunosum-moleculare in area CA1. The stratum radiatum was equally divided into three parts; proximal, middle and distal. The fluorescent signal was normalized to the mean signal in the stratum radiatum. The somatic signal ratio of GluR1 was significantly enhanced after A-type channel blockade (presynaptic protein staining: n = 8 for each, postsynaptic protein staining: TTX: n = 8, TTX + 4AP: n = 7) (scale bar = 100 μm) (*p < 0.05). (B) The enhancement of somatic GluR2/3 signal ratio after A-type channel blockade (n = 8 for each group) (scale bar = 100 μm) (*p < 0.05). (C) Co-treatment with a protein synthesis inhibitor, anisomycin (50 μM), blocked the somatic GluR1 signal enhancement produced by chronic A-type channel blockade (n = 4 for each) (scale bar = 100 μm).
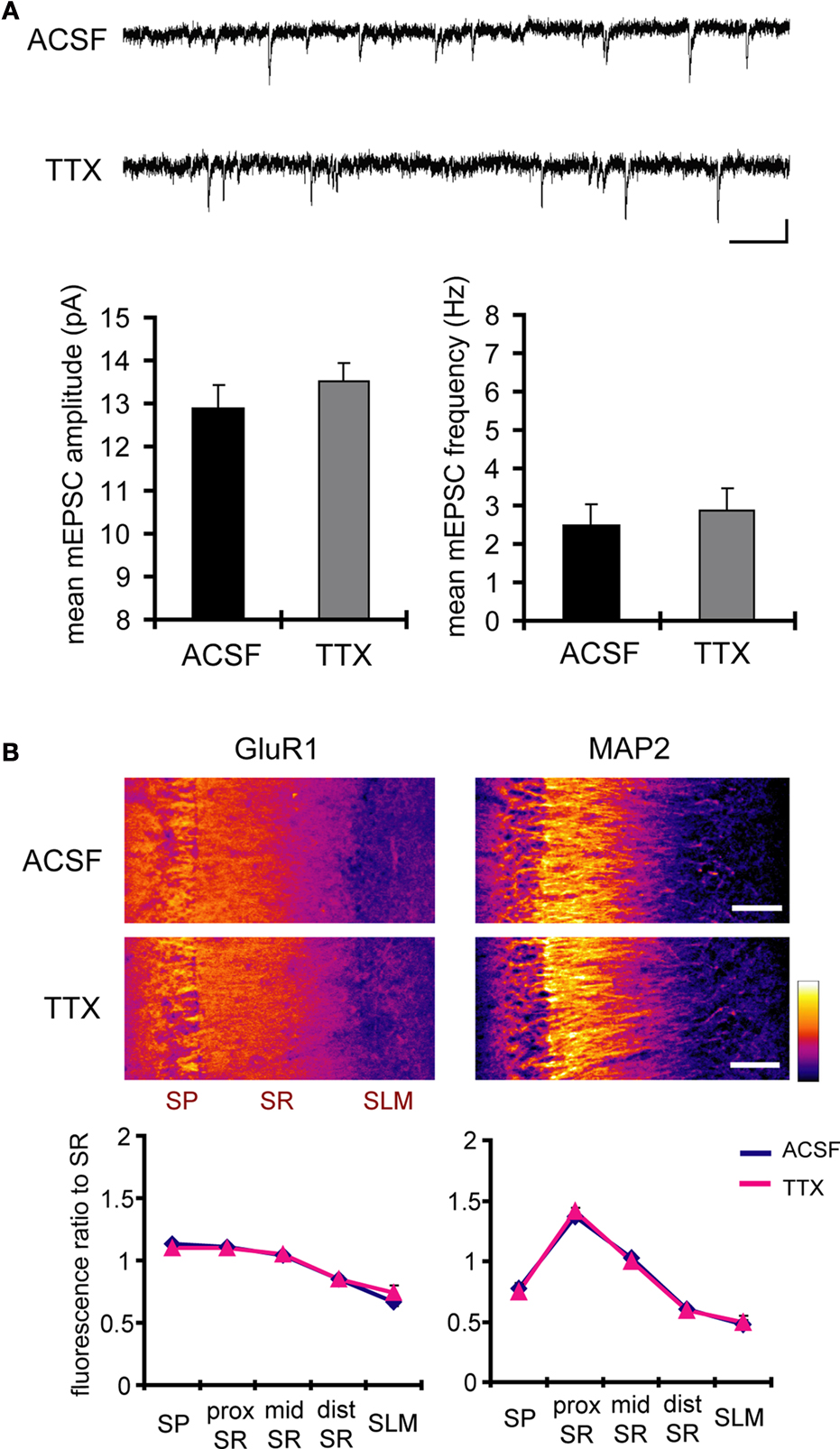
Figure 6. A 12-h treatment with TTX alone influenced neither mEPSC frequency/amplitude nor GluR1 distribution. (A) Representative mEPSCs recorded from the somata of CA1 pyramidal neurons. Hippocampal slices were treated either with or without TTX (2 μM) for 12 h. The TTX-treatment alone influenced neither the amplitude nor the frequency of mEPSCs (n = 9 for each group) (scale bar = 10 pA, 500 ms). No apparent difference in membrane properties was observed between experimental groups (Table 1
). (B) Immunohistochemistry of hippocampal slices after 12-h treatment either with or without TTX. The TTX-treatment alone did not influence GluR1 distribution along the dendritic axis (n = 9 for each group).
The change in AMPA receptor distribution following 4AP treatment suggests a postsynaptic mechanism for synaptic scaling. Our recording results, however, indicate that the primarily influence of 4AP treatment is to increase the frequency, but not the amplitude, of mEPSCs (Figure 1
). An analysis of the ranked mEPSC amplitude distribution, rather than the mean mEPSC amplitude, revealed that chronic 4AP treatment indeed enhances synaptic strength (Figures 7
A,B). How might an increase in postsynaptic AMPA receptors contribute to the observed increase in mEPSC frequency? One possible mechanism is that an increased expression of AMPA receptors may facilitate unsilencing of synapses, increasing a number of functional synapses (Kullmann, 2003
). Another possibility is that enhanced synaptic strength mediated by AMPA receptors may improve the detection of mEPSCs that are originally below our detection threshold, contributing to an increase in mEPSC frequency. Under our experimental conditions, changing the holding potential of the postsynaptic neuron is indeed sufficient to influence the mean frequency of mEPSCs (Figure 7
C).
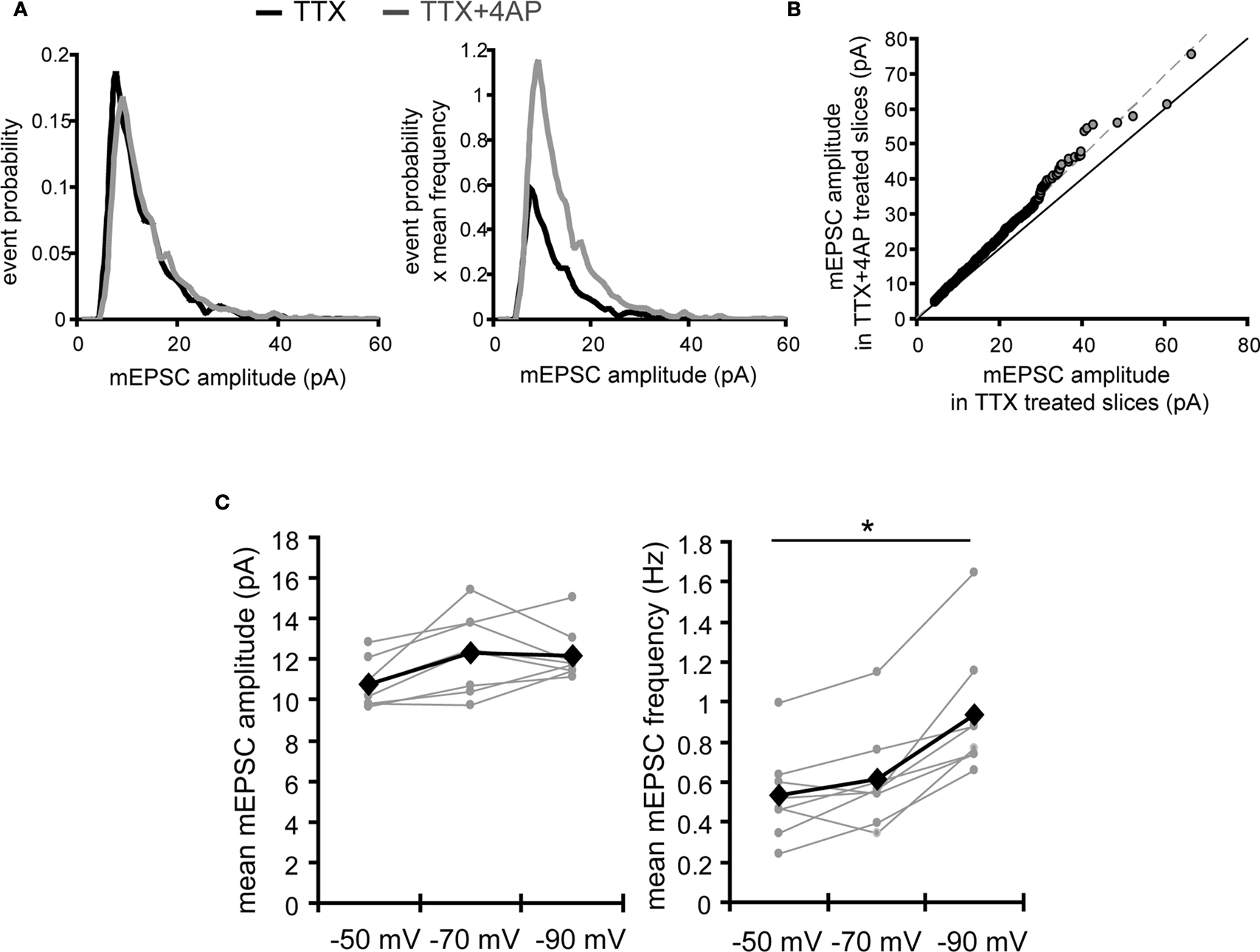
Figure 7. A postsynaptic mechanism may contribute to the enhancement of mEPSC frequency by chronic A-type channel blockade. (A) Analysis of mEPSC amplitude recorded from the soma in either TTX or TTX + 4AP-treated slices. The left figure shows the event probability of the mEPSC amplitude. In the right figure, the mean mEPSC frequency was multiplied by the event probability, thus the resulting traces are equivalent to the event histogram. Note that most events are of small amplitude, indicating that the detection threshold for mEPSC analysis can influence the recorded frequency of events under our experimental conditions (n = 2,080 mEPSCs chosen randomly from recording data used in Figure 1
A). (B) Ranked mEPSC amplitudes from TTX-treated slices were plotted against ranked mEPSC amplitudes from TTX + 4AP-treated slices. Data were subjected to a linear fit (gray dotted line; y = ax). The estimated line slope is 1.155 (between 1.153 and 1.156 with 95% confidence), which is significantly larger than a slope of 1 (black solid line), suggesting postsynaptic scaling. (C) An example showing the effect of postsynaptic membrane potential on mEPSC frequency. mEPSCs were recorded at different holding potentials (−50, −70 or −90 mV) from the same neuron. Data from individual experiments are represented by gray circles and black diamonds represent the mean. An ANOVA revealed a significant influence of the holding potential on mEPSC frequency, suggesting that postsynaptic membrane potential can influence the detection of mEPSCs under our experimental conditions probably by changing the driving force for the synaptic current (n = 8) (*p < 0.05).
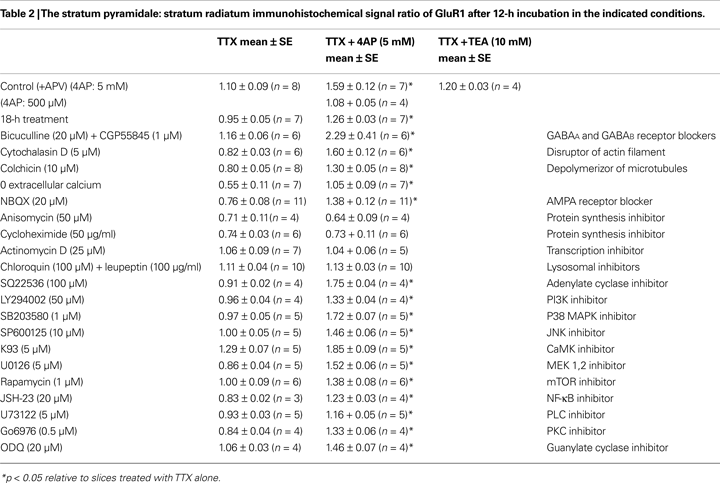
We next addressed potential cellular mechanisms for the 4AP-induced change in AMPA receptor distribution. The 4AP-induced change in GluR1 distribution was still observed under AMPA receptor blockade, GABA receptor blockade, a (nominally) zero extracellular calcium condition, or disruption of microtubules or microfilaments (Table 2
). Although recent studies have reported activity-dependent synaptic modulation by A-type potassium channels (Kim et al., 2007
; Andrasfalvy et al., 2008
; Jung et al., 2008
), the 4AP-mediated scaling we observed here is independent of synaptic activities, suggesting an involvement of a previously unexplored mechanism. The protein-synthesis inhibitors (anisomycin or cycloheximide) or a transcription inhibitor (actinomycin D), on the other hand, completely blocked the GluR1 redistribution induced by 4AP (Figure 5
C and Table 2
). Thus, our results suggest that A-type channels influence synaptic strength and GluR distribution along the somatic-dendritic axis by an activity- or calcium-independent, but protein synthesis and/or transcription-dependent mechanism.
Chronic External Electric Field-Application to Slices Mimics the Effect of A-Type Potassium Channel Blockade
How do A-type potassium channels influence synaptic strength by an activity- or calcium-independent mechanism? Because A-type channels can be activated near the resting membrane potential of neurons (Hoffman et al., 1997
), the distance-dependent distribution of A-type channels may differentially influence the resting membrane potential along the dendrites. To examine this idea, we directly measured the resting membrane potential either from the somata or dendrites of CA1 pyramidal neurons, using whole-cell recordings. In recordings conducted in control medium (ACSF + TTX), the resting membrane potential was significantly different between soma and dendrites (soma: −72.7 ± 0.44 mV, dendrites: −75.7 ± 0.65 mV; Figure 8
A). However, when 4AP was applied to the slices, the observed difference in resting membrane potential was abolished (soma: −73.3 ± 0.62 mV, dendrites: −73.6 ± 0.55 mV; Figure 8
A). Thus, A-type potassium channels appear to contribute to the voltage gradient along the dendrites, which may, in turn, modulate synaptic strength and GluR1 distribution.
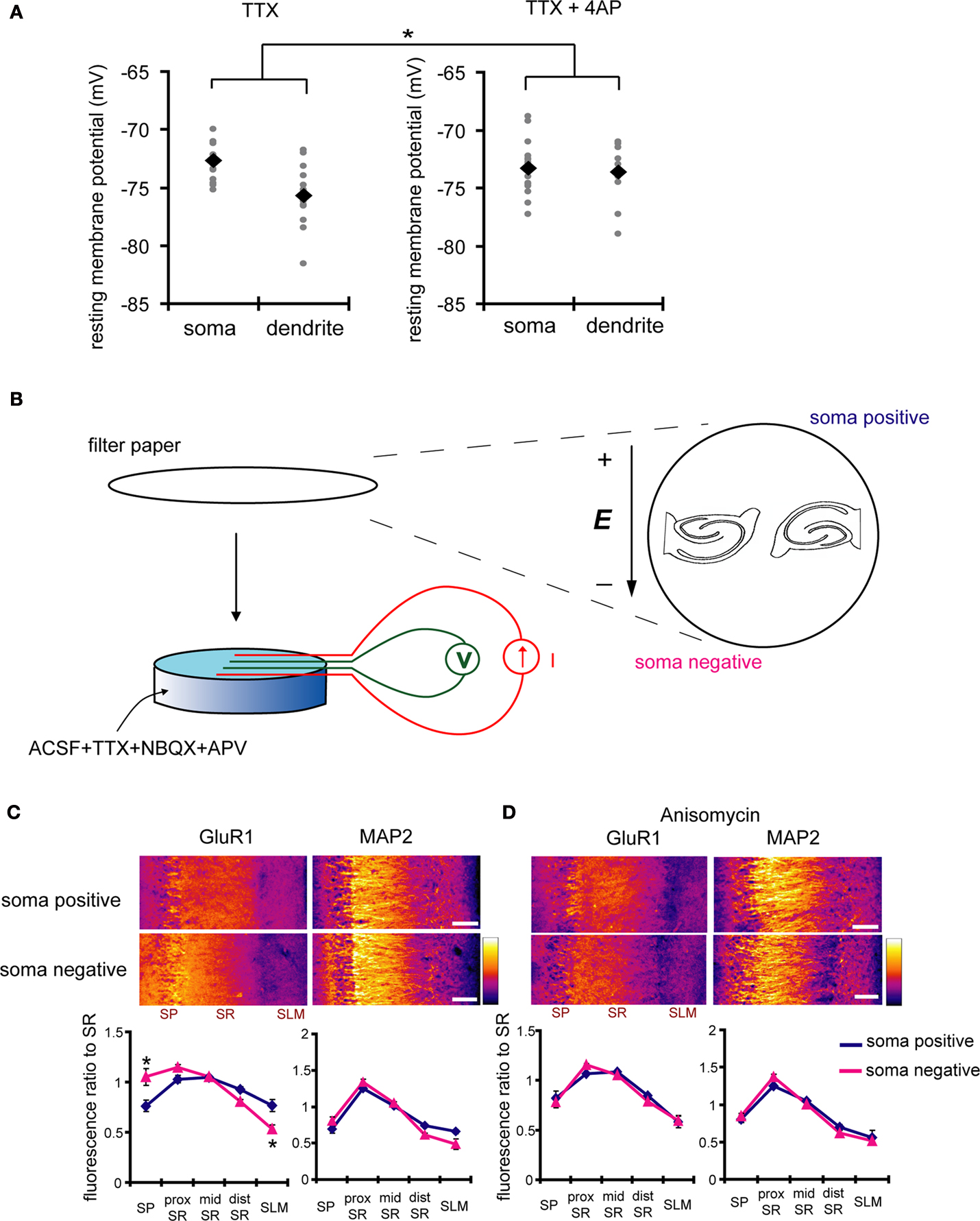
Figure 8. A chronically applied external electric field influences GluR1 distribution along the somatic-dendritic axis. (A) Resting membrane potentials measured from individual somata and dendrites of CA1 pyramidal neurons. The recording sites in dendrites were in the distal half of the stratum radiatum (approximately 300–350 μm away from soma). Data from individual experiments are represented by gray circles and black diamonds represent the mean. The application of 4AP (10 mM) abolished the voltage difference between soma and dendrites (n = 15 for each group) (*p < 0.05 in two-way ANOVA). (B) Scheme of apparatus used for the electric field-application to slices. The external electric field was applied to slices with a current source. The current amplitude was appropriately adjusted by monitoring the voltage difference between the two electrodes using a voltameter. The slices were treated with TTX (2 μM) + NBQX (20 μM) + APV (50 μM) to block neuronal activities. A chronic DC electric field (10 mV/mm) was applied to slices for 4 h. (C) The influence of the externally applied electric field on the GluR1 distribution. The relative somatic GluR1 signal intensity was significantly higher when the electric field was applied in a direction such that somatic side was negative relative to apical dendrites (n = 8 for each group) (*p < 0.05). (D) Slices were pretreated with a protein synthesis inhibitor, anisomycin (50 μM), beginning 30 min prior to the electric field-application. The external electric field was applied in the presence of TTX + NBQX + APV and anisomycin. Co-treatment with anisomycin abolished the GluR1 redistribution by the differentially applied chronic electric field (n = 7 for each group).
If a dendritic electric field generated by a voltage gradient along the dendritic axis regulates GluR1 distribution, then an externally applied electric field might be able to similarly alter GluR1 distribution. We tested this hypothesis by applying a chronic electric field, parallel to the main apical dendritic axis, to hippocampal slices (Figure 8
B; see Materials and Methods for detail) and then measuring GluR1 distribution using immunohistochemical techniques. The field was applied for 4 h, with the somatic region representing either the positive or negative pole. Following chronic field stimulation, we found that GluR1 distribution was significantly influenced in a direction-sensitive manner (signal ratio of s. pyramidale to s. radiatum: soma positive 0.76 ± 0.06, soma negative 1.05 ± 0.08; Figure 8
C). Corresponding to the GluR1 distribution, we found that mEPSC frequency was also significantly influenced by an external electric field-application in a direction-sensitive manner (mEPSC amplitude: soma positive 12.01 ± 0.40 pA, soma negative 12.52 ± 0.29 pA; mEPSC frequency: soma positive 0.83 ± 0.09 Hz, soma positive 1.15 ± 0.07 Hz; Figure 9
). Furthermore, this differential electric-field induced change in GluR1 distribution was completely abolished by the addition of a protein-synthesis inhibitor, anisomycin (soma positive: 0.82 ± 0.07, soma negative: 0.78 ± 0.06; Figure 8
D). Fluorescence-loss-in-photobleaching experiments in neurons expressing a GluR1-GFP fusion protein argue against a direct effect of electric field on GluR1 movement (Figure 10
). These results show that a modest change in the electric field along the dendritic axis is sufficient to elicit a protein-synthesis dependent change in the distribution of GluR1.
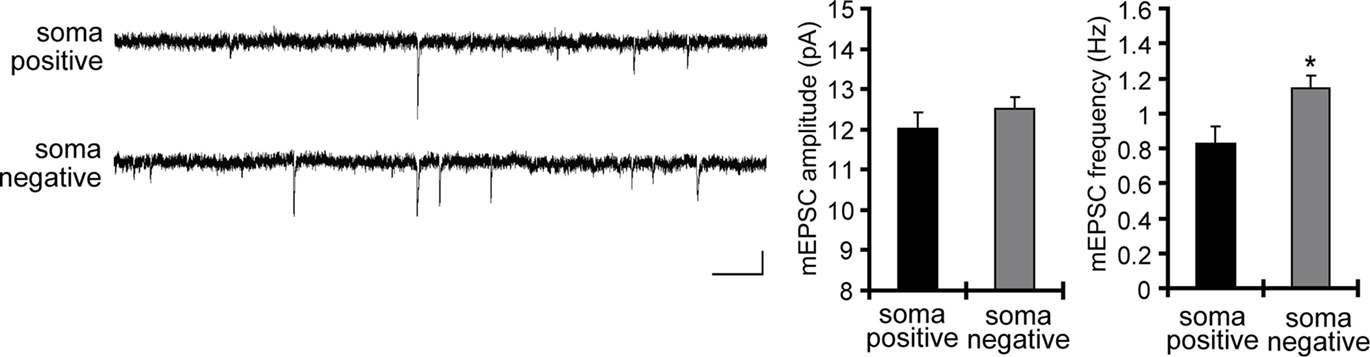
Figure 9. A chronically applied external electric field influences the frequency of mEPSCs. Representative mEPSCs recorded from the somata of CA1 pyramidal neurons after 4 h of external electric field-application (10 mV/mm) in the presence of TTX (2 μM) + APV (50 μM). The electric field-application significantly influenced mEPSC frequency in a direction-sensitive manner (*p < 0.05) (n = 14 for each group) (scale bar = 10 pA, 500 ms). No apparent difference in membrane properties was observed between experimental groups (Table 1
).
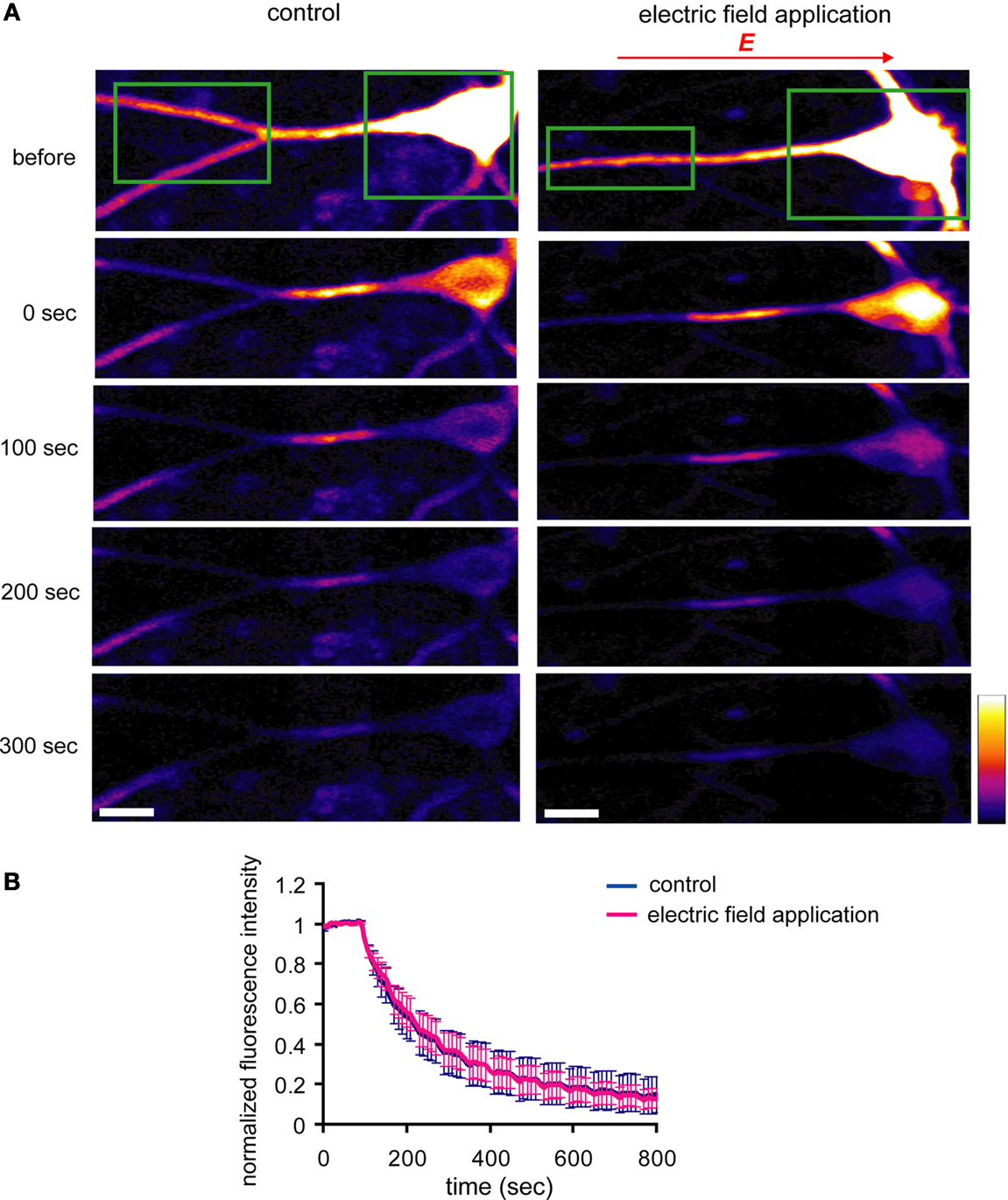
Figure 10. An externally applied electric field does not influence the movement of GluR1-GFP. (A) GluR1-GFP was expressed in hippocampal dissociated culture neurons by a Sindbis virus expression system. The images were taken every 10 s. After taking 10 baseline images, a region that included the soma and the distal dendrites (green boxes) was photobleached, leaving a 25 μm length segment in proximal dendrite. Then, the photobleaching was repeated after every 5 images acquired. For the experiments with the electric-field application, a 10 mV/mm of external electric field was continuously applied along the primary dendrites once the photobleaching started (scale bar = 12.5 μm). (B) Analysis of fluorescence decay in the 25 μm segment in proximal dendrite. No significant difference was observed in fluorescence decay under the external electric-field application, suggesting that the electric field does not influence GluR1-GFP movement, directly (n = 5).
The computational power of a single neuron arises from the large number of synaptic inputs distributed in its complex dendritic structure. These synaptic inputs are integrated in the dendrites and soma, shaping the neuron’s final output as action potentials. The strength of each synaptic input is dynamically modulated by activity-dependent synaptic plasticity, such as LTP or LTD, which will change the integrative pattern of the synaptic inputs. As such, the dynamics of synaptic plasticity at individual synapse allows for neurons to have an enormous number of synaptic integration patterns, giving rise to different types of computation.
It has been suggested, however, that activity-dependent synaptic plasticity poses a problem for neuronal stability (Miller and MacKay, 1994
; Miller, 1996
; Abbott and Nelson, 2000
). During LTP induction, strongly activated synapses achieve higher synaptic efficacy, resulting in a positive-feedback system. As strongly activated synapses continue to potentiate, synaptic strength will be eventually saturated and the output of a given neuron will be dominated by just a few strong synapses, sacrificing the computational power of the cell. Homeostatic synaptic plasticity can solve this issue, because it endows neurons with the ability to maintain their firing frequency in an appropriate range (Turrigiano et al., 1998
; Turrigiano, 2008
). The homeostatic mechanisms proposed thus far, however, do not address the synaptic balance problem. How is a distance-dependent increase in synaptic strength (i.e. proximal synapses are weaker than distal synapses) maintained during epochs of activity-dependent plasticity? The data presented here suggest that A-type potassium channels may be a key mediator of the distance-dependent scaling. Our results suggest that A-type potassium channels influence AMPA receptor distribution by influencing the voltage gradient along the dendrites.
A number of studies have examined the influence of an externally applied electric field on different neuronal functions, including neurite elongation (Patel and Poo, 1982
), receptor clustering (Orida and Poo, 1978
; Poo et al., 1978
), or spike initiation (Gluckman et al., 1996
; Francis et al., 2003
; Jefferys et al., 2003
). In addition, the distribution of several molecules can be directly influenced by electric fields. For example, the acetylcholine receptor (Young and Poo, 1983
) and epidermal growth factor receptor (Zhao et al., 2002
), accumulate at the cathodal side of electric fields. McLaughlin and Poo (1981)
suggested that charged macromolecules on the cell surface generate electro-osmotic fluxes during electric field-application to redistribute surface proteins. Externally applied electric fields can also influence the distribution of intracellular signaling molecules, including phosphoinositide 3-kinase (Zhao et al., 2006
), cAMP (Sebestikova et al., 2005
) and guanylate cyclase (Sato et al., 2009
). Thus, if A-type potassium channels create a voltage gradient via their differential distribution along the dendritic axis, it is possible that some signaling molecules or surface receptors are influenced by this self-generating electric field.
In our studies, the application of a weak DC electric field (10 mV/mm) changed the distribution GluR1 along the dendritic axis. This effect was not due to a direct influence of the electric field on GluR1 movement (Figure 10
). Electric fields may instead influence the transcription of GluR1 as several transcription factors, such as β-catenin or NF-κB, exist in synaptic regions (Meffert et al., 2003
; Abe and Takeichi, 2007
). Electric fields may influence the translocation of these transcription factors from synaptic regions to the nucleus.
Another possibility is that electric fields may influence the expression or distribution of proteins essential for GluR1 trafficking from the soma to the dendrites. A recent study demonstrated that the dendritic transport of GluR1 requires its phosphorylation or interactions with a GluR1-interacting protein, stargazin. In the absence of stargazin GluR1 is degraded in the lysosome (Kessels et al., 2009
). Electric fields may thus influence the proteins that control GluR1 transport along the dendrites. Supporting this idea, inhibitors of lysosomal degradation blocked the GluR1 distribution change either by 4AP- or external electric field-application (Figure 11
). Thus, the control of dendritic GluR1 distribution by electric fields likely requires the proper coordination of protein synthesis and degradation in the soma and dendrites.
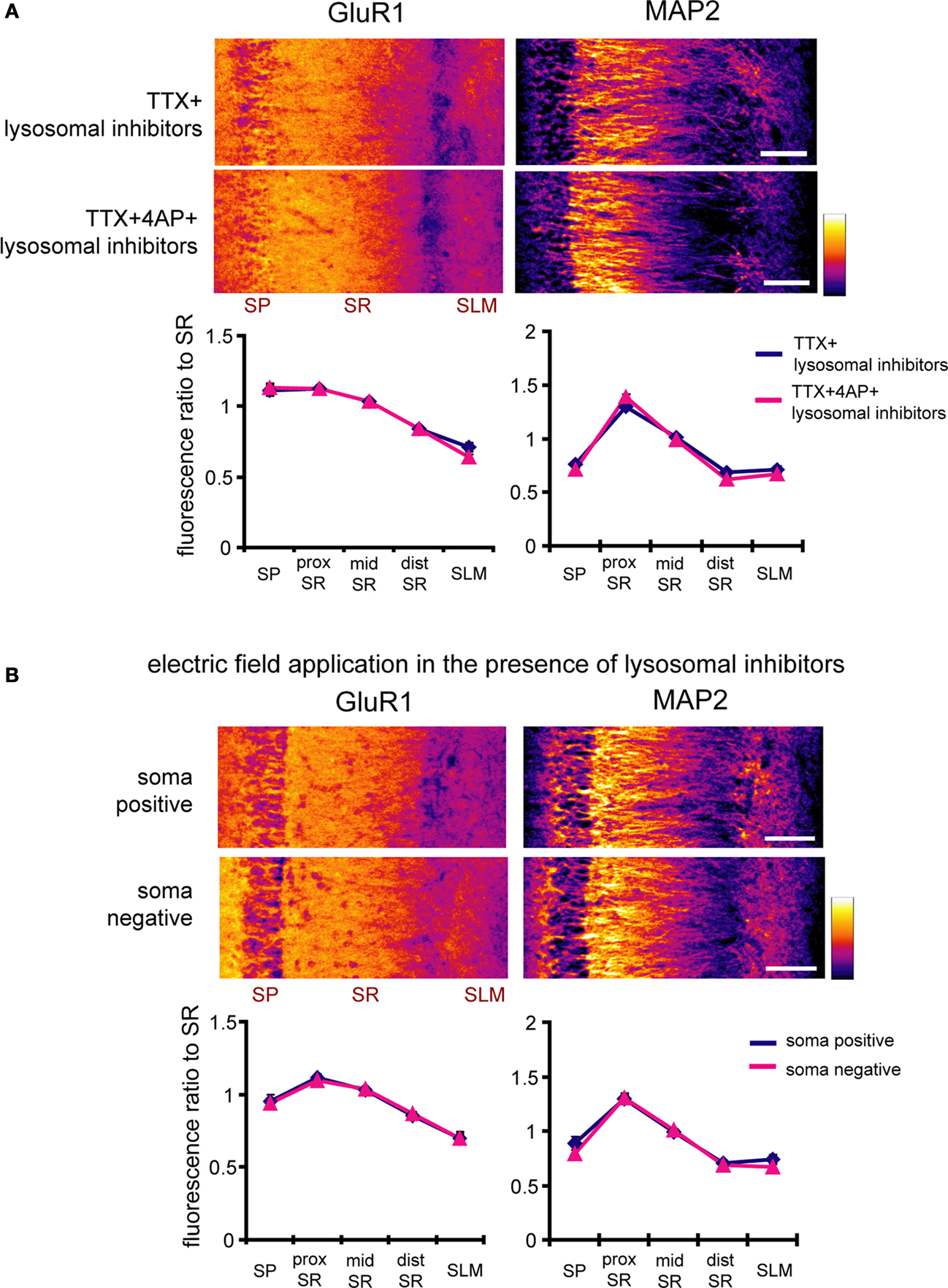
Figure 11. Inhibitors of lysosomal degradation block the enhancement of GluR1 signal ratio in the stratum pyramidale either by 4AP- or external electric field-application. (A) Immunohistochemistry of hippocampal slices was performed after a 12-h treatment with either TTX (2 μM) or TTX + 4AP (5 mM) in the presence of blockers of lysosomal degradation, chloroquine (100 μM) and leupeptin (100 μg/ml). The fluorescent signal was normalized to the mean signal in stratum radiatum. Inhibitors of lysosomal degradation blocked the GluR1 distribution change elicited by 4AP treatment (n = 10 for each group) (scale bar = 100 μm). (B) Hippocampal slices were pretreated with chloroquine (100 μM) and leupeptin (100 μg/ml) beginning 30 min prior to the electric field-application. The external DC electric field (10 mV/mm) was applied for 4 h in the presence of TTX + NBQX + APV, chloroquine and leupeptin. Inhibitors of lysosomal degradation blocked the GluR1 distribution change elicited by external electric field-application (soma positive: n = 8, soma negative: n = 9) (scale bar = 100 μm).
We demonstrated that A-type potassium channels play a key role in the distance-dependent scaling of AMPA receptors along the dendrites. Our results suggest a novel homeostatic scaling mechanism, which is mediated by a dendritic voltage gradient generated by differentially distributed A-type potassium channels. Neurons require a long-distance mechanism to control neurotransmitter receptor and ion channel density along the proximal to distal gradient of dendrites. The varied distribution of receptors and channels along this gradient may exceed the capacity of molecular diffusion. An electric field-mediated mechanism has the potential advantage to effect an enduring and long-range influence along the dendrites.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank members of the Schuman laboratory for discussions. Hiroshi T. Ito is supported by the Nakajima foundation. Erin M. Schuman is an Investigator of the Howard Hughes Medical Institute.
Sato, M. J., Kuwayama, H., van Egmond, W. N., Takayama, A. L., Takagi, H., van Haastert, P. J., Yanagida, T., and Ueda, M. (2009). Switching direction in electric-signal-induced cell migration by cyclic guanosine monophosphate and phosphatidylinositol signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 6667–6672.
Zhao, M., Song, B., Pu, J., Wada, T., Reid, B., Tai, G., Wang, F., Guo, A., Walczysko, P., Gu, Y., Sasaki, T., Suzuki, A., Forrester, J. V., Bourne, H. R., Devreotes, P. N., McCaig, C. D., and Penninger, J. M. (2006). Electrical signals control wound healing through phosphatidylinositol-3-OH kinase-gamma and PTEN. Nature 442, 457–460.