1
Centro de Neurociências e Biologia Celular, Universidade de Coimbra, Portugal
2
Departments of Neurology and Molecular and Integrative Physiology, University of Kansas School of Medicine, USA
3
Faculdade de Medicina, Universidade de Coimbra, Portugal
Parkinson’s disease (PD) is associated with perturbed mitochondria function and alpha-synuclein fibrillization. We evaluated potential mechanistic links between mitochondrial dysfunction and alpha-synuclein aggregation. We studied a PD cytoplasmic hybrid (cybrid) cell line in which platelet mitochondria from a PD subject were transferred to NT2 neuronal cells previously depleted of endogenous mitochondrial DNA. Compared to a control cybrid cell line, the PD line showed reduced ATP levels, an increased free/polymerized tubulin ratio, and alpha-synuclein oligomer accumulation. Taxol (which stabilizes microtubules) normalized the PD tubulin ratio and reduced alpha-synuclein oligomerization. A nexus exists between mitochondrial function, cytoskeleton homeostasis, and alpha-synuclein oligomerization. In our model, mitochondrial dysfunction triggers an increased free tubulin, which destabilizes the microtubular network and promotes alpha-synuclein oligomerization.
Sporadic Parkinson’s disease (PD) pathology is characterized by substantia nigra pars compacta neuronal loss, systemic dysfunction of the mitochondrial enzyme complex I, and intraneuronal alpha-synuclein (a-syn) aggregation (Cardoso et al., 2005
). While a-syn overexpression and mutation cause familial PD, the mechanisms that mediate this are unclear (Singleton et al., 2003
; Hope et al., 2004
). This is not surprising, as the normal function of a-syn is itself poorly characterized.
Recent data indicate a-syn interacts with microtubules to affect membrane stability and neuronal plasticity (Lee et al., 2006
). Microtubules are highly dynamic tubulin polymers that mediate organelle transport, cell motility, division, and morphology (Morris and Hollenbeck, 1995
). Conditions that favor incorporation of tubulin into microtubules reduce free/polymerized tubulin ratios, while conditions that retard microtubule formation increase this ratio.
The complex I inhibitors rotenone and 1-methyl-4-phenylpyridinium (MPP+) alter microtubule dynamics and increase free tubulin/polymerized tubulin ratios (Cappeleti et al., 2005
; Ren and Feng, 2007
). Tubulin, interestingly, induces a-syn fibrillization in yeast, rat brain, and human brain (Alim et al., 2002
; Kim et al., 2007
).
Because complex I dysfunction occurs in PD and may contribute to a-syn aggregation (Parker et al., 1989
; Schapira et al., 1989
; Lee, 2003
) we hypothesized a nexus between complex I dysfunction, ATP depletion, microtubule disassembly, and a syn oligomerization might exist. We tested this hypothesis using cytoplasmic hybrid (cybrid) cells prepared via the transfer of PD subject platelet mitochondria to mitochondrial DNA (mtDNA) depleted (ρ0) human NT2 teratocarcinoma cells. PD cybrids have been shown to model complex I dysfunction and its consequences, including increased oxidative stress, perturbed calcium homeostasis, and reduced ATP (Cassarino et al., 1997
; Sheehan et al., 1997
; Esteves et al., 2008
, 2009
).
Chemicals and Cell Media
Paclitaxel (taxol), was obtained from Sigma (St. Louis, MO, USA). Optimem medium was obtained from Gibco-Invitrogen. Non-dialyzed and dialyzed fetal bovine serum was obtained from Gibco-Invitrogen. NT2 ρ0 cell growth medium consisted of Optimem supplemented with 10% non-dialyzed fetal bovine serum, 200 μg/ml sodium pyruvate, 150 μg/ml uridine, and 100 IU/ml penicillin and 50 μg/ml streptomycin. NT2 cybrid selection medium consisted of Optimem supplemented with 10% dialyzed fetal bovine serum and penicillin–streptomycin. Cybrid expansion medium consisted of Optimem supplemented with 10% non-dialyzed fetal bovine serum and penicillin–streptomycin. Taxol medium consisted of cybrid expansion medium with 5 nM taxol, and was prepared using a DMSO-taxol stock solution. For all conditions tested, control experiments were performed using unsupplemented cybrid expansion medium.
Human Subjects
Subject participation was approved through the Institutional Review Board of the University Hospital of Coimbra. The two sporadic PD subjects, without any nuclear DNA mutation known to be relevant to PD, meet Gelb et al. (1999)
diagnostic criteria and two healthy, age-matched control subject provided 10 ml blood samples following written informed consent. Blood was drawn directly into tubes containing acid-citrate-dextrose.
Creation of Cybrid Cell Lines and Cell Culture
NT2 ρ0 cells were briefly agitated in polyethylene glycol with platelets from the human subjects (Cardoso et al., 2004
). Seven days after plating the resulting mixture in T75 flasks and ρ0 growth medium, the medium was changed to cybrid selection medium. NT2 ρ0 cells lack intact mtDNA, do not possess a functional electron transport chain (ETC), and are auxotrophic for pyruvate and uridine (Swerdlow et al., 1997
; Cardoso et al., 2004
). Maintaining cells in selection medium removes ρ0 cells that have not repopulated their mtDNA with platelet mtDNA. After selection was complete, the resultant cybrid cells were switched to cybrid expansion medium.
For taxol, experiments, cybrid cell lines were placed in media containing 5 nM taxol. Flasks were maintained in these media at 37°C, 5% CO2 for 24 h prior to harvesting.
Immunocytochemistry
Cybrid cell lines grown on coverslips in 12 well plates were washed twice with PBS and fixed for 30 min at room temperature using 4% paraformaldehyde. The fixed cells were washed again with PBS, permeabilized with 0.2% Triton X-100, and blocked with 3% BSA. The permeabilized cells were incubated with primary antibody (1:2,000 monoclonal anti-alpha-tubulin from Sigma; 1:400 monoclonal anti-a-syn from Santa Cruz Biotechnology, Inc., Santa Cruz, SA, USA, or 1:400 Polyclonal anti-A11 from BioSource International, Inc., Flynn Road, Camarillo, CA, USA) for 1 h and then with the appropriate secondary antibody (1:250 alexa fluor 594 and 488 from Molecular probes, Eugene, OR, USA). Cells were visualized by confocal microscopy.
Immunoblotting
Individual cybrid cell lines were scraped in buffer containing 25 mM HEPES, 1 mM EDTA, 1 mM EGTA, 2 mM MgCl2, protease inhibitors (commercial protease inhibitor cocktail from Sigma), 0.1 M PMSF (Sigma), 0.2 M DTT (Sigma), and 1% Triton X-100. Cell suspensions were frozen three times in liquid nitrogen and centrifuged at 20,000 g for 10 min. The resulting supernatants were removed and stored at −80°C. Protein concentrations were determined by the Bradford method and equal amounts of protein (30 or 60 μg) were used for immunoblotting. For the SDS-PAGE experiments samples were resolved by electrophoresis in SDS-polyacrylamide gels and transferred to PVDF membranes. For the PAGE experiments samples were not boiled and were diluted 1:2 in a sample buffer without SDS. Non-specific binding was blocked by gently agitating the membranes in 5% non-fat milk and 0.1% Tween in TBS for 1 h at room temperature. The blots were subsequently incubated with the respective primary antibodies overnight at 4°C with gentle agitation (1:1,000 Polyclonal anti a syn antibody from Cell Signaling Technology, Inc., Danvers, MA, USA; 1:2,500 monoclonal anti-GAPDH antibody from Chemicon International; or 1:10,000 monoclonal anti-alpha-tubulin antibody from SIGMA).
Blots were washed with TBS containing 0.1% non fat milk and 0.1% Tween three times (each time for 15 min), and then incubated with the appropriate horseradish peroxidase-conjugated secondary antibody for 2 h at room temperature with gentle agitation. After three washes specific bands of interest were detected by developing with an alkaline phosphatase enhanced chemical fluorescence reagent (ECF, GE Healthcare, Buckinghamshire, England). Fluorescence signals were detected using a Biorad Versa-Doc Imager, and band densities were determined using Quantity One Software.
Isolation of Soluble and Polymeric Tubulin
To prepare soluble and polymeric tubulin fractions, cells were very gently washed twice with a microtubule stabilizing buffer (0.1 M N-morpholinoethanesulfonic acid, pH 6.75; 1 mM MgSO4; 2 mM EGTA; 0.1 mM EDTA; 4 M glycerol). Soluble proteins were extracted for 4–6 min at 37°C in 100 μl of microtubule stabilizing buffer containing 0.1% Triton X-100. The remaining contents of the culture dish were scraped in 100 μl of 25 mM Tris (pH 6.8) and 0.5% SDS. This suspension was frozen three times in liquid nitrogen (Joshi and Cleveland, 1989
). Protein concentrations for each sample were quantified using the Bradford method.
Data Analysis
Each experimental endpoint for each cell line was run in duplicate, and data are expressed as the mean ± SEM from at least three different measurements. P-values were calculated by one-way ANOVA, followed by a post hoc Dunnett’s or Bonferroni’s t test when appropriate.
Alpha Synuclein Oligomerization and Protein Ubiquination were Increased in the PD Cybrid Cells
The PD cybrid cells and the control cybrid cells analyzed in this study were originally prepared and bioenergetically characterized as part of previously reported PD cybrid studies (Esteves et al., 2008
, 2009
). PD cybrid lines used in this current study show a decrease of 38% in complex I activity and of 28% in ATP levels when compared to the control cybrid lines (Esteves et al., 2008
). In order to confirm the presence of protein aggregates we used antibodies (A11) that detect oligomeric proteins. A11 antibody staining was much stronger in the PD cell cells, suggesting it contained more oligomeric protein or proteins (Figure 1
A). Moreover, we performed a PAGE western blot to detect if some of these oligomers were a-syn. We observed that the a-syn oligomer/monomer ratio was higher in the PD cybrid lines (Figures 1
B,C), and the PD cybrid lines showed more protein ubiquination levels (Figure 1
D). We also observed an increase in a-syn monomers levels detected by SDS-PAGE in PD cybrid lines as compared to CT cybrid cells (data not shown).
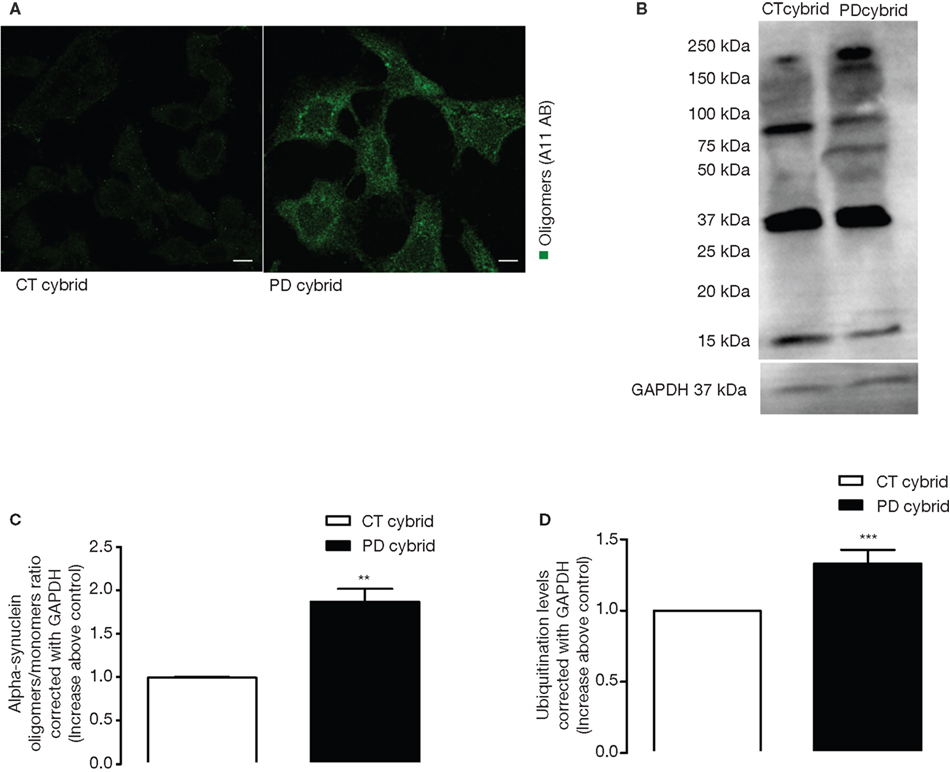
Figure 1. Protein oligomerization in PD cybrids. (A) A11 staining is greater in the PD cybrids. Bars, 10 μm. (B) A western blot of alpha-synuclein monomer and oligomers in the PD and CT cybrid lines. (C) Densitometry analysis of alpha-synuclein western blots showing the ratio between alpha-synuclein oligomer and monomer levels corrected for GAPDH. The ratio is increased in the PD cybrid line. (D) Ubiquitination densitometry analysis showed the PD cybrid cells had more ubiquitinated protein. **P < 0.01 and ***P < 0.001, significantly different when compared to the CT cybrid lines.
The Free Tubulin/Polymerized Tubulin Ratio was Increased in the Pd Cybrid
MT assembly requires ATP, therefore the reduction of PD cybrids ATP may interfere with the efficacy of the polymerization/depolymerization process of tubulin. For that reason we immunocytochemically evaluated MT network integrity. MT networks were less well defined in the PD cybrid cells (Figure 2
A), and the free/polymerized tubulin ratio was also elevated (Figures 2
B,C). In the CT cybrid cells approximately one-third of the tubulin was in the free form, and in the PD cybrid approximately one-half of the tubulin was in the free form (Table 1
). Because of the viscosity of the polymerized tubulin all samples were corrected with GAPDH antibody in order to eliminate loading errors (Figures 2
B,C).
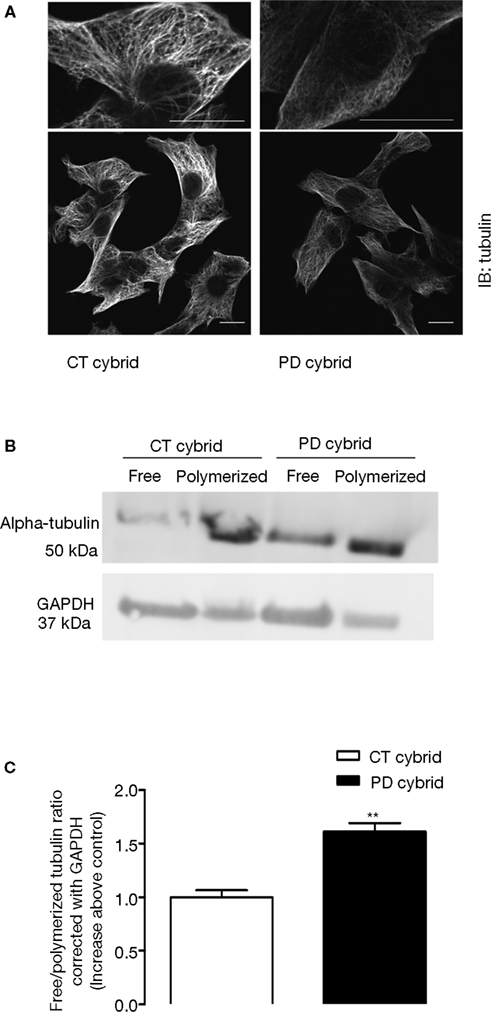
Figure 2. Tubulin alterations in the PD cybrid lines. (A) Tubulin immunocytochemistry reveals disruption of the microtubule network in the PD cybrid lines. (B) SDS-PAGE analysis shows that in the PD cybrids the free/polymerized tubulin ratio is elevated. (C) After correcting for GADPH content the PD cybrids free/polymerized tubulin ratio was elevated as compared to the control cybrid cell line. **P < 0.01, significantly different as compared to the CT cybrid lines. Bars, 10 μm.

Taxol Reduced Tubulin and Alpha Synuclein Alterations in PD Cybrid Cells
Taxol stabilizes MT assemblies and reduces free tubulin/polymerized tubulin ratios (Cragg and Newman, 2005
). Before testing its effects in our cybrid model (which consists of undifferentiated, rapidly dividing cells) we determined 5 nM taxol did not compromise cell viability (data not shown). 5 nM taxol reduced the free tubulin/polymerized tubulin ratio in the PD cybrid lines but not in the control cybrid lines (Figures 3
A,B). Taxol lowered a-syn oligomerization in PD cybrid cells but did not altered CT cybrid cells a-syn levels (Figures 3
C,D). These results support the idea that MT depolymerization may potentiate the formation of oligomeric a-syn.
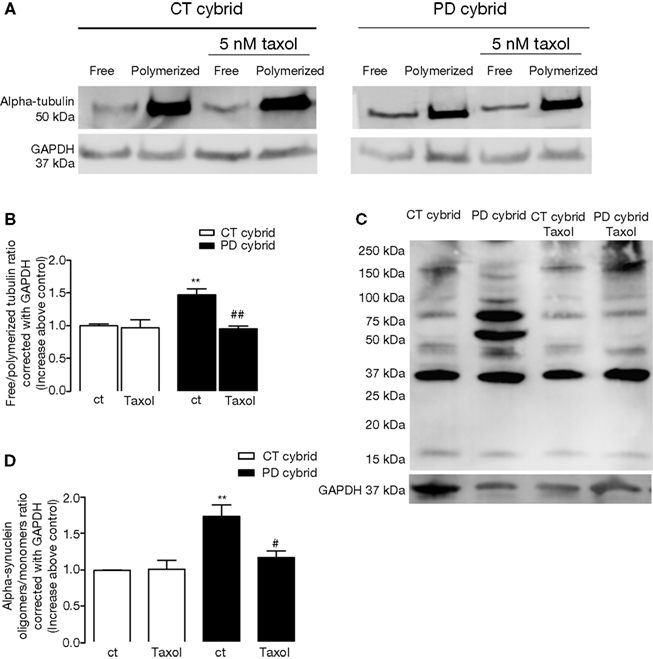
Figure 3. Effects of taxol on tubulin and alpha synuclein. (A) SDS-PAGE showing the effect of 5 nM taxol on free/polymerized tubulin ratios. (B) Densitometry analysis indicated after correcting for GADPH content, 5 nM taxol reduced the free/polymerized tubulin ratio in the PD but not the CT cybrid cells. (C) PAGE showing the effect of 5 nM taxol on alpha synuclein oligomer levels. (D) Densitometry analysis indicated 5 nM taxol reduced alpha synuclein oligomer levels in the PD cybrid cells. **P < 0.01, significantly different when compared to the untreated CT cybrid cells. #P < 0.05 and ##P < 0.01, significantly different as compared to the untreated PD cybrid line.
We recently reported that mitochondrial ROS potentiates an increase in free tubulin levels, which is followed by a significant increase of a-syn oligomers (Esteves et al., 2009
). In order to further clarify the relationship between mitochondrial metabolism and a-syn conformational change we now report studies performed using two carefully characterized PD and CT cybrid cell lines. Our results corroborate and extend our previous finding that PD cybrids mitochondrial dysfunction induces a-syn oligomerization, and suggest that microtubules may be a feasible therapeutic target to PD.
The cybrid approach has been used to model human disease-specific mitochondrial dysfunction in PD. It has been described that in cybrid models such as the one used in these experiments changes in cell physiology likely arise as a consequence of and in response to mitochondrial function (Swerdlow, 2007
).
Numerous cybrid studies using three different nuclear backgrounds have shown PD cybrids have a sustained reduction of complex I Vmax activity (Swerdlow et al., 1996
, 1998
; Gu et al., 1998
; Esteves et al., 2008
). In these studies biochemical data obtained from distinct, different cybrid cell lines were averaged and group means were comparable.
We fully characterized a-syn aggregation in two PD cybrid cell lines. We demonstrated that compared to cells from two control cybrid cell lines, the PD cybrid cells had increased a-syn oligomerization, ubiquitination, and fibrillization.
If mitochondrial dysfunction does in fact promote a-syn oligomerization, then it is necessary to elucidate the mechanisms that mediate this phenomenon. Our data support the hypothesis that free tubulin directly interacts with a-syn and promotes fibril formation (Alim et al., 2002
). While our data do not specifically elucidate how tubulin-a-syn interactions may physically promote oligomerization, they do provide a framework for developing further studies to address why a-syn oligomerization occurs in PD.
Taxol’s ability to reduce a-syn oligomerization in our experiments suggests free tubulin can promote a-syn oligomerization. This does not preclude the possibility that a-syn oligomerization might in turn disrupt tubulin polymerization (Chen et al., 2007
). It is therefore necessary to consider whether mitochondrial dysfunction supports a positive feedback loop in which mitochondrial dysfunction increases free tubulin levels, free tubulin increases a-syn oligomerization, and a-syn oligomers in turn further elevate the free/polymerized tubulin ratio. a-Syn fibrillization requires it to transition from a ‘natively unfolded’ random coil to a beta-sheet structure. The beta sheet form then aggregates into oligomers, protofibrils, and eventually amyloid fibrils with cross-beta structures (Shults, 2006
). a-Syn polymerization is nucleation-dependent and it is promoted by both tubulin and oxidative stress (Paik et al., 1999
; Giasson et al., 2000
; Souza et al., 2000
; Paxinou et al., 2001
; Sherer et al., 2002
; Esteves et al., 2009
).
Our data indicate for the first time that taxol prevents mitochondrial-mediated MT depolymerization and a-syn oligomerization in cells that have dysfunctional mitochondria. These in vitro observations were made in cell lines with a neuronal background, and it will be interesting to see whether these findings generalize to PD brains.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to Dr. Cristina Januário for identifying the PD patient, to Dr. Isabel Nunes Correia for cell culture support, and to Dr Luísa Cortes and Dr Carla Cardoso for Microscopy assistance. This work was supported by Fundação para a Ciência e a Tecnologia, Portugal (PhD grant: SFRH/BD/32470/2006); GAPI of Faculdade de Medicina da Universidade de Coimbra, Portugal; and the Parkinson’s Disease Foundation of the Heartland (USA).
Cassarino, D. S., Fall, C. P., Swerdlow, R. H., Smith, T. S., Halvorsen, E. M., Miller, S. W., Parks, J. K., Parker, W. D., and Bennet, J. P. (1997). Elevated reactive oxygen species and antioxidant enzyme activities in animal and cellular models of Parkinson’s disease. Biochim. Biophys. Acta 1362, 77–86.
Singleton, A. B., Farrer M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., Hulihan, M., Peuralinna, T., Dutra, A., Nussbaum, R., Lincoln, S., Crawley, A., Hanson, M., Maraganore, D., Adler, C., Cookson, M. R., Muenter, M., Baptista, M., Miller, D., Blancato, J., Hardy, J., and Gwinn-Hardy, K. (2003). Alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302, 841.
Souza, J. M., Giasson, B. I., Chen, Q., Lee, V. M., and Ischiropoulos, H. (2000). Dityrosine cross-linking promotes formation of stable alpha-synuclein polymers. Implications of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 275, 18344–18349.
Swerdlow, R. H., Parks, J. K., Davis, J. N., Cassarino, D. S. ., Trimmer, P. A., Currie, L. J., Dougherty, J., Bridges, W. S., Bennett, J. P., Wooten, G. F., and Parker, W. D. (1998). Matrilineal inheritance of complex I dysfunction in a multigenerational Parkinson’s disease family. Ann. Neurol. 44, 873–881.