Laboratory of Neurosciences, National Institute on Aging Intramural Research Program, Baltimore, MD, USA
Rodents that are insulin resistant and obese as the result of genetic factors, overeating and/or a sedentary lifestyle, exhibit cognitive deficits that worsen with advancing age compared to their more svelte counterparts. Data from epidemiological and clinical studies suggest similar adverse effects of excessive dietary energy intake and insulin resistance on cognition in humans. Our findings from studies of animal models suggest that dietary energy restriction can enhance neural plasticity and reduce the vulnerability of the brain to age-related dysfunction and disease. Dietary energy restriction may exert beneficial effects on the brain by engaging adaptive cellular stress response pathways resulting in the up-regulation of genes that encode proteins that promote neural plasticity and cell survival (e.g., neurotrophic factors, protein chaperones and redox enzymes). Two energy state-sensitive factors that are proving particularly important in regulating energy balance and improving/preserving cognitive function are brain-derived neurotrophic factor and glucagon-like peptide 1. Alternate day calorie restriction, novel insulin-sensitizing and neuroprotective agents, and drugs that activate adaptive stress response pathways, are examples of approaches for preserving cognitive function that show promise in preclinical studies.
Overeating and obesity are associated with increased risks for a range of diseases including cardiovascular disease, insulin resistance/diabetes and many types of cancer (Muoio and Newgard, 2006
). Emerging evidence suggests that excessive energy intake, together with a sedentary lifestyle, also places the brain at risk for the development of late life cognitive impairment and Alzheimer’s disease (Qiu et al., 2007
; Stranahan and Mattson, 2008
). This article is a mini-review of findings from animal and human studies that describe the effects of dietary energy intake on cognitive function, and elucidate the underlying cellular and molecular mechanisms. Recent reviews on one or more aspects of this topic can be found elsewhere (Parrott and Greenwood, 2007
; Taylor and MacQueen, 2007
; Stranahan and Mattson, 2008
).
The brain plays fundamental roles in the regulation of dietary energy (calorie) intake, with an evolutionary selection for individuals that are able to ‘outsmart’ their competitors in the acquisition of food (Mattson, 2002
). On the other hand, there is considerable evidence from studies of rats and mice and emerging evidence from studies of humans, that sustained excessive energy intake can adversely affect cognitive function by mechanisms described throughout this article. Accordingly, long-term reductions in energy intake (below the usual ad libitum levels) can enhance cognitive performance in rats and mice (Fontán-Lozano et al., 2007
; Adams et al., 2008
). Conversely, consumption of high energy diets (with elevated levels of simple sugars and saturated fats) negatively affects cognitive performance in animal models (Wu et al., 2004
; Stranahan et al., 2008a
–c
). Much of the experimental evidence linking excessive energy intake to cognitive decline comes from studies of the hippocampus, because the hippocampus a brain region that plays major roles in learning and memory processes; in humans the size of the hippocampus decreases during aging and more so in Alzheimer’s disease (Jack et al., 2005
; Driscoll et al., 2009
). The layered structural organization of the neurons and their synaptic connections permits reproducible electrophysiological measurements of synaptic transmission and long-term changes in synaptic strength. Moreover, the functional status of the hippocampus in cognition can be evaluated using well-characterized behavioral tests such as the water maze and novel object recognition tasks (Heldt et al., 2007
).
Studies using animal models are essential for understanding the impact of dietary energy intake on the brain at the molecular and cellular levels and, importantly, data obtained from animal experiments directs the design of clinical studies in humans. This article focuses mainly on findings from animal studies which have elucidated the effects of dietary energy restriction and excess on cognitive function in the absence of disease, and in experimental models of diabetes, Alzheimer’s disease, stroke and epilepsy. The collective literature on this topic leaves the reader with several take home messages including: (1) excessive energy intake and obesity adversely affect cognitive function; (2) diabetes also adversely affects cognitive function; (3) a sedentary lifestyle exacerbates the adverse effects of overeating and diabetes on cognitive function; (4) excessive energy intake renders neurons vulnerable to aging and neurodegenerative disorders including Alzheimer’s disease; (5) dietary energy restriction can enhance neural plasticity and cognitive function, and may protect against neurodegenerative disorders and improve the recovery from brain injuries. (6) excessive energy intake can impair, whereas energy restriction can enhance, adaptive cellular stress response pathways that protect neurons against aging, injury and disease; (7) among adaptive stress response pathways, those involving neurotrophic factors, protein chaperones and antioxidant enzymes may be particularly important.
The results of several epidemiological studies of human populations suggest that obesity and diabetes are associated with an increased risk for cognitive impairment and dementia (Nilsson and Nilsson, 2009
). For example, long-term obesity in adulthood is associated with lower cognitive scores in late mid-life (Sabia et al., 2009
). Others have reported that individuals with a high body mass index exhibit executive dysfunction (Gunstad et al., 2007
) and that patients with diabetes are impaired on multiple domains of cognitive function (Kodl and Seaquist, 2008
). However, not all subjects who are overweight or with diabetes are cognitively impaired, and some may even perform better than the average normal weight healthy subject (Kuo et al., 2006
).
In one study, male Long-Evans rats were maintained on a normal diet or a diet with elevated levels of saturated fats, and were then tested on a variable-interval delayed-alternation task (Greenwood and Winocur, 1996
). The rats on the high fat diet were impaired in their abilities to learn the basic alternation rule and their ability to remember trial-specific information over time. A diet with elevated levels of saturated fat and cholesterol caused rats to commit more working memory errors in the water radial arm maze, especially when the rats were challenged with high memory loads (Granholm et al., 2008
). Adverse effects of such ‘fast food’ diets on cognitive function may occur relatively quickly. For example, Murray et al. (2009)
recently reported that when male Wistar rats were fed a diet high in saturated fat for only 9 days, their physical performance on a treadmill and their cognitive performance on an 8-arm radial maze were impaired relative to rats on a control diet. Interestingly, a pro-obesity/diabetes diet during pregnancy may adversely affect the cognitive function of offspring, particularly when the offspring consume a similar high energy diet (White et al., 2009
). In the latter study the male progeny of dams that had been maintained on a diet high in saturated fat during pregnancy, and were themselves maintained on the same diet post-weaning, performed poorly in the memory retention component of the water maze test of spatial learning and memory when tested at 20 weeks of age compared to age-matched male rats from dams on a normal diet. Postmortem analysis of brain tissue demonstrated that markers of oxidative stress and inflammation were elevated in the rats from high fat diet dams, consistent with considerable evidence that oxidative stress and inflammation underlie many of the adverse health outcomes associated with excessive dietary energy intake. For example, levels of markers of oxidative stress (lipid peroxidation products, protein carbonyls and nitrotyrosine) and inflammation (tumor necrosis factor and leptin) were elevated systemically in overweight human subjects and alternate day caloric restriction (CR) reduced levels of all of these markers (Johnson et al., 2007
). A recent example from studies of the brain showed that markers of inflammation are reduced in several brain regions of mice maintained on an alternate day fasting (ADF) diet compared to mice fed the usual ad libitum diet (Arumugam et al., 2010
).
Additional mechanisms by which high fat/energy diets impair cognitive function are beginning to be elucidated and involve adverse effects on synaptic plasticity and neurogenesis (Figure 1
). High energy/fat diets may impair hippocampal plasticity by reducing the expression of brain-derived neurotrophic factor (BDNF). BDNF is a protein produced by neurons in an activity-dependent manner; two transcription factors that are known to induce the expression of the BDNF gene are cyclic AMP response element binding protein (CREB) and nuclear factor κ-B (NF-κB) (Lipsky and Marini, 2007
). BDNF activates a high-affinity receptor tyrosine kinase called trkB, which is located in the plasma membrane where it present in particularly high amounts in dendrites and presynaptic terminals. Activated trkB engages a signaling pathway involving PI3 kinase, Akt kinase and FOXO transcription factors; this pathway induces the expression of genes that enhance synaptic plasticity (glutamate receptor subunits and growth-associated protein 43, for example) and cell survival (the antioxidant enzyme superoxide dismutase 2, and the anti-apoptotic protein Bcl-2, for example) (Koponen et al., 2004
; Mattson et al., 2004
; Pang and Lu, 2004
; Bramham and Messaoudi, 2005
). In addition to enhancing synaptic plasticity and neuron survival, BDNF has been shown to stimulate neurogenesis (the differentiation of neurons from neural stem cells) (Cheng et al., 2003
; Schmidt and Duman, 2007
) which may contribute to the beneficial effects of BDNF on cognitive function.
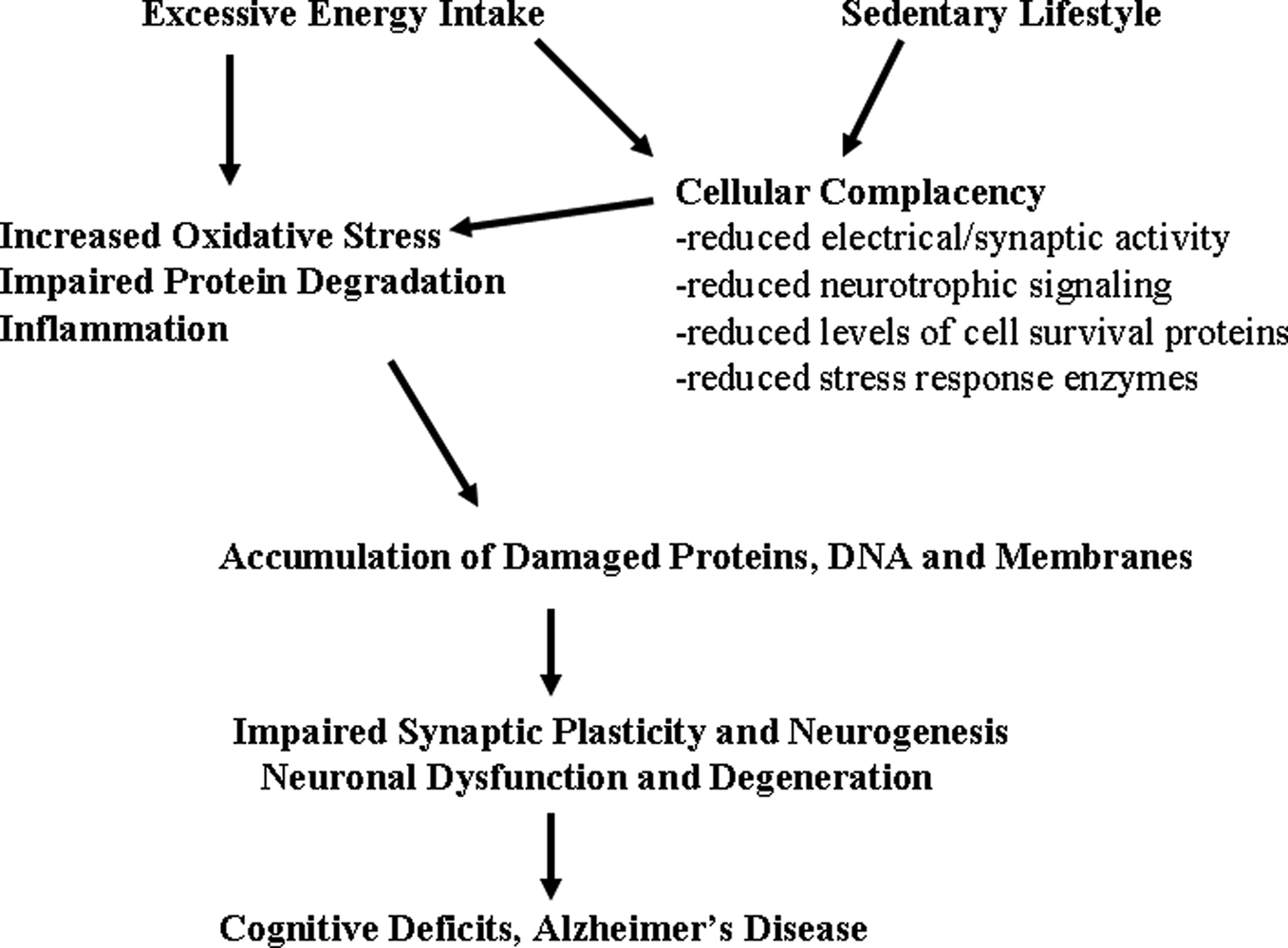
Figure 1. Mechanisms by which excessive energy intake and low levels of energy expenditure adversely affect synaptic plasticity and cognitive function. Overeating adversely affects the brain because of the increased oxidative stress resulting from increased levels of glucose which may result in increased generation of superoxide anion radical in the mitochondria and a nonenzymatic process called glycation. Also contributing to increased molecular damage is the reduction in the activation of adaptive cellular stress response pathways (cellular complacency) which manifests as reduced production of neurotorphic factors, protein chaperones and antioxidant enzymes. Physical inactivity exacerbates the effects of excessive energy intake because exercise activates adaptive cellular stress response pathways that can protect neurons against dysfunction and degeneration. Overeating and a sedentary lifestyle therefore promote the progressive accumulation of damaged proteins, nucleic acids and membranes in brain cells resulting in impaired synaptic function and neurogenesis; neuronal degeneration and death may ensue. In these ways a ‘couch potato’ lifestyle may place the individual on a trajectory towards premature cognitive impairment and Alzheimer’s disease.
When male Sprague-Dawley rats were maintained for 2 months on a high fat diet their cognitive function was impaired, and this behavioral abnormality was associated with decreased levels of BDNF and markers of synaptic function (Wu et al., 2004
). Dietary supplementation with vitamin E counteracted the cognitive and synaptic impairment otherwise caused by the high fat diet. In another study the authors showed that a saturated fat diet worsens the cognitive deficits caused by traumatic brain injury, and that this adverse effect of the diet is associated with decreased levels of BDNF in the hippocampus (Wu et al., 2003
). In one study we maintained rats on a high-fat, high-glucose diet supplemented with high-fructose corn syrup and then evaluated their performance on learning and memory tasks, and also quantified synaptic strength and dendritic spine density in hippocampal neurons (Stranahan et al., 2008a
). Male Sprague-Dawley rats maintained on this diet for 8 months exhibited impaired spatial learning ability, reduced hippocampal dendritic spine density, and reduced long-term potentiation at Schaffer collateral–CA1 synapses. These changes were associated with reductions in levels of BDNF in the hippocampus. Therefore, a diabetogenic high-calorie diet reduces hippocampal synaptic plasticity and impairs cognitive function, possibly by impairing BDNF-mediated effects on dendritic spines.
While most studies of the effects of energy intake on hippocampal plasticity have been performed in male animals, sex differences in susceptibility to high energy diet-induced impairment of synaptic plasticity and cognitive function have been reported. For example, male mice fed a high fat diet exhibited deficits in LTP and learning and memory, whereas these deficits did not occur in female mice on the same high fat diet (Hwang et al., 2009
).
It should be noted that certain types of learning and memory may be affected by dietary energy intake more than other types of learning and memory. For example, two different studies have shown that in the water maze test target crossing time is not impaired in mice whose dams were high-fat fed although distance and latency are impaired (Yu et al., 2009
). Similarly, a study in rats showed that high energy diet-fed progeny from high energy diet-fed dams exhibited impaired memory retention, but not acquisition, in the water maze (White et al., 2009
). Another study showed that obese mice exhibit impaired memory acquisition in three different cognitive tests (active avoidance T-maze, water maze and a food reward lever press task) (Farr et al., 2008
). Interestingly, cognition was improved and markers of oxidative stress were reduced in the obese mice by selectively lowering triglycerides, suggesting that elevated triglycerides mediate, at least in part, the adverse effects of a high energy/fat diet on cognition.
Several laboratories have provided evidence that excessive energy intake impairs adult hippocampal neurogenesis, an alteration associated with poorer cognitive outcomes. For example, Lindqvist et al. (2006)
showed that a high-fat diet impairs hippocampal neurogenesis in Sprague-Dawley rats. We reported that overeating, as the result of impaired leptin signaling, impairs hippocampal neurogenesis in mice, and is associated with deficits in synaptic plasticity at perforant path – dentate granule neuron synapses and impaired hippocampus-dependent memory (Stranahan et al., 2008b
). On the other hand, when normal mice that had been overfed were maintained on an energy-restricted diet, levels of hippocampal neurogenesis were increased (Lee et al., 2002
).
Interestingly, high energy diets in pregnant dams may adversely affect neurogenesis in their offspring (Walker et al., 2008
; Tozuka et al., 2009
). In the cases of studies where animals are maintained on high fat diets, the available evidence suggests that it is the elevated levels of energy intake rather than the fat molecules that adversely affects neurogenesis. For example, a high fat ketogenic diet that did not cause weight gain also did not impair hippocampal neurogenesis (Strandberg et al., 2008
).
To elucidate the molecular alterations by which dietary energy intake affects hippocampal plasticity, we performed a gene array analysis of the hippocampus in male and female Sprague-Dawley rats that had been maintained for 6 months on either the usual ad libitum diet, 20% CR, 40% CR, ADF or high fat/high glucose (HFG) diets (Martin et al., 2008
). The energy-restricted diets resulted in decreased levels of circulating glucose, insulin and leptin, whereas the HFG diet had the opposite effect on these markers of energy metabolism. Interestingly, the CR diets significantly increased the size of the hippocampus in female, but not male rats. The results of the gene expression analysis showed that: (1) the 20% CR diet down-regulated genes involved in mitochondrial energy metabolism and glycolysis in males, while these metabolic pathways were up-regulated in females; (2) the 40% CR diet up-regulated genes involved in glycolysis, protein deacetylation and mTor pathways in both males and females; (3) ADF down-regulated genes in males involved in protein degradation and apoptosis, whereas in females ADF up-regulated genes involved in cellular energy metabolism, cell cycle regulation and protein deacetylation; (4) the HFG diet altered the expression of genes involved in energy metabolism, oxidative stress responses and cell death similarly in males and females. Based upon these findings we hypothesize that the differential responses of the hippocampal transcriptomes of males and females to changes in dietary energy intake may underlie evolutionarily conserved sex-specific behavioral responses to food availability.
As part of the ‘atlas of gene expression in mouse aging project’ (AGEMAP; Zahn et al., 2007
), we measured relative expression levels of nearly 17,000 genes in five different regions of the central nervous system (CNS) (cerebral cortex, hippocampus, striatum, cerebellum and spinal cord) from young, middle-age and old C57BL/6 male and female mice that had been maintained since 6 weeks of age on either the usual ad libitum diet or a 40% CR diet (Xu et al., 2007
). Each CNS region exhibited a unique transcriptome fingerprint that was independent of age, gender and energy intake. The expression of most (>90%) of the genes was stable during aging, and most of the genes that were affected by age changed between middle and old age. Interestingly, the hippocampal transcriptome was remarkably responsive to energy restriction and gender in middle-aged mice. Functional groups of age- and dietary energy-sensitive genes that were responsive across CNS regions included those involved in cellular energy metabolism, protein degradation, cell fate determination (neurogenesis) and synaptic transmission. The data from this study (Xu et al., 2007
) provide many potential signaling pathways and homeostatic mechanisms involved in CNS aging and its modification by energy intake and sex-specific factors.
Multiple studies have provided evidence that diabetes is a risk factor for age-related cognitive impairment and Alzheimer’s disease (Kopf and Frölich, 2009
). Experimentally induced insulin deficiency, a model of type I diabetes, results in impaired hippocampal synaptic plasticity and cognitive impairment in rats (Biessels et al., 1998
; Stranahan et al., 2008b
). Because diabetes is often associated with hyperactivation of the hypothalamic-pituitary-adrenal axis, we designed experiments to elucidate the role of this neuroendocrine system in diabetes-induced cognitive dysfunction. We found that in both insulin-resistant (leptin receptor mutant) mice and insulin-deficient rats diabetes impairs hippocampus-dependent memory, dentate gyrus synaptic plasticity and neurogenesis (Stranahan et al., 2008b
). In both diabetes models, levels of corticosterone are elevated. When the adrenal glands are surgically removed and the animals given low (non-stress) levels of corticosterone, the adverse effects of diabetes on cognitive function, and hippocampal synaptic plasticity and neurogenesis are ameliorated. In a related study, we evaluated the performance of young diabetic rats in a hippocampus-dependent learning and memory task, the 14-unit T-maze (Stranahan et al., 2008c
). The analysis was performed on adrenally intact diabetic rats with elevated corticosterone levels and adrenalectomized rats with corticosterone levels maintained low by low-dose corticosterone replacement. The adrenally intact diabetic rats learned the maze poorly compared to rats with low corticosterone levels. Interestingly, the pattern of errors made by adrenally intact diabetic rats was similar to the pattern of errors made by old rats, suggesting that the adverse effects of aging and diabetes on hippocampal function are similar. Collectively, these findings suggest that cognitive impairment in diabetes results, at least in part, from glucocorticoid-mediated deficits in hippocampal cellular plasticity.
One mechanism by which elevated glucocorticoids may impair cognitive function is by suppressing BDNF production (Smith et al., 1995
; Schaaf et al., 2000
). To evaluate the role of BDNF in counteracting the adverse effects of diabetes on cognitive function, we examined the effects of voluntary running and dietary energy restriction on hippocampal neuron morphology and BDNF levels in leptin receptor mutant mice, a model of type 2 diabetes (Stranahan et al., 2009
). Running and dietary energy restriction increased levels of BDNF in the diabetic mice, and the combination of running and dietary energy restriction resulted in an additive increase in BDNF levels. Enhancement of hippocampal BDNF levels was correlated with increased dendritic spine density in dentate granule neurons. Our data show that diabetes has adverse effects on hippocampal synaptic structure which can be ameliorated by reducing energy intake and increasing energy expenditure.
Dietary Energy Restriction Protects Neurons in Animal Models of Age-Related Neurodegenerative Disorders
Several studies demonstrated that dietary restriction during adult life can attenuated age-related declines in the performance of rats in cognitive tests. For example, when begun in mid-life, energy restriction prevents the cognitive decline in mice (Means et al., 1993
) and long-term food restriction enhanced memory in a dry version of the water maze and in tests of passive avoidance (Hashimoto and Watanabe, 2005
). In rats, CR prevented age-related deficits in LTP and NMDA and AMPA glutamate receptor levels in the hippocampus (Eckles-Smith et al., 2000
; Shi et al., 2007
). However, dietary restriction has not always proven beneficial for cognitive function during aging. For example, Yanai et al. (2004)
reported that long-term dietary restriction resulted in worsened performance of the rats in the water maze when they were old. The reason for the latter result may be related to the rather severe amount of energy restriction.
We have obtained considerable evidence from studies of animal models to support the possibility that dietary energy restriction can enhance neural plasticity and reduce the vulnerability of the brain to age-related dysfunction and disease (Table 1
). More than a decade ago we reported that when rats are maintained for several months on an ADF dietary energy restriction diet, they exhibit increased resistance to excitotoxin-induced memory impairment (Bruce-Keller et al., 1999
). This beneficial effect of ADF was correlated with reduced vulnerability of hippocampal CA3 neurons to degeneration. In the same study we found that ADF ameliorates mitochondrial toxin-induced damage to striatal neurons and associated motor deficits in a rat model of Huntington’s disease (Bruce-Keller et al., 1999
). Subsequent experiments revealed neuroprotective effects of ADF in mouse (Duan and Mattson, 1999
) and monkey (Maswood et al., 2004
) models of Parkinson’s disease, in Huntingtin mutant mice (Duan et al., 2003a
) and in a rat model of ischemic stroke (Yu and Mattson, 1999
). We believe that an important mechanism underlying these neuroprotective effects of ADF involves activation of adaptive cellular stress response pathways that induce the expression of multiple neuroprotective proteins including neurotrophic factors (BDNF, GDNF and bFGF), protein chaperones (HSP-70 and GRP-78) and mitochondrial uncoupling proteins (UCPs) (Yu and Mattson, 1999
; Duan et al., 2001
; Lee et al., 2002
; Maswood et al., 2004
; Arumugam et al., 2010
).
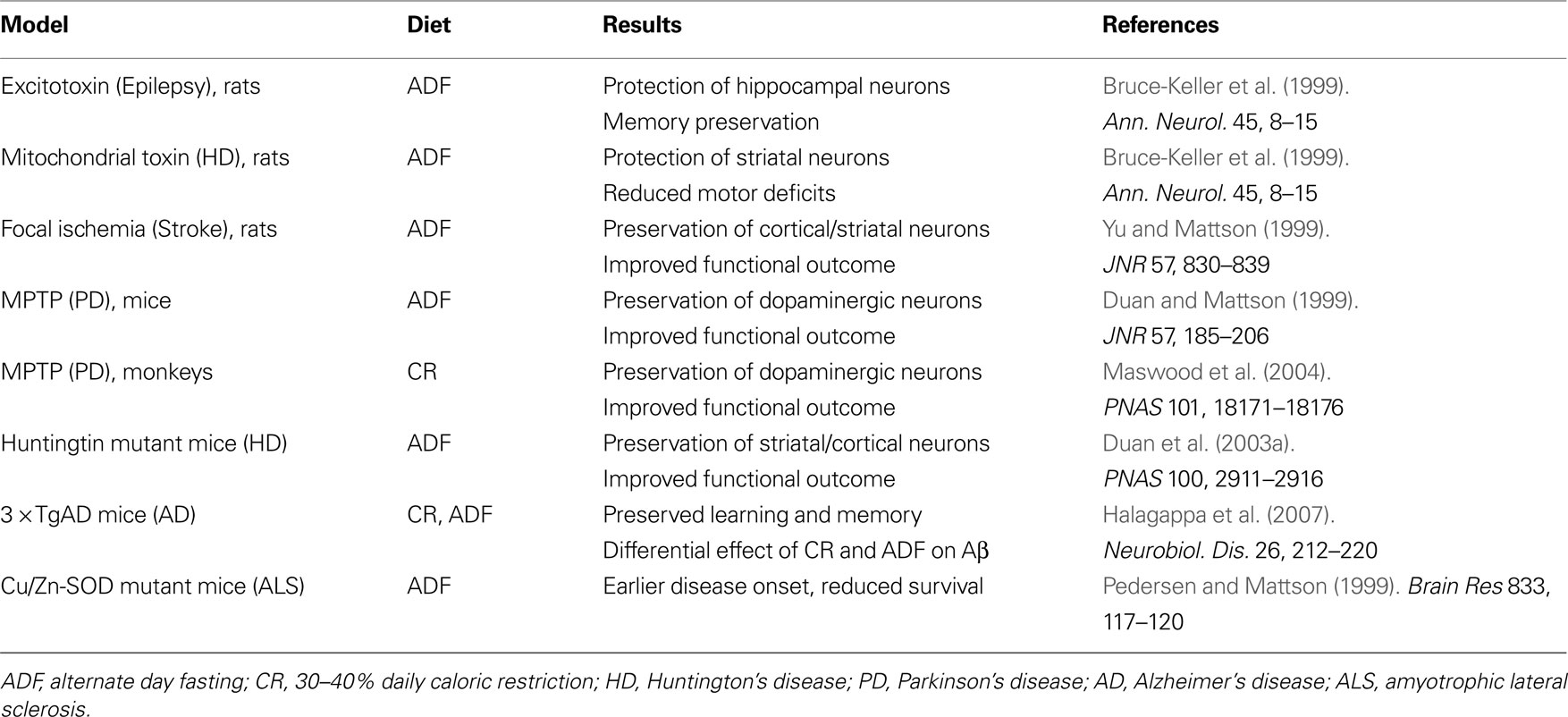
Table 1. Influence of dietary energy restriction on neuronal vulnerability in animal models of acute CNS injury and chronic neurodegenerative disorders.
To elucidate the influence of dietary energy restriction on cognitive function, we tested the effects of long-term controlled daily CR (40% reduction in calories) and ADF on learning and memory in 3 × TgAD mice, an animal model of Alzheimer’s disease (Halagappa et al., 2007
). 3 × TgAD mice develop progressive accumulation of amyloid β-peptide in their hippocampus and cerebral cortex, tau pathology in CA1 hippocampal neurons, and impaired hippocampal synaptic plasticity (Oddo et al., 2003
). Beginning at 5 months of age we maintained groups of 3 × TgAD mice on the usual ad libitum control diet, the 40% CR diet or the ADF diet for 12 months. As reference controls, we also included in the study a group of non-transgenic mice maintained on an ad libitum diet. The 3 × TgAD mice on the ad libitum diet exhibit impaired learning and memory in the water maze compared to non-transgenic mice, and both the CR and ADF diets ameliorated the cognitive deficit in the 3 × TgAD mice (Halagappa et al., 2007
). We found that 3 × TgAD mice on the CR diet exhibited reduced levels of amyloid β-peptide and hyperphosphorylated tau in their hippocampus compared to 3 × TgAD mice on the ad libitum diet. Interestingly, however, the ADF diet did not lessen the amyloid or tau pathologies. We hypothesize that, by activating adaptive cellular stress response pathways (BDNF, protein chaperones, etc.), ADF preserves synaptic function even in the presence of considerable amyloid and tau pathology (Figure 2
). Consistent with this hypothesis, Qin et al. (2008)
found that the transcription factor FOXO3a (which is known to mediate adaptive responses of neurons to stress) is involved in the mechanism by which CR suppresses amyloid pathology and memory deficits in a mouse model of Alzheimer’s disease.
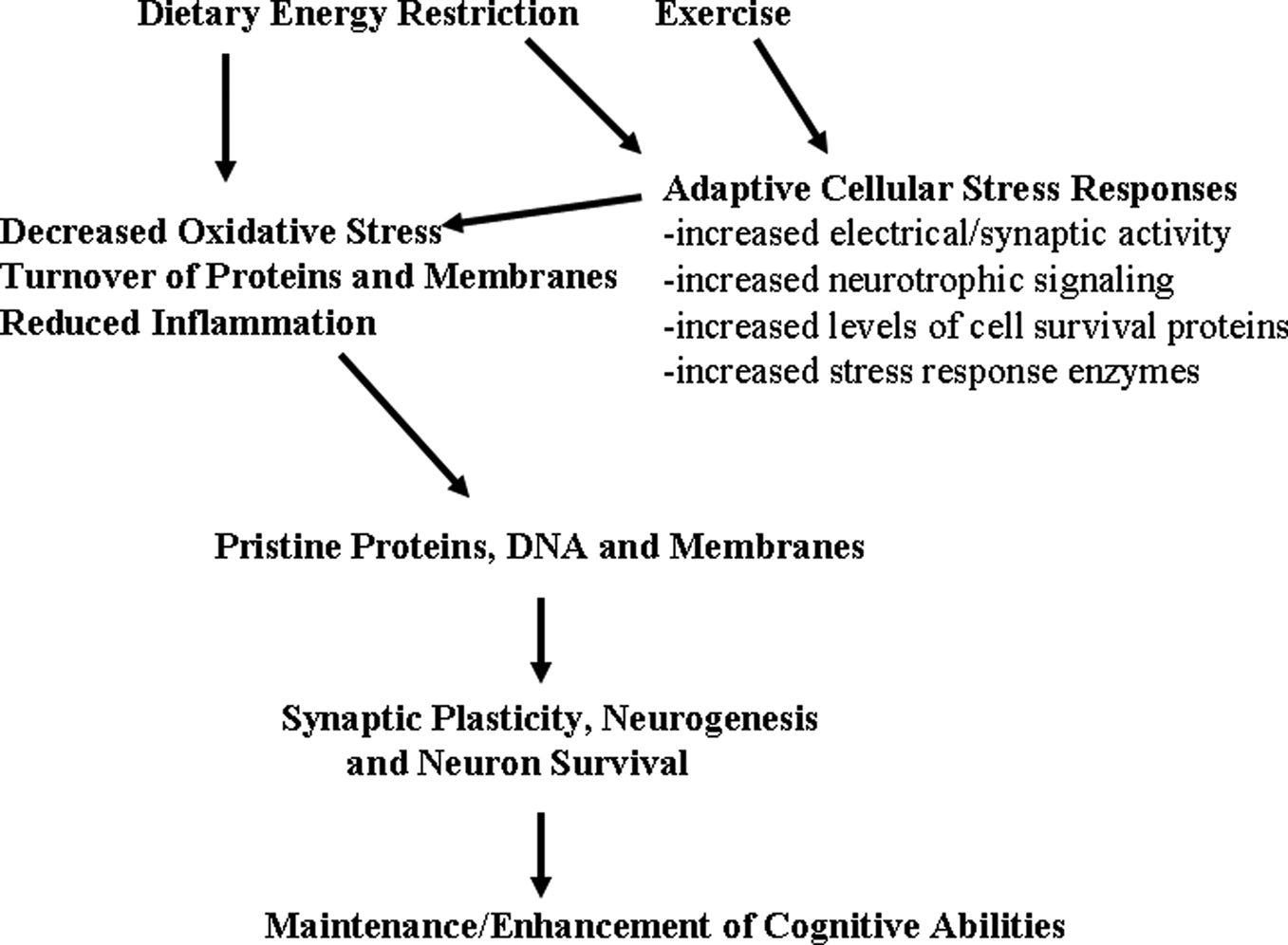
Figure 2. Hormetic mechanism of action of dietary energy restriction. A mild cellular stress occurs in neurons during periods of dietary energy restriction or vigorous physical exercise; this may result from increased electrical/synaptic activity and/or an energetic deficit. Adaptive cellular stress response signaling pathways are activated in brain cells including those that induce the expression of neurotrophic factors, protein chaperones and antioxidant enzymes. In this way, dietary energy restriction and exercise protect cells against oxidative and metabolic stress, and prevent the accumulation of damaged proteins, DNA and membranes in neurons. The increased survival and plasticity of brain cells is the reason why dietary moderation and exercise promote healthy brain aging.
Dietary energy restriction has also proven effective in reducing neuronal degeneration and cognitive deficits in models relevant to disorders other than Alzheimer’s disease. For example, the onset of prion disease was delayed by CR in wild type mice but not in mice lacking SIRT1, a deacetylase believed to retard aging (Chen et al., 2008
). Dietary energy restriction may counteract the effects of aging on fundamental molecular mechanisms involved in learning and memory. As evidence, CR prevented the age-related decline in glutamate receptor (NMDA and AMPA receptors) expression in the hippocampus of rats (Shi et al., 2007
). Cognitive impairment is a common outcome in stroke patients (Erkinjuntti, 2007
), and several recent studies have shown that cognitive impairment caused by cerebral ischemia can be ameliorated by reduced energy diets. Roberge et al. (2008)
reported that rats maintained on CR during a 70-day period after transient global forebrain ischemia exhibited better performance on radial arm maze tasks compared to rats fed ad libitum. In the latter study the reduced energy diet did not prevent loss of CA1 or CA3 neurons, suggesting an effect of dietary energy intake on plasticity of the remaining neurons. We found that ADF up-regulates the expression of the neuroprotective proteins bFGF, BDNF, HSP-70, GRP-78 and HO-1 in cortical brain cells of young mice, and is progressively less effective in doing so in middle-age and old mice (Arumugam et al., 2010
). ADF suppresses the expression of multiple pro-inflammatory cytokines (TNFα, IL-1β and IL-6), while increasing the expression of the anti-inflammatory cytokine IL-17, in young but not old mice. The increased expression of stress resistance proteins and decreased markers of inflammation in response to ADF were associated with protection of brain cells against focal ischemic stroke and improved functional outcome (Arumugam et al., 2010
). These findings suggest that high energy intakes may worsen stroke outcome, including cognitive deficits, whereas low energy intakes may improve outcome.
An important point to consider when extrapolating the now abundant data demonstrating quite striking neuroprotective effects of dietary energy restriction in rodent models is that the ‘control’ animals are overfed, sedentary and obese. Laboratory rats and mice live in small cages with constant access to food and so, in my opinion, should be considered equivalent to so-called ‘couch potato’ humans. It might therefore be predicted that dietary energy restriction would improve cognitive function, and protect neurons against age- and disease-related degeneration, in overweight sedentary human subjects. If, and to what extent, dietary energy restriction might benefit the brains of active normal weight humans remains to be determined.
Pivotal Roles for Adaptive Cellular Stress Response Pathways in the Beneficial Effects of Dietary Energy Intake on Neural Plasticity
My colleagues and I have recently reviewed the evidence that multiple environmental factors that promote synaptic plasticity, and that can protect against age-related cognitive impairment, activate adaptive cellular stress response signaling pathways in neurons (Arumugam et al., 2006
; Mattson, 2008
; Mattson and Calabrese, 2010
). These factors include exercise, intellectual challenges and dietary energy restriction. The general principle falls under the rubric of ‘hormesis’, a term used to describe biphasic dose responses of cells and organisms to a factor in which low doses induce a stimulatory beneficial response and high doses are inhibitory or detrimental. A widely experienced example of hormesis is the response of muscle cells to exercise. Vigorous exercise imposes a major stress on muscle cells including ion (Na+, Ca2+ and K+) fluxes, increased energy (ATP and NAD+) utilization and free radical (superoxide, hydrogen peroxide, hydroxyl radical) production. In response to this stress, several signaling pathways are activated that result in the production of proteins that serve to protect the cells against more severe or sustained stress. For example, exercise activates the transcriptional regulators nuclear respiratory factor 1 (Nrf-1) and nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) resulting in increased production of the antioxidant enzymes Mn-superoxide dismutase and heme oxygenase 1 (Baar, 2004
; Powers et al., 2009
).
Neurons involved in learning and memory respond to cognitive challenges, exercise and dietary energy restriction in a manner similar to the response of muscle cells to physical exercise. For example, levels of BDNF are increased in skeletal muscle cells in response to exercise (Gómez-Pinilla et al., 2001
; Ogborn and Gardiner, 2009
; Pedersen et al., 2009
) and in hippocampal neurons in response to exercise (Neeper et al., 1996
), cognitive stimulation (Broad et al., 2002
) and dietary energy restriction (Lee et al., 2002
). Protein chaperones, including heat-shock proteins (e.g., HSP-70) and endoplasmic reticulum chaperones (e.g., GRP-78) are increased in muscle cells in response to exercise (González et al., 2000
; Morton et al., 2009
) and in neurons in response to dietary energy restriction (Yu and Mattson, 1999
; Arumugam et al., 2010
) and exercise (Chen et al., 2007
). The antioxidant enzyme heme oxygenase-1 (HO-1) has also been reported to increase in muscle cells in response to exercise (Hildebrandt et al., 2003
) and in neurons in response to energy restriction (Arumugam et al., 2010
).
Another example of the generally similar hormetic effects of exercise on muscle cells and dietary energy restriction on neurons comes from studies of mitochondrial UCPs. Exercise induces the expression of UCP3 in skeletal muscle cells (Zhou et al., 2000
), and dietary energy restriction increases the expression of UCP4 in brain cells (Liu et al., 2006
). UCPs may protect cells against injury and disease by reducing free radical production and stabilizing cellular calcium homeostasis (Chan et al., 2006
; Liu et al., 2006
). Findings in the latter studies suggest a potential role for UCPs in mediating the neuroprotective effects of exercise and dietary energy restriction, thereby contributing to the maintenance of cognitive function during aging. In support of this possibility, Dietrich et al. (2008)
reported that voluntary exercise stimulates UCP2 mRNA expression in hippocampal cells, and that this up-regulation of UCP2 was associated with increases in mitochondrial number and dendritic spine synapses in dentate granule neurons and CA1 neurons. The exercise-induced increases in dendritic mitochondria and synapses did not occur in UCP2-deficient mice, indicating a pivotal role for UCP2 in the effects of exercise on hippocampal plasticity. Collectively, the accumulating evidence suggests that activation of adaptive cellular stress response pathways is fundamental to the beneficial effects of both dietary energy restriction and exercise on the survival, and structural and functional plasticity of neurons that mediate cognition.
In the brain, BDNF is produced mainly by neurons wherein its levels are increased considerably by electrical activity, and in response to exercise and dietary energy restriction (Mattson et al., 2004
). The transcription factors CREB (cyclic AMP response element binding protein) and NF-κB induce BDNF expression in response to neuronal activity and metabolic stress (Shieh and Ghosh, 1999
; Lipsky et al., 2001
). BDNF is released from neurons in an activity-dependent manner and binds to a high-affinity receptor tyrosine kinase called trkB which is expressed by neurons and glial cells throughout the nervous system (Reichardt, 2006
). The first clue that BDNF plays roles in regulating energy metabolism came when heterozygous BDNF knockout (BDNF+/−) were found to exhibit an obese phenotype as the result of excessive food intake (Kernie et al., 2000
). It turns out that BDNF acts upon a small group of neurons in the hypothalamus that control appetite. The obese diabetic phenotype of BDNF+/− mice can be reversed by maintaining the mice on an alternate day fasting dietary restriction regimen which increases BDNF expression in the brain (Duan et al., 2003b
).
BDNF may also act centrally to affect energy metabolism more directly and acutely. Infusion of BDNF into the brains of diabetic mice improved their peripheral insulin sensitivity, apparently by a mechanism independent of a change in food intake (Nonomura et al., 2001
). One possible mechanism by which this might occur involves modulation of the autonomic nervous system (Figure 3
). Indeed, by comparing the heart rate variability (HRV) of mice on ad libitum control diet with the HRV of mice on either ADF or limited daily feeding dietary restriction, we showed that dietary energy restriction results in a reduction in resting heart rate and an increase of HRV in rats (Mager et al., 2006
). Data in the latter study suggested that dietary energy restriction shifts the autonomic control of the heart such that parasympathetic (acetylcholine) activity is increased. More recently, we found that heart rate is reduced and HRV increased, within minutes of infusion of BDNF into the third ventricle of mice (author’s unpublished data).
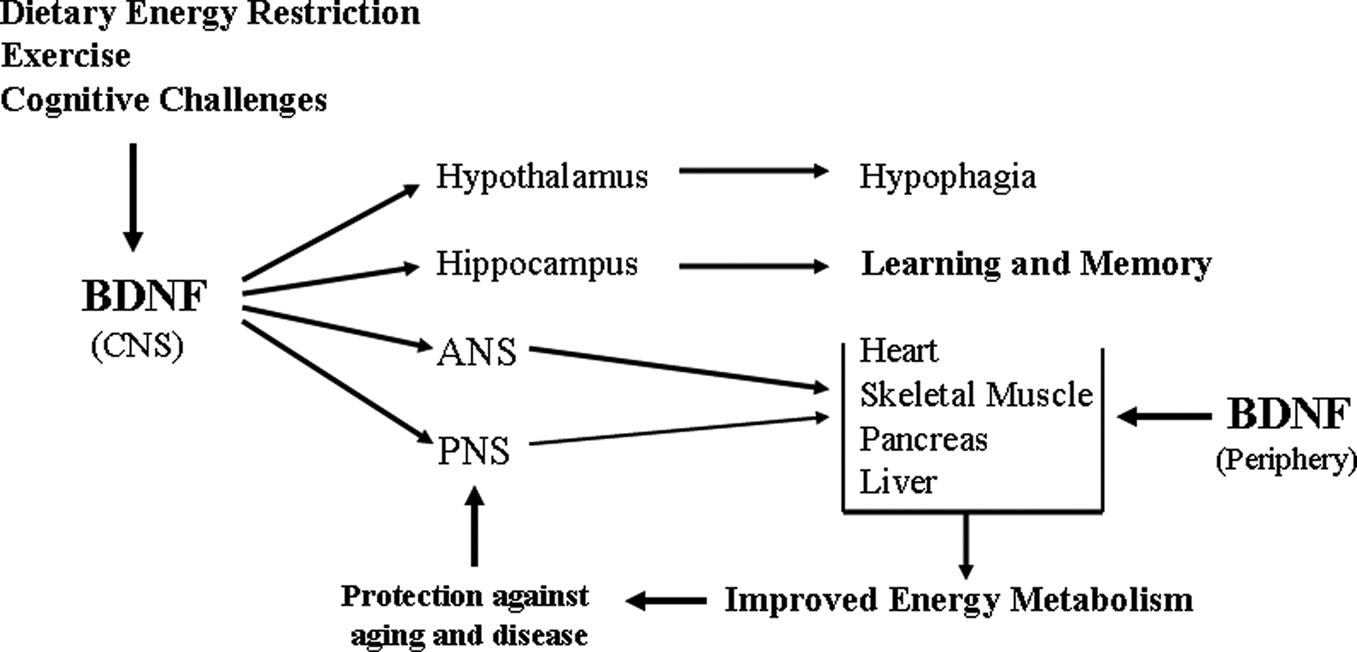
Figure 3. Pivotal roles for brain-derived neurotrophic factor in the integration of CNS neuroplasticity and peripheral energy metabolism. The production of BDNF by neurons in the central nervous system (CNS) is increased in response to the metabolic and electrochemical challenged imposed upon the neurons by dietary energy restriction, exercise and cognitive stimulation. BDNF acts a multiple levels of the nervous system to engage adaptive responses to the environmental demands. BDNF acts on: neurons in the hypothalamus to reduce appetite; cells in the hippocampus to enhance synaptic plasticity, neurogenesis and learning and memory ability; neurons in the autonomic nervous system (ANS) which innervate peripheral organs (heart, muscle, pancreas, liver and others); and neurons in the peripheral nervous system (e.g., lower motor neurons that innervate skeletal muscle cells). As of one or more of these actions, BDNF improves peripheral insulin sensitivity. By direct actions within the CNS, and by indirect effects on energy metabolism, BDNF may protect the nervous system against injury and disease. BDNF is also produced by peripheral cells and circulates in the blood, although its functions in the periphery are not known. PNS, peripheral nervous system.
While the mechanism by which BDNF signaling in the brain affects peripheral glucose metabolism remains to be determined, BDNF may also exert direct actions on peripheral tissues involved in energy metabolism including muscle (Matthews et al., 2009
) and pancreas (Hanyu et al., 2003
). Peripheral administration of BDNF results in improved blood glucose control in diabetic leptin receptor-deficient mice (Tonra et al., 1999
). Administration of BDNF is at least as effective as treatment with thiazolidinediones (diabetes drugs) in combating obesity and diabetes in mice (Yamanaka et al., 2007
). BDNF has been shown to preserve pancreatic β-cells in mouse models of diabetes (Yamanaka et al., 2006
). The roles for peripheral BDNF in physiological and pathological processes remains to be determined.
When food enters the gut, it (specifically sugars) stimulates enteroendocrine cells to release a peptide called glucagon-like peptide 1 (GLP-1) into the blood. GLP-1 binds and activates receptors on pancreatic β-cells resulting in the production and release of insulin (Wang et al., 1997
). In addition, GLP-1 enhances the sensitivity of muscle and liver cells to insulin, thereby exerting an anti-diabetic effect (Kim and Egan, 2008
). Moreover, GLP-1 can cross the blood-brain barrier and stimulate cells in the hypothalamus resulting in a suppression of appetite (anorexic effect). Because it acts at multiple sites to reduce circulating glucose levels, GLP-1 held considerable therapeutic potential for type 2 diabetes. However, the half-life of GLP-1 in the blood is very short (1–2 min) because it is cleaved and inactivated by a protease called DPP-IV (Palalau et al., 2009
) making it impractical for routine use in patients. This problem was circumvented by developing peptide analogs of GLP-1 that are resistant to inactivation by DPP-IV, and one such peptide called Exendin-4 (exenatide or Byetta) proved to be very effective in improving glucose regulation in patients with diabetes (Lovshin and Drucker, 2009
). Exendin-4 has been used in millions of patients with excellent outcomes.
Particularly exciting for neuroscientists has been the emerging evidence from studies in animal models suggesting that GLP-1 and Exendin-4 exert multiple actions on CNS cells that promote synaptic plasticity, neurogenesis, cell survival and resistance to injury. We first showed that GLP-1 and Exendin-4 can protect cultured neurons against excitotoxicity (Perry et al., 2002
). The latter study further demonstrated the ability of Exendin-4 to protect cholinergic neurons, a population of neurons critical for learning and memory and vulnerable in Alzheimer’s disease, against excitotoxic damage. Exendin-4 treatment also reduced damage to neurons and improved functional outcome in animal models of stroke and Parkinson’s disease (Li et al., 2009
). Huntington’s disease is an inherited neurodegenerative disorder caused by polyglutamine expansions in the huntingtin protein; the disease involves progressive degeneration of neurons in the striatum and cerebral cortex resulting in motor and cognitive dysfunction. Patients with Huntington’s disease exhibit abnormalities in energy metabolism, and huntingtin mutant mice are hyperglycemic (Duan et al., 2003a
). We found that treatment of huntingtin mutant mice with Exendin-4, beginning prior to the onset of neurological symptoms, normalizes peripheral glucose metabolism, delays disease onset and extends survival (Martin et al., 2009
).
Activation of GLP-1 receptors as a potential treatment for Alzheimer’s disease is also being pursued. Our preclinical studies showed that GLP-1 and Exendin-4 can protect cultured neurons from being damaged and killed by amyloid β-peptide (Perry et al., 2003
). More recently, we found that long-term treatment with Exendin-4 suppresses amyloid β-peptide accumulation in the brain in the 3 × TgAD mouse model of Alzheimer’s disease (Li et al., 2010
). Binding of GLP-1 or Exendin-4 to GLP-1 receptors results in the activation of the GTP-binding protein Gs and the membrane-associated enzyme adenylate cyclase which, in turn, generates the second messenger cyclic AMP. Cyclic AMP activates a specific kinase called PKA which then activates transcription factors including CREB (Skoglund et al., 2000
; Jhala et al., 2003
). Because CREB induces the expression of BDNF and mediates long-lasting changes in synaptic plasticity associated with learning and memory, it would be expected that GLP-1 would enhance synaptic plasticity. Indeed, GLP-1 receptor-deficient mice exhibit impaired hippocampus-dependent learning which is restored after viral vector-mediated expression of the GLP-1 receptor; in addition, overexpression of the GLP-1 receptor in the hippocampus enhanced learning and memory in rats (During et al., 2003
; Mattson et al., 2003
). More recent behavioral and electrophysiological studies of GLP-1 receptor-deficient mice have confirmed a role for GLP-1 signaling in hippocampal synaptic plasticity and learning and memory (Gault and Hölscher, 2008
; Abbas et al., 2009
). GLP-1 receptor activation enhances synaptic plasticity, while at the same time protecting neurons against excitotoxic overactivation of glutamate receptors (Gilman et al., 2003
).
Others have reported that Exendin-4 treatment increases neurogenesis in the subventricular zone and protects dopaminergic neurons in an animal model of Parkinson’s disease (Bertilsson et al., 2008
). In another study, Exendin-4 treatment proved effective in reversing the motor deficits caused by the dopaminergic neurotoxin 6-hydroxydopamine in a rat model of Parkinson’s disease (Harkavyi et al., 2008
).
Based upon the considerable data from studies of animal models described above, and the more limited data from studies of human subjects (Hendrickx et al., 2005
), it is evident that long-term excessive energy intake has adverse effects on neural plasticity and cognitive function. Moderation in dietary energy intake is therefore likely to protect the brain against age- and disease-related cognitive decline. It is not possible to make concrete recommendations as to what an ‘ideal’ energy intake is and, in any case, such an ideal energy intake will depend upon many factors including the activity level of the individual and their genetic constitution. A rough guideline would be 1800–2200 kcal/day for men and 1600–2000 calories for women if they are moderately active, and calorie intake may be adjusted to balance expenditure so as to maintain a body mass index within optimal range of 20–25 (Whitlock et al., 2009
). Other findings suggest that the adverse effects of overeating on hippocampal plasticity/cognition can be attenuated by regular aerobic exercise (30–60 day for at least 4 days/week) and muscle toning exercises (if not already part of one’s work) (van Praag, 2009
) as can regular social interactions and cognitively challenging work or hobbies (Bennett et al., 2006
). Specific dietary components may also affect cognitive function. For example, hippocampal plasticity and function may benefit from a minimization of dietary saturated and trans fats, and maximization of fish and other sources of omega-3 fatty acids, vegetables and fruits (Barberger-Gateau et al., 2007
). Other factors that may reduce the risk of cognitive impairment and Alzheimer’s disease include avoidance of head trauma (DeKosky et al., 2007
), control of blood pressure (Launer et al., 2000
), and minimization of chronic adverse stress and depression (Roose et al., 2007
).
There are undoubtedly genetic factors that can modify the impact of dietary energy intake on neural plasticity and cognitive function, and so one’s genetic background will contribute to the bottom line of whether cognition will decline prematurely, dramatically (Alzheimer’s disease) or negligibly in mid and late life. However, until future research identifies such genetic factors, the prudent tack for any individual is to assume that they have not inherited some rare gene that protects their brain against the adverse effects of overeating/obesity/diabetes.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Intramural Research Program of the National Institute on Aging.
Adams, M. M., Shi, L., Linville, M. C., Forbes, M. E., Long, A. B., Bennett, C., Newton, I. G., Carter, C. S., Sonntag, W. E., Riddle, D. R., and Brunso-Bechtold, J. K. (2008). Caloric restriction and age affect synaptic proteins in hippocampal CA3 and spatial learning ability. Exp. Neurol. 211, 141–149.
Bertilsson, G., Patrone, C., Zachrisson, O., Andersson, A., Dannaeus, K., Heidrich, J., Kortesmaa, J., Mercer, A., Nielsen, E., Rönnholm, H., and Wikström, L. (2008). Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease. J. Neurosci. Res. 86, 326–338.
Fontán-Lozano, A., Sáez-Cassanelli, J. L., Inda, M. C., de los Santos-Arteaga, M., Sierra-Domínguez, S. A., López-Lluch, G., Delgado-García, J. M., and Carrión, A. M. (2007). Caloric restriction increases learning consolidation and facilitates synaptic plasticity through mechanisms dependent on NR2B subunits of the NMDA receptor. J. Neurosci. 27, 10185–10195.
Hanyu, O., Yamatani, K., Ikarashi, T., Soda, S., Maruyama, S., Kamimura, T., Kaneko, S., Hirayama, S., Suzuki, K., Nakagawa, O., Nawa, H., and Aizawa, Y. (2003). Brain-derived neurotrophic factor modulates glucagon secretion from pancreatic alpha cells: its contribution to glucose metabolism. Diabetes Obes. Metab. 5, 27–37.
Hwang, L. L., Wang, C. H., Li, T. L., Chang, S. D., Lin, L. C., Chen, C. P., Chen, C. T., Liang, K. C., Ho, I. K., Yang, W. S., and Chiou, L. C. (2009). Sex differences in high-fat diet-induced obesity, metabolic alterations and learning, and synaptic plasticity deficits in mice. Obesity (Silver Spring). [Epub ahead of print].
Jack, C. R. Jr., Shiung, M. M., Weigand, S. D., O’Brien, P. C., Gunter, J. L., Boeve, B. F., Knopman, D. S., Smith, G. E., Ivnik, R. J., Tangalos, E. G., and Petersen, R. C. (2005). Brain atrophy rates predict subsequent clinical conversion in normal elderly and amnestic MCI. Neurology 65, 1227–1231.
Johnson, J. B., Summer, W., Cutler, R. G., Martin, B., Hyun, D. H., Dixit, V. D., Pearson, M., Nassar, M., Telljohann, R., Maudsley, S., Carlson, O., John, S., Laub, D. R., and Mattson, M. P. (2007). Alternate day calorie restriction improves clinical findings and reduces markers of oxidative stress and inflammation in overweight adults with moderate asthma. Free Radic. Biol. Med. 42, 665–674.
Kuo, H. K., Jones, R. N., Milberg, W. P., Tennstedt, S., Talbot, L., Morris, J. N., and Lipsitz, L. A. (2006). Cognitive function in normal-weight, overweight, and obese older adults: an analysis of the Advanced Cognitive Training for Independent and Vital Elderly cohort. J Am Geriatr Soc 54, 97–103.
Li, Y., Duffy, K., Ottinger, M. A., Balmiki, R., Bailey, J. A., Holloway, H. W., Tweedie, D., Mattson, M. P., Kapogiannis, D., Sambamurti, K., Lahiri, D. K., and Greig, N. H. (2010). GLP-1 receptor stimulation reduces amyloid β-peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J. Alzheimers Dis. In press.
Li, Y., Perry, T., Kindy, M. S., Harvey, B. K., Tweedie, D., Holloway, H. W., Powers, K., Shen, H., Egan, J. M., Sambamurti, K., Brossi, A., Lahiri, D. K., Mattson, M. P., Hoffer, B. J., Wang, Y., and Greig, N. H. (2009). GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc. Natl. Acad. Sci. U.S.A. 106, 1285–1290.
Liu, D., Chan, S. L., de Souza-Pinto, N. C., Slevin, J. R., Wersto, R. P., Zhan, M., Mustafa, K., de Cabo, R., and Mattson, M. P. (2006). Mitochondrial UCP4 mediates an adaptive shift in energy metabolism and increases the resistance of neurons to metabolic and oxidative stress. Neuromolecular Med. 8, 389–414.
Martin, B., Golden, E., Carlson, O. D., Pistell, P., Zhou, J., Kim, W., Frank, B. P., Thomas, S., Chadwick, W. A., Greig, N. H., Bates, G. P., Sathasivam, K., Bernier, M., Maudsley, S., Mattson, M. P., and Egan, J. M. (2009). Exendin-4 improves glycemic control, ameliorates brain and pancreatic pathologies, and extends survival in a mouse model of Huntington’s disease. Diabetes 58, 318–328.
Martin, B., Pearson, M., Brenneman, R., Golden, E., Keselman, A., Iyun, T., Carlson, O. D., Egan, J. M., Becker, K. G., Wood W. 3rd., Prabhu, V., de Cabo, R., Maudsley, S., and Mattson, M. P. (2008). Conserved and differential effects of dietary energy intake on the hippocampal transcriptomes of females and males. PLoS ONE 2008 3, e2398. doi: 10.1371/journal.pone.0002398.
Maswood, N., Young, J., Tilmont, E., Zhang, Z., Gash, D. M., Gerhardt, G. A., Grondin, R., Roth, G. S., Mattison, J., Lane, M. A., Carson, R. E., Cohen, R. M., Mouton, P. R., Quigley, C., Mattson, M. P., and Ingram, D. K. (2004). Caloric restriction increases neurotrophic factor levels and attenuates neurochemical and behavioral deficits in a primate model of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 18171–18176.
Matthews, V. B., Aström, M. B., Chan, M. H., Bruce, C. R., Krabbe, K. S., Prelovsek, O., Akerström, T., Yfanti, C., Broholm, C., Mortensen, O. H., Penkowa, M., Hojman, P., Zankari, A., Watt, M. J., Bruunsgaard, H., Pedersen, B. K., and Febbraio, M. A. (2009). Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 52, 1409–1418.
Prospective Studies Collaboration, Whitlock, G., Lewington, S., Sherliker, P., Clarke, R., Emberson, J., Halsey, J., Qizilbash, N., Collins, R., and Peto, R. (2009). Body-mass index and cause-specific mortality in 900 000 adults: collaborative analyses of 57 prospective studies. Lancet 373, 1083–1096.
Qin, W., Zhao, W., Ho, L., Wang, J., Walsh, K., Gandy, S., and Pasinetti, G. M. (2008). Regulation of forkhead transcription factor FoxO3a contributes to calorie restriction-induced prevention of Alzheimer’s disease-type amyloid neuropathology and spatial memory deterioration. Ann. N. Y. Acad. Sci. 1147, 335–347.
White, C. L., Pistell, P. J., Purpera, M. N., Gupta, S., Fernandez-Kim, S. O., Hise, T. L., Keller, J. N., Ingram, D. K., Morrison, C. D., and Bruce-Keller, A. J. (2009). Effects of high fat diet on Morris maze performance, oxidative stress, and inflammation in rats: contributions of maternal diet. Neurobiol. Dis. 35, 3–13.
Xu, X., Zhan, M., Duan, W., Prabhu, V., Brenneman, R., Wood, W., Firman, J., Li, H., Zhang, P., Ibe, C., Zonderman, A. B., Longo, D. L., Poosala, S., Becker, K. G., and Mattson, M. P. (2007). Gene expression atlas of the mouse central nervous system: impact and interactions of age, energy intake and gender. Genome Biol. 8, R234.
Zahn, J. M., Poosala, S., Owen, A. B., Ingram, D. K., Lustig, A., Carter, A., Weeraratna, A. T., Taub, D. D., Gorospe, M., Mazan-Mamczarz, K., Lakatta, E. G., Boheler, K. R., Xu, X., Mattson, M. P., Falco, G., Ko, M. S., Schlessinger, D., Firman, J., Kummerfeld, S. K., Wood, W. H. 3rd, Zonderman, A. B., Kim, S. K., and Becker, K. G. (2007). AGEMAP: a gene expression database for aging in mice. PLoS Genet. 3, e201. doi: 10.1371/journal.pgen.0030201.