 Jesús Avila
Jesús Avila Alberto Gómez-Ramos
Alberto Gómez-Ramos Eduardo Soriano
Eduardo Soriano- 1Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED), ISCIII, Madrid, Spain
- 2Centro de Biología Molecular Severo Ochoa (CSIC-UAM), Neurobiology Laboratory, Madrid, Spain
- 3Department of Cell Biology, Faculty of Biology, University of Barcelona, Developmental Neurobiology and Regeneration Lab, Parc Científic de Barcelona, Barcelona, Spain
- 4Vall d’Hebrón Institut de Recerca (VHIR), Barcelona, Spain
It is assumed that DNA sequences are conserved in the diverse cell types present in a multicellular organism like the human being. Thus, in order to compare the sequences in the genome of DNA from different individuals, nucleic acid is commonly isolated from a single tissue. In this regard, blood cells are widely used for this purpose because of their availability. Thus blood DNA has been used to study genetic familiar diseases that affect other tissues and organs, such as the liver, heart, and brain. While this approach is valid for the identification of familial diseases in which mutations are present in parental germinal cells and, therefore, in all the cells of a given organism, it is not suitable to identify sporadic diseases in which mutations might occur in specific somatic cells. This review addresses somatic DNA variations in different tissues or cells (mainly in the brain) of single individuals and discusses whether the dogma of DNA invariance between cell types is indeed correct. We will also discuss how single nucleotide somatic variations arise, focusing on the presence of specific DNA mutations in the brain.
Introduction
DNA molecules have been described as the conserved stores of genetic information (Cech, 2012). Whole-genome and exome sequencing are powerful research tools with which to analyze the molecular basis of many human diseases of familial origin (Drmanac, 2012). However, during development or in adulthood, somatic mutations may appear in specific tissues of a human being (or other organism).
With the sequencing data in hand, the bottleneck lies in the bioinformatic analysis (Brunham and Hayden, 2012) required to obtain reliable results (it has been reported that whole-genome sequencing technology has an accuracy of only one false single nucleotide variation per 500 kbp (Roach et al., 2011)).
High-throughput sequencing technologies, like those from Illumina Life Technologies, Ion Torrent, and Roche Diagnostics, are widely used. Other sequence detection methods are based on magnetic tweezers (Linnarsson, 2012) or on other approaches, such as nanopore sequencing analysis, in which single molecules of DNA can be deciphered as they pass through a tiny channel (Pennisi, 2012). All these techniques have facilitated whole-genome or exome sequencing at an unprecedented scale, thus allowing the launch of initiatives such as the 1000 Genomes Project, which seeks to analyze DNA variations in human populations (Clarke et al., 2012). It has been suggested that each genome contains 1.5 105 new single nucleotide variants (SNVs) that are not present in the dbSNP database (Pelak et al., 2010). These variants are present in different genome regions, like the exome, and may be related to genes involved in human diseases (MacArthur et al., 2012).
In this review, we will comment on somatic DNA mutations occurring mainly in the brain.
How to Obtain Reliable Data in Genome (Exome) Sequencing
Sequence analysis can be done from homogeneous (from a single cell type) or heterogeneous DNA samples. In the first case, Sanger’s method may still be the most accurate analysis to obtain reliable results. However, samples containing heterogeneous (more than five types) DNA molecules cannot be sequenced by this technique, thus requiring alternative methods like Illumina sequencing. However, these massive sequencing techniques can generate artifacts. A preliminary approach to identify these artifacts is through examining the genome database to determine whether an identified variant is already present (thus suggesting that is not an artifact). Another approach, based on the old idea that the DNA sequence in a single organism is identical in every cell of the organism, is to look for possible Mendelian errors. To distinguish false from true variants in the genome of a human being, the sequences of the DNA of his/her parents will indicate the presence of a new variant present in the child but that is absent in the parents. This Mendelian error may suggest a false variant when somatic mutations are not taken into account (Patel et al., 2014). However, this possible false variant may also originate from a somatic mutation occurring specifically in the DNA of the child. This issue can be addressed by obtaining 50–200 reads of each base to confirm the presence of a SNV. Nevertheless, it cannot be concluded that a variant found only at low number of reads is not a true variant. When a heterogeneous sample contains more than one cell type, the difference among the proportion of DNA molecules containing a variant correlates with the proportion of cells in which these molecules are present. If the change in sequence in the somatic cells took place during development, more cells are likely to contain this variation. However, if the change occurred late in adult life, only a small proportion of the cells will hold the DNA modification. It may be difficult to validate such DNA somatic sequences by methods other than those of Illumina. This is a limitation of current next-generation sequencing techniques, which have the sensitivity required for the analysis of heterogeneous DNA samples but generate an (low) error rate.
Somatic DNA Mutations
The development of a human starts from one cell that divides into two, followed by further cell divisions until the organism has generated more than 1013 cells in a precisely controlled ontogenetic process (Frank, 2010). During development, failures in DNA replication (or repair) during cell proliferation may occur and result in the appearance of somatic mutations. Afterwards, during adulthood, additional somatic mutations may arise, and genomic variability may encode distinct cell lineages in different tissues (Frumkin et al., 2005). In fact, studies have pointed to the presence of multiple different genomes in a single human being (Lupski, 2013), and differences in DNA sequences have been reported in a variety of tissues (Frank, 2010; Pelak et al., 2010; Clarke et al., 2012; MacArthur et al., 2012; Pennisi, 2012) from the same individual. Depending on the circumstances, the appearance of somatic mutations differs depending on the tissue type. One of these circumstances is the presence of distinct types and amounts of DNA polymerases or repair enzymes in tissues. Low fidelity DNA polymerases can introduce not only nucleotide substitutions but also tandem mutations, as is the case of DNA polymerase ζ (Saribasak et al., 2012). Various forms of mutations may arise, such as sense and non-sense base substitutions (SNP), deletions, and insertions or through other mechanisms like the movement of transportable elements (Lynch, 2010; Vogel, 2011). These variations could change depending on the cell origin. Moreover, some types of cell are more sensitive to DNA damage or changes than others. This is reflected by germinal and somatic cells. The protection of the genome is critical for germ line development. A mechanism to ensure this protection has been described in Drosophila (Rangan et al., 2011). This mechanism does not act on somatic cells and consequently this cell type is more susceptible to mutations. However, to facilitate genome maintenance, in response to DNA damaging agents somatic cells have a DNA-damage checkpoint-signaling pathway involving DNA-repair scaffolding proteins like SlX4 (Ohouo et al., 2013). Moreover, specific double breaks are essential for the proper function of specific cells, like sperm cells, because these DNA breaks are required for the correct exchange of DNA (Kauppi et al., 2013).
At the cellular level, considerable efforts have been devoted to minimizing genomic insults in cultured pluripotent stem cells (Weissbein et al., 2014). Additionally, in the whole organism, clonal mosaicism for large chromosomal anomalies, from birth to old age, has been reported (Jacobs et al., 2012; Laurie et al., 2012). At cell cycle level, differences in DNA repair between the phases of the cycle have been described. For instance, during mitosis, DNA double-strand breaks (DSBs) are not repaired. It has been suggested that these mitotic DBSs cause end to end chromosome fusions and that they promote aberrant chromosome segregation (Orthwein et al., 2014).
Also, at the molecular level, not all the bases in the human genome are equally prone to chance mutations (Ponting, 2012). Mutations are more frequently observed in three types of sequences, namely simple repeats, DNAse hypersensitive sites in embryonic stem cells, and some trinucleotide sequences (Michaelson et al., 2012). Also, the expansion of trinucleotide repeats causes some disorders, mainly in neurons and myopathies, which could be caused by a slippage that involves DNA polymerases β and δ (Chan et al., 2013). In contrast, for some cells, like neuroblastoma cells, sensitivity to DNA damage depends on their state of differentiation. Undifferentiated human SH-SY5Y neuroblastoma cells are less sensitive to DNA damage than differentiated cells, in part because they show more efficient base excision repair mechanisms (Sykora et al., 2013).
Somatic DNA Mutation with Aging
Some tissues show an increased rate of DNA mutations with aging (Kong et al., 2012). The appearance of somatic mutations, which increases with age, is known to cause or increase susceptibility to diseases like cancer (Moskalev et al., 2012). For example, an age effect on the repair of DNA strand breaks in blood mononuclear cells has been reported (Garm et al., 2013). Aging is also proposed to be a risk factor for neurodegenerative disorders. Indeed, an increasing number of somatic mutations are being associated with neurological diseases (Poduri et al., 2013; Madabhushi et al., 2014; Singleton, 2014).
Furthermore, specific changes in mitochondrial brain DNA have been reported (Bender et al., 2006; Kraytsberg et al., 2006). Mitochondrial DNA (mtDNA) is not protected by histones and is therefore more sensitive to external damage. Indeed, mtDNA deletions are abundant in aged substantia nigra neurons (Bender et al., 2006; Kraytsberg et al., 2006) and in peripheral tissues (Baines et al., 2014). Curiously, mtDNA damage in a mouse model of Alzheimer disease (AD) decreases amyloid beta plaque formation (Pinto et al., 2013). Moreover, DNA ligase activity, which is probably involved in DNA repair, is lower in mitochondrial extracts from AD patients than in matched non-demented controls (Canugovi et al., 2014).
Mechanisms for DNA Sequence Variations
Briefly, we will comment on several mechanisms that can give rise to somatic DNA sequence variations. As mentioned, not all the nucleotides in the human genome are equally prone to chance mutations, with exonic sequences and GpG-rich sequences showing greater susceptibility (Michaelson et al., 2012). CpG-rich sequences can also be methylated (or demethylated), and such changes in methylation in neurons affect memory formation (Kaas et al., 2013).
Furthermore, CpG-rich sequences are present in the promoters involved in divergent (on both sides with opposite orientations) transcription. Changes in such sequences may alter gene expression (Wu and Sharp, 2013).
Opening of the double helix facilitates DNA damage. This opening occurs not only during DNA replication, a process in which lesions may occur anywhere in the genome, but also during the active transcription of regions. High levels of transcription induce genomic instability (Wu and Sharp, 2013). A mouse model deficient in DNA repair and transcription shows an increase in DNA damage, which results in premature aging (de Boer et al., 2002). This observation suggests that the opening of the DNA double helix during DNA transcription also facilitates DNA damage, as proposed for replication (Marchesi, 2011). Indeed, during transcription, the coding DNA strand is exposed as a single DNA strand, whereas the non-coding strand is base-paired and protected by the nascent RNA (Aguilera and García-Muse, 2012; Wu and Sharp, 2013). Also, the unwinding of the DNA helix during transcription sometimes generates topological perturbations that can affect genome stability (Bermejo et al., 2012). In addition, somatic hypermutation of immunoglobulin genes has been related to changes in RNA polymerase II progression (Kodgire et al., 2013). Also, it has been described that RNA polymerase and the protein UvrD, a helicase, cooperate to target a damaged DNA site for repair (Epshtein et al., 2014).
DNA Damage Repair Mechanisms
There are several DNA damage repair mechanisms, these involving base or nucleotide excision repair, mismatch repair, non-homologous end joining or homologous recombination. In base excision repair, it has been proposed that damaged DNA bases are removed through a major pathway involving DNA glycosylases (Caldecott, 2003; Jackson and Bartek, 2009). For example, 7,8-dihydro-8-oxoguanine (8-OHdG), which arises by oxidative damage, is hydrolyzed by MuTYH DNA glycosylases (Markkanen et al., 2013). Also, Nei13 DNA glycosylase may participate in this pathway (Regnell et al., 2012). Recently, the effects of DNA sequence context on the glycosylase activity of human 8-oxoguanine DNA glycosylase have been reported (Sassa et al., 2012).
In addition, the modified base can be identified by a DNA glycosylase and removed by an endonuclease, resulting in a gap in the DNA sequence that is filled by a DNA polymerase and sealed by a DNA ligase (Caldecott, 2003; Maiti et al., 2012). When this repair mechanism is not working properly, the DNA sequence undergoes a mutation (see also Deng et al., 2014) for other repair mechanisms that can result in the introduction of mutations into the genome). The probability of the appearance of a mutation increases in the aged brain when there is an accumulation of 8-OHdG (Wolf et al., 2005). It should be noted that some DNA glycosylases involved in base excision repair contain a Fe-S cluster essential for their activity (Cunningham et al., 1989). Furthermore, oxidative damage specifically in neuronal DNA can also occur. Indeed, the brain has an enhanced cellular metabolism compared to other tissues, and this may result in the formation of free radicals that damage the DNA of brain cells (Hofman, 1991). In this regard, impaired repair of oxidative DNA damage may influence the clinical manifestation of AD (Silva et al., 2014).
Also, the deregulation of the DNA damage response has been described upon intercellular contact in non-neuronal cells (Kang et al., 2012). Nothing is known about whether synaptic neuronal activity facilitates the deregulation of DNA damage. Also, it has been established that ribonucleotides contaminate DNA in some circumstances. Okazaki fragments play an important role in replication. These fragments consist of a short sequence of about 10 ribonucleotides followed by a sequence (about 300 nucleotides) of deoxyribonucleotides. The ribonucleotides must be removed and replaced by deoxyribonucleotides during DNA replication, but if complete removal is impaired, ribonucleotides can be incorporated into nascent DNA. This aberrant incorporation results in DNA damage (Caldecott, 2014), directly or through the formation of DNA single-strand breaks (SSBs), which in turn have been linked to neurodegeneration (Caldecott, 2008). In some conditions, like when the number of rNTPs exceeds that of dNTPs, replicative DNA polymerases incorporate ribonucleotides into DNA (Reijns et al., 2011). This aberrant incorporation can be repaired by enzymatic removal of ribonucleotides from DNA, to preserve genome integrity (Reijns et al., 2012). A mammalian RNAse, RNAse H2, removes ribonucleotides from DNA to maintain genome integrity (Hiller et al., 2012). Also, the structural localization of DNA lesions in nucleosome core particles influences accessibility to base excision repair enzymes (Rodriguez and Smerdon, 2013). In addition, it is postulated that the histone mark H3K36me3 regulates human DNA mismatch repair (Li et al., 2013).
As previously indicated, DNA damage can result in SSBs or DSBs. When occurring in non-dividing cells, such as neurons, these breaks promote premature aging of the cell (Ames et al., 1993), thereby accelerating their demise. The repair of SSBs or DSBs has been extensively reviewed (Ciccia and Elledge, 2010). Single nucleotide excision repair can take place anywhere in the genome (Ciccia and Elledge, 2010), even in actively transcribing regions, as the transcribing RNA polymerase acts as a damage sensor (de Laat et al., 1999; Dogliotti et al., 2001). The result can be the appearance of a mutation. The consequences of mutations will differ depending on whether they take place in introns, upstream sequences, promotors, codifying sequences (exons), etc., since they will affect the regulation of the amount of RNA expressed, RNA stability, and the presence of silent or non-silent mutations in the translated proteins. SSBs can be corrected by various DNA repair mechanisms, and the DNA damage associated with these breaks induces p53 target genes to repair DNA (Smith and Seo, 2002).
DNA DSBs are generated throughout cell life and can result in irreversible damage. To avoid this damage, the cell can generate a new DNA replicate in order to produce homologous templates once again (homology-directed repair) (Jones and Petermann, 2012). In the absence of new DNA replication (a feature of non-dividing cells like neurons), a fast process known as non-homologous end-joining occurs, which repairs DNA breaks by shielding the DNA ends (Sale et al., 2012). In proliferating cells, homology-directed repair is blocked when non-homologous end-joining is active (Lukas and Lukas, 2013). Also, in several neurodegenerative disorders, slippage during replication of repetitive sequences may occur and thus require repair of the newly synthesized strand (Schofield and Hsieh, 2003). These mechanisms are not discussed in this review. However, they can occur in neurons and do not result in the appearance of SNVs but in insertions (or deletions) (indels). Neither will we discuss the regulation of DNA damage responses by ubitiquin and SUMO (Jackson and Durocher, 2013). Moreover, DNA damage can arise by reversal apoptosis in a mechanism that rescues cells from a critical stage named “anastasis” (Tang et al., 2012).
DNA Variations in Somatic Cells
All the above-mentioned processes related to changes in the DNA sequence of somatic cells may act in a different way in tissues of different origins. Preliminary results from the comparison of DNA sequences of exomes from tissues with an endoderm, mesoderm or exoderm origin support this notion (Gómez-Ramos et al., 2014). Furthermore, somatic mosaicism has been described in human skin (Abyzov et al., 2012).
In addition, in the same tissue, for example the central nervous system, different DNA sequence variations may occur in distinct cell types.
Variations in Brain DNA Sequences
Little is known about specific DNA mutations occurring specifically in brain cells. The adult human brain comprises two main cell types, neurons and glia cells, in a roughly 50/50 proportion. The main difference between glia cells and neurons is that the former are proliferating cells that are renewed many times throughout adult life, while the latter It is assumed that DNA sequences are conserved in the diverse cell types present in a multicellular organism like the human being. Thus, in order to compare the sequences in the genome of DNA from different individuals, nucleic acid is commonly isolated from a single tissue. In this regard, blood cells are widely used for this purpose because of their availability. Thus blood DNA has been used to study genetic familiar diseases that affect other tissues and organs, such as the liver, heart, and brain. While this approach is valid for the identification of familial diseases in which mutations are present in parental germinal cells and, therefore, in all the cells of a given organism, it is not suitable to identify sporadic diseases in which mutations might occur in specific somatic cells. This review addresses somatic DNA variations in different tissues or cells (mainly in the brain) of single individuals and discusses whether the dogma of DNA invariance between cell types is indeed correct. We will also discuss how single nucleotide somatic variations arise, focusing on the presence of specific DNA mutations in the brain.are mainly terminally differentiated, non-dividing cells, usually of the same age as the host organism.
Some DNA variations (or mutations) can be present in all the cell types of an organism, as is the case of neurodegenerative disorders with a familial origin. In these conditions, germinal cells are responsible for ensuring that the mutations appear in the DNA of every cell of the organism. Also, if the mutation takes place in precursor-dividing cells early in development, both glia and neuron cells carry the same variation in their DNA. However, when it occurs later during development or in adIt is assumed that DNA sequences are conserved in the diverse cell types present in a multicellular organism like the human being. Thus, in order to compare the sequences in the genome of DNA from different individuals, nucleic acid is commonly isolated from a single tissue. In this regard, blood cells are widely used for this purpose because of their availability. Thus blood DNA has been used to study genetic familiar diseases that affect other tissues and organs, such as the liver, heart, and brain. While this approach is valid for the identification of familial diseases in which mutations are present in parental germinal cells and, therefore, in all the cells of a given organism, it is not suitable to identify sporadic diseases in which mutations might occur in specific somatic cells. This review addresses somatic DNA variations in different tissues or cells (mainly in the brain) of single individuals and discusses whether the dogma of DNA invariance between cell types is indeed correct. We will also discuss how single nucleotide somatic variations arise, focusing on the presence of specific DNA mutations in the brain.ulthood, this variation may be specific for a cell type or lineage (Figure 1). Also, in adult cells, somatic DNA variations may be specific for a cell type, and the mechanism for these variations may differ in glia and neurons.
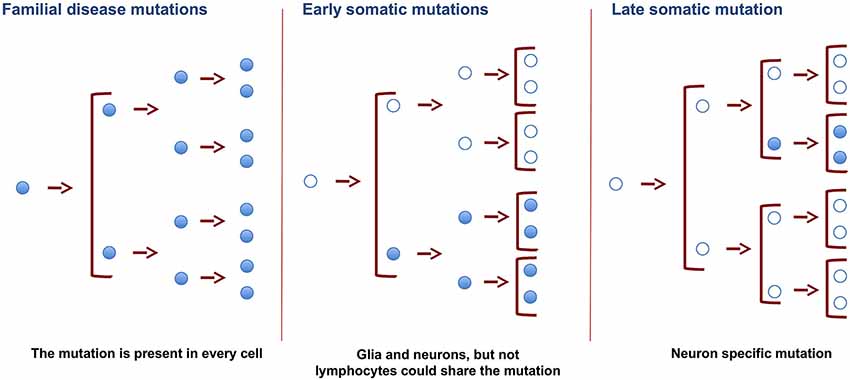
Figure 1. Changes in DNA of germinal and somatic cells. SNVs in germinal cells, present in the zygote, will result in the appearance of these SNVs in all the cells of the organism. Changes resulting in SNVs at earlier developmental stages will be present in more cells than those occurring in late adulthood. Late somatic mutations in brain cells may promote specific changes in DNA. These changes may cause the appearance of neurodegenerative disorders.
Limitations to Detect SNVs in Neuronal Populations
The presence of various cell types in a single tissue hinders the detection of somatic cell variations using some techniques (with a low sensitivity), like Sanger’s sequencing. Regarding the presence of a DNA mutation in specific cells within brain tissue, when a mutation is present exclusively in neurons, a maximum of only 50% of the cells will show the DNA change; however, when the variation occurs in a specific neuronal population, this percentage is much lower. This scenario impedes reliable results because dye-terminator sequencing (with a higher sensitivity) data is usually validated by Sanger’s method (with a lower sensitivity), which is not suitable for these types of sample. Moreover, dye-terminator sequencing can generate errors because of low signal to noise ratios and non- or mis-detection of the fluorescent signal (Breslow et al., 2008). However, the problem can be partially solved by increasing the number of reads of the DNA regions containing the variations during dye-terminator sequencing. Also, quality filtering techniques may improve Illumina sequencing results (Bokulich et al., 2013).
One of the major difficulties of making such analysis in living human brains is that the taking of human samples from brains must not be done because is a very invasive method. Thus, samples should be taken from autopsies. On the other hand there is a high cellular complexity (many cell types). Recently reported methods of single-cell sequencing are promising for the detection of individual variations in a single cell but they are not fully developed, and the extensive PCR-based amplification used in this method might interfere with the resolution of this approach (Eberwine et al., 2014).
Brain Cells and DNA Sequences
During DNA replication in glia cells, erroneous insertions of a base in the newly synthesized strand, which does not match the parental strand, can generate a mutation if the mismatch is not repaired. This type of mutation does not arise in the case of non-proliferating cells like neurons. In neurons (and also in glia cells), changes in the DNA sequence can be associated with other forms of DNA damage. Damage promoted by the extracellular environment can be solved, or not, depending on the DNA repair mechanism present in each cell type. These repair mechanisms involve various proteins, polymerases, ligases, nucleases, and helicases. Also, small RNAs have been implicated in DNA repair (Wei et al., 2012), and other factors like ubiquitin and SUMO may also participate in this process (Ulrich, 2012), as previously described. Histone ubiquitylation, a process regulated by various E3 ligases, is a main response to DSBs (Gudjonsson et al., 2012). The amount of messenger RNAs expressing these repair proteins in brain cell types differs (see Allen Brain Atlas (Hawrylycz et al., 2012)). Thus these differences, together with other distinct transcription levels in different neuronal types, can result in damage to specific neurons, which may lead to the onset of neurodegeneration. Indeed, our preliminary data indicate the presence of a number of SNVs in hippocampal neurons that are absent in cerebellar neurons (Parcerisas et al., 2014). The DNA damage response may involve many molecular changes to ensure correct DNA repair. Some of these changes have been studied in depth (Beli et al., 2012).
Aging and Oxidative Damage to DNA
Aging is a major risk for DNA damage. Aging can bring about an increase in neuronal DNA lesions (Sedelnikova et al., 2004), which activate the Ras signaling pathway (Boldogh et al., 2012). In neurons, it may result in defects in dendritic spine development and synapse formation (Yang et al., 2013). The damage can be due, at least in part, to the high levels of reactive oxygen species (ROS) and the low levels of anti-oxidant defenses in these brain cells (Finkel and Holbrook, 2000).
ROS-induced DNA damage can lead to the formation of 8-OHdG (see below).
Also, it has been reported that post-mitotic neurons develop a p21-dependent senescence phenotype driven by a DNA damage response (Jurk et al., 2012). On the other hand, it has been found that DNA methylation declines with age (Heyn et al., 2012). However, in this short review, we will not address changes in the epigenome but only briefly comment that chromatin actively participates in the DNA damage response (Soria et al., 2012). Although DNA replication takes place in S phase, the DNA damage response occurs at any point of the cell cycle and in differentiated, non-proliferating cells like neurons. This response occurs in the context of chromatin in eukaryotic cells, where DNA is wrapped with histone proteins, and there are some variations in proteins, like H2A-X, whose phosphorylation is increased in the DNA damage response (Hiller et al., 2012).
The DNA damage response is characterized by the activation of a kinase such as H2A-X (as previously indicated), which modifies histones, or by the induction of proteins like p53 or p21, which contribute to cell cycle arrest. Also, the acetylation of some proteins is involved in this response (Robert et al., 2011), and specific acetylation of p53 by HDAC inhibition prevents DNA damage in neurons (Brochier et al., 2013). Furthermore, DNA damage, mainly in the form of DSBs, may lead to cellular senescence.
Regarding new neuronal cells, adult neurogenesis requires a stringent genomic maintenance program to ensure the correct transmission of the genetic program to newborn neurons. A factor in this program is TopBP1, a protein linked to DNA replication that is essential for the maintenance of genome integrity during the proliferation of neuronal precursors. A failure in this protein may promote a modification of DNA in the affected neuronal precursors (Lee et al., 2012). Also, regarding transposons, it has been reported that some small RNAs trigger the formation of a class of small RNAs that silence transposon targets (Xiol and Pillai, 2012).
More recently, it has been described that brain activity causes DNA DSBs in neurons and that these breaks are exacerbated by the presence of amyloid beta (Suberbielle et al., 2013). It is not clear yet whether this exacerbation is due to an increase in the transcription of specific genes.
Mechanisms for DNA Sequence Variations in Brain
Neuron DNA damage, resulting in the appearance of SNVs, can favor neurodegeneration and cognitive decline in diseases like AD (Brasnjevic et al., 2008; Moreira et al., 2008; Suberbielle et al., 2013).
These SNVs are present after DNA opening, DNA damage, and/or inefficient repair. Opening of the DNA double helix occurs by two mechanisms, namely DNA replication and DNA transcription. Various proteins, present in different amounts, related to DNA replication transcription or reparation are expressed in neurons and glia cells, as clearly indicated in the Allen Brain Atlas (Hawrylycz et al., 2012). For neurons, the opening of the helix takes place during transcription. Gene expression in the adult mammalian brain is complex. It has been suggested that at least 80% of all genes are expressed in the central nervous system (Lein et al., 2007; Hawrylycz et al., 2012). For glia, DNA opening occurs during both DNA replication and transcription. It has been hypothesized that transcription may be involved mainly in the appearance of DNA variations in neuronal cells, late in development or in the adult organism. We have been examining the SNVs of 200 genes in hippocampal tissue (see legend of Figure 2). Based on the data of the Human Protein Atlas and on the basis of the transcription/translation level of these genes, we have divided them into high, moderate, low, or undetected expression. Thus, we have tested the percentage of SNVs in genes expressed at different levels in neurons and glia cells. We found a higher percentage of DNA changes in cells with high expression of neuronal genes than those with low expression (Figure 2). These observations point to a greater relationship between transcription level and DNA damage in neuronal cells, while DNA damage in glia cells is caused mainly by DNA replication.
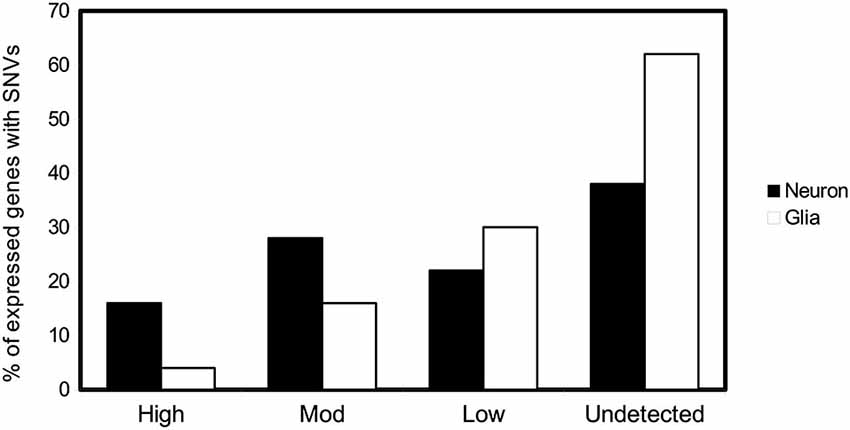
Figure 2. SNVs in neuron and glia. Possible relationship with DNA transcription or DNA replication. Percentage of genes with at least one SNV expressed in hippocampal neuron and glial cells (Parcerisas et al., 2014) that are translated into proteins according to the expression levels provided by the database The Human Protein Atlas.1 The levels of expression of the first 200 genes containing the most hippocampal-specific SNVs (according to a Fisher test respect to the SNVs found in blood), as shown by immunological detection, were checked one by one in the database and their expression in this tissue was annotated.
Regarding a possible relation between DNA transcription and DNA damage, a ubiquitin-driven link between gene expression and the DNA damage response has been put forward (Shiloh et al., 2011). On the other hand, the presence of DNA loops during transcription may facilitate oxidation, mainly of deoxyguanosine, by ROS (Cadet et al., 1997), yielding the formation of 8-OHdG. Indeed, it has been proposed that oxidative DNA damage initiates neurodegenerative diseases (Perry et al., 2001). In this regard, the damage is not exerted only at the DNA molecule, but unassembled DNA bases can also be oxidized, and these modified bases can be misincorporated into DNA (Luo et al., 2010). However, there is a mechanism—through the enzyme MTH1—that hydrolyzes oxidized DNA bases, thus preventing misincorporation (Dominissini and He, 2014).
Variations in Neuron DNA Amount and Sequences and their Possible Association with Neurological Diseases
Although most neurons are diploid, a small population of these cells surpasses the diploid level (Fischer et al., 2012). These hyperploid neurons are selectively affected by cell death at early stages of AD (Arendt et al., 2010). More recently, an increase in X chromosome aneuploidy has been reported in brain cells of female AD patients (Sugiyama et al., 2014).
By testing DNA from hippocampus, researchers recently revealed the presence of SNVs or insertions/deletions (indels) in AD patients but not in demented controls (Parcerisas et al., 2014). Little is known about the mechanism underlying indels, although a process similar to that of RNA-guided human genome engineering via cas9 has been proposed (Cho et al., 2013; Mali et al., 2013). However, whether this type of mechanism takes place in aging or in AD remains to be elucidated. Finally, transposons are found in brain DNA (Singer et al., 2010; Vogel, 2011), although no association has been made with AD. In addition, aging and AD are characterized by a decreased capacity for DNA repair as a result of a reduction in DNA end-joining activity (a process that calls for DNA-dependent protein kinase activity) required to repair DSBs (Kanungo, 2013).
Variations in DNA Sequences of Brain Cells Obtained from AD Patients
The most predominant neurodegenerative disorder is AD (Selkoe, 2011). This disease has been divided into two types, the familial type of predominantly early onset, and the sporadic type, with no familial association, of later onset (Bertram et al., 2010). For familial cases (FAD), genome sequence analysis of DNA from the patients’ lymphocytes have indicated that the disease is caused by mutations in three genes (APP, PS-1 and PS-2) (Selkoe, 2011). To look for possible genetic risk factors in sporadic AD (SAD), genome-wide association studies (GWAS) have been carried out in patients with this disease (Manolio et al., 2009; Lambert et al., 2013; Ridge et al., 2013; Zhang et al., 2013), also using DNA from lymphocytes. Also, a higher frequency of DNA damage in blood lymphocytes of SAD patients compared with age-matched controls has been reported (Zivković et al., 2013). This observation could be attributable to the presence of a different DNA damage repair mechanism (Leandro et al., 2013).
Early work revealed that the ε4 allele of APOE is a strong risk factor for AD. Other risk factors include the presence of SNVs as specific sequences in genes like: ABCS7, BIN1, CD33, CD2AP, CLU, CR1, EPHA1, MS4A4E/MS4A6A, PICALM and SORL1 (Schellenberg and Montine, 2012). In addition, two new susceptibility genes for AD have been reported (Escott-Price et al., 2014), and recently an AD-associated polymorphism in human OGG1 that sensitizes cells to DNA damage has been described (Jacob et al., 2013).
However, a significant proportion of the possible genetic defects related to the development of SAD remains unexplained. This missing defect has been named the “dark matter” of GWAS (Manolio et al., 2009), in an attempt to explain the “missing heritability” by means of GWAS analysis using DNA from lymphocytes (Manolio et al., 2009). Furthermore, little has been reported about the presence of a specific type of mutation, through insertions or deletions (indels), in AD.
Our hypothesis is that some genetic defects of SAD are present only in somatic mutations in neurons but not in peripheral cells like lymphocytes or in the germ line, as is the case for FAD patients (Figure 1). These somatic defects, which are specific to neuronal tissue, are postulated to favor the appearance of late onset dementia in SAD patients.
Preliminary data have indicated the presence of specific mutations in brain tissue from AD patients that are not present in the blood of these patients (Parcerisas et al., 2014). The proteins expressed by these brain genes include transcription factors, ion channels, and proteins related to lipid transport and metabolism, to the cytoskeleton, etc… The possible relation between variations in these genes and neurological disorders deserves further attention. Although mainly non-silent exome DNA variations have been characterized, the analysis of silent exome DNA SNVs could also be of interest due to the consequences of the codon bias in gene expression (Plotkin and Kudla, 2011).
Role of Beta Amyloid and Tau Protein in DNA Damage
About a century ago, Alzheimer described the presence of two aberrant structures, senile plaques and neurofibrillary tangles, in the brain of a demented patient (Alzheimer, 1907). We now know that the main component of the plaques is beta amyloid peptide and that of tangles is tau protein.
It has been proposed that Aβ peptide exerts DNA nicking activity that promotes DNA damage and that such damage may occur in AD patients (Gupta et al., 2013). Brain activity causes DNA DSBs in neurons, and these breaks are exacerbated by amyloid-β (Suberbielle et al., 2013). It has recently been shown that NAD attenuates oxidative DNA damage induced by Aβ peptide in cultured cortical neurons (Wu et al., 2014).
Furthermore, mutations in the MAPT gene cause chromosome instability and can introduce copy number variation in the genome (Rossi et al., 2013) (see also ref Rossi et al., 2008). Tau protein is not only a cytoskeletal protein but it is found in the nucleus of neurons (Wang et al., 1993; Brady et al., 1995; Lambert et al., 1995; Sultan et al., 2011). Indeed, tau is a DNA-binding protein (Corces et al., 1980; Krylova et al., 2005; Wei et al., 2008; Camero et al., 2014) and it plays a major role in neuronal DNA protection (Violet et al., 2014). Also it has been described that a high level of cytoplasm tau protein promotes neurodegeneration via DNA damage, heterochromatin relaxation, and piwi-like RNA-mediated gene silencing 1 (PIWIL-1), thus facilitating cell cycle re-entry (Frost et al., 2014). The differences between the two effects exerted by tau can be explained by its form and distribution. Dephosphorylated tau localizes in the nucleus, playing a protective role. In contrast, phosphorylated tau, present in the cytoplasm, interacts with mitochondrial protein DRP1, impairing mitochondrial function and facilitating the production of ROS and DNA damage (DuBoff et al., 2012; Manczak and Reddy, 2012; Camero et al., 2014).
In summary, brain-specific DNA changes occur. Thus brain DNA rather than blood DNA should be used to analyze somatic DNA variations linked to neurological diseases and present in brain cells.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by grants from the BBVA Foundation and MICINN-MINECO.
We also wish to Acknowledgments the help of the Reina Sofia Foundation, the CIEN Foundation, CIBERNED (ISCIII), and grants BFU2008-3980 (ES) and SAF2011-24841 (JA) from MICINNN-MINECO.
Glossary
De novo mutation: new mutation. Alteration in a gene that appears for the first time in a member of a family as a result of a mutation produced in a germ cell (egg or sperm) or in the fertilized egg. Germinal mutation: mutation that occurs in a germ cell (egg or sperm) and can be transmitted to the offspring. Somatic mutation: mutation that is not inherited from parents and not transferred to the offspring. They can occur in any of the cells except the germ cells. Exome: part of the genome composed by the exons, the portions of the genes containing the information to synthesize the proteins. Mosaicism: it describes a condition in which one individual who has developed from a single fertilized egg, has two or more populations of cells with different genotypes. If these different populations of cells are in a somatic tissue, we talk about somatic mosaicism. We refer to germline mosaicism when some gametes (oocytes or sperm) carry a mutation in an individual cell, but the rest of cells are normal.
Footnotes
References
Abyzov, A., Mariani, J., Palejev, D., Zhang, Y., Haney, M. S., Tomasini, L., et al. (2012). Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature 492, 438–442. doi: 10.1038/nature11629
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aguilera, A., and García-Muse, T. (2012). R loops: from transcription byproducts to threats to genome stability. Mol. Cell 46, 115–124. doi: 10.1016/j.molcel.2012.04.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Alzheimer, A. (1907). Uber eine eigenartige erkrankung der hirnrinde. Allgemeine. J. Psychiatry and Forensic Medicine - phychish, (Berlin) 64, 146–148.
Ames, B. N., Shigenaga, M. K., and Hagen, T. M. (1993). Oxidants, antioxidants and the degenerative diseases of aging. Proc. Natl. Acad. Sci. U S A 90, 7915–7922. doi: 10.1073/pnas.90.17.7915
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arendt, T., Brückner, M. K., Mosch, B., and Lösche, A. (2010). Selective cell death of hyperploid neurons in Alzheimer’s disease. Am. J. Pathol. 177, 15–20. doi: 10.2353/ajpath.2010.090955
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Baines, H. L., Turnbull, D. M., and Greaves, L. C. (2014). Human stem cell aging: do mitochondrial DNA mutations have a causal role? Aging Cell 13, 201–205. doi: 10.1111/acel.12199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beli, P., Lukashchuk, N., Wagner, S. A., Weinert, B. T., Olsen, J. V., Baskcomb, L., et al. (2012). Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 46, 212–225. doi: 10.1016/j.molcel.2012.01.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bender, A., Krishnan, K. J., Morris, C. M., Taylor, G. A., Reeve, A. K., Perry, R. H., et al. (2006). High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38, 515–517. doi: 10.1038/ng1769
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bermejo, R., Lai, M. S., and Foiani, M. (2012). Preventing replication stress to maintain genome stability: resolving conflicts between replication and transcription. Mol. Cell 45, 710–718. doi: 10.1016/j.molcel.2012.03.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bertram, L., Lill, C. M., and Tanzi, R. E. (2010). The genetics of Alzheimer disease: back to the future. Neuron 68, 270–281. doi: 10.1016/j.neuron.2010.10.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I., Knight, R., et al. (2013). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59. doi: 10.1038/nmeth.2276
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boldogh, I., Hajas, G., Aguilera-Aguirre, L., Hegde, M. L., Radak, Z., Bacsi, A., et al. (2012). Activation of ras signaling pathway by 8-oxoguanine DNA glycosylase bound to its excision product, 8-oxoguanine. J. Biol. Chem. 287, 20769–20773. doi: 10.1074/jbc.c112.364620
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brady, R. M., Zinkowski, R. P., and Binder, L. I. (1995). Presence of tau in isolated nuclei from human brain. Neurobiol. Aging 16, 479–486. doi: 10.1016/0197-4580(95)00023-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brasnjevic, I., Hof, P. R., Steinbusch, H. W. M., and Schmitz, C. (2008). Accumulation of nuclear DNA damage or neuron loss: molecular basis for a new approach to understanding selective neuronal vulnerability in neurodegenerative diseases. DNA Repair (Amst) 7, 1087–1097. doi: 10.1016/j.dnarep.2008.03.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Breslow, D. K., Cameron, D. M., Collins, S. R., Schuldiner, M., Stewart-Ornstein, J., Newman, H. W., et al. (2008). A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat. Methods 5, 711–718. doi: 10.1038/nmeth.1234
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brochier, C., Dennis, G., Rivieccio, M. A., McLaughlin, K., Coppola, G., Ratan, R. R., et al. (2013). Specific acetylation of p53 by HDAC inhibition prevents DNA damage-induced apoptosis in neurons. J. Neurosci. 33, 8621–8632. doi: 10.1523/jneurosci.5214-12.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brunham, L. R., and Hayden, M. R. (2012). Medicine. Whole-genome sequencing: the new standard of care? Science 336, 1112–1113. doi: 10.1126/science.1220967
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cadet, J., Berger, M., Douki, T., and Ravanat, J. L. (1997). Oxidative damage to DNA: formation, measurement and biological significance. Rev. Physiol. Biochem. Pharmacol. 131, 1–87. doi: 10.1007/3-540-61992-5_5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caldecott, K. W. (2003). Protein-protein interactions during mammalian DNA single-strand break repair. Biochem. Soc. Trans. 31, 247–251. doi: 10.1042/bst0310247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caldecott, K. W. (2008). Single-strand break repair and genetic disease. Nat. Rev. Genet. 9, 619–631. doi: 10.1038/nrg2380
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caldecott, K. W. (2014). Molecular biology. Ribose–an internal threat to DNA. Science 343, 260–261. doi: 10.1126/science.1248234
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Camero, S., Benítez, M. J., Barrantes, A., Ayuso, J. M., Cuadros, R., Avila, J., et al. (2014). Tau protein provides DNA with thermodynamic and structural features which are similar to those found in histone-DNA complex. J. Alzheimers Dis. 39, 649–660. doi: 10.3233/JAD-131415
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Canugovi, C., Shamanna, R. A., Croteau, D. L., and Bohr, V. A. (2014). Base excision DNA repair levels in mitochondrial lysates of Alzheimer’s disease. Neurobiol. Aging 35, 1293–1300. doi: 10.1016/j.neurobiolaging.2014.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cech, T. R. (2012). The RNA worlds in context. Cold Spring Harb. Perspect. Biol. 4:a006742. doi: 10.1101/cshperspect.a006742
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chan, N. L. S., Guo, J., Zhang, T., Mao, G., Hou, C., Yuan, F., et al. (2013). Coordinated processing of 3’ slipped (CAG)n/(CTG)n hairpins by DNA polymerases β and δ preferentially induces repeat expansions. J. Biol. Chem. 288, 15015–15022. doi: 10.1074/jbc.M113.464370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cho, S. W., Kim, S., Kim, J. M., and Kim, J.-S. (2013). Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 230–232. doi: 10.1038/nbt.2507
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ciccia, A., and Elledge, S. J. (2010). The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204. doi: 10.1016/j.molcel.2010.09.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clarke, L., Zheng-Bradley, X., Smith, R., Kulesha, E., Xiao, C., Toneva, I., et al. (2012). The 1000 genomes project: data management and community access. Nat. Methods 9, 459–462. doi: 10.1038/nmeth.1974
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Corces, V. G., Manso, R., De La Torre, J., Avila, J., Nasr, A., and Wiche, G. (1980). Effects of DNA on microtubule assembly. Eur. J. Biochem. 105, 7–16. doi: 10.1111/j.1432-1033.1980.tb04468.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cunningham, R. P., Asahara, H., Bank, J. F., Scholes, C. P., Salerno, J. C., Surerus, K., et al. (1989). Endonuclease III is an iron-sulfur protein. Biochemistry 28, 4450–4455. doi: 10.1021/bi00436a049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Boer, J., Andressoo, J. O., de Wit, J., Huijmans, J., Beems, R. B., van Steeg, H., et al. (2002). Premature aging in mice deficient in DNA repair and transcription. Science 296, 1276–1279. doi: 10.1126/science.1070174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
de Laat, W. L., Jaspers, N. G., and Hoeijmakers, J. H. (1999). Molecular mechanism of nucleotide excision repair. Genes Dev. 13, 768–785. doi: 10.1101/gad.13.7.768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deng, S. K., Gibb, B., de Almeida, M. J., Greene, E. C., and Symington, L. S. (2014). RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat. Struct. Mol. Biol. 21, 405–412. doi: 10.1038/nsmb.2786
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dogliotti, E., Fortini, P., Pascucci, B., and Parlanti, E. (2001). The mechanism of switching among multiple BER pathways. Prog. Nucleic Acid Res. Mol. Biol. 68, 3–27. doi: 10.1016/s0079-6603(01)68086-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dominissini, D., and He, C. (2014). Cancer: damage prevention targeted. Nature 508, 191–192. doi: 10.1038/nature13221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Drmanac, R. (2012). Medicine. The ultimate genetic test. Science 336, 1110–1112. doi: 10.1126/science.1221037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
DuBoff, B., Götz, J., and Feany, M. B. (2012). Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 75, 618–632. doi: 10.1016/j.neuron.2012.06.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eberwine, J., Sul, J.-Y., Bartfai, T., and Kim, J. (2014). The promise of single-cell sequencing. Nat. Methods 11, 25–27. doi: 10.1038/nmeth.2769
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Epshtein, V., Kamarthapu, V., McGary, K., Svetlov, V., Ueberheide, B., Proshkin, S., et al. (2014). UvrD facilitates DNA repair by pulling RNA polymerase backwards. Nature 505, 372–377. doi: 10.1038/nature12928
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Escott-Price, V., Bellenguez, C., Wang, L.-S., Choi, S.-H., Harold, D., Jones, L., et al. (2014). Gene-wide analysis detects two new susceptibility genes for Alzheimer’s disease. PloS One 9:e94661. doi: 10.1371/journal.pone.0094661
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Finkel, T., and Holbrook, N. J. (2000). Oxidants, oxidative stress and the biology of ageing. Nature 408, 239–247. doi: 10.1038/35041687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fischer, H.-G., Morawski, M., Brückner, M. K., Mittag, A., Tarnok, A., and Arendt, T. (2012). Changes in neuronal DNA content variation in the human brain during aging. Aging Cell 11, 628–633. doi: 10.1111/j.1474-9726.2012.00826.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frank, S. A. (2010). Evolution in health and medicine sackler colloquium: somatic evolutionary genomics: mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc. Natl. Acad. Sci. U S A 107(Suppl. 1 ), 1725–1730. doi: 10.3410/f.1346962.818060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frost, B., Hemberg, M., Lewis, J., and Feany, M. B. (2014). Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 17, 357–366. doi: 10.1038/nn.3639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Frumkin, D., Wasserstrom, A., Kaplan, S., Feige, U., and Shapiro, E. (2005). Genomic variability within an organism exposes its cell lineage tree. PLoS Comput. Biol. 1:e50. doi: 10.1371/journal.pcbi.0010050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garm, C., Moreno-Villanueva, M., Bürkle, A., Petersen, I., Bohr, V. A., Christensen, K., et al. (2013). Age and gender effects on DNA strand break repair in peripheral blood mononuclear cells. Aging Cell 12, 58–66. doi: 10.1111/acel.12019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gómez-Ramos, A., Sanchez-Sanchez, R., Muhaisen, A., Rábano, A., Soriano, E., and Avila, J. (2014). Similarities and differences between exome sequences found in a variety of tissues from the same individual. PloS One 9:e101412. doi: 10.1371/journal.pone.0101412
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gudjonsson, T., Altmeyer, M., Savic, V., Toledo, L., Dinant, C., Grøfte, M., et al. (2012). TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell 150, 697–709. doi: 10.1016/j.cell.2012.06.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gupta, V. B., Monica, F. S., Berrocal, R., Rao, K. S., and Rao, K. S. J. (2013). Studies on the mechanism of the DNA nicking property of amyloid-β40: implications in Alzheimer’s disease. J. Alzheimers Dis. 33, 1059–1071. doi: 10.3233/JAD-121249
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hawrylycz, M. J., Lein, E. S., Guillozet-Bongaarts, A. L., Shen, E. H., Ng, L., Miller, J. A., et al. (2012). An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399. doi: 10.1038/nature11405
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heyn, H., Li, N., Ferreira, H. J., Moran, S., Pisano, D. G., Gómez, A., et al. (2012). Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. U S A 109, 10522–10527. doi: 10.1073/pnas.1120658109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hiller, B., Achleitner, M., Glage, S., Naumann, R., Behrendt, R., and Roers, A. (2012). Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J. Exp. Med. 209, 1419–1426. doi: 10.1084/jem.20120876
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hofman, M. A. (1991). From here to eternity: brain aging in an evolutionary perspective. Neurobiol. Aging 12, 338–340; discussion 352–355. doi: 10.1016/0197-4580(91)90014-b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. doi: 10.1038/nature08467
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jackson, S. P., and Durocher, D. (2013). Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 49, 795–807. doi: 10.1016/j.molcel.2013.01.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jacob, K. D., Noren Hooten, N., Tadokoro, T., Lohani, A., Barnes, J., and Evans, M. K. (2013). Alzheimer’s disease-associated polymorphisms in human OGG1 alter catalytic activity and sensitize cells to DNA damage. Free Radic. Biol. Med. 63, 115–125. doi: 10.1016/j.freeradbiomed.2013.05.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jacobs, K. B., Yeager, M., Zhou, W., Wacholder, S., Wang, Z., Rodriguez-Santiago, B., et al. (2012). Detectable clonal mosaicism and its relationship to aging and cancer. Nat. Genet. 44, 651–658. doi: 10.1038/ng.2270
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jones, R. M., and Petermann, E. (2012). Replication fork dynamics and the DNA damage response. Biochem. J. 443, 13–26. doi: 10.1042/bj20112100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jurk, D., Wang, C., Miwa, S., Maddick, M., Korolchuk, V., Tsolou, A., et al. (2012). Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11, 996–1004. doi: 10.1111/j.1474-9726.2012.00870.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaas, G. A., Zhong, C., Eason, D. E., Ross, D. L., Vachhani, R. V., Ming, G.-L., et al. (2013). TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription and memory formation. Neuron 79, 1086–1093. doi: 10.1016/j.neuron.2013.08.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kang, M. A., So, E.-Y., and Ouchi, T. (2012). Deregulation of DNA damage response pathway by intercellular contact. J. Biol. Chem. 287, 16246–16255. doi: 10.1074/jbc.m111.337212
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kanungo, J. (2013). DNA-dependent protein kinase and DNA repair: relevance to Alzheimer’s disease. Alzheimers Res. Ther. 5:13. doi: 10.1186/alzrt167
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kauppi, L., Barchi, M., Lange, J., Baudat, F., Jasin, M., and Keeney, S. (2013). Numerical constraints and feedback control of double-strand breaks in mouse meiosis. Genes Dev. 27, 873–886. doi: 10.1101/gad.213652.113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kodgire, P., Mukkawar, P., Ratnam, S., Martin, T. E., and Storb, U. (2013). Changes in RNA polymerase II progression influence somatic hypermutation of Ig-related genes by AID. J. Exp. Med. 210, 1481–1492. doi: 10.1084/jem.20121523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kong, A., Frigge, M. L., Masson, G., Besenbacher, S., Sulem, P., Magnusson, G., et al. (2012). Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475. doi: 10.1038/nature11396
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kraytsberg, Y., Kudryavtseva, E., McKee, A. C., Geula, C., Kowall, N. W., and Khrapko, K. (2006). Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 38, 518–520. doi: 10.1038/ng1778
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krylova, S. M., Musheev, M., Nutiu, R., Li, Y., Lee, G., and Krylov, S. N. (2005). Tau protein binds single-stranded DNA sequence specifically–the proof obtained in vitro with non-equilibrium capillary electrophoresis of equilibrium mixtures. FEBS Lett. 579, 1371–1375. doi: 10.1016/j.febslet.2005.01.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lambert, M. P., Sabo, S., Zhang, C., Enam, S. A., and Klein, W. L. (1995). Constitutive Alzheimer’s-type tau epitopes in a neuritogenic rat CNS cell line. Neurobiol. Aging 16, 583–589. doi: 10.1016/0197-4580(95)00042-d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laurie, C. C., Laurie, C. A., Rice, K., Doheny, K. F., Zelnick, L. R., McHugh, C. P., et al. (2012). Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat. Genet. 44, 642–650. doi: 10.1038/ng.2271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leandro, G. S., Lobo, R. R., Oliveira, D. V. N. P., Moriguti, J. C., and Sakamoto-Hojo, E. T. (2013). Lymphocytes of patients with Alzheimer’s disease display different DNA damage repair kinetics and expression profiles of DNA repair and stress response genes. Int. J. Mol. Sci. 14, 12380–12400. doi: 10.3390/ijms140612380
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, Y., Katyal, S., Downing, S. M., Zhao, J., Russell, H. R., and McKinnon, P. J. (2012). Neurogenesis requires TopBP1 to prevent catastrophic replicative DNA damage in early progenitors. Nat. Neurosci. 15, 819–826. doi: 10.1038/nn.3097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lein, E. S., Hawrylycz, M. J., Ao, N., Ayres, M., Bensinger, A., Bernard, A., et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. doi: 10.1038/nature05453
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, F., Mao, G., Tong, D., Huang, J., Gu, L., Yang, W., et al. (2013). The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSα. Cell 153, 590–600. doi: 10.1016/j.cell.2013.03.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Linnarsson, S. (2012). Magnetic sequencing. Nat. Methods 9, 339–341. doi: 10.1038/nmeth.1934
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lukas, J., and Lukas, C. (2013). Molecular biology. Shielding broken DNA for a quick fix. Science 339, 652–653. doi: 10.1126/science.1234602
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luo, M., He, H., Kelley, M. R., and Georgiadis, M. M. (2010). Redox regulation of DNA repair: implications for human health and cancer therapeutic development. Antioxid. Redox Signal. 12, 1247–1269. doi: 10.1089/ars.2009.2698
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lupski, J. R. (2013). Genetics. Genome mosaicism–one human, multiple genomes. Science 341, 358–359. doi: 10.1126/science.1239503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lynch, M. (2010). Rate, molecular spectrum and consequences of human mutation. Proc. Natl. Acad. Sci. U S A 107, 961–968. doi: 10.1073/pnas.0912629107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
MacArthur, D. G., Balasubramanian, S., Frankish, A., Huang, N., Morris, J., Walter, K., et al. (2012). A systematic survey of loss-of-function variants in human protein-coding genes. Science 335, 823–828. doi: 10.1126/science.1215040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Madabhushi, R., Pan, L., and Tsai, L.-H. (2014). DNA damage and its links to neurodegeneration. Neuron 83, 266–282. doi: 10.1016/j.neuron.2014.06.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maiti, A., Noon, M. S., MacKerell, A. D. Jr., Pozharski, E., and Drohat, A. C. (2012). Lesion processing by a repair enzyme is severely curtailed by residues needed to prevent aberrant activity on undamaged DNA. Proc. Natl. Acad. Sci. U S A 109, 8091–8096. doi: 10.1073/pnas.1201010109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., et al. (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. doi: 10.1126/science.1232033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Manczak, M., and Reddy, P. H. (2012). Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 21, 2538–2547. doi: 10.1093/hmg/dds072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., et al. (2009). Finding the missing heritability of complex diseases. Nature 461, 747–753. doi: 10.1038/nature08494
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marchesi, V. T. (2011). Alzheimer’s dementia begins as a disease of small blood vessels, damaged by oxidative-induced inflammation and dysregulated amyloid metabolism: implications for early detection and therapy. FASEB J. 25, 5–13. doi: 10.1096/fj.11-0102ufm
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Markkanen, E., Dorn, J., and Hübscher, U. (2013). MUTYH DNA glycosylase: the rationale for removing undamaged bases from the DNA. Front. Genet. 4:18. doi: 10.3389/fgene.2013.00018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Michaelson, J. J., Shi, Y., Gujral, M., Zheng, H., Malhotra, D., Jin, X., et al. (2012). Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 151, 1431–1442. doi: 10.1016/j.cell.2012.11.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moreira, P. I., Nunomura, A., Nakamura, M., Takeda, A., Shenk, J. C., Aliev, G., et al. (2008). Nucleic acid oxidation in Alzheimer disease. Free Radic. Biol. Med. 44, 1493–1505. doi: 10.1016/j.freeradbiomed.2008.01.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Moskalev, A. A., Smit-McBride, Z., Shaposhnikov, M. V., Plyusnina, E. N., Zhavoronkov, A., Budovsky, A., et al. (2012). Gadd45 proteins: relevance to aging, longevity and age-related pathologies. Ageing Res. Rev. 11, 51–66. doi: 10.1016/j.arr.2011.09.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ohouo, P. Y., Bastos de Oliveira, F. M., Liu, Y., Ma, C. J., and Smolka, M. B. (2013). DNA-repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature 493, 120–124. doi: 10.1038/nature11658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Orthwein, A., Fradet-Turcotte, A., Noordermeer, S. M., Canny, M. D., Brun, C. M., Strecker, J., et al. (2014). Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 344, 189–193. doi: 10.1126/science.1248024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Parcerisas, A., Rubio, S. E., Muhaisen, A., Gómez-Ramos, A., Pujadas, L., Puiggros, M., et al. (2014). Somatic signature of brain-specific single nucleotide variations in Sporadic Alzheimer Disease. J. Alzheimers Dis. 42, 1357–1382. doi: 10.3233/JAD-140891
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Patel, Z. H., Kottyan, L. C., Lazaro, S., Williams, M. S., Ledbetter, D. H., Tromp, H., et al. (2014). The struggle to find reliable results in exome sequencing data: filtering out Mendelian errors. Front. Genet. 5:16. doi: 10.3389/fgene.2014.00016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pelak, K., Shianna, K. V., Ge, D., Maia, J. M., Zhu, M., Smith, J. P., et al. (2010). The characterization of twenty sequenced human genomes. PLoS Genet. 6:e1001111. doi: 10.1371/journal.pgen.1001111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pennisi, E. (2012). Genome sequencing. Search for pore-fection. Science 336, 534–537. doi: 10.1126/science.336.6081.534
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Perry, G., Avila, J., Espey, M. G., Wink, D. A., Atwood, C. S., and Smith, M. A. (2001). Biochemistry of neurodegeneration. Science 291, 595–597. doi: 10.1126/science.291.5504.595c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pinto, M., Pickrell, A. M., Fukui, H., and Moraes, C. T. (2013). Mitochondrial DNA damage in a mouse model of Alzheimer’s disease decreases amyloid beta plaque formation. Neurobiol. Aging 34, 2399–2407. doi: 10.1016/j.neurobiolaging.2013.04.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Plotkin, J. B., and Kudla, G. (2011). Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 12, 32–42. doi: 10.1038/nrg2899
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Poduri, A., Evrony, G. D., Cai, X., and Walsh, C. A. (2013). Somatic mutation, genomic variation and neurological disease. Science 341:1237758. doi: 10.1126/science.1237758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ponting, C. P. (2012). Loaded dice for human genome mutation. Cell 151, 1399–1400. doi: 10.1016/j.cell.2012.12.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rangan, P., Malone, C. D., Navarro, C., Newbold, S. P., Hayes, P. S., Sachidanandam, R., et al. (2011). piRNA production requires heterochromatin formation in Drosophila. Curr. Biol. 21, 1373–1379. doi: 10.1016/j.cub.2011.06.057
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Regnell, C. E., Hildrestrand, G. A., Sejersted, Y., Medin, T., Moldestad, O., Rolseth, V., et al. (2012). Hippocampal adult neurogenesis is maintained by Neil3-dependent repair of oxidative DNA lesions in neural progenitor cells. Cell Rep. 2, 503–510. doi: 10.1016/j.celrep.2012.08.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reijns, M. A. M., Bubeck, D., Gibson, L. C. D., Graham, S. C., Baillie, G. S., Jones, E. Y., et al. (2011). The structure of the human RNase H2 complex defines key interaction interfaces relevant to enzyme function and human disease. J. Biol. Chem. 286, 10530–10539. doi: 10.1074/jbc.m110.177394
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reijns, M. A. M., Rabe, B., Rigby, R. E., Mill, P., Astell, K. R., Lettice, L. A., et al. (2012). Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149, 1008–1022. doi: 10.1016/j.cell.2012.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ridge, P. G., Mukherjee, S., Crane, P. K., Kauwe, J. S. K., and Alzheimer’s Disease Genetics Consortium (2013). Alzheimer’s disease: analyzing the missing heritability. PloS One 8:e79771. doi: 10.1371/journal.pone.0079771
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Roach, J. C., Glusman, G., Hubley, R., Montsaroff, S. Z., Holloway, A. K., Mauldin, D. E., et al. (2011). Chromosomal haplotypes by genetic phasing of human families. Am. J. Hum. Genet. 89, 382–397. doi: 10.1016/j.ajhg.2011.07.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Robert, T., Vanoli, F., Chiolo, I., Shubassi, G., Bernstein, K. A., Rothstein, R., et al. (2011). HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 471, 74–79. doi: 10.1038/nature09803
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rodriguez, Y., and Smerdon, M. J. (2013). The structural location of DNA lesions in nucleosome core particles determines accessibility by base excision repair enzymes. J. Biol. Chem. 288, 13863–13875. doi: 10.1074/jbc.m112.441444
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rossi, G., Conconi, D., Panzeri, E., Redaelli, S., Piccoli, E., Paoletta, L., et al. (2013). Mutations in MAPT gene cause chromosome instability and introduce copy number variations widely in the genome. J. Alzheimers Dis. 33, 969–982. doi: 10.3233/JAD-2012-121633
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rossi, G., Dalprà, L., Crosti, F., Lissoni, S., Sciacca, F. L., Catania, M., et al. (2008). A new function of microtubule-associated protein tau: involvement in chromosome stability. Cell Cycle 7, 1788–1794. doi: 10.4161/cc.7.12.6012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sale, J. E., Lehmann, A. R., and Woodgate, R. (2012). Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 13, 141–152. doi: 10.1038/nrm3289
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saribasak, H., Maul, R. W., Cao, Z., Yang, W. W., Schenten, D., Kracker, S., et al. (2012). DNA polymerase ζ generates tandem mutations in immunoglobulin variable regions. J. Exp. Med. 209, 1075–1081. doi: 10.1084/jem.20112234
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sassa, A., Beard, W. A., Prasad, R., and Wilson, S. H. (2012). DNA sequence context effects on the glycosylase activity of human 8-oxoguanine DNA glycosylase. J. Biol. Chem. 287, 36702–36710. doi: 10.1074/jbc.m112.397786
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schellenberg, G. D., and Montine, T. J. (2012). The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol. 124, 305–323. doi: 10.1007/s00401-012-0996-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schofield, M. J., and Hsieh, P. (2003). DNA mismatch repair: molecular mechanisms and biological function. Annu. Rev. Microbiol. 57, 579–608. doi: 10.1146/annurev.micro.57.030502.090847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sedelnikova, O. A., Horikawa, I., Zimonjic, D. B., Popescu, N. C., Bonner, W. M., and Barrett, J. C. (2004). Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat. Cell Biol. 6, 168–170. doi: 10.1038/ncb1095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Selkoe, D. J. (2011). Alzheimers disease. Cold Spring Harb. Perspect. Biol. 3:a004457. doi: 10.1101/cshperspect.a004457
Shiloh, Y., Shema, E., Moyal, L., and Oren, M. (2011). RNF20-RNF40: a ubiquitin-driven link between gene expression and the DNA damage response. FEBS Lett. 585, 2795–2802. doi: 10.1016/j.febslet.2011.07.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Silva, A. R. T., Santos, A. C. F., Farfel, J. M., Grinberg, L. T., Ferretti, R. E. L., Campos, A. H. J. F. M., et al. (2014). Repair of oxidative DNA damage, cell-cycle regulation and neuronal death may influence the clinical manifestation of Alzheimer’s disease. PloS One 9:e99897. doi: 10.1371/journal.pone.0099897
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Singer, T., McConnell, M. J., Marchetto, M. C. N., Coufal, N. G., and Gage, F. H. (2010). LINE-1 retrotransposons: mediators of somatic variation in neuronal genomes? Trends Neurosci. 33, 345–354. doi: 10.1016/j.tins.2010.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Singleton, A. B. (2014). Genetics. A unified process for neurological disease. Science 343, 497–498. doi: 10.1126/science.1250172
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smith, M. L., and Seo, Y. R. (2002). p53 regulation of DNA excision repair pathways. Mutagenesis 17, 149–156. doi: 10.1093/mutage/17.2.149
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Soria, G., Polo, S. E., and Almouzni, G. (2012). Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol. Cell 46, 722–734. doi: 10.1016/j.molcel.2012.06.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suberbielle, E., Sanchez, P. E., Kravitz, A. V., Wang, X., Ho, K., Eilertson, K., et al. (2013). Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat. Neurosci. 16, 613–621. doi: 10.1038/nn.3356
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sugiyama, T., Saito, M., Nishigori, H., Nagase, S., Yaegashi, N., Sagawa, N., et al. (2014). Comparison of pregnancy outcomes between women with gestational diabetes and overt diabetes first diagnosed in pregnancy: a retrospective multi-institutional study in Japan. Diabetes Res. Clin. Pract. 103, 20–25. doi: 10.1016/j.diabres.2013.10.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sultan, A., Nesslany, F., Violet, M., Bégard, S., Loyens, A., Talahari, S., et al. (2011). Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 286, 4566–4575. doi: 10.1074/jbc.m110.199976
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sykora, P., Yang, J.-L., Ferrarelli, L. K., Tian, J., Tadokoro, T., Kulkarni, A., et al. (2013). Modulation of DNA base excision repair during neuronal differentiation. Neurobiol. Aging 34, 1717–1727. doi: 10.1016/j.neurobiolaging.2012.12.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tang, H. L., Tang, H. M., Mak, K. H., Hu, S., Wang, S. S., Wong, K. M., et al. (2012). Cell survival, DNA damage and oncogenic transformation after a transient and reversible apoptotic response. Mol. Biol. Cell 23, 2240–2252. doi: 10.1091/mbc.e11-11-0926
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ulrich, H. D. (2012). Ubiquitin and SUMO in DNA repair at a glance. J. Cell Sci. 125, 249–254. doi: 10.1242/jcs.091801
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Violet, M., Delattre, L., Tardivel, M., Sultan, A., Chauderlier, A., Caillierez, R., et al. (2014). A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 8:84. doi: 10.3389/fncel.2014.00084
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vogel, G. (2011). Retrotransposons. Do jumping genes spawn diversity? Science 332, 300–301. doi: 10.1126/science.332.6027.300
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, Y., Loomis, P. A., Zinkowski, R. P., and Binder, L. I. (1993). A novel tau transcript in cultured human neuroblastoma cells expressing nuclear tau. J. Cell Biol. 121, 257–267. doi: 10.1083/jcb.121.2.257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wei, W., Ba, Z., Gao, M., Wu, Y., Ma, Y., Amiard, S., et al. (2012). A role for small RNAs in DNA double-strand break repair. Cell 149, 101–112. doi: 10.1016/j.cell.2012.03.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wei, Y., Qu, M.-H., Wang, X.-S., Chen, L., Wang, D.-L., Liu, Y., et al. (2008). Binding to the minor groove of the double-strand, tau protein prevents DNA from damage by peroxidation. PloS One 3:e2600. doi: 10.1371/journal.pone.0002600
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weissbein, U., Benvenisty, N., and Ben-David, U. (2014). Quality control: genome maintenance in pluripotent stem cells. J. Cell Biol. 204, 153–163. doi: 10.1083/jcb.201310135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wolf, F. I., Fasanella, S., Tedesco, B., Cavallini, G., Donati, A., Bergamini, E., et al. (2005). Peripheral lymphocyte 8-OHdG levels correlate with age-associated increase of tissue oxidative DNA damage in Sprague-Dawley rats. Protective effects of caloric restriction. Exp. Gerontol. 40, 181–188. doi: 10.1016/j.exger.2004.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, X., and Sharp, P. A. (2013). Divergent transcription: a driving force for new gene origination? Cell 155, 990–996. doi: 10.1016/j.cell.2013.10.048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wu, M.-F., Yin, J.-H., Hwang, C.-S., Tang, C.-M., and Yang, D.-I. (2014). NAD attenuates oxidative DNA damages induced by amyloid beta-peptide in primary rat cortical neurons. Free Radic. Res. 48, 794–805. doi: 10.3109/10715762.2014.907889
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xiol, J., and Pillai, R. S. (2012). Molecular biology. Outsourcing genome protection. Science 337, 529–530. doi: 10.1126/science.1227095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, K., Cao, F., Sheikh, A. M., Malik, M., Wen, G., Wei, H., et al. (2013). Up-regulation of Ras/Raf/ERK1/2 signaling impairs cultured neuronal cell migration, neurogenesis, synapse formation and dendritic spine development. Brain Struct. Funct. 218, 669–682. doi: 10.1007/s00429-012-0420-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, B., Gaiteri, C., Bodea, L.-G., Wang, Z., McElwee, J., Podtelezhnikov, A. A., et al. (2013). Integrated systems approach identifies genetic nodes and networks in late-onset alzheimer’s disease. Cell 153, 707–720. doi: 10.1016/j.cell.2013.03.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zivković, L., Spremo-Potparević, B., Siedlak, S. L., Perry, G., Plećaš-Solarović, B., Milićević, Z., et al. (2013). DNA damage in Alzheimer disease lymphocytes and its relation to premature centromere division. Neurodegener. Dis. 12, 156–163. doi: 10.1159/000346114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: SNV, somatic mutations, sequence analysis, DNA, brain diseases, Alzheimer disease
Citation: Avila J, Gómez-Ramos A and Soriano E (2014) Variations in brain DNA. Front. Aging Neurosci. 6:323. doi: 10.3389/fnagi.2014.00323
Received: 03 July 2014; Accepted: 06 November 2014;
Published online: 25 November 2014.
Edited by:
Jean Mariani, Universite Pierre et Marie Curie, FranceReviewed by:
Yunfei Huang, Albany Medical College, USANibaldo C. Inestrosa, Pontifical Catholic University of Chile, Chile
Copyright © 2014 Avila, Gómez-Ramos and Soriano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jesús Avila, Centro de Biología Molecular Severo Ochoa (CSIC-UAM), Neurobiology Laboratory, 208, C/ Nicolás Cabrera no. 1, Madrid, 28049, Spain e-mail:amVzdXMuYXZpbGFAY3NpYy5lcw==;
Eduardo Soriano, Department of Cell Biology, Faculty of Biology, University of Barcelona, Developmental Neurobiology and Regeneration Lab, Parc Científic de Barcelona, Baldiri i Reixac, 10, Barcelona 08028, Spain e-mail:ZWR1YXJkb3Nvcmlhbm8udWJAZ21haWwuY29t