 Caroline Haikal
Caroline Haikal Graham M. Winston
Graham M. Winston Michael G. Kaplitt
Michael G. Kaplitt- 1Weill Cornell Medicine, Department of Neurological Surgery, New York, NY, United States
- 2Aligning Science Across Parkinson’s Collaborative Research Network, Chevy Chase, MD, United States
Cognitive impairments are a common feature of synucleinopathies such as Parkinson’s Disease Dementia and Dementia with Lewy Bodies. These pathologies are characterized by accumulation of Lewy bodies and Lewy neurites as well as neuronal cell death. Alpha-synuclein is the main proteinaceous component of Lewy bodies and Lewy neurites. To model these pathologies in vivo, toxins that selectively target certain neuronal populations or different means of inducing alpha-synuclein aggregation can be used. Alpha-synuclein accumulation can be induced by genetic manipulation, viral vector overexpression or the use of preformed fibrils of alpha-synuclein. In this review, we summarize the cognitive impairments associated with different models of synucleinopathies and relevance to observations in human diseases.
Introduction
Parkinson’s Disease Dementia (PDD) and Dementia with Lewy Bodies (DLB) are two of the most common pathologic causes of dementia. DLB is the second most common pathologic cause of dementia, affecting up to 5% of the general population and up to 30% of dementia cases (Zaccai et al., 2005; Mueller et al., 2017). PDD is highly prevalent among individuals with Parkinson’s disease (PD), with estimates suggesting that up to 75% of PD patients surviving 10 years and 83% of those who survive 20 years develop PDD (Aarsland et al., 2003; Hely et al., 2008; Aarsland and Kurz, 2010). The prevalence of dementia in PD patients is thought to be 3–6 times higher than the general population (Aarsland et al., 2003; Emre et al., 2007).
These disorders share many clinical, anatomic, and neuropathologic characteristics (Gomperts, 2016; Koga et al., 2021). Patients with each disorder suffer from executive dysfunction, cognitive impairment, hallucinations, and parkinsonism. Both diseases are defined neuropathologically by Lewy bodies (LB), cytoplasmic neuronal inclusions containing α-synuclein (α-syn), in the limbic and cortical regions of the brain. The similarities between the diseases have led some to suggest that they may exist on a common pathophysiologic spectrum.
Modeling the diseases and validating animal models for human Lewy Body Dementia pose a challenge for two main reasons. First, establishing a model of PD or DLB that recapitulates the full spectrum of the disease remains elusive. As will be described in detail below, there is often great heterogeneity in both pathological findings and nervous system regions affected among individual patients with either disorder, while pathogenesis can also be multifactorial. Second, testing cognition in animals with the inability to perform the complex functions that are impaired in humans as well as a lacking of anatomical complexity necessitates rigorous development of animal behavioral paradigms. In this review, we will first describe the current theories for cognitive decline in human Lewy-Body Dementia. We will then explore progress to date in recreating pathology in animals to model cognitive phenotypes seen in humans.
Part I: understanding cognitive decline in human lewy-body dementia
Clinical phenotypes
While there is heterogeneity in the cognitive profile of PDD patients, a typical pattern of executive dysfunction, deficits in concentration and attention, memory impairment, and flawed visuo-spatial abilities exists (Emre et al., 2007; Kalbe et al., 2008; Irwin et al., 2013). Specifically, executive function impairment leads to worsened task planning, task initiation, pattern recognition, and concept and rule forming abilities (Litvan et al., 1991; Paolo et al., 1995; Emre et al., 2007; Irwin et al., 2013). Attention and concentration deficits manifest as impaired spontaneous and focused attention, difficulty maintaining concentration, and characteristically fluctuates throughout the day (Ballard et al., 2002; Noe et al., 2004; Kemps et al., 2005; Emre et al., 2007; Kalbe et al., 2008). Poor executive function is thought to contribute to a predominantly recall-related memory impairment in PDD, (Kemps et al., 2005, Kalbe et al., 2008, Godefroy et al., 2010) though memory encoding and storage are affected as well (Higginson et al., 2005; Emre et al., 2007). Considerable evidence reveals that visuo-spatial and perceptive abilities are also impaired in PDD patients (Hovestadt et al., 1987; Bradley et al., 1989; Waterfall and Crowe, 1995; Mosimann et al., 2004; Irwin et al., 2013). The variability in clinical presentation among PDD patients may be reflective of the heterogeneity in neuropathologic patterns in PD (Hurtig et al., 2000; Jellinger and Korczyn, 2018).
Patients with DLB share many clinical characteristics with PDD patients, though there are a few notable differences. The most apparent difference between the two is the timing of cognitive decline, as DLB patients experience cognitive decline at the time of diagnosis or shortly after motor symptom development (usually within 1 year), while PDD patients usually have motor symptoms for a substantial period before cognitive decline onset (McKeith and Burn, 2000; McKeith et al., 2005; Emre et al., 2007). Subtle differences in the cognitive profiles of the patients also exist, as DLB patients have been shown to perform worse than PDD patients in attention tasks (measured by verbal memory and delayed recall) and executive function tasks (measured by the cognitive interference “Stroop” test) (Park et al., 2011). The overall rate of cognitive decline is thought to be faster in DLB than either PDD or Alzheimer’s Disease (AD) (Rongve et al., 2016; Kramberger et al., 2017). Furthermore, hallucinations are more frequent in DLB and can occur any time after disease onset, while PDD patients typically develop hallucinations after levodopa replacement therapy (Jellinger and Korczyn, 2018). Rest tremor is less frequent and balance is less affected in DLB than in PDD, though other motor manifestations, such as bradykinesia and rigidity, are similar between entities (Fritz et al., 2016; Jellinger and Korczyn, 2018).
Neuropathological phenotypes
α synuclein patterns
While neurodegeneration of the nigrostriatal basal ganglia circuitry may be responsible for the motor symptoms of PD, the neuropathologic underpinnings of cognitive decline in DLB and PDD remain less clear, as studies have highlighted the importance of cortical LB and Lewy-neurite pathology, concurrent Alzheimer’s disease-like pathology, and involvement in the brain’s cholinergic system in the development of cognitive decline (Braak et al., 2003; Compta et al., 2011; Irwin et al., 2013). Parkinson’s disease is traditionally characterized by neurodegeneration and accumulation of α-syn1 in the substantia nigra, and α-syn pathology can be observed throughout the brain (Braak et al., 2003; Bohnen and Albin, 2011; Irwin et al., 2013). Post-mortem neuropathologic studies have revealed the importance of cortical LBs in development of PDD, (Hurtig et al., 2000, Mattila et al., 2000, Harding and Halliday, 2001, Kövari et al., 2003) with specific involvement of the entorhinal cortex, temporal neocortex, anterior cingulate gyrus, and the pre-frontal cortices (Irwin et al., 2012, 2013). Cortical LB burden has also been shown to predict dementia onset and severity in DLB patients (Ruffmann et al., 2016; Ferman et al., 2018). Studies of α-syn aggregates in animal models have long raised the increasingly accepted concept of a prion-like ability for aggregates to spread throughout the nervous system and even from the periphery, although mechanisms of spread are an active area of research (Luk et al., 2012; Masuda-Suzukake et al., 2013; Kovacs et al., 2017; Henderson et al., 2019). Meticulous pathologic analysis of the brains of PD patients, conducted by Braak and others, have led to theories that PD pathogenesis may follow a caudal-to-rostral pattern of spread in at least some patients (Braak et al., 2003). In these patients, pathology was found to start in the brainstem, spread to limbic, sub-cortical structures, before eventually propagating to the cerebral cortex. In fact, the original description of this abnormality by Lewy noted considerable pathology in the basal forebrain, where cholinergic neurons project to regions responsible for many cognitive functions (Lewy, 1913; Bohnen and Albin, 2011) (Box 1). A challenge for modeling these forms of human disease is thus to recreate where possible this concept of pathology propagation in a manner that is consistent with human observations.
| BOX 1 Definition of terms. |
| Dopaminergic Neurons: Neurons that produce dopamine as their primary neurotransmitter, characterized by expression of tyrosine hydroxylase (TH), dopamine transporter (DAT), GIRK2 (a G-Protein coupled receptor), and others. These neurons are found in various regions of the brain, including the substantia nigra and the ventral tegmental area. |
| Cholinergic Neurons: Neurons that release acetylcholine (ACh) as their primary neurotransmitter, characterized by expression of choline acetyl-transferase (ChAT), acetylcholine esterase (AChE), and others. While they are found throughout the brain, they are typical of basal forebrain structures. |
| Cortical Neurons: Neurons located in the cerebral cortex, the outer layer of the brain responsible for higher cognitive functions. Cortical neurons are morphologically diverse and classified into various types based on their structure, function, and connectivity within the cortical circuitry. |
Alzheimer’s disease co-pathology
The presence of concurrent AD-like pathology, extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tau-tangles, are predictive of PDD (Kalaitzakis et al., 2008; Compta et al., 2011) and LBD (Jellinger and Attems, 2008; Gomperts, 2016). In fact, it is estimated that between one-quarter and one-half of PDD patients have enough AD pathology to meet post-mortem pathologic criteria for diagnosis (Mattila et al., 1998; Galvin et al., 2006; Sabbagh et al., 2009). Some have demonstrated that time onset to dementia in parkinsonian patients is correlated with cortical deposition of both α-syn and Aβ (Ballard et al., 2006). Both α-syn and AD-pathology are likely involved in development of the Lewy-Body Dementia, possibly in an additive or synergistic manner (Irwin et al., 2013; Ferman et al., 2018).
Cholinergic degeneration
In addition to these pathologic hallmarks of neurodegeneration in DLB and PDD, impairment of the brain’s cholinergic systems may play a significant role in development of the diseases. Specifically, cholinergic neurons of the basal forebrain (BF), which have projections to the hippocampus, olfactory bulb, cortical mantle, and amygdala, (Bohnen and Albin, 2011; Pasquini et al., 2021) are implicated in PDD and DLB. Neurodegeneration of BF cholinergic neurons (Arendt et al., 1983; Candy et al., 1983; Nakano and Hirano, 1984; Braak et al., 2003) and their targets in the hippocampus (Perry et al., 1994; Tiraboschi et al., 2002; Hall et al., 2014) have been associated with PDD and DLB. This has been supported by positron-emission tomography (PET) studies revealing decrease in cortical cholinergic activity, (Asahina et al., 1995; Bohnen et al., 2003; Hilker et al., 2005; Shimada et al., 2009; Kanel et al., 2020) and the fact that acetylcholinesterase inhibitors, which prevent acetylcholine degradation, have been shown to improve cognitive outcomes in PDD and DLB (McKeith et al., 2000; Reading et al., 2001; Bergman and Lerner, 2002; Fabbrini et al., 2002; Giladi et al., 2003; Beversdorf et al., 2004; Dujardin et al., 2006; van Laar et al., 2011; Dubois et al., 2012; Mori et al., 2012; Wang et al., 2015). While the exact relationship between cholinergic neurodegeneration and the other neuropathologic findings of Lewy Body Dementia is unknown, there is likely a complex interplay of factors resulting in the ultimate clinical phenotype.
PDD-DLB pathologic discrepancies
While there is certainly overlap in the pathology seen in PDD and DLB, subtle differences may explain the disparities in cognitive and overall clinical presentation. There is less nigral neurodegeneration in DLB patients compared to PDD patients, which may explain the motor differences seen between the disorders (Tsuboi and Dickson, 2005). Compared with PDD patients, DLB patients have higher α-syn burden in the hippocampus and amygdala (Kalaitzakis et al., 2009; Jellinger and Korczyn, 2018). Additionally, DLB patients have an increase in cortical and subcortical Aβ when compared to PDD patients, specifically with increased Aβ load in the entorhinal cortex, amygdala, and putamen (Hepp et al., 2016). DLB patients also have higher amounts of cortical atrophy compared to PDD patients, most notably in the temporal, parietal, and occipital lobes (Beyer et al., 2007). Collectively, these may account for the cognitive differences seen between patients with PDD and DLB. These findings also emphasize the challenges faced when attempting to model cognitive dysfunction from synucleiopathies in animal models, since not only must the specific mechanism of generating α-syn pathology be considered but also co-pathologies can be critical to the models and may differ in consequences depending upon both levels of expression and location of expression within specific brain regions.
Part II: toward animal models of synucleinopathies
Cognitive testing in rodents
Any attempt to model cognitive decline first requires thorough evaluation and validation of behavioral paradigms. The most commonly used tests for gauging cognitive ability in rodents include the Y-maze, the novel object recognition (NOR) test, the Morris Water Maze (MWM), and fear conditioning (Taylor et al., 2010; Tanila, 2018). In the Y-maze, animals are allowed to freely explore a three-armed maze. An unimpaired animal’s inclination to explore novel areas results in a tendency for alternation between the maze’s arms, while an animal with short-term memory impairment would explore the arms of the maze in an uneven manner. In the NOR test, an animal’s tendency to explore unfamiliar objects is again evaluated. The time spent examining a novel object compared to a familiar one is measured over 2 days. Therefore the NOR test can be used as a measure of both short- and long-term memory. In the MWM, animals are trained to find a hidden platform in a pool of water using visual cues. With extended training, a cognitively unimpaired animal would display a decrease in the latency to find the platform and an increase in the time spent around the platform. The MWM is accepted as a measure of spatial-visual memory.2 In fear conditioning, animals are conditioned to associate a neutral stimulus with an aversive stimulus. The development, persistence in a new context and unlearning of the conditioned fear response can all be measured. The facets of cognition that these tests measure and their clinical counterparts are listed in Table 1.
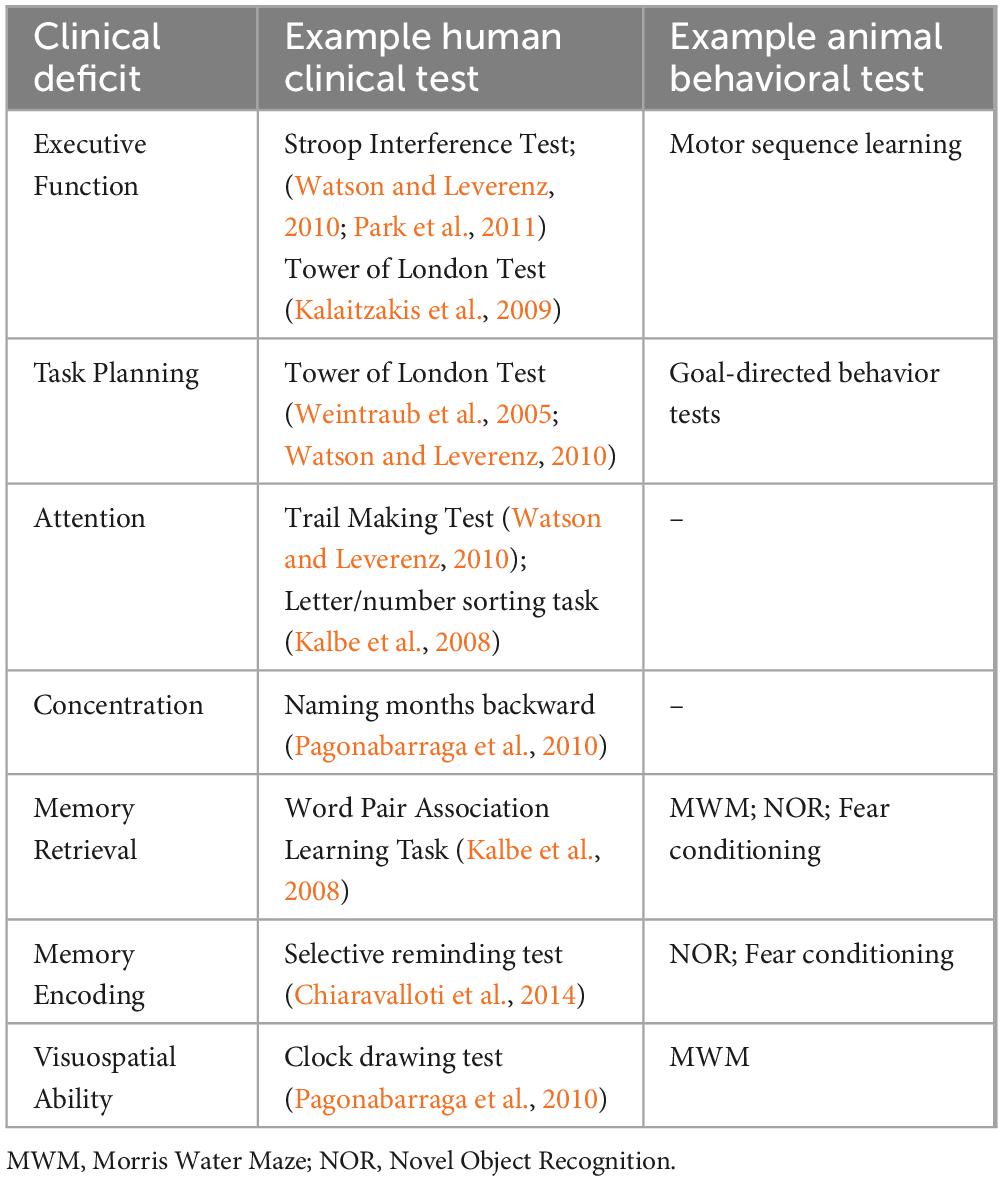
Table 1. Representative human clinical and animal behavioral tests for various cognitive impairments in Lewy Body Dementia.
Besides the technical challenge in performing these tests, the tests themselves may not be specific to cognitive ability. For example, the Y-maze, MWM, and NOR can be skewed by visual or olfactory dysfunction. Additionally, the MWM and fear conditioning are heavily influenced by an animal’s sensitivity to aversive stimuli and anxiety, which can be heavily affected by pathology in the limbic system, a region known to be affected in synucleinopathies. Furthermore, these tests rely on movement of animals in a behavioral context due to the inability to communicate with rodents, so any motor deficits could confound interpretation of cognitive dysfunction in these models. Finally, many cognitive tests can only be performed once and so choosing a time where the cognitive impairment will be detectable is critical. The order in which cognitive tests are performed also matters, as stress-inducing tests can confound subsequent performance (Rodriguiz and Wetsel, 2006). As such, it may be difficult to assess the order of symptom development as compared to humans.
Modeling aspects of lewy body dementia in rodents
When it comes to modeling synucleinopathy in rodents, the two aspects of the disease primarily focused on are the loss of dopaminergic innervation in the midbrain and the progressive accumulation of both α-syn and pSyn. To replicate these fundamental pathologic features, researchers have undertaken a multitude of innovative approaches.
Toxin models
To date, the most efficient way of achieving dopaminergic denervation is through the use of the catecholaminergic selective toxins MPTP and 6-hydroxydopamine (6-OHDA). When administered systemically (MPTP) or intracerebrally (6-OHDA), these toxins cause an accumulation of oxidative species resulting in death of dopaminergic neurons. While not involving Lewy pathology specifically, due to their selective targeting of the dopaminergic pathway, these models capture the motor impairments associated with midbrain degeneration well. Toxin-treated mice fail to remain on the rotarod as long as their sham treated littermates and display amphetamine desensitization. When 6-OHDA is injected unilaterally, mice display rotation behaviors as an effect of the asymmetric burden of neurodegeneration. These toxin models are also useful in understanding the contribution of the nigro-striatal dopaminergic pathway to cognition. While both models have been shown to induce deficits in memory and attention, there is some evidence that MPTP is better at impairing short-term memory, and that 6-OHDA primarily affects long-term memory (Fan et al., 2021). One drawback to these models, however, is that the cognitive impairment induced by the toxins is not stable long-term, with animals showing recovery a few months post injection (p.i.) (Bezard et al., 2000; Fan et al., 2021). This could indicate the measured cognitive decline is related to the brain’s inflammatory response rather than the neurodegeneration itself. In a rotenone study, depleting or inactivating microglia (with PLX397 or minocycline, respectively) attenuated the cognitive impairments as measured by the MWM, NOR, and passive avoidance tests. The microglial inhibition however, also attenuated the pSyn pathology and neuronal loss, making it difficult to ascertain whether the neuroinflammation itself caused the impairment (Zhang D. et al., 2021). In contrast, inducing neuroinflammation in wildtype mice by systemic administration of lipopoylysaccharide (LPS), activates microglia and causes hippocampal neuronal loss as well as sickness behavior and impairments in the MWM and passive avoidance tests (Zhao et al., 2019).
Viral vector models
Viral vector-mediated overexpression of SNCA, the gene coding for α-syn, has been shown to result in a local, progressive accumulation of the protein and severe neurotoxicity around the area of injection (Huntington and Srinivasan, 2021). The use of viral vectors to stably overexpress the protein allows for control of the types of cells affected and the degree of pathology by modulating the injection site, serotype, volume and titre of virus delivered as well as promoter and α-syn form (human vs mouse; full-length vs truncated; wildtype vs mutated) expressed. Even though these models have been reported to display cognitive and motor impairments, (Fan et al., 2021) the focal overexpression, the acute neurotoxicity, and the ensuing inflammatory response are not thought to be representative of the neuropathology of synucleinopathies (Volpicelli-Daley et al., 2016). These models have therefore fallen out of favor in recent years. Nonetheless, if the level of α-syn expression influences susceptibility to pathology or co-pathologies, the viral model still can be attractive if dosing, promoter strength or regulation and cell-type specificity can be adjusted to specific applications.
Transgenic models
In contrast, transgenic mouse models overexpressing some form of SNCA, often display a time-dependent accumulation of α-syn aggregates which more closely mimic the disease. These models rarely achieve levels of neurodegeneration comparable to the toxin models, but they do reflect a genetic form of human disease, characterized by triplication or mutations of the SNCA gene leading to overexpression and eventual pathogenesis. Depending on the promoter and SNCA variant used, the spatial and neuropathologic signature of the accumulation can be manipulated (Giasson et al., 2002; Freichel et al., 2007; Zhou et al., 2008). The M83 line, which overexpresses the human familial PD-associated A53T form of SNCA under the control of the prion promoter, is one of the most popular transgenic models of synucleinopathy (Giasson et al., 2002). When bred homozygous, these mice develop severe motor phenotypes such as trembling, limb paralysis and inability to stand. Prior to motor symptom onset, which is typically described at 7–8 months of age, these mice display diminished nesting behavior and impaired spatial memory as tested by the Y-maze (Paumier et al., 2013). These mice, however, accumulate α-syn in CNS regions not typically associated with human synucleinopathy, such as the cerebellum and the spinal cord, which may be explained by the use of the prion promoter.
Another well characterized transgenic mouse line, line 61, which overexpresses human full-length wild-type SNCA under the control of the Thy-1 promoter, also displays progressive accumulation of α-syn, with the highest fold increase described in the thalamus when compared to wildtype littermates (Chesselet et al., 2012). These mice display reduced motility in the open-field test at 14-months of age, around which time a 40% decrease in striatal TH levels are observed. Prior to motor symptom onset, these mice display mild cognitive impairments as measured by the Y-maze and NOR tests. Similar to many other transgenic mouse models, these mice do not display TH cell loss in the nigra, even at 22-months of age. Similarly, other Thy-1-asyn mice also display cognitive impairments in the NOR (Subramaniam et al., 2018).
Another approach, in an effort to more closely mimic accumulation of α-syn in relevant brain regions, is to overexpress the human form of the protein under the control of the mouse’s endogenous SNCA promoter, on either a wildtype (Hansen et al., 2013) or mouse SNCA knockout background (Janezic et al., 2013). These mice also display a progressive accumulation of α-syn, very mild neurodegeneration, and progressive motor and cognitive phenotypes starting at around 9-months of age. Besides SNCA, other genes have been implicated in PD and LBD in GWAS studies. Among the most commonly reported are PINK1 and LRRK2. PINK1 KO mice have been developed and though they show impaired synaptic plasticity, (Kitada et al., 2007) cognitive impairment hasn’t been as widely reported (Gispert et al., 2009). LRRK2G2019S mice on the other hand, have been reported to show impairments in executive function in the touchscreen operant task tests (Cheng et al., 2022; Hussein et al., 2022). A comprehensive review of transgenic models used and the cognitive impairments reported can be found elsewhere (Fan et al., 2021).
Preformed fibril models
A model that has been gaining popularity in recent years is the preformed fibril (PFF) model, in which preformed fibrils of human or mouse α-syn are injected into mice (Chung et al., 2019). Alpha-synuclein can be recombinantly expressed and fibrillization induced, commonly by shaking at 37°C. The resulting α-syn fibrils closely resemble those found in Lewy bodies when examined by electron microscopy (Cremades et al., 2012; Marotta et al., 2021). When added to cells or injected into animals, PFFs have been shown to rapidly induce the templating of endogenous α-syn, resulting in a spatio-temporally defined spread of α-syn aggregates (Polinski et al., 2018). PFFs also cause neurotoxicity, with the severity of the toxicity determined by the α-syn PFF ‘strain’ (Bousset et al., 2013) and cellular subtype, (Alegre-Abarrategui et al., 2019) with nigral dopaminergic neurons showing particular vulnerability. One main advantage of this model is that the targeted pathways and the signature of α-syn pathology can be altered with the injection site. As such, the contributions of different pathways to behavioral deficits can be dissected. Summaries of the cognitive impairments induced with PFF injections are listed in Figure 1 and Table 2.
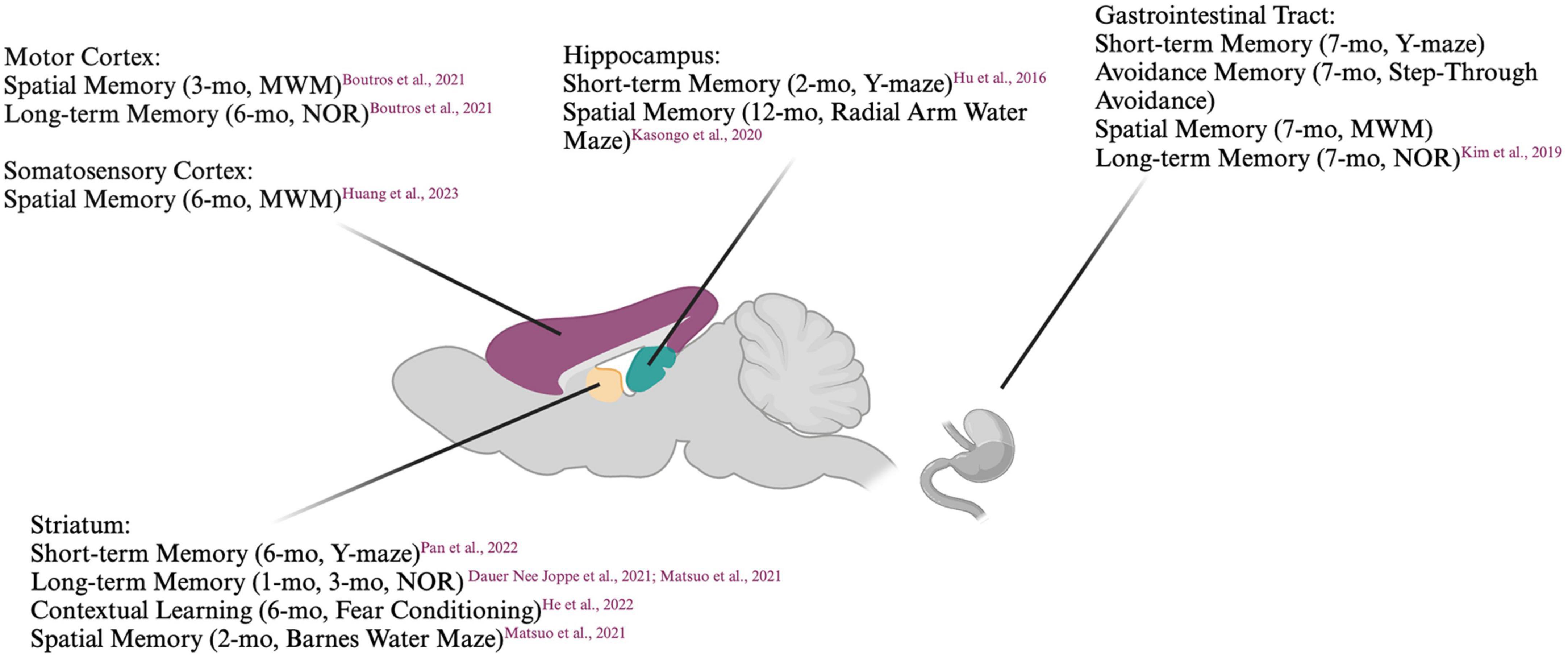
Figure 1. Summary of cognitive deficits elicited based on location of preformed fibril injection. MWM, Morris Water Maze; NOR, Novel Object Recognition. Created with BioRender.com.

Table 2. Summary of conditions and cognitive impairments in rodent preformed fibril models.
Striatum
Mice injected unilaterally in the striatum have been reported to display impaired short-term memory as measured by the Y-maze (Pan et al., 2022) and impaired long-term memory in the NOR test, (Dauer Née Joppe et al., 2021) albeit several months p.i.. When injected bilaterally into the striatum, mouse PFFs have been shown to induce accumulation of pSyn not only in the striato-nigral pathway but also in connected areas such as the prefrontal cortex and amygdala. Six months p.i., these mice have been reported to display diminished social dominance and impairments in fear conditioning in some (Stoyka et al., 2020) but not all studies (Stoyka et al., 2021) despite a loss of tyrosine hydroxylase (TH) immunoreactivity. The coordinates targeted within the striatum may modulate the development of pathology, as mice injected in the dorsolateral but not dorsomedial striatum showed impaired learning even though both groups of mice displayed impaired goal-directed behavior. When the substantia nigra was targeted, impairments in sequence learning were already apparent at 1-month p.i. (He et al., 2022). The NOR test and the Barnes maze test (another spatial memory test) have been shown to detect impairments in mice injected in the dorsal striatum as early as 60 days p.i., at which point the Y-maze did not. This cognitive impairment was attributed to a decrease in septal GABAergic neurons (Matsuo et al., 2021). This is surprising, as a previous study suggested that the septo-hippocampal pathway was not susceptible to pSyn accumulation post PFF injection into the hippocampus (Luk et al., 2012). Interestingly, two other studies have demonstrated a particular resistance of the (cholinergic) medial septal, but not (GABAergic and glutamatergic) lateral septal neurons to α-syn accumulation when PFFs were injected into the hippocampus (Nouraei et al., 2018; Dues et al., 2023).
Hippocampus
Mice with PFFs injected bilaterally in the hippocampus displayed pSyn accumulation near the site of injection, in the dentate gyrus. Two months p.i., a reduction in Y-maze alteration was described, which was attributed to aberrant adenosine A2A receptor signaling, Hu et al. (2016) a pathway involved in both glutamatergic and dopaminergic regulation (He et al., 2022). Similarly, rats with bilateral hippocampal PFF injections displayed accumulation of pSyn in the injected and connected areas with concomitant cognitive impairment detected by the two radial arm water maze (a combination of the Y-maze and MWM), but not with the MWM (Espa et al., 2019; Kasongo et al., 2020).
Cortex
Cortical structures have also been targeted with PFFs. Bilateral injection of PFFs into the motor cortex induced impairment of spatial memory as measured by MWM at 3-months p.i. After 6-months, these same mice displayed impairment in memory as measured by NOR (Boutros et al., 2021). While no habituation was observed in the open field test, this may be explained by hyperactivity, which has previously been reported in PD-mouse models (Freichel et al., 2007). When PFFs were injected bilaterally into the medial prefrontal cortex 6-months after AAV-induced overexpression of human SNCA in the medial prefrontal cortex, ventral tegmental area, and basal forebrain, pSyn accumulation was induced not only in the PFF-injected areas, but also in the striatum, substantia nigra, and entorhinal cortex. Six months after the PFF injections, however, the mice displayed no differences in interval timing compared to controls. Although it seemed the PFF injected mice displayed a slight decrease in mobility and discrimination in the open field and NOR tests, respectively, these differences did not reach statistical significance (Zhang Q. et al., 2021). Six months after human PFFs were unilaterally injected into the right somatosensory cortex and dorsal striatum, mice displayed cognitive impairments as measured by the MWM (Huang et al., 2023). In contrast, pSyn in the hippocampus and frontal cortex induced by a unilateral injection of PFFs in the medial forebrain bundle in rats did not result in a measurable cognitive impairment in the MWM up to 120 days p.i. (Pang et al., 2022).
Peripheral injections
In an attempt to model a “body first” synucleinopathy, mouse PFFs injected in the gut have been shown to produce a progressive, caudal-rostral accumulation of pSyn in the brainstem, cortex, hippocampus and striatum (Kim et al., 2019). This was accompanied by cognitive impairments as measured by the MWM, Y-maze, NOR, and step-through passive-avoidance test 7-months p.i.
Alpha-synuclein co-pathologies
Considering the prevalence of co-pathology in patients, another strategy for PFF-induction of synucleinopathy is the use of α-syn PFFs in Aβ, tau, or APOE mouse models. Injecting A53T PFFs unilaterally into the right frontal cortex has been shown to improve the spatial memory deficits observed in APP/PS1 mice with Aβ accumulation. Surprisingly, in that same study, wildtype mice injected with the PFFs also showed a statistically significant improvement in the MWM compared to PBS-injected controls (Hao et al., 2018). In contrast, in a study using a different model of Aβ pathology, 5xFAD mice injected unilaterally in the hippocampus with mouse α-syn PFFs displayed cognitive impairment, as measured by the Y-maze, starting at 3-months p.i. and continued to worsen over time. These mice showed a progressive increase in pSyn accumulation compared to wildtype mice injected with the PFFs. While the pSyn was primarily contained to the hippocampus, hypothalamus, and cortex at 9-months p.i. in wildtype mice, the pSyn load was already ubiquitous throughout the brain at 4.5-months p.i. in the 5xFAD mice. The PFF injected 5xFAD mice also displayed an increase Aβ and hyperphosphorylated tau burden compared to PBS injected 5xFAD mice (Bassil et al., 2020). Even though tau has been shown to co-localize with α-syn at presynaptic terminals, there have been conflicting reports on the neuroprotective effects of knocking out tau in the PFF model (Stoyka et al., 2021; Pan et al., 2022) with one study suggesting a unidirectional relationship with α-syn as a modulator of tau pathology (Bassil et al., 2020). In the A53T transgenic model, knocking out tau not only reduced neuronal loss but attenuated the memory deficits associated with the model (Singh et al., 2019). Both phosphorylated tau and pSyn have been shown to be upregulated in the MPTP model of neurodegeneration (Hu et al., 2020). Finally, two separate studies have demonstrated a modulatory effect of APOE genotype on α-syn pathology. While APOE2 showed a neuroprotective effect compared to APOE3 and even APOE KO, APOE4 exacerbated α-syn pathology burden, both in the A53T mouse (Davis et al., 2020) and in the AAV-induced overexpression model (Zhao et al., 2020). The α-syn APOE4 mice also demonstrated a faster cognitive decline compared to the other APOE genotypes.
Confounding factors and potential explanations of discrepancies
The discrepancies in results between studies may be attributed to one of several factors. The strain of PFFs used, which is dictated by the aggregating conditions and specific α-syn variant (mouse vs human, wildtype vs familial-associated point-mutations (A53T, A30P, etc.), may influence outcome. Although many groups use the same behavioral tests, the protocols for these tests (for instance, whether they are performed during the light or dark cycle) and therefore also the readouts vary between groups. The number and sex of animals used also affects whether any potential impairment reaches statistical significance, which in turn affects how differences in behavior are reported. Additionally, the timepoint at which the cognitive behaviors are evaluated can influence what is observed. If a late timepoint is used, many areas of the brain may be burdened with pSyn and as such conclusions about the studied pathway can be confounded. If an early timepoint is used, however, the pSyn pathology may be restricted to the area of injection and closely connected pathways, but the acute inflammation accompanying the injections may confound any observed cognitive impairment. While cholinergic pathology is thought to underlie cognitive impairments in humans, many mouse models have failed to recapitulate cholinergic degeneration in mice. This could be due to the key differences in cholinergic neurons in terms of distribution, functionality and structural roles in rodents as compared to primates (Disney and Robert, 2019). Acute systemic inflammation is known to cause reversible cognitive impairments (Skelly et al., 2019) but inflammation has also been suggested as a key driver in α-syn pathology (Kim et al., 2022).
Conclusion
In this review, we have explored the clinical manifestations and underlying pathologic mechanisms of Lewy Body Dementia, encompassing both Parkinson’s Disease Dementia and Dementia with Lewy Bodies. Additionally, we have assessed the different strategies by which researchers have attempted to emulate the cognitive deficits of these diseases in animal models. The cognitive decline in Lewy Body Dementia emerges from a complex interplay of α-syn propagation, concomitant AD-like Aβ and tau pathology, and loss of neurotransmitter tone, which may work in an additive or synergistic manner. While these pathologic hallmarks are diffuse by the time cognitive decline becomes evident, careful investigations highlight pivotal regions that drive the progression to dementia, including the pre-frontal cortex, temporal neo-cortex, hippocampus, and cholinergic neurons of the basal forebrain.
While the discrepancies in reported pathology in the different animal models pose challenges to researchers, it is important to remember that the symptomatology of PDD and LBD patients can also vary widely. An enhanced understanding of the pathophysiology of these diseases as well as the now large repertoire of animal models available to researchers should aide in developing breakthrough treatments if used correctly. Improved testing will also be key in elucidating relevant cognitive impairments and potential therapies for them. Tests for higher executive cognitive function, such as attentional set shifting tasks and odor span tasks, have not been routinely performed but would be interesting in light of the specific executive dysfuntion seen in patients.
As of now, there is no ultimate animal model—the choice to be made depends on the question to be answered with each model having a unique profile. The tools to study these challenging disorders continue to evolve, and the emergence of multiple hit models offer the promise to better replicate PDD and LBD pathology in a near-physiologic manner.
Author contributions
GW: Conceptualization, Data curation, Formal analysis, Methodology, Project administration, Writing – original draft, Writing – review and editing. CH: Conceptualization, Data curation, Formal analysis, Methodology, Project administration, Writing – original draft, Writing – review and editing. MK: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The study was funded by the joint efforts of The Michael J. Fox Foundation for Parkinson’s Research (MJFF) and the Aligning Science Across Parkinson’s (ASAP) initiative. MJFF administers the grant ASAP-020608 on behalf of ASAP and itself. MK was also supported by funding from the JPB Foundation for Medical Research.
Acknowledgments
IP rights notice: For the purpose of open access, the author has applied a CC-BY public copyright license to the author Accepted Manuscript (AAM) version arising from this submission. Figures in text were generated using BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
- ^ Recent mass spectroscopy-based studies have shown that modified α-syn with phosphorylation of serine residue 129 (pSyn) is the dominant version of the protein within LBs and may in fact be responsible for aggregation and pathogenesis in PDD and LBD.
- ^ The Barnes maze test is a similar test of spatial memory and navigation, though involves a circular platform with several holes, one of which leads to escape from an elevated platform.
References
Aarsland, D., and Kurz, M. (2010). The epidemiology of dementia associated with Parkinson disease. J. Neurol. Sci. 289, 18–22. doi: 10.1016/j.jns.2009.08.034
Aarsland, D., Andersen, K., Larsen, J., Lolk, A., and Kragh-Sørensen, P. (2003). Prevalence and characteristics of dementia in Parkinson disease: An 8-year prospective study. Arch. Neurol. 60, 387–392. doi: 10.1001/archneur.60.3.387
Alegre-Abarrategui, J., Brimblecombe, K., Roberts, R., Velentza-Almpani, E., Tilley, B., Bengoa-Vergniory, N., et al. (2019). Selective vulnerability in α-synucleinopathies. Acta Neuropathol. 138, 681–704. doi: 10.1007/s00401-019-02010-2
Anderson, J., Walker, D., Goldstein, J., de Laat, R., Banducci, K., Caccavello, R., et al. (2006). Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 281, 29739–29752. doi: 10.1074/jbc.M600933200
Arendt, T., Bigl, V., Arendt, A., and Tennstedt, A. (1983). Loss of neurons in the nucleus basalis of Meynert in Alzheimer’s disease, paralysis agitans and Korsakoff’s Disease. Acta Neuropathol. 61, 101–108. doi: 10.1007/BF00697388
Asahina, M., Shinotoh, H., Hirayama, K., Suhara, T., Shishido, F., Inoue, O., et al. (1995). Hypersensitivity of cortical muscarinic receptors in Parkinson’s disease demonstrated by PET. Acta Neurol. Scand. 91, 437–443. doi: 10.1111/j.1600-0404.1995.tb00443.x
Ballard, C., Aarsland, D., McKeith, I., O’Brien, J., Gray, A., Cormack, F., et al. (2002). Fluctuations in attention: PD dementia vs DLB with parkinsonism. Neurology 59, 1714–1720. doi: 10.1212/01.wnl.0000036908.39696.fd
Ballard, C., Ziabreva, I., Perry, R., Larsen, J., O’Brien, J., McKeith, I., et al. (2006). Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology 67, 1931–1934. doi: 10.1212/01.wnl.0000249130.63615.cc
Bassil, F., Brown, H., Pattabhiraman, S., Iwasyk, J., Maghames, C., Meymand, E., et al. (2020). Amyloid-Beta (Aβ) Plaques Promote Seeding and Spreading of Alpha-Synuclein and Tau in a Mouse Model of Lewy Body Disorders with Aβ Pathology. Neuron 105:260–275.e6. doi: 10.1016/j.neuron.2019.10.010
Bergman, J., and Lerner, V. (2002). Successful use of donepezil for the treatment of psychotic symptoms in patients with Parkinson’s disease. Clin. Neuropharmacol. 25, 107–110. doi: 10.1097/00002826-200203000-00009
Beversdorf, D., Warner, J., Davis, R., Sharma, U., Nagaraja, H., and Scharre, D. (2004). Donepezil in the treatment of dementia with Lewy bodies. Am. J. Geriatr. Psychiatry 12, 542–544. doi: 10.1176/appi.ajgp.12.5.542
Beyer, M., Larsen, J., and Aarsland, D. (2007). Gray matter atrophy in Parkinson disease with dementia and dementia with Lewy bodies. Neurology 69, 747–754. doi: 10.1212/01.wnl.0000269666.62598.1c
Bezard, E., Dovero, S., Imbert, C., Boraud, T., and Gross, C. (2000). Spontaneous long-term compensatory dopaminergic sprouting in MPTP-treated mice. Synapse 38, 363–368. doi: 10.1002/1098-2396(20001201)38:3<363::AID-SYN16>3.0.CO;2-A
Bohnen, N., and Albin, R. (2011). The cholinergic system and Parkinson disease. Behav. Brain Res. 221, 564–573. doi: 10.1016/j.bbr.2009.12.048
Bohnen, N., Kaufer, D., Ivanco, L., Lopresti, B., Koeppe, R., Davis, J., et al. (2003). Cortical cholinergic function is more severely affected in Parkinsonian dementia than in Alzheimer disease: An in vivo positron emission tomographic study. Arch. Neurol. 60, 1745–1748. doi: 10.1001/archneur.60.12.1745
Bousset, L., Pieri, L., Ruiz-Arlandis, G., Gath, J., Jensen, P., Habenstein, B., et al. (2013). Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 4:2575. doi: 10.1038/ncomms3575
Boutros, S., Raber, J., and Unni, V. (2021). Effects of alpha-synuclein targeted antisense oligonucleotides on Lewy body-like pathology and behavioral disturbances induced by injections of pre-formed fibrils in the mouse motor cortex. J. Parkinsons Dis. 11, 1091–1115. doi: 10.3233/JPD-212566
Braak, H., Del Tredici, K., Rüb, U., de Vos, R., Jansen Steur, E., and Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 24, 197–211. doi: 10.1016/s0197-4580(02)00065-9
Bradley, V., Welch, J., and Dick, D. (1989). Visuospatial working memory in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 52, 1228–1235. doi: 10.1136/jnnp.52.11.1228
Candy, J., Perry, R., Perry, E., Irving, D., Blessed, G., Fairbairn, A., et al. (1983). Pathological changes in the nucleus of Meynert in Alzheimer’s and Parkinson’s diseases. J. Neurol. Sci. 59, 277–289. doi: 10.1016/0022-510x(83)90045-x
Cheng, X., Wu, X., Zhang, Y., Li, W., Feng, L., You, H., et al. (2022). LRRK2 deficiency aggravates sleep deprivation-induced cognitive loss by perturbing synaptic pruning in mice. Brain Sci. 12:1200. doi: 10.3390/brainsci12091200
Chesselet, M., Richter, F., Zhu, C., Magen, I., Watson, M., and Subramaniam, S. R. (2012). A progressive mouse model of Parkinson’s disease: The Thy1-aSyn (“Line 61”) mice. Neurotherapeutics 9, 297–314. doi: 10.1007/s13311-012-0104-2
Chiaravalloti, N., Ibarretxe-Bilbao, N., DeLuca, J., Rusu, O., Pena, J., García-Gorostiaga, I., et al. (2014). The source of the memory impairment in Parkinson’s disease: Acquisition versus retrieval. Mov Disord. 29, 765–771. doi: 10.1002/mds.25842
Chung, H., Ho, H., Pérez-Acuña, D., and Lee, S. (2019). Modeling α-synuclein propagation with preformed fibril injections. J. Mov. Disord. 12, 139–151. doi: 10.14802/jmd.19046
Compta, Y., Parkkinen, L., O’Sullivan, S., Vandrovcova, J., Holton, J., Collins, C., et al. (2011). Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: Which is more important? Brain 134, 1493–1505. doi: 10.1093/brain/awr031
Cremades, N., Cohen, S., Deas, E., Abramov, A., Chen, A., Orte, A., et al. (2012). Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 149, 1048–1059. doi: 10.1016/j.cell.2012.03.037
Dauer Née Joppe, K., Tatenhorst, L., Caldi, G. L, Zhang, S., Parvaz, M., Carboni, E., et al. (2021). Brain iron enrichment attenuates α-synuclein spreading after injection of preformed fibrils. J. Neurochem. 159, 554–573. doi: 10.1111/jnc.15461
Davis, A., Inman, C., Wargel, Z., Dube, U., Freeberg, B., Galluppi, A., et al. (2020). APOE genotype regulates pathology and disease progression in synucleinopathy. Sci. Transl. Med. 12:eaay3069. doi: 10.1126/scitranslmed.aay3069
Disney, A., and Robert, J. (2019). Translational implications of the anatomical nonequivalence of functionally equivalent cholinergic circuit motifs. Proc. Natl. Acad. Sci. U.S.A. 116, 26181–26186. doi: 10.1073/pnas.1902280116
Dubois, B., Tolosa, E., Katzenschlager, R., Emre, M., Lees, A., Schumann, G., et al. (2012). Donepezil in Parkinson’s disease dementia: A randomized, double-blind efficacy and safety study. Mov. Disord. 27, 1230–1238. doi: 10.1002/mds.25098
Dues, D., Nguyen, A., Becker, K., Ma, J., and Moore, D. (2023). Hippocampal subfield vulnerability to α-synuclein pathology precedes neurodegeneration and cognitive dysfunction. NPJ Parkinsons Dis. 9:125. doi: 10.1038/s41531-023-00574-1
Dujardin, K., Devos, D., Duhem, S., Destée, A., Marié, R., Durif, F., et al. (2006). Utility of the Mattis dementia rating scale to assess the efficacy of rivastigmine in dementia associated with Parkinson’s disease. J. Neurol. 253, 1154–1159. doi: 10.1007/s00415-006-0175-2
Emre, M., Aarsland, D., Brown, R., Burn, D., Duyckaerts, C., Mizuno, Y., et al. (2007). Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov. Disord. 22, 1689–707; quiz1837. doi: 10.1002/mds.21507
Espa, E., Clemensson, E., Luk, K., Heuer, A., Björklund, T., and Cenci, M. (2019). Seeding of protein aggregation causes cognitive impairment in rat model of cortical synucleinopathy. Mov. Disord. 34, 1699–1710. doi: 10.1002/mds.27810
Fabbrini, G., Barbanti, P., Aurilia, C., Pauletti, C., Lenzi, G., and Meco, G. (2002). Donepezil in the treatment of hallucinations and delusions in Parkinson’s disease. Neurol. Sci. 23, 41–43. doi: 10.1007/s100720200022
Fan, Y., Han, J., Zhao, L., Wu, C., Wu, P., Huang, Z., et al. (2021). Experimental models of cognitive impairment for use in Parkinson’s disease research: The distance between reality and ideal. Front. Aging Neurosci. 13:745438. doi: 10.3389/fnagi.2021.745438
Ferman, T., Aoki, N., Crook, J., Murray, M., Graff-Radford, N., van Gerpen, J., et al. (2018). The limbic and neocortical contribution of α-synuclein, tau, and amyloid β to disease duration in dementia with Lewy bodies. Alzheimers Dement. 14, 330–339. doi: 10.1016/j.jalz.2017.09.014
Freichel, C., Neumann, M., Ballard, T., Müller, V., Woolley, M., Ozmen, L., et al. (2007). Age-dependent cognitive decline and amygdala pathology in alpha-synuclein transgenic mice. Neurobiol. Aging 28, 1421–1435. doi: 10.1016/j.neurobiolaging.2006.06.013
Fritz, N., Kegelmeyer, D., Kloos, A., Linder, S., Park, A., Kataki, M., et al. (2016). Motor performance differentiates individuals with Lewy body dementia, Parkinson’s and Alzheimer’s disease. Gait Post. 50, 1–7. doi: 10.1016/j.gaitpost.2016.08.009
Galvin, J., Pollack, J., and Morris, J. (2006). Clinical phenotype of Parkinson disease dementia. Neurology 67, 1605–1611. doi: 10.1212/01.wnl.0000242630.52203.8f
Giasson, B., Duda, J., Quinn, S., Zhang, B., Trojanowski, J., and Lee, V. (2002). Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34, 521–533. doi: 10.1016/s0896-6273(02)00682-7
Giladi, N., Shabtai, H., Gurevich, T., Benbunan, B., Anca, M., and Korczyn, A. (2003). Rivastigmine (Exelon) for dementia in patients with Parkinson’s disease. Acta Neurol. Scand. 108, 368–373. doi: 10.1034/j.1600-0404.2003.00211.x
Gispert, S., Ricciardi, F., Kurz, A., Azizov, M., Hoepken, H., Becker, D., et al. (2009). Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS One 4:e5777. doi: 10.1371/journal.pone.0005777
Godefroy, O., Azouvi, P., Robert, P., Roussel, M., LeGall, D., Meulemans, T., et al. (2010). Dysexecutive syndrome: Diagnostic criteria and validation study. Ann Neurol. 68, 855–864. doi: 10.1002/ana.22117
Gomperts, S. (2016). Lewy body dementias: Dementia with Lewy bodies and parkinson disease dementia. Continuum 22, 435–463. doi: 10.1212/CON.0000000000000309
Hall, H., Reyes, S., Landeck, N., Bye, C., Leanza, G., Double, K., et al. (2014). Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson’s disease. Brain 137, 2493–2508. doi: 10.1093/brain/awu193
Hansen, C., Björklund, T., Petit, G., Lundblad, M., Murmu, R., Brundin, P., et al. (2013). A novel α-synuclein-GFP mouse model displays progressive motor impairment, olfactory dysfunction and accumulation of α-synuclein-GFP. Neurobiol. Dis. 56, 145–155. doi: 10.1016/j.nbd.2013.04.017
Hao, Y., Lu, Q., Zhai, Y., Wang, H., Wu, M., Hu, M., et al. (2018). Cerebral inoculation of human A53T α-synuclein reduces spatial memory decline and amyloid-β aggregation in APP/PS1 transgenic mice of Alzheimer’s disease. Brain Res. Bull. 143, 116–122. doi: 10.1016/j.brainresbull.2018.10.003
Harding, A., and Halliday, G. (2001). Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol. 102, 355–363. doi: 10.1007/s004010100390
He, Y., Huang, L., Wang, K., Pan, X., Cai, Q., Zhang, F., et al. (2022). α-Synuclein selectively impairs motor sequence learning and value sensitivity: Reversal by the adenosine a2a receptor antagonists. Cereb. Cortex 32, 808–823. doi: 10.1093/cercor/bhab244
Hely, M., Reid, W., Adena, M., Halliday, G., and Morris, J. (2008). The Sydney multicenter study of Parkinson’s disease: The inevitability of dementia at 20 years. Mov. Disord. 23, 837–844. doi: 10.1002/mds.21956
Henderson, M., Trojanowski, J., and Lee, V. (2019). α-Synuclein pathology in Parkinson’s disease and related α-synucleinopathies. Neurosci. Lett. 709:134316. doi: 10.1016/j.neulet.2019.134316
Hepp, D. H., Vergoossen, D. L. E., Huisman, E., Lemstra, A. W., Berendse, H. W., Rozemuller, A. J., et al. (2016). Distribution and load of amyloid-β pathology in Parkinson disease and dementia with Lewy bodies. J. Neuropathol. Exp. Neurol. 75, 936–945. doi: 10.1093/jnen/nlw070
Higginson, C., Wheelock, V., Carroll, K., and Sigvardt, K. (2005). Recognition memory in Parkinson’s disease with and without dementia: Evidence inconsistent with the retrieval deficit hypothesis. J. Clin. Exp. Neuropsychol. 27, 516–528. doi: 10.1080/13803390490515469
Hilker, R., Thomas, A., Klein, J., Weisenbach, S., Kalbe, E., Burghaus, L., et al. (2005). Dementia in Parkinson disease: Functional imaging of cholinergic and dopaminergic pathways. Neurology 65, 1716–1722. doi: 10.1212/01.wnl.0000191154.78131.f6
Hovestadt, A., de Jong, G., and Meerwaldt, J. (1987). Spatial disorientation as an early symptom of Parkinson’s disease. Neurology 37, 485–487. doi: 10.1212/wnl.37.3.485
Hu, Q., Ren, X., Liu, Y., Li, Z., Zhang, L., Chen, X., et al. (2016). Aberrant adenosine A2A receptor signaling contributes to neurodegeneration and cognitive impairments in a mouse model of synucleinopathy. Exp. Neurol. 283, 213–223. doi: 10.1016/j.expneurol.2016.05.040
Hu, S., Hu, M., Liu, J., Zhang, B., Zhang, Z., Zhou, F., et al. (2020). Phosphorylation of Tau and α-Synuclein Induced neurodegeneration in MPTP mouse model of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 16, 651–663. doi: 10.2147/NDT.S235562
Huang, R., Gao, Y., Duan, Q., Zhang, Q., He, P., Chen, J., et al. (2023). Endothelial LRP1-ICD accelerates cognition-associated alpha-synuclein pathology and neurodegeneration through PARP1 activation in a mouse model of Parkinson’s disease. Mol. Neurobiol. 60, 979–1003. doi: 10.1007/s12035-022-03119-4
Huntington, T., and Srinivasan, R. (2021). Adeno-associated virus expression of α-synuclein as a tool to model Parkinson’s disease: Current understanding and knowledge gaps. Aging Dis. 12, 1120–1137. doi: 10.14336/AD.2021.0517
Hurtig, H., Trojanowski, J., Galvin, J., Ewbank, D., Schmidt, M., Lee, V., et al. (2000). Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology 54, 1916–1921. doi: 10.1212/wnl.54.10.1916
Hussein, A., Tielemans, A., Baxter, M., Benson, D., and Huntley, G. (2022). Cognitive deficits and altered cholinergic innervation in young adult male mice carrying a Parkinson’s disease Lrrk2G2019S knockin mutation. Exp. Neurol. 355:114145. doi: 10.1016/j.expneurol.2022.114145
Irwin, D., Lee, V., and Trojanowski, J. (2013). Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 14, 626–636. doi: 10.1038/nrn3549
Irwin, D., White, M., Toledo, J., Xie, S., Robinson, J., Van Deerlin, V., et al. (2012). Neuropathologic substrates of Parkinson disease dementia. Ann. Neurol. 72, 587–598. doi: 10.1002/ana.23659
Janezic, S., Threlfell, S., Dodson, P., Dowie, M., Taylor, T., Potgieter, D., et al. (2013). Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc. Natl. Acad. Sci. U.S.A. 110, E4016–E4025. doi: 10.1073/pnas.1309143110
Jellinger, K., and Attems, J. (2008). Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol. 115, 427–436. doi: 10.1007/s00401-008-0347-5
Jellinger, K., and Korczyn, A. (2018). Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease? BMC Med. 16:34. doi: 10.1186/s12916-018-1016-8
Kalaitzakis, M., Graeber, M., Gentleman, S., and Pearce, R. (2008). Striatal beta-amyloid deposition in Parkinson disease with dementia. J. Neuropathol. Exp. Neurol. 67, 155–161. doi: 10.1097/NEN.0b013e31816362aa
Kalaitzakis, M., Pearce, R., and Gentleman, S. (2009). Clinical correlates of pathology in the claustrum in Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 461, 12–15. doi: 10.1016/j.neulet.2009.05.083
Kalbe, E., Calabrese, P., Kohn, N., Hilker, R., Riedel, O., Wittchen, H., et al. (2008). Screening for cognitive deficits in Parkinson’s disease with the Parkinson neuropsychometric dementia assessment (PANDA) instrument. Parkinsonism Relat. Disord. 14, 93–101. doi: 10.1016/j.parkreldis.2007.06.008
Kanel, P., Müller, M., van der Zee, S., Sanchez-Catasus, C., Koeppe, R., Frey, K., et al. (2020). Topography of cholinergic changes in dementia with Lewy bodies and key neural network hubs. J. Neuropsychiatry Clin. Neurosci. 32, 370–375. doi: 10.1176/appi.neuropsych.19070165
Kasongo, D., de Leo, G., Vicario, N., Leanza, G., and Legname, G. (2020). Chronic α-Synuclein Accumulation in Rat Hippocampus Induces Lewy Bodies Formation and Specific Cognitive Impairments. eNeuro 7, ENEURO.0009–ENEURO.20. doi: 10.1523/ENEURO.0009-20.2020
Kemps, E., Szmalec, A., Vandierendonck, A., and Crevits, L. (2005). Visuo-spatial processing in Parkinson’s disease: Evidence for diminished visuo-spatial sketch pad and central executive resources. Parkinsonism Relat. Disord. 11, 181–186. doi: 10.1016/j.parkreldis.2004.10.010
Kim, S., Kwon, S., Kam, T., Panicker, N., Karuppagounder, S., Lee, S., et al. (2019). Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron 103:627–641.e7. doi: 10.1016/j.neuron.2019.05.035
Kim, T., Bae, E., Jung, B., Choi, M., Shin, S., Park, S., et al. (2022). Inflammation promotes synucleinopathy propagation. Exp. Mol. Med. 54, 2148–2161. doi: 10.1038/s12276-022-00895-w
Kitada, T., Pisani, A., Porter, D., Yamaguchi, H., Tscherter, A., Martella, G., et al. (2007). Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 104, 11441–11446. doi: 10.1073/pnas.0702717104
Koga, S., Sekiya, H., Kondru, N., Ross, O., and Dickson, D. (2021). Neuropathology and molecular diagnosis of Synucleinopathies. Mol. Neurodegener. 16:83. doi: 10.1186/s13024-021-00501-z
Kovacs, G., Lee, V., and Trojanowski, J. (2017). Protein astrogliopathies in human neurodegenerative diseases and aging. Brain Pathol. 27, 675–690. doi: 10.1111/bpa.12536
Kövari, E., Gold, G., Herrmann, F., Canuto, A., Hof, P., Bouras, C., et al. (2003). Lewy body densities in the entorhinal and anterior cingulate cortex predict cognitive deficits in Parkinson’s disease. Acta Neuropathol. 106, 83–88. doi: 10.1007/s00401-003-0705-2
Kramberger, M., Auestad, B., Garcia-Ptacek, S., Abdelnour, C., Olmo, J., Walker, Z., et al. (2017). Long-term cognitive decline in dementia with Lewy bodies in a large multicenter, international cohort. J. Alzheimers Dis. 57, 787–795. doi: 10.3233/JAD-161109
Lewy, F. (1913). Zue pathologischen anatomie der paralysis agitans. Detsch Z. Nervenhaeilkunde 50:50.
Litvan, I., Mohr, E., Williams, J., Gomez, C., and Chase, T. (1991). Differential memory and executive functions in demented patients with Parkinson’s and Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 54, 25–29. doi: 10.1136/jnnp.54.1.25
Luk, K., Kehm, V., Carroll, J., Zhang, B., O’Brien, P., Trojanowski, J., et al. (2012). Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953. doi: 10.1126/science.1227157
Marotta, N., Ara, J., Uemura, N., Lougee, M., Meymand, E., Zhang, B., et al. (2021). Alpha-synuclein from patient Lewy bodies exhibits distinct pathological activity that can be propagated in vitro. Acta Neuropathol. Commun. 9:188. doi: 10.1186/s40478-021-01288-2
Masuda-Suzukake, M., Nonaka, T., Hosokawa, M., Oikawa, T., Arai, T., Akiyama, H., et al. (2013). Prion-like spreading of pathological α-synuclein in brain. Brain 136, 1128–1138. doi: 10.1093/brain/awt037
Matsuo, K., Yabuki, Y., Melki, R., Bousset, L., Owada, Y., and Fukunaga, K. (2021). Crucial role of FABP3 in αSyn-induced reduction of septal GABAergic neurons and cognitive decline in mice. Int. J. Mol. Sci. 22:400. doi: 10.3390/ijms22010400
Mattila, P., Rinne, J., Helenius, H., Dickson, D., and Röyttä, M. (2000). Alpha-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson’s disease. Acta Neuropathol. 100, 285–290. doi: 10.1007/s004019900168
Mattila, P., Röyttä, M., Torikka, H., Dickson, D., and Rinne, J. (1998). Cortical Lewy bodies and Alzheimer-type changes in patients with Parkinson’s disease. Acta Neuropathol. 95, 576–582. doi: 10.1007/s004010050843
McKeith, I. G., and Burn, D. (2000). SPECTRUM OF PARKINSON’S DISEASE, PARKINSON’S DEMENTIA, AND LEWY BODY DEMENTIA. Neurol. Clin. 18, 865–883. doi: 10.1016/S0733-8619(05)70230-9
McKeith, I., Del Ser, T., Spano, P., Emre, M., Wesnes, K., Anand, R., et al. (2000). Efficacy of rivastigmine in dementia with Lewy bodies: A randomised, double-blind, placebo-controlled international study. Lancet 356, 2031–2036. doi: 10.1016/S0140-6736(00)03399-7
McKeith, I., Dickson, D., Lowe, J., Emre, M., O’Brien, J., Feldman, H., et al. (2005). Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 65, 1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1
Mori, E., Ikeda, M., and Kosaka, K. (2012). Donepezil for dementia with Lewy bodies: A randomized, placebo-controlled trial. Ann. Neurol. 72, 41–52. doi: 10.1002/ana.23557
Mosimann, U., Mather, G., Wesnes, K., O’Brien, J., Burn, D., and McKeith, I. (2004). Visual perception in Parkinson disease dementia and dementia with Lewy bodies. Neurology 63, 2091–2096. doi: 10.1212/01.wnl.0000145764.70698.4e
Mueller, C., Ballard, C., Corbett, A., and Aarsland, D. (2017). The prognosis of dementia with Lewy bodies. Lancet Neurol. 16, 390–398. doi: 10.1016/S1474-4422(17)30074-1
Nakano, I., and Hirano, A. (1984). Parkinson’s disease: Neuron loss in the nucleus basalis without concomitant Alzheimer’s disease. Ann Neurol. 15, 415–418. doi: 10.1002/ana.410150503
Noe, E., Marder, K., Bell, K., Jacobs, D., Manly, J., and Stern, Y. (2004). Comparison of dementia with Lewy bodies to Alzheimer’s disease and Parkinson’s disease with dementia. Mov. Disord. 19, 60–67. doi: 10.1002/mds.10633
Nouraei, N., Mason, D., Miner, K., Carcella, M., Bhatia, T., Dumm, B., et al. (2018). Critical appraisal of pathology transmission in the α-synuclein fibril model of Lewy body disorders. Exp. Neurol. 299, 172–196. doi: 10.1016/j.expneurol.2017.10.017
Pagonabarraga, J., Kulisevsky, J., Llebaria, G., García-Sánchez, C., Pascual-Sedano, B., Martinez-Corral, M., et al. (2010). PDD-Short Screen: A brief cognitive test for screening dementia in Parkinson’s disease. Mov. Disord. 25, 440–446. doi: 10.1002/mds.22877
Pan, L., Li, C., Meng, L., Tian, Y., He, M., Yuan, X., et al. (2022). Tau accelerates α-synuclein aggregation and spreading in Parkinson’s disease. Brain 145, 3454–3471. doi: 10.1093/brain/awac171
Pang, C., Sørensen, M., Lee, K., Luk, K., Trojanowski, J., Lee, V., et al. (2022). Investigating key factors underlying neurodegeneration linked to alpha-synuclein spread. Neuropathol. Appl. Neurobiol. 48:e12829. doi: 10.1111/nan.12829
Paolo, A., Tröster, A., Glatt, S., Hubble, J., and Koller, W. (1995). Differentiation of the dementias of Alzheimer’s and Parkinson’s disease with the dementia rating scale. J. Geriatr. Psychiatry Neurol. 8, 184–188. doi: 10.1177/089198879500800308
Park, K., Kim, H., Cheon, S., Cha, J., Kim, S., and Kim, J. (2011). Dementia with Lewy bodies versus Alzheimer’s disease and Parkinson’s disease dementia: A comparison of cognitive profiles. J. Clin. Neurol. 7, 19–24. doi: 10.3988/jcn.2011.7.1.19
Pasquini, J., Brooks, D., and Pavese, N. (2021). The cholinergic brain in Parkinson’s disease. Mov. Disord. Clin. Pract. 8, 1012–1026. doi: 10.1002/mdc3.13319
Paumier, K., Sukoff Rizzo, S., Berger, Z., Chen, Y., Gonzales, C., Kaftan, E., et al. (2013). Behavioral characterization of A53T mice reveals early and late stage deficits related to Parkinson’s disease. PLoS One 8:e70274. doi: 10.1371/journal.pone.0070274
Perry, E., Haroutunian, V., Davis, K., Levy, R., Lantos, P., Eagger, S., et al. (1994). Neocortical cholinergic activities differentiate Lewy body dementia from classical Alzheimer’s disease. Neuroreport 5, 747–749. doi: 10.1097/00001756-199403000-00002
Polinski, N., Volpicelli-Daley, L., Sortwell, C., Luk, K., Cremades, N., Gottler, L., et al. (2018). Best practices for generating and using alpha-synuclein pre-formed fibrils to model Parkinson’s disease in rodents. J. Parkinsons Dis. 8, 303–322. doi: 10.3233/JPD-171248
Reading, P., Luce, A., and McKeith, I. (2001). Rivastigmine in the treatment of parkinsonian psychosis and cognitive impairment: Preliminary findings from an open trial. Mov. Disord. 16, 1171–1174. doi: 10.1002/mds.1204
Rodriguiz, R. M., and Wetsel, W. C. (2006) Assessments of Cognitive Deficits in Mutant Mice. In: Levin, ED, Buccafusco JJ, (eds) Animal Models of Cognitive Impairment. Boca Raton (FL): CRC Press/Taylor & Francis.
Rongve, A., Soennesyn, H., Skogseth, R., Oesterhus, R., Hortobágyi, T., Ballard, C., et al. (2016). Cognitive decline in dementia with Lewy bodies: A 5-year prospective cohort study. BMJ Open 6:e010357. doi: 10.1136/bmjopen-2015-010357
Ruffmann, C., Calboli, F., Bravi, I., Gveric, D., Curry, L., de Smith, A., et al. (2016). Cortical Lewy bodies and Aβ burden are associated with prevalence and timing of dementia in Lewy body diseases. Neuropathol. Appl. Neurobiol. 42, 436–450. doi: 10.1111/nan.12294
Sabbagh, M., Adler, C., Lahti, T., Connor, D., Vedders, L., Peterson, L., et al. (2009). Parkinson disease with dementia: Comparing patients with and without Alzheimer pathology. Alzheimer Dis. Assoc. Disord. 23, 295–297. doi: 10.1097/WAD.0b013e31819c5ef4
Shimada, H., Hirano, S., Shinotoh, H., Aotsuka, A., Sato, K., Tanaka, N., et al. (2009). Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology 73, 273–278. doi: 10.1212/WNL.0b013e3181ab2b58
Singh, B., Covelo, A., Martell-Martínez, H., Nanclares, C., Sherman, M., Okematti, E., et al. (2019). Tau is required for progressive synaptic and memory deficits in a transgenic mouse model of α-synucleinopathy. Acta Neuropathol. 138, 551–574. doi: 10.1007/s00401-019-02032-w
Skelly, D., Griffin, ÉW., Murray, C. L., Harney, S., O’Boyle, C., Hennessy, E., et al. (2019). Acute transient cognitive dysfunction and acute brain injury induced by systemic inflammation occur by dissociable IL-1-dependent mechanisms. Mol. Psychiatry 24, 1533–1548. doi: 10.1038/s41380-018-0075-8
Stoyka, L., Arrant, A., Thrasher, D., Russell, D., Freire, J., Mahoney, C., et al. (2020). Behavioral defects associated with amygdala and cortical dysfunction in mice with seeded α-synuclein inclusions. Neurobiol. Dis. 134:104708. doi: 10.1016/j.nbd.2019.104708
Stoyka, L., Mahoney, C., Thrasher, D., Russell, D., Cook, A., Harris, A., et al. (2021). Templated α-synuclein inclusion formation is independent of endogenous tau. eNeuro 8, ENEURO.458–ENEURO.420. doi: 10.1523/ENEURO.0458-20.2021
Subramaniam, S., Magen, I., Bove, N., Zhu, C., Lemesre, V., Dutta, G., et al. (2018). Chronic nicotine improves cognitive and social impairment in mice overexpressing wild type α-synuclein. Neurobiol. Dis. 117, 170–180. doi: 10.1016/j.nbd.2018.05.018
Tanila, H. (2018). Testing cognitive functions in rodent disease models: Present pitfalls and future perspectives. Behav. Brain Res. 352, 23–27. doi: 10.1016/j.bbr.2017.05.040
Taylor, T., Greene, J., and Miller, G. (2010). Behavioral phenotyping of mouse models of Parkinson’s disease. Behav. Brain Res. 211, 1–10. doi: 10.1016/j.bbr.2010.03.004
Tiraboschi, P., Hansen, L., Alford, M., Merdes, A., Masliah, E., Thal, L., et al. (2002). Early and widespread cholinergic losses differentiate dementia with Lewy bodies from Alzheimer disease. Arch. Gen. Psychiatry 59, 946–951. doi: 10.1001/archpsyc.59.10.946
Tsuboi, Y., and Dickson, D. (2005). Dementia with Lewy bodies and Parkinson’s disease with dementia: Are they different? Parkinsonism Relat. Disord. 11, S47–S51. doi: 10.1016/j.parkreldis.2004.10.014
van Laar, T., De Deyn, P., Aarsland, D., Barone, P., and Galvin, J. (2011). Effects of cholinesterase inhibitors in Parkinson’s disease dementia: A review of clinical data. CNS Neurosci. Ther. 17, 428–441. doi: 10.1111/j.1755-5949.2010.00166.x
Volpicelli-Daley, L., Kirik, D., Stoyka, L., Standaert, D., and Harms, A. (2016). How can rAAV-α-synuclein and the fibril α-synuclein models advance our understanding of Parkinson’s disease? J. Neurochem. 139, 131–155. doi: 10.1111/jnc.13627
Wang, H., Yu, J., Tang, S., Jiang, T., Tan, C., Meng, X., et al. (2015). Efficacy and safety of cholinesterase inhibitors and memantine in cognitive impairment in Parkinson’s disease, Parkinson’s disease dementia, and dementia with Lewy bodies: Systematic review with meta-analysis and trial sequential analysis. J. Neurol. Neurosurg. Psychiatry 86, 135–143. doi: 10.1136/jnnp-2014-307659
Waterfall, M., and Crowe, S. (1995). Meta-analytic comparison of the components of visual cognition in Parkinson’s disease. J. Clin. Exp. Neuropsychol. 17, 759–772. doi: 10.1080/01688639508405165
Watson, G., and Leverenz, J. (2010). Profile of cognitive impairment in Parkinson’s disease. Brain Pathol. 20, 640–645. doi: 10.1111/j.1750-3639.2010.00373.x
Weintraub, D., Moberg, P., Culbertson, W., Duda, J., Katz, I., and Stern, M. (2005). Dimensions of executive function in Parkinson’s disease. Dement. Geriatr. Cogn. Disord. 20, 140–144. doi: 10.1159/000087043
Zaccai, J., McCracken, C., and Brayne, C. A. (2005). Systematic review of prevalence and incidence studies of dementia with Lewy bodies. Age Ageing 34, 561–566. doi: 10.1093/ageing/afi190
Zhang, D., Li, S., Hou, L., Jing, L., Ruan, Z., Peng, B., et al. (2021). Microglial activation contributes to cognitive impairments in rotenone-induced mouse Parkinson’s disease model. J. Neuroinflammn. 18:4. doi: 10.1186/s12974-020-02065-z
Zhang, Q., Abdelmotilib, H., Larson, T., Keomanivong, C., Conlon, M., Aldridge, G., et al. (2021). Cortical alpha-synuclein preformed fibrils do not affect interval timing in mice. Neurosci. Lett. 765:136273. doi: 10.1016/j.neulet.2021.136273
Zhao, J., Bi, W., Xiao, S., Lan, X., Cheng, X., Zhang, J., et al. (2019). Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 9:5790. doi: 10.1038/s41598-019-42286-8
Zhao, N., Attrebi, O., Ren, Y., Qiao, W., Sonustun, B., Martens, Y., et al. (2020). APOE4 exacerbates α-synuclein pathology and related toxicity independent of amyloid. Sci. Transl. Med. 12:eaay1809. doi: 10.1126/scitranslmed.aay1809
Keywords: lewy body dementia, Parkinson’s disease, Parkinson’s disease dementia, dementia with lewy bodies, preformed fibril, animal models
Citation: Haikal C, Winston GM and Kaplitt MG (2024) Cognitive dysfunction in animal models of human lewy-body dementia. Front. Aging Neurosci. 16:1369733. doi: 10.3389/fnagi.2024.1369733
Received: 12 January 2024; Accepted: 18 June 2024;
Published: 22 July 2024.
Edited by:
Ines Moreno-Gonzalez, University of Malaga, SpainReviewed by:
Insup Choi, Icahn School of Medicine at Mount Sinai, United StatesFiona E. N. LeBeau, Newcastle University, United Kingdom
Copyright © 2024 Haikal, Winston and Kaplitt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael G. Kaplitt, bWlrMjAwMkBtZWQuY29ybmVsbC5lZHU=
†These authors share first authorship
‡ORCID: Caroline Haikal, orcid.org/0000-0002-9066-5940; Graham M. Winston, orcid.org/0000-0002-3878-992X; Michael G. Kaplitt, orcid.org/0000-0002-8979-2459