 Chaowen Huang1†Huilin Xiao2†Yang Yang3†Jiankun Luo2Yixi Lai2Shizhen Liu2
Chaowen Huang1†Huilin Xiao2†Yang Yang3†Jiankun Luo2Yixi Lai2Shizhen Liu2 Kanmin Mao4*
Kanmin Mao4* Jialong Chen2*Liling Wang3*
Jialong Chen2*Liling Wang3*- 1Department of Respiratory Medicine, Jiangmen Central Hospital Affiliated Jiangmen Hospital of Sun Yat-sen University, Jiangmen, China
- 2School of Public Health, Guangdong Medical University, Dongguan, China
- 3Department of Rehabilitation, Affiliated Shenzhen Baoan Central Hospital Group of Guangdong Medical University, Shenzhen, China
- 4Hubei Key Laboratory of Food Nutrition and Safety and the Ministry of Education (MOE) Key Laboratory of Environment and Health, Department of Nutrition and Food Hygiene, School of Public Health, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
Adenosine diphosphate-ribosylation (ADPRylation) is a reversible posttranslational modification that plays a crucial role in cellular homeostasis and disease development. ADPRylation is produced via nicotinamide adenine dinucleotide hydrolysis and modifies proteins via corresponding transferases, mainly poly(ADP-ribose) polymerases (PARPs), the inhibitors of which have been used in the clinical treatment of cancer. ADPRylation is involved in various physiological processes, including pathogen infection, inflammation, DNA repair, and neurological disorders. In neurodegenerative diseases (NDs), dysregulated ADPRylation contributes to protein aggregation, neuroinflammation, and metabolic disturbances, while targeted modulation shows therapeutic potential. ADPRylation differentially regulates neurodegenerative processes, and PARP inhibitors can reduce neuroinflammation, oxidative stress, and metabolic dysfunction. However, challenges such as poor blood–brain barrier penetration and cell type-specific responses limit clinical translation. This review summarizes recent findings on the role of ADPRylation and PARPs in NDs, highlighting their involvement in protein aggregation and cellular signaling. It emphasizes the importance of ADPRylation in neuronal cells and supports the development of precision therapies targeting this pathway to address current treatment challenges in NDs.
1 Introduction
Cellular activity is fundamental to life and is primarily influenced by protein function and associated posttranslational modifications (PTMs) (Karve and Cheema, 2011). ADP-ribosylation (ADPRylation) is a reversible PTM that is widely involved in multiple critical biological processes, such as DNA damage, stress responses and cell proliferation, inflammation and cell death (Li et al., 2019; Kliza et al., 2021). Based on the number of ADP-ribose units attached to a target, the ADP-ribosylation modification is classified into mono-ADP-ribosylation (MARylation) and poly-ADP-ribosylation (PARylation), both of which are regulated primarily by poly(ADP-ribose) (PAR) polymerase family proteins (Groslambert et al., 2021). The human poly(ADP-ribose) polymerase (PARP) family consists of 17 members (Vyas et al., 2014; McGurk et al., 2019): (1) PARP-3, PARP-4, PARP-6, PARP-7, PARP-8, PARP-10, PARP-11, PARP-12, PARP-14, PARP-15 and PARP-16 are mono-ADPRylation transferases that transfer a single ADP-ribose from nicotinamide adenine dinucleotide (NAD+) to a target molecule; (2) PARP-1, PARP-2 and PARP-5 (tankyrase) add successive ADP-ribose units (2–200 units) to specific amino acids of proteins to form linear or branched polymer PARs (Aberle et al., 2020; Hendriks et al., 2021); and (3) PARP-9 and PARP-13, which have been reported to be enzymatically inactive, specific roles of which remain to be clarified. ADPRylation plays a crucial role in DNA damage repair and cancer therapy. Specifically, ADPRylation is involved in the regulation of DNA damage response pathways, where Mono-ADP-ribosylhydrolase 2 haploinsufficiency has been shown to impair the catalytic activity of PARP-1, resulting in chromosome instability and promoting tumorigenesis (Sakthianandeswaren et al., 2018). Additionally, nicotinamide adenine dinucleotide phosphate functions as an endogenous inhibitor of ADPRylation, suppressing PARP-1-mediated DNA repair and enhancing the sensitivity of tumor cells to PARP inhibitors (Bian et al., 2019). In the case of DNA damage repair, poly(ADP-ribose) polymerases (PARPs) function as DNA damage sensors, catalyzing ADPRylation at DNA lesions and functioning as docking platforms for DNA repair factors (Ray and Nussenzweig, 2017). PARP-1 has been reported to be the major PARP family member that is activated by single-strand DNA Breaks and double-strand DNA Breaks (DSBs) and interacts with X-ray repair cross-complementing protein 1 (XRCC1) to recruit the SSB repair machinery (Wu et al., 2021) or initiate the DNA-PKcs-KU70/80 signaling pathways to promote the repair of DSBs (Beck et al., 2014). PARs are removed from a protein by ADPRylation hydrolases. Each hydrolase carries specific domains and substrates. Poly(ADP-ribose) glycohydrolase (PARG) cleaves a PAR molecule that has bound proteins to free it or a single ADP-ribose moiety via its endoglycosidase or exoglycosidase activity (Houl et al., 2019). Furthermore, ADP-ribosylhydrolase 3 (ARH3) hydrolyzes the O-glycosidic bonds of short PAR chains or a single ADP-ribose moiety conjugated to a serine residue, in contrast to PARG, which less efficiently hydrolyzes a terminal ADP-ribose moiety (McGurk et al., 2019; Prokhorova et al., 2021). Humans express other ADPRylation hydrolases: Mono-ADP-ribosylhydrolase 1 and Mono-ADP-ribosylhydrolase 2 catalyze remove an ADP-ribose from acidic residues, and TADG1 ADP-ribosylhydrolase 1 (TARG1), Nudix Hydrolase 16 (NUDT16) and Ectonucleotide Pyrophosphatase/Phosphodiesterase 1 (ENPP1) reverse the MARylation or PARylation of proteins (Rosenthal et al., 2013; Sharifi et al., 2013; Palazzo et al., 2016; Zhang et al., 2020). Interestingly, hydrolases protect cells from excessive PARylation, which induces high levels of cytotoxicity. In fact, a PAR chain can be shuttled between the nucleus and cytoplasm, and cytoplasmic PAR is a fundamental factor in the abrogation of nuclear energy induced by BCL-2-Associated X Protein (BAX)-apoptosis inducing factor pathways, thereby linking PARylation to cellular apoptosis, also known as parthanatos (Fatokun et al., 2014; Mashimo et al., 2021). Hence, the dynamic and reversible progress of ADPRylation needs to be strictly controlled.
In recent years, the PARP-ADPRylation field has been further expanded by an increasing number of studies on aging-related diseases, especially neurodegenerative diseases (NDs), and it is now clear that PARylation of pathogenic proteins such as α-synuclein(α-syn) and β-amyloid (Aβ) can affect the phase separation and aggregation of these proteins, thereby accelerating disease progression (Strosznajder et al., 2012; Kam et al., 2018; Liu and Fang, 2019). In response to excessive PARP activity/ADPRylation that is evident in most brain regions and cerebrospinal fluid in patients with NDs, inhibition of PARPs attenuates mitochondrial damage and abnormal protein degradation systems (Lehmann et al., 2016; Mao et al., 2020), supporting the possibility that PARP/ADPRylation inhibitors protect neurons from pathological attack and contribute to normal brain function maintenance (Figure 1). Hence, this review is organized to present an overview of PARP/ADPRylation biochemistry and the known contribution of the ADPRylation modification to cellular signaling pathway activity in NDs, and then, future uses of PARP inhibitors as disease-targeting agents is discussed.
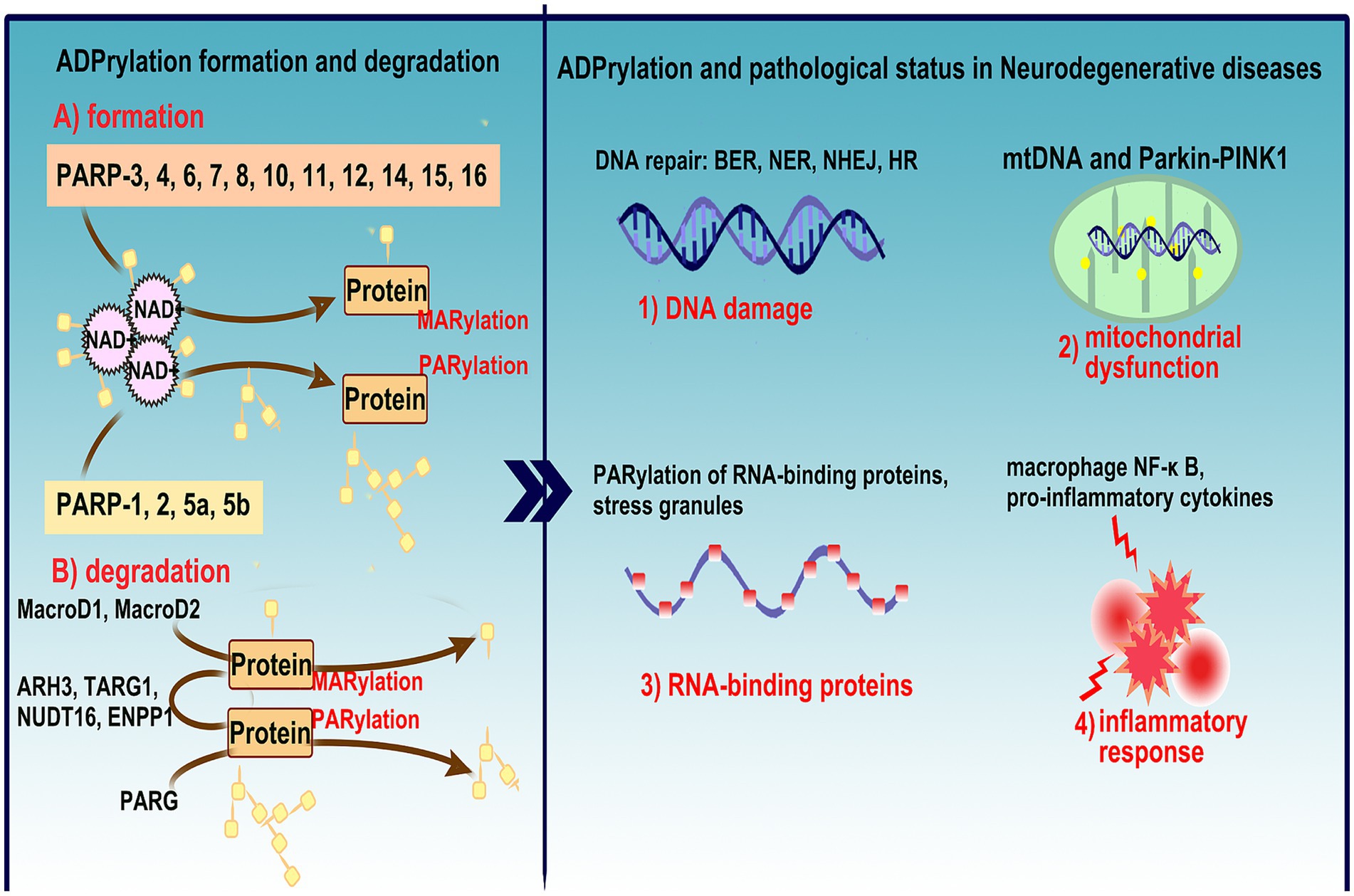
Figure 1. Overview of ADPRylation mechanisms and their role in NDs. This figure illustrates the formation and degradation of ADPRylation, a critical post-translational modification involved in various biological processes including DNA damage response, cell death, and inflammation. (A) Depicts the enzymatic activity of PARP family members (PARP-1, 2, 5a, 5b, and PARP-3, 4, 6, 7, 8, 10, 11, 12, 14, 15, 16), which transfer ADP-ribose units from NAD+ to target proteins via PARylation and MARylation, and the role of key hydrolases (MacroD1, MacroD2, ARH3, TARG1, NUDT16, ENPP1, PARG) in reversing these modifications, highlighting the dynamic and reversible nature of ADPRylation. (B) Links ADPRylation to the pathophysiology of neurodegenerative disease, discussing its impact on DNA repair mechanisms (BER, NER, NHEJ, HR), mitochondrial dysfunction via the mtDNA and Parkin-PINK1 pathways, and the implications for neuronal health. It also details the involvement of PARylated RNA-binding proteins in stress granule formation, activation of macrophage NF-κB pathways, and the release of pro-inflammatory cytokines, underscoring the comprehensive role of ADPRylation in exacerbating inflammatory responses within the nervous system. This figure encapsulates the complex interplay between ADPRylation and neurodegenerative disease progression, emphasizing its potential as a therapeutic target through the use of PARP inhibitors.
2 ADPRylation and DNA damage in neurodegenerative disease
Due to high oxygen demand and neuroelectrical activity, neurons are particularly vulnerable to reactive oxygen species (ROS) attack, leading to severe DNA damage (Belanger et al., 2011; Kharrati-Koopaee et al., 2021). Accumulating evidence suggests that DNA repair and genomic stability play crucial roles in aging-related NDs, including Alzheimer’s disease (AD) (Hou et al., 2018), Parkinson’s disease (PD) (Gonzalez-Hunt and Sanders, 2021) and amyotrophic lateral sclerosis (ALS) (Penndorf et al., 2018). Throughout their lives, organisms trigger a series of mechanisms to prevent genomes from being poisoned by endogenous and exogenous factors, and multiple monitoring and repair mechanisms have been acquired to repair DNA lesions in a timely manner. Many different NDs are characterized by significant overlap in signaling pathways and pathogenic patterns. Among neuronal DNA repair, base excision repair (BER), nucleotide excision repair (NER) and nonhomologous end joining (NHEJ) repair are considered major types, while homologous recombination (HR) is not typically induced (Lautrup et al., 2019; Mitra and Hegde, 2019; Gupta et al., 2021). However, recent studies have shown that HR, traditionally linked to the cell cycle, is also activated in post-mitotic neurons via RNA strand sensors, such as the RNA recognition motif of proteins like Fused in Sarcoma (FUS). This RNA-templated repair mechanism aids in fixing double-strand breaks using nascent RNA templates during transcription (Welty et al., 2018; Mamontova et al., 2023). PARP-1 plays a key role in regulating various DNA damage repair modalities, and its activation can hinder the pathological progression of NDs induced by environmental toxins. Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is commonly used to establish animal models of PD (Przedborski et al., 2000; Cao et al., 2008). After MPTP exposure, mice show increased ROS and DNA damage together with hyperactivated PARP-1 in SNpc neurons (Wang et al., 2003). PARP-1 is involved in both classical NHEJ and alternative NHEJ repair mechanisms, as well as HR through the DNA-PKcs-KU70/80 signaling pathway (Wei and Yu, 2016; Noordermeer and van Attikum, 2019), whereas a portion of PAR molecules are labile in the cytoplasm, leading to apoptosis (Wang et al., 2016). The tumor suppressor p53 is heavily involved in the response to DNA damage. Upon activation, p53 accelerates cell cycle progression and induces apoptosis through the regulation of downstream genes like Bax, Fas, RB1, and Cdkn1a (Scoumanne et al., 2009; Jenkins et al., 2012; Iida et al., 2013). Interestingly, p53 itself is regulated by post-translational modifications, including phosphorylation and PARylation, which stabilize the protein and activate its tumor-suppressive functions (Fischbach et al., 2018). In the context of NDs, such as PD, p53 has been shown to be heavily PARylated by PARP-1 (Mandir et al., 2002), indicating a critical interplay between p53, DNA damage, and ADPRylation in neurons. In MPTP-induced PD models, the presence of p53 PARylation is consistent with the activation of cell death pathways and neuronal degeneration following DNA damage (Gonzalez-Hunt and Sanders, 2021). Moreover, familial PD is closely associated with mutations in the Synuclein Alpha (SNCA) gene encoding α-syn. SNCA A53T (Chen et al., 2020), A30P (Paiva et al., 2018), A30G (Liu H. et al., 2021), or E46K (Zhao et al., 2020) mutant mice present aggregated α-syn and Lewy bodies in dopaminergic (DA) neurons, disrupting the normal function of neurons and even leading to neuronal death. Recent evidence suggests that α-syn can directly interact with DNA, generating DNA strand breaks and activating PARP-1, further promoting neurodegeneration through the amplification of α-syn aggregation (Schaser et al., 2019). The subsequent DNA damage induces PARP-1 activation, and activated PARP-1 increases PAR recruitment to α-syn, thereby accelerating α-syn aggregation via the generation of additional toxic fibrotic structures. However, mono-ADP ribose does not interact with α-syn and exerts no toxic effect on α-syn (Kam et al., 2018). Collectively, these data indicate that the activation of PARP-1 in response to DNA damage and the subsequent PARylation of p53 appear to be central to the progression of several NDs, highlighting the importance of targeting PARP-1 and the ADPRylation pathway for therapeutic intervention. In addition, PARP-2, PARP-3 and PARP-10 have also been reported to be involved in DNA damage repair via ADPRylation (Beck et al., 2014; Nicolae et al., 2014), and whether they are involved in the development of NDs remains to be further investigated. Notably, mitochondrial DNA (mtDNA) can also be abrogated in response to damaged nuclear DNA, and this mitochondrial response is associated with NDs (Antonyova et al., 2020; Martin-Jimenez et al., 2020). Through nuclear-mitochondrial cross talk, PARP and ADPRylation are involved in mitochondrial NAD+ metabolism (Pang et al., 2018). PAR chain synthesis mediated through mitochondrial PARP-1 (Brunyanszki et al., 2014; Basello and Scovassi, 2015) supports the notion that the PARylation of mitochondrial proteins involved in mtDNA lesion repair contributes to organelle homeostasis.
Huntington’s disease (HD) is an autosomal dominant disorder caused mainly by mutations in the Huntingtin (HTT) gene on Chromosome 4 (Bates, 2003). In neurons, the mutated HTT protein is widely expressed and aggregates, affecting the normal function of neurons and ultimately leading to death (Steffan et al., 2000). Relevant data show that HD patients carry mutations in Exon 1 of the HTT gene, thereby amplifying CAG repeats, which is associated with DNA repair mechanisms and enzymes that are similar to telomerase (Neueder et al., 2017; Gao et al., 2019). Mutations in DNA damage senor proteins, a class of proteins that monitor and detect DNA damage within cells, have been shown to reduce the stability of core checkpoint proteins (Mec1, Rad 9, and Rad 53), resulting in abnormalities in the recognition of CAG repeat sequences cells, leading to the increased frequency of CAG repeats (Massey and Jones, 2018; Maiuri et al., 2019). In addition, HTT can interact with a variety of transcription factors and thus affect protein expression (Thomas et al., 2013; Goodliffe et al., 2020). Cyclic AMP response element-binding protein (CREB) mediates the expression of cyclic Adenosine Monophosphate and calcium-dependent genes by binding to CREB-binding protein (CBP) (Screaton et al., 2004). However, aggregated HTT recruits CBP without CREB, leading to the deacetylation of the transcriptional complex and resulting in the failure of downstream gene expression (Chaturvedi et al., 2012; Jeong et al., 2012). Recently, PARP-1 inhibitors have been confirmed to modulate the localization and activation of phosphorylated CREB and CBP in distinct striatal subregions (Pramanik et al., 2014; Cardinale et al., 2015), which indicates their potential as effective treatments for HD (Figure 2).
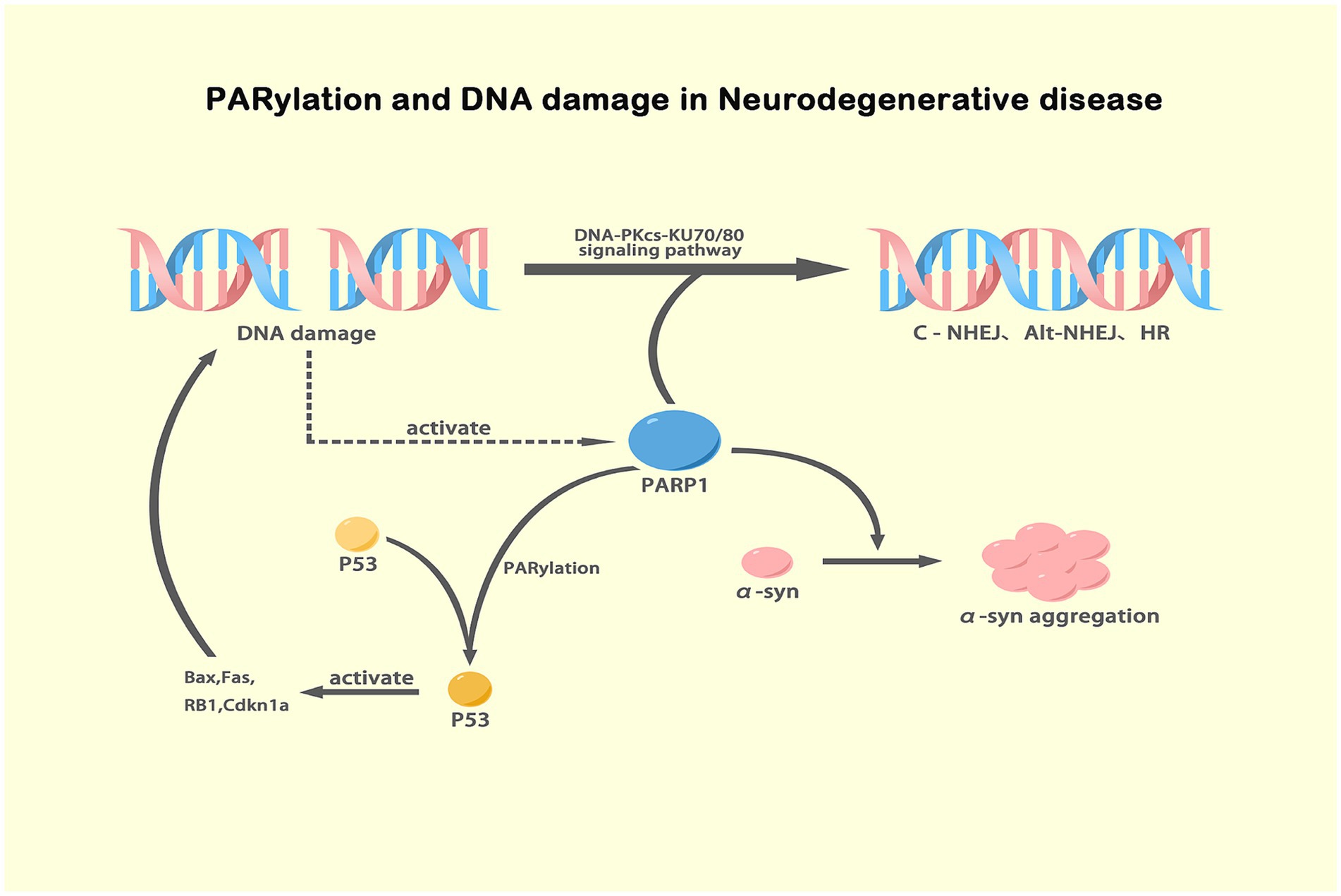
Figure 2. Mechanisms of PARP-1 activation and its effects in NDs. This illustration depicts the role of PARP-1 in DNA damage response where it activates DNA repair pathways including DNA-PKcs-KU70/80 signaling for classic and alternative NHEJ and HR. It also shows how PARP-1-mediated PARylation of p53 stabilizes and activates this protein, leading to the activation of apoptotic genes such as Bax, Fas, RB1, and Cdkn1a. Additionally, the diagram connects excessive PARylation of α-syn, driven by PARP-1 in response to DNA damage, to its aggregation, a key pathological event in PD. This comprehensive view underscores the dual impacts of PARylation in DNA repair and protein aggregation, highlighting the therapeutic potential of PARP inhibitors in NDs. Created with BioRender.
3 ADPRylation and damaged mitochondria in neurodegenerative diseases
Mitochondrial dysfunction is the main cause of many neurodegenerative disorders. In both sporadic PD and familial PD, mitochondrial dysfunction is linked to damaged neurons and aggravated pathological progression (Moon and Paek, 2015). After MPTP or rotenone poisoning, mitochondria are profoundly affected by global oxidative stress and DNA damage (Sanders and Greenamyre, 2013; Cheng et al., 2021; Dionisio et al., 2021). Mitochondrial oxidative phosphorylation plays a crucial role in energy metabolism, and neurons are highly energy dependent (Sheng and Cai, 2012; Markham et al., 2014). The link between mitochondria and brain disorders has been extensively studied but it has not been clarified completely. In postmortem brain tissue from PD and AD patients, defective mitochondrial genome integrity, mtDNA depletion and mtDNA rearrangements have been identified (Pinto et al., 2013; Gurunathan et al., 2019). Moreover, an animal model of damaged mtDNA displayed DA neurodegeneration accompanied by motor disorder (Song et al., 2017), suggesting that NDs may be results of neuronal mitochondrial damage. Indeed, polymeric mtDNA mutation may result in the depletion of mitochondrial NAD+, thereby eliciting energy transfer chain breakdown (Lauritzen et al., 2021). As a major consumer of cellular NAD+ and a responder to DNA damage, mitochondrial PARP involvement in mtDNA repair and metabolism is undisputed. It has been shown that inhibiting PARP-1 reduces the integrity of the mtDNA genome and the expression of Cyclooxygenase 1, Cyclooxygenase 2 and NADH Dehydrogenase Subunit 2, subunits of the respiratory complex encoded by mitochondrial genes (Mohammed et al., 2020). PARylation helps regulate the expression of mtRNA repair proteins such as UNG1 and MYH1, as PARP-1 is located at the promoter of the genes that encode them (Lapucci et al., 2011; Mohammed et al., 2020). PARP-1 deletion may disrupt the epigenetic regulation of nuclear genes involved in mtDNA repair, thereby impairing mitochondrial homeostasis. In addition, it has been proposed that activated PARP-1 in patients with AD can block mitochondrial respiration and impair mitochondrial function by lowering the expression of peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α). NAD+ plays an important role in mitochondrial redox balance and the tricarboxylic acid cycle (Lu et al., 2019). Two consumers of NAD+ are PARP-1 and Sirtuin 1 (SIRT1), which function antagonistically (Krishnakumar and Kraus, 2010). PGC-1α is a core regulator of mitochondrial function and acts as a transcriptional coactivator with transcriptional activity and is subject to acetylation, which is tightly regulated by SIRT1 (Cui et al., 2021). Therefore, overactivated PARP-1 induces NAD+ deficiency, blocking SIRT1 deacetylase activity, which affects the acetylation status of PGC-1α and mitochondrial function. Familial AD is associated with aggregated toxic Aβ is also strongly associated with NAD+ consumption and PARP-1 activation (Martire et al., 2013; Yu et al., 2021). Studies by Yu YZ on Drosophila overexpressing Aβ showed that PARP gene mutation or pharmacological supplementation with NAD+ precursors rescued mitochondrial dysfunction, supporting the idea that PARP polymorphisms and NAD+ bioavailability are linked to Aβ toxicity and AD severity (Yu et al., 2021).
Mitochondrial damage is closely related to altered mitochondrial morphology and abnormal mitochondrial dynamics, including processes such as mitochondrial fission, fusion, biogenesis and mitophagy (mitochondrial autophagy) (Lee et al., 2020). Among these processes, mitophagy clears and degrades damaged mitochondria and is a key mechanism in cellular homeostasis. Under pathological conditions, the mitochondrial membrane potential decreases, resulting in the inability of the mitochondrial kinase PTEN Induced Kinase 1 (PINK1) (Park6) to be properly sheared and degraded, and therefore, accumulates in the mitochondria. Aggregated PINK1 recruits the ubiquitin ligase E3 Ubiquitin Ligase (Parkin) (Park2) to the outer mitochondrial membrane, causing the degradation of mitochondrial surface proteins, which is required for the subsequent clearance of damaged mitochondria. This process is known as mitophagy (Jin and Youle, 2012; Lin et al., 2020). Dysregulated mitophagy, especially in long-lived cells such as neurons, promotes the development and progression of NDs. In addition, PINK1 and Parkin are known genetic factors in PD pathology. Mutations in PINK1 and Parkin result in impaired mitophagy and defective mitochondrial mass monitoring (Pickrell and Youle, 2015). Recently, inhibition of PARP has been shown to increase NAD+ levels and mitochondrial function in PINK1 or Parkin mutants (Yu et al., 2021). In Parkin-deficient models of mitochondrial dysfunction, decreasing PARP activity or increasing NAD+ bioavailability increased mitochondrial function and protected neurons from degeneration (Lehmann et al., 2016). Furthermore, in PINK1 mutants, increased oxidative stress and NAD+ depletion were associated with PARP activation. In contrast, mutation of PARP greatly attenuated the increase in protein PARylation and reversed the mitochondrial morphology defects and △ψm in the PINK1 mutant. PARP mutation is neuroprotective in the Drosophila PINK1 mutation model (Lehmann et al., 2017). Collectively, inhibiting PARP-1 may protect neurons from degeneration by regulating mtDNA and mitochondrial function (Figure 3).
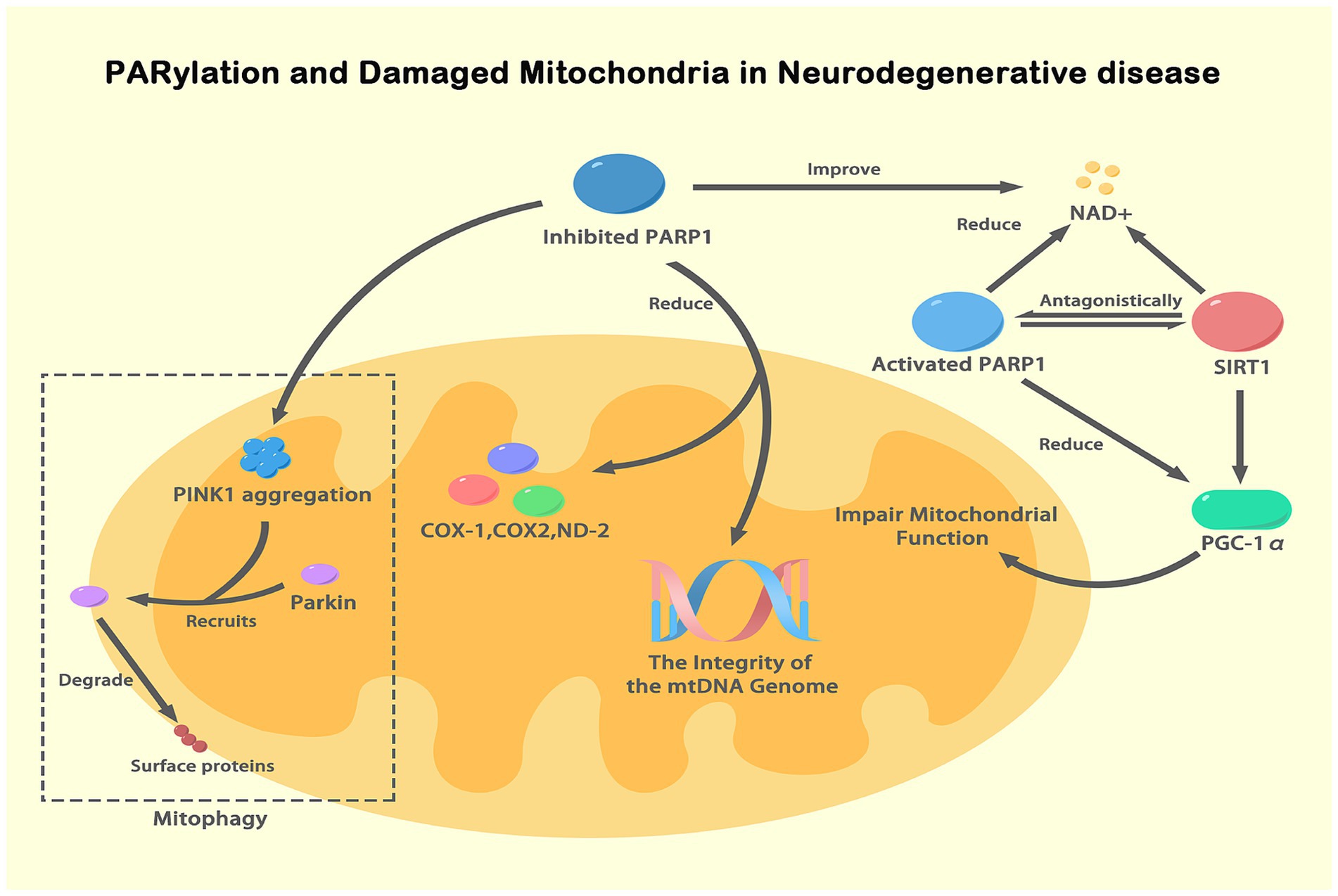
Figure 3. PARylation and damaged mitochondria in NDs. This figure illustrates the role of PARylation (a specific form of ADPRylation) in mitochondrial dysfunction associated with NDs. Activated PARP-1, in response to DNA damage, depletes NAD+ levels, impairing SIRT1 activity and the deacetylation of PGC-1α, thus reducing mitochondrial biogenesis and function. Inhibition of PARP-1 improves NAD+ availability, enhancing mitochondrial function and maintaining mtDNA integrity. NAD+ serves as a substrate for both PARP-1 and SIRT1, functioning antagonistically. PINK1 accumulates on the outer mitochondrial membrane under mitochondrial dysfunction, recruiting Parkin, which leads to the degradation of mitochondrial surface proteins and mitophagy. This figure highlights how PARylation, particularly through PARP-1, regulates mitochondrial function and integrity, contributing to neurodegenerative disease pathogenesis by influencing NAD+ levels and key mitochondrial processes.
4 ADPRylation and RNA-binding proteins in neurodegenerative diseases
ALS, also known as motor neuron disease (MND), is characterized by progressive muscle atrophy, feebleness and ultimately breathing failure (Al-Sarraj et al., 2011). To date, the most severely affected motor neurons in ALS are in the cortex, brainstem and spinal cord (Peters et al., 2015). Currently, the etiology of ALS is unknown, but 10–20% of cases may be associated with hereditary and genetic defects (Machado et al., 2019). More than 30 genes have been linked to ALS, with FUS, Superoxide Dismutase 1 and TAR DNA-binding Protein (TARDBP) genes being the three most common ALS-associated genes (Hou et al., 2016; Nakamura et al., 2020). When DNA damage occurs, FUS localizes to the site of the damaged DNA, is subsequently PARylated and then contributes to DNA damage repair, but FUS mislocalization expedites ALS progression in a PARP-1-dependent manner (Rulten et al., 2014). ADP-ribosylation has been confirmed to regulate several disease signaling pathways involved in the pathological progression of ALS, with the PARylation of RNA-binding proteins (RBPs) notable (Duan et al., 2019; McGurk et al., 2019). Some RBPs have one or more conserved RNA-binding domains (RBDs) and PAR-binding motifs (PBMs), explaining why PARP or PARylation impacts RNA homeostasis, such as through translocation and turnover (Forman et al., 2021). Notably, FUS is an RBP, and mutations in its PBMs may lead to aberrant phase separation and affect dynamic RNA interactions (Niaki et al., 2020). In addition, other pathological RBPs localized to ribonucleoprotein (RNP) granules in ALS are hnRNP A1 with a PARylation site at K298 and containing a PBM (Farina et al., 2021). Unexpectedly, changes in PARylation rates and PAR-binding levels of hnRNP A1 were found to be related, and hnRNP A1 mediated its own cytoplasmic translocation and association with stress granules based on these changes (Duan et al., 2019). Notably, PAR thus sequesters nontranslating mRNAs and increases aberrant protein aggregation by regulating disease-related RBPs.
Furthermore, the normal nuclear RNA-binding protein involved in transactive response (TAR) DNA-binding protein 43 (TDP-43, encoded by TARDBP) is a general pathogenic factor in ALS that is mislocalized to the cytoplasm, forming a ubiquitin-positive complex that is with hyperphosphorylated (Prasad et al., 2019; Chen, 2021). When DNA damage occurs, TDP-43 is recruited to the DNA lesion, facilitating the repair of the damaged DNA (Wood et al., 2020). Therefore, loss of nuclear TDP-43 and the subsequent increase in DNA damage is an early symptom of ALS. However, it remains to be determined how pathological TDP-43 is translocated to the cytoplasm. Leeanne McGurk reported that TDP-43 binds noncovalently to PAR at PAR-binding motifs, which are among the nuclear localization sequences om TDP-43; thus, PAR promotes TDP-43 accumulation by stimulating stress granule formation and liquid–liquid phase separation (McGurk et al., 2018a). In particular, under sustained stress, TDP-43 is phosphorylated and combines with other binding proteins, such as FUS, Atain 2 and hnRNPA1, to form stress granules that initiate pathological aggregation and promote progressive degeneration (Dao et al., 2019; Gasset-Rosa et al., 2019). Moreover, PARylation facilitates the progress of stress granule formation by mediating the condensation of the RNA–protein complex in a dynamic droplet, a process known as liquid–liquid separation (Leung, 2020; Jin et al., 2021). Finally, a small-molecule inhibitor of PARP-1/PARP-2 can block cytoplasmic TDP-43 foci and neurodegeneration in ALS (McGurk et al., 2018a). Similar to PARP-1/2, PARP-5a/5b activation-induced PARylation has also been revealed to aggravate TDP-43 expression in the cytoplasm and neurotoxicity (McGurk et al., 2018b). Collectively, the disease-linked pathologies related to FUS, hnRNP A1 and TDP-43 all highly rely on PARP-mediated nucleoplasmic transport and stress granules; therefore, agents that inhibit PARylation or PAR chain formation are therapeutic tools for ALS (Figure 4).
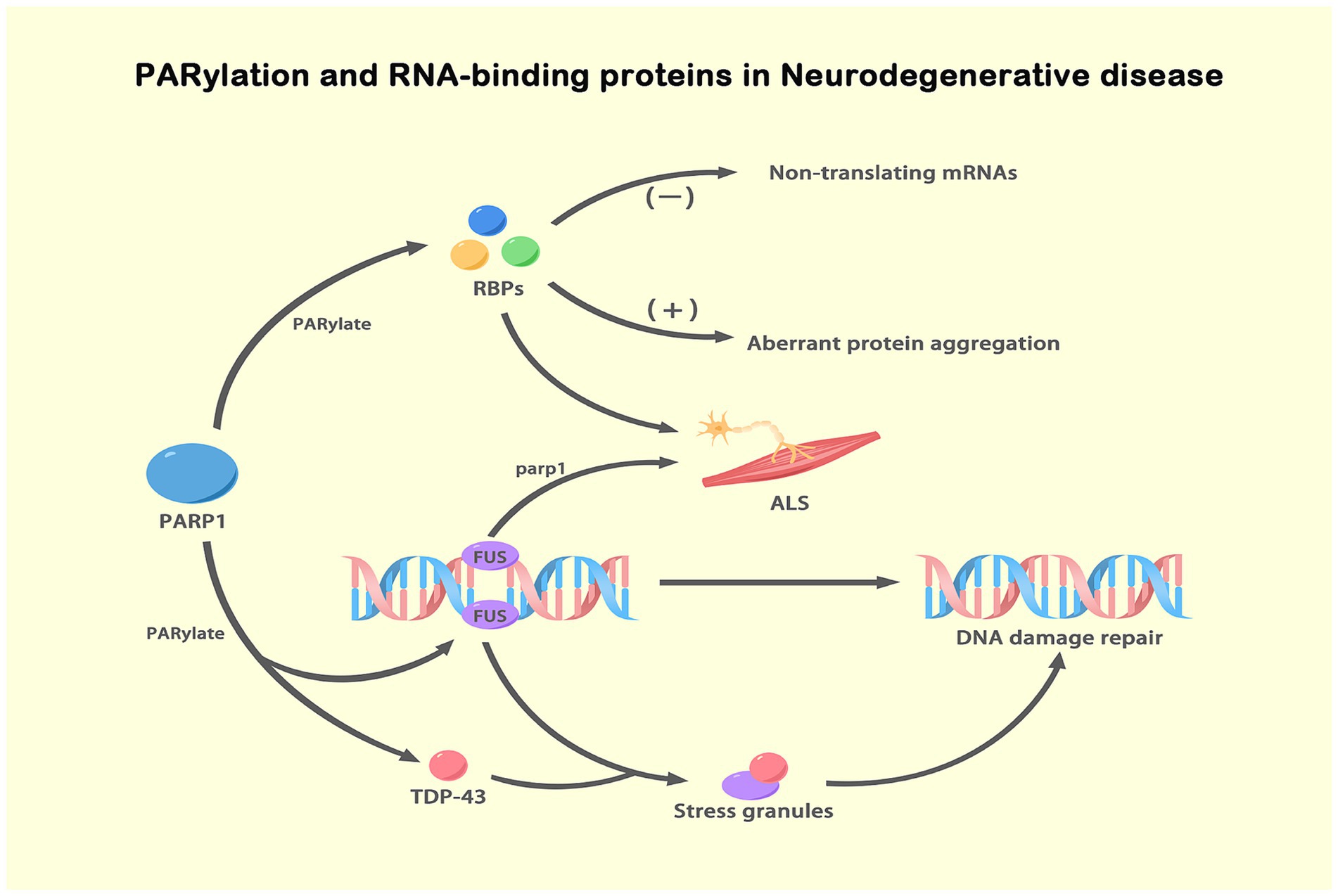
Figure 4. PARylation and RBPs in NDs. This figure illustrates the role of ADPRylation, specifically PARylation, in the regulation of RBPs and their impact on NDs, particularly ALS. PARylation, a specific form of ADP-ribosylation mediated by PARP1, modifies RBPs such as FUS and TDP-43. Upon DNA damage, FUS is PARylated by PARP-1 and recruited to DNA damage sites, facilitating DNA repair. Simultaneously, TDP-43 is PARylated and localizes to stress granules under stress conditions. This PARylation modification impacts RNA metabolism and protein aggregation, leading to aberrant protein interactions and the formation of toxic aggregates. These processes contribute to ALS pathogenesis. PARylation, through the regulation of RBPs, plays a critical role in maintaining genomic stability and regulating stress responses. The figure underscores the importance of PARylation in modulating key cellular processes involved in neurodegenerative disease progression.
5 ADPRylation and inflammatory response
The PARP catalytic domain transfers the ADP ribose portion from NAD+ to the amino acid residues of a target protein, resulting in mono- or poly-ADP-ribosylation. Therefore, PARP members are “writers” of ADP ribose. Accumulating evidence shows that chronic inflammation is related to various pathological processes, including NDs (Gelders et al., 2018; Kwon and Koh, 2020) and aging (Bektas et al., 2018). A series of studies have shown that signal transduction pathways participate in the inflammatory response and are regulated by posttranslational modifications of proteins, such as ubiquitination, phosphorylation, methylation and acetylation (Chen et al., 2018; Francis et al., 2019; Leng et al., 2021). This section focuses on the relationship between ADPRylation and the inflammatory response and summarizes their roles in NDs.
Some evidence suggests a potential connection between ADPRylation and inflammation. PARP-1 is associated with a proinflammatory response. In some cases, PARP-1 promotes an increase in the expression proinflammatory mediators, such as NF-κB, which do not depend on enzymatic activity, suggesting that PARP-1 is involved in the mechanisms underlying inflammation (Kauppinen and Swanson, 2007; Kim and Kang, 2019). Neuroinflammation plays an important role in the etiology of different nervous system diseases. Inflammatory responses in the nervous system are mediated by the inflammasome, activated immune molecules, and glial cells (Morimoto and Nakajima, 2019). However, chronic activation of inflammation can accelerates the progression of neurodegeneration (Beck et al., 2014). Alberto Chiarug reported that PARP-1 enzyme activity and PARylation are important to glial cell transcriptional activation and proved that PARP-1 activity-dependent regulation of NF-κB is a potential strategy for attenuating neuroinflammation and neurodegeneration (Komirishetty et al., 2017). In addition, PARP can regulate the levels of high mobility group box 1 (HMGB1). HMGB1 in the extracellular environment can activate microglia and increase the expression of inflammatory cytokines, which contribute to PD. Notably, anti-HMGB1 antibodies have been used to treat PD in mice (Yang et al., 2014; Sasaki et al., 2016). Moreover, the overactivation of PARP-1 in microglia directly impaired glutamate uptake, which is related to chronic neuroinflammation in NDs (Suh et al., 2007; Doaee et al., 2019). Various published reports have shown that PARP-1 inhibitors reduce the expression of proinflammatory cytokines, such as tumor necrosis factor α (TNF-α), interleukin-1 (IL-1) and IL-6, which manifests as a reduction in the mRNA levels of these inflammatory mediators (Liu Z. et al., 2021; Wasyluk and Zwolak, 2021). A recent study by Kunze et al. reported that ADP-ribosyl transferase diphosphate toxin like 1 (ARTD1) increased the lipopolysaccharide (LPS)-induced transcriptional upregulation of a series of genes, such as IL-12, TNF-a, and IL-6. These results emphasize the proinflammatory potential of PARP-1 (Love et al., 1999; Kunze et al., 2019). It has been shown that PARP interacts with Aβ and Tau in the brains of AD patients (Bagyinszky et al., 2020). Studies have confirmed that PARP-1 inhibition can slow the progression of AD (Salech et al., 2017; Hu et al., 2018). PARP knockout and treatment with PARP inhibitors can effectively reduce the inflammatory response caused by microglial activation and neurodegeneration in AD (Stoica et al., 2014; Mao and Zhang, 2021). Moreover, Abby L. Olsen proposed a PARP-1 inhibitor as a potential drug for PD treatment, but its mechanism of action is not clear (Olsen and Feany, 2019). The PARP-1 level is increased in PD models, including TgCRND8 mice with double Swedish (KM670/671/NL) and Indiana (V717F) mutations. Activated PARP-1 causes mitochondrial damage and directly modifies α-syn into more toxic strains, which accelerates the progression of PD (Kam et al., 2018). Recently, our team found that poly (ADP-ribose) polymerase 1 inhibition promoted α-syn degradation via transcription factor EB-dependent autophagy in a mutant α-synA53T model of Parkinson’s disease (Mao et al., 2020). Hitherto, extensive research on the relationship between ADPRylation and inflammation is still lacking, and we are actively working to make advancements in this field. As shown in our published data, we found that PARP led to the activation of NOD-Like Receptor Protein 3 by inhibiting autophagy, which exacerbated the progression of PD (Zhang et al., 2023; Figure 5).
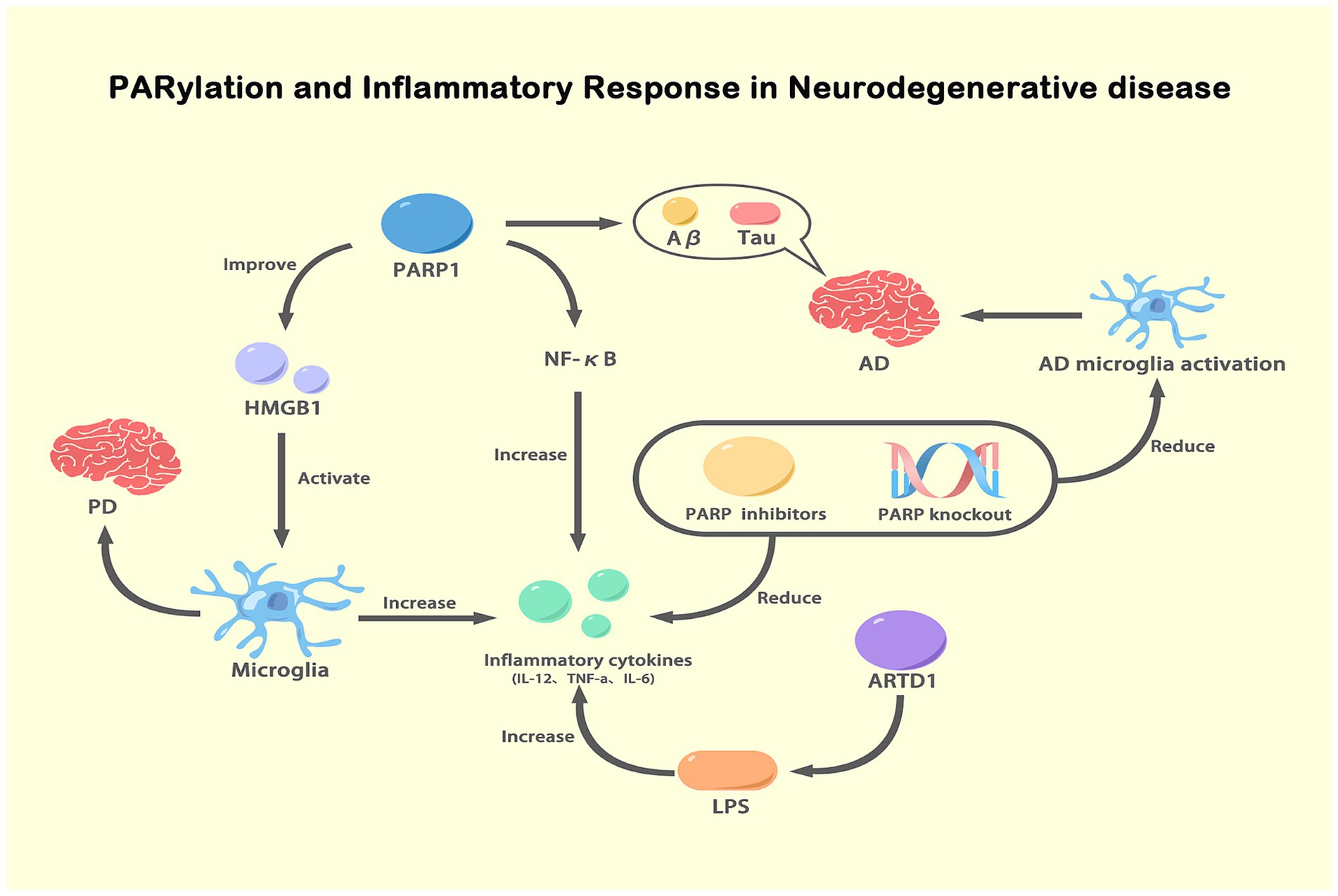
Figure 5. PARylation and inflammatory response in NDs. This figure illustrates the role of PARylation in modulating inflammatory responses in NDs. Activated PARP-1 enhances the expression and activity of NF-κB, leading to increased production of pro-inflammatory cytokines (IL-12, TNF-α, IL-6). These cytokines exacerbate inflammation and are involved in the pathogenesis of AD and PD. PARP-1 activation also promotes the release of HMGB1, which activates microglia, further increasing inflammation. In AD, PARP-1 activation is associated with the accumulation of Aβ and tau proteins, which activate microglia and contribute to disease progression. Inhibition or knockout of PARP reduces NF-κB activity and pro-inflammatory cytokine levels, thereby decreasing microglial activation and inflammation. Additionally, ARTD1, another ADP-ribosyltransferase, increases inflammatory cytokine expression in response to LPS stimulation. This figure highlights the critical role of PARylation in regulating inflammatory responses and its potential as a therapeutic target in NDs.
6 Conclusion and future direction
In recent years, researchers have consistently suggested PARP activation plays an extremely important role in NDs, but its potential mechanism remains to be explored. In NDs, PARP-1 is activated due to the physiological characteristics of neurons and DNA damage, and α-syn undergoes pathological aggregation, disrupting the normal function of neurons and even leading to neuronal death. Mitochondrial oxidative phosphorylation plays a crucial role in energy metabolism, and neurons are highly energy dependent. Selective PARP-1 inhibitors, such as Olaparib, have been shown to reduce α-syn aggregation in PD models (Kam et al., 2018). However, it is important to note that PARP-5a/5b inhibitors may exacerbate the cytoplasmic localization of TDP-43 (McGurk et al., 2018b), highlighting that subtype selectivity is crucial in drug development. These findings suggest that PARylation or PARP-1 is involved in a widely altered DNA damage response in PD, indicating the therapeutic value of PARP-1 inhibitors. Mitochondria in brain tissue from PD and AD patients, defective mitochondrial genome integrity, mtDNA depletion and mtDNA rearrangements have been identified in patients with NDs. In addition, RBPs and the inflammatory response also play important roles in ADPRylation. It has been shown that inhibiting PARP-1 reduces the integrity of the mtDNA genome and the aggregation of pathological α-syn. In addition, it has been proposed that activation of PARP-1 is necessary for the formation of long-term memory, which is related to the integrity of nucleoli, rRNA and synaptic plasticity (Cohen-Armon et al., 2004; Goldberg et al., 2009). In the nuclei of hippocampus cells in AD patients, PARP-1 is lost, indicating that PARP-1 may be impaired in the function of the nucleoli. Some studies have suggested that PARP-1 causes cognitive impairment in AD patients for two reasons, one of which is because the loss of neuronal nucleoli PARP-1 may lead to synaptic plasticity defects, resulting in cognitive impairment; in the other possibility, in late stages of AD, PARP-1 may be overactivated and cause brain cell death (Zeng et al., 2016; Mao and Zhang, 2021). PARP-1 inhibitors can block the formation of long-term memory (Visochek et al., 2016), but treatment of AD patients with PARP-1 inhibitors has not been extensively studied. Recent advancements in PARP inhibitor development have demonstrated their growing potential for NDs treatment. Pharmaceutical companies and academic laboratories have identified novel brain-penetrant PARP inhibitors, further emphasizing the applicability of these compounds as potential therapeutics and imaging probes for NDs (Lengyel-Zhand et al., 2022). As a result, it is now timely to explore the possibility of repurposing these inhibitors for use beyond cancer treatment, particularly in the context of central nervous system (CNS) disorders where DNA damage and inflammation play significant roles. Specifically, parthanatos, a regulated form of necrosis associated with PARP-1 overactivation, is gaining attention as a critical target for intervention in CNS injuries (Mekhaeil et al., 2022). Parthanatos has been implicated in NDs, where the overactivation of PARP-1 leads to excessive ADPRylation, mitochondrial dysfunction, and neuronal cell death, particularly in response to oxidative stress and excitotoxicity (Zhang et al., 2025). As recent studies suggest, inhibiting PARP-1 in models of CNS injury, including traumatic brain injury and spinal cord injury, has demonstrated therapeutic promise by mitigating cellular damage and promoting tissue preservation (Berger et al., 2017). Therefore, integrating insights from parthanatos research into PARP inhibitor development could offer a new avenue for treating CNS disorders, positioning these inhibitors as dual-purpose agents for both therapeutic and diagnostic applications in NDs. However, the exact molecular mechanisms by which PARP-1 contributes to CNS development or to the progression of various NDs are incompletely understood. Due to its multifunctional nature, the biological pathways involving PARP-1 do not operate in isolation; instead, they run alongside other molecular processes, suggesting a complex interplay in regulating cell behavior and determining cellular outcomes. The comprehensive and in-depth study of the molecular mechanism underlying PARP-1 action is beneficial to the exploration of drugs in the treatment of NDs. PARP inhibitors have gradually shown potential for use in the treatment of NDs. The combination of further molecular mechanism exploration and clinical trials will provide strong evidence for their practical application in NDs treatments.
Author contributions
CH: Conceptualization, Funding acquisition, Investigation, Writing – original draft. HX: Supervision, Writing – review & editing. YY: Supervision, Writing – review & editing. JL: Supervision, Writing – review & editing. YL: Supervision, Writing – review & editing. SL: Visualization, Writing – review & editing. KM: Project administration, Supervision, Writing – review & editing. JC: Supervision, Writing – review & editing, Conceptualization, Funding acquisition, Resources. LW: Supervision, Writing – review & editing, Project administration.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was funded by the National Natural Science Foundation of China (82103879), GuangDong Basic and Applied Basic Research Foundation (2021B1515140032, 2022A1515140173, 2023A1515140164), Guangdong Medical University Clinical and Basic Technology Innovation Special Program (GDMULCJC2024152, GDMULCJC2024153, GDMULCJC140) and Dongguan Social Development Science and Technology (Key) Project (Dongguan Shipai Hospital) (20211800905332).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aberle, L., Kruger, A., Reber, J. M., Lippmann, M., Hufnagel, M., Schmalz, M., et al. (2020). PARP1 catalytic variants reveal branching and chain length-specific functions of poly(ADP-ribose) in cellular physiology and stress response. Nucleic Acids Res. 48, 10015–10033. doi: 10.1093/nar/gkaa590
Al-Sarraj, S., King, A., Troakes, C., Smith, B., Maekawa, S., Bodi, I., et al. (2011). p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 122, 691–702. doi: 10.1007/s00401-011-0911-2
Antonyova, V., Kejik, Z., Brogyanyi, T., Kaplanek, R., Pajkova, M., Talianova, V., et al. (2020). Role of mtDNA disturbances in the pathogenesis of Alzheimer's and Parkinson's disease. DNA Repair 91-92:102871. doi: 10.1016/J.Dnarep.2020.102871
Bagyinszky, E., Giau, V. V., and An, S. A. (2020). Transcriptomics in Alzheimer’s disease: aspects and challenges. Int. J. Mol. Sci. 21:3517. doi: 10.3390/ijms21103517
Basello, D. A., and Scovassi, A. I. (2015). Poly(ADP-ribosylation) and neurodegenerative disorders. Mitochondrion 24, 56–63. doi: 10.1016/j.mito.2015.07.005
Bates, G. (2003). Huntingtin aggregation and toxicity in Huntington's disease. Lancet 361, 1642–1644. doi: 10.1016/S0140-6736(03)13304-1
Beck, C., Robert, I., Reina-San-Martin, B., Schreiber, V., and Dantzer, F. (2014). Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 329, 18–25. doi: 10.1016/j.yexcr.2014.07.003
Bektas, A., Schurman, S. H., Sen, R., and Ferrucci, L. (2018). Aging, inflammation and the environment. Exp. Gerontol. 105, 10–18. doi: 10.1016/j.exger.2017.12.015
Belanger, M., Allaman, I., and Magistretti, P. J. (2011). Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 14, 724–738. doi: 10.1016/j.cmet.2011.08.016
Berger, N. A., Besson, V. C., Boulares, A. H., Bürkle, A., Chiarugi, A., Clark, R. S., et al. (2017). Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br. J. Pharmacol. 175, 192–222. doi: 10.1111/bph.13748
Bian, C. J., Zhang, C., Luo, T., Vyas, A., Chen, S. H., Liu, C., et al. (2019). NADP(+) is an endogenous PARP inhibitor in DNA damage response and tumor suppression. Nat. Commun. 10:693. doi: 10.1038/s41467-019-08530-5
Brunyanszki, A., Olah, G., Coletta, C., Szczesny, B., and Szabo, C. (2014). Regulation of mitochondrial poly(ADP-ribose) polymerase activation by the beta-adrenoceptor/cAMP/protein kinase a Axis during oxidative stress. Mol. Pharmacol. 86, 450–462. doi: 10.1124/mol.114.094318
Cao, X. Q., Wada, M., Hayakawa, H., Ren, Y. R., Yasuda, T., Hattori, N., et al. (2008). The analysis of PD mouse model induced by continuous MPTP infusion. Neurosci. Res. 61:S211. doi: 10.1016/j.neures.2008.05.002
Cardinale, A., Paldino, E., Giampa, C., Bernardi, G., and Fusco, F. R. (2015). PARP-1 inhibition is neuroprotective in the R6/2 mouse model of Huntington's disease. PLoS One 10:e0134482. doi: 10.1371/journal.pone.0134482
Chaturvedi, R. K., Hennessey, T., Johri, A., Tiwari, S. K., Mishra, D., Agarwal, S., et al. (2012). Transducer of regulated CREB-binding proteins (TORCs) transcription and function is impaired in Huntingtons disease. Hum. Mol. Genet. 21, 3474–3488. doi: 10.1093/hmg/dds178
Chen, L. (2021). The important functional role of TDP-43 plays in amyotrophic lateral sclerosis-frontotemporal dementia. Neural Regen. Res. 16, 682–683. doi: 10.4103/1673-5374.293142
Chen, V., Moncalvo, M., Tringali, D., Tagliafierro, L., Shriskanda, A., Ilich, E., et al. (2020). The mechanistic role of alpha-synuclein in the nucleus: impaired nuclear function caused by familial Parkinson's disease SNCA mutations. Hum. Mol. Genet. 29, 3107–3121. doi: 10.1093/hmg/ddaa183
Chen, H. L., Tan, H. L., Yang, J., Wei, Y. Y., and Hu, T. J. (2018). Sargassum polysaccharide inhibits inflammatory response in PCV2 infected-RAW264.7 cells by regulating histone acetylation. Carbohydr. Polym. 200, 633–640. doi: 10.1016/j.carbpol.2018.06.060
Cheng, Q., Chen, J., Guo, H., Lu, J. L., Zhou, J., Guo, X. Y., et al. (2021). Pyrroloquinoline quinone promotes mitochondrial biogenesis in rotenone-induced Parkinson's disease model via AMPK activation. Acta Pharmacol. Sin. 42, 665–678. doi: 10.1038/s41401-020-0487-2
Cohen-Armon, M., Visochek, L., Katzoff, A., Levitan, D., Susswein, A. J., Klein, R., et al. (2004). Long-term memory requires polyADP-ribosylation. Science 304, 1820–1822. doi: 10.1126/science.1096775
Cui, W. X., Wu, X., Shi, Y. W., Guo, W., Luo, J. N., Liu, H. X., et al. (2021). 20-HETE synthesis inhibition attenuates traumatic brain injury-induced mitochondrial dysfunction and neuronal apoptosis via the SIRT1/PGC-1 alpha pathway: a translational study. Cell Prolif. 54:e12964. doi: 10.1111/cpr.12964
Dao, T. P., Martyniak, B., Canning, A. J., Lei, Y. N., Colicino, E. G., Cosgrove, M. S., et al. (2019). ALS-linked mutations affect UBQLN2 oligomerization and phase separation in a position- and amino acid-dependent manner. Structure 27:937. doi: 10.1016/j.str.2019.03.012
Dionisio, P. A., Amaral, J. D., and Rodrigues, C. M. P. (2021). Oxidative stress and regulated cell death in Parkinson's disease. Ageing Res. Rev. 67:101263. doi: 10.1016/j.arr.2021.101263
Doaee, P., Rajaei, Z., Roghani, M., Alaei, H., and Kamalinejad, M. (2019). Effects of Boswellia serrata resin extract on motor dysfunction and brain oxidative stress in an experimental model of Parkinson's disease. Avicenna J Phytomed 9, 281–290. doi: 10.22038/AJP.2019.12403
Duan, Y. J., Du, A. Y., Gu, J. G., Duan, G., Wang, C., Gui, X. R., et al. (2019). PARylation regulates stress granule dynamics, phase separation, and neurotoxicity of disease-related RNA-binding proteins. Cell Res. 29, 233–247. doi: 10.1038/s41422-019-0141-z
Farina, S., Esposito, F., Battistoni, M., Biamonti, G., and Francia, S. (2021). Post-translational modifications modulate Proteinopathies of TDP-43, FUS and hnRNP-A/B in amyotrophic lateral sclerosis. Front. Mol. Biosci. 8:693325. doi: 10.3389/fmolb.2021.693325
Fatokun, A. A., Dawson, V. L., and Dawson, T. M. (2014). Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 171, 2000–2016. doi: 10.1111/bph.12416
Fischbach, A., Kruger, A., Hampp, S., Assmann, G., Rank, L., Hufnagel, M., et al. (2018). The C-terminal domain of p53 orchestrates the interplay between non-covalent and covalent poly(ADP-ribosyl)ation of p53 by PARP1. Nucleic Acids Res. 46, 804–822. doi: 10.1093/nar/gkx1205
Forman, T. E., Dennison, B. J. C., and Fantauzzo, K. A. (2021). The role of RNA-binding proteins in vertebrate neural crest and craniofacial development. J Dev. Biol. 9:34. doi: 10.3390/jdb9030034
Francis, M., Pandya, M., Gopinathan, G., Lyu, H. L., Ma, W., Foyle, D., et al. (2019). Histone methylation mechanisms modulate the inflammatory response of periodontal ligament progenitors. Stem Cells Dev. 28, 1015–1025. doi: 10.1089/scd.2019.0125
Gao, R., Chakraborty, A., Geater, C., Pradhan, S., Gordon, K. L., Snowden, J., et al. (2019). Mutant huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription. eLife 8:e42988. doi: 10.7554/eLife.42988
Gasset-Rosa, F., Lu, S., Yu, H. Y., Chen, C., Melamed, Z., Guo, L., et al. (2019). Cytoplasmic TDP-43 De-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron 102:339. doi: 10.1016/j.neuron.2019.02.038
Gelders, G., Baekelandt, V., and Van der Perren, A. (2018). Linking Neuroinflammation and neurodegeneration in Parkinson's disease. J Immunol Res 2018:4784268. doi: 10.1155/2018/4784268
Goldberg, S., Visochek, L., Giladi, E., Gozes, I., and Cohen-Armon, M. (2009). PolyADP-ribosylation is required for long-term memory formation in mammals. J. Neurochem. 111, 72–79. doi: 10.1111/j.1471-4159.2009.06296.x
Gonzalez-Hunt, C. P., and Sanders, L. H. (2021). DNA damage and repair in Parkinson's disease: recent advances and new opportunities. J. Neurosci. Res. 99, 180–189. doi: 10.1002/jnr.24592
Goodliffe, J., Rubakovic, A., Chang, W. E., Pathak, D., and Luebke, J. (2020). Structural and functional features of medium spiny neurons in the BACHD Delta N17 mouse model of Huntington's disease. PLoS One 15:e0234394. doi: 10.1371/journal.pone.0234394
Groslambert, J., Prokhorova, E., and Ahel, I. (2021). ADP-ribosylation of DNA and RNA. DNA Repair 105:103144. doi: 10.1016/j.dnarep.2021.103144
Gupta, S., You, P. N., SenGupta, T., Nilsen, H., and Sharma, K. (2021). Crosstalk between different DNA repair pathways contributes to neurodegenerative diseases. Biology 10:163. doi: 10.3390/biology10020163
Gurunathan, S., Jeyaraj, M., Kang, M. H., and Kim, J. H. (2019). Mitochondrial peptide Humanin protects silver nanoparticles-induced neurotoxicity in human neuroblastoma Cancer cells (SH-SY5Y). Int. J. Mol. Sci. 20:4439. doi: 10.3390/ijms20184439
Hendriks, I. A., Buch-Larsen, S. C., Prokhorova, E., Elsborg, J. D., Rebak, A., Zhu, K., et al. (2021). The regulatory landscape of the human HPF1- and ARH3-dependent ADP-ribosylome. Nat. Commun. 12:5893. doi: 10.1038/s41467-021-26172-4
Hou, L. H., Jiao, B., Xiao, T. T., Zhou, L., Zhou, Z. F., Du, J., et al. (2016). Screening of SOD1, FUS and TARDBP genes in patients with amyotrophic lateral sclerosis in Central-Southern China. Sci. Rep. 6:32478. doi: 10.1038/srep32478
Hou, Y. J., Lautrup, S., Cordonnier, S., Wang, Y., Croteau, D. L., Zavala, E., et al. (2018). NAD(+) supplementation normalizes key Alzheimer's features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 115, E1876–E1885. doi: 10.1073/pnas.1718819115
Houl, J. H., Ye, Z., Brosey, C. A., Balapiti-Modarage, L. P. F., Namjoshi, S., Bacolla, A., et al. (2019). Selective small molecule PARG inhibitor causes replication fork stalling and cancer cell death. Nat. Commun. 10:5654. doi: 10.1038/s41467-019-13508-4
Hu, X.-M., Dong, W., Cui, Z.-W., Gao, C.-Z., Yu, Z.-J., Yuan, Q., et al. (2018). In silico identification of AChE and PARP-1 dual-targeted inhibitors of Alzheimer’s disease. J. Mol. Model. 24:151. doi: 10.1007/s00894-018-3696-6
Iida, N., Dzutsev, A., Stewart, C. A., Smith, L., Bouladoux, N., Weingarten, R. A., et al. (2013). Commensal Bacteria control Cancer response to therapy by modulating the tumor microenvironment. Science 342, 967–970. doi: 10.1126/science.1240527
Jenkins, L. M. M., Durell, S. R., Mazur, S. J., and Appella, E. (2012). p53 N-terminal phosphorylation: a defining layer of complex regulation. Carcinogenesis 33, 1441–1449. doi: 10.1093/carcin/bgs145
Jeong, H., Cohen, D. E., Cui, L. B., Supinski, A., Savas, J. N., Mazzulli, J. R., et al. (2012). Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat. Med. 18, 159–165. doi: 10.1038/nm.2559
Jin, X. J., Cao, X. L., Liu, S. K., and Liu, B. D. (2021). Functional roles of poly(ADP-ribose) in stress granule formation and dynamics. Fronti. Cell Dev. Biol. 9:671780. doi: 10.3389/fcell.2021.671780
Jin, S. M., and Youle, R. J. (2012). PINK1-and Parkin-mediated mitophagy at a glance. J. Cell Sci. 125, 795–799. doi: 10.1242/jcs.093849
Kam, T. I., Mao, X., Park, H., Chou, S. C., Karuppagounder, S. S., Umanah, G. E., et al. (2018). Poly(ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson's disease. Science 362:eaat8407. doi: 10.1126/science.aat8407
Karve, T. M., and Cheema, A. K. (2011). Small changes huge impact: the role of protein posttranslational modifications in cellular homeostasis and disease. J. Amino Acids 2011, 1–13. doi: 10.4061/2011/207691
Kauppinen, T. M., and Swanson, R. A. (2007). The role of poly(ADP-ribose) polymerase-1 in CNS disease. Neuroscience 145, 1267–1272. doi: 10.1016/j.neuroscience.2006.09.034
Kharrati-Koopaee, H., Ebrahimie, E., Dadpasand, M., Niazi, A., Tian, R. G., and Esmailizadeh, A. (2021). Gene network analysis to determine the effect of hypoxia-associated genes on brain damages and tumorigenesis using an avian model. J. Genet. Eng. Biotechnol. 19:100. doi: 10.1186/s43141-021-00184-5
Kim, J. E., and Kang, T. C. (2019). PKC, AKT and ERK1/2-mediated modulations of PARP1, NF-kappaB and PEA15 activities distinctly regulate regional specific Astroglial responses following status epilepticus. Front. Mol. Neurosci. 12:180. doi: 10.3389/fnmol.2019.00180
Kliza, K. W., Liu, Q., Roosenboom, L. W. M., Jansen, P. W. T. C., Filippov, D. V., and Vermeulen, M. (2021). Reading ADP-ribosylation signaling using chemical biology and interaction proteomics. Mol. Cell 81:4552. doi: 10.1016/j.molcel.2021.08.037
Komirishetty, P., Areti, A., Gogoi, R., Sistla, R., and Kumar, A. (2017). Combination strategy of PARP inhibitor with antioxidant prevent bioenergetic deficits and inflammatory changes in CCI-induced neuropathy. Neuropharmacology 113, 137–147. doi: 10.1016/j.neuropharm.2016.09.027
Krishnakumar, R., and Kraus, W. L. (2010). The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol. Cell 39, 8–24. doi: 10.1016/j.molcel.2010.06.017
Kunze, F. A., Bauer, M., Komuczki, J., Lanzinger, M., Gunasekera, K., Hopp, A. K., et al. (2019). ARTD1 in myeloid cells controls the IL-12/18-IFN-gamma Axis in a model of sterile Sepsis, chronic bacterial infection, and Cancer. J. Immunol. 202, 1406–1416. doi: 10.4049/jimmunol.1801107
Kwon, H. S., and Koh, S. H. (2020). Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl. Neurodegeneration 9:42. doi: 10.1186/s40035-020-00221-2
Lapucci, A., Pittelli, M., Rapizzi, E., Felici, R., Moroni, F., and Chiarugi, A. (2011). Poly(ADP-ribose) Polymerase-1 is a nuclear epigenetic regulator of mitochondrial DNA repair and transcription. Mol. Pharmacol. 79, 932–940. doi: 10.1124/mol.110.070110
Lauritzen, K. H., Olsen, M. B., Ahmed, M. S., Yang, K., Rinholm, J. E., Bergersen, L. H., et al. (2021). Instability in NAD(+) metabolism leads to impaired cardiac mitochondrial function and communication. eLife 10:e59828. doi: 10.7554/eLife.59828
Lautrup, S., Sinclair, D. A., Mattson, M. P., and Fang, E. F. (2019). NAD(+) in brain aging and neurodegenerative disorders. Cell Metab. 30, 630–655. doi: 10.1016/j.cmet.2019.09.001
Lee, D., Jo, M. G., Kim, S. Y., Chung, C. G., and Lee, S. B. (2020). Dietary antioxidants and the mitochondrial quality control: their potential roles in Parkinson's disease treatment. Antioxidants 9:1056. doi: 10.3390/antiox9111056
Lehmann, S., Costa, A. C., Celardo, I., Loh, S. H., and Martins, L. M. (2016). Parp mutations protect against mitochondrial dysfunction and neurodegeneration in a PARKIN model of Parkinson's disease. Cell Death Dis. 7:e2166. doi: 10.1038/cddis.2016.72
Lehmann, S., Loh, S. H. Y., and Martins, L. M. (2017). Enhancing NAD(+) salvage metabolism is neuroprotective in a PINK1 model of Parkinson's disease. Biol. Open 6, 141–147. doi: 10.1242/bio.022186
Leng, Y., Zhang, Y., Li, X., Wang, Z., Zhuang, Q., and Lu, Y. (2021). Receptor interacting protein kinases 1/3: the potential therapeutic target for cardiovascular inflammatory diseases. Front. Pharmacol. 12:762334. doi: 10.3389/fphar.2021.762334
Lengyel-Zhand, Z., Puentes, L. N., and Mach, R. H. (2022). PARkinson's: from cellular mechanisms to potential therapeutics. Pharmacol. Ther. 230:107968. doi: 10.1016/j.pharmthera.2021.107968
Leung, A. K. L. (2020). Poly(ADP-ribose): a dynamic trigger for biomolecular condensate formation. Trends Cell Biol. 30, 370–383. doi: 10.1016/j.tcb.2020.02.002
Li, M., Tang, Y., Li, Q. S., Xiao, M., Yang, Y. Y., and Wang, Y. L. (2019). Mono-ADP-ribosylation of H3R117 traps 5mC hydroxylase TET1 to impair demethylation of tumor suppressor gene TFPI2. Oncogene 38, 3488–3503. doi: 10.1038/s41388-018-0671-8
Lin, J. J., Chen, K., Chen, W. F., Yao, Y. Z., Ni, S. W., Ye, M. N., et al. (2020). Paradoxical Mitophagy regulation by PINK1 and TUFm. Mol. Cell 80:607. doi: 10.1016/j.molcel.2020.10.007
Liu, C., and Fang, Y. (2019). New insights of poly(ADP-ribosylation) in neurodegenerative diseases: a focus on protein phase separation and pathologic aggregation. Biochem. Pharmacol. 167, 58–63. doi: 10.1016/j.bcp.2019.04.028
Liu, H., Koros, C., Strohaker, T., Schulte, C., Bozi, M., Varvaresos, S., et al. (2021). A novel SNCA A30G mutation causes familial Parkinson's disease. Mov. Disord. 36, 1624–1633. doi: 10.1002/mds.28534
Liu, Z., Wang, H., Wang, S., Gao, J., and Niu, L. (2021). PARP-1 inhibition attenuates the inflammatory response in the cartilage of a rat model of osteoarthritis. Bone Joint Res. 10, 401–410. doi: 10.1302/2046-3758.107.BJR-2020-0200.R2
Love, S., Barber, R., and Wilcock, G. K. (1999). Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer's disease. Brain 122, 247–253. doi: 10.1093/brain/122.2.247
Lu, P., Hogan-Cann, A. D., Kamboj, A., Chowdhury, S. K. R., Aghanoori, M. R., Fernyhough, P., et al. (2019). Poly(ADP-ribose) polymerase-1 inhibits mitochondrial respiration by suppressing PGC-1 alpha activity in neurons. Neuropharmacology 160:107755. doi: 10.1016/j.neuropharm.2019.107755
Machado, C. B., Pluchon, P., Harley, P., Rigby, M., Sabater, V. G., Stevenson, D. C., et al. (2019). In vitro modeling of nerve-muscle connectivity in a compartmentalized tissue culture device. Adv. Biosyst. 3:800307. doi: 10.1002/adbi.201800307
Maiuri, T., Suart, C. E., Hung, C. L. K., Graham, K. J., Bazan, C. A. B., and Truant, R. (2019). DNA damage repair in Huntington's disease and other neurodegenerative diseases. Neurotherapeutics 16, 948–956. doi: 10.1007/s13311-019-00768-7
Mamontova, E. M., Clément, M.-J., Sukhanova, M. V., Joshi, V., Bouhss, A., Rengifo-Gonzalez, J. C., et al. (2023). FUS RRM regulates poly(ADP-ribose) levels after transcriptional arrest and PARP-1 activation on DNA damage. Cell Rep. 42:113199. doi: 10.1016/j.celrep.2023.113199
Mandir, A. S., Simbulan-Rosenthal, C. M., Poitras, M. F., Lumpkin, J. R., Dawson, V. L., Smulson, M. E., et al. (2002). A novel in vivo post-translational modification of p53 by PARP-1 in MPTP-induced parkinsonism. J. Neurochem. 83, 186–192. doi: 10.1046/j.1471-4159.2002.01144.x
Mao, K., Chen, J., Yu, H., Li, H., Ren, Y., Wu, X., et al. (2020). Poly (ADP-ribose) polymerase 1 inhibition prevents neurodegeneration and promotes alpha-synuclein degradation via transcription factor EB-dependent autophagy in mutant alpha-synucleinA53T model of Parkinson's disease. Aging Cell 19:e13163. doi: 10.1111/acel.13163
Mao, K., and Zhang, G. (2021). The role of PARP1 in neurodegenerative diseases and aging. FEBS J. 289, 2013–2024. doi: 10.1111/febs.15716
Markham, A., Bains, R., Franklin, P., and Spedding, M. (2014). Changes in mitochondrial function are pivotal in neurodegenerative and psychiatric disorders: how important is BDNF? Br. J. Pharmacol. 171, 2206–2229. doi: 10.1111/bph.12531
Martin-Jimenez, R., Lurette, O., and Hebert-Chatelain, E. (2020). Damage in mitochondrial DNA associated with Parkinson's disease. DNA Cell Biol. 39, 1421–1430. doi: 10.1089/dna.2020.5398
Martire, S., Fuso, A., Rotili, D., Tempera, I., Giordano, C., De Zottis, I., et al. (2013). PARP-1 modulates amyloid Beta peptide-induced neuronal damage. PLoS One 8:e72169. doi: 10.1371/journal.pone.0072169
Mashimo, M., Onishi, M., Uno, A., Tanimichi, A., Nobeyama, A., Mori, M., et al. (2021). The 89-kDa PARP1 cleavage fragment serves as a cytoplasmic PAR carrier to induce AIF-mediated apoptosis. J. Biol. Chem. 296:100046. doi: 10.1074/jbc.RA120.014479
Massey, T. H., and Jones, L. (2018). The central role of DNA damage and repair in CAG repeat diseases. Dis. Model. Mech. 11:dmm031930. doi: 10.1242/dmm.031930
McGurk, L., Gomes, E., Guo, L., Mojsilovic-Petrovic, J., Tran, V., Kalb, R. G., et al. (2018a). Poly(ADP-ribose) prevents pathological phase separation of TDP-43 by promoting liquid Demixing and stress granule localization. Mol. Cell 71:703. doi: 10.1016/j.molcel.2018.07.002
McGurk, L., Mojsilovic-Petrovic, J., Van Deerlin, V. M., Shorter, J., Kalb, R. G., Lee, V. M., et al. (2018b). Nuclear poly(ADP-ribose) activity is a therapeutic target in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 6:84. doi: 10.1186/s40478-018-0586-1
McGurk, L., Rifai, O. M., and Bonini, N. M. (2019). Poly(ADP-Ribosylation) in age-related neurological disease. J. Cell Biol. 35, 601–613. doi: 10.1083/jcb.20190207410.1016/j.tig.2019.05.004
Mekhaeil, M., Dev, K. K., and Conroy, M. J. (2022). Existing evidence for the repurposing of PARP-1 inhibitors in rare demyelinating diseases. Cancers 14:687. doi: 10.3390/cancers14030687
Mitra, J., and Hegde, M. L. (2019). A commentary on TDP-43 and DNA damage response in amyotrophic lateral sclerosis. J. Exp. Neurosci. 13:1179069519880166. doi: 10.1177/1179069519880166
Mohammed, S. A., Ambrosini, S., Luscher, T., Paneni, F., and Costantino, S. (2020). Epigenetic control of mitochondrial function in the vasculature. Front. Cardiovasc. Med. 7:28. doi: 10.3389/fcvm.2020.00028
Moon, H. E., and Paek, S. H. (2015). Mitochondrial dysfunction in Parkinson’s disease. Exp. Neurobiol. 24, 103–116. doi: 10.5607/en.2015.24.2.103
Morimoto, K., and Nakajima, K. (2019). Role of the immune system in the development of the central nervous system. Front. Neurosci. 13:916. doi: 10.3389/fnins.2019.00916
Nakamura, R., Misawa, K., Tohnai, G., Nakatochi, M., Furuhashi, S., Atsuta, N., et al. (2020). A multi-ethnic meta-analysis identifies novel genes, including ACSL5, associated with amyotrophic lateral sclerosis. Commun. Biol. 3:526. doi: 10.1038/s42003-020-01251-2
Neueder, A., Landles, C., Ghosh, R., Howland, D., Myers, R. H., Faull, R. L. M., et al. (2017). The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington's disease patients. Sci. Rep. 7:1307. doi: 10.1038/s41598-017-01510-z
Niaki, A. G., Sarkar, J., Cai, X. Y., Rhine, K., Vidaurre, V., Guy, B., et al. (2020). Loss of dynamic RNA interaction and aberrant phase separation induced by two distinct types of ALS/FTD-linked FUS mutations. Mol. Cell 77:82. doi: 10.1016/j.molcel.2019.09.022
Nicolae, C. M., Aho, E. R., Vlahos, A. H. S., Choe, K. N., De, S., Karras, G. I., et al. (2014). The ADP-ribosyltransferase PARP10/ARTD10 interacts with proliferating cell nuclear antigen (PCNA) and is required for DNA damage tolerance. J. Biol. Chem. 289, 13627–13637. doi: 10.1074/jbc.M114.556340
Noordermeer, S. M., and van Attikum, H. (2019). PARP inhibitor resistance: a tug-of-war in BRCA-mutated cells. Trends Cell Biol. 29, 820–834. doi: 10.1016/j.tcb.2019.07.008
Olsen, A. L., and Feany, M. B. (2019). PARP inhibitors and Parkinson's disease. N. Engl. J. Med. 380, 492–494. doi: 10.1056/NEJMcibr1814680
Paiva, I., Jain, G., Lazaro, D. F., Jercic, K. G., Hentrich, T., Kerimoglu, C., et al. (2018). Alpha-synuclein deregulates the expression of COL4A2 and impairs ER-Golgi function. Neurobiol. Dis. 119, 121–135. doi: 10.1016/j.nbd.2018.08.001
Palazzo, L., Daniels, C. M., Nettleship, J. E., Rahman, N., McPherson, R. L., Ong, S. E., et al. (2016). ENPP1 processes protein ADP-ribosylation in vitro. FEBS J. 283, 3371–3388. doi: 10.1111/febs.13811
Pang, Y., Lu, Y., Caisova, V., Liu, Y., Bullova, P., Huynh, T. T., et al. (2018). Targeting NAD(+)/PARP DNA repair pathway as a novel therapeutic approach to SDHB-mutated cluster I Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 24, 3423–3432. doi: 10.1158/1078-0432.CCR-17-3406
Penndorf, D., Witte, O. W., and Kretz, A. (2018). DNA plasticity and damage in amyotrophic lateral sclerosis. Neural Regen. Res. 13, 173–180. doi: 10.4103/167-5374.226377
Peters, O. M., Ghasemi, M., and Brown, R. H. (2015). Emerging mechanisms of molecular pathology in ALS (vol 125, pg 1767, 2015). J. Clin. Invest. 125:2548. doi: 10.1172/JCI82693
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, Parkin, and mitochondrial Fidelity in Parkinson's disease. Neuron 85, 257–273. doi: 10.1016/j.neuron.2014.12.007
Pinto, M., Pickrell, A. M., Fukui, H., and Moraes, C. T. (2013). Mitochondrial DNA damage in a mouse model of Alzheimer's disease decreases amyloid beta plaque formation. Neurobiol. Aging 34, 2399–2407. doi: 10.1016/j.neurobiolaging.2013.04.014
Pramanik, K. C., Fofaria, N. M., Gupta, P., and Srivastava, S. K. (2014). CBP-mediated FOXO-1 acetylation inhibits pancreatic tumor growth by targeting SirT. Mol. Cancer Ther. 13, 687–698. doi: 10.1158/1535-7163.MCT-13-0863
Prasad, A., Bharathi, V., Sivalingam, V., Girdhar, A., and Patel, B. K. (2019). Molecular mechanisms of TDP-43 Misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 12:25. doi: 10.3389/fnmol.2019.00025
Prokhorova, E., Agnew, T., Wondisford, A. R., Tellier, M., Kaminski, N., Beijer, D., et al. (2021). Unrestrained poly-ADP-ribosylation provides insights into chromatin regulation and human disease. Mol. Cell 81:2640. doi: 10.1016/j.molcel.2021.04.028
Przedborski, S., Jackson-Lewis, V., Djaldetti, R., Liberatore, G., Vila, M., Vukosavic, S., et al. (2000). The parkinsonian toxin MPTP: action and mechanism. Restor. Neurol. Neurosci. 16, 135–142. doi: 10.3233/RNN-2000-00132
Ray, C. A., and Nussenzweig, A. (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 18, 610–621. doi: 10.1038/nrm.2017.53
Rosenthal, F., Feijs, K. L. H., Frugier, E., Bonalli, M., Forst, A. H., Imhof, R., et al. (2013). Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat. Struct. Mol. Biol. 20:502. doi: 10.1038/nsmb.2521
Rulten, S. L., Rotheray, A., Green, R. L., Grundy, G. J., Moore, D. A. Q., Gomez-Herreros, F., et al. (2014). PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 42, 307–314. doi: 10.1093/nar/gkt835
Sakthianandeswaren, A., Parsons, M. J., Mouradov, D., MacKinnon, R. N., Catimel, B., Liu, S., et al. (2018). MACROD2 Haploinsufficiency impairs catalytic activity of PARP1 and promotes chromosome instability and growth of intestinal tumors. Cancer Discov. 8, 988–1005. doi: 10.1158/2159-8290.CD-17-0909
Salech, F., Ponce, D. P., SanMartín, C. D., Rogers, N. K., Chacón, C., Henríquez, M., et al. (2017). PARP-1 and p53 regulate the increased susceptibility to oxidative death of Lymphocytes from MCI and AD patients. Front. Aging Neurosci. 9:310. doi: 10.3389/fnagi.2017.00310
Sanders, L. H., and Greenamyre, J. T. (2013). Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radic. Biol. Med. 62, 111–120. doi: 10.1016/j.freeradbiomed.2013.01.003
Sasaki, T., Liu, K., Agari, T., Yasuhara, T., Morimoto, J., Okazaki, M., et al. (2016). Anti-high mobility group box 1 antibody exerts neuroprotection in a rat model of Parkinson's disease. Exp. Neurol. 275, 220–231. doi: 10.1016/j.expneurol.2015.11.003
Schaser, A. J., Osterberg, V. R., Dent, S. E., Stackhouse, T. L., Wakeham, C. M., Boutros, S., et al. (2019). Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 9:10919. doi: 10.1038/s41598-019-47227-z
Scoumanne, A., Zhang, J., and Chen, X. (2009). PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res. 37, 4965–4976. doi: 10.1093/nar/gkp516
Screaton, R. A., Conkright, M. D., Katoh, Y., Best, J. L., Canettieri, G., Jeffries, S., et al. (2004). The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell 119, 61–74. doi: 10.1016/j.cell.2004.09.015
Sharifi, R., Morra, R., Appel, C. D., Tallis, M., Chioza, B., Jankevicius, G., et al. (2013). Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. EMBO J. 32, 1225–1237. doi: 10.1038/emboj.2013.51
Sheng, Z.-H., and Cai, Q. (2012). Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 13, 77–93. doi: 10.1038/nrn3156
Song, L. Y., McMackin, M., Nguyen, A., and Cortopassi, G. (2017). Parkin deficiency accelerates consequences of mitochondrial DNA deletions and parkinsonism. Neurobiol. Dis. 100, 30–38. doi: 10.1016/j.nbd.2016.12.024
Steffan, J. S., Kazantsev, A., Spasic-Boskovic, O., Greenwald, M., Zhu, Y. Z., Gohler, H., et al. (2000). The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 97, 6763–6768. doi: 10.1073/pnas.100110097
Stoica, B. A., Loane, D. J., Zhao, Z., Kabadi, S. V., Hanscom, M., Byrnes, K. R., et al. (2014). PARP-1 inhibition attenuates neuronal loss, microglia activation and neurological deficits after traumatic brain injury. J. Neurotrauma 31, 758–772. doi: 10.1089/neu.2013.3194
Strosznajder, J. B., Czapski, G. A., Adamczyk, A., and Strosznajder, R. P. (2012). Poly(ADP-ribose) Polymerase-1 in amyloid Beta toxicity and Alzheimer's disease. Mol. Neurobiol. 46, 78–84. doi: 10.1007/s12035-012-8258-9
Suh, S. W., Aoyama, K., Alano, C. C., Anderson, C. M., Hamby, A. M., and Swanson, R. A. (2007). Zinc inhibits astrocyte glutamate uptake by activation of poly(ADP-ribose) polymerase-1. Mol. Med. 13, 344–349. doi: 10.2119/2007-00043.Suh
Thomas, B., Matson, S., Chopra, V., Sun, L. P., Sharma, S., Hersch, S., et al. (2013). A novel method for detecting 7-methyl guanine reveals aberrant methylation levels in Huntington disease. Anal. Biochem. 436, 112–120. doi: 10.1016/j.ab.2013.01.035
Visochek, L., Grigoryan, G., Kalal, A., Milshtein-Parush, H., Gazit, N., Slutsky, I., et al. (2016). A PARP1-ERK2 synergism is required for the induction of LTP. Sci. Rep. 6:24950. doi: 10.1038/srep24950
Vyas, S., Matic, I., Uchima, L., Rood, J., Zaja, R., Hay, R. T., et al. (2014). Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat. Commun. 5:4426. doi: 10.1038/ncomms5426
Wang, Y. F., An, R., Umanah, G. K., Park, H., Nambiar, K., Eacker, S. M., et al. (2016). A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 354:aad6872. doi: 10.1126/science.aad6872
Wang, H. M., Shimoji, M., Yu, S. W., Dawson, T. M., and Dawson, V. L. (2003). Apoptosis inducing factor and PARP-mediated injury in the MPTP mouse model of Parkinson's disease. Park. Dis. 991, 132–139. doi: 10.1111/j.1749-6632.2003.tb07471.x
Wasyluk, W., and Zwolak, A. (2021). PARP inhibitors: An innovative approach to the treatment of inflammation and metabolic disorders in Sepsis. J. Inflamm. Res. 14, 1827–1844. doi: 10.2147/JIR.S300679
Wei, H. T., and Yu, X. C. (2016). Functions of PARylation in DNA damage repair pathways. Genomics Proteomics Bioinformatics 14, 131–139. doi: 10.1016/j.gpb.2016.05.001
Welty, S., Teng, Y. Q., Liang, Z. B., Zhao, W. X., Sanders, L. H., Greenamyre, J. T., et al. (2018). RAD52 is required for RNA-templated recombination repair in post-mitotic neurons. J. Biol. Chem. 293, 1353–1362. doi: 10.1074/jbc.M117.808402
Wood, M., Quinet, A., Lin, Y. L., Davis, A. A., Pasero, P., Ayala, Y. M., et al. (2020). TDP-43 dysfunction results in R-loop accumulation and DNA replication defects. J. Cell Sci. 133:jcs244129. doi: 10.1242/jcs.244129
Wu, W., Hill, S. E., Nathan, W. J., Paiano, J., Callen, E., Wang, D. P., et al. (2021). Neuronal enhancers are hotspots for DNA single-strand break repair. Nature 593:440-+. doi: 10.1038/s41586-021-03468-5
Yang, Z., Li, L., Chen, L., Yuan, W., Dong, L., Zhang, Y., et al. (2014). PARP-1 mediates LPS-induced HMGB1 release by macrophages through regulation of HMGB1 acetylation. J. Immunol. 193, 6114–6123. doi: 10.4049/jimmunol.1400359
Yu, Y. Z., Fedele, G., Celardo, I., Loh, S. H. Y., and Martins, L. M. (2021). Parp mutations protect from mitochondrial toxicity in Alzheimer's disease. Cell Death Dis. 12:651. doi: 10.1038/s41419-021-03926-y
Zeng, J., Libien, J., Shaik, F., Wolk, J., and Hernández, A. I. (2016). Nucleolar PARP-1 expression is decreased in Alzheimer's disease: consequences for epigenetic regulation of rDNA and cognition. Neural Plast. 2016:8987928. doi: 10.1155/2016/8987928
Zhang, J., Hu, X., Geng, Y., Xiang, L., Wu, Y., Li, Y., et al. (2025). Exploring the role of parthanatos in CNS injury: molecular insights and therapeutic approaches. J. Adv. Res. 70, 271–286. doi: 10.1016/j.jare.2024.04.031
Zhang, F., Lou, L. H., Peng, B., Song, X. T., Reizes, O., Almasan, A., et al. (2020). Nudix hydrolase NUDT16 regulates 53BP1 protein by reversing 53BP1 ADP-Ribosylation. Cancer Res. 80, 999–1010. doi: 10.1158/0008-5472.CAN-19-2205
Zhang, H., Xie, Z., Peng, Y., Xie, A., Fu, C., Zheng, D., et al. (2023). PARP1 promotes NLRP3 activation via blocking TFEB-mediated autophagy in rotenone-induced neurodegeneration. Ecotoxicol. Environ. Saf. 252:114630. doi: 10.1016/j.ecoenv.2023.114630
Keywords: adenosine diphosphate-ribosylation, neurodegenerative diseases, proteins function, PARP, post-translational modification
Citation: Huang C, Xiao H, Yang Y, Luo J, Lai Y, Liu S, Mao K, Chen J and Wang L (2025) Adenosine diphosphate-ribosylation greatly affects proteins function: a focus on neurodegenerative diseases. Front. Aging Neurosci. 17:1575204. doi: 10.3389/fnagi.2025.1575204
Edited by:
Robert Henry Mach, University of Pennsylvania, United StatesReviewed by:
Jason Cai, Yale University, United StatesNootan Pandey, University of Pennsylvania, United States
Copyright © 2025 Huang, Xiao, Yang, Luo, Lai, Liu, Mao, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liling Wang, d2FuZ2xpbGluZ0AxNjMuY29t; Jialong Chen, Y2hlbmppYWxvbmdhYUAxNjMuY29t; Kanmin Mao, bWttOTQwNzIxQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship