 Koji Hoshino
Koji Hoshino- Department of Anesthesiology, Hokkaido University Hospital, Sapporo, Japan
Sepsis-associated encephalopathy (SAE) is a frequent yet underrecognized complication of sepsis that significantly contributes to long-term cognitive dysfunction in survivors. Despite advances in sepsis management, there is currently no established therapy targeting SAE, and translational gaps between basic and clinical research persist. Rodent models of sepsis suffer from variability in immune responses and poor translational fidelity. Moreover, behavioral tests commonly used to assess cognition in animal models are often confounded by sepsis-induced sickness behaviors and depression-like phenotypes, especially during the acute phase. Given these limitations, targeting synaptic plasticity—both mechanistically and therapeutically—has emerged as a promising approach. Accumulating evidence indicates that SAE arises from neuroinflammation triggered by systemic inflammation, in which activated microglia and subsequent cytokine signaling contribute to neuronal dysfunction and lead to impaired hippocampal long-term potentiation (LTP), a fundamental mechanism of learning and memory. Importantly, electrophysiological studies have shown that LTP impairment occurs within hours to days after sepsis onset, highlighting its potential as an early and sensitive biomarker for SAE. Recent experimental interventions, including low-intensity exercise, environmental enrichment, and modulation of gut microbiota, have shown beneficial effects on SAE. These findings underscore the need for integrative, multimodal strategies that address the complex pathophysiology of SAE. Synaptic plasticity, particularly LTP, may serve not only as a functional readout of neuroinflammatory damage but also as a modifiable target for early intervention. This review highlights the translational challenges in current SAE research and advocates for a paradigm shift toward mechanism-driven and plasticity-focused therapeutic development.
1 Introduction
Sepsis remains a global health burden, accounting for nearly 20% of all deaths worldwide, although its incidence and mortality have declined substantially over the past 25 years (Rudd et al., 2020). The link between sepsis and cognitive dysfunction has long been recognized. A historical cohort study involving approximately one million individuals aged 65 years or older in the United Kingdom reported that sepsis nearly doubles the risk of subsequent dementia (Muzambi et al., 2021). Sepsis-associated encephalopathy (SAE) refers to brain dysfunction occurring in the context of sepsis, without direct central nervous system infection. There is currently no standardized diagnostic criterion for SAE. Eidelman et al. (1996) first proposed the utility of the Glasgow Coma Scale (GCS) in defining SAE. Since then, numerous studies have defined SAE using criteria such as impaired consciousness with a GCS score <15 (typically <13–14) or the presence of delirium as assessed by tools like the Confusion Assessment Method for the Intensive Care Unit (CAM-ICU; Thy et al., 2025).
SAE presents with a broad clinical spectrum, ranging from mild disorientation to coma, and affects nearly half of all septic patients. Even mild consciousness disturbances (e.g., GCS 13–14) are associated with increased mortality (Sonneville et al., 2017). Beyond its acute impact, SAE can lead to persistent cognitive impairments. Although more than 30% of patients experience resolution of cognitive symptoms within 1 month of sepsis onset, some individuals exhibit long-term deficits (Manabe and Heneka, 2021). As a result, sepsis management is no longer focused solely on survival alone, and SAE must be recognized as a worthy therapeutic target. Despite numerous clinical trials, including those with vitamin C and thiamine (Park et al., 2020), no effective treatments have been established, and basic research on SAE has not yet been translated into clinical practice. This review aims to delineate the challenges faced in basic SAE research and explore future directions that may facilitate therapeutic development.
2 Pathophysiology of SAE
In the past 10–15 years, major progress has been made in elucidating the pathophysiological mechanisms of SAE. It is now widely accepted that systemic inflammation initiates neuroinflammation, which subsequently leads to cognitive impairment (Denver and Cunningham, 2025). There are four principal pathways through which peripheral inflammation induces neuroinflammation (Dantzer et al., 2008; Widmann and Heneka, 2014; Barbosa-Silva et al., 2021): (1) Increased blood-brain barrier (BBB) permeability, allowing cytokines and immune cells to infiltrate the brain parenchyma. BBB disruption is associated with dysfunction of the glial vascular unit, involving astrocytes, microglia, endothelial cells, and pericytes (Hu et al., 2025). (2) Interleukin (IL)-1 receptor-mediated signaling from perivascular macrophages and endothelial cells initiates localized inflammatory responses. (3) Humoral signaling through circumventricular organs, which lack a complete BBB. Macrophage-like cells expressing toll-like receptors release cytokines that diffuse into the brain via volume diffusion or active transport. (4) Neural signaling via the vagus and trigeminal nerves, which detect peripheral IL-1β and transmit signals to the brain.
All these pathways converge on microglial activation, which sustains the neuroinflammatory state. Downstream consequences of activated microglia include enhanced expression of pro-inflammatory cytokines, mitochondrial dysfunction, and oxidative stress. These factors contribute to the formation of the nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing protein 3 (NLRP3) inflammasome, leading to the production of mature IL-1β (Manabe and Heneka, 2021; Moraes et al., 2021). Recent studies suggest that persistent activation of the NLRP3-IL-1β pathway may underlie chronic SAE (Beyer et al., 2020; Zhao et al., 2020). Clinically, advanced age and pre-existing cognitive decline have been associated with prolonged SAE (Sonneville et al., 2017). Our group has demonstrated elevated hippocampal IL-1β levels during the acute phase of sepsis in senescence-accelerated mice, suggesting a role for the NLRP3-IL-1β pathway in the chronicity of SAE (Hoshino et al., 2021).
Recently, Gao et al. (2024) reported that N-acetyltransferase 10 (NAT10) is upregulated in hippocampal neurons of the dentate gyrus as a downstream mechanism following microglial activation. NAT10 promotes the acetylation of gamma-aminobutyric acid B receptor 1 (GABABR1) mRNA, enhancing its expression and increasing inhibitory synaptic currents, thereby contributing to impaired neurotransmission and learning deficits. Additionally, excessive microglia-mediated synaptic pruning has been reported. For instance, decreased hippocampal PSD-95 expression at 24 h post-lipopolysaccharide (LPS) injection (Song et al., 2020) and reduced neocortical spine density at 8 weeks (Kondo et al., 2011) have been documented. These findings underscore the complex, multifactorial nature of SAE and support the need for multipronged therapeutic strategies.
3 Challenges in basic research on SAE
3.1 Limitations of rodent models in sepsis research
One persistent criticism is that basic research on sepsis has failed to yield translational benefits (Cavaillon et al., 2020). Rodent models are limited by their intrinsic resistance to endotoxins and robust recovery from infections (Warren et al., 2010). Moreover, genetic differences among inbred strains lead to variable immune responses (Sellers et al., 2012). While humanized mice have been proposed as a potential solution (Laudanski, 2021), their complexity and cost hinder widespread adoption.
Common sepsis models include bacterial infection, intraperitoneal injection of LPS (endotoxemia model), cecal ligation and puncture (CLP), and cecal slurry. A recently proposed alternative is the fecal suspension and intraperitoneal injection model (Tsuchida et al., 2022). Each has strengths and limitations (Cai et al., 2023). Endotoxemia models poorly replicate clinical sepsis, and LPS results vary depending on type and lot (Beyer et al., 2020). Even CLP, the so-called “gold standard,” is subject to variability based on cecum size, needle gauge, extruded fecal volume, and fat handling (Buras et al., 2005; Niiyama et al., 2016). As Gonnert et al. (2011) emphasized, standardized models using clinical severity scores are essential for reproducible original laboratory research.
3.2 Limitations of behavioral testing in sepsis animal model
In contrast to humans, encephalopathy in animals cannot be assessed using GCS or CAM-ICU. As a result, behavioral tests evaluating cognition—such as learning and memory—are employed. The hippocampus is considered the most vulnerable brain region in sepsis (Semmler et al., 2005), and hippocampus-dependent tasks are preferred. However, contextual fear conditioning and the Morris water maze require equivalent baseline locomotor activity between groups—a condition not met in sepsis models. Sepsis induces profound behavioral alterations, including sickness behavior during the acute phase, marked by reduced activity, food intake, weight loss, and sleep disturbance. This is followed by depression-like behavior, attributed to cytokine-induced activation of indoleamine 2,3-dioxygenase (IDO), leading to anhedonia and helplessness (Dantzer et al., 2008; Pereira De Souza Goldim et al., 2020). These overlapping behavioral states complicate data interpretation (Dantzer et al., 2008). Indeed, even endotoxemia models show that such behaviors persist for up to 14 days after intraperitoneal LPS injections (Sohroforouzani et al., 2022), corresponding to the clinical ICU phase, during which behavioral testing may not reliably reflect cognitive function. Furthermore, general limitations of behavioral tests include experimenter bias, stress induction, low ecological-validity, and circadian mismatches (Lang et al., 2023). When coupled with sepsis-specific behavioral abnormalities, these factors introduce substantial confounds.
4 SAE and the pathophysiological role of synaptic plasticity
4.1 The impact of SAE on long-term potentiation
Neuroinflammation resulting from sepsis leads to various forms of synaptic dysfunction, including neurotransmitter imbalance, synaptic deficiency, myelin damage, and inhibition of synaptic plasticity (Tang et al., 2022; Yang et al., 2025). Synaptic plasticity is broadly categorized into structural and functional types. Among the latter, long-term potentiation (LTP)—first described by Bliss and Collingridge (1993)—is regarded as a cellular substrate for learning and memory (Takeuchi et al., 2014).
Historically, SAE research relied heavily on behavioral assessments. However, due to limitations in such tests during the acute phase of sepsis, electrophysiological approaches have gained traction. In 2000, Vereker et al. (2000) demonstrated impaired LTP in the hippocampal Schaffer collateral (SC)–CA1 pathway 3 h after intraperitoneal LPS injection in rats. Imamura et al. (2011) later reported reduced LTP in the CA1 region 24 h post-CLP.
Table 1 summarizes key studies evaluating hippocampal LTP in rodent SAE models. Many studies report reduced LTP during the acute phase (3 h−14 days post-insult), highlighting its utility as a surrogate marker for SAE. However, outcomes vary depending on stimulation protocols and animal age. Chapman et al. (2010) found no reduction in early-phase LTP (E-LTP) following 100 Hz stimulation in either young (3 months) or aged (24 months) rats 4–5 days after intraperitoneal E. coli infection. Interestingly, only aged rats exhibited reduced late-phase LTP (L-LTP) in response to theta-burst stimulation (TBS) 8 days after E. coli infection (Tanaka et al., 2018), suggesting that the impairment is both age-specific and dependent on the stimulation protocol. Kakizaki et al. (2023) showed that immature rats (4–5 weeks old) subjected to CLP displayed no significant reduction in E-LTP at 8 or 16 h post-insult. These findings emphasize the need to account for stimulation protocol and animal age when interpreting LTP outcomes in SAE models. Although most studies summarized in Table 1 have been conducted exclusively in male rodents, recent evidence indicates that hippocampal synaptic plasticity exhibits sex-dependent mechanisms. For instance, N-methyl-D-aspartate (NMDA) receptor contributions to LTP and spatial memory differ between sexes even in juvenile animals (Narattil and Maroun, 2024), and estrogen receptor signaling critically regulates LTP in females (Wang et al., 2018). Future studies should address sex as a biological variable to enhance translational relevance.
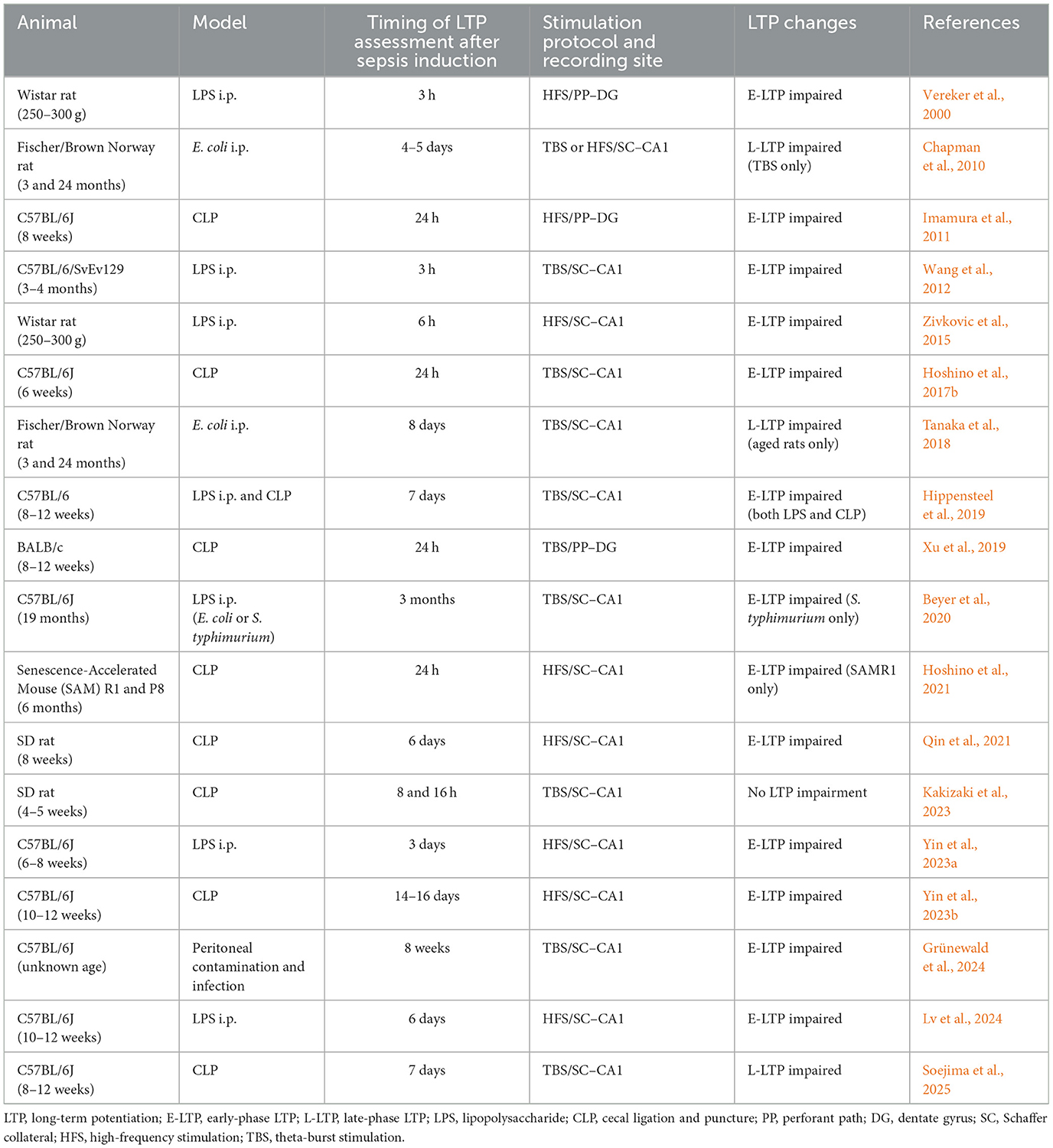
Table 1. Overview of experimental studies on sepsis models and hippocampal LTP (chronological order).
4.2 Mechanisms of LTP impairment in SAE
In NMDA receptor-dependent LTP, as seen in hippocampal SC–CA1 and perforant path (PP)–dentate gyrus (DG) pathways, calcium influx via NMDA receptors—elicited by TBS or high-frequency stimulation (HFS)—activates calcium–calmodulin-dependent protein kinase II (CaMKII) and enhances alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) trafficking, thereby potentiating synaptic transmission (Nicoll and Roche, 2013).
A key mechanism by which SAE impairs LTP involves the activation of the NLRP3–IL-1β pathway in microglia. Although physiological levels of IL-1β are thought to be important for the maintenance of hippocampal LTP (Ross et al., 2003), its overexpression has long been known to disrupt hippocampal LTP (Bellinger et al., 1993), particularly the NMDA receptor-dependent form (Hoshino et al., 2017a). This impairment is mediated via activation of p38 mitogen-activated protein kinase (MAPK; Coogan et al., 1999). IL-1β also promotes the neuronal surface expression of GABAA receptors—especially those containing the α5 subunit—leading to increased tonic inhibitory currents and further LTP impairment through p38 MAPK-dependent mechanisms (Serantes et al., 2006; Wang et al., 2012). Additionally, IL-1β suppresses NMDA-induced outward currents via p38 MAPK (Zhang et al., 2010), offering another potential mechanism of LTP disruption.
Furthermore, microglia-driven synaptic pruning, discussed earlier in the context of SAE pathophysiology, may also impair hippocampal plasticity, warranting further investigation (Yang et al., 2025).
4.3 LTP as a therapeutic target in SAE
Grünewald et al. (2024) reported significant downregulation of Arc/Arg3.1—a master regulator of synaptic plasticity (Shepherd and Bear, 2011)—in the hippocampus of mice subjected to peritoneal contamination and infection. Hippocampal overexpression of Arc with adeno-associated virus vectors restored impaired E-LTP. Similarly, environmental enrichment during the post-septic period enhanced hippocampal Arc expression via the brain-derived neurotrophic factor (BDNF)/TrkB pathway, rescuing LTP and spatial memory. These findings suggest that directly targeting impaired LTP could offer therapeutic benefits.
In clinical studies, cerebrospinal fluid (CSF) from infected patients with delirium showed reduced levels of proteins involved in synapse formation and function, compared to non-delirious patients (Peters Van Ton et al., 2020), lending further support to this hypothesis. Moreover, postoperative cognitive dysfunction—believed to share pathophysiological features with SAE—has been linked to impaired E-LTP to L-LTP transformation mediated by hippocampal BDNF downregulation (Liu et al., 2025), underscoring the growing relevance of LTP-focused interventions. To date, no clinical studies have directly examined the relationship between hippocampal LTP and standard cognitive assessments such as the Mini-Mental State Examination (MMSE) or Montreal Cognitive Assessment (MoCA) in patients with sepsis or SAE. However, indirect evidence from other neurological conditions supports a potential link between LTP-related plasticity and cognitive performance. For instance, in patients with multiple sclerosis, reduced transcranial magnetic stimulation (TMS)-induced cortical plasticity—considered a human proxy for LTP—was associated with both lower CSF amyloid-β1 − 42 levels and poorer attention and concentration scores on cognitive tests (Mori et al., 2011). Although these data reflect neocortical rather than hippocampal plasticity, they suggest that LTP-like measures may serve as functional indicators of cognitive integrity. Given the role of hippocampal LTP in memory formation, future studies are warranted to determine whether electrophysiological markers of hippocampal plasticity in animal models of SAE correspond to clinical memory assessments such as MMSE or MoCA in human patients. Establishing such links would enhance the translational value of LTP as a surrogate marker and may provide a measurable endpoint for therapeutic trials targeting cognitive dysfunction in SAE.
5 Discussion
Numerous compounds have demonstrated therapeutic potential in preclinical models of SAE (Barichello et al., 2019; Catarina et al., 2021). Nevertheless, no pharmacologic agent has yet shown clinical efficacy. The use of standardized, reproducible animal models with clinical severity scoring systems is essential. Given the behavioral confounds during the acute-to-subacute phase of sepsis, LTP offers a reliable and interpretable surrogate marker. As SAE pathophysiology is inherently multifaceted, monotherapies are unlikely to effectively address its complexity. Multimodal interventions—including pharmacologic, nutritional, and physical therapies typical of intensive care—should be integrated into future preclinical research to foster successful translation to clinical care. Indeed, previous studies have shown that environmental enrichment following endotoxemia improved spatial learning in mice (Keymoradzadeh et al., 2020), and that low-intensity exercise during the acute phase of sepsis restored hippocampal synaptic plasticity (Soejima et al., 2025). Moreover, recent research has also highlighted a potential link between sepsis-induced gut dysbiosis and the development of SAE (Fang et al., 2022). In translating therapeutic interventions such as environmental enrichment and low-intensity exercise from rodent models to clinical practice, practical barriers remain. Environmental enrichment paradigms in rodents typically involve access to toys, running wheels, and group housing, which do not directly map onto human hospital settings. For example, in stroke care, enriched environments have included cognitive stimulation through music, games, and communal spaces, yet their definitions and applications remain inconsistent and context-dependent (Anåker et al., 2024). To date, there have been no clinical trials assessing environmental enrichment in patients with sepsis or SAE. This highlights the need for research to define and translate rodent-based enrichment paradigms into feasible and effective interventions for human patients. Similarly, early rehabilitation has shown promise in improving physical function and inducing systemic anti-inflammatory effects in sepsis patients (Kayambu et al., 2015). Rodent studies often employ short-term, low-intensity treadmill protocols, but their clinical counterparts must account for patient variability, resource limitations, and safety considerations. Such translational gaps call for research frameworks that integrate both experimental rigor and clinical practicality. Future efforts should focus on developing mechanistically informed, multimodal strategies—guided by insights into synaptic plasticity—that can serve both as measurable biomarkers and modifiable targets for restoring cognitive function.
Author contributions
KH: Investigation, Conceptualization, Writing – review & editing, Writing – original draft, Funding acquisition.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by a grant from JSPS KAKENHI (Grant no. 25K12223).
Acknowledgments
I would like to express my sincere gratitude to Dr. Takashi Soejima for his continuous support of my research.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Anåker, A., Kevdzija, M., and Elf, M. (2024). Enriched environments in stroke units: defining characteristics and limitations. HERD 17, 344–359. doi: 10.1177/19375867231224972
Barbosa-Silva, M. C., Lima, M. N., Battaglini, D., Robba, C., Pelosi, P., Rocco, P. R. M., et al. (2021). Infectious disease-associated encephalopathies. Crit. Care 25:236. doi: 10.1186/s13054-021-03659-6
Barichello, T., Sayana, P., Giridharan, V. V., Arumanayagam, A. S., Narendran, B., Della Giustina, A., et al. (2019). Long-term cognitive outcomes after sepsis: a translational systematic review. Mol. Neurobiol. 56, 186–251. doi: 10.1007/s12035-018-1048-2
Bellinger, F. P., Madamba, S., and Siggins, G. R. (1993). Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 628, 227–234. doi: 10.1016/0006-8993(93)90959-Q
Beyer, M. M. S., Lonnemann, N., Remus, A., Latz, E., Heneka, M. T., and Korte, M. (2020). Enduring changes in neuronal function upon systemic inflammation are NLRP3 inflammasome dependent. J. Neurosci. 40, 5480–5494. doi: 10.1523/JNEUROSCI.0200-20.2020
Bliss, T. V., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. doi: 10.1038/361031a0
Buras, J. A., Holzmann, B., and Sitkovsky, M. (2005). Animal models of sepsis: setting the stage. Nat. Rev. Drug Discov. 4, 854–865. doi: 10.1038/nrd1854
Cai, L., Rodgers, E., Schoenmann, N., and Raju, R. P. (2023). Advances in rodent experimental models of sepsis. Int. J. Mol. Sci. 24:9578. doi: 10.3390/ijms24119578
Catarina, A. V., Branchini, G., Bettoni, L., De Oliveira, J. R., and Nunes, F. B. (2021). Sepsis-associated encephalopathy: from pathophysiology to progress in experimental studies. Mol. Neurobiol. 58, 2770–2779. doi: 10.1007/s12035-021-02303-2
Cavaillon, J. M., Singer, M., and Skirecki, T. (2020). Sepsis therapies: learning from 30 years of failure of translational research to propose new leads. EMBO Mol. Med. 12:e10128. doi: 10.15252/emmm.201810128
Chapman, T. R., Barrientos, R. M., Ahrendsen, J. T., Maier, S. F., and Patterson, S. L. (2010). Synaptic correlates of increased cognitive vulnerability with aging: peripheral immune challenge and aging interact to disrupt theta-burst late-phase long-term potentiation in hippocampal area CA1. J. Neurosci. 30, 7598–7603. doi: 10.1523/JNEUROSCI.5172-09.2010
Coogan, A. N., O'neill, L. A., and O'connor, J. J. (1999). The P38 mitogen-activated protein kinase inhibitor SB203580 antagonizes the inhibitory effects of interleukin-1beta on long-term potentiation in the rat dentate gyrus in vitro. Neuroscience 93, 57–69. doi: 10.1016/S0306-4522(99)00100-1
Dantzer, R., O'connor, J. C., Freund, G. G., Johnson, R. W., and Kelley, K. W. (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56. doi: 10.1038/nrn2297
Denver, P., and Cunningham, C. (2025). Microglial activation and neuroinflammation in acute and chronic cognitive deficits in sepsis. Neuropharmacology 267:110285. doi: 10.1016/j.neuropharm.2024.110285
Eidelman, L. A., Putterman, D., Putterman, C., and Sprung, C. L. (1996). The spectrum of septic encephalopathy. Definitions, etiologies, and mortalities. JAMA 275, 470–473. doi: 10.1001/jama.1996.03530300054040
Fang, H., Wang, Y., Deng, J., Zhang, H., Wu, Q., He, L., et al. (2022). Sepsis-induced gut dysbiosis mediates the susceptibility to sepsis-associated encephalopathy in mice. mSystems 7:e0139921. doi: 10.1128/msystems.01399-21
Gao, S., Shen, R., Li, J., Jiang, Y., Sun, H., Wu, X., et al. (2024). N-acetyltransferase 10 mediates cognitive dysfunction through the acetylation of GABA(B)R1 mRNA in sepsis-associated encephalopathy. Proc. Natl. Acad. Sci. U.S.A. 121:e2410564121. doi: 10.1073/pnas.2410564121
Gonnert, F. A., Recknagel, P., Seidel, M., Jbeily, N., Dahlke, K., Bockmeyer, C. L., et al. (2011). Characteristics of clinical sepsis reflected in a reliable and reproducible rodent sepsis model. J. Surg. Res. 170, e123–134. doi: 10.1016/j.jss.2011.05.019
Grünewald, B., Wickel, J., Hahn, N., Rahmati, V., Rupp, H., Chung, H. Y., et al. (2024). Targeted rescue of synaptic plasticity improves cognitive decline in sepsis-associated encephalopathy. Mol. Ther. 32, 2113–2129. doi: 10.1016/j.ymthe.2024.05.001
Hippensteel, J. A., Anderson, B. J., Orfila, J. E., Mcmurtry, S. A., Dietz, R. M., Su, G., et al. (2019). Circulating heparan sulfate fragments mediate septic cognitive dysfunction. J. Clin. Invest. 129, 1779–1784. doi: 10.1172/JCI124485
Hoshino, K., Hasegawa, K., Kamiya, H., and Morimoto, Y. (2017a). Synapse-specific effects of IL-1beta on long-term potentiation in the mouse hippocampus. Biomed. Res. 38, 183–188. doi: 10.2220/biomedres.38.183
Hoshino, K., Hayakawa, M., and Morimoto, Y. (2017b). Minocycline prevents the impairment of hippocampal long-term potentiation in the septic mouse. Shock 48, 209–214. doi: 10.1097/SHK.0000000000000847
Hoshino, K., Uchinami, Y., Uchida, Y., Saito, H., and Morimoto, Y. (2021). Interleukin-1β modulates synaptic transmission and synaptic plasticity during the acute phase of sepsis in the senescence-accelerated mouse hippocampus. Front. Aging Neurosci. 13:637703. doi: 10.3389/fnagi.2021.637703
Hu, J., Xie, S., Chen, T., Liao, Y., Qian, Z., and Zhang, L. (2025). Glial vascular unit as a bridge between blood-brain barrier and glymphatic system: roles in sepsis-associated encephalopathy. Neuroscience 570, 68–71. doi: 10.1016/j.neuroscience.2025.02.039
Imamura, Y., Wang, H., Matsumoto, N., Muroya, T., Shimazaki, J., Ogura, H., et al. (2011). Interleukin-1beta causes long-term potentiation deficiency in a mouse model of septic encephalopathy. Neuroscience 187, 63–69. doi: 10.1016/j.neuroscience.2011.04.063
Kakizaki, R., Narimatsu, E., Kasai, T., and Nomura, K. (2023). Sepsis-induced modulation of long-term potentiation induced by theta burst stimulation in the rat hippocampus. Front. Neurosci. 17:1296391. doi: 10.3389/fnins.2023.1296391
Kayambu, G., Boots, R., and Paratz, J. (2015). Early physical rehabilitation in intensive care patients with sepsis syndromes: a pilot randomised controlled trial. Intensive Care Med. 41, 865–874. doi: 10.1007/s00134-015-3763-8
Keymoradzadeh, A., Hedayati Ch, M., Abedinzade, M., Gazor, R., Rostampour, M., and Taleghani, B. K. (2020). Enriched environment effect on lipopolysaccharide-induced spatial learning, memory impairment and hippocampal inflammatory cytokine levels in male rats. Behav. Brain Res. 394:112814. doi: 10.1016/j.bbr.2020.112814
Kondo, S., Kohsaka, S., and Okabe, S. (2011). Long-term changes of spine dynamics and microglia after transient peripheral immune response triggered by LPS in vivo. Mol. Brain 4:27. doi: 10.1186/1756-6606-4-27
Lang, B., Kahnau, P., Hohlbaum, K., Mieske, P., Andresen, N. P., Boon, M. N., et al. (2023). Challenges and advanced concepts for the assessment of learning and memory function in mice. Front. Behav. Neurosci. 17:1230082. doi: 10.3389/fnbeh.2023.1230082
Laudanski, K. (2021). Humanized mice as a tool to study sepsis-more than meets the eye. Int. J. Mol. Sci. 22:2403. doi: 10.3390/ijms22052403
Liu, Q., Wang, H. B., Lin, J. T., Jiao, X. H., Liu, Y. P., Li, T. Z., et al. (2025). Role of brain-derived neurotrophic factor in dysfunction of short-term to long-term memory transformation after surgery and anaesthesia in older mice. Br. J. Anaesth. 134, 1134–1145. doi: 10.1016/j.bja.2024.11.045
Lv, X., Jia, M., Feng, X., Jian, J. X., Yang, J. J., Ma, D. Q., et al. (2024). STING driving synaptic phagocytosis of hippocampal microglia/macrophages contributes to cognitive impairment in sepsis-associated encephalopathy in mice. CNS Neurosci. Ther. 30:e70166. doi: 10.1111/cns.70166
Manabe, T., and Heneka, M. T. (2021). Cerebral dysfunctions caused by sepsis during ageing. Nat. Rev. Immunol. 22, 1–15. doi: 10.1038/s41577-021-00643-7
Moraes, C. A., Zaverucha-Do-Valle, C., Fleurance, R., Sharshar, T., Bozza, F. A., and D'avila, J. C. (2021). Neuroinflammation in sepsis: molecular pathways of microglia activation. Pharmaceuticals 14:416. doi: 10.3390/ph14050416
Mori, F., Rossi, S., Sancesario, G., Codecà, C., Mataluni, G., Monteleone, F., et al. (2011). Cognitive and cortical plasticity deficits correlate with altered amyloid-β CSF levels in multiple sclerosis. Neuropsychopharmacology 36, 559–568. doi: 10.1038/npp.2010.187
Muzambi, R., Bhaskaran, K., Smeeth, L., Brayne, C., Chaturvedi, N., and Warren-Gash, C. (2021). Assessment of common infections and incident dementia using UK primary and secondary care data: a historical cohort study. Lancet Healthy Longev. 2, e426–e435. doi: 10.1016/S2666-7568(21)00118-5
Narattil, N. R., and Maroun, M. (2024). Differential role of NMDA receptors in hippocampal-dependent spatial memory and plasticity in juvenile male and female rats. Hippocampus 34, 564–574. doi: 10.1002/hipo.23631
Nicoll, R. A., and Roche, K. W. (2013). Long-term potentiation: peeling the onion. Neuropharmacology. 74, 18–22. doi: 10.1016/j.neuropharm.2013.02.010
Niiyama, S., Takasu, O., Sakamoto, T., and Ushijima, K. (2016). Intraperitoneal adipose tissue is strongly related to survival rate in a mouse cecal ligation and puncture model. Clin. Transl. Immunol. 5:e64. doi: 10.1038/cti.2016.3
Park, J. E., Shin, T. G., Jo, I. J., Jeon, K., Suh, G. Y., Park, M., et al. (2020). Impact of vitamin C and thiamine administration on delirium-free days in patients with septic shock. J. Clin. Med. 9:193. doi: 10.3390/jcm9010193
Pereira De Souza Goldim, M., Della Giustina, A., Mathias, K., De Oliveira Junior, A., Fileti, M. E., De Carli, R., et al. (2020). Sickness behavior score is associated with neuroinflammation and late behavioral changes in polymicrobial sepsis animal model. Inflammation 43, 1019–1034. doi: 10.1007/s10753-020-01187-z
Peters Van Ton, A. M., Verbeek, M. M., Alkema, W., Pickkers, P., and Abdo, W. F. (2020). Downregulation of synapse-associated protein expression and loss of homeostatic microglial control in cerebrospinal fluid of infectious patients with delirium and patients with Alzheimer's disease. Brain Behav. Immun. 89, 656–667. doi: 10.1016/j.bbi.2020.06.027
Qin, Z., Zhou, C., Xiao, X., and Guo, C. (2021). Metformin attenuates sepsis-induced neuronal injury and cognitive impairment. BMC Neurosci. 22:78. doi: 10.1186/s12868-021-00683-8
Ross, F. M., Allan, S. M., Rothwell, N. J., and Verkhratsky, A. (2003). A dual role for interleukin-1 in LTP in mouse hippocampal slices. J. Neuroimmunol. 144, 61–67. doi: 10.1016/j.jneuroim.2003.08.030
Rudd, K. E., Johnson, S. C., Agesa, K. M., Shackelford, K. A., Tsoi, D., Kievlan, D. R., et al. (2020). Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study. Lancet 395, 200–211. doi: 10.1016/S0140-6736(19)32989-7
Sellers, R. S., Clifford, C. B., Treuting, P. M., and Brayton, C. (2012). Immunological variation between inbred laboratory mouse strains: points to consider in phenotyping genetically immunomodified mice. Vet. Pathol. 49, 32–43. doi: 10.1177/0300985811429314
Semmler, A., Okulla, T., Sastre, M., Dumitrescu-Ozimek, L., and Heneka, M. T. (2005). Systemic inflammation induces apoptosis with variable vulnerability of different brain regions. J. Chem. Neuroanat. 30, 144–157. doi: 10.1016/j.jchemneu.2005.07.003
Serantes, R., Arnalich, F., Figueroa, M., Salinas, M., Andrés-Mateos, E., Codoceo, R., et al. (2006). Interleukin-1beta enhances GABAA receptor cell-surface expression by a phosphatidylinositol 3-kinase/Akt pathway: relevance to sepsis-associated encephalopathy. J. Biol. Chem. 281, 14632–14643. doi: 10.1074/jbc.M512489200
Shepherd, J. D., and Bear, M. F. (2011). New views of Arc, a master regulator of synaptic plasticity. Nat. Neurosci. 14, 279–284. doi: 10.1038/nn.2708
Soejima, T., Hoshino, K., and Morimoto, Y. (2025). The effects of treadmill exercise on the recovery of synaptic plasticity in septic mice: a focus on brain-derived neurotrophic factor/tropomyosin-related kinase B signaling. Anesth. Analg. doi: 10.1213/ANE.0000000000007572. [Epub ahead of print].
Sohroforouzani, A. M., Shakerian, S., Ghanbarzadeh, M., and Alaei, H. (2022). Effect of forced treadmill exercise on stimulation of BDNF expression, depression symptoms, tactile memory and working memory in LPS-treated rats. Behav. Brain Res. 418:113645. doi: 10.1016/j.bbr.2021.113645
Song, Z., Shen, F., Zhang, Z., Wu, S., and Zhu, G. (2020). Calpain inhibition ameliorates depression-like behaviors by reducing inflammation and promoting synaptic protein expression in the hippocampus. Neuropharmacology 174:108175. doi: 10.1016/j.neuropharm.2020.108175
Sonneville, R., De Montmollin, E., Poujade, J., Garrouste-Orgeas, M., Souweine, B., Darmon, M., et al. (2017). Potentially modifiable factors contributing to sepsis-associated encephalopathy. Intensive Care Med. 43, 1075–1084. doi: 10.1007/s00134-017-4807-z
Takeuchi, T., Duszkiewicz, A. J., and Morris, R. G. (2014). The synaptic plasticity and memory hypothesis: encoding, storage and persistence. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 369:20130288. doi: 10.1098/rstb.2013.0288
Tanaka, N., Cortese, G. P., Barrientos, R. M., Maier, S. F., and Patterson, S. L. (2018). Aging and an immune challenge interact to produce prolonged, but not permanent, reductions in hippocampal L-LTP and mBDNF in a rodent model with features of delirium. eNeuro 5:ENEURO.0009-18. doi: 10.1523/ENEURO.0009-18.2018
Tang, C., Jin, Y., and Wang, H. (2022). The biological alterations of synapse/synapse formation in sepsis-associated encephalopathy. Front. Synaptic Neurosci. 14:1054605. doi: 10.3389/fnsyn.2022.1054605
Thy, M., Sonneville, R., Ruckly, S., Mourvillier, B., Schwebel, C., Cohen, Y., et al. (2025). Early systemic insults following severe sepsis-associated encephalopathy of critically ill patients: association with mortality and awakening-an analysis of the OUTCOMEREA database. J. Intensive Care 13:5. doi: 10.1186/s40560-024-00773-9
Tsuchida, T., Wada, T., Mizugaki, A., Oda, Y., Kayano, K., Yamakawa, K., et al. (2022). Protocol for a sepsis model utilizing fecal suspension in mice: fecal suspension intraperitoneal injection model. Front. Med. 9:765805. doi: 10.3389/fmed.2022.765805
Vereker, E., Campbell, V., Roche, E., Mcentee, E., and Lynch, M. A. (2000). Lipopolysaccharide inhibits long term potentiation in the rat dentate gyrus by activating caspase-1. J. Biol. Chem. 275, 26252–26258. doi: 10.1074/jbc.M002226200
Wang, D. S., Zurek, A. A., Lecker, I., Yu, J., Abramian, A. M., Avramescu, S., et al. (2012). Memory deficits induced by inflammation are regulated by alpha5-subunit-containing GABAA receptors. Cell Rep. 2, 488–496. doi: 10.1016/j.celrep.2012.08.022
Wang, W., Le, A. A., Hou, B., Lauterborn, J. C., Cox, C. D., Levin, E. R., et al. (2018). Memory-related synaptic plasticity is sexually dimorphic in rodent hippocampus. J. Neurosci. 38, 7935–7951. doi: 10.1523/JNEUROSCI.0801-18.2018
Warren, H. S., Fitting, C., Hoff, E., Adib-Conquy, M., Beasley-Topliffe, L., Tesini, B., et al. (2010). Resilience to bacterial infection: difference between species could be due to proteins in serum. J. Infect. Dis. 201, 223–232. doi: 10.1086/649557
Widmann, C. N., and Heneka, M. T. (2014). Long-term cerebral consequences of sepsis. Lancet Neurol. 13, 630–636. doi: 10.1016/S1474-4422(14)70017-1
Xu, X. E., Liu, L., Wang, Y. C., Wang, C. T., Zheng, Q., Liu, Q. X., et al. (2019). Caspase-1 inhibitor exerts brain-protective effects against sepsis-associated encephalopathy and cognitive impairments in a mouse model of sepsis. Brain Behav. Immun. 80, 859–870. doi: 10.1016/j.bbi.2019.05.038
Yang, L., Li, J., Liu, F., Chai, X., Fang, Z., and Zhang, X. (2025). The biological changes of synaptic plasticity in the pathological process of sepsis-associated encephalopathy. Curr. Neuropharmacol. 23, 359–374. doi: 10.2174/1570159X23666241028105746
Yin, L., Zhang, J., Ma, H., Zhang, X., Fan, Z., Yang, Y., et al. (2023a). Selective activation of cholinergic neurotransmission from the medial septal nucleus to hippocampal pyramidal neurones improves sepsis-induced cognitive deficits in mice. Br. J. Anaesth. 130, 573–584. doi: 10.1016/j.bja.2023.01.019
Yin, X. Y., Tang, X. H., Wang, S. X., Zhao, Y. C., Jia, M., Yang, J. J., et al. (2023b). HMGB1 mediates synaptic loss and cognitive impairment in an animal model of sepsis-associated encephalopathy. J. Neuroinflamm. 20:69. doi: 10.1186/s12974-023-02756-3
Zhang, R., Sun, L., Hayashi, Y., Liu, X., Koyama, S., Wu, Z., et al. (2010). Acute p38-mediated inhibition of NMDA-induced outward currents in hippocampal CA1 neurons by interleukin-1beta. Neurobiol. Dis. 38, 68–77. doi: 10.1016/j.nbd.2009.12.028
Zhao, Z., Wang, Y., Zhou, R., Li, Y., Gao, Y., Tu, D., et al. (2020). A novel role of NLRP3-generated IL-1β in the acute-chronic transition of peripheral lipopolysaccharide-elicited neuroinflammation: implications for sepsis-associated neurodegeneration. J. Neuroinflamm. 17:64. doi: 10.1186/s12974-020-1728-5
Keywords: sepsis-associated encephalopathy, synaptic plasticity, long-term potentiation, hippocampus, neuroinflammation
Citation: Hoshino K (2025) Targeting synaptic plasticity to bridge translational gaps in sepsis-associated encephalopathy. Front. Aging Neurosci. 17:1616736. doi: 10.3389/fnagi.2025.1616736
Received: 23 April 2025; Accepted: 26 May 2025;
Published: 10 June 2025.
Edited by:
Daniel Ortuño-Sahagún, University of Guadalajara, MexicoReviewed by:
Yukio Imamura, Kyoto University, JapanCopyright © 2025 Hoshino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Koji Hoshino, aG9zaGlub2tvQG1lZC5ob2t1ZGFpLmFjLmpw