 Sofiia Baranykova
Sofiia Baranykova- Laboratory of Neurodegeneration, International Institute of Molecular and Cell Biology in Warsaw, Warsaw, Poland
Ca2+ homeostasis is essential for glial cell activity and normal neuronal function, and store-operated Ca2+ entry (SOCE) is one mechanism that maintains it. The present review discusses the interplay between Ca2+ dysregulation and microglial activation in glaucomatous retinal degeneration. We examine the impact of Ca2+ homeostasis and SOCE on microglial function and their potential role in retinal ganglion cell degeneration and present the hypothesis that SOCE dysregulation may underlie glaucomatous pathology. This review suggests that targeting Ca2+ pathways in microglial cells can be a potential treatment for glaucoma.
Introduction
Glaucoma is a pathology that is still not fully understood, has different subtypes, is often undiagnosed, and needs new treatment approaches. Some of its subtypes, such as retinal degeneration without intraocular pressure (IOP), need early diagnostic markers.
Glaucoma has a comprehensive mechanism that involves various cell types and pathways. The present review concentrates on Ca2+ signaling in microglial cells because of their involvement in inflammation and neurodegenerative processes (Heo et al., 2015; Michaelis et al., 2015). These observations are supported by our data. The information that is presented in this mini-review indicates that proteins that are involved in Ca2+ homeostasis in microglial cells may be used as diagnostic markers and may be potential targets for therapeutic strategies for glaucoma.
Definition and types of glaucoma
Glaucoma comprises a group of diseases that affect approximately 100 million people worldwide, often leading to vision loss. Its major features are optic nerve degeneration and the loss or dysfunction of retinal ganglion cells (RGCs) (Crish and Calkins, 2011; Zhang et al., 2021). In addition to these factors, mitochondrial dysfunction and neuroinflammation are also observed (Saccà and Izzotti, 2014).
Glaucoma can be classified based on age (congenital, juvenile or adult), etiology (primary, with no identifiable cause or secondary, resulting from previous ocular trauma), and the location of obstruction in the drainage system of the eye (primary open angle or primary angle closure glaucoma) (Pang and Clark, 2020; Ekici and Moghimi, 2023).
In most cases, glaucoma is primarily associated with elevated intraocular pressure (IOP), which results from dysfunction in outflow pathways of aqueous humor from the eye (Jayaram et al., 2023). However, it can also occur without an elevation of IOP, which is referred to as normal-tension glaucoma or low-tension glaucoma and can be caused by vascular dysregulation, impairments in blood flow, glymphatic system failure, or other factors (Shinozaki et al., 2024).
The majority of all these types of glaucoma have common players, such as microglial cells (Duarte, 2021; Wang et al., 2014; Somerville et al., 2025), mostly because of their involvement in neuroinflammation and retinal ganglion cell degeneration. The present review discusses both findings from the literature and our own data, emphasizing that microglial activity during retinal neurodegeneration may be significantly influenced by Ca2+ homeostasis, particularly the store-operated Ca2+ entry (SOCE) pathway. Additionally, zebrafish with stim2 gene knockout may be a promising animal model of glaucomatous pathology.
Pathophysiology and models of glaucoma
Environmental factors, such as air pollution and an inappropriate diet (Almarzouki, 2024), may be risk factors for glaucoma development. However, there is no clinically proven evidence that cigarette smoking, alcohol consumption, or excessive caffeine intake can cause this disease (Stuart et al., 2023). The most crucial factor could be an imbalance in the consumption of essential fatty acids or frequent salt intake. Glaucoma has been shown to be strongly associated with chronic stress and anxiety (Stuart et al., 2023). However, many different factors contribute to the development of glaucoma (Stuart et al., 2023).
To gain a complete picture of glaucoma's pathogenesis, researchers must employ various animal models that reflect its multifaceted origins. Most models are based on IOP pathology and are called ocular hypertension models (Reinehr et al., 2024). They are induced by blocking drainage pathways, injecting hypertonic saline, or introducing mutations that block aqueous humor outflow. Proinflammatory pathways during glaucoma can be activated by high IOP, along with other factors (e.g., oxidative stress), which in turn lead to the release of signaling molecules by glial cells and RGCs that contribute to cellular damage and death (Bugara et al., 2024).
Hyperactivation of the 2′3′-cyclic guanosine monophosphate–adenosine monophosphate–stimulator of interferon gene (cGAS–STING) signaling pathway in microglial cells is responsible for RGC loss and vision deterioration (Liu et al., 2024). cGAS–STING signaling proteins are located in the endoplasmic reticulum and Golgi apparatus. In a glaucoma model in mice, 95% of STING was shown to colocalize with microglia markers (Liu et al., 2024). However, microglial activation does not depend on this signaling. cGAS–STING signaling is responsible for activation of the innate immune response by sensing double-stranded DNA (dsDNA) in the cytoplasm. Such dsDNA is observed in RGCs during their degeneration. When cGAS–STING is activated, it induces the expression of cytokines and chemokines by microglia and other innate immune cells. The ablation of this pathway has been shown to prevent the apoptosis of RGCs and visual deterioration (Zhang et al., 2024; Wu et al., 2023). Additionally, cGAS–STING signaling has been proposed to be highly connected to cellular stress (Schmid et al., 2024).
Our recent ultrastructural analyses revealed mitochondrial damage and cristae loss in photoreceptors in stim2 knockout zebrafish larvae (Baranykova et al., 2024), which are hallmarks of cellular stress. Considering the connection between STING signaling, mitochondrial stress, and glaucomatous neurodegeneration, these data support a possible link between retinal neurodegeneration and Ca2+ dysregulation. Overall, our evidence suggests that SOCE dysfunction, particularly attributable to the loss of stromal interaction molecule 2 (STIM2) protein, may play an important role in the pathogenesis of glaucoma.
Other genetic models of IOP-dependent glaucoma include mutations of the Prss56 gene in E50Ktg mice, which mimics the angle closure subtype (i.e., normotensive glaucoma with RGC loss) (Amato, 2022). The most widely used and common models are DBA/2J mice and myocilin mutant mice, which carry a point mutation in Tyr423His in the Prss56 gene and exhibit an increase in IOP and age-dependent glaucoma progression (Amato, 2022). Studies of age-dependent eye diseases show a significant increase in the pathogenic activity of microglia with age, which in turn can cause chronic inflammation (Ahmad and Subramani, 2022).
Models to study IOP-independent cases usually involve excitotoxicity, immune modulation, ischemia or reperfusion, optic nerve crush, and gene modification (Tsai et al., 2024). In some IOP-independent rat models, the peak of the microglial response appears at initial stages (i.e., day 3 in the case of N-methyl-D-aspartate-induced retinal degeneration) (Kuehn et al., 2017). The activity of microglial cells decreases thereafter (Kuehn et al., 2017).
None of the existing models provide a complete picture of glaucoma. Multiple model systems are needed to obtain a more comprehensive picture of the disease (Bugara et al., 2024). Most available treatments to improve vision in glaucoma are designed to decrease IOP, but there is a need to focus on possible neuroprotective pathways in IOP-independent cases (Bugara et al., 2024).
Given the multifactorial nature of glaucoma and limitations of current IOP-based models, there is an increasing need to investigate IOP-independent mechanisms, especially those that are related to neuroinflammation and cellular stress pathways. Among these, Ca2+ homeostasis and its regulators appear to be critical factors that influence RGC survival and microglial activation. Thus, SOCE and its key components (i.e., STIM1, STIM2, and Ca2+ release-activated Ca2+ channel protein [ORAI]) deserve further investigation. The next section discusses the role of STIM2-dependent SOCE in neurodegenerative processes, highlighting its potential contribution to glaucomatous retinal damage.
STIM2-dependent SOCE: role of STIM2 in neurodegenerative mechanisms
SOCE is potentially an important mechanism for Ca2+ regulation in all metazoan cells (Prakriya and Lewis, 2015; Korshunov and Prakriya, 2025). It is a major Ca2+ influx pathway in non-excitable cells (Wegierski and Kuznicki, 2018). The main components of SOCE are STIM1 and STIM2 (which are located in the endoplasmic reticulum and trigger Ca2+ influx when endoplasmic reticulum Ca2+ levels drop) and ORAI1, ORAI2, and ORAI3 (which are located in the plasma membrane and enable Ca2+ ions to enter). Additional components include transient receptor potential canonical channels (TRPCs; which modulate SOCE), SARAF (store-operated Ca2+ entry-associated regulatory factor which prevents Ca2+ overload), and Ca2+ release-activated channel regulator 2A (CRACR2A; a modulator of immune cells) (Srikanth et al., 2010; Wegierski and Kuznicki, 2018; Prakriya and Lewis, 2015). When Ca2+ in the endoplasmic reticulum is sufficient, STIM remains inactive. After Ca2+ levels decrease to a particular level, STIM changes its conformation, oligomerizes, migrates to endoplasmic reticulum—plasma membrane junctions, and binds to ORAI, which allows Ca2+ to enter the cytoplasm.
Previous studies (Baranykova et al., 2024; Wasilewska et al., 2023) have shown that stim2 knockout in Danio rerio leads to a glaucoma-like phenotype. Mutant zebrafish exhibit signs of activated microglia (i.e., an increase in the anxa3a gene), a loss of RGCs and their dendrite numbers, a decrease in γ-aminobutyric acid-ergic cells and photoreceptors, downregulation of the ins gene, and mitochondrial disruptions. These findings strongly suggest that STIM2 is essential for retinal homeostasis and microglial function. A previous study (Wasilewska et al., 2023) showed that stim2-deficient zebrafish exhibit impairments in visual behavior and greater sensitivity to hypoxia, which supports the hypothesis that STIM2 might regulate neuroprotective mechanisms in the retina.
Recent evidence has shown a link between SOCE and STING activation in neurons. Woo et al. (2024) demonstrated that STIM1 directly interacts with STING, in which it helps keep STING in the endoplasmic reticulum when the cell is in a resting state. During inflammation, STIM1 detaches from STING, followed by STING activation and neurotoxic signaling. Interestingly, SOCE activation serves as a trigger for this process. In neurons that lack STIM1 protein, STING activation increases. These findings suggest that the dysregulation of STIM1 or STIM2 may make neurons more sensitive to STING-related stress, which can explain how Ca2+ imbalance and nerve cell loss occur in such diseases as glaucoma (Figure 1).
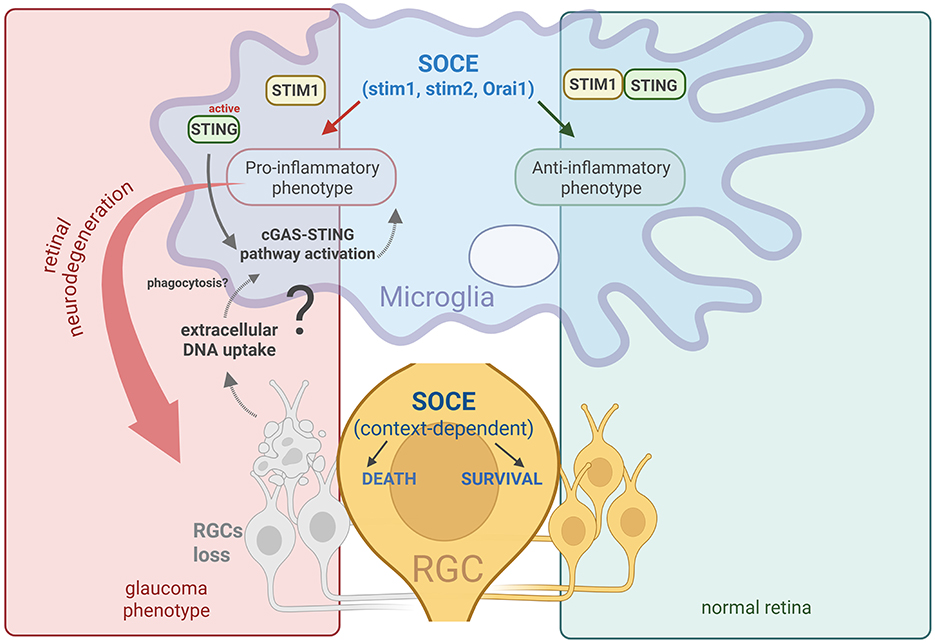
Figure 1. Schematic illustration of the possible role of SOCE in microglial cells and the progression of RGC loss during glaucomatous neurodegeneration.
Based on prior data (Baranykova et al., 2024; Wasilewska et al., 2023), the disruption of STIM2 is hypothesized to cause microglial cell activation, which in turn leads to RGC loss (Figure 1). Still not fully understood is how SOCE, particularly STIM2, is involved in glaucomatous neurodegeneration. Nevertheless, our recent studies provide some new evidence of a link between SOCE dysregulation and RGC loss in the retina (Baranykova et al., 2024; Wasilewska et al., 2023).
Ca2+ homeostasis in retinal ganglion cells
The survival of RGCs during axon injury depends on baseline Ca2+ levels. Cells with lower baseline Ca2+ levels degenerate more frequently during neuropathology than those with higher levels (McCracken et al., 2023). This phenomenon means that Ca2+ homeostasis determines the survival and resilience of RGCs. SOCE participates in the regulation of baseline Ca2+ levels; thus, a reasonable assumption is that its components regulate RGC survival during neuropathology (McCracken et al., 2023; Molnar et al., 2012). During glaucomatous neurodegeneration, Ca2+ homeostasis is significantly altered in RGCs, particularly light-driven Ca2+ dynamics (Quintero et al., 2022). These data support a regulatory role for Ca2+ in RGCs. However, unclear is how Ca2+ signals influence RGC death and survival (McCracken et al., 2023).
One suggestion is that Ca2+ may be involved in IOP-dependent and -independent glaucoma (Kastner et al., 2023). The involvement of Ca2+ in IOP-dependent diseases may occur through mitogen-activated protein kinase/extracellular signal-regulated kinase and nuclear factor-κB pathways (Wang et al., 2023). The TRPC5 Ca2+ channel, which regulates Ca2+ levels, is a negative regulator of RGC axonal outgrowth (Oda et al., 2020).
In summary, correct Ca2+ regulation is essential for the survival and function of RGCs, especially under stress conditions during glaucoma. Disruptions of Ca2+ signaling, particularly through SOCE, appear to be involved in RGC degeneration. However, the exact mechanisms by which Ca2+ imbalance causes RGC death (i.e., either directly or through interactions with other retinal cell types like microglia) remain to be explored. Further studies of these interconnected pathways are necessary to develop more advanced treatment strategies for glaucoma.
Ca2+ homeostasis in microglial activation
Several studies have shown that SOCE might influence the activity of microglial cells (Heo et al., 2015; Wegierski and Kuznicki, 2018). Changes in expression of the SOCE channels Orai1 and TRPC1 have been shown to regulate the transformation of microglia toward either a proinflammatory or antiinflammatory state (Nascimento Da Conceicao et al., 2021). For example, the antiinflammatory phenotype can be achieved through Orai1 activation. Because Orai1 is functionally connected to STIM2, any changes in STIM2 levels or its structure can be expected to alter microglial function.
Mechanistically, SOCE regulates microglial functions primarily through Ca2+-dependent phagocytosis and motility. STIM1 and STIM2 sense endoplasmic reticulum Ca2+ depletion and activate Orai1 channels, generating Ca2+ influx that sustains essential microglial processes. Blocking or knocking down STIM1, STIM2, or Orai1 impairs uridine diphosphate-induced phagocytosis and FcγR-mediated phagocytosis, demonstrating the dependence of microglial clearance functions on SOCE (Michaelis et al., 2015; Heo et al., 2015; Sogkas et al., 2015). STIM1 also facilitates Ca2+-dependent phagosome–endoplasmic reticulum interactions, and its ablation decreases phagocytic efficiency (Nunes et al., 2012). The disruption of STIM2 has been shown to reduce inflammation and apoptosis, indicating a broader role in regulating Ca2+-dependent cellular responses (Ye et al., 2024). Additionally, acute microglial motility is Ca2+-dependent and requires STIM1 (Lim et al., 2017). These findings indicate that proper SOCE activity is essential for microglial surveillance and debris clearance (Hallett, 2020).
Disruption of the STIM/Orai system can have downstream consequences for neuronal survival. For example, in zebrafish, stim2a and stim2b double knockouts exhibit RGC loss that resembles glaucoma (Baranykova et al., 2024). This suggests that Ca2+ dysregulation may contribute to both microglial dysfunction and neuronal damage.
Both RGC resilience and microglial function are interconnected with, and depend on, Ca2+ levels. Different scenarios can be assumed: (i) microglial activation or other functional changes that are attributable to Ca2+ pathways promote RGC damage, (ii) RGC loss can appear independently of microglial activation through Ca2+ dysregulation in RGCs, or (iii) RGC degradation (through Ca2+ dysregulation) triggers microglial activation. It is difficult to establish which event occurs first and which is secondary. It is supposed that they are interdependent, in which Ca2+ dysregulation plays a significant role.
Dual role of microglia in glaucoma: beneficial or detrimental?
Microglia perform various functions, including immune surveillance, synaptic refinement, neurotrophic support, and debris clearance. During neurodegenerative disorders, microglia are activated and increase the secretion of cytokines and phagocytic activity. Under normal conditions, the retina contains 0.3–1% microglial cells, which are located mostly in the inner plexiform layer or outer plexiform layer. In glaucoma, microglial cells undergo significant changes in morphology, cytokine synthesis, number, and distribution (Noailles et al., 2014). Activated microglia have been detected in retinitis pigmentosa, age-related macular degeneration, diabetic retinopathy, uveitis, and glaucoma. In all of these diseases, there is a loss of photoreceptor cells and/or RGCs (Wang and Cepko, 2022; Ramirez et al., 2017; Wang et al., 2021).
During retinal degeneration, an increase in the number of immunoglobulin G deposits and an increase in microglial activity have been detected. Microglial cells were observed near the site of immunoglobulin G deposition, and their increase was observed in the RGC layer and inner plexiform layer. The mechanism that leads to RGC loss appears to be connected to microglia activation and inflammatory processes (Joachim et al., 2012).
Microglia are highly interconnected with other glial cells (e.g., Müller cells) (Carpi-Santos et al., 2022). Hu et al. (2021) showed that experimental glaucoma is observed during the activation of microglia and Müller cells. Müller cell activation occurs through the adenosine triphosphate/P2X7 receptor pathway (Xu et al., 2022). Inflammation during glaucoma is suggested to be intensified by an interplay between microglia and Müller cells.
The role of microglia in glaucoma is not fully understood; it can be either detrimental or ameliorative. The depletion of microglia decreases cell survival (Ishikawa et al., 2023; Diemler et al., 2024) and worsens the process of regeneration or aggravates neurodegeneration (Diemler et al., 2024). Other studies reported that the depletion of microglia had no significant effect on visual function during glaucoma (Tan et al., 2021) or was a promising approach for neuroprotection (Ishikawa et al., 2023). Microglia fix visual circuits by engulfing synaptic elements during development (Schafer et al., 2012), interacting with dendritic spines in an activity-dependent manner (Tremblay et al., 2010) and mediating ocular dominance plasticity (Sipe et al., 2016), with roles in adult synapse remodeling (Wang et al., 2016). In glaucoma, these functions may deteriorate, potentially promoting synapse and RGC loss.
One hypothetical model of events in the retina during glaucoma was proposed to explain these inconsistencies (Ramirez et al., 2017). According to this model, microglial activation in the outer plexiform layer, inner plexiform layer, ganglion cell layer, and nerve fiber layer during initial stages of the disease plays mostly a protective role. However, in the case of chronic disease, activated microglial cells are mostly detrimental, and their blockade can reduce pathological progression (Ramirez et al., 2017).
These observations indicate that whether microglia are detrimental or beneficial depends on such factors as disease stage and the presence of mutations or dysregulated cell pathways (Ahmad and Subramani, 2022). Therefore, radical therapeutic approaches, such as microglia depletion, blockade, or activation, cannot be considered. Treatments should focus on correcting intracellular microglial pathways, such as Ca2+ homeostasis, because of its significant regulatory role in these cells. For example, blocking a change in the proinflammatory phenotype to the antiinflammatory phenotype can be achieved by modifying Ca2+ channel activity (Nascimento Da Conceicao et al., 2021).
Therapeutic implications
The immune system plays a crucial role in the pathogenesis of glaucoma. Much data point to autoimmune aspects of this disease. However, the role of microglial cells in glaucoma is far from being fully understood (Tsai et al., 2024). Some glaucoma patients have specific antibodies against retinal antigens. Because some retinal diseases exhibit microglial activation, some treatments are designed to target microglia (Wang and Cepko, 2022). There are different strategies to target microglia in the eye, such as depleting microglia with specific chemicals or radiation, reprogramming microglia, and blocking cytokine activity or phagocytosis (Wang and Cepko, 2022). The behavior of microglial cell populations varies in various locations of the eye (Wang and Cepko, 2022). One way to deplete microglial cells is to use pexidartinib. This chemical inhibits colony stimulating factor 1 receptors that are expressed in microglia. However, several experiments have reported no benefits of such depletion (Wang et al., 2019).
Recently, Zhou et al. (2024) performed experiments using a mouse chronic ocular hypertension model to understand how non-coding RNA molecules that play role in regulating inflammation can influence glaucoma. Downregulation of the interleukin-1 receptor-associated kinase 1/tumor necrosis factor receptor-associated factor 6/nuclear factor-κB signaling pathway in microglia after transfection with miR-146a-5p reduced neuroinflammation and increased the survival of RGCs, suggesting that microglia may be a viable therapeutic target for glaucoma (Joachim et al., 2012).
Summary
The loss of RGCs in glaucoma can have multiple causes. As described above, microglial cell dysfunction and Ca2+ homeostasis dysregulation can be linked to this disease. Microglia participate in glaucoma development via inflammation, phagocytosis, and other processes. In RGCs, proper Ca2+ levels determine survival, whereas in microglia, Ca2+ levels dictate cellular activity and whether it exhibits an anti- or proinflammatory phenotype. Changes in Ca2+ levels influence both microglial cells and RGCs, thereby complicating the identification of which processes initiate the pathology. For example, can activated microglia change Ca2+ homeostasis in RGCs to induce their neurodegeneration? It is suggested that proteins involved in Ca2+ homeostasis in microglial cells could be targets for anti-degeneration treatment.
Author contributions
SB: Writing – original draft, Visualization, Writing – review & editing.
Funding
The author declares that financial support was received for the research and/or publication of this article. This work was supported by Preludium Grant NCN 2023/49/N/NZ3/02921.
Acknowledgments
The author expresses gratitude to Professor Jacek Kuźnicki for his guidance throughout this work, to Dr. Łukasz Majewski for his valuable suggestions that improved this review, and to Michael Arends for his assistance with language editing of the manuscript.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author declares that Gen AI was used in the creation of this manuscript. Generative AI tools were used for grammar correction and language refinement.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmad, I., and Subramani, M. (2022). Microglia: friends or foes in glaucoma? A developmental perspective. Stem Cells Transl. Med. 11, 1210–1218. doi: 10.1093/stcltm/szac077
Almarzouki, N. (2024). Impact of environmental factors on glaucoma progression: a systematic review. Clin. Ophthalmol. 18, 2705–2720. doi: 10.2147/OPTH.S484855
Amato, R. (2022). In vivo murine models for the study of glaucoma pathophysiology: procedures, analyses, and typical outcomes. Ann. Eye Sci. 7:30. doi: 10.21037/aes-21-48
Baranykova, S., Gupta, R. K., Kajdasz, A., Wasilewska, I., Macias, M., Szybinska, A., et al. (2024). Loss of Stim2 in zebrafish induces glaucoma-like phenotype. Sci. Rep. 14:24442. doi: 10.1038/s41598-024-74909-0
Bugara, K., Pacwa, A., and Smedowski, A. (2024). Molecular pathways in experimental glaucoma models. Front. Neurosci. 18:1363170. doi: 10.3389/fnins.2024.1363170
Carpi-Santos, R., de Melo Reis, R. A., Gomes, F. C. A., and Calaza, K. C. (2022). Contribution of Müller cells in the diabetic retinopathy development: focus on oxidative stress and inflammation. Antioxidants 11:617. doi: 10.3390/antiox11040617
Crish, S. D., and Calkins, D. J. (2011). Neurodegeneration in glaucoma: progression and calcium-dependent intracellular mechanisms. Neuroscience 176, 1–11. doi: 10.1016/j.neuroscience.2010.12.036
Diemler, C. A., MacLean, M., Heuer, S. E., Hewes, A. A., Marola, O. J., Libby, R. T., et al. (2024). Microglia depletion leads to increased susceptibility to ocular hypertension-dependent glaucoma. Front. Aging Neurosci. 16:1396443. doi: 10.3389/fnagi.2024.1396443
Duarte, J. N. (2021). Neuroinflammatory mechanisms of mitochondrial dysfunction and neurodegeneration in glaucoma. J. Ophthalmol. 2021:4581909. doi: 10.1155/2021/4581909
Ekici, E., and Moghimi, S. (2023). Advances in understanding glaucoma pathogenesis: a multifaceted molecular approach for clinician scientists. Mol. Aspects Med. 94:101223. doi: 10.1016/j.mam.2023.101223
Heo, D. K., Lim, H. M., Nam, J. H., Lee, M. G., and Kim, J. Y. (2015). Regulation of phagocytosis and cytokine secretion by store-operated calcium entry in primary isolated murine microglia. Cell Signal 27, 177–186. doi: 10.1016/j.cellsig.2014.11.003
Hu, X., Zhao, G. L., Xu, M. X., Zhou, H., Li, F., Miao, Y., et al. (2021). Interplay between Müller cells and microglia aggravates retinal inflammatory response in experimental glaucoma. J. Neuroinflammation 18:303. doi: 10.1186/s12974-021-02366-x
Ishikawa, M., Izumi, Y., Sato, K., Sato, T., Zorumski, C. F., Kunikata, H., et al. (2023). Glaucoma and microglia-induced neuroinflammation. Front. Ophthalmol. 3:1132011. doi: 10.3389/fopht.2023.1132011
Jayaram, H., Kolko, M., Friedman, D. S., and Gazzard, G. (2023). Glaucoma: now and beyond. Lancet 402, 1788–1801. doi: 10.1016/S0140-6736(23)01289-8
Joachim, S. C., Gramlich, O. W., Laspas, P., Schmid, H., Beck, S., von Pein, H. D., et al. (2012). Retinal ganglion cell loss is accompanied by antibody depositions and increased levels of microglia after immunization with retinal antigens. PLoS ONE 7:e40616. doi: 10.1371/journal.pone.0040616
Kastner, A., Stuart, K. V., Montesano, G., De Moraes, C. G., Kang, J. H., Wiggs, J. L., et al. (2023). Calcium channel blocker use and associated glaucoma and related traits among UK biobank participants. JAMA Ophthalmol. 141, 956–964. doi: 10.1001/jamaophthalmol.2023.3877
Korshunov, K. S., and Prakriya, M. (2025). Store-operated calcium channels in the nervous system. Annu. Rev. Physiol. 87, 173–199. doi: 10.1146/annurev-physiol-022724-105330
Kuehn, S., Rodust, C., Stute, G., Grotegut, P., Meißner, W., Reinehr, S., et al. (2017). Concentration-dependent inner retina layer damage and optic nerve degeneration in a NMDA model. J. Mol. Neurosci. 63, 283–299. doi: 10.1007/s12031-017-0978-x
Lim, H. M., Woon, H., Han, J. W., Baba, Y., Kurosaki, T., Lee, M. G., et al. (2017). UDP-induced phagocytosis and ATP-stimulated chemotactic migration are impaired in STIM1(–/–) microglia in vitro and in vivo. Mediators Inflamm. 2017:8158514. doi: 10.1155/2017/8158514
Liu, Y., Wang, A., Chen, C., Zhang, Q., Shen, Q., Zhang, D., et al. (2024). Microglial cGAS–STING signaling underlies glaucoma pathogenesis. Proc. Nat. Acad. Sci. 121:e2409493121. doi: 10.1073/pnas.2409493121
McCracken, S., Fitzpatrick, M. J., Hall, A. L., Wang, Z., Kerschensteiner, D., Morgan, J. L., et al. (2023). Diversity in homeostatic calcium set points predicts retinal ganglion cell survival following optic nerve injury in vivo. Cell Rep. 42:113165. doi: 10.1016/j.celrep.2023.113165
Michaelis, M., Nieswandt, B., Stegner, D., Eilers, J., and Kraft, R. (2015). STIM1, STIM2, and Orai1 regulate store-operated calcium entry and purinergic activation of microglia. Glia 63, 652–663. doi: 10.1002/glia.22775
Molnar, T., Barabas, P., Birnbaumer, L., Punzo, C., Kefalov, V., and KriŽaj, D. (2012). Store-operated channels regulate intracellular calcium in mammalian rods. J. Physiol. 590, 3465–3481. doi: 10.1113/jphysiol.2012.234641
Nascimento Da Conceicao, V., Sun, Y., Ramachandran, K., Chauhan, A., Raveendran, A., Venkatesan, M., et al. (2021). Resolving macrophage polarization through distinct Ca2+ entry channel that maintains intracellular signaling and mitochondrial bioenergetics. iScience 24:103339. doi: 10.1016/j.isci.2021.103339
Noailles, A., Fernández-Sánchez, L., Lax, P., and Cuenca, N. (2014). Microglia activation in a model of retinal degeneration and TUDCA neuroprotective effects. J. Neuroinflammation 11:186. doi: 10.1186/s12974-014-0186-3
Nunes, P., Cornut, D., Bochet, V., Hasler, U., Oh-Hora, M., Waldburger, J. M., et al. (2012). STIM1 juxtaposes ER to phagosomes, generating Ca2+ hotspots that boost phagocytosis. Curr. Biol. 22, 1990–1997. doi: 10.1016/j.cub.2012.08.049
Oda, M., Yamamoto, H., Matsumoto, H., Ishizaki, Y., and Shibasaki, K. (2020). TRPC5 regulates axonal outgrowth in developing retinal ganglion cells. Lab. Invest. 100, 297–310. doi: 10.1038/s41374-019-0347-1
Pang, I-. H., and Clark, A. F. (2020). Inducible rodent models of glaucoma. Prog. Retin. Eye Res. 75:100799. doi: 10.1016/j.preteyeres.2019.100799
Prakriya, M., and Lewis, R. S. (2015). Store-operated calcium channels. Physiol. Rev. 95, 1383–1436. doi: 10.1152/physrev.00020.2014
Quintero, H., Shiga, Y., Belforte, N., Alarcon-Martinez, L., El Hajji, S., Villafranca-Baughman, D., et al. (2022). Restoration of mitochondria axonal transport by adaptor Disc1 supplementation prevents neurodegeneration and rescues visual function. Cell Rep. 40:111324. doi: 10.1016/j.celrep.2022.111324
Ramirez, A. I., de Hoz, R., Salobrar-Garcia, E., Salazar, J. J., Rojas, B., Ajoy, D., et al. (2017). The role of microglia in retinal neurodegeneration: Alzheimer's disease, Parkinson, and Glaucoma. Front. Aging Neurosci. 9:214. doi: 10.3389/fnagi.2017.00214
Reinehr, S., Köseoğlu, A., Qin, W., Tsai, T., and Joachim, S. (2024). Mechanisms of age-related ocular diseases: a comprehensive review with an emphasis on glaucoma. Aging Adv. 1. doi: 10.4103/AGINGADVANCES.AGINGADV-D-24-00001
Saccà, S., and Izzotti, A. (2014). “Glaucoma: an overview,” in Handbook of Nutrition, Diet, and the Eye (Amsterdam: Elsevier), 29–41.
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Schmid, M., Fischer, P., Engl, M., Widder, J., Kerschbaum-Gruber, S., and Slade, D. (2024). The interplay between autophagy and cGAS-STING signaling and its implications for cancer. Front. Immunol. 15:1356369. doi: 10.3389/fimmu.2024.1356369
Shinozaki, Y., Namekata, K., Guo, X., and Harada, T. (2024). Glial cells as a promising therapeutic target of glaucoma: beyond the IOP. Front. Ophthalmol. 3:1310226. doi: 10.3389/fopht.2023.1310226
Sipe, G., Lowery, R., Tremblay, M. È., Kelly, E. A., Lamantia, C. E., and Majewska, A. K. (2016). Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 7:10905. doi: 10.1038/ncomms10905
Sogkas, G., Stegner, D., Syed, S. N., Vögtle, T., Rau, E., Gewecke, B., et al. (2015). Cooperative and alternate functions for STIM1 and STIM2 in macrophage activation and in the context of inflammation. Immun. Inflamm. Dis. 3, 154–170. doi: 10.1002/iid3.56
Somerville, M. M., Garner, M. A., Girkin, C. A., and Gross, A. K. (2025). Role of microglia in glaucomatous pathology. Adv. Exp. Med. Biol. 1468, 149–153. doi: 10.1007/978-3-031-76550-6_25
Srikanth, S., Jung, H. J., Kim, K. D., Souda, P., Whitelegge, J., and Gwack, Y. (2010). A novel EF-hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells. Nat. Cell Biol. 12, 436–446. doi: 10.1038/ncb2045
Stuart, K. V., Pasquale, L. R., Kang, J. H., Foster, P. J., and Khawaja, A. P. (2023). Towards modifying the genetic predisposition for glaucoma: an overview of the contribution and interaction of genetic and environmental factors. Mol. Aspects Med. 93:101203. doi: 10.1016/j.mam.2023.101203
Tan, Z., Guo, Y., Shrestha, M., Gregory-Ksander, M. S., and Jakobs, T. C. (2021). Depletion of optic nerve microglia does not improve visual function in experimental glaucoma. Invest. Ophthalmol. Vis. Sci. 62, 2383–2383. doi: 10.1167/iovs.62.8.2383
Tremblay, M. È., Lowery, R. L., and Majewska, A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 8:e1000527. doi: 10.1371/journal.pbio.1000527
Tsai, T., Reinehr, S., Deppe, L., Strubbe, A., Kluge, N., Dick, H. B., et al. (2024). Glaucoma animal models beyond chronic IOP increase. Int. J. Mol. Sci. 25:906. doi: 10.3390/ijms25020906
Wang, H. N., Qian, W. J., Zhao, G. L., Li, F., Miao, Y. Y., Lei, B., et al. (2023). L- and T-type Ca2+ channels dichotomously contribute to retinal ganglion cell injury in experimental glaucoma. Neural Regen. Res. 18, 1106–1111. doi: 10.4103/1673-5374.360277
Wang, K., Peng, B., and Lin, B. (2014). Fractalkine receptor regulates microglial neurotoxicity in an experimental mouse glaucoma model. Glia 62, 1943–1954. doi: 10.1002/glia.22715
Wang, S. K., and Cepko, C. L. (2022). Targeting microglia to treat degenerative eye diseases. Front. Immunol. 13:843558. doi: 10.3389/fimmu.2022.843558
Wang, S. K., Xue, Y., and Cepko, C. L. (2021). Augmentation of CD47/SIRPα signaling protects cones in genetic models of retinal degeneration. JCI Insight 6:e150796. doi: 10.1172/jci.insight.150796
Wang, S. K., Xue, Y., Rana, P., Hong, C. M., and Cepko, C. L. (2019). Soluble CX3CL1 gene therapy improves cone survival and function in mouse models of retinitis pigmentosa. Proc. Natl. Acad. Sci. U. S. A. 116, 10140–10149. doi: 10.1073/pnas.1901787116
Wang, X., Zhao, L., Zhang, J., Fariss, R. N., Ma, W., Kretschmer, F., et al. (2016). Requirement for microglia for the maintenance of synaptic function and integrity in the mature retina. J. Neurosci. 36, 2827–2842. doi: 10.1523/JNEUROSCI.3575-15.2016
Wasilewska, I., Majewski, Ł., Adamek-Urbańska, D., Mondal, S. S., Baranykova, S., Gupta, R. K., et al. (2023). Lack of Stim2 affects vision-dependent behavior and sensitivity to hypoxia. Zebrafish 20, 146–159. doi: 10.1089/zeb.2022.0068
Wegierski, T., and Kuznicki, J. (2018). Neuronal calcium signaling via store-operated channels in health and disease. Cell Calcium 74, 102–111. doi: 10.1016/j.ceca.2018.07.001
Woo, M. S., Mayer, C., Binkle-Ladisch, L., Sonner, J. K., Rosenkranz, S. C., Shaposhnykov, A., et al. (2024). STING orchestrates the neuronal inflammatory stress response in multiple sclerosis. Cell 187, 4043–4060.e4030. doi: 10.1016/j.cell.2024.05.031
Wu, X., Yu, N., Ye, Z., Gu, Y., Zhang, C., Chen, M., et al. (2023). Inhibition of cGAS-STING pathway alleviates neuroinflammation-induced retinal ganglion cell death after ischemia/reperfusion injury. Cell Death Dis. 14:615. doi: 10.1038/s41419-023-06140-0
Xu, M. X., Zhao, G. L., Hu, X., Zhou, H., Li, S. Y., Li, F., et al. (2022). P2X7/P2X4 Receptors mediate proliferation and migration of retinal microglia in experimental glaucoma in mice. Neurosci. Bull. 38, 901–915. doi: 10.1007/s12264-022-00833-w
Ye, X., Chen, Q., Gong, X., Zhou, C., Yuan, T., Wang, X., et al. (2024). STIM2 suppression blocks glial activation to alleviate brain ischemia reperfusion injury via inhibition of inflammation and pyroptosis. Mol. Biotechnol. 66, 2046–2063. doi: 10.1007/s12033-023-00823-x
Zhang, N., Wang, J., Li, Y., and Jiang, B. (2021). Prevalence of primary open angle glaucoma in the last 20 years: a meta-analysis and systematic review. Sci. Rep. 11:13762. doi: 10.1038/s41598-021-92971-w
Zhang, Q., Xiong, Y., Li, R., Wang, X., Lin, X., and Tong, Y. (2024). Targeting cGAS-STING signaling protects retinal ganglion cells from DNA damage-induced cell loss and promotes visual recovery in glaucoma. Aging 16, 9813–9823. doi: 10.18632/aging.205900
Keywords: store-operated calcium entry (SOCE), STIM2, calcium homeostasis, microglia, retinal ganglion cells, retina, glaucoma
Citation: Baranykova S (2025) Role of store-operated Ca2+ entry and STIM2 in retinal neurodegeneration during glaucoma. Front. Aging Neurosci. 17:1657590. doi: 10.3389/fnagi.2025.1657590
Received: 01 July 2025; Accepted: 11 September 2025;
Published: 25 September 2025.
Edited by:
R. M. Damian Holsinger, The University of Sydney, AustraliaReviewed by:
Ricardo Augusto De Melo Reis, Federal University of Rio de Janeiro, BrazilLidawani Lambuk, Universiti Sains Malaysia, Malaysia
Copyright © 2025 Baranykova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sofiia Baranykova, c29maWFiYXJhbnlrb3ZhQGdtYWlsLmNvbQ==