 Horacio Uri Saragovi
Horacio Uri Saragovi Alba Galan1,2
Alba Galan1,2 Leonard A. Levin
Leonard A. Levin- 1Lady Davis Institute, Montreal, QC, Canada
- 2Jewish General Hospital, Montreal, QC, Canada
- 3Department of Ophthalmology and Visual Sciences, McGill University, Montreal, QC, Canada
- 4McGill University Health Centre, Montreal, QC, Canada
- 5Montreal Neurological Institute, Mcgill University, Montreal, QC, Canada
Neurotrophins (NTs) are a subset of the neurotrophic factor family. These growth factors were originally named based on the nerve growth functional assays used to identify them. NTs act as paracrine or autocrine factors for cells expressing NT receptors. The receptors and their function have been studied primarily in cells of the nervous system, but are also present in the cardiovascular, endocrine, and immune systems, as well as in many neoplastic cells. The signals activated by NTs can be varied, depending on cellular stage and context, healthy or disease states, and depending on whether the specific NTs and their receptors are expressed in the relevant cells. In the healthy central and peripheral adult nervous systems, NTs drive neuronal survival, phenotype, synaptic maintenance, and function. Deficiencies of the NT/NT receptor axis are causally associated with disease onset or disease progression. Paradoxically, NTs can also drive synaptic loss and neuronal death. In the embryonic stage this activity is essential for proper developmental pruning of the nervous system, but in the adult it can be associated with neurodegenerative disease. Given their key role in neuronal survival and death, NTs and NT receptors have long been considered therapeutic targets to achieve neuroprotection. The first neuroprotective approaches consisted of enhancing neuronal survival signals using NTs. Later strategies selectively targeted receptors to induce survival signals specifically, while avoiding activation of death signals. Recently, the concept of selectively targeting receptors to reduce neuronal death signals has emerged. Here, we review the rationale of each neuroprotective strategy with respect to the complex cell biology and pharmacology of each target receptor.
Introduction
Neurotrophins (NTs) are a family of growth factors that include nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin 4/5 (NT-4/5). Neurotrophin mRNAs are translated into larger precursors named pro-NTs, which are then processed into mature NTs by enzymatic cleavage. The pro-NTs and the mature NTs activate different and sometimes opposing signals and physiological pathways, acting through different receptors (Teng, and Hempstead, 2004).
Mature NTs preferentially activate Trk receptors, which are generally associated with survival signals, whereas pro-NTs activate p75 receptors, which are generally associated with death signals (Chao et al., 2006). Neurodegenerative diseases often exhibit imbalances either in NT (e.g., poor processing of pro-NTs to the mature state or poor transport of NTs to the site, where they are needed) or imbalances in NT receptors (e.g., decreases in pro-survival Trks and increases in pro-death p75). This summary is a simplification, and there are additional issues, such as positive and negative functional cross-regulation of Trks and p75, when these receptors are co-expressed. In certain cellular contexts the receptors may also have altered functions, for example in neoplasia.
Clinical trials using NTs as drugs to promote neuroprotection have consistently failed, in large part due to poor receptor selectivity (e.g., binding to Trks and p75), their pleiotropic pro-survival and pro-death activities, short half-lives, poor pharmacokinetics and bioavailability (Yuen and Mobley, 1996; Thoenen and Sendtner, 2002), or because pro-death p75 receptors may be up-regulated in disease (Rudzinski et al., 2004; Ibanez and Simi, 2012), thus negating the full benefit of Trk activation (Josephy-Hernandez et al., 2017). In addition, in some cases Trk receptors can also promote neuronal death (Tauszig-Delamasure et al., 2007), resulting in a confusing pattern of in vivo physiology and hard-to-predict pharmacology.
Many excellent reviews of the NT field discuss the biology and physiology of each factor and receptor (Ibanez and Simi, 2012; Bothwell, 2016), and postulate reasons to explain clinical failures (Yuen and Mobley, 1996; Thoenen and Sendtner, 2002). Other comprehensive reviews describe compounds reported to activate/inactivate Trks and p75 receptors, some of which were used as proof-of-concept therapeutics [reviewed in (Longo and Massa, 2004; Josephy-Hernandez et al., 2017)].
Here, we present a reassessment of neuroprotection strategies and their challenges, focusing on the paradoxes of receptor pharmacology and signals. We distinguish strategies that promote survival and strategies that reduce neurotoxicity, as separate but complementary approaches. Each strategy faces challenges which must be addressed for successful translation into clinically effective neuroprotective therapies.
Neurotrophin Receptors and Roles in Disease
Neurotrophins (NTs) act through two distinct receptor families: three receptor tyrosine kinases named TrkA TrkB and TrkC, and a receptor named p75. NGF binds TrkA, BDNF and NT-4 bind TrkB, and NT-3 prefers TrkC but also can bind TrkA and TrkB (Figure 1).
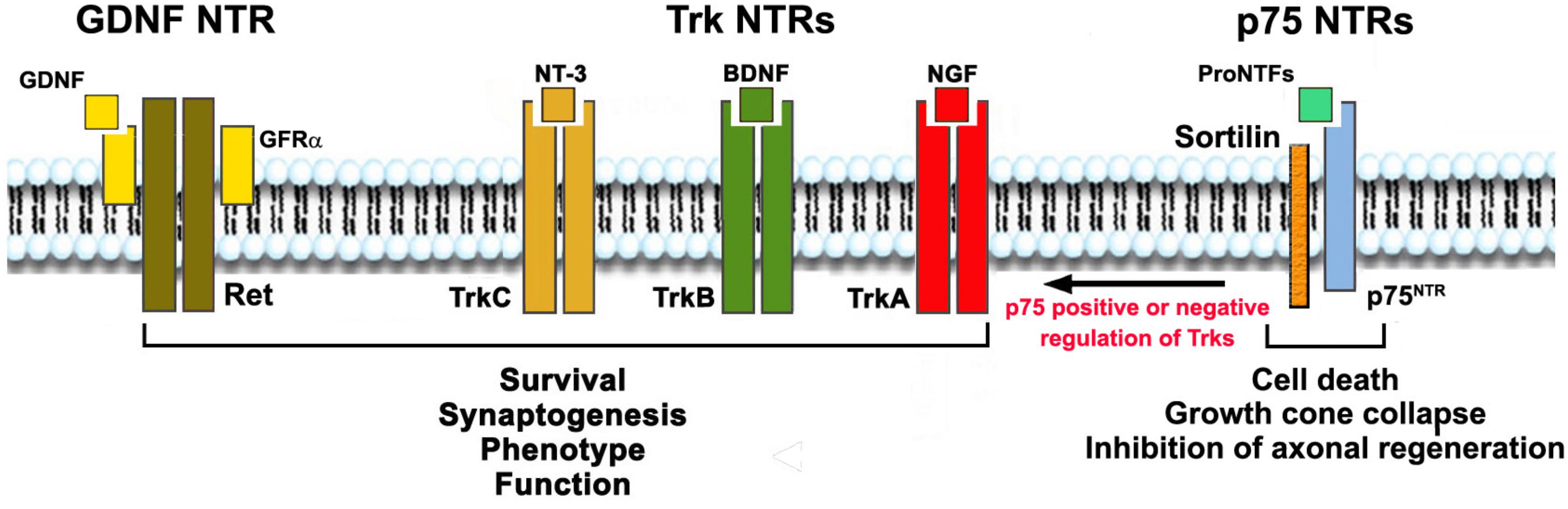
Figure 1. Neurotrophic Factors, their receptors (NTRs) and functions. NTs activate signaling receptors TrkA, TrkB, TrkC, and p75NTR. Each NT receptor (NTR) can act alone, and all Trks can also cooperate positively or negatively with p75NTR. GDNF acts through RET but binds the glycosyl-phosphatidylinositol-anchored co-receptor (GFRα1–4) subunits. Mature NTs (NGF, BDNF, NT-3) bind, respectively, TrkA, TrkB and TrkC with some selectivity. All NTs and the precursor pro-neurotrophin proteins bind to p75NTR. Growth factor activation of Trks or RET promote survival, growth, differentiation and synaptogenesis. Trk receptors may be present in truncated forms that are not protective (not shown). Growth factor activation of p75NTR/sortilin complex in neurons leads to apoptosis, growth cone collapse, and inhibition of axonal regeneration; and in glia, activation of p75NTR leads to production of pro-inflammatory and neurotoxic factors. p75NTR can also regulate the action of Trk receptors positively or negatively, depending on its binding partners and other factors.
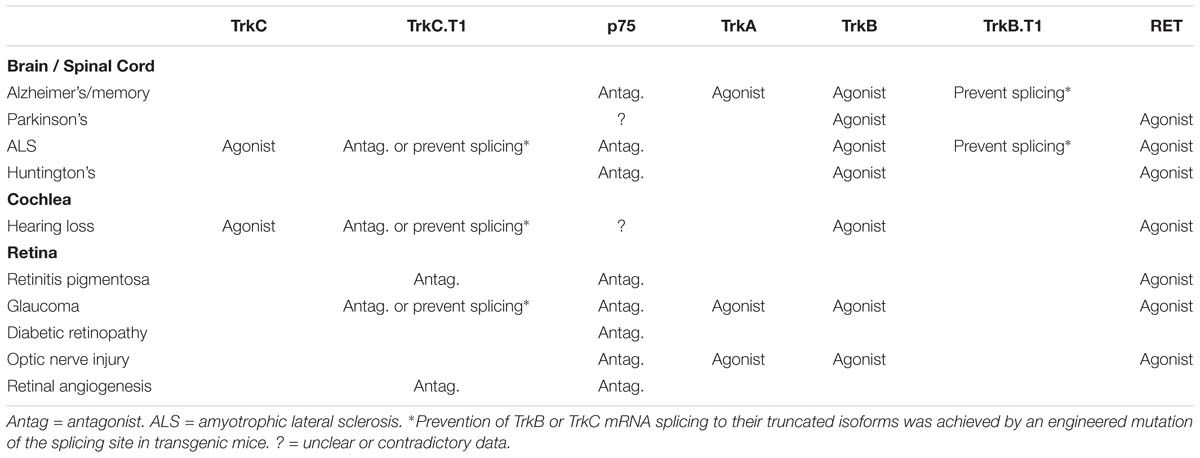
Table 1. Experimental (and in some cases clinical) validation of the indicated receptor targets as “proof-of-concept” in neurodegenerative diseases affecting the CNS.
Ligands binding to Trks drive the activation of the receptor tyrosine kinase enzymatic activity and the tyrosine phosphorylation of intracellular proteins (e.g., PLCγ, PI3K, Ras and Raf/MEK/Erk1) to initiate signaling pathways (Saragovi and Gehring, 2000; Miller and Kaplan, 2001). These intracellular signaling pathways are usually associated with neuronal survival, maintenance and function in the peripheral and central nervous systems (CNS), and the survival of stressed neurons (Huang and Reichardt, 2003). Therefore, activation of Trks or their signaling cascades has been sought as a mechanism to thwart neuronal degeneration (Saragovi and Gehring, 2000; Saragovi et al., 2009).
All NTs, including the pro-NTs, also bind to p75, a member of the Tumor Necrosis Factor-α (TNFα) receptor superfamily. NTs binding to p75 lead to the activation of complex cascades that are stage- or tissue-specific. Generally, p75 is believed to signal neuronal atrophy, synaptic loss, loss of function, and cell death (Zaccaro et al., 2001; Hempstead, 2002, 2006; Saragovi and Zaccaro, 2002; Ivanisevic et al., 2003; Chao et al., 2006; Saragovi et al., 2009), and is commonly upregulated in neurodegenerative diseases. In pathological states (Table 1), pro-NTs binding to p75 promotes the production of inflammatory cytokines TNFα and α2 Macroglobulin (α2M) by glial cells, and increase levels of pro-NGF itself, thus perpetuating the deleterious activation of p75. All of these factors are neurotoxic at increased levels, and result in a vicious cycle of neurotoxic events (Barcelona and Saragovi, 2015).
Adding another layer of complexity to this signaling axis, the biological function of p75 depends on several factors. The p75 receptor cooperates with other proteins, such as the sortilin family of receptors, which are integral to p75 signaling (Vaegter et al., 2011; Skeldal et al., 2012). Additionally, co-expression of Trks can influence p75 biology, as can the type of cell and its stage of maturation/differentiation. As mentioned above, the p75 receptor also has multiple ligands (all NTs as well as all pro-NTs) (Dechant and Barde, 2002; Nykjaer et al., 2005; Al-Shawi et al., 2007), and co-expression of p75 and Trks regulates ligand binding and affinity (Hempstead and Chao, 1997), as well as functional signaling (Zaccaro et al., 2001; Saragovi and Zaccaro, 2002) in a positive or negative manner (Figure 1). Due to the complexity of the signaling pathways, the consequences of pharmacological modulation of p75 in vivo are difficult to predict.
Yet another level of complexity in Trk receptor signaling emerges from the presence of Trk receptor isoforms. Trks can be expressed as full-length Trk tyrosine kinase receptors (Trk-FL) or as truncated isoforms without a kinase domain. The truncated isoforms are generated by alternative mRNA splicing, gaining a new and distinct intracellular sequence while lacking intrinsic tyrosine kinase activity (Brahimi et al., 2016). Both truncated TrkB.T1 and TrkC.T1 are able to mediate signals, either by inhibiting full-length tyrosine kinase as a dominant-negative mechanism (Valenzuela et al., 1993; Palko et al., 1999; Das et al., 2000) or by being activated in a ligand-dependent manner (Baxter et al., 1997; Esteban et al., 2006). Pathological upregulation of TrkC.T1 induces neuronal cell death through activation of Rac1 GTPase and pERK signaling pathways, with subsequent increase of TNFα production to toxic levels (Esteban et al., 2006; Galán et al., 2017b). Expression of TrkB.T1 and TrkC.T1 receptors increases in many neurodegenerative diseases (Table 1; Bai et al., 2010b; Yanpallewar et al., 2012; Galán et al., 2017b). Given that truncated isoforms are increased in disease and can be activated by NTs to cause toxicity, it would seem counterintuitive to use NTs as therapeutic agents in these conditions.
Neurotrophic Factors in Clinical Development
Neurotrophins (NTs) are part of a larger family of neurotrophic factors (NTFs). Among the NTFs, glial-derived neurotrophic factor (GDNF), ciliary-derived neurotrophic factor (CNTF), and insulin-like growth factor-1 (IGF-1) are key determinants of neuronal health (Sakowski et al., 2009; Pasquin et al., 2015) and were tested for clinical efficacy (Yuen and Mobley, 1996; Thoenen and Sendtner, 2002).
Glial-derived neurotrophic factor (GDNF) signals by binding to a co-receptor known as GFRα1, leading to the activation of the tyrosine kinase Ret. Similarly to its counterparts in the Trk family, Ret induces survival through the PI3K/Akt, Ras/Erk, Src and PLCγ signaling pathways (Ibanez, 2013). The absolute requirement of GFRα co-receptors for GDNF activation of Ret limits the number of neurons targeted by GDNF.
The Evolution of the Concept of NT-Based Neuroprotection
The translational potential of NTs as drugs and NT receptors as targets for neuroprotection has long been recognized, and several clinical trials were carried out in the 1990’s, none resulting in regulatory approval, the main problems attributed to lack of specific targeting, not achieving an effective dose, and failure to avoid side effects (Thoenen and Sendtner, 2002). The failure of these trials to achieve their endpoints led to a re-evaluation of the validity and druggability of these targets in disease, and particularly the scrutiny of the physiological basis for their pharmacology.
Below we present evolving concepts of NT receptor biology and receptor physiology relevant to their roles in disease. We discuss 4 generations of neuroprotection strategies. Neuroprotection strategy 1.0 refers to the use of NTs or NT-inducing agents regardless of receptor selectivity, pharmacokinetics, or receptor expression patterns. Neuroprotection strategy 2.0 refers to the selective activation of Trk receptors without p75 binding and activation, particularly in diseases, where p75 is upregulated. Neuroprotection strategy 3.0 refers to inhibition of p75-mediated signals, which are pro-inflammatory and neurotoxic. Neuroprotection strategy 4.0 refers to improved Trk-activation in a selective manner, not only circumventing p75 activation but also the activation of truncated Trk isoforms that mediate neurotoxicity (Figure 2). Conceivably, these 4 strategies may be combined to achieve synergy, given that they have different mechanisms of action.
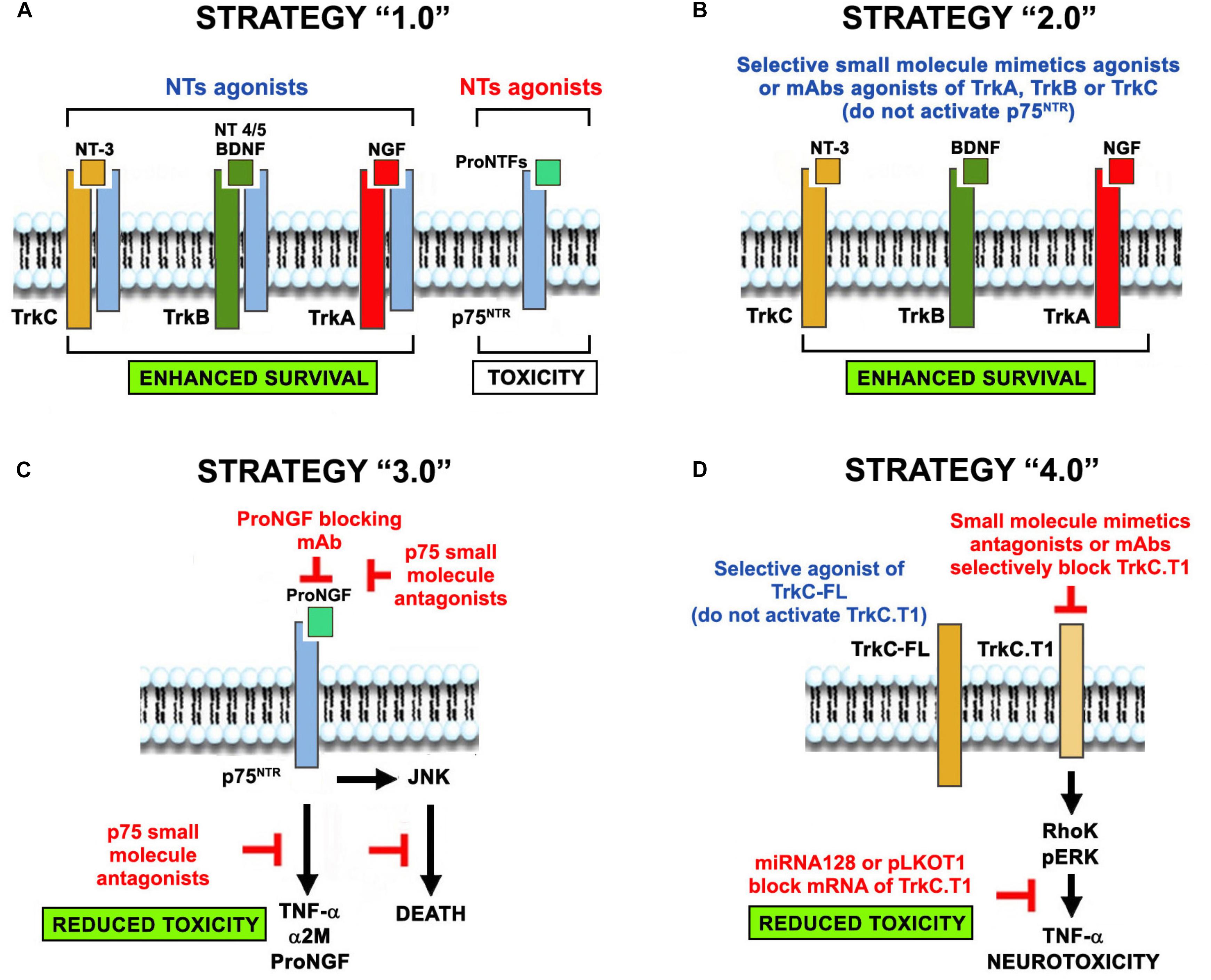
Figure 2. The evolution of the concept of NT-based neuroprotection. Four neuroprotective strategies evolved from the use of NTs-based therapies in neurodegenerative diseases. (A) Neuroprotection strategy 1.0 was the original use of NTs regardless of receptor selectivity, causing unintended signaling through p75NTR or truncated Trk isoforms that are up-regulated in neurodegenerative pathologies. (B) Neuroprotection strategy 2.0 overcomes some of the drawbacks by using selective agonists of Trk receptors (small molecules, mAbs, and mutant NTs that do not bind p75), circumventing p75NTR activation. (C) Neuroprotection strategy 3.0 addresses p75NTR neurotoxicity using selective blocking antibodies and small molecules antagonists. (D) Neuroprotection strategy 4.0 addresses the toxic function of truncated Trk isoforms by using selective Trk full-length agonists (that do not activate the truncated forms) or selective antagonists of truncated isoforms (small molecules, mAbs, miRNAs and shRNA vectors). Co-expression of p75 and TrkC.T1 in glia may exacerbate neurotoxicity. We predict that combinations of these strategies (e.g., strategy 2 + strategy 3) may be synergistic because of their complementary mechanisms of action.
Neuroprotection Strategy 1.0: Activating NT Receptors
Neurotrophins (NTs) normally drive neuronal survival, maintenance of phenotype and synapses, and function. Ligand-dependent activation of the Trk receptors is associated with those survival signals (Sendtner et al., 1996; Bechade et al., 2002; Wan et al., 2014; Brahimi et al., 2016). As mentioned earlier, deficits in the activation of Trk receptor tyrosine kinases (for instance, impaired cellular transport, decreased receptor expression, or agonist deficiency) are linked to early stages of neurodegeneration, and precede neuronal death and symptoms. These data supported the rationale that Trk-agonism may be therapeutic, and NTs were evaluated in multiple experimental models with the expectation that they would solely act as Trk agonists.
Nerve growth factor (NGF) was studied therapeutically to activate TrkA in models of Alzheimer disease (AD) (Schindowski et al., 2008; Cuello et al., 2010), and ageing (Mufson et al., 2000; Bruno et al., 2004; Saragovi, 2005), or Down syndrome (Sendera et al., 2000; Dorsey et al., 2006), models with cholinergic deficits and memory impairment. BDNF and GDNF were studied therapeutically in models of Parkinson disease (PD) (Rangasamy et al., 2010) and Huntington disease (HD) (Alberch et al., 2004), with loss of neurons that express TrkB and RET. NT-3 was studied therapeutically in models of amyotrophic lateral sclerosis (ALS) with loss of spinal cord motor neurons that express TrkC (Ekestern, 2004). Ophthalmic neurodegenerative diseases such as glaucoma (Rudzinski et al., 2004; Nafissi and Foldvari, 2016), retinitis pigmentosa (RP) (Thanos and Emerich, 2005), and diabetic retinopathy (DR) (Bikbova et al., 2014) were studied using NTs as therapeutic agents. In most of these diseases, growth factors other than NTs were explored, including GDNF, CNTF, and IGF-1 (Kaspar et al., 2003; Dupraz et al., 2013; Bassil et al., 2014; Ma et al., 2015; Agostinone et al., 2018) [reviewed in (Gould and Oppenheim, 2011)].
Clinical trials using exogenous delivery of these factors or cells secreting these factors, designed to agonize Trk or RET or IGF-1 receptors, have been consistently unsuccessful. Reasons include the poor pharmacokinetics and pharmacodynamics of NTs, short half-lives, undesirable high potency and pleiotropic effects, inability to penetrate tissue barriers, and difficulty in delivery of these large proteins across the BBB, requiring increasingly sophisticated and risky methods of administration (Weissmiller and Wu, 2012; Rafii et al., 2018), with consequent limitations in reaching the relatively high doses required for efficacy in experimental studies.
Notably, the reasons thought to be responsible for failure are not exclusive to NTs: they generally affect most protein-based therapies, and the issues are resolvable. But there are problems specific to NTs. These include poor receptor selectivity and unpredictable in vivo pharmacology, particularly off-target effects on unintended activation of p75 or truncated Trk receptors (Saragovi and Gehring, 2000; Josephy-Hernandez et al., 2017). The expression and activity of these receptors are increased in neurodegenerative states, and given that they are neurotoxic, they invalidate the benefit of Trk activation, decrease the therapeutic effect, and create a poor risk/benefit ratio (Figure 2A; Peleshok and Saragovi, 2006; Josephy-Hernandez et al., 2017).
Neuroprotection Strategy 2.0: Targeting Full Length Trk Receptors and Avoiding p75 Activation
Based on the many failures of neuroprotection strategy 1.0, we hypothesized that selective Trk-activating agents which circumvent p75 binding and activation would be neuroprotective. To test this idea, we produced a wide range of TrkA-, TrkB-, and TrkC-selective agonists and tested them in different neurodegenerative pathologies (Figure 2B). GDNF agonists are also briefly summarized.
TrkA-, TrkB-, or TrkC-Selective Agonists
We generated small molecule mimetics of NTs (LeSauteur et al., 1995, 1996a; Debeir et al., 1999; Maliartchouk et al., 2000a,b; Pattarawarapan et al., 2002; Zaccaro et al., 2005; Chen et al., 2009), agonistic mAbs (LeSauteur et al., 1996b, Saragovi et al., 1998; Bai et al., 2010c; Guillemard et al., 2010), small molecule mimetics of the mAbs (Brahimi et al., 2009, 2010, 2014; Chen et al., 2009), and mutant NTs (Saragovi et al., 1998; Ivanisevic et al., 2007; Bai et al., 2010a; Aboulkassim et al., 2011). We then tested them in animal models of neurodegenerative disease, demonstrating effectiveness in vivo.
For instance, a TrkA-selective NGF mutant that does not activate p75 (Bai et al., 2010a) and the selective small molecule TrkA agonist D3 (Shi et al., 2007) rescued retinal ganglion cells (RGCs) in glaucoma and optic nerve axotomy models. A selective agonistic mAb that binds TrkB delayed RGC death and preserved the structure of retinal layers from degenerating in optic nerve axotomy and glaucoma (Bai et al., 2010c). A mutant NT-3 selective for TrkC, and an agonistic mAb that activates TrkC (Guillemard et al., 2010) selectively protected motor neurons in an ALS model (Enomoto et al., 2013; Brahimi et al., 2016). None of these ligands bind or activate p75. Many of these agents were very effective in vivo in disease states and paradigms, where the native NTs were ineffective unless p75 was concomitantly silenced or neutralized (Bruno et al., 2004; Shi et al., 2007; Lebrun-Julien et al., 2009b, 2010).
Later, other groups validated the concept by generating TrkB small molecule agonists and TrkB and TrkC agonistic mAbs that were protective in the MPTP neurotoxicity mouse model of PD (Berezov et al., 2002; Jang et al., 2010; Devi and Ohno, 2012; Bollen et al., 2013; Coles et al., 2014; Chitranshi et al., 2015; Nie et al., 2015), HD (Jiang et al., 2013; Simmons et al., 2013), and ALS. These agents were effective in experimental paradigms, where the wild type NTs were ineffective, whether endogenously produced or added as therapeutic agents. Yet the wild type NTs became effective when p75 expression was concomitantly silenced or inhibited [reviewed in Josephy-Hernandez et al. (2017)].
With respect to translation to clinical use, one of our TrkA-selective small molecule agonists is currently in Phase 3 clinical trials for an ophthalmic indication, a TrkC-selective mAb agonist is in pre-clinical studies for ALS, and Trk-selective agonistic mAbs and NT mutants are under investigation for neurosensory hearing loss. Other translational efforts with Trk-selective agonists are in progress in PD, ALS, AD, and HD.
GDNF/Ret Agonists
The GDNF/Ret/GFRα1 axis plays an important neuroprotective role in the retina and other anatomical sites. GDNF causes upregulation of the glutamate aspartate transporter in glial cells, and therefore counteracts the excitotoxic environment in the degenerating retina (Delyfer et al., 2005). GDNF also stimulates the secretion of osteopontin and basic fibroblast growth factor (bFGF), which have been shown to prolong rod survival (Hauck et al., 2006; Del Rio et al., 2011). Following the initial evaluation of GDNF therapies (Strategy 1.0), new efforts have led to the generation of small-molecule agonists with GDNF-like activity or Ret modulatory activity (Sidorova et al., 2017).
Norgestrel is a small molecule related to progesterone that has been demonstrated to be neuroprotective in the rd10 model of RP (Doonan et al., 2011), possibly through the upregulation of bFGF. XIB4035t is a small molecule with effects on GFRα1/Ret signaling. Originally, it was inappropriately characterized as a GFRα1 agonist (Tokugawa et al., 2003), but is nowadays considered to be a GFRα1 modulator, able to potentiate signaling in the presence of GDNF (Hedstrom et al., 2014; Sidorova et al., 2017; Ivanova et al., 2018).
Neuroprotection Strategy 3.0: Inhibiting p75 Receptors
In the adult, the p75 receptor is expressed at low levels in healthy states (Tomellini et al., 2014), but is upregulated in disease. This injury-induced expression recapitulates the role of p75 in development (Ibanez and Simi, 2012), where p75 is expressed at high levels and modulates synaptic pruning and the death of unwanted neurons (Lebrun-Julien et al., 2009b; Bai et al., 2010b). The upregulated p75 receptor is generally associated with neuronal death and is activated primarily by proNGF (Figure 2C; Teng et al., 2005).
In neurons, activation of p75 by proNGF triggers apoptotic death, decreases synaptic function (Jansen et al., 2007), and reduces the neuroprotective effect associated with agonists of full-length Trk receptors (Zaccaro et al., 2001; Saragovi and Zaccaro, 2002; Ivanisevic et al., 2003; Saragovi et al., 2009).
In the vasculature, p75 activation causes pericyte dysfunction (Siao et al., 2013; Barcelona et al., 2016) and breakdown of blood–tissue barriers, causing vascular permeability and edema, and leading to vascular endothelial cell death and hypoxia (Shanab et al., 2015). Consequent vaso-obliteration and hypoxia induce VEGF, angiogenic remodeling, and pathological neovascularization (Siao et al., 2013; Barcelona et al., 2016). Thus, p75 on pericytes is relevant to deficits after cardiac hypoxia associated with cardiac injury, retinal neovascularization in DR, and choroidal neovascularization in the wet form of age-related macular degeneration.
In glia, p75 enhances production of the inflammatory mediators TNFα (Srinivasan et al., 2004; Nakazawa et al., 2006; Lebrun-Julien et al., 2009a, Lebrun-Julien et al., 2010; Bai et al., 2010b; Barcelona et al., 2016; Galan et al., 2017a; Platon-Corchado et al., 2017), proNGF (Srinivasan et al., 2004; Lim et al., 2008; Lebrun-Julien et al., 2009b, 2010; Xu et al., 2009; Barcelona et al., 2016), and α2M (Shi et al., 2008; Bai et al., 2010a,b; Barcelona et al., 2016; Galan et al., 2017a; Platon-Corchado et al., 2017). Each of these factors is neurotoxic, and they cooperate to synergistically worsen pathology (Mysona et al., 2013; Barcelona and Saragovi, 2015). Mechanistically, α2M extends the half-lives of TNFα and proNGF (Barcelona and Saragovi, 2015), and p75-driven production of proNGF generates an autocrine loop, resulting in its persistent activation.
Notably, TNFα, proNGF, and α2M are each validated therapeutic targets. Inhibition of TNFα (Nakazawa et al., 2006; Roh et al., 2012), proNGF (Barcelona et al., 2013; Barcelona and Saragovi, 2015), or α2M (Shi et al., 2008; Bai et al., 2010a,b) as monotherapy affords moderate efficacy in retinal neurodegeneration, and other pathologies. Thus, p75 overexpression in disease creates an unfavorable environment by driving at least three neurotoxic proteins, which then feeds back in an autocrine-mediated vicious cycle. Expression and activity of p75 in disease disrupts neuro-glia-vascular homeostasis, with progressive pathology, whereby inflammation causes neuronal death, and neuronal death causes more inflammation and vascular pathology.
These observations in animal models are relevant to human neurodegenerative and vascular diseases because increased levels of p75, TNFα, proNGF, and α2M, alone or in combination, have been documented in ALS, cardiac hypoxia, RP, DR, and others. Inhibition of p75 should also prevent direct neuronal death, vascular deficits, acute inflammation, and chronic production of TNFα, proNGF, and α2M.
We developed a family of drug-like agents that selectively inhibit p75 activity (Bai et al., 2010a; Josephy-Hernandez et al., 2017). These p75 antagonists were therapeutic in models of glaucoma and optic nerve injury (Bai et al., 2010a), RP (Galán et al., 2017b; Platon-Corchado et al., 2017), and neovascularization in DR (Barcelona et al., 2016; Galan et al., 2017a), even when applied as monotherapy after disease onset. Other small p75 ligands reduced tau-related pathology in AD (Massa et al., 2002; Nguyen et al., 2014) and p75-induced motor neuron cell death in ALS (Pehar et al., 2006).
Neuroprotection Strategy 4.0: Inhibiting Truncated Trk Receptors
As described above, the full length Trk receptors have a tyrosine kinase intracellular domain that phosphorylates and activates survival signals such as Akt or PLC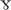 pathways in a ligand-dependent manner. The “full length” (FL) receptor activity is critical to the survival and function of neurons, where they are expressed. For instance, TrkC-FL is necessary for the physiology of motor neurons and cochlear neurons (Sendtner et al., 1996; Bechade et al., 2002; Mellado Lagarde et al., 2014; Wan et al., 2014; Wan and Corfas, 2015; Brahimi et al., 2016; Suzuki et al., 2016), whereas TrkB-FL is necessary for brain dopaminergic neurons (Nie et al., 2015).
pathways in a ligand-dependent manner. The “full length” (FL) receptor activity is critical to the survival and function of neurons, where they are expressed. For instance, TrkC-FL is necessary for the physiology of motor neurons and cochlear neurons (Sendtner et al., 1996; Bechade et al., 2002; Mellado Lagarde et al., 2014; Wan et al., 2014; Wan and Corfas, 2015; Brahimi et al., 2016; Suzuki et al., 2016), whereas TrkB-FL is necessary for brain dopaminergic neurons (Nie et al., 2015).
However, there are truncated Trk receptors which arise from alternative mRNA splicing of Trk-FL mRNAs. The resulting truncated isoforms lack the kinase domain, and signal differently. The most common truncated TrkC and TrkB isoforms in humans and rodents are TrkC.T1 (Tessarollo, 1998; Palko et al., 1999) and TrkB.T1, respectively, (Figure 2D; Yanpallewar et al., 2012).
Truncated TrkC Isoform (TrkC.T1)
TrkC.T1 arises from alternative mRNA splicing of TrkC-FL mRNA, resulting in deletion of the kinase domain, and gain of a new intracellular domain with a unique sequence from a spliced-in exon. For this reason, the TrkC-FL and TrkC.T1 isoforms only differ at the intracellular domain primary sequences, but retain the same extracellular primary sequence. Both TrkC-FL and TrkC.T1 bind NT-3 equally well and both signal in an NT-3–dependent manner (Esteban et al., 2006).
TrkC.T1 acts by activation of Rho kinase-Erk pathways (Esteban et al., 2006; Galán et al., 2017b), and via tamalin (Esteban et al., 2006). One physiological consequence of TrkC.T1 activity is an NT-3–dependent increase in TNFα, which is neurotoxic (Bai et al., 2010b; Brahimi et al., 2016; Galán et al., 2017b). Hence there must be a balance of TrkC-FL and TrkC.T1 signals in health and disease. In healthy adult tissue TrkC.T1 is low or undetectable, but it is significantly upregulated shortly after injury, but before degeneration and detectable symptoms, in neurodegenerative diseases such as glaucoma, RP, and ALS (Bai et al., 2010b; Brahimi et al., 2016; Galán et al., 2017b), as well as in noise-induced hearing loss (Saragovi, unpublished).
TrkC.T1 mRNA is produced constitutively and is immediately degraded by miRNA128. miRNA128 is reduced and TrkC.T1 mRNA is increased in neurodegenerative diseases (Brahimi et al., 2016). Hence, at the onset of neurodegenerative diseases TrkC.T1 protein is upregulated, but without a decrease in TrkC-FL until very late in disease, when neurons die (Bai et al., 2010b; Brahimi et al., 2016; Galán et al., 2017b).
TrkC-FL is present mainly in neurons, whereas in disease states TrkC.T1 is mainly in glia and astrocytes. Hence the TrkC.FL/TrkC.T1 ratio is reduced in diseased tissues with a difference in cellular distribution that is relevant to the mechanisms of action. In glia, TrkC.T1 activity promotes TNFα production in a pErk–dependent manner, and in vivo all TrkC.T1 mRNA co-localizes in cells expressing TNFα mRNA (Brahimi et al., 2016; Galan et al., 2017a; Galán et al., 2017b). The TrkC.T1–dependent increase in TNFα is neurotoxic and neurodegenerative, and relevant to the etiology of glaucoma and RP (Bai et al., 2010b; Galán et al., 2017b).
NT-3 binds TrkC-FL and TrkC.T1, and therefore paradoxically activates both neuroprotective and neurodegenerative signals. In this context, we postulated that NT-3 would be therapeutically useful if administered before disease onset (e.g., before TrkC.T1 upregulation), whereas neurotoxicity would predominate when NT-3 was applied after disease onset (e.g., after TrkC.T1 upregulation) (Corse et al., 1999; Park et al., 2009; Wyatt et al., 2011; Suzuki et al., 2016). Indeed, this paradox has been reported in motor neuron degeneration (such as SOD1 mutant rodent models of ALS) (Brahimi et al., 2016), in the death of retinal ganglion cell neurons in a model of RP (Galán et al., 2017b), and may be germane to the death of spiral ganglion neurons in a noise-induced hearing loss model (NIHL).
To directly test the hypothesis that the therapeutic failure of NT-3 is due in part to the unintended activation of TrkC.T1, we developed selective agonists of TrkC-FL. These agents activate TrkC-FL but do not bind or activate TrkC.T1, and therefore do not stimulate TNFα production (Guillemard et al., 2010; Brahimi et al., 2016). In an animal model of ALS these selective TrkC-FL agonists protect motor neuron health and significantly prolong life-span, even when injected after disease onset (Brahimi et al., 2016).
Additionally, we validated this concept by showing that in disease models of neurodegeneration (glaucoma causing RGC neuronal death (Bai et al., 2010b) and a genetically driven model of RP causing photoreceptor neuronal death (Galán et al., 2017b), reduced TrkC.T1 expression had significantly reduced disease progression. reduced levels of TNFα, lower activation of pErk in glia, and reduced neuronal death and neurodegeneration.
With respect to pharmacological inhibition, we have also developed highly selective inhibitors of TrkC.T1 expression (miRNA128 and shRNA vectors) which silence or inhibit TrkC.T1 and prevent induction of TNFα by NT-3 in vitro (Brahimi et al., 2016) and ex vivo in organotypic cultures (Galán et al., 2017b). Published small molecule inhibitors of TrkC (Brahimi et al., 2010, Brahimi et al., 2014, 2016; Liu et al., 2010) are non-selective between TrkC-FL and TrkC.T1, but significantly decrease TNFα levels and neuronal cell death in a mouse model of glaucoma (Bai et al., 2010b). The non-selective TrkC antagonists used in glaucoma were useful because virtually all the TrkC in the glaucomatous retina is TrkC.T1. However, selective TrkC.T1 inhibitors would be preferable, and we have developed and evaluated such agents (Brahimi et al., 2009, Brahimi et al., 2016).
Truncated TrkB Isoform (TrkB.T1)
TrkB.T1 (T1) is the main TrkB isoform in the mature brain (Dorsey et al., 2006) and its function was studied in vivo (Ferrer et al., 1999). Similar to TrkC.T1, TrkB.T1 inhibits TrkB.FL signaling acting as a dominant-negative receptor, thereby decreasing the effects of BDNF on neuronal survival, differentiation, and plasticity. TrkB.T1 also has BDNF-independent functions and regulates Rho GTPase activity (Ohira et al., 2006) and may stimulate PLCγ and MAPK signaling (Ohira et al., 2007).
TrkB.T1 is upregulated in neurodegenerative diseases such as AD (Ferrer et al., 1999) and ALS (Yanpallewar et al., 2012). Genetic deletion of TrkB.T1 in the SOD mouse model of ALS significantly delayed the onset of motor neuron degeneration (Yanpallewar et al., 2012) and restored cognitive abnormalities (Quarta et al., 2018).
In summary, TrkC.T1 (along with p75, TNFα, proNGF, and α2M) and TrkB.T1 are upregulated in animal and in human diseases (Sanchez et al., 2007; Bai et al., 2011; Roh et al., 2012; Yanpallewar et al., 2012; Siao et al., 2013; Shepheard et al., 2014) and are relevant to pathophysiology, making them excellent therapeutic targets.
Conclusion
It has been 25 years since the first clinical trials of NTs in CNS neurodegenerative disorders. These and subsequent studies failed, due not only to poor pharmacokinetics, short half-lives and/or poor bioavailability of the drugs, but also due to the complex biology of NTs and their receptors. Our understanding of the finely tuned physiology of NT receptors is critical to developing strategies for selectively restoring the balance between neuroprotective and neurotoxic signals. Over the years, we have learned that failures of NTs (neuroprotection strategy 1.0) were related to their pleiotropic effects, poor selectivity and off-target effects on unintended p75 or truncated Trk isoforms. More specific and successful strategies were developed using specific Trk-activating agents that circumvented p75 (neuroprotection strategy 2.0) and therefore potentiated neuroprotection, or by specifically inhibiting p75 receptors (neuroprotection strategy 3.0) or truncated Trk isoforms (neuroprotection strategy 4.0), both signaling neurotoxicity.
NT-based neuroprotection is still an evolving concept, and we can expect the development of even more focused strategies in coming years, which should deal with the complex nature of NT receptor physiology. We envision a future neuroprotection strategy 5.0 based on combining strategies, for example a synergistic neuroprotective and anti-neurotoxic combination, which might finally provide successful translation for treatment of chronic neurodegenerative diseases.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by grants from the Canadian Institutes of Health Research (HS and LL), NIH (LL), and Canada Research Chairs (LL). HS has licensed products for neuroprotection, and is a consultant to Otonomy Inc. LL is a consultant to Aerie, Eyevensys, Galimedix, Prilenia, Quark, and Regenera on neuroprotection.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aboulkassim, T., Tong, X. K., Chung Tse, Y., Wong, T. P., Woo, S. B., Neet, K. E., et al. (2011). Ligand-dependent TrkA activity in brain differentially affects spatial learning and long-term memory. Mol. Pharmacol. 80, 498–508. doi: 10.1124/mol.111.071332
Agostinone, J., Alarcon-Martinez, L., Gamlin, C., Yu, W. Q., Wong, R. O. L., and Di Polo, A. (2018). Insulin signalling promotes dendrite and synapse regeneration and restores circuit function after axonal injury. Brain 141, 1963–1980. doi: 10.1093/brain/awy142
Alberch, J., Perez-Navarro, E, and Canals, J. M. (2004). Neurotrophic factors in Huntington’s disease. Prog. Brain Res. 146, 195–229.
Al-Shawi, R., Hafner, A., Chun, S., Raza, S., Crutcher, K., Thrasivoulou, C., et al. (2007). ProNGF, sortilin, and age-related neurodegeneration. Ann. N. Y. Acad. Sci. 1119, 208–215. doi: 10.1196/annals.1404.024
Bai, Y., Dergham, P., Nedev, H., Xu, J., Galan, A., Rivera, J. C., et al. (2010a). Chronic and acute models of retinal neurodegeneration TrkA activity are neuroprotective whereas p75NTR activity is neurotoxic through a paracrine mechanism. J. Biol. Chem. 285, 39392–39400. doi: 10.1074/jbc.M110.147801
Bai, Y., Shi, Z., Zhuo, Y., Liu, J., Malakhov, A., Ko, E., et al. (2010b). In glaucoma the upregulated truncated TrkC.T1 receptor isoform in glia causes increased TNF-alpha production, leading to retinal ganglion cell death. Invest. Ophthalmol. Vis. Sci. 51, 6639–6651. doi: 10.1167/iovs.10-5431
Bai, Y., Sivori, D., Woo, S. B., Neet, K. E., Lerner, S. F., Saragovi, H. U. et al. (2011). During glaucoma, alpha2-macroglobulin accumulates in aqueous humor and binds to nerve growth factor, neutralizing neuroprotection. Invest. Ophthalmol. Vis. Sci. 52, 5260–5265. doi: 10.1167/iovs.10-6691
Bai, Y., Xu, J., Brahimi, F., Zhuo, Y., Marinko Sarunic, V., and Saragovi, H. U. et al. (2010c). An agonistic anti-TrkB mAb, but not BDNF, causes sustained TrkB activation, delays RGC death, and protects the retinal structure in optic nerve axotomy and in glaucoma. Invest. Ophthalmol. Vis. Sci. 51, 4722–4731.
Barcelona, P. F., Ortiz, S. G., Chiabrando, G. A., and Sanchez, M. C. (2013). alpha2-Macroglobulin induces glial fibrillary acidic protein expression mediated by low-density lipoprotein receptor-related protein 1 in Muller cells. Invest. Ophthalmol. Vis. Sci. 52, 778–786. doi: 10.1167/iovs.10-5759
Barcelona, P. F., Sitaras, N., Galan, A., Esquiva, G., Jmaeff, S., Jian, Y., et al. (2016). p75NTR and its ligand ProNGF activate paracrine mechanisms etiological to the vascular, inflammatory, and neurodegenerative pathologies of diabetic retinopathy. J. Neurosci. 36, 8826–8841. doi: 10.1523/JNEUROSCI.4278-15.2016
Barcelona, P. F and Saragovi, H. U. (2015). A pro-nerve growth factor (proNGF) and NGF binding protein, alpha2-macroglobulin, differentially regulates p75 and TrkA receptors and is relevant to neurodegeneration ex vivo and in vivo. Mol. Cell. Biol. 35, 3396–3408. doi: 10.1128/mcb.00544-15
Bassil, F., Fernagut, P. O., Bezard, E., and Meissner, W. G. (2014). Insulin, IGF-1 and GLP-1 signaling in neurodegenerative disorders: targets for disease modification? Prog. Neurobiol. 118, 1–18. doi: 10.1016/j.pneurobio.2014.02.005
Baxter, G. T., Radeke, M. J., Kuo, R. C., Makrides, V., Hinkle, B., Hoang, R., et al. (1997). Signal transduction mediated by the truncated trkB receptor isoforms, trkB.T1 and trkB.T2. J. Neurosci. 17, 2683–2690. doi: 10.1523/jneurosci.17-08-02683.1997
Bechade, C., Mallecourt, C., Sedel, F., Vyas, S., and Triller, A. (2002). Motoneuron-derived neurotrophin-3 is a survival factor for PAX2-expressing spinal interneurons. J. Neurosci. 22, 8779–8784. doi: 10.1523/jneurosci.22-20-08779.2002
Berezov, A., Chen, J., Liu, Q., Zhang, H. T., Greene, M. I., Murali, R. et al. (2002). Disabling receptor ensembles with rationally designed interface peptidomimetics. J. Biol. Chem. 277, 28330–28339. doi: 10.1074/jbc.m202880200
Bikbova, G., Oshitari, T., Baba, T., and Yamamoto, S. (2014). Neurotrophic factors for retinal ganglion cell neuropathy - with a special reference to diabetic neuropathy in the retina. Curr. Diabetes Rev. 10, 166–176. doi: 10.2174/1573399810666140508121927
Bollen, E., Vanmierlo, T., Akkerman, S., Wouters, C., Steinbusch, H. M., Prickaerts, J. et al. (2013). 7,8-Dihydroxyflavone improves memory consolidation processes in rats and mice. Behav. Brain Res. 257, 8–12. doi: 10.1016/j.bbr.2013.09.029
Bothwell, M. (2016). Recent advances in understanding neurotrophin signaling. F1000Res. 5:F1000 Faculty Rev-1885.
Brahimi, F., Ko, E., Malakhov, A., Burgess, K., and Saragovi, H. U. (2014). Combinatorial assembly of small molecules into bivalent antagonists of TrkC or TrkA receptors. PLoS One 9:e89617. doi: 10.1371/journal.pone.0089617
Brahimi, F., Liu, J., Malakhov, A., Chowdhury, S., Purisima, E. O., Ivanisevic, L., et al. (2010). A monovalent agonist of TrkA tyrosine kinase receptors can be converted into a bivalent antagonist. Biochim. Biophys. Acta 1800, 1018–1026. doi: 10.1016/j.bbagen.2010.06.007
Brahimi, F., Maira, M., Barcelona, P. F., Galan, A., Aboulkassim, T., Teske, K., et al. (2016). The Paradoxical Signals of Two TrkC Receptor Isoforms Supports a Rationale for Novel Therapeutic Strategies in ALS. PLoS One 11:e0162307. doi: 10.1371/journal.pone.0162307
Brahimi, F., Malakhov, A., Lee, H. B., Pattarawarapan, M., Ivanisevic, L., Burgess, K., et al. (2009). A peptidomimetic of NT-3 acts as a TrkC antagonist. Peptides 30, 1833–1839. doi: 10.1016/j.peptides.2009.07.015
Bruno, M. A., Clarke, P. B., Seltzer, A., Quirion, R., Burgess, K., Cuello, A. C., et al. (2004). Long-lasting rescue of age-associated deficits in cognition and the CNS cholinergic phenotype by a partial agonist peptidomimetic ligand of TrkA. J. Neurosci. 24, 8009–8018. doi: 10.1523/jneurosci.1508-04.2004
Chao, M. V., Rajagopal, R., and Lee, F. S. (2006). Neurotrophin signalling in health and disease. Clin. Sci. 110, 167–173. doi: 10.1042/cs20050163
Chen, D., Brahimi, F., Angell, Y., Li, Y. C., Moscowicz, J., Saragovi, H. U., et al. (2009). Bivalent peptidomimetic ligands of TrkC are biased agonists and selectively induce neuritogenesis or potentiate neurotrophin-3 trophic signals. ACS Chem. Biol. 4, 769–781. doi: 10.1021/cb9001415
Chitranshi, N., Gupta, V., Kumar, S., and Graham, S. L. (2015). Exploring the molecular interactions of 7,8-dihydroxyflavone and its derivatives with TrkB and VEGFR2 proteins. Int. J. Mol. Sci. 16, 21087–21108. doi: 10.3390/ijms160921087
Coles, C. H., Mitakidis, N., Zhang, P., Elegheert, J., Lu, W., Stoker, A. W., et al. (2014). Structural basis for extracellular cis and trans RPTPsigma signal competition in synaptogenesis. Nat. Commun. 5:5209. doi: 10.1038/ncomms6209
Corse, A. M., Bilak, M. M., Bilak, S. R., Lehar, M., Rothstein, J. D., Kuncl, R. W. et al. (1999). Preclinical testing of neuroprotective neurotrophic factors in a model of chronic motor neuron degeneration. Neurobiol. Dis. 6, 335–346. doi: 10.1006/nbdi.1999.0253
Cuello, A. C., Bruno, M. A., Allard, S., Leon, W., and Iulita, M. F. (2010). Cholinergic involvement in Alzheimer’s disease. A link with NGF maturation and degradation. J. Mol. Neurosci. 40, 230–235. doi: 10.1007/s12031-009-9238-z
Das, I., Sparrow, J. R., Lin, M. I., Shih, E., Mikawa, T., Hempstead BL. (2000). Trk C signaling is required for retinal progenitor cell proliferation [published erratum appears. J. Neurosci. 20, 2887–2895. doi: 10.1523/jneurosci.20-08-02887.2000
Debeir, T., Saragovi, H. U., and Cuello, A. C. (1999). A nerve growth factor mimetic TrkA antagonist causes withdrawal of cortical cholinergic boutons in the adult rat. Proc. Natl. Acad. Sci. U.S.A. 96, 4067–4072. doi: 10.1073/pnas.96.7.4067
Dechant, G., and Barde, Y. A. (2002). The neurotrophin receptor p75(NTR): novel functions and implications for diseases of the nervous system. Nat. Neurosci.refvol5, 1131–1136. doi: 10.1038/nn1102-1131
Del Rio, P., Irmler, M., Arango-Gonzalez, B., Favor, J., Bobe, C., Bartsch, U., et al. (2011). GDNF-induced osteopontin from Muller glial cells promotes photoreceptor survival in the Pde6brd1 mouse model of retinal degeneration. Glia 59, 821–832. doi: 10.1002/glia.21155
Delyfer, M. N., Forster, V., Neveux, N., Picaud, S., Leveillard, T., Sahel, J. A. et al. (2005). Evidence for glutamate-mediated excitotoxic mechanisms during photoreceptor degeneration in the rd1 mouse retina. Mol. Vis. 11, 688–696.
Devi, L, and Ohno, M. (2012). 7,8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 37, 434–444. doi: 10.1038/npp.2011.191
Doonan, F., O’Driscoll, C., Kenna, P., and Cotter, T. G. (2011). Enhancing survival of photoreceptor cells in vivo using the synthetic progestin Norgestrel. J. Neurochem. 118, 915–927. doi: 10.1111/j.1471-4159.2011.07354.x
Dorsey, S. G., Renn, C. L., Carim-Todd, L. Barrick, C. A., Bambrick, L., Krueger, B. K., et al. (2006). In vivo restoration of physiological levels of truncated TrkB.T1 receptor rescues neuronal cell death in a trisomic mouse model. Neuron 51, 21–28. doi: 10.1016/j.neuron.2006.06.009
Dupraz, S., Grassi, D., Karnas, D., Nieto, A. F., Guil, D., Hicks, D et al. (2013). The insulin-like growth factor 1 receptor is essential for axonal regeneration in adult central nervous system neurons. PLoS One 8:e54462. doi: 10.1371/journal.pone.0054462
Ekestern, E. (2004). Neurotrophic factors and amyotrophic lateral sclerosis. Neurodegener. Dis. 1, 88–100. doi: 10.1159/000080049
Enomoto, M., Bunge, M. B., and Tsoulfas, P. (2013). A multifunctional neurotrophin with reduced affinity to p75NTR enhances transplanted Schwann cell survival and axon growth after spinal cord injury. Exp. Neurol. 248, 170–182. doi: 10.1016/j.expneurol.2013.06.013
Esteban, P. F., Yoon, H. Y., Becker, J., Dorsey, S. G., Caprari, P., Palko, M. E., et al. (2006). A kinase-deficient TrkC receptor isoform activates Arf6-Rac1 signaling through the scaffold protein tamalin. J. Cell Biol. 173, 291–299. doi: 10.1083/jcb.200512013
Ferrer, I., Marin, C., Rey, M. J., Ribalta, T., Goutan, E., Blanco, R., et al. (1999). BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J. Neuropathol. Exp. Neurol. 58, 729–739. doi: 10.1097/00005072-199907000-00007
Galan, A., Barcelona, P. F., Nedev, H., Sarunic, M. V., Jian, Y., Saragovi, H. U., et al. (2017a). Subconjunctival delivery of p75NTR antagonists reduces the inflammatory, vascular, and neurodegenerative pathologies of diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 58, 2852–2862. doi: 10.1167/iovs.16-20988
Galán, A., Jmaeff, S., Barcelona, P. F., Brahimi, F., Sarunic, M. V., and Saragovi, H. U. (2017b). In retinitis pigmentosa TrkC.T1-dependent vectorial Erk activity upregulates glial TNF-alpha, causing selective neuronal death. Cell Death Dis. 8, 3222. doi: 10.1038/s41419-017-0074-8
Gould, T. W, and Oppenheim, R. W. (2011). Motor neuron trophic factors: therapeutic use in ALS? Brain Res. Rev. 67, 1–39. doi: 10.1016/j.brainresrev.2010.10.003
Guillemard, V., Ivanisevic, L., Garcia, A. G., Scholten, V., Lazo, O. M., Bronfman, F. C., et al. (2010). An agonistic mAb directed to the TrkC receptor juxtamembrane region defines a trophic hot spot and interactions with p75 coreceptors. Dev. Neurobiol. 70, 150–164. doi: 10.1002/dneu.20776
Hauck, S. M., Kinkl, N., Deeg, C. A., Swiatek-de Lange, M., Schoffmann, S., and Ueffing, M. (2006). GDNF family ligands trigger indirect neuroprotective signaling in retinal glial cells. Mol. Cell. Biol. 26, 2746–2757. doi: 10.1128/mcb.26.7.2746-2757.2006
Hedstrom, K. L., Murtie, J. C., Albers, K., Calcutt, N. A., and Corfas, G. (2014). Treating small fiber neuropathy by topical application of a small molecule modulator of ligand-induced GFRalpha/RET receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 111, 2325–2330. doi: 10.1073/pnas.1308889111
Hempstead, B. L. (2002). The many faces of p75NTR. Curr. Opin. Neurobiol. 12, 260–267. doi: 10.1016/s0959-4388(02)00321-5
Hempstead, B. L. (2006). Dissecting the diverse actions of pro- and mature neurotrophins. Curr. Alzheimer Res. 3, 19–24. doi: 10.2174/156720506775697061
Hempstead, B. L, and Chao, M. V. (1997). Entering the domain of neurotrophin binding. Nat. Biotechnol. 15, 623–624. doi: 10.1038/nbt0797-623
Huang, E. J, and Reichardt, L. F. (2003). Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642. doi: 10.1146/annurev.biochem.72.121801.161629
Ibanez, C. F. (2013). Structure and physiology of the RET receptor tyrosine kinase. Cold Spring Harb. Perspect. Biol. 5:a009134. doi: 10.1101/cshperspect.a009134
Ibanez, C. F, and Simi, A. (2012). p75 neurotrophin receptor signaling in nervous system injury and degeneration: paradox and opportunity. Trends Neurosci. 35, 431–440. doi: 10.1016/j.tins.2012.03.007
Ivanisevic, L., Banerjee, K., and Saragovi, H. U. (2003). Differential cross-regulation of TrkA and TrkC tyrosine kinase receptors with p75. Oncogene 22, 5677–5685. doi: 10.1038/sj.onc.1206864
Ivanisevic, L., Zheng, W., Woo, S. B., Neet, K. E., Saragovi, H. U. (2007). TrkA receptor hot spots for binding of NT-3 as a heterologous ligand. J. Biol. Chem. 282, 16754–16763. doi: 10.1074/jbc.m701996200
Ivanova, L., Tammiku-Taul, J., Garcia-Sosa, A. T., Sidorova, Y., Saarma, M., and Karelson, M. (2018). Molecular Dynamics Simulations of the Interactions between Glial Cell Line-Derived Neurotrophic Factor Family Receptor GFRalpha1 and Small-Molecule Ligands. ACS Omega 3, 11407–11414. doi: 10.1021/acsomega.8b01524
Jang, S. W., Liu, X., Yepes, M., Shepherd, K. R., Miller, G. W., Liu, Y., et al. (2010). A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. U.S.A. 107, 2687–2692. doi: 10.1073/pnas.0913572107
Jansen, P., Giehl, K., Nyengaard, J. R., Teng, K., Lioubinski, O., Sjoegaard, S. S., et al. (2007). Roles for the pro-neurotrophin receptor sortilin in neuronal development, aging and brain injury. Nat. Neurosci. 10, 1449–1457. doi: 10.1038/nn2000
Jiang, M., Peng, Q., Liu, X., Jin, J., Hou, Z., Zhang, J., et al. (2013). Small-molecule TrkB receptor agonists improve motor function and extend survival in a mouse model of Huntington’s disease. Hum. Mol. Genet. 22, 2462–2470. doi: 10.1093/hmg/ddt098
Josephy-Hernandez, S., Jmaeff, S., Pirvulescu, I., Aboulkassim, T., and Saragovi, H. U. (2017). Neurotrophin receptor agonists and antagonists as therapeutic agents: an evolving paradigm. Neurobiol. Dis. 97(Pt. B), 139–155. doi: 10.1016/j.nbd.2016.08.004
Kaspar, B. K., Llado, J., Sherkat, N., Rothstein, J. D., and Gage, F. H. (2003). Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science 301, 839–842. doi: 10.1126/science.1086137
Lebrun-Julien, F., Bertrand, M. J., De Backer, O., Stellwagen, D., Morales, C. R., Di Polo, A, and Barker, P. A. (2010). ProNGF induces TNFalpha-dependent death of retinal ganglion cells through a p75NTR non-cell-autonomous signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 107, 3817–3822. doi: 10.1073/pnas.0909276107
Lebrun-Julien, F., Duplan, L., Pernet, V., Osswald, I., Sapieha, P., Bourgeois, P., et al. (2009a). Excitotoxic death of retinal neurons in vivo occurs via a non-cell-autonomous mechanism. J. Neurosci. 29, 5536–5545. doi: 10.1523/JNEUROSCI.0831-09.2009
Lebrun-Julien, F., Morquette, B., Douillette, A., Saragovi, H. U., and Di Polo, A. (2009b). Inhibition of p75(NTR) in glia potentiates TrkA-mediated survival of injured retinal ganglion cells. Mol. Cell. Neurosci. 40, 410–420. doi: 10.1016/j.mcn.2008.12.005
LeSauteur, L., Cheung, N. K., Lisbona, R., and Saragovi, H. U. (1996a). Small molecule nerve growth factor analogs image receptors in vivo. Nat. Biotechnol. 14, 1120–1122.
LeSauteur, L., Maliartchouk, S., Le Jeune, H., Quirion, R., and Saragovi, H. U. (1996b). Potent human p140-TrkA agonists derived from an anti-receptor monoclonal antibody. J. Neurosci. 16, 1308–1316. doi: 10.1523/jneurosci.16-04-01308.1996
LeSauteur, L., Wei, L., Gibbs, B. F., and Saragovi, H. U. (1995). Small peptide mimics of nerve growth factor bind TrkA receptors and affect biological responses. J. Biol. Chem. 270, 6564–6569. doi: 10.1074/jbc.270.12.6564
Lim, Y. S., McLaughlin, T., Sung, T. C., Santiago, A., Lee, K. F., and O’Leary, D. D. (2008). p75(NTR) mediates ephrin-A reverse signaling required for axon repulsion and mapping. Neuron 59, 746–758. doi: 10.1016/j.neuron.2008.07.032
Liu, J., Brahimi, F., Saragovi, H. U., and Burgess, K. (2010). Bivalent diketopiperazine-based tropomysin receptor kinase C (TrkC) antagonists. J. Med. Chem. 53, 5044–5048. doi: 10.1021/jm100148d
Longo, F. M, and Massa, S. M. (2004). Neurotrophin-based strategies for neuroprotection. J. Alzheimers Dis. 6:S13–S17.
Ma, J., Guo, C., Guo, C., Sun, Y., Liao, T., Beattie, U., et al. (2015). Transplantation of human neural progenitor cells expressing IGF-1 enhances retinal ganglion cell survival. PLoS One 10:e0125695. doi: 10.1371/journal.pone.0125695
Maliartchouk, S., Debeir, T., Beglova, N., Cuello, A. C., Gehring, K., and Saragovi, H. U. (2000a). Genuine monovalent ligands of TrkA nerve growth factor receptors reveal a novel pharmacological mechanism of action. J. Biol. Chem. 275, 9946–9956. doi: 10.1074/jbc.275.14.9946
Maliartchouk, S., Feng, Y., Ivanisevic, L., Debeir, T., Cuello, A. C., Burgess, K., et al. (2000b). A designed peptidomimetic agonistic ligand of TrkA nerve growth factor receptors. Mol. Pharmacol. 57, 385–391.
Massa, S. M., Xie, Y., and Longo, F. M. (2002). Alzheimer’s therapeutics: neurotrophin small molecule mimetics. J. Mol. Neurosci. 19, 107–111. doi: 10.1007/s12031-002-0019-1
Mellado Lagarde, M. M., Wan, G., Zhang, L., Gigliello, A. R., McInnis, J. J., Zhang, Y., et al. (2014). Spontaneous regeneration of cochlear supporting cells after neonatal ablation ensures hearing in the adult mouse. Proc. Natl. Acad. Sci. U.S.A. 111, 16919–16924. doi: 10.1073/pnas.1408064111
Miller, F. D, and Kaplan, D. R. (2001). Neurotrophin signalling pathways regulating neuronal apoptosis. Cell. Mol. Life Sci. 58, 1045–1053. doi: 10.1007/pl00000919
Mufson, E. J., Ma, S. Y., Cochran, E. J., Bennett, D. A., Beckett, L. A., Jaffar, S., et al. (2000). Loss of nucleus basalis neurons containing trkA immunoreactivity in individuals with mild cognitive impairment and early Alzheimer’s disease. J. Comp. Neurol. 427, 19–30. doi: 10.1002/1096-9861(20001106)427:1<19::aid-cne2>3.0.co;2-a
Mysona, B. A., Al-Gayyar, M. M., Matragoon, S., Abdelsaid, M. A., El-Azab, M. F., Saragovi, H. U., et al. (2013). Modulation of p75(NTR) prevents diabetes- and proNGF-induced retinal inflammation and blood-retina barrier breakdown in mice and rats. Diabetologia 56, 2329–2339. doi: 10.1007/s00125-013-2998-6
Nafissi, N., and Foldvari, M. (2016). Neurotrophin signalling pathways regulating neuronal apoptosis. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 8, 240–254.
Nakazawa, T., Nakazawa, C., Matsubara, A., Noda, K., Hisatomi, T., She, H., et al. (2006). Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J. Neurosci. 26, 12633–12641. doi: 10.1523/jneurosci.2801-06.2006
Nguyen, T. V., Shen, L., Vander Griend, L., Quach, L. N., Belichenko, N. P., Saw, N., et al. (2014). Small molecule p75NTR ligands reduce pathological phosphorylation and misfolding of tau, inflammatory changes, cholinergic degeneration, and cognitive deficits in AbetaPPL/S transgenic mice. J. Alzheimers Dis. 42, 459–483. doi: 10.3233/JAD-140036
Nie, S., Xu, Y., Chen, G., Ma, K., Han, C., Guo, Z., et al. (2015). Small molecule TrkB agonist deoxygedunin protects nigrostriatal dopaminergic neurons from 6-OHDA and MPTP induced neurotoxicity in rodents. Neuropharmacology 99, 448–458. doi: 10.1016/j.neuropharm.2015.08.016
Nykjaer, A., Willnow, T. E., and Petersen, C. M. (2005). p75NTR–live or let die. Curr. Opin. Neurobiol. 15, 49–57. doi: 10.1016/j.conb.2005.01.004
Ohira, K., Funatsu, N., Homma, K. J., Sahara, Y., Hayashi, M., Kaneko, T., et al. (2007). Truncated TrkB-T1 regulates the morphology of neocortical layer I astrocytes in adult rat brain slices. Eur. J. Neurosci. 25, 406–416. doi: 10.1111/j.1460-9568.2007.05282.x
Ohira, K., Homma, K. J., Hirai, H., Nakamura, S., and Hayashi, M. (2006). TrkB-T1 regulates the RhoA signaling and actin cytoskeleton in glioma cells. Biochem. Biophys. Res. Commun. 342, 867–874. doi: 10.1016/j.bbrc.2006.02.033
Palko, M. E., Coppola, V., and Tessarollo, L. (1999). Evidence for a role of truncated trkC receptor isoforms in mouse development. Neurosci. J. 19, 775–782. doi: 10.1523/jneurosci.19-02-00775.1999
Park, S., Kim, H. T., Yun, S., Kim, I. S., Lee, J., Lee, I. S., et al. (2009). Growth factor-expressing human neural progenitor cell grafts protect motor neurons but do not ameliorate motor performance and survival in ALS mice. Exp. Mol. Med. 41, 487–500. doi: 10.3858/emm.2009.41.7.054
Pasquin, S., Sharma, M., and Gauchat, J. F. (2015). Ciliary neurotrophic factor (CNTF): new facets of an old molecule for treating neurodegenerative and metabolic syndrome pathologies. Cytokine Growth Factor Rev. 26, 507–515. doi: 10.1016/j.cytogfr.2015.07.007
Pattarawarapan, M., Zaccaro, M. C., Saragovi, U. H., and Burgess, K. (2002). New templates for syntheses of ring-fused, C(10) beta-turn peptidomimetics leading to the first reported small-molecule mimic of neurotrophin-3. J. Med. Chem. 45, 4387–4390. doi: 10.1021/jm0255421
Pehar, M., Cassina, P., Vargas, M. R., Xie, Y., Beckman, J. S., Massa, S. M., et al. (2006). Modulation of p75-dependent motor neuron death by a small non-peptidyl mimetic of the neurotrophin loop 1 domain. Eur. J. Neurosci. 24, 1575–1580. doi: 10.1111/j.1460-9568.2006.05040.x
Peleshok, J., and Saragovi, H. U. (2006). Functional mimetics of neurotrophins and their receptors. Biochem. Soc. Trans. 34(Pt. 4), 612–617. doi: 10.1042/bst0340612
Platon-Corchado, M., Barcelona, P. F., Jmaeff, S., Marchena, M., Hernandez-Pinto, A. M., Hernandez- Sanchez, C., et al. (2017). p75NTR antagonists attenuate photoreceptor cell loss in murine models of retinitis pigmentosa. Cell Death Dis. 8:e2922. doi: 10.1038/cddis.2017.306
Quarta, E., Fulgenzi, G., Bravi, R., Cohen, E. J., Yanpallewar, S., Tessarollo, L., et al. (2018). Deletion of the endogenous TrkB.T1 receptor isoform restores the number of hippocampal CA1 parvalbumin-positive neurons and rescues long-term potentiation in pre-symptomatic mSOD1(G93A) ALS mice. Mol. Cell. Neurosci. 89, 33–41. doi: 10.1016/j.mcn.2018.03.010
Rafii, M. S., Tuszynski, M. H., Thomas, R. G., Barba, D., Brewer, J. B., Rissman, R. A., et al. (2018). Adeno-associated viral vector (Serotype 2)-nerve growth factor for patients with Alzheimer disease: a randomized clinical trial. JAMA Neurol. 75, 834–841. doi: 10.1001/jamaneurol.2018.0233
Rangasamy, S. B., Soderstrom, K., Bakay, R. A., and Kordower, J. H. (2010). Neurotrophic factor therapy for Parkinson’s disease. Prog. Brain Res. 184, 237–264. doi: 10.1016/S0079-6123(10)84013-0
Roh, M., Zhang, Y., Murakami, Y., Thanos, A., Lee, S. C., Vavvas, D. G., et al. (2012). Etanercept, a widely used inhibitor of tumor necrosis factor-alpha (TNF-alpha), prevents retinal ganglion cell loss in a rat model of glaucoma. PLoS One 7:e40065. doi: 10.1371/journal.pone.0040065
Rudzinski, M., Wong, T. P., and Saragovi, H. U. (2004). Changes in retinal expression of neurotrophins and neurotrophin receptors induced by ocular hypertension. J. Neurobiol. 58, 341–354. doi: 10.1002/neu.10293
Sakowski, S. A., Schuyler, A. D., and Feldman, E. L. (2009). Insulin-like growth factor-I for the treatment of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 10, 63–73. doi: 10.1080/17482960802160370
Sanchez, M. C., Luna, J. D., Barcelona, P. F., Gramajo, A. L., Juarez, P. C., Riera, C. M., et al. (2007). Effect of retinal laser photocoagulation on the activity of metalloproteinases and the alpha(2)-macroglobulin proteolytic state in the vitreous of eyes with proliferative diabetic retinopathy. Exp. Eye Res. 85, 644–650. doi: 10.1016/j.exer.2007.07.018
Saragovi, H. U. (2005). Progression of age-associated cognitive impairment correlates with quantitative and qualitative loss of TrkA receptor protein in nucleus basalis and cortex. J. Neurochem. 95, 1472–1480. doi: 10.1111/j.1471-4159.2005.03479.x
Saragovi, H. U., Hamel, E., and Di Polo A. (2009). A neurotrophic rationale for the therapy of neurodegenerative disorders. Curr. Alzheimer Res. 6, 419–423. doi: 10.2174/156720509789207912
Saragovi, H. U., Zheng, W., Maliartchouk, S., DiGugliemo, G. M., Mawal, Y. R., Kamen, A., et al. (1998). A TrkA-selective, fast internalizing nerve growth factor-antibody complex induces trophic but not neuritogenic signals. Biol. J. Chem. 273, 34933–34940. doi: 10.1074/jbc.273.52.34933
Saragovi, H. U, and Zaccaro, M. C. (2002). Small molecule peptidomimetic ligands of neurotrophin receptors, identifying binding sites, activation sites and regulatory sites. Curr. Pharm. Des. 8, 2201–2216. doi: 10.2174/1381612023393215
Saragovi, H. U., and Gehring, K. (2000). Development of pharmacological agents for targeting neurotrophins and their receptors. Trends Pharmacol. Sci.refvol 21, 93–98. doi: 10.1016/s0165-6147(99)01444-3
Schindowski, K., Belarbi, K., and Buee, L. (2008). Neurotrophic factors in Alzheimer’s disease: role of axonal transport. Genes Brain Behav. 7, 43–56. doi: 10.1111/j.1601-183X.2007.00378.x
Sendera, T. J., Ma, S. Y., Jaffar, S., Kozlowski, P. B., Kordower, J. H., Mawal, Y., et al. (2000). Reduction in TrkA-immunoreactive neurons is not associated with an overexpression of galaninergic fibers within the nucleus basalis in Down’s syndrome. Neurochem. J. 74, 1185–1196. doi: 10.1046/j.1471-4159.2000.741185.x
Sendtner, M., Holtmann, B., and Hughes, R. A. (1996). The response of motoneurons to neurotrophins. Neurochem. Res. 21, 831–841. doi: 10.1007/bf02532307
Shanab, A. Y., Mysona, B. A., Matragoon, S., and El-Remessy, A. B. (2015). Silencing p75(NTR) prevents proNGF-induced endothelial cell death and development of acellular capillaries in rat retina. Mol. Ther. Methods Clin. Dev. 2:15013. doi: 10.1038/mtm.2015.13
Shepheard, S. R., Chataway, T., Schultz, D. W., Rush, R. A., and Rogers, M. L. (2014). The extracellular domain of neurotrophin receptor p75 as a candidate biomarker for amyotrophic lateral sclerosis. PLoS One 9:e87398. doi: 10.1371/journal.pone.0087398
Shi, Z., Birman, E., and Saragovi, H. U. (2007). Neurotrophic rationale in glaucoma: a TrkA agonist, but not NGF or a p75 antagonist, protects retinal ganglion cells in vivo. Dev. Neurobiol. 67, 884–894. doi: 10.1002/dneu.20360
Shi, Z., Rudzinski, M., Meerovitch, K., Lebrun-Julien, F., Birman, E., Di, A., et al. (2008). Alpha2-macroglobulin is a mediator of retinal ganglion cell death in glaucoma. J. Biol. Chem. 283, 29156–29165. doi: 10.1074/jbc.M802365200
Siao, C. J., Lorentz, C. U., Kermani, P., Marinic, T., Carter, J., McGrath, K., et al. (2013). ProNGF, a cytokine induced after myocardial infarction in humans, targets pericytes to promote microvascular damage and activation. J. Exp. Med. 209, 2291–2305. doi: 10.1084/jem.20111749
Sidorova, Y. A., Bespalov, M. M., Wong, A. W., Kambur, O., Jokinen, V., Lilius, T. O., et al. (2017). A novel small molecule GDNF receptor RET agonist, BT13, promotes neurite growth from sensory neurons in vitro and attenuates experimental neuropathy in the rat. Front. Pharmacol. 8:365. doi: 10.3389/fphar.2017.00365
Simmons, D. A., Belichenko, N. P., Yang, T., Condon, C., Monbureau, M., Shamloo, M., et al. (2013). A small molecule TrkB ligand reduces motor impairment and neuropathology in R6/2 and BACHD mouse models of Huntington’s disease. J. Neurosci. 33, 18712–18727. doi: 10.1523/JNEUROSCI.1310-13.2013
Skeldal, S., Sykes, A. M., Glerup, S., Matusica, D., Palstra, N., Autio, H., et al. (2012). Mapping of the interaction site between sortilin and the p75 neurotrophin receptor reveals a regulatory role for the sortilin intracellular domain in p75 neurotrophin receptor shedding and apoptosis. J. Biol. Chem. 287, 43798–43809. doi: 10.1074/jbc.M112.374710
Srinivasan, B., Roque, C. H., Hempstead, B. L., Al-Ubaidi, M. R., and Roque, R. S. (2004). Microglia-derived pronerve growth factor promotes photoreceptor cell death via p75 neurotrophin receptor. J. Biol. Chem.refvol 279, 41839–41845. doi: 10.1074/jbc.m402872200
Suzuki, J., Corfas, G., and Liberman, M. C. (2016). Round-window delivery of neurotrophin 3 regenerates cochlear synapses after acoustic overexposure. Sci. Rep. 6:24907. doi: 10.1038/srep24907
Tauszig-Delamasure, S., Yu, L. Y., Cabrera, J. R., Bouzas-Rodriguez, J., Mermet-Bouvier, C., Guix, C., et al. (2007). The TrkC receptor induces apoptosis when the dependence receptor notion meets the neurotrophin paradigm. Proc. Natl. Acad. Sci. U.S.A. 104, 13361–13366. doi: 10.1073/pnas.0701243104
Teng, H. K., Teng, K. K., Lee, R., Wright, S., Tevar, S., Almeida, R. D., et al. (2005). ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 25, 5455–5463. doi: 10.1523/jneurosci.5123-04.2005
Teng K. K., and Hempstead, B. L. (2004). Neurotrophins and their receptors: signaling trios in complex biological systems. Cel Mol. Life Sci. 61, 35–48. doi: 10.1007/s00018-003-3099-3
Tessarollo, L. (1998). Pleiotropic functions of neurotrophins in development. Cytokine Growth Factor Rev. 9, 125–137. doi: 10.1016/s1359-6101(98)00003-3
Thanos C. and Emerich, D. (2005). Delivery of neurotrophic factors and therapeutic proteins for retinal diseases. Expert Opin. Biol. Ther.refvol 5, 1443–1452. doi: 10.1517/14712598.5.11.1443
Thoenen H. and Sendtner, M. (2002). Neurotrophins: from enthusiastic expectations through sobering experiences to rational therapeutic approaches. Nat. Neurosci.refvol 5 1046–1050. doi: 10.1038/nn938
Tokugawa, K., Yamamoto, K., Nishiguchi, M., Sekine, T., Sakai, M., Ueki, T., et al. (2003). XIB4035, a novel nonpeptidyl small molecule agonist for GFRalpha-1. Neurochem. Int. 42, 81–86. doi: 10.1016/s0197-0186(02)00053-0
Tomellini, E., Lagadec, C., Polakowska, R., and Le Bourhis, X. (2014). Role of p75 neurotrophin receptor in stem cell biology: more than just a marker. Cell. Mol. Life Sci. 71, 2467–2481. doi: 10.1007/s00018-014-1564-9
Vaegter, C. B., Jansen, P., Fjorback, A. W., Glerup, S., Skeldal, S., Kjolby, M., et al. (2011). Sortilin associates with Trk receptors to enhance anterograde transport and neurotrophin signaling. Nat. Neurosci. 14, 54–61. doi: 10.1038/nn.2689
Valenzuela, D. M., Maisonpierre, P. C., Glass, D. J., Rojas, E., Nunez, L., Kong, Y., et al. (1993). Alternative forms of rat TrkC with different functional capabilities. Neuron 10, 963–974. doi: 10.1016/0896-6273(93)90211-9
Wan, G., and Corfas, G. (2015). No longer falling on deaf ears: mechanisms of degeneration and regeneration of cochlear ribbon synapses. Hear. Res. 329, 1–10. doi: 10.1016/j.heares.2015.04.008
Wan, G., Gomez-Casati, M. E., Gigliello, A. R., Liberman, M. C., and Corfas, G. (2014). Neurotrophin-3 regulates ribbon synapse density in the cochlea and induces synapse regeneration after acoustic trauma. eLife 3:e03564.
Weissmiller, A. M, and Wu, C. (2012). Current advances in using neurotrophic factors to treat neurodegenerative disorders. Transl. Neurodegener. 1:14. doi: 10.1186/2047-9158-1-14
Wyatt, T. J., Rossi, S. L., Siegenthaler, M. M., Frame, J., Robles, R., Nistor, G., et al. (2011). Human motor neuron progenitor transplantation leads to endogenous neuronal sparing in 3 models of motor neuron loss. Stem Cells Int. 2011:207230. doi: 10.4061/2011/207230
Xu, F., Wei, Y., Lu, Q., Zheng, D., Zhang, F., Gao, E., et al. (2009). Immunohistochemical localization of sortilin and p75(NTR) in normal and ischemic rat retina. Neurosci. Lett. 454, 81–85. doi: 10.1016/j.neulet.2009.02.036
Yanpallewar, S. U., Barrick, C. A., Buckley, H., Becker, J., and Tessarollo, L. (2012). Deletion of the BDNF truncated receptor TrkB.T1 delays disease onset in a mouse model of amyotrophic lateral sclerosis. PLoS One 7:e39946. doi: 10.1371/journal.pone.0039946
Yuen, E. C, and Mobley, W. C. (1996). Therapeutic potential of neurotrophic factors for neurological disorders. Ann. Neurol. 40, 346–354. doi: 10.1002/ana.410400304
Zaccaro, M. C., Ivanisevic, L., Perez, P., Meakin, S. O., and Saragovi, H. U. (2001). p75 Co-receptors regulate ligand-dependent and ligand-independent Trk receptor activation, in part by altering Trk docking subdomains. J. Biol. Chem. 276, 31023–31029. doi: 10.1074/jbc.m104630200
Keywords: small molecule, mimetic, antibody, growth factor, neurotrophin, receptor, neurodegeneration, therapy
Citation: Saragovi HU, Galan A and Levin LA (2019) Neuroprotection: Pro-survival and Anti-neurotoxic Mechanisms as Therapeutic Strategies in Neurodegeneration. Front. Cell. Neurosci. 13:231. doi: 10.3389/fncel.2019.00231
Received: 31 January 2019; Accepted: 08 May 2019;
Published: 06 June 2019.
Edited by:
Mario Eduardo Guido, Center for Research in Biological Chemistry Córdoba (CIQUIBIC), ArgentinaReviewed by:
Christopher von Bartheld, University of Nevada, Reno, United StatesCarlos Bas Orth, Universität Heidelberg, Germany
Copyright © 2019 Saragovi, Galan and Levin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Horacio Uri Saragovi, dXJpLnNhcmFnb3ZpQG1jZ2lsbC5jYQ==