 Xiao Mao1,2†
Xiao Mao1,2† Nadine Bruneau3†
Nadine Bruneau3† Quwen Gao4†Hélène Becq3Zhengjun Jia1,2Hui Xi1,2Li Shu1,2Hua Wang1,2*
Quwen Gao4†Hélène Becq3Zhengjun Jia1,2Hui Xi1,2Li Shu1,2Hua Wang1,2* Pierre Szepetowski3*
Pierre Szepetowski3* Laurent Aniksztejn3*
Laurent Aniksztejn3*- 1Hunan Provincial Maternal and Child Health Care Hospital, Changsha, China
- 2NHC Key Laboratory of Birth Defects Research, Prevention and Treatment, Changsha, China
- 3INSERM, Aix-Marseille University, INMED, UMR1249, Marseille, France
- 4Department of Epilepsy, General Hospital of Southern Theater Command, Guangzhou, China
The epilepsy of infancy with migrating focal seizures (EIMFS; previously called Malignant migrating partial seizures of infancy) are early-onset epileptic encephalopathies (EOEE) that associate multifocal ictal discharges and profound psychomotor retardation. EIMFS have a genetic origin and are mostly caused by de novo mutations in the KCNT1 gene, and much more rarely in the KCNT2 gene. KCNT1 and KCNT2 respectively encode the KNa1.1 (Slack) and KNa1.2 (Slick) subunits of the sodium-dependent voltage-gated potassium channel KNa. Functional analyses of the corresponding mutant homomeric channels in vitro suggested gain-of-function effects. Here, we report two novel, de novo truncating mutations of KCNT2: one mutation is frameshift (p.L48Qfs43), is situated in the N-terminal domain, and was found in a patient with EOEE (possibly EIMFS); the other mutation is nonsense (p.K564*), is located in the C-terminal region, and was found in a typical EIMFS patient. Using whole-cell patch-clamp recordings, we have analyzed the functional consequences of those two novel KCNT2 mutations on reconstituted KNa1.2 homomeric and KNa1.1/KNa1.2 heteromeric channels in transfected chinese hamster ovary (CHO) cells. We report that both mutations significantly impacted on KNa function; notably, they decreased the global current density of heteromeric channels by ~25% (p.K564*) and ~55% (p.L48Qfs43). Overall our data emphasize the involvement of KCNT2 in EOEE and provide novel insights into the role of heteromeric KNa channel in the severe KCNT2-related epileptic phenotypes. This may have important implications regarding the elaboration of future treatment.
Introduction
Channelopathies represent an important cause of neurological disorders (Kumar et al., 2016). Dysfunction of potassium channels has notably been involved in various types of epileptic encephalopathies, including epilepsy of infancy with migrating focal seizures (EIMFS), previously known as malignant migrating partial seizures of infancy. EIMFS are rare, neonatal epilepsies characterized by onset before the age of 6 months, and usually during the first weeks of life, by continuous migrating polymorphous focal seizures with corresponding multifocal ictal electroencephalographic (EEG) discharges associated with progressive deterioration of psychomotor development (Coppola et al., 1995). EIMFS have a genetic origin and can be caused by de novo mutations in the KCNT1 gene encoding the KNa1.1 subunit (Slack or Slo2.2) of KNa channels (Barcia et al., 2012; Ishii et al., 2013; McTague et al., 2013; Rizzo et al., 2016). More recently, two pathogenic mutations in the KCNT2 gene encoding the KNa1.2 subunit (Slick or Slo2.1) have been reported (Gururaj et al., 2017; Ambrosino et al., 2018). KNa channels are voltage-gated potassium channels that are activated by an increase of cytoplasmic Na+ concentration. They contribute to the slow afterhyperpolarization that follows a train of the action potential in several neuronal populations of the brain (Stafstrom et al., 1985; Kim and McCormick, 1998; Budelli et al., 2009; Hage and Salkoff, 2012; Kaczmarek, 2013; Kaczmarek et al., 2016). These subunits co-assemble to form homo or tetra-heteromeric KNa channels. Each subunit is composed of six transmembrane segments and of two intracellular N and C terminal domains (Figure 1). These two subunits display structural differences notably regarding their distal C-terminal region, their electrophysiological properties, their responses to neuromodulators, and their sensitivities to changes in cell volume (Bhattacharjee et al., 2003; Santi et al., 2006; Kaczmarek, 2013; Tejada et al., 2017). The C terminal part contains consensus sites for Na+ within the RCK2 (regulator of conductance of K+) domain and interaction sites for cytoplasmic proteins (e.g., protein kinase C). In KNa1.2 but not KNa1.1, the C-terminus also harbors a binding site for ATP, which function remains elusive (Bhattacharjee et al., 2003; Berg et al., 2007; Kaczmarek, 2013; Garg and Sanguinetti, 2014; Kaczmarek et al., 2016; Gururaj et al., 2017). In heterologous cells, functional analysis of mutant channels associated with EIMFS mostly revealed gain of function effects: potassium current was increased in cells expressing homomeric KNa1.1 channels harboring either of the p.Val271Phe, p.Gly288Ser, p.Arg398Gln, p.Arg428Gln, p.Arg474His, p.Met516Val, p.Lys629Asn, p.Ile760Met, p.Pro924Leu or p.Ala934Thr missense mutations, and in cells expressing homomeric KNa1.2 channels harboring either of the p.Arg190His or p.Arg190Pro missense mutations (Barcia et al., 2012; Rizzo et al., 2016; Villa and Combi, 2016; Ambrosino et al., 2018). A change in channel function has also been described in cells expressing the KNa1.2 subunit harboring the p.Phe240Leu missense mutation: the mutant channel lost its selectivity to K+ ions and gained permissiveness to Na+ ions (Gururaj et al., 2017).
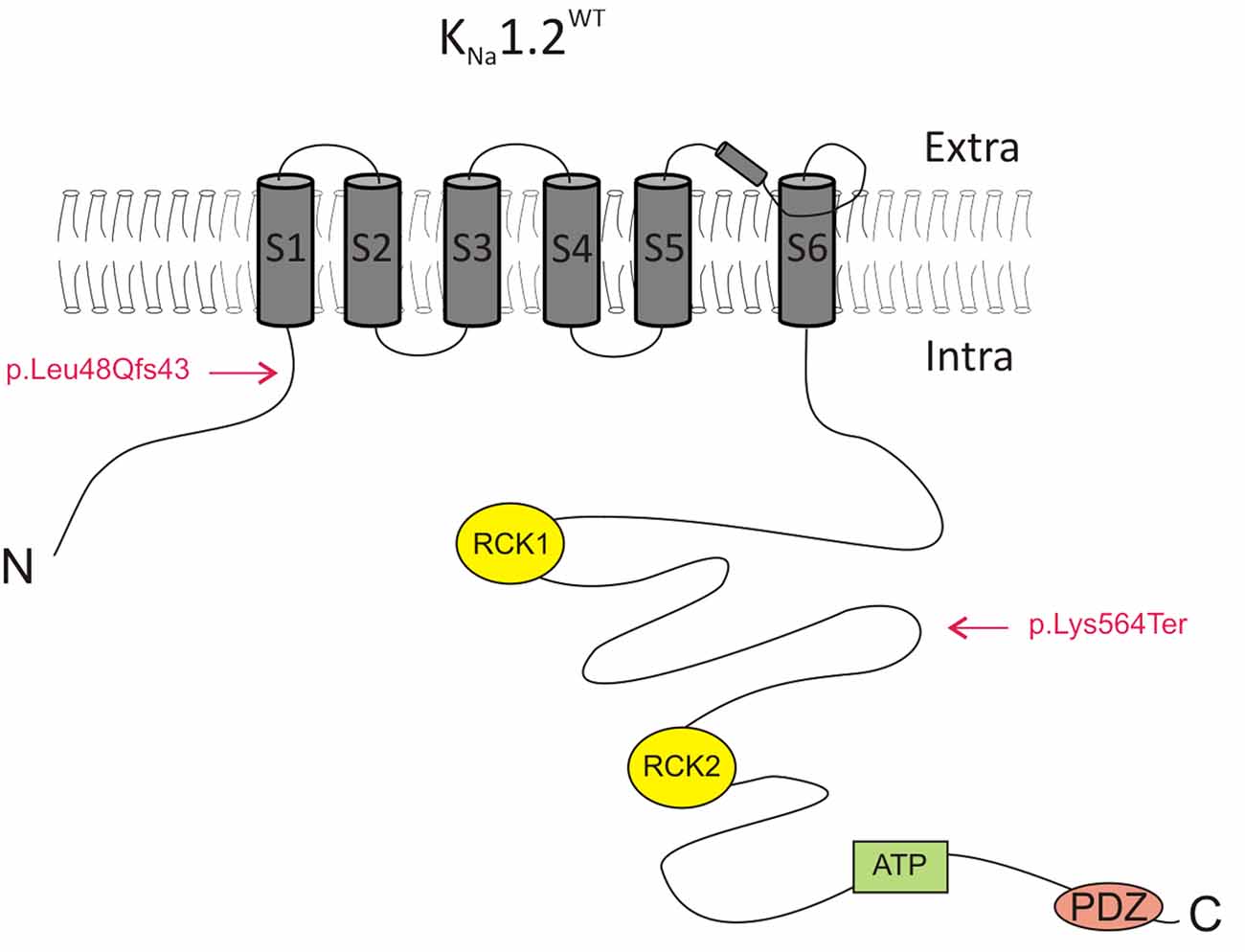
Figure 1. Schematic representation of the KNa1.2 subunit. The location of the two mutations identified in the present study (p.K564* and p.L48Qfs43) is indicated. ATP, adenosine triphosphate binding domain; Extra, extracellular milieu; Intra, intracellular milieu. RCK1 and RCK2, regulator of conductance of K+ domains 1 and 2.
Here, we have used exome sequencing to identify two novel de novo nonsense and frameshift mutations of the KCNT2 gene in two patients with ascertained EIMFS and with EIMFS-like early-onset epileptic encephalopathies (EOEE), respectively. We have investigated the functional consequences of the two mutations in heterologous cells expressing heteromeric channels and showed that both mutations reduced whole-cell potassium current. Therefore, EIMFS may be caused not only by an increase but also by a decrease in the function of KNa.
Materials and Methods
Patients
The two patients with KCNT2 mutations were recruited at Hunan Provincial Maternal and Child Health Care Hospital. Leukocyte DNA was extracted from peripheral blood stored in EDTA tubes by the phenol-chloroform method. Clinical information was collected by experienced neurologists. Patients’ parents had informed consents and the study was approved by the Ethics Committee of Hunan Provincial Maternal and Child Health Care Hospital.
Exome Sequencing
Patients’ DNAs were analyzed by next-generation sequencing with the whole-exome sequencing (WES) approach. DNA fragments were sequenced on the HiSeq2500 system (Illumina, San Diego, CA, USA) with a mean depth of 100×. A preliminary processing of WES data (data alignment and filter) followed pipelines as previously reported (Wang et al., 2011).
The annotated data by ANNOVAR (version 20160201; Wang et al., 2011) was used for further data analyses. Public databases (1000Genome, ESP6500, ExAC, dbSNP, and gnomAD) were used to filter known variants with minor allele frequencies (MAFs) over 0.001. Bioinformatics software (PolyPhen, SIFT, CADD or Mutation Taster) were used to predict the pathogenicity of single-nucleotide variations (SNVs). The loss-of-function variants (nonsense variants, frameshift variants, and splicing variants) and predicted pathogenic SNVs were retained. ACMG guidelines were finally used to evaluate the pathogenicity of the variants (Richards et al., 2015).
Sanger sequencing was performed on patients’ DNA to validate the findings of WES and on parents’ DNA to study familial inheritance. Sequencing primers were designed according to the sequences of the detected variants and polymerase chain reaction amplification was carried out for Sanger sequencing.
Constructs and Site-Directed Mutagenesis
The human KCNT1 cDNA construct (Genecopoeia EX-Y5001-M02, thereafter designated as pKCNT1) was used for the expression of wild-type human KNa1.1 subunit (NM_020822). Two human KCNT2 cDNA constructs (Genecopoeia EX-Y5628-M61and EX-Y5628-M83, thereafter designated as pKCNT2-ires-GFP and pKCNT2-ires-mCherry, respectively) were used for expression of mutant or wild-type human KNa1.2 subunits (NM_198503) coupled with internal ribosome entry site (IRES)-driven independent expression of eGFP (green fluorescent protein) or mcherry protein, respectively.
Mutant KCNT2 constructs were generated from their wild-type counterpart by using QuikChange Lightning Site-Directed Mutagenesis Kit according to the manufacturer’s protocol (Agilent Technologies) and the following forward and reverse primers: KCNT2-A1690T: 5′-tctctgctggtcttggttttaaaatgctgaattctcttctttg and 5′-caaagaagagaattcagcattttaaaaccaagaccagcagaga (KNa1.2K564*); KCNT2-del143-144: 5′-cttgatctctggttttttatgaaaaataatttgtctttctttaaatgtattttcattcatatag and 5′-ctatatgaatgaaaatacatttaaagaaagacaaattatttttcataaaaaaccagagatcaag(KNa1.2L48Qfs43). KCNT2 sequences from wild-type and mutant constructs were all verified by Sanger sequencing (GATC Biotech).
Cell Cultures and Transfections
Chinese hamster ovary (CHO) cells were cultured at 37°C in a humidified atmosphere with 5% CO2 with an F-12 Nutrient Mixture (Life Technologies) supplemented with 10% FBS (Fetal Bovine Serum) and 100 units/ml antibiotics/antimycotics (Life Technologies). These cells were transiently transfected using the Neon® Transfection System (Life Technologies) according to the manufacturer’s protocol. Briefly, 105 cells in suspension were transfected with a total amount of 2 μg of DNA. Non-recombinant pcDNA3.1 was added if necessary and concentrations were adjusted to get a total amount of 2 μg of DNA. Electroporation configuration was 1,400 V, 1 pulse, 20 ms. Cells were transiently co-transfected with pKCNT1 and either of pKCNT2-Mutant-ires-GFP or pKCNT2-WT-ires-mCherry or both (see below). Following electroporation, cells were cultured on pre-coated glass coverslips and maintained at 37°C and 5% CO2 with a complete medium for 2 days before recordings. Combinations of plasmids used in this study are shown in Table 1. The eGFP and mCherry fluorescent dyes were used to ascertain the efficacy of transfection assays and select cells for recordings.
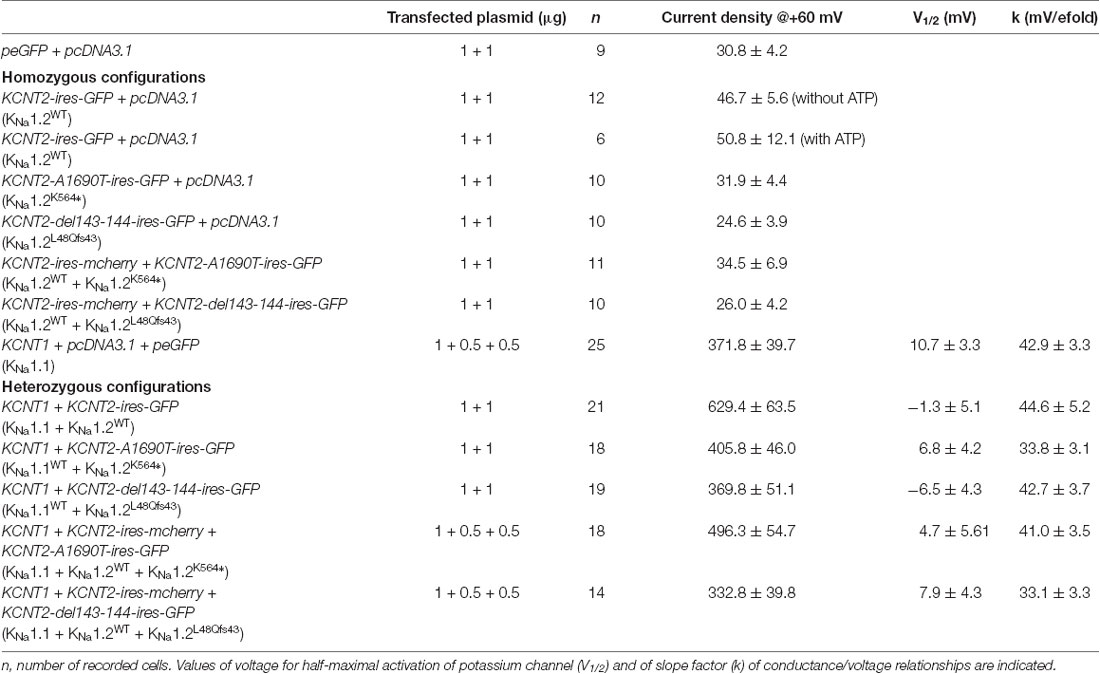
Table 1. Biophysical properties of currents recorded in chinese hamster ovary (CHO) cells transfected with the following plasmid combinations.
Electrophysiology
CHO cells were perfused at 1–2 ml/min with the following solution (in mM): 135 NaCl, 3.5 KCl, 5 NaHCO3, 0.5 NaH2PO4, 1 MgCl2, 1.5 CaCl2, 10 HEPES, 10 glucose, and pH 7.3 adjusted with NaOH. Whole-cell patch-clamp recordings were performed with microelectrodes (borosilicate glass capillaries GC 150F-15, Harvard apparatus) filled with a solution containing (in mM): 135 KCl, 0.1 CaCl2, 1.1 EGTA, 10 HEPES, 3 Mg2+ATP, 0.3 Na+GTP, 4 phosphocreatine, pH 7.3 adjusted with KOH and a resistance of 4–6 MΩ. In some experiments, ATP and GTP were omitted from the internal pipette solution. Data were sampled at 10 kHz and filtered with a cut-off frequency of 3 kHz using an EPC-10 amplifier (HEKA Electronik). An hyperpolarizing voltage step of 10 mV during 500 ms followed by incremental depolarizing voltage steps command of 10 mV was applied from a holding potential of −90 mV and up to +110 mV in order to analyze current densities and the conductance–voltage (G–V) relationships. Current densities (expressed in pA/pF) were calculated by measuring current amplitude at the end of the voltage step divided by the capacitance (Cm). G values were obtained from peak amplitudes of the slow outward current divided by the driving force for K+ ions with EK ~−93 mV and normalized to the maximal conductance. Plotted points were fitted with a Boltzmann function: G/Gmax = 1/[1 + exp(V1/2 − Vm)/k] to yield the voltage for half-maximum activation (Vhalf) and the slope factor (k) values. Currents were analyzed using Origin 8.0 software. Analyses were performed after offline leak current subtraction. Membrane potentials were corrected for liquid junction potential (~5 mV).
Statistical Analyses
Data are represented as means ± SEM. Two-way ANOVA with Tukey’s correction for multiple testing or Kruskal–Wallis test were used to assess statistical significance; *adjusted p < 0.05; **adjusted p < 0.01; ***adjusted p < 0.001.
Results
Identification of Two Novel de novo KCNT2 Defects in Patients With Early-Onset Epileptic Encephalopathies
Patient A, female, was born at 42 weeks of gestation after normal pregnancy and delivery. She is the first child of healthy non-consanguineous parents. At 2 months of age, she started to have seizures characterized by twitches of the eyelids, tonic elevation of a single limb or both limbs, and perioral cyanosis. The seizures usually lasted several minutes and occurred in clusters with an increasing frequency of more than 20 seizures per day at 3 months. Neurologic examination revealed generalized hypotonia and severe neurologic impairment with the poor visual following. Seizures were refractory to various antiepileptic drugs including valproate, lamotrigine and levetiracetam. Brain magnetic resonance imaging (MRI) was normal. EEG showed a symmetric slow background pattern, multifocal spikes and seizures arising from different regions independently and migrating from one hemisphere to the other at times (Figure 2A).
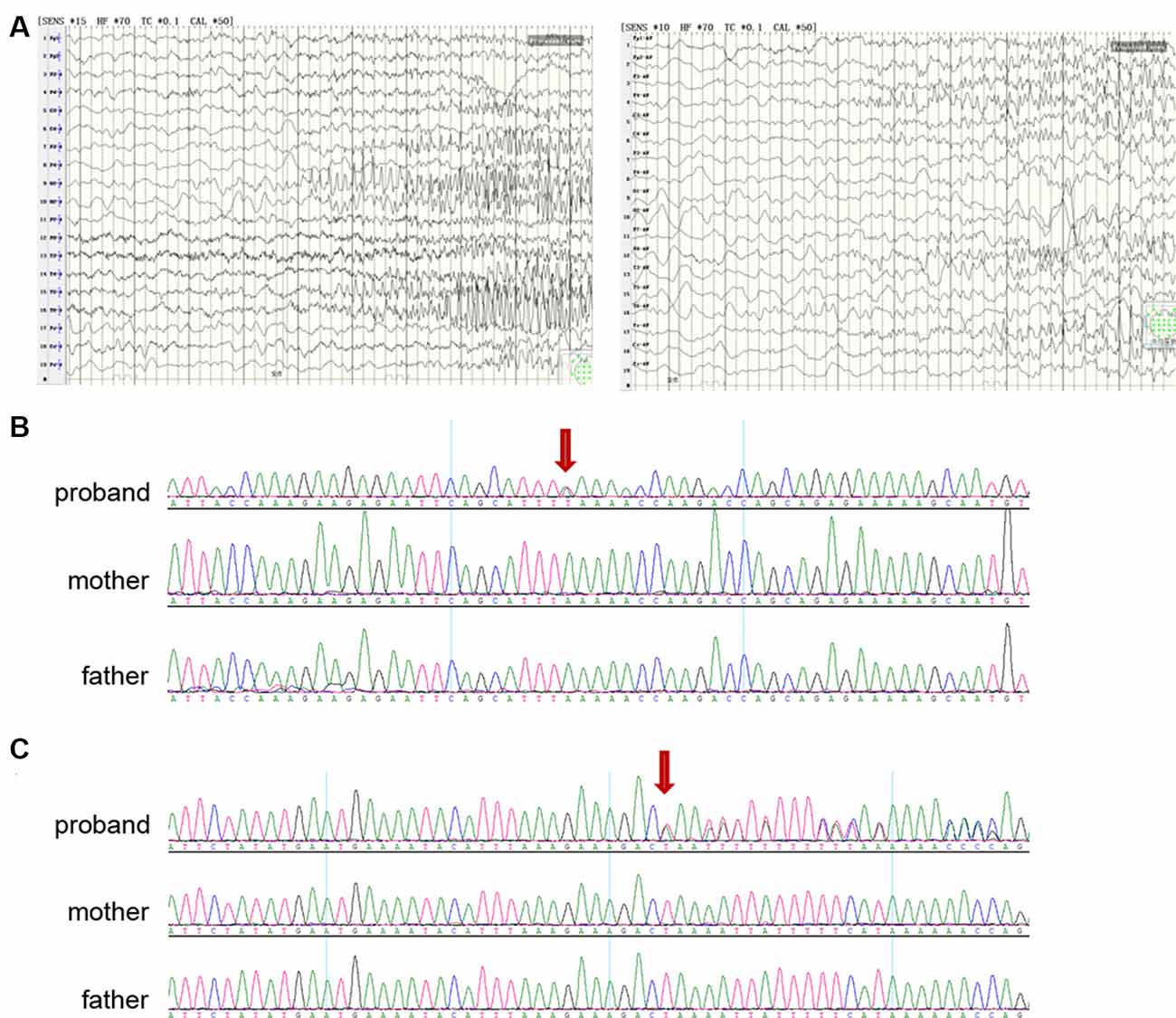
Figure 2. Clinical and genetic data. (A) Electroencephalographic (EEG) of a proband with epilepsy of infancy with migrating focal seizures (EIMFS; patient A), showing seizures arising from different hemispheres (left: left occipital lobe; right: right frontal lobe). (B) Sanger sequencing showing (red arrow) nonsense variant p.K564* (NM_198503.2:c.1690A>T) in KCNT2 in the proband (patient A) and not in the parents. (C) Sanger sequencing showing (red arrow) frameshift variant p.L48Qfs43 (NM_198503.2:c.143-144 delTA) in KCNT2 in the proband (patient B) and not in the parents.
DNA from patient A were screened by WES and analyzed by Clinical Sequencing Analyzer (CSA of WuXiNextCODE). After applying filtering methods, Sanger sequencing was performed to exclude false-positive and examine inheritance. We identified a de novo nonsense variant p.K564* (NM_198503.2:c.1690A>T; ClinVar accession number: VCV000695093.1) in KCNT2 which was absent from controls in ExAC, gnomAD, 1000 Genomes, ESP6500 and dbSNP databases, and compound heterozygous variants in ABCC2 (NM_000392.3:c.1018C>A and c.1313T>G). No other variant of interest was identified in other genes including known epilepsy genes. Pathogenic variants in ABCC2 can cause Dubin-Johnson syndrome, a benign autosomal recessive disorder characterized by hyperbilirubinemia with no clinical feature shared with our patients. In the course of the present study, Gururaj et al. (2017) and Ambrosino et al. (2018) identified two de novo KCNT2 missense variants in patients with epileptic encephalopathy. Despite the fact that the KCNT2 gene would not be highly intolerant to loss-of-function mutations, as a few nonsense variants have been detected in control individuals and the pLI score (the probability of being loss of function intolerant) is at 0.67 only (see the ExAC database at: http://exac.broadinstitute.org/), we considered the de novo nonsense variant p.K564* as the most plausible genetic cause (Figure 2B).
We then searched additional KCNT2 variants in our in-house WES database of a cohort of more than 200 patients with early-onset epileptic encephalopathy (EOEE). We found a de novo frameshift variant p.L48Qfs43 (NM_198503.2:c.143-144 delTA; ClinVar accession number: VCV000695094.1) in KCNT2 in an EOEE patient (Patient B) and validated this variant by Sanger sequencing (Figure 2C). This frameshift variant was absent from controls in control databases and no other variant of interest was found in known causative epilepsy genes including EIMFS. Patient B is a 29 years old female who showed mild intellectual disability and seizures. According to her parents and to the medical records, the patient began to have seizure attacks when she was 4 months old. Seizures were mainly focal and migrating, which likely corresponded to EIMFS. However, due to the fact that this is an aged case and considering the relatively low medical level in China almost 30 years ago, diagnosis cannot be firmly ascertained.
Functional Analysis of Wild Type and Mutant Homomeric Human KNa1.2 Channels
To investigate the functional consequences of the KNa1.2 (KCNT2) mutations, CHO cells were first transfected with plasmids encoding the wild type KNa1.2 (KNa1.2WT) subunit. Two days later, cells were recorded with a KCl filled pipette solution that did not contain ATP, as this nucleotide which binds the C-terminal domain of KNa1.2 inhibits channel activity (Bhattacharjee et al., 2003; but see Berg et al., 2007; Garg and Sanguinetti, 2014; Gururaj et al., 2017). We observed that whole-cell current density was slightly but significantly higher than in cells transfected with control plasmid encoding GFP only (n = 12 and 9 cells respectively, Figure 3; Table 1). Values were close to those reported recently in HEK cells (Ambrosino et al., 2018). Similar recordings with pipette solution containing ATP yielded the same results (n = 6 cells, Table 1). This suggested that low expression of KNa1.2 channel-mediated current in CHO cells is independent of the presence or absence of ATP.
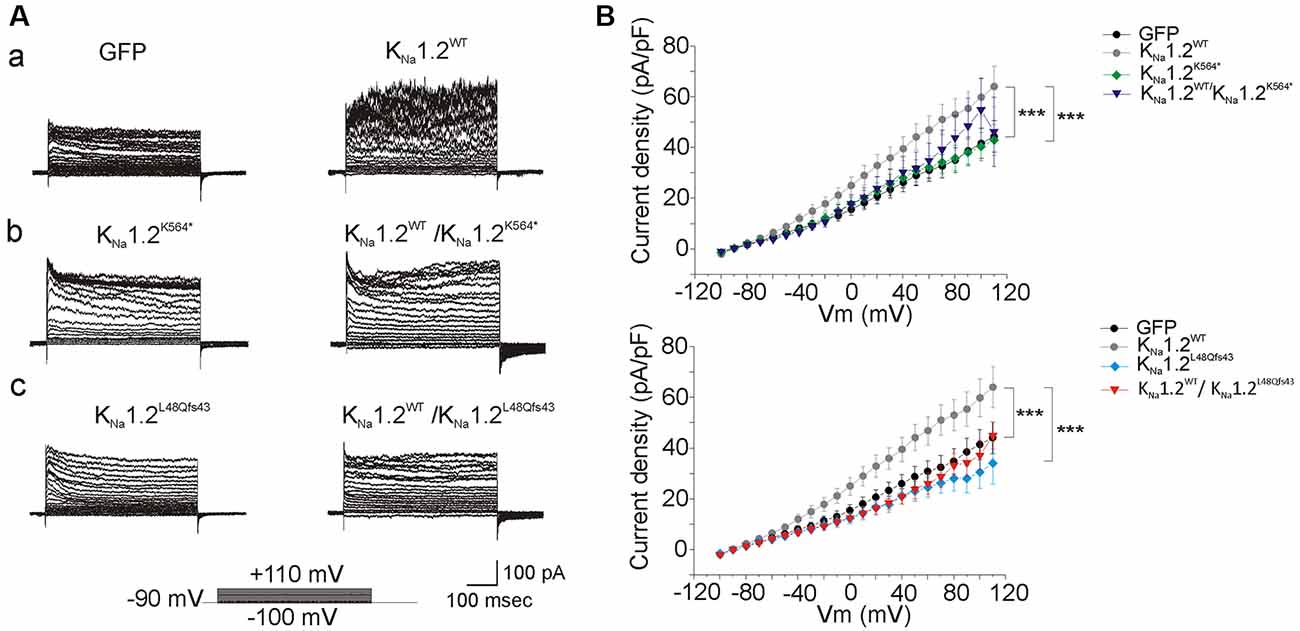
Figure 3. Functional analysis of homomeric wild type and mutant human KNa1.2 channels in chinese hamster ovary (CHO) cells. (A) Current response to 10 milliVolts (mV) voltage steps command from −90 mV to +110 mV for 500 ms in cells transfected with plasmids encoding: (a) green fluorescent protein (GFP, left traces), wild-type KNa1.2 (KNa1.2WT, right traces) subunits; (b) the mutant p.K564* (KNa1.2K564*, left traces), both KNa1.2WT and KNa1.2K564* subunits (right traces); (c) the mutant p.L48Qfs43 (KNa1.2L48Qfs43, left traces), both KNa1.2WT and KNa1.2L48Qfs43 subunits (right traces). (B) Upper graph: current density expressed in picoAmperes/picofarads (pA/pF) measured at membrane potentials (Vm) as indicated in abscissa, in cells transfected with plasmids encoding GFP, KNa1.2WT, KNa1.2K564*, both KNa1.2WT, and KNa1.2K564* subunits. Bottom graph: same measurements in cells transfected with plasmids encoding GFP, KNa1.2WT, KNa1.2L48Qfs43, both KNa1.2WT and KNa1.2L48Qfs43 subunits. Corresponding symbols are shown on the right of each graph. Two-way ANOVA with Tukey’s correction for multiple comparisons. ***p < 0.001.
CHO cells were then transfected with plasmids encoding either the human KNa1.2 p.K564* (KNa1.2K564*; n = 10 cells) or the KNa1.2 p.L48Qfs43 (KNa1.2L48Qfs43; n = 10 cells) mutant subunits. p.K564* is a nonsense mutation localized in the C-terminal part of KNa1.2 and situated between the RCK1 and RCK2 domains (Figure 1). This mutation leads to a truncated protein lacking the RCK2 domain and the ATP and PDZ binding sites. p.L48Qfs43 is a frameshift mutation localized in the N-terminal domain of the protein. This mutation leads to a protein composed of the N-terminal domain and the first transmembrane segment S1. For both mutations, depolarizing voltage steps elicited significantly smaller currents compared to cells transfected with KNa1.2WT plasmids (n = 12 cells), and responses were similar to those obtained in cells expressing only GFP. Same results were obtained in cells co-transfected with plasmids encoding KNa1.2WT and KNa1.2K564* subunits (n = 11 cells), or KNa1.2WT and KNa1.2L48Qfs43 subunits (n = 10 cells, Figure 3). Although the current mediated by KNa1.2WT was very small, these data suggested that in contrast with other previously reported KCNT2 mutations (Ambrosino et al., 2018), p.K564* and p.L48Qfs43 decreased KNa1.2-mediated currents.
Functional Analysis of Wild Type and Mutants Heteromeric KNa1.1/KNa1.2 Channels
Immunohistochemical studies performed in rodent brain have shown that KNa1.1 and KNa1.2 subunits exhibited distinct expression patterns but could also co-localize (Bhattacharjee et al., 2005; Chen et al., 2009; Rizzi et al., 2016). Moreover, biochemical and electrophysiological studies performed in heterologous cells have demonstrated that rat KNa1.1 and rat KNa1.2 subunits can form heteromeric channels (Chen et al., 2009). The co-assembly of the two subunits enhances channel expression to the plasma membrane, leading to the global current density that is higher than with KNa1.1 or KNa1.2 alone (Chen et al., 2009). We thus decided to study if the same properties would also characterize the human KNa subunits, and if so, to analyze the impact of the two pathogenic mutations in this heteromeric condition.
To this aim, CHO cells were first transfected with plasmid encoding KNa1.1 and recorded with a KCl filled pipette solution containing ATP. These cells responded to depolarizing voltage steps by large outwardly rectifying currents (n = 25 cells, Figures 4A,B). Currents were abolished in cells superfused with bepridil 10 μM (n = 5 cells), or in cells recorded with a Na+-free internal pipette solution (n = 5 cells, Figures 4A,B). These data confirmed that the outward rectifying current was mediated by activation of the Na+-dependent potassium KNa channels. Boltzmann analysis of the conductance/voltage curve showed that Vhalf was at 10.7 ± 3.3 mV, a mean value close to the one reported previously (Rizzo et al., 2016), and the slope factor was at 42.9 ± 3.3 mV/e fold (n = 17 cells, Table 1), a mean value higher than the one reported by the same authors but which indicated the low voltage sensitivity of KNa channels (Salkoff et al., 2006).
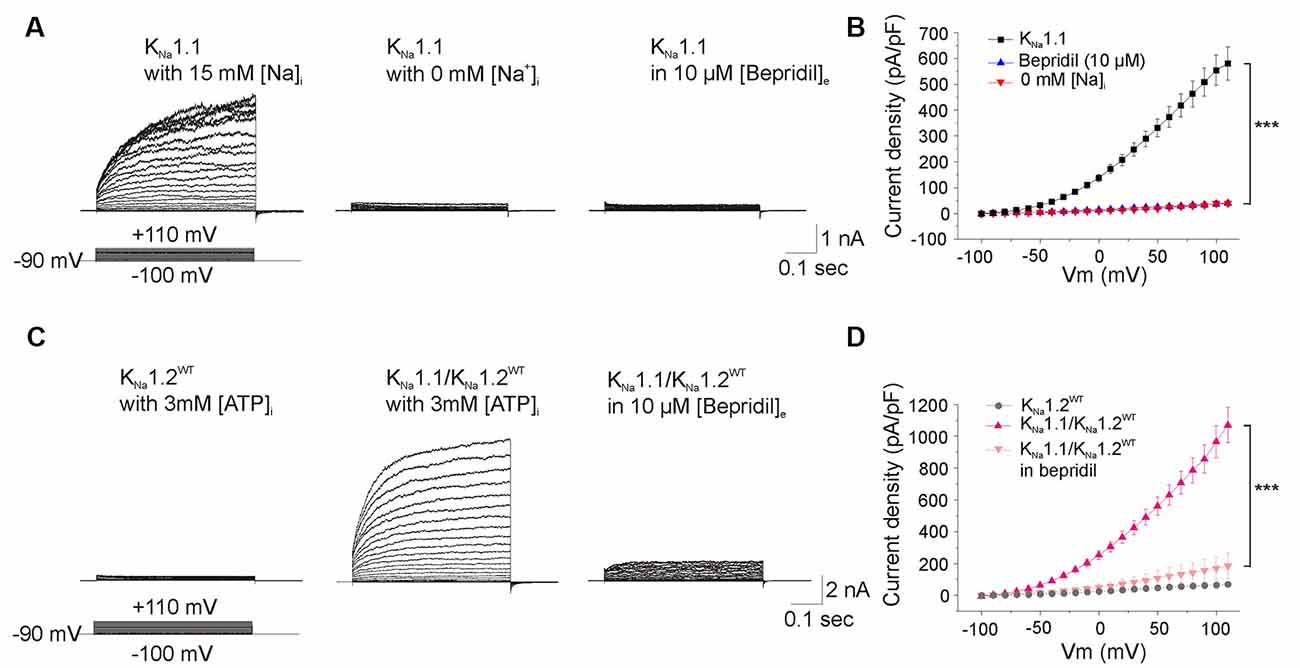
Figure 4. Functional analysis of KNa1.1 and heteromeric KNa1.1/KNa1.2 channels. (A) Current traces evoked by voltage steps in cells expressing KNa1.1 subunit and recorded with pipette solution containing either 15 mM Na+ (left traces), or no Na+ (middle traces), or 15 mM Na+ and an extracellular medium containing 10 μM bepridil (right traces). (B) Current densities measured in the three recording conditions as in (A). (C) Current traces recorded with pipette solution containing 3 mM ATP/0.3 mM GTP in cells expressing the KNa1.2WT subunit (left traces), both KNa1.1 and KNa1.2WT subunits (middle traces) and both KNa1.1 and KNa1.2WT subunits in the presence of 10 μM bepridil in the extracellular medium (right traces). (D) Current densities measured in the three conditions as depicted in (C). Two-way ANOVA with Tukey’s correction for multiple comparisons. ***p < 0.001.
CHO cells were then transfected with plasmids encoding the KNa1.1 and KNa1.2WT subunits (n = 21 cells). We observed that whole-cell current was almost twice higher than that generated by KNa1.1 alone (n = 25 cells, Figures 4C,D), without any significant change in Vhalf and in the slope factor of the conductance/voltage relationship (Figure 5, Table 1). This current was also dramatically reduced by bepridil 10 μM (n = 5 cells). Thus, like for rat KNa1.1 and rat KNa1.2, the two human subunits might co-assemble in CHO cells to form heteromeric channels with larger currents.
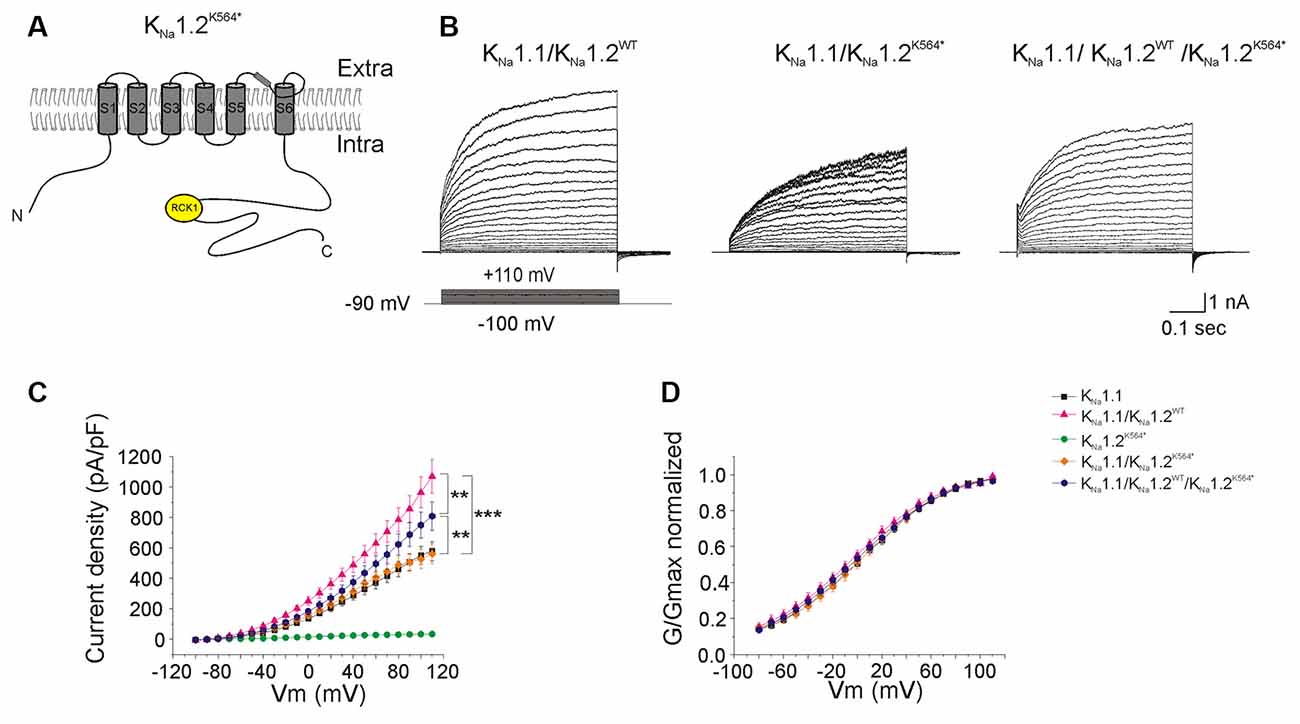
Figure 5. Functional consequences of the p.K564* mutation on heteromeric channels. (A) Schematic representation of the expected KNa1.2K564* mutant subunit. The nonsense mutation locates in the C-terminal part of the protein between the RCK1 and RCK2 domains, leading to a predicted truncated protein. (B) Representative current responses to depolarizing voltage steps in CHO cells transfected with KNa1.1 + KNa1.2WT (left traces); KNa1.1 + KNa1.2K564* (middle traces); KNa1.1 + KNa1.2WT + KNa1.2K564* (right traces) plasmids. (C) Current densities measured in these three conditions but also including KNa1.1 and KNa1.2WT for comparison. (D) Conductance-voltage relationship of wild type homomeric KNa1.1; wild type heteromeric KNa1.1 + KNa1.2; heteromeric mutant KNa1.1 + KNa1.2K564* and heteromeric mutant KNa1.1 + KNa1.2WT+ KNa1.2K564* channels normalized. Two-way ANOVA with Tukey’s correction for multiple comparison. ***p < 0.001; **p < 0.01.
We then evaluated the consequences of each of the two mutant KNa1.2 subunits on heteromeric KNa1.1/KNa1.2 channels, either in a homozygous state, or in a heterozygous state to mimic the patient’s situation. In cells co-transfected with KNa1.1 and KNa1.2K564* plasmids (homozygous mutant state, n = 18 cells), the level of whole-cell current was lower as compared to the wild-type situation, and was similar to the current recorded in cells expressing KNa1.1 only (Figures 5A,B). The reduction of global current density was not associated with any significant change in the conductance-voltage relationship (n = 15 cells). In cells co-transfected with KNa1.1/KNa1.2WT/KNa1.2K564* plasmids (heterozygous mutant state, n = 18 cells), the level of whole-cell current was significantly increased as compared with cells expressing KNa1.1 channels only and was significantly decreased as compared with cells expressing heteromeric wild-type KNa1.1/KNa1.2WT channels (Figures 5B–D, Table 1).
As with the p.K564* mutation, whole-cell current measured in cells co-transfected with KNa1.1 and mutant KNa1.2L48Qfs43 plasmids (homozygous mutant state, n = 19 cells, Figures 6A,B) was identical to the current measured in cells transfected with KNa1.1 plasmid only (n = 25 cells), again without any significant change in the conductance/voltage relationship. Interestingly, currents measured in cells co-transfected with KNa1.1/KNa1.2WT/KNa1.2L48Qfs43 plasmids (n = 14 cells) were identical to currents measured in cells expressing KNa1.1 only (Figures 6B–D, Table 1). This indicated a possible dominant-negative effect for p.L48Qfs43.
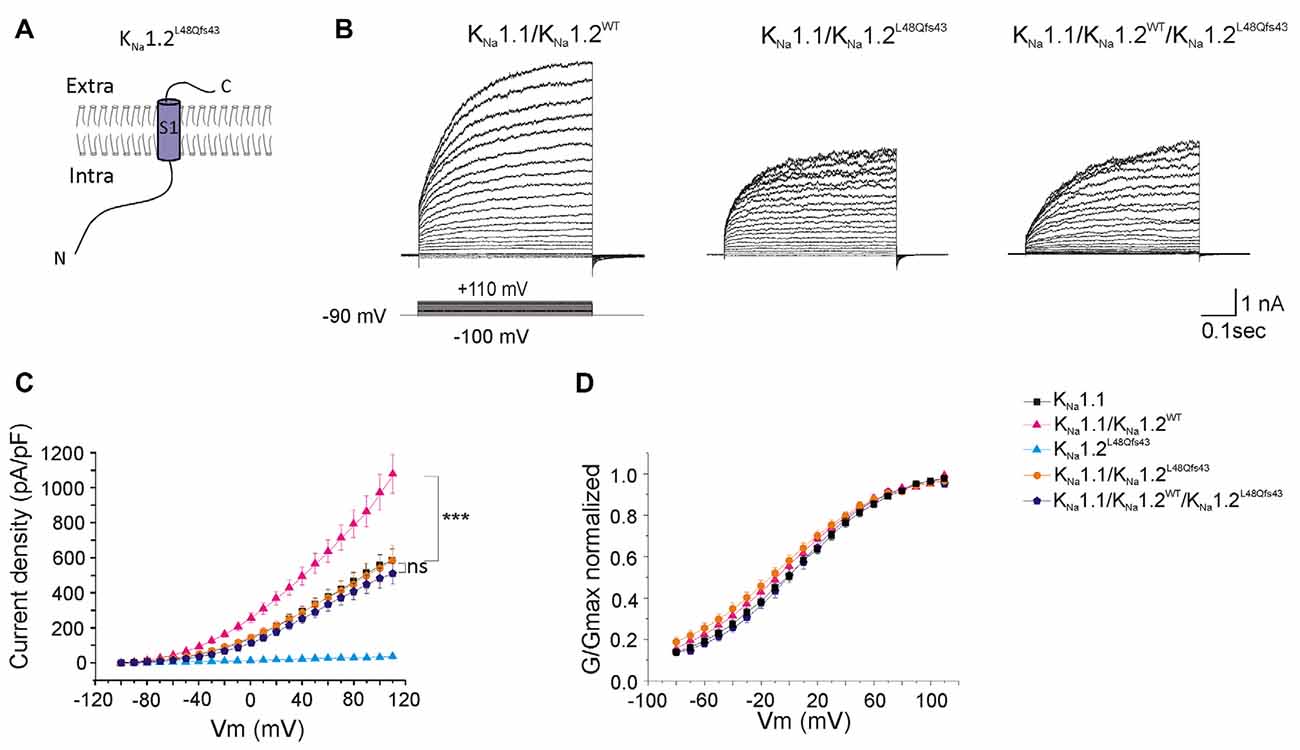
Figure 6. Functional consequences of the p.L48Qfs43 mutation on heteromeric channels. (A) Schematic representation of the expected KNa1.2L48Qfs43 mutant subunit. The frameshift mutation locates in the N-terminal part of the protein, leading to predicted protein composed of the N-terminal domain and of transmembrane segment S1. (B) Representative current responses to depolarizing voltage steps in CHO cells transfected with KNa1.1 + KNa1.2WT (left traces); KNa1.1 + KNa1.2L48Qfs43 (middle traces); KNa1.1 + KNa1.2WT+ KNa1.1L48Qfs43 (right traces) plasmids. (C) Current densities measured in these three conditions but also including KNa1.1 and KNa1.2WT for comparison. Corresponding symbols are indicated on the top of the graph. (D) Conductance-voltage relationship of wild type homomeric KNa1.1; wild type heteromeric KNa1.1 + KNa1.2; heteromeric mutant KNa1.1 + KNa1.2L48Qfs43 and heteromeric mutant KNa1.1 + KNa1.2WT+ KNa1.2L48Qfs43 channels normalized. Two-way ANOVA with Tukey’s correction for multiple comparisons. ***p < 0.001; ns, not significant.
Discussion
Here, we report two patients diagnosed as EIMFS and EIMFS-like EOEE, respectively, and carrying two novel de novo variants in the KCNT2 gene. Patient A in our study fulfilled the diagnostic criterion for EIMFS (Coppola et al., 1995) including age at the onset before 6 months of age, migrating focal motor seizures, seizures refractory to antiepileptic drugs, and severe psychomotor delay. EEG of patient A also showed the typical “jumping” areas at onset between two hemispheres. According to the clinical manifestations and the EEGs, diagnosis of EIMFS in patient A can be ascertained. We also diagnosed patient B as probably having EIMFS, based on the age at onset and characteristics of her seizure attacks.
EIMFS is a severe, drug-resistant, early-onset epilepsy encephalopathy in which variants in the KCNT1, SCN1A, SCN8A, SCN2A, PLCB1, KCNT1, SLC25A22, TBC1D24 and SLC12A5 genes as well as 16p11.2 duplication have been reported. While de novo gain of function KCNT1 (KNa1.1) variants are the most common cause of EIMFS (Bedoyan et al., 2010; Carranza Rojo et al., 2011; Barcia et al., 2012; Poduri et al., 2012, 2013; Ishii et al., 2013; McTague et al., 2013; Milh et al., 2013; Ohba et al., 2014; Howell et al., 2015; Stödberg et al., 2015; Rizzo et al., 2016; Villa and Combi, 2016), it was recently reported that de novo mutations in the KCNT2 (KNa1.2) gene also caused EOEE, including EIMFS. Two gain-of-function KCNT2 mutations were identified in a patient with West syndrome that evolved to a Lennox-Gastaut syndrome, and in a patient with EIMFS, respectively (Ambrosino et al., 2018). A third KCNT2 mutation leading to KNa1.2 channels that were no more selective for K+ ions and that became permeable to Na+ ions was identified in a patient with multi-focal epileptogenic activity or hypsarrhythmia (Gururaj et al., 2017). Here, we have identified in two patients with EIMFS and EIMFS-like phenotypes, two novel de novo mutations in KCNT2 (KNa1.2), respectively localized in the N-terminal (p.L48Qfs43) and C-terminal (p.K564*) domains of the protein and which both led to significantly reduced activity of heteromeric KNa channels in vitro. To reach this conclusion, we have analyzed the macroscopic current in CHO cells co-transfected with wild type and mutant KNa1.2 subunits, notably in the heterozygous configuration to mimic the patient’s situation. To the best of our knowledge, the functional impact of KNa1.2 pathogenic mutations on currents generated by human heteromeric KNa channels had never been tested in such a configuration, although immunohistochemical studies performed in adult rodent brain provided evidence that KNa1.1 and KNa1.2 subunits can co-localize and potentially form heteromeric channels (Bhattacharjee et al., 2005; Chen et al., 2009; Rizzi et al., 2016). KNa channels are also very likely to be formed by homomeric KNa1.1 or KNa1.2 subunits in another large subset of neuronal cells. Here, we observed that human KNa1.2WT produced a very small current either in the presence or absence of ATP. The same difficulty to detect current in CHO cells was mentioned by Gururaj et al. (2017). In cells co-transfected with KNa1.1 and KNa1.2 plasmids, the current was larger as compared to cells transfected either with KNa1.1 or with KNa1.2 plasmids alone, indicating that both subunits are expressed and would co-assemble to form a heteromeric channel. It is possible that in spite of the internal dialysis due to whole-cell recording, the endogenous concentration of ATP in CHO cells is high enough to exert its inhibitory effect on the KNa1.2 subunit (Bhattacharjee et al., 2003)—although the action of ATP has been challenged in other studies (Berg et al., 2007; Garg and Sanguinetti, 2014; Gururaj et al., 2017). Another possibility is that the intracellular concentration of Na+ (15 mM) is not high enough to activate the channel. The reliable current was observed in HEK cells expressing human KNa1.2 subunit but with a patch pipette solution containing 70 mM Na+ concentration (Berg et al., 2007). In fact, KNa1.2 channels might be less sensitive to [Na+]i than KNa1.1 (Bhattacharjee et al., 2003; Kaczmarek, 2013). The small current produced by human KNa1.2 contrasts with the large whole-cell current produced in the same cells by rat KNa1.2 subunit (Gururaj et al., 2017). There are slight differences in amino acids sequence between the rat and human KNa1.2 subunits (~2%; Bhattacharjee et al., 2003): some of these amino acids may be instrumental for the functional discrepancy between rat and human KNa1.2 subunit, as shown for the rodent and human Kv7.3 subunit of Kv7 potassium channels (Etxeberria et al., 2004). Thus, the role of homomeric KNa1.2 channel in human neurons might even be questioned if, like in CHO cells, they mediated a very small current only. Hence and apart from the gain of function mutations reported recently (Ambrosino et al., 2018), the role of KNa1.2 and the functional consequences of the two mutants reported here would be exerted in human neurons co-expressing KNa1.1 and KNa1.2.
In the present study, we did not investigate the cellular mechanisms that would account for the effects of the two mutations. First, we showed that both mutant subunits failed to generate significant current. This was expected for KNa1.2L48Qfs43 as the mutant subunit would not contain a pore domain—if not degraded. This was also not surprising for KNa1.2K564* as the truncated part of the C-terminal domain includes the RCK2 domain which contains coordination motif for Na+ interaction (Thomson et al., 2015). Second, cells co-transfected with plasmids encoding KNa1.1 and either of the mutant KNa1.2 subunits in a homozygous state exhibited whole-cell currents that were similar to the currents measured in cells transfected with the KNa1.1 plasmid alone. This suggested either that the mutant subunits are rapidly degraded or lead to a non-functional heteromeric channel, or that the mutations prevented the assembly of KNa1.2 with KNa1.1. Indeed, the N-terminal domain of KNa1.1 plays a key role in heteromerization and channel trafficking (Chen et al., 2009). As channel formation (assembly, stabilization, trafficking) generally involves multiple inter-subunit association sites (Deutsch, 2002), it is also possible that the lack of the C-terminal part of mutant KNa1.2 subunits plays instrumental role.
In the KNa1.1/KNa1.2WT/KNa1.2K564* mutant heterozygous configuration mimicking the patient situation, global current density was intermediate between that of cells expressing KNa1.1 alone and that of cells expressing both KNa1.1/KNa1.2 subunits. Whether the moderate alteration (~25% decrease) of KNa current density observed with the p.K564* mutation would be sufficient to cause EIFMS remains to be firmly established. Indeed, KCNT2 does not look that intolerant to heterozygous loss of function mutations, as can be inferred from databases of control individuals. On the one hand and although very unlikely, we cannot firmly exclude that the identification of the de novo p.K564* mutation in an EFMIS patient was coincidental by chance only. On the other hand, nonsense mutations in other epilepsy genes (e.g., DEPDC5) have also been detected in control individuals, and pLI scores should be interpreted with caution (Fuller et al., 2019). Also, different nonsense mutations in a given gene might be differently subjected to nonsense-mediated mRNA decay (NMD) which in turn sustains compensatory effects (El-Brolosy et al., 2019). Moreover, the actual impact of a deleterious ion channel mutation might be better seen in the genetic context of variants in other ion channels (Klassen et al., 2011). Interestingly, while the typical mutations of KCNT1 leading to EIFMS are gain of function and KCNT1 would be even more tolerant than KCNT2 to loss-of-function variants (pLI score at 0.01 at the ExAC database, http://exac.broadinstitute.org/), a Phe932Ile loss of function variant in KCNT1 was reported in a patient with severe epilepsy, delayed myelination and leukoencephalopathy (Vanderver et al., 2014; Evely et al., 2017), further indicating that decreased activity of KNa channels can indeed be associated with severe neurological manifestations including epilepsy. The other mutation found here in KCNT2, p.L48Qfs43, had more dramatic effects than p.K564*: in the KNa1.1/KNa1.2WT/KNa1.2 L48Qfs43 configuration, current global density was more severely affected than with p.K564* and was similar to that of KNa1.1 alone, consistent with a dominant-negative effect of p.L48Qfs43. Overall this indicates how important these channels are to control neuronal excitability at early developmental stages. That similar phenotypes were observed in patient B carrying the p.L48Qfs43 mutation (~55% decrease KNa current density) or in a patient carrying the gain of function p.R190P mutation (Ambrosino et al., 2018) suggests that KNa channels efficiency should be tightly regulated during brain development and that any alteration, whatever its direction, would deeply impact on cortical networks activities.
There is now evidence that gain or loss of function mutations of a given ion channel may both lead to epileptic encephalopathies—although differences in phenotypes may exist (see above). This has been well documented for Kv7.2 de novo mutations (Miceli et al., 2013, 2015; Orhan et al., 2014; Abidi et al., 2015; Devaux et al., 2016; Mulkey et al., 2017). We now show that this is also the case for KNa1.2 mutations. This may have practical implications as drugs inhibiting/reducing KNa1.1 channel activity such as quinidine have been used to improve the EEG and background activity in a subset of the patients. Different hypotheses have been proposed to explain how an increase or a decrease in the function of a given ion channel may have similar consequences on network activity. Notably different sensitivity of a mutant channel in pyramidal cells and in interneurons has been suggested, creating an imbalance between excitation and inhibition or favoring neuronal synchronization (Miceli et al., 2015; Niday and Tzingounis, 2018). The development of animal models carrying loss and gain of function mutations is needed to solve this apparent paradox. This is particularly important for the KNa1.2 subunit, whose exact role in neuronal activity remains to be addressed.
Data Availability Statement
The raw data of WES were deposited at the Sequence Read Archive (SRA) public database at NCBI (https://www.ncbi.nlm.nih.gov/sra; accession numbers: PRJNA592898; release date 2020-01-31, and PRJNA593942, release date 2019-12-08). The KCNT2 variants were deposited at the ClinVar public database at NCBI https://www.ncbi.nlm.nih.gov/clinvar/; c.1690A>T: accession number VCV000695093.1; c.143-144 delTA: accession number VCV000695094.1).
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Hunan Provincial Maternal and Child Health Care Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
XM designed and performed clinical investigations and genetic analyzes. NB designed and performed cell biology experiments (expression constructs, cell cultures and transfections). HB participated in the electrophysiological experiments. QG, ZJ, and HW collected clinical data. HX and LS performed data analysis. PS coordinated and participated in the design of the overall study. LA designed, performed, analyzed and coordinated the electrophysiological experiments and wrote the article with the help of XM, NB, and PS.
Funding
This work was supported by INSERM (Institut National de la Santé et de la Recherche Médicale), by the European Union Seventh Framework Programme FP7/2007–2013 under the project DESIRE (grant agreement n°602531), by the National Natural Science Foundation of China (grant number 81801136), and by the Natural Science Foundation of Hunan Province, China (grant number 2017JJ3142).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abidi, A., Devaux, J. J., Molinari, F., Alcaraz, G., Michon, F.-X., Sutera-Sardo, J., et al. (2015). A recurrent KCNQ2 pore mutation causing early onset epileptic encephalopathy has a moderate effect on M current but alters subcellular localization of Kv7 channels. Neurobiol. Dis. 80, 80–92. doi: 10.1016/j.nbd.2015.04.017
Ambrosino, P., Soldovieri, M. V., Bast, T., Turnpenny, P. D., Uhrig, S., Biskup, S., et al. (2018). De novo gain-of-function variants in KCNT2 as a novel cause of developmental and epileptic encephalopathy. Ann. Neurol. 83, 1198–1204. doi: 10.1002/ana.25248
Barcia, G., Fleming, M. R., Deligniere, A., Gazula, V. R., Brown, M. R., Langouet, M., et al. (2012). De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat. Genet. 44, 1255–1259. doi: 10.1038/ng.2441
Bedoyan, J. K., Kumar, R. A., Sudi, J., Silverstein, F., Ackley, T., Iyer, R. K., et al. (2010). Duplication 16p11.2 in a child with infantile seizure disorder. Am. J. Med. Genet. 152A, 1567–1574. doi: 10.1002/ajmg.a.33415
Berg, A. P., Sen, N., and Bayliss, D. A. (2007). TrpC3/C7 and Slo2.1 are molecular targets for metabotropic glutamate receptor signaling in rat striatal cholinergic interneurons. J. Neurosci. 27, 8845–8856. doi: 10.1523/JNEUROSCI.0551-07.2007
Bhattacharjee, A., Joiner, W. J., Wu, M., Yang, Y., Sigworth, F. J., and Kaczmarek, L. K. (2003). Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J. Neurosci. 23, 11681–11691. doi: 10.1523/JNEUROSCI.23-37-11681.2003
Bhattacharjee, A., von Hehn, C. A. A., Mei, X., and Kaczmarek, L. K. (2005). Localization of the Na+-activated K+ channel slick in the rat central nervous system. J. Comp. Neurol. 484, 80–92. doi: 10.1002/cne.20462
Budelli, G., Hage, T. A., Wei, A., Rojas, P., Jong, Y. J., O’Malley, K., et al. (2009). Na+-activated K+ channels express a large delayed outward current in neurons during normal physiology. Nat. Neurosci. 12, 745–750. doi: 10.1038/nn.2313
Carranza Rojo, D., Hamiwka, L., McMahon, J. M., Dibbens, L. M., Arsov, T., Suls, A., et al. (2011). De novo SCN1A mutations in migrating partial seizures of infancy. Neurology 77, 380–383. doi: 10.1212/WNL.0b013e318227046d
Chen, H., Kronengold, J., Yan, Y., Gazula, V.-R., Brown, M. R., Ma, L., et al. (2009). The N-terminal domain of slack determines the formation and trafficking of slick/slack heteromeric sodium-activated potassium channels. J. Neurosci. 29, 5654–5665. doi: 10.1523/JNEUROSCI.5978-08.2009
Coppola, G., Plouin, P., Chiron, C., Robain, O., and Dulac, O. (1995). Migrating partial seizures in infancy: a malignant disorder with developmental arrest. Epilepsia 36, 1017–1024. doi: 10.1111/j.1528-1157.1995.tb00961.x
Deutsch, C. (2002). Potassium channel ontogeny. Annu. Rev. Physiol. 64, 19–46. doi: 10.1146/annurev.physiol.64.081501.155934
Devaux, J., Abidi, A., Roubertie, A., Molinari, F., Becq, H., Lacoste, C., et al. (2016). A Kv7.2 mutation associated with early onset epileptic encephalopathy with suppression-burst enhances Kv7/M channel activity. Epilepsia 57, e87–e93. doi: 10.1111/epi.13366
El-Brolosy, M. A., Kontarakis, Z., Rossi, A., Kuenne, C., Günther, S., Fukuda, N., et al. (2019). Genetic compensation triggered by mutant mRNA degradation. Nature 568, 193–197. doi: 10.1038/s41586-019-1064-z
Etxeberria, A., Santana-Castro, I., Regalado, M. P., Aivar, P., and Villarroel, A. (2004). Three mechanisms underlie KCNQ2/3 heteromeric potassium M-channel potentiation. J. Neurosci. 24, 9146–9152. doi: 10.1523/JNEUROSCI.3194-04.2004
Evely, K. M., Pryce, K. D., and Bhattacharjee, A. (2017). The Phe932Ile mutation in KCNT1 channels associated with severe epilepsy, delayed myelination and leukoencephalopathy produces a loss-of-function channel phenotype. Neuroscience 351, 65–70. doi: 10.1016/j.neuroscience.2017.03.035
Fuller, Z. L., Berg, J. J., Mostafavi, H., Sella, G., and Przeworski, M. (2019). Measuring intolerance to mutation in human genetics. Nat. Genet. 51, 772–776. doi: 10.1038/s41588-019-0383-1
Garg, P., and Sanguinetti, M. C. (2014). Intracellular ATP does not inhibit Slo2.1 K+ channels. Physiol. Rep. 2:e12118. doi: 10.14814/phy2.12118
Gururaj, S., Palmer, E. E., Sheehan, G. D., Kandula, T., Macintosh, R., Ying, K., et al. (2017). A de novo mutation in the sodium-activated potassium channel KCNT2 alters ion selectivity and causes epileptic encephalopathy. Cell Rep. 21, 926–933. doi: 10.1016/j.celrep.2017.09.088
Hage, T. A., and Salkoff, L. (2012). Sodium-activated potassium channels are functionally coupled to persistent sodium currents. J. Neurosci. 32, 2714–2721. doi: 10.1523/JNEUROSCI.5088-11.2012
Howell, K. B., McMahon, J. M., Carvill, G. L., Tambunan, D., Mackay, M. T., Rodriguez-Casero, V., et al. (2015). SCN2A encephalopathy: a major cause of epilepsy of infancy with migrating focal seizures. Neurology 85, 958–966. doi: 10.1212/WNL.0000000000001926
Ishii, A., Shioda, M., Okumura, A., Kidokoro, H., Sakauchi, M., Shimada, S., et al. (2013). A recurrent KCNT1 mutation in two sporadic cases with malignant migrating partial seizures in infancy. Gene 531, 467–471. doi: 10.1016/j.gene.2013.08.096
Kaczmarek, L. K. (2013). Slack, slick, and sodium-activated potassium channels. ISRN Neurosci. 2013:354262. doi: 10.1155/2013/354262
Kaczmarek, L. K., Aldrich, R. W., Chandy, K. G., Grissmer, S., Wei, A. D., and Wulff, H. (2016). International union of basic and clinical pharmacology. C. Nomenclature and properties of calcium-activated and sodium-activated potassium channels. Pharmacol. Rev. 69, 1–11. doi: 10.1124/pr.116.012864
Kim, U., and McCormick, D. A. (1998). Functional and ionic properties of a slow afterhyperpolarization in ferret perigeniculate neurons in vitro. J. Neurophysiol. 80, 1222–1235. doi: 10.1152/jn.1998.80.3.1222
Klassen, T., Davis, C., Goldman, A., Burgess, D., Chen, T., Wheeler, D., et al. (2011). Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell 145, 1036–1048. doi: 10.1016/j.cell.2011.05.025
Kumar, P., Kumar, D., Jha, S. K., Jha, N. K., and Ambasta, R. K. (2016). Ion Channels in Neurological Disorders. 1st Edn. Elsevier Inc.
McTague, A., Appleton, R., Avula, S., Cross, H., King, M. D., Jacques, T. S., et al. (2013). Migrating partial seizures of infancy: expansion of the electroclinical, radiological and pathological disease spectrum. Brain 136, 1578–1591. doi: 10.1093/brain/awt073
Miceli, F., Soldovieri, M. V., Ambrosino, P., Barrese, V., Migliore, M., Cilio, M. R., et al. (2013). Genotype-phenotype correlations in neonatal epilepsies caused by mutations in the voltage sensor of Kv7.2 potassium channel subunits. Proc. Natl. Acad. Sci. U S A 110, 4386–4391. doi: 10.1073/pnas.1216867110
Miceli, F., Soldovieri, M. V., Ambrosino, P., De Maria, M., Migliore, M., Migliore, R., et al. (2015). Early-onset epileptic encephalopathy caused by gain-of-function mutations in the voltage sensor of Kv7.2 and Kv7.3 potassium channel subunits. J. Neurosci. 35, 3782–3793. doi: 10.1523/JNEUROSCI.4423-14.2015
Milh, M., Falace, A., Villeneuve, N., Vanni, N., Cacciagli, P., Assereto, S., et al. (2013). Novel compound heterozygous mutations in TBC1D24 cause familial malignant migrating partial seizures of infancy. Hum. Mutat. 34, 869–872. doi: 10.1002/humu.22318
Mulkey, S. B., Ben-Zeev, B., Nicolai, J., Carroll, J. L., Grønborg, S., Jiang, Y. H., et al. (2017). Neonatal nonepileptic myoclonus is a prominent clinical feature of KCNQ2 gain-of-function variants R201C and R201H. Epilepsia 58, 436–445. doi: 10.1111/epi.13676
Niday, Z., and Tzingounis, A. V. (2018). Potassium channel gain of function in epilepsy: an unresolved paradox. Neuroscientist 24, 368–380. doi: 10.1177/1073858418763752
Ohba, C., Kato, M., Takahashi, S., Lerman-Sagie, T., Lev, D., Terashima, H., et al. (2014). Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia 55, 994–1000. doi: 10.1111/epi.12668
Orhan, G., Bock, M., Schepers, D., Ilina, E. I., Reichel, S. N., Löffler, H., et al. (2014). Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann. Neurol. 75, 382–394. doi: 10.1002/ana.24080
Poduri, A., Chopra, S. S., Neilan, E. G., Elhosary, P. C., Kurian, M. A., Meyer, E., et al. (2012). Homozygous PLCB1 deletion associated with malignant migrating partial seizures in infancy. Epilepsia 53, e146–e150. doi: 10.1111/j.1528-1167.2012.03538.x
Poduri, A., Heinzen, E. L., Chitsazzadeh, V., Lasorsa, F. M., Elhosary, P. C., LaCoursiere, C. M., et al. (2013). SLC25A22 is a novel gene for migrating partial seizures in infancy. Ann. Neurol. 74, 873–882. doi: 10.1002/ana.23998
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rizzi, S., Knaus, H. G., and Schwarzer, C. (2016). Differential distribution of the sodium-activated potassium channels slick and slack in mouse brain. J. Comp. Neurol. 524, 2093–2116. doi: 10.1002/cne.23934
Rizzo, F., Ambrosino, P., Guacci, A., Chetta, M., Marchese, G., Rocco, T., et al. (2016). Characterization of two de novo KCNT1 mutations in children with malignant migrating partial seizures in infancy. Mol. Cell. Neurosci. 72, 54–63. doi: 10.1016/j.mcn.2016.01.004
Salkoff, L., Butler, A., Ferreira, G., Santi, C., and Wei, A. (2006). High-conductance potassium channels of the SLO family. Nat. Rev. Neurosci. 7, 921–931. doi: 10.1038/nrn1992
Santi, C. M., Ferreira, G., Yang, B., Gazula, V. R., Butler, A., Wei, A., et al. (2006). Opposite regulation of slick and slack K+ channels by neuromodulators. J. Neurosci. 26, 5059–5068. doi: 10.1523/JNEUROSCI.3372-05.2006
Stafstrom, C. E., Schwindt, P. C., Chubb, M. C., and Crill, W. E. (1985). Properties of persistent sodium conductance and calcium conductance of layer V neurons from cat sensorimotor cortex in vitro. J. Neurophysiol. 53, 153–170. doi: 10.1152/jn.1985.53.1.153
Stödberg, T., McTague, A., Ruiz, A. J., Hirata, H., Zhen, J., Long, P., et al. (2015). Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat. Commun. 6:8038. doi: 10.1038/ncomms9038
Tejada, M. A., Hashem, N., Calloe, K., and Klaerke, D. A. (2017). Heteromeric Slick/Slack K+ channels show graded sensitivity to cell volume changes. PLoS One 12:e0169914. doi: 10.1371/journal.pone.0169914
Thomson, S. J., Hansen, A., and Sanguinetti, M. C. (2015). Identification of the intracellular Na+ sensor in Slo2.1 potassium channels. J. Biol. Chem. 290, 14528–14535. doi: 10.1074/jbc.m115.653089
Vanderver, A., Simons, C., Schmidt, J. L., Pearl, P. L., Bloom, M., Lavenstein, B., et al. (2014). Identification of a novel de novo p.Phe932Ile KCNT1 mutation in a patient with leukoencephalopathy and severe epilepsy. Pediatr. Neurol. 50, 112–114. doi: 10.1016/j.pediatrneurol.2013.06.024
Villa, C., and Combi, R. (2016). Potassium channels and human epileptic phenotypes: an updated overview. Front. Cell. Neurosci. 10:81. doi: 10.3389/fncel.2016.00081
Keywords: epilepsy of infancy with migrating focal seizures, KNa channels, KCNT genes, epilepsy, encephalopathy
Citation: Mao X, Bruneau N, Gao Q, Becq H, Jia Z, Xi H, Shu L, Wang H, Szepetowski P and Aniksztejn L (2020) The Epilepsy of Infancy With Migrating Focal Seizures: Identification of de novo Mutations of the KCNT2 Gene That Exert Inhibitory Effects on the Corresponding Heteromeric KNa1.1/KNa1.2 Potassium Channel. Front. Cell. Neurosci. 14:1. doi: 10.3389/fncel.2020.00001
Received: 11 July 2019; Accepted: 06 January 2020;
Published: 24 January 2020.
Edited by:
Eleonora Palma, Sapienza University of Rome, ItalyReviewed by:
Amy McTague, University College London, United KingdomTerence Hébert, McGill University, Canada
Copyright © 2020 Mao, Bruneau, Gao, Becq, Jia, Xi, Shu, Wang, Szepetowski and Aniksztejn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Wang, d2FuZ2h1YV8yMTNAaG90bWFpbC5jb20=; Pierre Szepetowski, cGllcnJlLnN6ZXBldG93c2tpQGluc2VybS5mcg==; Laurent Aniksztejn, bGF1cmVudC5hbmlrc3p0ZWpuQGluc2VybS5mcg==
† These authors have contributed equally to this work