 Gabriele Di Pasquale1
Gabriele Di Pasquale1 Jacopo Colella1Carola P. Di Cataldo1
Jacopo Colella1Carola P. Di Cataldo1 Miguel A. Soler2Sara Fortuna3Emma Mizrahi-Powell4Mathilde Nizon5,6
Miguel A. Soler2Sara Fortuna3Emma Mizrahi-Powell4Mathilde Nizon5,6 Benjamin Cognè5,6Valentina Turchetti7Giuseppe D. Mangano8
Benjamin Cognè5,6Valentina Turchetti7Giuseppe D. Mangano8 Francesco F. Comisi9
Francesco F. Comisi9 Corrado Cecchetti10Alessandra Giliberti11
Corrado Cecchetti10Alessandra Giliberti11 Rosaria Nardello11Piero Pavone12
Rosaria Nardello11Piero Pavone12 Raffaele Falsaperla13Gabriella Di Rosa14Gilad D. Evrony4,15
Raffaele Falsaperla13Gabriella Di Rosa14Gilad D. Evrony4,15 Maurizio Delvecchio1
Maurizio Delvecchio1 Mariasavina Severino16Andrea Accogli17,18
Mariasavina Severino16Andrea Accogli17,18 Alessandro Vittori10*
Alessandro Vittori10* Vincenzo Salpietro1,19*
Vincenzo Salpietro1,19*- 1Department of Biotechnological and Applied Clinical Sciences, Academic Unit of Pediatrics, University of L'Aquila, L'Aquila, Italy
- 2Department of Mathematics, Computer Science and Physics, University of Udine, Udine, Italy
- 3Italian Institute of Technology (IIT), Genova, Italy
- 4Center for Human Genetics and Genomics, New York University Grossman School of Medicine, New York, NY, United States
- 5Nantes Université, CHU Nantes, Service de Génétique Médicale, Nantes, France
- 6Nantes Université, CNRS, INSERM, l'Institut du Thorax, Nantes, France
- 7Department of Neuromuscular Disorders, UCL Queen Square Institute of Neurology, London, United Kingdom
- 8Department of Medicine and Surgery, University of Enna Kore, Enna, Italy
- 9Pediatric Clinic and Rare Diseases, Microcitemico Hospital "A. Cao", University of Cagliari, Cagliari, Italy
- 10Department of Anesthesia, Critical Care and Pain Medicine, ARCO, Ospedale Pediatrico Bambino Gesù IRCCS, Rome, Italy
- 11Department of Health Promotion, Mother and Child Care, Internal Medicine and Medical Specialities "G. D'Alessandro", University of Palermo, Palermo, Italy
- 12Department of Pediatrics, University of Catania, Catania, Italy
- 13Department of Pediatrics, University of Ferrara, Ferrara, Italy
- 14Unit of Child Neurology and Psychiatry, Maternal-Infantile Department, University of Messina, Messina, Italy
- 15Department of Pediatrics, Department of Neuroscience and Physiology, Institute for Systems Genetics, Perlmutter Cancer Center and Neuroscience Institute, New York University Grossman School of Medicine, New York, NY, United States
- 16Neuroradiology Unit, IRCCS Istituto Giannina Gaslini, Genoa, Italy
- 17Division of Medical Genetics, Department of Medicine, McGill University Health Center, Montreal, QC, Canada
- 18Department of Human Genetics, McGill University, Montreal, QC, Canada
- 19European Brain Research Institute “Rita Levi-Montalcini” Viale Regina Elena, Rome, Italy
Introduction: Microtubules are essential components of the neuronal cytoskeleton. The α- and β-tubulins, variably expressed in the central nervous system, play key roles in neurogenesis and brain development. Pathogenic variants in TUBB2A have recently been identified as an ultra-rare cause of pediatric neurodevelopmental disorders (NDDs). However, the neurological and behavioral manifestations, genotype–phenotype correlations, and underlying disease mechanisms remain poorly understood due to the limited number of reported families.
Methods: We describe a cohort of families presenting with microcephaly, global developmental delay, speech impairment, seizures and/or EEG abnormalities, movement disorders and severe behavioral disorders. Clinical assessments and brain imaging studies were conducted over a 10-year follow-up period. Genetic analysis was performed via whole-exome sequencing (WES), and structural modeling was used to investigate the functional impact of the identified variants.
Results: WES revealed a novel recurrent heterozygous pathogenic variant in TUBB2A (NM_001069.3:c.1172G > A; NP_001060.1:p.Arg391His), identified as the cause of disease in multiple affected individuals from unrelated families. Comparative analysis with previously reported TUBB2A de novo variants confirmed that this novel recurrent mutation affects a highly conserved Arg391 residue within the longitudinal E-site heterodimer interface. Computational modeling demonstrated that the variant disrupts α/β-tubulin heterodimer formation, impairing binding stability at this critical interaction site.
Discussion: Our findings expand the phenotypic and genotypic spectrum of TUBB2A-related disorders and identify Arg391 as a mutational hotspot linked to severe brain developmental disorders due to aberrant tubulin dynamics, highlighting the disruption of the α/β-tubulin heterodimer formation as the disease mechanism associated to this novel hotspot variant. These results provide new insights into disease mechanisms and offer a foundation for potential future therapeutic approaches aimed at stabilizing α/β-tubulin interactions.
1 Introduction
Microtubules are critical structural and functional components of the neuronal cytoskeleton, orchestrating a wide array of processes essential for brain development. These dynamic polymers are composed of α- and β-tubulin heterodimers, which assemble into highly organized arrays that support neurogenesis, neuronal migration, differentiation, and the formation of synaptic architecture and connectivity (Janke and Magiera, 2020; Breuss et al., 2017). The functional diversity of microtubules is further modulated by the differential expression of tubulin isotypes and a range of post-translational modifications, which together fine-tune neuronal migration and intracellular dynamics, leading to the correct development of the brain (Moutin et al., 2021; Wloga et al., 2017; Tantry and Santhakumar, 2023). Proper microtubule function underpins essential developmental processes, including the proliferation of neural progenitors, guidance of immature neurons from periventricular zones to the cortical plate, stabilization of axonal projections, and the formation of synaptic networks (Tantry and Santhakumar, 2023; Kumar et al., 2010; Maillard et al., 2023). To date, at least ten α-tubulin isotypes (i.e., TUBA1A, TUBA1B, TUBA1C, TUBA4A, TUBA4B, TUBA3C, TUBA3D, TUBA3E, TUBA8, and TUBAL3) and seven β-tubulin isotypes (i.e., TUBB, TUBB2A, TUBB2B, TUBB3, TUBB4A, TUBB4B, and TUBB6) have been identified (Gonçalves et al., 2018; Park et al., 2021). Their interactions with key microtubule-associated proteins—including DCX, LIS1, DYNC1H1, FLNA, and Tau—are indispensable for cytoskeletal integrity and neuronal polarity.
Neurodevelopmental disorders (NDDs) comprise a heterogeneous group of frequently overlapping conditions, including developmental delay (DD), intellectual disability (ID), speech and motor impairments, behavioral dysregulation, and autism spectrum disorder (ASD) (Salpietro et al., 2019; de Masfrand et al., 2024; Kotchetkov et al., 2023). Despite the advent of next-generation sequencing (NGS), the molecular underpinnings of NDDs remain unresolved in a substantial proportion of patients (Salpietro et al., 2018; Iacomino et al., 2024). A deeper understanding of the genetic architecture and disrupted biological pathways is essential to improve diagnostic precision and inform targeted interventions.
In recent years, pathogenic variants in tubulin-encoding genes have emerged as causative factors in a broad spectrum of neurodevelopmental and neurological disorders. Given their fundamental roles in neuroanatomical organization, tubulin gene mutations have been strongly linked to malformations of cortical development (MCDs), defining a growing group of disorders collectively termed tubulinopathies (Severino et al., 2020; Bahi-Buisson et al., 2014; Romaniello et al., 2018). The resultant phenotypes reflect diverse pathogenic mechanisms, including impaired neuroblast proliferation, aberrant neuronal migration, defective axonal pathfinding, and disrupted synaptogenesis (Breuss et al., 2017; Bahi-Buisson et al., 2014; Oegema et al., 2015; Breuss et al., 2017). Clinically, tubulinopathies encompass a broad neurodevelopmental spectrum including microcephaly, variable neurodevelopmental impairment including motor delay, intellectual disability, speech difficulties and broad behavior abnormalities, as well as frequent movement disorders, stereotypies and epilepsy (Brock et al., 2021; Schmidt et al., 2021). Neuroimaging typically reveals characteristic abnormalities, such as polymicrogyria, lissencephaly-pachygyria, dysgyria, agenesis of the corpus callosum, basal ganglia dysmorphism, brainstem asymmetry, and cerebellar hypoplasia or dysplasia (Breuss et al., 2017; Patel et al., 2017; Cushion et al., 2014; Ejaz et al., 2017).
While variants in TUBA1A, TUBB2B, and TUBB3 account for the majority (>90%) of known tubulinopathy-associated mutations (Gonçalves et al., 2018; Bahi-Buisson et al., 2014), Pathogenic variants in other tubulin genes (including TUBB2A) are considerably rarer and less well characterized. Although initial reports suggested stereotyped phenotypes for mutations in specific tubulin genes, subsequent studies have challenged this notion, revealing marked inter- and intra-genic variability (Bahi-Buisson et al., 2014; Amrom et al., 2014; Zanni et al., 2013; Guerrini et al., 2012; Cushion et al., 2013).
TUBB2A encodes a β-tubulin isotype predominantly expressed in the developing brain, although its relative expression appears lower compared to other isoforms (Breuss et al., 2012). Pathogenic variants in TUBB2A are extremely rare and currently account for only a small fraction of tubulinopathy cases (Schmidt et al., 2021). To date, only 24 individuals have been reported with TUBB2A-related disease, showing wide variability in both neurodevelopmental trajectories and neuroimaging findings, ranging from near-normal cortical architecture to severe cortical malformations (Cushion et al., 2014; Ejaz et al., 2017; Brock et al., 2021; Schmidt et al., 2021; Cai et al., 2020; Sferra et al., 2018; Rodan et al., 2017; Lee et al., 2014).
Here, we expand the phenotypic and mutational spectrum of TUBB2A-associated tubulinopathy by presenting four additional individuals harboring a novel heterozygous missense variant (NM_001069.3:c.1172G > A; NP_001060.1:p.Arg391His). This variant affects a highly conserved residue in the C-terminal domain of β-tubulin and highlights a mutational hotspot with significant implications for microtubule dynamics and α/β-tubulin heterodimer formation, leading to disorders in brain development and function.
2 Materials and methods
2.1 Patient recruitment and clinical evaluation
Four individuals (from three unrelated families) harboring the same novel TUBB2A heterozygous missense variant (NM_001069.3:c.1172G > A; NP_001060.1:p.Arg391His) were enrolled in this study. Two were siblings (Individuals 1 and 2), followed longitudinally at an Italian tertiary care center, and two were unrelated patients (Individuals 3 and 4) identified via international collaborative networks among clinicians, clinical geneticists, researchers and neuroradiologists. Individual 4 was enrolled into the Pediatric Undiagnosed Diseases Program (UDP) at the NYU Grossman School of Medicine. Detailed medical histories, physical and neurological examinations, electroencephalograms (EEGs), and brain magnetic resonance imaging (MRI) were collected for all individuals participating in this study. Neurodevelopmental trajectories were assessed through a structured deep phenotyping protocol, including standardized neuropsychiatric evaluation and follow-up spanning multiple years. Peripheral blood samples were collected from affected individuals and their relatives for genetic testing. DNA extraction was performed using standard protocols at each participating center. Whole-exome sequencing (WES) was conducted to identify potential disease-causing variants. Written informed consent was obtained from legal guardians and the study and associated research protocols received approval from the Review Boards and Bioethics Committees at University College London Hospital (project06/N076).
2.2 Whole exome sequencing
Genomic DNA was extracted from peripheral blood samples of affected individuals and their parents. Whole exome sequencing (WES) was performed using the 3B-EXOME platform (3billion Inc., Seoul, South Korea) or similar platforms, which targets the protein-coding regions (exons) of the genome, as well as mitochondrial DNA (mtDNA). Sequencing for families 1 and 2 was carried out on the Illumina NovaSeq X platform with an average depth of coverage of ~100×, ensuring that >95% of targeted regions were covered at ≥20×. The exomes were analyzed using a pipeline designed to detect single nucleotide variants (SNVs), small insertions and deletions (INDELs), and copy number variants (CNVs). Family 3 underwent research reanalysis of clinical WES. Variants were annotated, filtered, and classified using ACMG guidelines. Confirmatory testing for candidate variants was performed using Sanger sequencing where applicable.
2.3 Literature review
A systematic literature review was conducted to compile all published cases of TUBB2A-associated neurodevelopmental disorders. Clinical, genetic, and radiological features from previously reported individuals were extracted and summarized in Table 1, allowing phenotypic comparison with the newly described cases.
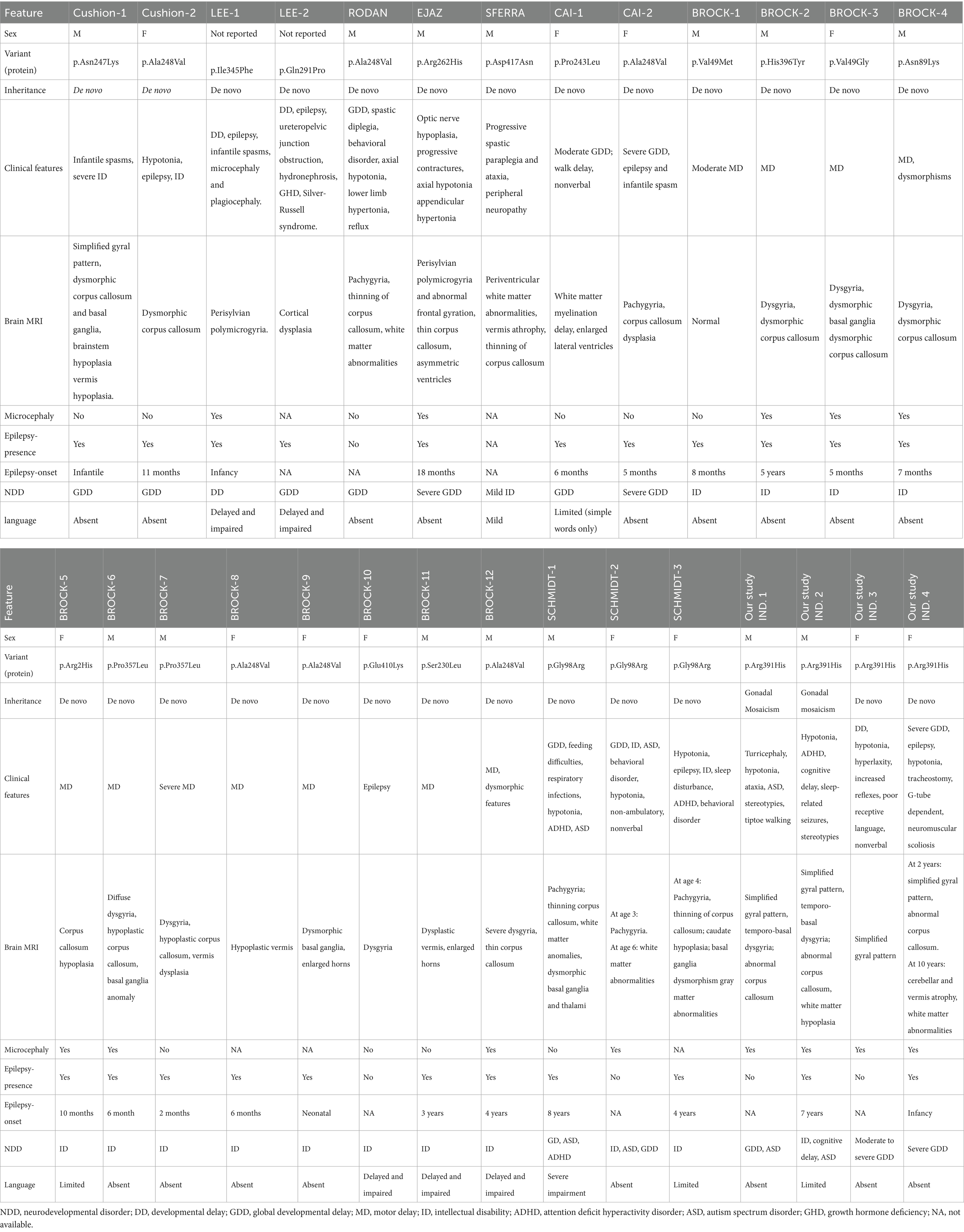
Table 1. Clinical, EEG and brain MRI features of TUBB2A-related neurodevelopmental disorders.
2.4 Computational studies
To assess evolutionary conservation of the TUBB2A protein and evaluate the potential functional relevance of the p.Arg391His variant, a multiple sequence alignment was performed using Clustal Omega.1
To explore the relevance of TUBB2A expression to neurodevelopmental and behavioral phenotypes observed in our cohort, we analyzed transcriptomic data from the Human Protein Atlas Brain Atlas.2 Normalized RNA expression levels (nTPM) across 13 major human brain regions were reviewed. This dataset provided insights into the spatial distribution of TUBB2A expression, supporting its functional relevance in brain regions implicated in the observed clinical features.
2.5 Molecular modeling of the Arg391His variant
To model the structural impact of the TUBB2A p.Arg391His variant, heterodimer complexes involving TUBB2A and three major α-tubulin isotypes (TUBA1A, TUBA1C, and TUBA4A) were constructed using cryo-EM structures from the Protein Data Bank (PDB IDs: 8ixa, 8ixd, and 8ixf) (Diao et al., 2023). The chains corresponding to the α/β-tubulin interface and the associated hydrolyzed GTP molecule were extracted to replicate the non-seam binding regions of the microtubule polymer.
The p.Arg391His mutation was introduced using UCSF Chimera, with the most probable rotamer selected to avoid steric clashes (Pettersen et al., 2004; Shapovalov and Dunbrack, 2011). The protonation state of His391 at physiological pH (7.0) was estimated using the PypKa server (Reis et al., 2020; Reis et al., 2024), which predicted a neutral imidazole ring across all heterodimeric models. Molecular dynamics simulations were prepared using the AMBER99SB force field and TIP3P water model. Ligand parameters were generated using Antechamber with BCC-derived GAFF/AMBER topologies (Maier et al., 2015). Systems were solvated in cubic boxes with a 1.5 nm water buffer and prepared using tleap. GROMACS-compatible topologies were obtained through ACPYPE conversion (Sousa da Silva and Vranken, 2012). Energy minimization was carried out in GROMACS v2021 using the steepest descent algorithm under periodic boundary conditions (Abraham et al., 2015). Electrostatics were computed using the Particle Mesh Ewald method until convergence at a maximum force threshold of 1000.0 kJ/mol/nm. Electrostatic binding free energies and electrostatic complementarity were evaluated using bluues_cplx (Soler et al., 2025), which employs Generalized Born radii (Bashford and Case, 2000; Onufriev and Case, 2019) derived from molecular surface integrals generated by Nanoshaper (Decherchi and Rocchia, 2013).
3 Results
3.1 Identification of a recurrent TUBB2A missense variant
WES of four individuals from three unrelated families (Figure 1) identified a shared heterozygous missense variant in TUBB2A (NM_001069.3:c.1172G > A; NP_001060.1:p.Arg391His). In the familial case (Individuals 1 and 2), the variant was absent in both parents, as confirmed by Sanger sequencing, suggesting the possibility of parental germline mosaicism. However, further investigations using additional tissues or sensitive mosaicism detection assays were not conducted, in accordance with the decision of Family 1.
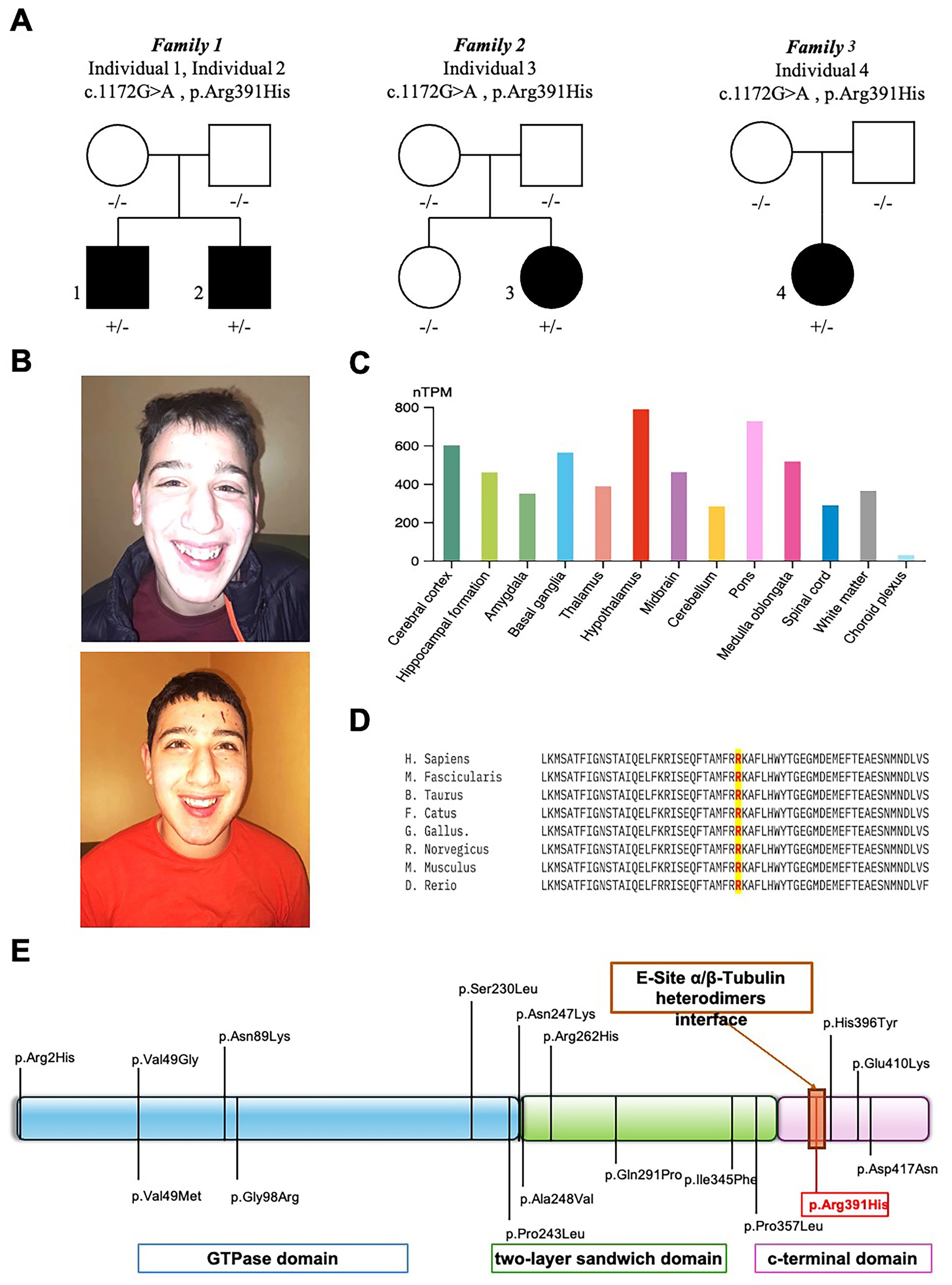
Figure 1. Clinical, molecular, and bioinformatic characterization of TUBB2A-related disorder. (A) Family pedigrees of affected individuals carrying the heterozygous TUBB2A c.1172G > A (p.Arg391His) variant. (B) Representative craniofacial features of TUBB2A-mutated individuals 1 (upper panel) and 2 (lower panel) showing microcephaly, bitemporal narrowing, and a pointed chin. (C) Normalized TUBB2A RNA expression levels (nTPM) across 13 human brain regions, based on data from the Human Protein Atlas. Each bar represents the subregion with the highest expression within its region; color coding corresponds to the respective brain region. Notably, the hypothalamus exhibited the highest expression levels. (D) Interspecies protein sequence alignment demonstrating complete conservation of the affected residue (Arg391) across vertebrate species, underscoring its evolutionary and functional importance. (E) Schematic diagram of the TUBB2A protein, illustrating previously reported pathogenic variants in relation to structural domains and highlighting the position of Arg391.
The same heterozygous p.Arg391His variant in TUBB2A was also detected in two additional unrelated individuals (Individuals 3 and 4 from Family 2 and 3, Figure 1A) and confirmed de novo in trio analyses. The identified hotspot variant p.Arg391His is absent from the gnomAD exome and genome databases3 and is predicted to be deleterious by multiple in silico tools, with a high CADD-Phred score of 32. This variant is also highly conserved, as demonstrated by interspecies alignment (Figure 1D) and a GERP++ score of 5.06.
No additional pathogenic or likely pathogenic variants were identified in other known disease-associated and/or NDD-associated genes.
3.2 Expression analysis and evolutionary conservation
Transcriptomic data from the Human Protein Atlas revealed TUBB2A expression across multiple brain regions, with the highest normalized RNA expression levels (nTPM) observed in the hypothalamus (Figure 1). Notably, TUBB2A was expressed in both cortical and subcortical regions, consistent with the widespread neurodevelopmental abnormalities observed in affected individuals. Interspecies protein alignment using Clustal Omega demonstrated that the affected Arg391 residue is fully conserved across vertebrate species, indicating strong evolutionary constraint and potential functional importance.
3.3 Structural modeling and functional impact of p.Arg391His
To investigate the structural consequences of the p.Arg391His substitution, we modeled three human α/β-tubulin heterodimers (TUBA1A–TUBB2A, TUBA1C–TUBB2A, and TUBA4A–TUBB2A) based on cryo-EM structures (PDBs: 8ixa, 8ixd, and 8ixf). In the wild-type state, Arg391 forms a conserved interaction network at the longitudinal E-site interface with α-tubulin residues Tyr262, Trp346, and Glu434, stabilizing the heterodimer complex.
Introduction of the Arg391His variant disrupted this interface across all three models. Electrostatic calculations of the interfaces revealed increased binding free energies in the mutant dimers compared to wild type, reflecting reduced heterodimer stability (Figure 2). Electrostatic complementarity, assessed via Pearson correlation of electrostatic potential surfaces, was consistently lower in mutant complexes, confirming loss of favorable electrostatic interactions. While minor gains in solvation energy were observed, these did not offset the destabilizing effects of the Arg391His substitution on α/β-tubulin dimerization.
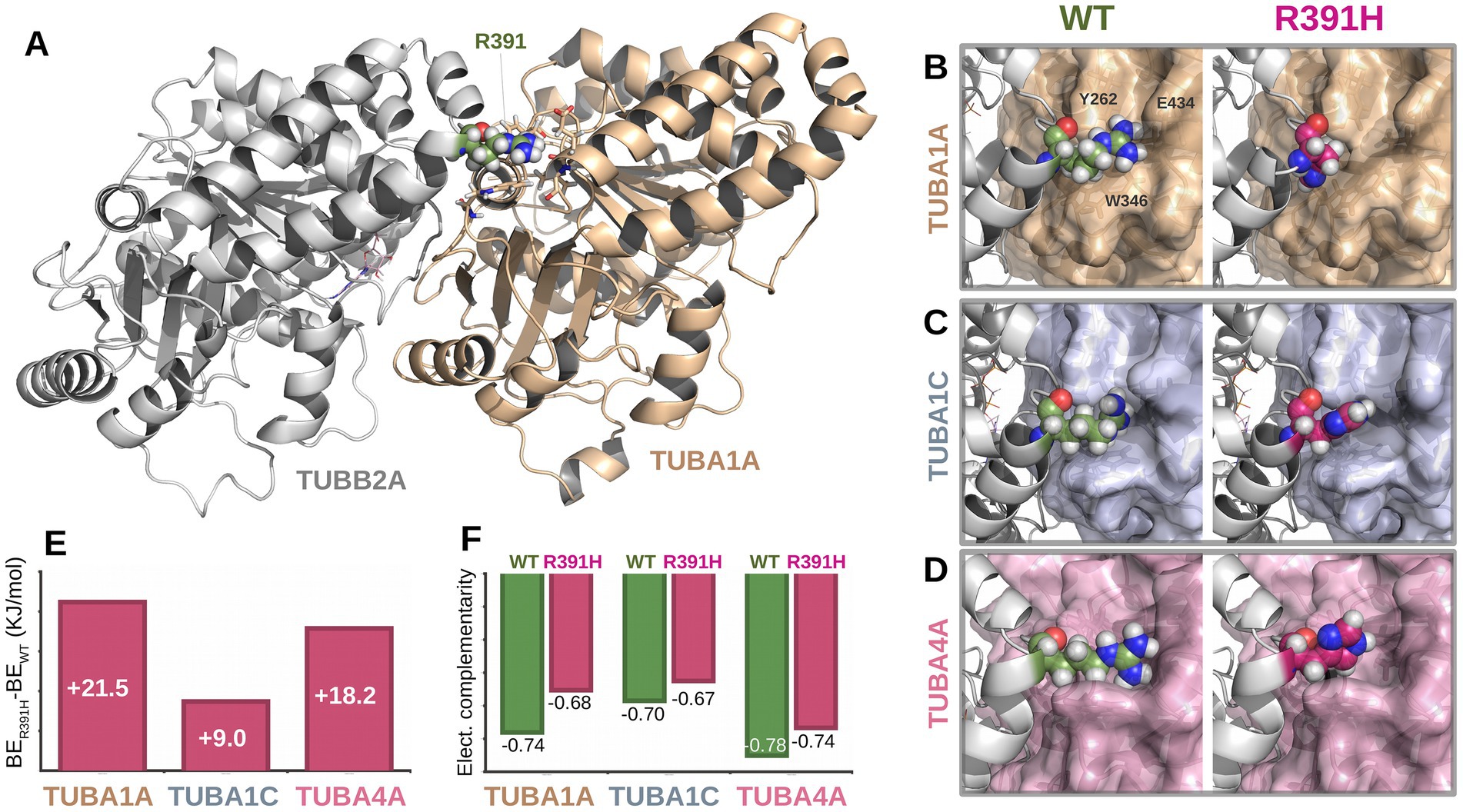
Figure 2. Structural and energetic impact of the TUBB2A p.Arg391His variant on α/β-tubulin heterodimerization. (A) Structural model of the TUBA1A–TUBB2A heterodimer showing the location of residue Arg391 (green) at the longitudinal E-site interface. (B–D) Close-up views of the Arg391 interface in wild-type (left) and mutant p. Arg391His (right) configurations across three α-tubulin isoforms: TUBA1A (B), TUBA1C (C), and TUBA4A (D). In the wild-type complex, Arg391 forms conserved interactions with Tyr262, Trp346, and Glu434 (shown for TUBA1A). These interactions are disrupted upon substitution with histidine. (E) Binding energy differences (ΔBE = BE_mut – BE_WT) showing increased free energy in all mutant complexes, indicating reduced binding affinity. (F) Electrostatic complementarity at the α/β interface, measured by Pearson correlation coefficients of electrostatic surface potentials. In all cases, mutant dimers (R391H) exhibit lower complementarity compared to their wild-type counterparts.
3.4 Clinical presentations
3.4.1 Individual 1
A 15-year-old boy, born at term following an uneventful pregnancy, exhibited postnatal microcephaly and turricephaly during early infancy. Early neurodevelopment was marked by global delay, reduced muscle tone, impaired primitive reflexes, and poor verbal output limited to non-purposeful syllables. By age 2 years, exaggerated deep tendon reflexes and ataxic gait emerged. Formal neuropsychiatric evaluation confirmed features consistent with ASD, including impaired communication and relational functioning. On his examination at our department at the age of 4 years, microcephaly was documented at -2SD and behavioral stereotypies, sensory-seeking behaviors, and tiptoe walking were also documented. ADOS-2 confirmed ASD diagnosis, and Risperidone was introduced with partial behavioral benefit. Follow-up at the age of 10 years revealed improved receptive language but persistent absence of expressive speech. Brain MRI at this age showed microcephaly with disproportionally large corpus callosum, simplified gyral pattern, bilateral temporo-basal dysgyria, and cerebellar foliar anomalies (Figure 3). Array-CGH identified a paternally inherited 16p13.11 duplication of uncertain clinical significance. WES revealed a novel TUBB2A variant (c.1172G > A; p.Arg391His). The variant was absent in the parents.
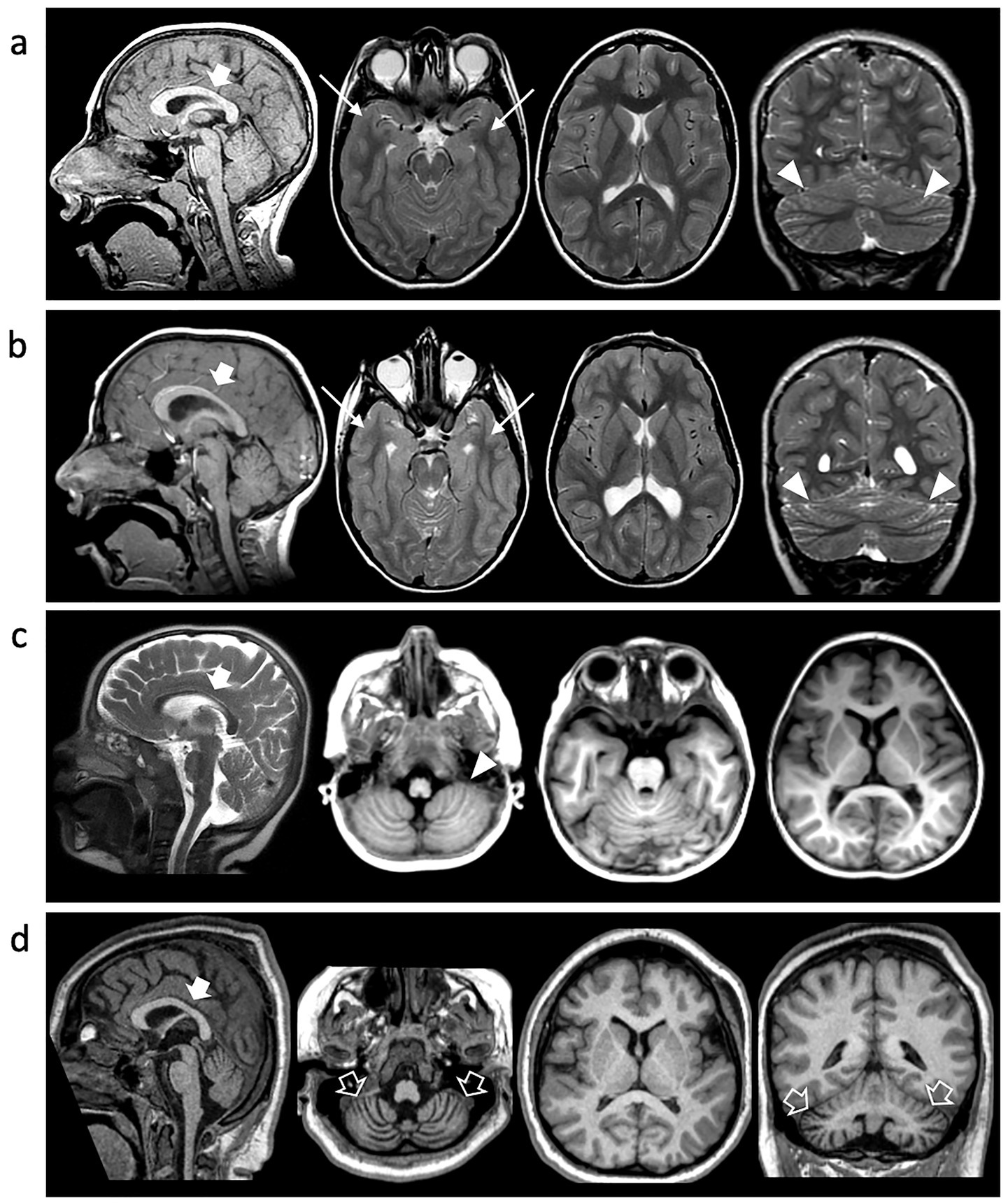
Figure 3. Neuroimaging findings. Brain MRI of Individual 1 at 10y (A), Individual 2 at 7 y (B) and Individual 4 at 2 y (C) and 10y (D). Sagittal T1 or T2-weighted images (first column), axial images (second and third columns), and coronal images (last column). Microcephaly with disproportionally large corpus callosum (thick arrows), simplified gyral pattern and cerebellar foliar anomalies are noted in all cases (arrowheads). There is bilateral temporo-basal dysgyria in Individual 1 and Individual 2 (arrows). Progressive cerebellar atrophy is detected in Individual 3 over the course of 8 years (empty arrows).
3.4.2 Individual 2
The younger brother of Individual 1, currently 12 years old, also presented with global developmental delay and hypotonia. Motor milestones were severely delayed; unsupported walking began at 20 months. Language development was significantly impaired, with first intelligible words spoken at 4 years. By age 6, he exhibited hyperactivity, motor stereotypies, poor comprehension, and limited verbal output. Diagnosed with moderate ASD and ID, he required behavioral management with Risperidone.
At age 7, he developed sleep-related seizures characterized by myoclonus and tonic posturing. Seizure control was achieved with Clobazam after failure of Valproic Acid. At 9 years, EEG showed mild anterior-predominant abnormalities. Brain MRI revealed microcephaly with disproportionally large corpus callosum, simplified gyral pattern, temporo-basal dysgyria, thinning of the periventricular white matter with symmetric ventriculomegaly, and cerebellar foliar anomalies (Figure 3). Genetic testing was negative by array-CGH. WES confirmed the same heterozygous TUBB2A p.Arg391His variant identified in his brother. The variant was absent in the parents.
3.4.3 Individual 3
Individual 3 is a 7-year-old female, born full-term with congenital hip dislocation, exhibited postnatal microcephaly (−1 SD at birth, progressing to −3.5 SD at 5 years). Gross motor milestones were profoundly delayed; independent walking was achieved at 6 years, although she primarily crawled or scooted. Examination revealed generalized hyperlaxity and increased reflexes without spasticity. Expressive language was absent, though receptive comprehension and social engagement (e.g., eye contact, smiling) were preserved. EEGs at 1 and 4 years were normal. MRI at age 1 revealed anterior-predominant simplified gyration without subcortical or white matter abnormalities. Array-CGH was normal. WES identified a de novo TUBB2A c.1172G > A (p.Arg391His) variant.
3.4.4 Individual 4
Individual 4 is a 14-year-old female born to unrelated parents. The patient has microcephaly and global developmental delay with progressive loss of motor milestones; she initially was able to sit without support and stand with support, but by the age of 4 was no longer able to sit unsupported. The patient is currently non-verbal and with limited communication. She developed refractory epilepsy with multiple seizure types and subsequently manifested hyperkinetic movement disorders including choreoathetosis. Respiratory compromise emerged at the age of 10 years, necessitating tracheostomy and G-tube feeding due to neuromuscular dysphagia. She also developed progressive (neuromuscular) scoliosis and is wheelchair-dependent requiring adaptive equipment. Neuroimaging at age 2 showed microcephaly with disproportionally large corpus callosum, simplified gyral pattern, and cerebellar foliar anomalies, and by age 10, marked cerebellar atrophy and periatrial white matter signal changes were evident. WES identified the same de novo TUBB2A variant (c.1172G > A; p.Arg391His).
4 Discussion
The assembly of α- and β-tubulin isotypes into microtubules is a fundamental process underlying neuronal development and connectivity. These microtubule networks mediate diverse cellular events, including neurogenesis, neuronal migration, axonal guidance, and the formation of both cortical and subcortical brain architecture (Bahi-Buisson et al., 2014; Cushion et al., 2013; Breuss et al., 2012). Perturbations in tubulin dynamics are increasingly recognized as the molecular basis of a broad and clinically heterogeneous group of neurodevelopmental disorders termed tubulinopathies (Bahi-Buisson et al., 2014; Romaniello et al., 2018). These disorders arise from heterozygous pathogenic variants in genes encoding α- or β-tubulins, and are commonly associated with a spectrum of malformations of cortical development (MCD), as well as anomalies affecting subcortical structures such as the basal ganglia, corpus callosum, and cerebellum (Bahi-Buisson et al., 2014; Cushion et al., 2014; Cushion et al., 2013; Brock et al., 2021; Schmidt et al., 2021).
In this study, we describe four individuals carrying a novel recurrent TUBB2A missense variant (NM_001069.3:c.1172G > A; NP_001060.1:p.Arg391His), expanding the mutational and phenotypic spectrum associated with TUBB2A-related tubulinopathy. Importantly, the recurrence of the p.Arg391His variant across unrelated families raises the possibility of a mutational hotspot or, alternatively, low-level germline mosaicism, which may underlie its repeated occurrence.
Interestingly, all subjects presented with microcephaly and a simplified gyral pattern at the time of presentation. Temporo-basal dysgyria was observed in two individuals, and cerebellar foliar anomalies were noted in three cases. The corpus callosum appeared disproportionately large relative to the degree of microcephaly. Notably, none of the patients exhibited the hallmark features of tubulinopathy, such as asymmetric ventriculomegaly and basal ganglia dysmorphisms. Moreover, dysgyria was noted only in two cases and was limited to the temporo-basal regions. One individual demonstrated a progressive phenotype, developing cerebellar atrophy over an eight-year period. Cerebellar atrophy is, in fact, a recognized feature in certain tubulinopathies, particularly those linked to mutations in the TUBB4A gene, such as Hypomyelination with Atrophy of the Basal Ganglia and Cerebellum.
We systematically compared these individuals with the 24 previously reported cases in the literature, resulting in a combined cohort of 28 patients (Table 1) (Cushion et al., 2013; Brock et al., 2021; Schmidt et al., 2021; Cai et al., 2020; Sferra et al., 2018; Rodan et al., 2017; Lee et al., 2014). Analysis of this cohort reveals a highly penetrant core phenotype characterized by neurodevelopmental delay or intellectual disability in all individuals (100%), predominantly of moderate-to-severe severity (89.2%, 25/28). Language impairment was universally present (100%), with complete absence of speech in 64.2% (18/28). Autism spectrum disorder (ASD) or ASD traits were noted in 39.2% (11/28), reinforcing the neuropsychiatric vulnerability associated with TUBB2A dysfunction.
Postnatal microcephaly was present in nearly half of the patients (46.4%, 13/28), underscoring the role of TUBB2A in early neurogenesis and brain volume regulation. Epilepsy was a prominent comorbidity, affecting 78.5% (22/28), with 63.6% of cases manifesting seizures before 12 months of age. Seizure onset varied widely, ranging from infancy to mid-childhood. Motor deficits were reported in 82.1% (23/28), including axial hypotonia (35.7%), spastic diplegia or paraplegia (7.1%), and marked delays in achieving motor milestones (75%). Dysmorphic facial features were present in 28.5% (8/28), and visual impairment in 14.2% (4/28), though these features were not uniformly observed. Neuroimaging findings further support the pathogenic role of TUBB2A variants. MRI abnormalities were present in all individuals, with 67.8% (19/28) exhibiting cortical malformations. Pachygyria (21.4%) and diffuse dysgyria (32.1%) were the most common cortical anomalies, while dysgenesis or thinning of the corpus callosum was observed in 64.2% (18/28). Additional structural changes included cerebellar vermis or hemispheric hypoplasia (21.4%), basal ganglia dysplasia (21.4%), white matter abnormalities (25%), and ventricular enlargement (10.7%). These imaging findings emphasize the widespread disruption of both cortical and subcortical development resulting from TUBB2A dysfunction.
Among our newly described individuals, two (Individuals 1 and 2) are siblings born to unaffected, non-consanguineous parents. Sanger sequencing confirmed the absence of the variant in both parents, indicating germline mosaicism as the likely mechanism of transmission although additional tests were nor performed based on parents’ decision. This is the first documented case of TUBB2A-related tubulinopathy due to gonadal mosaicism, contrasting with all previously published TUBB2A variants, which have arisen de novo. This finding underscores the importance of considering recurrence risk in familial settings, even when initial testing suggests de novo origin. The identified variant, p.Arg391His, affects a highly conserved residue within the C-terminal domain of β-tubulin, localized at the longitudinal E-site heterodimer interface—a critical surface for α/β-tubulin dimerization (Figure 1E). To assess the structural and energetic consequences of this mutation, we employed high-resolution molecular modeling based on cryo-electron microscopy structures of human α/β-tubulin heterodimers (PDB IDs: 8ixa, 8ixd, 8ixf for TUBA1A-TUBB2A, TUBA1C-TUBB2A, and TUBA4A-TUBB2A, respectively). In all three models, Arg391 of β-tubulin forms conserved electrostatic and hydrophobic interactions with Tyr262, Trp346, and Glu434 of the α-tubulin subunit (Figures 2A–2D), stabilizing the heterodimer interface. The introduction of the Arg391His substitution was found to disrupt this conserved interaction network. The imidazole side chain of histidine is unable to replicate the native arginine’s interaction profile, resulting in altered side-chain packing and reduced electrostatic complementarity. Binding free energy calculations revealed a consistent energetic penalty across all three heterodimer models, with higher binding free energies in mutant complexes, indicating weakened dimerization (Figure 2E). Further analysis of the electrostatic surface potential revealed loss of electrostatic complementarity in all mutant complexes, with reduced Pearson correlation coefficients relative to wild-type pairs (Figure 2F). While solvation energies were slightly more favorable in mutant dimers (data not shown), this minor gain did not compensate for the loss of stabilizing electrostatic interactions. These findings strongly support a destabilizing effect of the Arg391His mutation on α/β-tubulin heterodimer formation, which may underlie the observed neurodevelopmental phenotype by compromising microtubule integrity and dynamics during early brain development (Soler et al., 2025).
Taken together, the convergence of clinical, neuroimaging, genetic, and computational data reinforces the role of Arg391 as a mutational hotspot in TUBB2A, with key structural and functional implications for microtubule assembly.
This study provides a comprehensive clinical and molecular characterization of TUBB2A-related neurodevelopmental disorders, incorporating four newly described patients—including the first familial recurrence due to gonadal mosaicism—and a detailed comparison with previously reported cases. The identification of a novel pathogenic variant, p.Arg391His, at a highly conserved and structurally critical interface, provides important insights into genotype–phenotype relationships and disease mechanisms. Through integrative structural modeling, we demonstrate that Arg391 plays a key role in maintaining heterodimer stability across multiple α-tubulin isoforms. Its substitution by histidine disrupts a conserved binding network, weakening α/β-tubulin dimerization and, by extension, microtubule function. These findings add to growing evidence that TUBB2A mutations can produce a spectrum of tubulinopathy phenotypes through impaired microtubule assembly.
However, our study has certain limitations. The lack of functional in vivo evidence—such as experiments using cultured cells or patient-derived neuronal models—restricts our ability to fully evaluate the physiological relevance of the p.Arg391His variant in microtubule dynamics. Notably, Luscan and colleagues conducted insightful in vivo studies on the same residue (c.1172G > A [p.Arg391His]; c.1171C > T [p.Arg391Cys]) in TUBB4B, employing COS-7 cell transfection and patient fibroblast assays. Their findings demonstrated deleterious effects of these substitutions on microtubule repolymerization dynamics and growth rates (Luscan et al., 2017). Given the high structural homology among tubulin subunit genes, these results provide valuable context for interpreting our observations.
Future analogous studies should aim to elucidate the impact of TUBB2A mutations in neuronal models and to investigate whether therapeutic stabilization of dimer interfaces might offer a viable intervention strategy. The recurrent involvement of Arg391 positions this site as a candidate target for future precision medicine approaches, with implications for both diagnosis and therapy in tubulinopathy-related neurodevelopmental disorders.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://zenodo.org/records/15869597, 15869597.
Ethics statement
The studies involving humans were approved by Review Boards and Bioethics Committees at University College London Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s)/’s next of kin for the publication of any identifiable images or data included in this article.
Author contributions
GP: Conceptualization, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing, Data curation, Funding acquisition, Project administration, Resources, Software, Supervision, Validation, Visualization. JC: Conceptualization, Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing, Formal analysis, Funding acquisition, Project administration, Resources, Software, Supervision, Validation, Visualization. CaC: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization. MiS: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization. SF: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization. EM-P: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization. MN: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization. BC: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization. VT: Writing – original draft, Writing – review & editing. GM: Writing – original draft, Writing – review & editing. FC: Writing – original draft, Writing – review & editing. CoC: Writing – original draft, Writing – review & editing. AG: Writing – original draft, Writing – review & editing. RN: Writing – original draft, Writing – review & editing. PP: Writing – original draft, Writing – review & editing. RF: Writing – original draft, Writing – review & editing. GR: Writing – original draft, Writing – review & editing. GE: Writing – original draft, Writing – review & editing. MD: Writing – original draft, Writing – review & editing. MaS: Writing – original draft, Writing – review & editing. AA: Writing – original draft, Writing – review & editing. AV: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. VS: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported also by the Italian Ministry of Health with “Current Research funds.” MS contribution was supported by the Italian Ministry of Health (codice Ricerca finalizzata - project RF GR-2019-12370042).
Acknowledgments
The authors would like to thank the families for their enthusiastic participation in this research study. MiS and SF acknowledge ISCRA Award N. HP10CP2SN9, 2023, for granting the project access to the LEONARDO supercomputer, owned by the EuroHPC Joint Undertaking, hosted by CINECA (Italy). The New York University Undiagnosed Diseases Program (EM-P and GE) was supported by the Jacob Goldfield Foundation. MS contribution was supported by the Italian Ministry of Health (codice Ricerca finalizzata - project RF GR-2019-12370042).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor EB declared a shared affiliation with the authors CoC & AV at the time of review.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
1. ^https://www.ebi.ac.uk/jdispatcher/msa/clustalo
2. ^https://www.proteinatlas.org/ENSG00000137267-TUBB2A/brain
References
Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., et al. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25. doi: 10.1016/j.softx.2015.06.001
Amrom, D., Tanyalçin, I., Verhelst, H., Deconinck, N., Brouhard, G. J., Décarie, J. C., et al. (2014). Polymicrogyria with dysmorphic basal ganglia? Think tubulin! Clin. Genet. 85, 178–183. doi: 10.1111/cge.12141
Bahi-Buisson, N., Poirier, K., Fourniol, F., Saillour, Y., Valence, S., Lebrun, N., et al. (2014). The wide spectrum of tubulinopathies: what are the key features for the diagnosis? Brain 137, 1676–1700. doi: 10.1093/brain/awu082
Bashford, D., and Case, D. A. (2000). Generalized born models of macromolecular solvation effects. Annu. Rev. Phys. Chem. 51, 129–152. doi: 10.1146/annurev.physchem.51.1.129
Breuss, M., Heng, J. I. T., Poirier, K., Tian, G., Jaglin, X. H., Qu, Z., et al. (2012). Mutations in the β-tubulin gene TUBB5 cause microcephaly with structural brain abnormalities. Cell Rep. 2, 1554–1562. doi: 10.1016/j.celrep.2012.11.017
Breuss, M. W., Leca, I., Gstrein, T., Hansen, A. H., and Keays, D. A. (2017). Tubulins and brain development - the origins of functional specification. Mol. Cell. Neurosci. 84, 58–67. doi: 10.1016/j.mcn.2017.03.002
Breuss, M. W., Nguyen, T., Srivatsan, A., Leca, I., Tian, G., Fritz, T., et al. (2017). Uner Tan syndrome caused by a homozygous TUBB2B mutation affecting microtubule stability. Hum. Mol. Genet. 26, 258–269. doi: 10.1093/hmg/ddw383
Brock, S., Vanderhasselt, T., Vermaning, S., Keymolen, K., Régal, L., Romaniello, R., et al. (2021). Defining the phenotypical spectrum associated with variants in TUBB2A. J. Med. Genet. 58, 33–40. doi: 10.1136/jmedgenet-2019-106740
Cai, S., Li, J., Wu, Y., and De Jiang, Y. (2020). Novo mutations of TUBB2A cause infantile-onset epilepsy and developmental delay. J. Hum. Genet. 65, 601–608. doi: 10.1038/s10038-020-0739-5
Cushion, T. D., Dobyns, W. B., Mullins, J. G. L., Stoodley, N., Chung, S. K., Fry, A. E., et al. (2013). Overlapping cortical malformations and mutations in TUBB2B and TUBA1A. Brain 136, 536–548. doi: 10.1093/brain/aws338
Cushion, T. D., Paciorkowski, A. R., Pilz, D. T., Mullins, J. G. L., Seltzer, L. E., Marion, R. W., et al. (2014). De novo mutations in the beta-tubulin gene TUBB2A cause simplified gyral patterning and infantile-onset epilepsy. Am. J. Hum. Genet. 94, 634–641. doi: 10.1016/j.ajhg.2014.03.009
de Masfrand, S., Cogné, B., Nizon, M., Deb, W., Goldenberg, A., Lecoquierre, F., et al. (2024). Penetrance, variable expressivity and monogenic neurodevelopmental disorders. Eur. J. Med. Genet. 69:104932. doi: 10.1016/j.ejmg.2024.104932
Decherchi, S., and Rocchia, W. (2013). A general and robust ray-casting-based algorithm for triangulating surfaces at the nanoscale. PLoS One 8:e59744. doi: 10.1371/journal.pone.0059744
Diao, L., Zheng, W., Zhao, Q., Liu, M., Fu, Z., Zhang, X., et al. (2023). Cryo-EM of α-tubulin isotype-containing microtubules revealed a contracted structure of α4A/β2A microtubules. Acta Biochim. Biophys. Sin. Shanghai 55, 1551–1560. doi: 10.3724/abbs.2023130
Ejaz, R., Lionel, A. C., Blaser, S., Walker, S., Scherer, S. W., Babul-Hirji, R., et al. (2017). De novo pathogenic variant in TUBB2A presenting with arthrogryposis multiplex congenita, brain abnormalities, and severe developmental delay. Am. J. Med. Genet. A 173, 2725–2730. doi: 10.1002/ajmg.a.38352
Gonçalves, F. G., Freddi, T. A. L., Taranath, A., Lakshmanan, R., Goetti, R., Feltrin, F. S., et al. (2018). Tubulinopathies. Top. Magn. Reson. Imaging 27, 395–408. doi: 10.1097/RMR.0000000000000188
Guerrini, R., Mei, D., Cordelli, D. M., Pucatti, D., Franzoni, E., and Parrini, E. (2012). Symmetric polymicrogyria and pachygyria associated with TUBB2B gene mutations. Eur. J. Hum. Genet. 20, 995–998. doi: 10.1038/ejhg.2012.21
Iacomino, M., Houerbi, N., Fortuna, S., Howe, J., Li, S., Scorrano, G., et al. (2024). Allelic heterogeneity and abnormal vesicle recycling in PLAA-related neurodevelopmental disorders. Front. Mol. Neurosci. 17:1268013. doi: 10.3389/fnmol.2024.1268013
Janke, C., and Magiera, M. M. (2020). The tubulin code and its role in controlling microtubule properties and functions. Nat. Rev. Mol. Cell Biol. 21, 307–326. doi: 10.1038/s41580-020-0214-3
Kotchetkov, P., Blakeley, N., and Lacoste, B. (2023). Involvement of brain metabolism in neurodevelopmental disorders. Int. Rev. Neurobiol. 173, 67–113. doi: 10.1016/bs.irn.2023.08.004
Kumar, R. A., Pilz, D. T., Babatz, T. D., Cushion, T. D., Harvey, K., Topf, M., et al. (2010). TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum. Mol. Genet. 19, 2817–2827. doi: 10.1093/hmg/ddq182
Lee, H., Deignan, J. L., Dorrani, N., Strom, S. P., Kantarci, S., Quintero-Rivera, F., et al. (2014). Clinical exome sequencing for genetic identification of rare mendelian disorders. JAMA 312, 1880–1887. doi: 10.1001/jama.2014.14604
Luscan, R., Mechaussier, S., Paul, A., Tian, G., Gérard, X., Defoort-Dellhemmes, S., et al. (2017). Mutations in TUBB4B cause a distinctive sensorineural disease. Am. J. Hum. Genet. 101, 1006–1012. doi: 10.1016/j.ajhg.2017.10.010
Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., and Simmerling, C. (2015). ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713. doi: 10.1021/acs.jctc.5b00255
Maillard, C., Roux, C. J., Charbit-Henrion, F., Steffann, J., Laquerriere, A., Quazza, F., et al. (2023). Tubulin mutations in human neurodevelopmental disorders. Semin. Cell Dev. Biol. 137, 87–95. doi: 10.1016/j.semcdb.2022.07.009
Moutin, M.-J., Bosc, C., Peris, L., and Andrieux, A. (2021). Tubulin post-translational modifications control neuronal development and functions. Dev. Neurobiol. 81, 253–272. doi: 10.1002/dneu.22774
Oegema, R., Cushion, T. D., Phelps, I. G., Chung, S. K., Dempsey, J. C., Collins, S., et al. (2015). Recognizable cerebellar dysplasia associated with mutations in multiple tubulin genes. Hum. Mol. Genet. 24, 5313–5325. doi: 10.1093/hmg/ddv250
Onufriev, A. V., and Case, D. A. (2019). Generalized born implicit solvent models for biomolecules. Annu. Rev. Biophys. 48, 275–296. doi: 10.1146/annurev-biophys-052118-115325
Park, K., Hoff, K. J., Wethekam, L., Stence, N., Saenz, M., and Moore, J. K. (2021). Kinetically stabilizing mutations in Beta tubulins create isotype-specific brain malformations. Front. Cell Dev. Biol. 9:765992. doi: 10.3389/fcell.2021.765992
Patel, R. M., Liu, D., Gonzaga-Jauregui, C., Jhangiani, S., Lu, J. T., Sutton, V. R., et al. (2017). An exome sequencing study of moebius syndrome including atypical cases reveals an individual with CFEOM3A and a TUBB3 mutation. Cold Spring Harb Mol Case Stud 3:a000984. doi: 10.1101/mcs.a000984
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Reis, P. B. P. S., Clevert, D.-A., and Machuqueiro, M. (2024). PypKa server: online pKa predictions and biomolecular structure preparation with precomputed data from PDB and AlphaFold DB. Nucleic Acids Res. 52, W294–W298. doi: 10.1093/nar/gkae255
Reis, P. B. P. S., Vila-Viçosa, D., Rocchia, W., and Machuqueiro, M. (2020). PypKa: a flexible Python module for Poisson-Boltzmann-based pKa calculations. J. Chem. Inf. Model. 60, 4442–4448. doi: 10.1021/acs.jcim.0c00718
Rodan, L. H., el Achkar, C. M., Berry, G. T., Poduri, A., Prabhu, S. P., Yang, E., et al. (2017). De novo TUBB2A variant presenting with anterior temporal pachygyria. J. Child Neurol. 32, 127–131. doi: 10.1177/0883073816672998
Romaniello, R., Arrigoni, F., Fry, A. E., Bassi, M. T., Rees, M. I., Borgatti, R., et al. (2018). Tubulin genes and malformations of cortical development. Eur. J. Med. Genet. 61, 744–754. doi: 10.1016/j.ejmg.2018.07.012
Salpietro, V., Malintan, N. T., Llano-Rivas, I., Spaeth, C. G., Efthymiou, S., Striano, P., et al. (2019). Mutations in the neuronal vesicular SNARE VAMP2 affect synaptic membrane fusion and impair human neurodevelopment. Am. J. Hum. Genet. 104, 721–730. doi: 10.1016/j.ajhg.2019.02.016
Salpietro, V., Manole, A., Efthymiou, S., and Houlden, H. (2018). A review of copy number variants in inherited neuropathies. Curr. Genomics 19, 412–419. doi: 10.2174/1389202919666180330153316
Schmidt, L., Wain, K. E., Hajek, C., Estrada-Veras, J. I., Guillen Sacoto, M. J., Wentzensen, I. M., et al. (2021). Expanding the phenotype of TUBB2A-related Tubulinopathy: three cases of a novel, heterozygous TUBB2A pathogenic variant p.Gly98Arg. Mol Syndromol 12, 33–40. doi: 10.1159/000512160
Severino, M., Geraldo, A. F., Utz, N., Tortora, D., Pogledic, I., Klonowski, W., et al. (2020). Definitions and classification of malformations of cortical development: practical guidelines. Brain 143, 2874–2894. doi: 10.1093/brain/awaa174
Sferra, A., Fattori, F., Rizza, T., Flex, E., Bellacchio, E., Bruselles, A., et al. (2018). Defective kinesin binding of TUBB2A causes progressive spastic ataxia syndrome resembling sacsinopathy. Hum. Mol. Genet. 27, 1892–1904. doi: 10.1093/hmg/ddy096
Shapovalov, M. V., and Dunbrack, R. L. (2011). A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 19, 844–858. doi: 10.1016/j.str.2011.03.019
Soler, M. A., Yakout, R. B. A., Ozkilinc, O., Esposito, G., Rocchia, W., Klein, C., et al. (2025). Bluues_cplx: electrostatics at protein-protein and protein-ligand interfaces. Molecules 30:159. doi: 10.3390/molecules30010159
Sousa da Silva, A. W., and Vranken, W. F. (2012). ACPYPE - AnteChamber PYthon parser interfacE. BMC. Res. Notes 5:367. doi: 10.1186/1756-0500-5-367
Tantry, M. S. A., and Santhakumar, K. (2023). Insights on the role of α- and β-tubulin isotypes in early brain development. Mol. Neurobiol. 60, 3803–3823. doi: 10.1007/s12035-023-03302-1
Wloga, D., Joachimiak, E., and Fabczak, H. (2017). Tubulin post-translational modifications and microtubule dynamics. Int. J. Mol. Sci. 18:2207. doi: 10.3390/ijms18102207
Keywords: TUBB2A, neurodevelopmental disorders, tubulinopathies, intellectual disability, behavioral disorders, microcephaly, protein modeling
Citation: Di Pasquale G, Colella J, Di Cataldo CP, Soler MA, Fortuna S, Mizrahi-Powell E, Nizon M, Cognè B, Turchetti V, Mangano GD, Comisi FF, Cecchetti C, Giliberti A, Nardello R, Pavone P, Falsaperla R, Di Rosa G, Evrony GD, Delvecchio M, Severino M, Accogli A, Vittori A and Salpietro V (2025) A mutational hotspot in TUBB2A associated with impaired heterodimer formation and severe brain developmental disorders. Front. Cell. Neurosci. 19:1664953. doi: 10.3389/fncel.2025.1664953
Edited by:
Enrico Bertini, Ospedale Pediatrico Bambino Gesu’, IRCCS, ItalyReviewed by:
Filippo M. Santorelli, Stella Maris Foundation (IRCCS), ItalyFrancesca Bartolini, Columbia University, United States
Copyright © 2025 Di Pasquale, Colella, Di Cataldo, Soler, Fortuna, Mizrahi-Powell, Nizon, Cognè, Turchetti, Mangano, Comisi, Cecchetti, Giliberti, Nardello, Pavone, Falsaperla, Di Rosa, Evrony, Delvecchio, Severino, Accogli, Vittori and Salpietro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandro Vittori, YWxleHZpdHRvcmk4MkBnbWFpbC5jb20=; Vincenzo Salpietro, di5zYWxwaWV0cm9AZWJyaS5pdA==