 Guang Yang
Guang Yang Xiao Xu1†
Xiao Xu1† Yan Zhao
Yan Zhao Ying Xu
Ying Xu- 1Department of Physiology, School of Integrative Medicine, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 2Shanghai Institute of Traditional Chinese Medicine for Mental Health, Shanghai, China
Microglia, the innate immune cells of the central nervous system (CNS), play essential roles in maintaining neural homeostasis through dynamic interactions with neurons and other brain structures. While their protective functions are well-established, recent studies have illuminated the detrimental consequences of sustained microglial activation in the context of neurodegeneration. In particular, overactivated microglia contribute to neuroinflammation and induce synaptic alterations through the release of pro-inflammatory cytokines and engagement of specific receptors. These interactions disrupt synaptic structure and function, compromising connectivity, plasticity, and cognitive processes. Notably, neuronal synapses are primary targets of such inflammation-driven dysfunction, where prolonged exposure to cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α), and signaling via receptor systems including cluster of differentiation-200 (CD200)/CD200 receptor (CD200R), C-X3-C motif chemokine ligand 1 (CX3CL1)/CX3C receptor 1 (CX3CR1), colony-stimulating factor 1 (CSF1)/CSF1 receptor (CSF1R), and interferon-γ (IFN-γ)/IFN-γ receptor (IFN-γR), lead to impaired learning, excitotoxicity, and neurodegenerative progression. This review synthesizes emerging evidence on the mechanisms by which microglia-mediated immune responses regulate synaptic remodeling, emphasizing the roles of pro-inflammatory cytokines and their receptors in neurodegenerative disorders.
1 Introduction
Microglia, the resident immune cells of the brain, originate from primitive c-kit(+) erythromyeloid precursors in the yolk sac and migrate into the brain parenchyma during early development. This population is self-sustaining under normal conditions, with peripheral macrophages contributing only in pathological states when the blood–brain barrier is compromised (Ginhoux et al., 2010). Beyond their well-established immunological functions in maintaining central nervous system (CNS) homeostasis, microglia have gained recognition for their critical involvement in synaptic modification, both during neural development and in the pathogenesis of neurodegenerative diseases.
Microglia typically display two phenotypic states: “resting” and “activated.” The term “resting” is somewhat misleading, as these cells are highly dynamic even under physiological conditions. Characterized by small somata and extensively branched processes, resting microglia continuously survey the CNS microenvironment and interact with surrounding neural elements, particularly neurons and synapses. Through their motile processes, they engage in synaptic remodeling, support CNS repair, and respond to immune challenges arising from peripheral insults (Figure 1). In pathological contexts, such as Alzheimer’s disease (AD) (Hopp et al., 2018), Parkinson’s disease (PD) (Rabaneda-Lombarte et al., 2021), amyotrophic lateral sclerosis (ALS) (Cox et al., 2022), multiple sclerosis (MS) (Cignarella et al., 2020), stroke (He et al., 2019), and chronic pain (Zhou et al., 2019), microglia undergo activation in response to injury or inflammatory stimuli. This transformation encompasses a series of biological processes, including local proliferation, morphological changes, migration, antigen presentation, phagocytosis, and the release of a wide array of signaling molecules (Kaur et al., 2019).
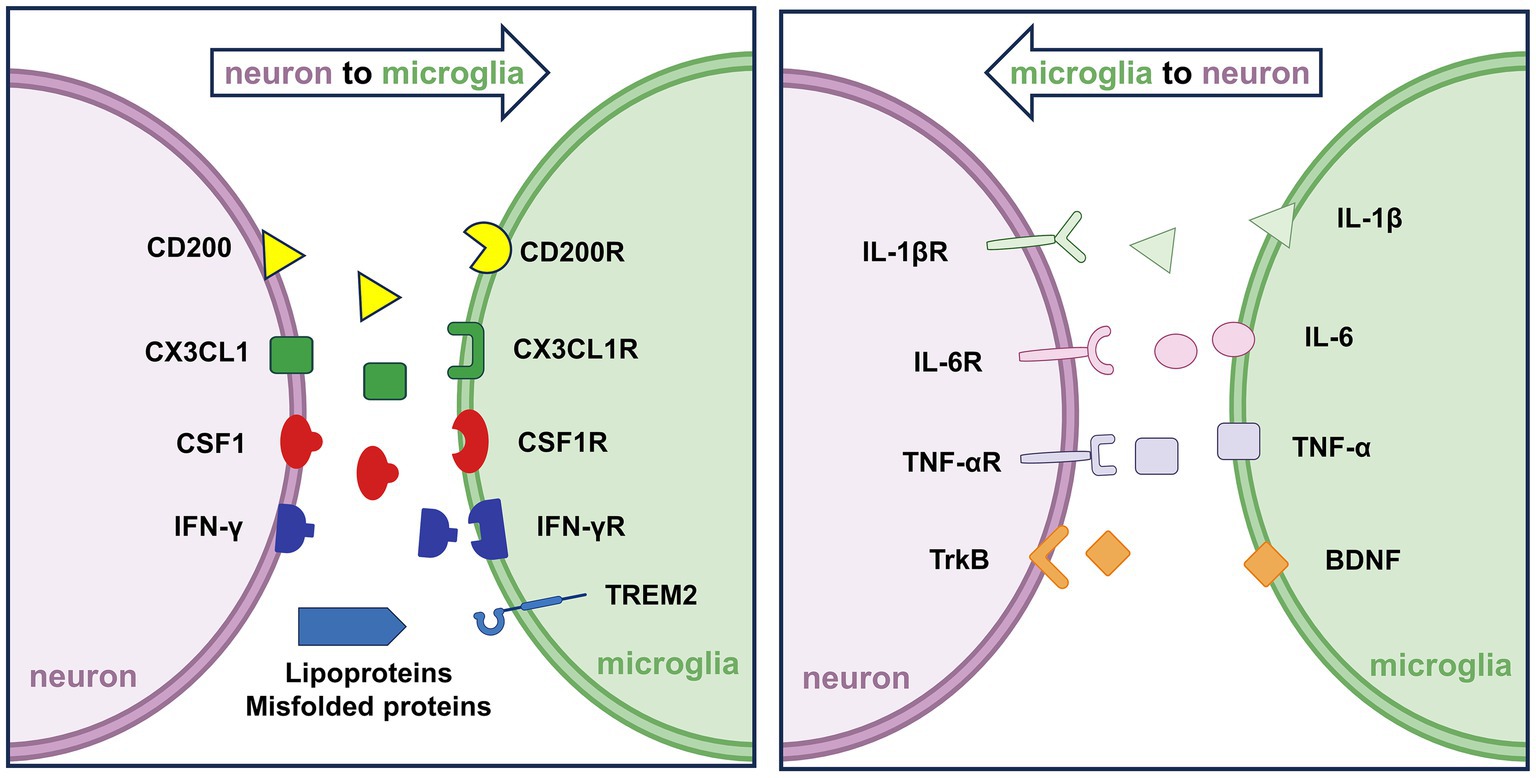
Figure 1. Neuron–microglia interactions under physiological conditions. (Left) Neurons secrete signaling molecules such as CX3CL1, CSF1, and IFN-γ, which bind to their respective microglial receptors, CX3CL1R, CSF1R, and IFN-γR, to regulate microglial surveillance and activation. Additionally, neuronal CD200 interacts with microglial CD200R to maintain microglial quiescence. (Right) Activated microglia release pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α, which bind to neuronal receptors IL-1βR, IL-6R, and TNF-αR, respectively. These bidirectional interactions coordinate immune homeostasis and contribute to the maintenance of neuronal and synaptic integrity.
Activated microglia may exert either protective or deleterious effects, depending on the nature and duration of the stimuli. In the early stages of CNS injury, they clear debris, protein aggregates, and apoptotic cells, while releasing chemokines, cytokines, and neurotrophic factors that support immune regulation and tissue repair. However, persistent microglial activation can contribute to neuropathology. Chronically active microglia release pro-inflammatory cytokines and engage receptor-mediated signaling cascades that disrupt synaptic function and plasticity. Notably, elevated levels of interleukin-1β (IL-1β), interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), cluster of differentiation-200 (CD200)/CD200 receptor (CD200R), C-X3-C motif chemokine ligans 1 (CX3CL1)/CX3C receptor 1 (CX3CR1), colony-stimulating factor 1 (CSF1)/CSF1 receptor (CSF1R), and interferon-γ (IFN-γ)/IFN-γ receptor 1 (IFNGR1)-IFN-γ receptor 2 (IFNGR2) have been implicated in neuroinflammation (Rabaneda-Lombarte et al., 2021; Yang et al., 2023; van der Maten et al., 2017; Yang et al., 2018; Panagiotakopoulou et al., 2020). Such dysregulated signaling interferes with key processes like dendritic spine remodeling (Huang et al., 2021), synaptic phagocytosis (Wang et al., 2021), and neurotransmission (Yang et al., 2018), ultimately contributing to excitotoxicity and the progression of neurodegeneration.
Despite increasing research interest, the precise mechanisms through which microglial cytokines and receptors influence synaptic structures remain incompletely understood, and controversy persists regarding the dualistic role of microglia in disease. This review highlights recent advances in our understanding of overactive microglia and their contribution to neuroinflammation-driven synaptic dysfunction in neurodegenerative disorders. In particular, we focus on how pro-inflammatory cytokines and their receptors orchestrate microglia-mediated signaling networks that influence synaptic structure and function under pathological conditions. By synthesizing emerging mechanistic insights, this review aims to elucidate the complex interplay between microglial signaling and synaptic pathology in neurodegeneration.
2 Microglia-mediated immune responses regulate synaptic modification
As the principal immune cells of the CNS, microglia are fundamental to maintaining brain homeostasis under physiological conditions. Rather than fitting into binary activation states, recent high-dimensional transcriptomic and functional studies reveal that microglia exist along a dynamic and context-dependent continuum of states closely shaped by their microenvironment (Paolicelli et al., 2022). This updated framework better captures the remarkable heterogeneity and plasticity of microglia in both health and disease. This raises a critical question: how do activated microglia contribute to neuronal damage, particularly following CNS injury? While the mechanisms are multifactorial and not yet fully defined, recent research highlights the intricate crosstalk between microglial immune responses, neuroinflammation, and synaptic function.
Microglia and immune signaling pathways interact dynamically with synaptic structures under both normal and pathological conditions. Neuroinflammation, often referred to as a “double-edged sword,” plays a central role in the development and progression of neurodegenerative diseases. On one hand, it serves to eliminate harmful agents such as pathogens or cellular debris; on the other, it contributes to cytotoxicity and accelerates neuronal damage. Synaptic alterations are not only hallmark features of neurodegenerative diseases but also critical drivers of disease progression (Overk and Masliah, 2014). Inflammatory stimuli following neural injury can modulate synaptic efficacy and plasticity, leading to either long-term depression or potentiation depending on the molecular context (Stefano et al., 2025). Notably, chronic CNS inflammation is associated with sustained production of deleterious pro-inflammatory mediators that impair synaptic plasticity, ultimately contributing to maladaptive forms of synaptic remodeling (Cianciulli et al., 2022).
Under physiological conditions, microglia actively participate in the formation, elimination, and functional modulation of synapses. While some synaptic alterations are cell-autonomous, mounting evidence points to non-cell-autonomous mechanisms wherein microglia exert regulatory control over synaptic architecture and function. A key mechanism involves immune-related molecules, including cytokines and their receptors, that mediate communication between activated microglia and synaptic elements (Wake et al., 2019). These molecules are rapidly induced in response to disease, trauma, or infection and drive synaptic structural remodeling. In turn, altered synaptic states can further activate microglia, thereby creating a feed-forward loop that reinforces inflammation and disrupts synaptic homeostasis. This bidirectional interaction forms a mechanistic link between microglial overactivation and synaptic dysfunction.
The following sections examine current evidence that supports the contribution of microglia to inflammation-related synaptic dysregulation in neurodegenerative disorders.
3 Pro-inflammatory cytokines from activated microglia alter synaptic modification
Neuroinflammation plays a pivotal role in the pathogenesis of neurodegenerative disorders. Elevated levels of pro-inflammatory cytokines, particularly IL-1β, IL-6, and TNF-α, have been observed in cognitive-related brain regions such as the hippocampus, underscoring their relevance in modulating synaptic structure and function (Yang et al., 2018). Upon encountering immune challenges, microglia become activated and secrete these cytokines, thereby contributing to synaptic dysregulation and the progression of neurodegenerative diseases (Figure 2). This section reviews the specific roles of pro-inflammatory cytokines in mediating synaptic modification in pathological conditions, emphasizing their mechanistic involvement in neuronal plasticity and degeneration.
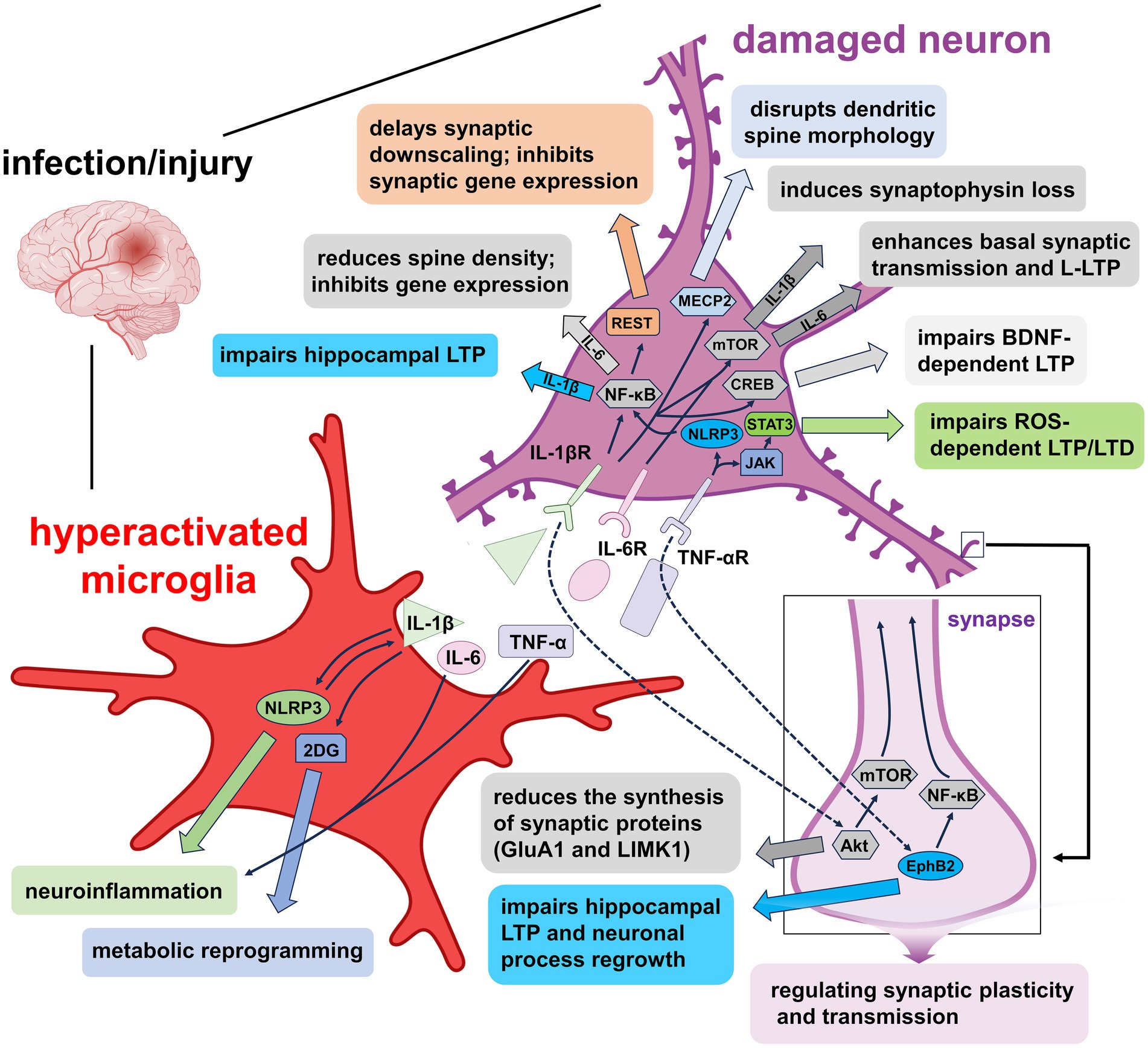
Figure 2. Mechanisms of pro-inflammatory factors-induced synaptic plasticity impairment under pathological conditions. Overactivated microglia release IL-1β, IL-6, and TNF-α, each disrupting synaptic function via distinct signaling pathways. IL-1β activates NLRP3 inflammasomes and the 2-DG metabolic pathway, promoting inflammation and metabolic reprogramming that further enhance microglial activation. Binding of IL-1β to IL-1βR engages signaling cascades such as NF-κB, NF-κB/REST, MECP2, mTOR, and CREB, leading to impaired synaptic downscaling, altered dendritic spine morphology, hippocampal LTP suppression, and reduced gene expression. Additionally, IL-1β disrupts synaptic protein synthesis through the Akt/mTOR pathway. IL-6 signaling via IL-6R facilitates basal synaptic transmission and L-LTP through mTOR activation. TNF-α, via TNF-αR, impairs hippocampal LTP and LTD through NLRP3/NF-κB and JAK/STAT3 pathways, while also affecting LTP and neuronal regeneration via the EphB2/NF-κB axis. Collectively, these cytokine-mediated mechanisms contribute to synaptic dysfunction and neurodegeneration by disrupting neuronal and microglial signaling homeostasis.
3.1 IL-1β
IL-1β, a key pro-inflammatory cytokine generated via the proteolytic cleavage of pro-IL-1β, is central to both immune signaling and synaptic modulation. Under physiological conditions, IL-1β contributes to normal cognitive function and neural development by modulating synaptic plasticity, promoting memory consolidation, and regulating activity-dependent synaptic scaling (Yirmiya and Goshen, 2011). Activated microglia release IL-1β, which profoundly influences synaptic plasticity, including Hebbian plasticity and synaptic scaling (Stampanoni Bassi et al., 2020; Buffolo et al., 2021). Its elevated expression in the cortex, striatum, and hippocampus is closely associated with cognitive deficits (Yang et al., 2018; Zhao et al., 2019; Hoshino et al., 2021; Lin et al., 2019), prompting intensive investigations into its functional roles in neuroplasticity.
Impairment of long-term potentiation (LTP) by IL-1β has been widely documented as a mechanism underlying memory deficits. Rodent studies demonstrate that increased IL-1β levels suppress hippocampal LTP and impair learning and memory (Rizzo et al., 2018). Intracerebral injection of lipopolysaccharide (LPS) elevates IL-1β expression and disrupts LTP in the CA3-CA1 Schaffer collaterals (Golia et al., 2019; Yang et al., 2019; Li et al., 2024). Similar inhibitory effects are seen when IL-1β is applied to hippocampal slices (Hoshino et al., 2017; York et al., 2021) or isolated synaptosomes (Prieto et al., 2019). These deficits can be reversed by IL-1 receptor antagonists such as IL-1RA or anakinra (Hoshino et al., 2021; York et al., 2021; Prieto et al., 2019; Tong et al., 2018; Guo et al., 2020; Han et al., 2016). In delirium models, a negative correlation has been established between IL-1β levels and both LTP and hippocampus-dependent memory (Tanaka et al., 2018).
Mechanistically, IL-1β modulates neuroplasticity through multiple pathways. It activates immune signaling cascades such as the NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome (Yu et al., 2023), impairs excitatory synapses in sepsis-associated encephalopathy (SAE) models (Moraes et al., 2022), and alters LTP via synaptic hyperexcitability in MS (Stampanoni Bassi et al., 2020). IL-1β interferes with N-methyl-D-aspartate receptor (NMDAR)-mediated calcium signaling, selectively impairing NMDAR-dependent LTP (Rizzo et al., 2018; Hoshino et al., 2017), and enhances Ca2+ influx in dorsal root ganglion neurons, contributing to pain processing (Noh et al., 2019). Anakinra has been shown to restore NMDAR-dependent hippocampal plasticity and reduce seizure susceptibility in prion disease models (Bertani et al., 2017).
IL-1β also influences synaptic scaling through RE1-silencing transcription factor (REST) activation, which delays synaptic downscaling at excitatory synapses (Buffolo et al., 2021). Additionally, IL-1β-mediated metabolic reprogramming of microglia impairs LTP, an effect reversible by glycolytic inhibition with 2-deoxyglucose (2DG) (York et al., 2021). In AD models, mitochondrial dynamics are disrupted by IL-1β through the activation of NF-κB and NLRP3 signaling pathways, resulting in Drp1-dependent fragmentation, excessive ROS production, and ATP depletion (Batista et al., 2021). It has been shown that these alterations impair synaptic energy metabolism and hippocampal LTP, thereby linking IL-1β-mediated mitochondrial dysfunction to cognitive decline (Batista et al., 2021). These findings suggest that IL-1β mediated bioenergetic imbalance represents a crucial pathogenic pathway in the progression of neurodegenerative diseases.
At the structural level, IL-1β impairs dendritic spine formation, alters spine morphology, and induces synaptic loss. These effects involve suppression of mechanistic target of rapamycin (mTOR) signaling and downregulation of key synaptic proteins such as GluA1 and LIMK1 (Prieto et al., 2019; Tong et al., 2018), activation of MECP2 and nuclear factor κB (NF-κB) pathways (Tomasoni et al., 2017; Fan et al., 2018), and inhibition of axonal growth via p38-MAPK (Han et al., 2017). IL-1β also promotes synaptic displacement through microglial engulfment mechanisms (You et al., 2024) and reduces synaptic protein expression via the PI3K/Akt/mTOR axis (Sheppard et al., 2019; Xiao et al., 2017). It induces tau hyperphosphorylation, which disrupts synaptic integrity (Ising et al., 2019; Jiang et al., 2021), and differentially modulates excitatory and inhibitory neurotransmission (Patel et al., 2019; Yan et al., 2019; Sung et al., 2017). Moreover, IL-1β-driven dysfunction involves the miR-142-3p/GLAST axis, underscoring the regulatory role of microglia in synaptic remodeling (Mandolesi et al., 2017).
Beyond direct synaptic effects, IL-1β interferes with BDNF signaling. It disrupts BDNF-Akt/CREB pathways, reducing LTP and contributing to synaptic degeneration (Li et al., 2024; Dong et al., 2018; Carlos et al., 2017). Knockdown of IL-1β restores inflammation-induced downregulation of BDNF and VGF (Li et al., 2017). Interestingly, BDNF can also stimulate IL-1β and TNF-α release from glial cells, creating a feedback loop that amplifies neuroinflammation (Ding et al., 2020). Depending on context, IL-1β-induced microglial activation may either exacerbate or mitigate neuronal injury by modulating BDNF release (Sung et al., 2017; Todd et al., 2019; Abd-El-Basset et al., 2020). Inhibition of IL-1β alleviates cognitive deficits in neuroinflammatory models (Taoro-Gonzalez et al., 2019). Collectively, these findings reveal IL-1β’s dual role in mediating microglial activation and neuroplasticity.
3.2 IL-6
IL-6, a pleiotropic pro-inflammatory cytokine primarily secreted by activated microglia and astrocytes, exerts multifaceted effects on various CNS cell types through both membrane-bound IL-6 receptor (IL-6Rα) and soluble IL-6 receptor (sIL-6R) pathways (Gruol et al., 2023). Beyond its pro-inflammatory roles, IL-6 also contributes to normal CNS physiology by modulating neuronal survival, synaptic function, and cognitive processes essential for brain development (Erta et al., 2012). Both IL-6 and its receptors are expressed in the hippocampus and play critical roles in CNS pathophysiology (Lin et al., 2019; Li et al., 2016; Willis et al., 2020; Onufriev et al., 2017). Under physiological conditions, IL-6 is present at low levels; however, its expression markedly increases during infections, injuries, and neurodegenerative or psychiatric disorders, implicating IL-6 in the regulation of hippocampus-dependent synaptic plasticity and memory.
Experimental evidence supports deleterious effects on synaptic function. Neonatal pro-inflammatory stress leads to impaired LTP and elevated IL-6 in rat hippocampal slices (Onufriev et al., 2017), while developmental IL-6 elevation disrupts connectivity through excessive excitatory synaptogenesis (Mirabella et al., 2021). In vivo studies further confirm that LPS-induced IL-6 elevation alters synaptic structure in the hippocampus (Lin et al., 2019), and IL-6 knockout in mice confers protection against age-related cognitive decline (Bialuk et al., 2020). In hippocampal slices from mice, IL-6 impairs LTP without affecting baseline synaptic excitability, an effect also observed in cerebrospinal fluid of relapsing–remitting multiple sclerosis (RR-MS) patients following paired associative stimulation (PAS) (Stampanoni Bassi et al., 2019).
IL-6 signaling is also implicated in amyloid-beta (Aβ) and tau pathologies. In AD mouse models (Tg2576, 3xTg-AD mice), inhibition of IL-6 trans-signaling reduces Aβ burden in both cortex and hippocampus (Escrig et al., 2019). Tau phosphorylation discussed in Section 3.1, is another key event influenced by IL-6. Chronic alcohol exposure and sevoflurane anesthesia elevate IL-6 levels, leading to tau hyperphosphorylation, mitochondrial dysfunction, and synaptic loss marked by reduced levels of postsynaptic density protein 95 (PSD-95), synaptophysin, N-cadherin, and total synapse number (Jiang et al., 2021; Zhang et al., 2020). These changes are accompanied by increased reactive oxygen species (ROS), decreased mitochondrial membrane potential (MMP), and reduced ATP levels, suggesting a mitochondrial pathway of IL-6-mediated neurotoxicity.
Microglial processing and release of tau may further drive IL-6 elevation and reactivation of microglia (Hopp et al., 2018), establishing a pathological loop. ROS-mediated synaptic pruning, although essential for physiological development, can become maladaptive under excessive mitochondrial ROS conditions, resulting in aberrant synapse elimination (Cobley, 2018; Sidlauskaite et al., 2018). IL-6 exacerbates this process by inducing mitochondrial dysfunction and oxidative stress, thereby contributing to synaptic degradation (Omoyinmi et al., 2016; Liu et al., 2017). These findings highlight the potential of IL-6-mitochondria-ROS interactions as therapeutic targets in neurodegenerative diseases.
In addition to structural changes, IL-6 modulates synaptic excitability and transmission, predominantly via trans-signaling (Rothaug et al., 2016). In genetically modified mice (GFAP-sgp130Fc, TG), inhibition of IL-6 trans-signaling enhances seizure susceptibility and increases synaptic excitability, evidenced by changes in paired-pulse ratios and prolonged seizures after pentylenetetrazole (PTZ) challenge (Cuevas-Olguin et al., 2017). IL-6 signaling also alters plasticity in a context-dependent manner: in social defeat models, it promotes resilience-associated plasticity but suppresses excitability in stress-susceptible animals (Esquivel-Rendón et al., 2019). Electrophysiological data from IL-6-overexpressing transgenic mice (IL-6 tg) demonstrate enhanced basal synaptic transmission and long-lasting LTP (L-LTP), yet rapamycin, a mTOR inhibitor, attenuates these effects, indicating a negative regulatory role of chronic IL-6 via mTOR signaling (Olde Engberink et al., 2017).
Behavioral outcomes also link IL-6 to synaptic dysfunction. Blocking IL-6 trans-signaling improves sociability in BTBR T+ Itpr3tf (BTBR) autism model mice, likely through enhanced cortical glutamate release (Wei et al., 2016), while cerebral IL-6 infusion impairs NMDA receptor-mediated synaptic currents and learning (Wang et al., 2019). Furthermore, IL-6 elevation contributes to depressive-like behaviors through reduced neuronal excitability and impaired GABAergic transmission (Roberts et al., 2019). In Fragile X syndrome (FXS) models, aberrant synapse formation and increased excitatory synapses are associated with elevated IL-6, emphasizing its role in excitatory/inhibitory imbalance (Krasovska and Doering, 2018).
Importantly, IL-6 may exert protective effects under specific pathological conditions (Stefano et al., 2025). In traumatic brain injury (TBI) models, IL-6 administration improves active place avoidance (APA) performance and promotes neurogenesis, suggesting a context-dependent neuroprotective role (Willis et al., 2020). Similarly, IL-6 produced by repopulated microglia aids in neuronal repair. These findings underscore the dual nature of IL-6 in the CNS and highlight the need for a nuanced understanding of its functions in neuroinflammation and synaptic modulation.
3.3 TNF-α
TNF-α is a pivotal pro-inflammatory cytokine present in both transmembrane (tmTNF-α) and soluble (sTNF-α) forms, the latter generated via TNF-α-converting enzyme (TACE). Its biological effects are primarily mediated through two receptors: TNFR1 and TNFR2. In the CNS, microglia serve as the principal source of TNF-α, though astrocytes and neurons also contribute. Moreover, microglial TNF-α has been shown to play a physiological role in regulating synaptic plasticity and supporting normal cognitive function under non-pathological conditions (Lewitus et al., 2016). Elevated TNF-α levels have been reported in various neurodegenerative and cognitive disorders, correlating with impaired hippocampal LTP, neuronal excitotoxicity, microglial activation, and neuroinflammation (Yang et al., 2019; Mashhadizadeh et al., 2017; Xu et al., 2019; Yang et al., 2021). Experimental studies demonstrate that exogenous TNF-α application reduces LTP in hippocampal slices and synaptosomes, while TNF-α neutralization or inhibition of microglial activation restores LTP and synaptic function (Prieto et al., 2019; Yang et al., 2021; Zhou and Bickler, 2017; Figueiredo et al., 2019). Furthermore, TNF-α mediates Aβ-induced LTP suppression via TNFR and downstream IKK/NF-κB signaling, as evidenced in TREM2R47H knock-in (KI) models where excessive TNF-α disrupted synaptic transmission and promoted Aβ production (Samidurai et al., 2018; Ren et al., 2020b).
TNF-α exhibits concentration-dependent bidirectional effects on synaptic plasticity: low concentrations facilitate synaptic potentiation, whereas elevated levels suppress it (Kleidonas et al., 2023). It preferentially targets excitatory synapses over inhibitory ones and regulates homeostatic synaptic scaling (Rizzo et al., 2018; Kleidonas and Vlachos, 2021). Studies in the mouse visual cortex revealed that TNF-α modulates dendritic spine size in a spatially restricted manner, aligning with synaptic scaling mechanisms. However, spine loss occurred independently of TNF-α, suggesting a role for Hebbian-like plasticity (Barnes et al., 2017). Collectively, these findings establish TNF-α as a crucial immune regulator of both Hebbian and homeostatic synaptic modifications.
Beyond classical plasticity, TNF-α also acts as a metaplasticity modulator, wherein previous synaptic activity shapes future plastic responses. This has implications for neurodegenerative pathologies. At Schaffer collateral-CA1 synapses, TNF-α was shown to modulate LTP in a concentration-dependent manner through mechanisms involving intracellular calcium stores and the actin-associated protein synaptopodin (Maggio and Vlachos, 2018). In APP/PS1 AD models, aberrant TNF-α release triggered pathological metaplasticity, impairing synaptic strengthening (Singh et al., 2019), further highlighting TNF-α’s central role in regulating synaptic adaptability.
TNF-α is also implicated in microglia-mediated synaptic pruning and structural remodeling. In primary cortical neurons, TNF-α (10 ng/mL) reduced neurite outgrowth and synaptic protein colocalization-effects reversed by tricyclic antidepressants (O'Neill et al., 2016). Interestingly, in human neuronal cultures under HIV-1-induced stress, TNF-α promoted neurite regrowth through Ephrin receptor B2 (EphB2)/NF-κB signaling, suggesting context-dependent duality in its actions (Pozniak et al., 2016). Additionally, TNF-α has been associated with neuropathic pain through modulation of synaptic strength and LTP. Intrathecal TNF-α mimicked vincristine-induced LTP at C-fiber synapses via upregulation of NR2B-containing NMDA receptors, while microglial inhibition or TNFR1 deletion attenuated TNF-α-driven synaptic changes and pain behaviors (Wigerblad et al., 2017; Xu et al., 2017; Liu et al., 2017). These data underscore the pathogenic synergy between TNF-α and microglia in both pain and synaptic dysfunction.
During neurodevelopment, TNF-α-related mechanisms also shape synaptic refinement. In Spinocerebellar Ataxia Type 1 (SCA1), NF-κB inhibition in Purkinje cells exacerbated motor deficits by interfering with microglial proliferation and TNF-α production, emphasizing the nuanced role of TNF-α signaling in neuronal circuitry formation (Ferro et al., 2018). Poly(I:C)-induced neuroinflammation further supports this, where increased TNF-α disrupted dendritic spine remodeling and perineuronal net (PNN) integrity, impairing synaptic and electrophysiological properties in hippocampal neurons via microglia-derived factors (Garre et al., 2017; Wegrzyn et al., 2021). In the auditory system, TNF-α-induced synaptopathy was reversible with etanercept, implicating TNF-α in broader sensory network dysfunctions (Hassan et al., 2017; Katsumi et al., 2019).
Emerging studies reveal that TNF-α facilitates pericyte-microglia interactions through NF-κB and tyrosine kinase (JAK)/STAT3 signaling, enhancing IL-6 release and sustaining neuroinflammation (Matsumoto et al., 2018). At the neuromuscular junction, TNF-α serves as a synaptotoxic regulator of synapse elimination (Fu et al., 2020). Moreover, in amyloid-based models of AD, TNF-α elevation promotes glutamatergic hyperactivity and the excitatory/inhibitory (E/I) imbalance in hippocampal circuits, with TNF-α blockade by XPro1595 preventing early synaptic deficits (Cavanagh et al., 2016) Similar hyperexcitability and LTP impairment were observed in TREM2R47H KI rats, linking microglial dysfunction to TNF-α-mediated synaptic alterations (Ren et al., 2020a). Cocaine exposure and exogenous TNF-α infusion further support its role in glutamatergic transmission dysregulation and excitotoxicity in limbic regions (Wang et al., 2019; Lewitus et al., 2016).
Collectively, these studies establish TNF-α as a multifaceted modulator of synaptic plasticity and structure across development, health, and disease. Its involvement in synaptic transmission, pruning, metaplasticity, and neuroimmune interactions underscores its central role in neuroinflammatory-driven synaptic pathologies.
4 Receptors on activated microglia modulate synaptic modification
Microglia express a broad array of cytokine and chemokine receptors, enabling dynamic communication with surrounding neurons and glial cells beyond their traditional immune functions. Among these, cytokine/cytokine receptor signaling has emerged as a key modulator of synaptic structure and plasticity, linking immune activation to neuronal dysfunction. This section focuses on the role of microglia-expressed receptors, particularly under pathological conditions, in disrupting synaptic modification and contributing to neurodegenerative progression.
4.1 CD200/CD200R
CD200, a type I transmembrane glycoprotein, and its receptor CD200R are critical for maintaining microglial quiescence and immune homeostasis in the CNS. CD200 is primarily expressed on neurons, including in somas, axons, dendrites, and synapses, while CD200R is predominantly localized on microglia, with CD200 receptor 1 (CD200R1) showing the highest binding affinity among its receptor family members (Manich et al., 2019; Yang et al., 2023). CD200/CD200R signaling serves as an inhibitory axis that restrains microglial activation and suppresses excessive inflammatory responses. Multiple studies have demonstrated that activation of this pathway through the administration of CD200 fusion protein (CD200Fc), a CD200R1 agonist, alleviates neuroinflammatory responses and limits microglial activation in various disease models (Rabaneda-Lombarte et al., 2021; Wang et al., 2020; Hernangomez et al., 2016; Jiang et al., 2016). For instance, CD200Fc ameliorated Aβ-induced LTP deficits in the CA1 region of mouse hippocampal slices (Ngwa and Liu, 2019), while inhibition of CD200R impaired the anti-inflammatory phenotype of microglia under excitotoxic conditions (Yi et al., 2016). Clinically, downregulation of CD200R1 has been observed in the cerebrospinal fluid of patients with AD and delirium (Peters van Ton et al., 2020), and neuronal CD200 expression negatively correlates with hallmark AD pathologies including neurofibrillary tangles and amyloid plaques (Walker et al., 2017).
Functionally, CD200/CD200R signaling contributes to synaptic integrity by modulating microglial activity. CD200 engagement suppresses ATP release and microglial activation via ATP-sensitive potassium (KATP) channel opening, thereby reducing inflammatory cytokine secretion and protecting neurons (Ren et al., 2016). Notably, Wang et al. (2020) and Yi et al. (2016) reported that CD200R1 expression was significantly reduced in microglia following systemic LPS and α-synuclein challenge. CD200 knockout exacerbated microglial activation in the midbrain, leading to dopaminergic neuron loss in the substantia nigra, whereas CD200Fc administration reversed these changes. Interestingly, despite LPS inducing a nearly tenfold increase in cytokine release compared to α-synuclein, CD200R1 downregulation was comparable, CD200/CD200R1 signaling may influence microglia–neuron interactions beyond inflammation. Reduced CD200R1 expression in Parkinson’s models has been associated with microglial overactivation and synaptic dysfunction, supporting its role in maintaining synaptic stability and preventing maladaptive remodeling (Wang et al., 2020).
Supporting its synaptic role, CD200 has been identified in both pre- and post-synaptic compartments of excitatory synapses. In CD200-deficient mice, a significant reduction in synapse number was observed in the visual thalamus, implying a role in synaptic refinement (Loh et al., 2016). In a depression-like rat model, reduced CD200R expression in microglia was associated with aberrant synaptic plasticity (Wang et al., 2017). In APP/PS1 transgenic mice, hippocampal overexpression of CD200 via APP/PS1 mice, intrahippocampal injection of CD200 (AAV2/9-syn-CD200-mCherry) improved cognitive performance, restored synaptic function, and prevented synaptic loss by limiting microglial activation and secretion. Conversely, CD200 deletion exacerbated synaptic dysfunction and cognitive impairment (Feng et al., 2019). Similar findings were observed in a stroke model, where intracerebroventricular injection of CD200Fc restored synaptic integrity and motor function post- middle cerebral artery occlusion (MCAO), while CD200R antibody blockade worsened these outcomes (Sun et al., 2020). In a kainic acid-induced epilepsy model, upregulation of microglial CD200R by minocycline restored synaptic protein levels and dendritic morphology in the hippocampus, underscoring the receptor’s role in structural plasticity (Yang et al., 2023).
Interestingly, CD200 deficiency may also alter microglial phagocytic function. In Aβ-challenged CD200 knockout mice, enhanced phagocytosis and lysosomal activity reduced Aβ accumulation despite elevated microglial activation (Lyons et al., 2017). This paradoxical finding suggests a context-dependent role for CD200/CD200R signaling, wherein suppression of inflammatory activation must be balanced against potential impairments in Aβ clearance. Together, these studies establish the CD200/CD200R axis as a critical regulatory mechanism at the intersection of neuroinflammation and synaptic modification.
4.2 CX3CL1/CX3CR1
CX3CL1, also known as fractalkine (FKN), is a unique chemokine predominantly expressed by neurons and inducible in astrocytes under pro-inflammatory conditions (Luo et al., 2019). It exists in both membrane-bound and soluble forms, both of which are biologically active and signal through CX3CR1, a receptor selectively expressed on microglia in the CNS (Luo et al., 2019). The CX3CL1-CX3CR1 axis represents a critical molecular interface for neuron–microglia communication, modulating microglial activation and synaptic function in health and disease.
Recent evidence highlights the multifaceted role of this signaling pathway in regulating neuroinflammatory responses and synaptic remodeling (Wang et al., 2023). For instance, reduced CX3CL1 levels have been observed in schizophrenia, accompanied by strong correlations between CX3CR1 expression and presynaptic protein SNAP-25 (Hill et al., 2020). In the auditory system, CX3CR1 mutations alter synaptic organization, particularly in regions involved in complex sensory processing (Milinkeviciute et al., 2021). In PD models, CX3CR1 is downregulated at early stages and is sensitive to LPS or α-synuclein in primary microglia, indicating that inflammatory triggers dynamically regulate its expression (Wang et al., 2020). In the hippocampus, CX3CR1 maintains normal glutamatergic transmission; its disruption leads to impaired synaptic function and abnormal synapse morphology (Basilico et al., 2021).
Ischemic brain injury models further demonstrate that CX3CL1 downregulation promotes microglial-mediated inflammation and neuronal autophagy (Zhu et al., 2019). In parallel, CX3CR1 modulates microglial morphology during this process, indicating its importance in inflammatory microglial phenotypes (van der Maten et al., 2017). In AD models (APP/PS1), mesenchymal stem cells (MSCs) overexpressing CX3CL1 reduced TNF-α levels and upregulated synaptic proteins, though cognitive benefits were observed only when CX3CL1 was co-expressed with Wnt3a (Li et al., 2020). Membrane-bound CX3CL1 also appears to maintain microglia in a surveying, non-reactive state. CX3CR1-deficient (CX3CR1−/−) mice exhibit excessive microglial activation, increased extracellular matrix deposition, and impaired synaptic integration in dentate gyrus neurons, along with reductions in dendritic spine density, axonal terminal area, and overall synaptic function. Behaviorally, female CX3CR1−/− mice display hyperactivity, reduced anxiety, and depressive-like behaviors (Bolós et al., 2018).
Further studies confirm that CX3CR1 is essential for activity-dependent synaptic plasticity. In the motor cortex, its absence disrupts learning and synaptic remodeling under direct current stimulation (Gellner et al., 2023). In the mouse barrel cortex, CX3CR1/CX3CL1/ADAM10 signaling mediates synaptic engulfment by microglia during whisker deprivation, indicating its involvement in experience-dependent synaptic elimination (Gunner et al., 2019). Similarly, the pathway contributes to synapse pruning and the maintenance of newly integrated neurons (Reshef et al., 2017), supporting its fundamental role in homeostatic synaptic regulation.
However, CX3CL1/CX3CR1 signaling can exert context-dependent and even opposing effects on synaptic plasticity. In AD models, CX3CR1 deletion enhances Aβ clearance in a gene dose-dependent manner (Merino et al., 2016) and appears to protect synapses from stress-induced (Milior et al., 2016; Schubert et al., 2018) or HIV gp120-induced neuronal injury (Ru et al., 2019). Elevated CSF CX3CL1 levels in delirium and AD patients are associated with decreased synapse-associated proteins (Peters van Ton et al., 2020), suggesting disease-specific alterations in signaling dynamics. Mechanistically, CX3CL1 inhibits CA1 hippocampal LTP via activation of adenosine A3 receptors (A3R) (Poniatowski et al., 2017). Under hypoxic conditions, CX3CL1/CX3CR1 signaling promotes microglial phagocytic activity, accelerating synaptic loss (Wang et al., 2023).
Interestingly, the effects of CX3CL1/CX3CR1 signaling appear to be region- and disease stage-specific. For example, in the visual system, CX3CL1 deletion does not affect microglial morphology or synaptic turnover (Lowery et al., 2017), while in stroke models, CX3CR1 deficiency does not alter brain injury outcomes (van der Maten et al., 2017). Similarly, in CA3 pyramidal neurons, GABAergic transmission remains unchanged in CX3CR1−/− mice, although a notable reduction in giant depolarizing potential (GDP) frequency is observed (Bertot et al., 2019). Collectively, these findings underscore the complexity of the CX3CL1/CX3CR1 axis, which exerts diverse, and sometimes contradictory, effects on synaptic structure and plasticity depending on the brain region, pathological condition, and cellular context.
4.3 CSF1/CSF1R
CSF1, also known as macrophage colony-stimulating factor (MCSF), is a cytokine essential for the development and maintenance of cells in the mononuclear phagocyte system. Its receptor, CSF1R (c-Fms/CD115), belongs to the type III receptor tyrosine kinase family. In the CNS, CSF1 is mainly expressed by neurons, whereas CSF1R is selectively expressed on microglia (Chitu et al., 2016). CSF1/CSF1R signaling governs multiple microglial processes, including migration, proliferation, survival, differentiation, and polarization, and plays a fundamental role in regulating neuroinflammation. However, excessive activation of this pathway has been linked to pathological neuroinflammatory states, thereby positioning CSF1R inhibition as a potential therapeutic strategy (El-Gamal et al., 2018).
In a lumbar disc puncture model using CX3CR1GFP/+ mice, upregulation of CSF1/CSF1R signaling intensified microglial activation after disc degeneration (Yang et al., 2018), suggesting a link between this axis and maladaptive neuroplasticity. CSF1R inhibitors such as PLX5622 suppress microglial activation and neuroinflammation while preserving synaptic structure and function. Low-dose PLX5622 has been shown to prevent sepsis induced synaptic loss and cognitive impairment, underscoring the role of CSF1R signaling in inflammation related synaptic remodeling (Mein et al., 2023). Guan et al. (2016) demonstrated that deletion of CSF1 from sensory neurons reduced microglia-mediated activation and mechanical hypersensitivity after nerve injury, likely through disruption of the CSF1R/DAP12 pathway. Conversely, exogenous CSF1 administration had the opposite effect. Similar microglial responses were observed in neuropathic pain (Okubo et al., 2016) and autoimmune encephalomyelitis models (Gushchina et al., 2018), in which CSF1 expression correlated with microglial proliferation and enhanced neuroinflammation. Intrathecal injection of anti-CSF1 antibodies also reversed high-frequency stimulation (HFS)-induced spinal LTP at C-fiber synapses and attenuated CSF1R expression and microglial activation, thereby highlighting the contribution of this pathway to microglia-driven synaptic plasticity (Zhou et al., 2019).
Beyond pain and inflammatory disorders, CSF1/CSF1R signaling plays a critical role in microglia-mediated synaptic pruning. In chronic unpredictable stress (CUS) models of cognitive impairment, increased CSF1/CSF1R activity drove microglial engulfment of neuronal components and spine loss in the medial prefrontal cortex (mPFC). These effects were reversed by neuronal CSF1 knockdown (Wohleb et al., 2018), glucocorticoid receptor antagonism (RU486) (Horchar and Wohleb, 2019), or diazepam treatment (Bollinger et al., 2020), indicating that CSF1-dependent microglial remodeling contributes to stress-induced behavioral and synaptic deficits. In SAE models, inhibition of CSF1R by PLX5622 reduced C1q-tagged synapses and microglial phagocytosis, providing further evidence that this pathway regulates synaptic elimination under inflammatory conditions (Chung et al., 2023). Prolonged CSF1R blockade using small molecules such as pexidartinib has been shown to attenuate neuroinflammatory damage and stabilize synaptic integrity, potentially facilitating neuronal recovery (Weyer et al., 2024). Similar neuroprotective effects were reported in models of MS, where CSF1R inhibition prevented synaptic loss in the context of cortical inflammation (Ding et al., 2021).
Emerging data also suggest a pathogenic role for CSF1 in early-stage AD (Klein et al., 2020). In APP/PS1 mice, administration of the tyrosine kinase inhibitor (GW2580) improved memory and behavior by suppressing microglial proliferation and synaptic degeneration (Olmos-Alonso et al., 2016). Short-term CSF1R inhibition in 5xFAD mice led to reduced neuroinflammation and enhanced autophagy in residual microglia, which was associated with mitigation of synaptic loss (Kodali et al., 2024). Interestingly, withdrawal of CSF1R inhibitors facilitated microglial repopulation, which promoted brain recovery and reduced inflammation following injury (Rice et al., 2017). Similar observations were made in TBI models, where repopulated microglia restored synaptic and neuronal function while reducing long-term inflammation (Wang et al., 2022). Furthermore, neuronal CSF1 was found to modulate depression-like behaviors via microglial STAT3-dependent regulation of synaptic transmission (Kwon et al., 2017). In models of neuropathic pain, CSF1 increased excitatory input to excitatory neurons in a BDNF-dependent manner and concurrently reduced excitatory input to inhibitory neurons via a BDNF-independent mechanism, thereby shifting the excitatory/inhibitory balance (Boakye et al., 2019).
CSF1R signaling is also developmentally regulated. In the olfactory bulb, microglial depletion via PLX5622 reduced both synapse formation and elimination in young, but not mature, adult-born granule cells (abGCs), indicating a time-specific requirement for microglia in synaptic remodeling (Reshef et al., 2017). Similarly, in a retinitis pigmentosa (RP) model, treatment with the CSF1R inhibitor PLX3397 reduced microglia-mediated synaptic pruning (He et al., 2019). Collectively, these findings underscore the dual and context-dependent role of CSF1/CSF1R signaling in shaping synaptic architecture under both physiological and pathological conditions.
4.4 IFN-γ/IFN-γR
IFN-γ, the sole type II interferon, is a soluble pro-inflammatory cytokine primarily produced by T helper cell type 1 (Th1) lymphocytes, CD8+ cytotoxic T cells, natural killer (NK) cells, and microglia within the CNS (Monteiro et al., 2017). It exerts its biological effects via binding to the IFNGR1/IFNGR2 receptor complex expressed on both microglia and neurons (Monteiro et al., 2017). IFN-γ is a potent activator of microglia, inducing phenotypic polarization, receptor upregulation, morphological changes, and elevated expression of inflammatory mediators, including autocrine IFN-γ release. This, in turn, initiates transcriptional programs that drive classical activation (Vergara et al., 2019; Lewen et al., 2020; Ta et al., 2019).
Functionally, IFN-γ-activated microglia modulate synaptic integrity and plasticity. In hippocampal slice cultures, IFN-γ stimulation enhances microglial activation and proliferation, accompanied by morphological remodeling and increased production of nitric oxide and pro-inflammatory cytokines (Lewen et al., 2020; Ta et al., 2019). These changes underlie heightened synaptic pruning and impaired neuroplasticity. For instance, systemic delivery of IFN-γ upregulates major histocompatibility complex class I (MHCI) in the mPFC, promoting synapse elimination (Sobue et al., 2018). IFN-γ-deficient mice display improved hippocampal synaptic architecture, neuronal morphology, and cognitive performance, suggesting that basal IFN-γ negatively regulates synaptic plasticity (Monteiro et al., 2016). Mechanistically, IFN-γ activates STAT1 signaling in hippocampal microglia, priming them for excessive pruning and disrupting social memory via aberrant microglia–neuron interactions (He et al., 2024).
Elevated IFN-γ has also been implicated in synaptic degeneration in disease models. In experimental autoimmune encephalomyelitis (EAE), neutralization of IFN-γ alleviated dendritic spine loss in the cortex (Huang et al., 2021). Similarly, genetic ablation of CD8 + T cells or microglial IFN-γ signaling conferred resistance to virus-induced synapse loss and neuronal apoptosis in West Nile virus (WNV) and Zika virus (ZIKV) models (Garber et al., 2019). In PD models, IFN-γ exacerbates neuronal susceptibility by enhancing the pathological leucine-rich repeat kinase 2 (LRRK2)-G2019S pathway, thereby impairing Akt signaling and nuclear factor of activated T-cells (NFAT) activation in both neurons and microglia (Panagiotakopoulou et al., 2020). In the rostral ventrolateral medulla (RVLM), IFN-γ deficiency compromised microglial synaptic engulfment, highlighting its role in modulating synaptic density and excitability with implications for hypertension pathophysiology (Tong et al., 2023).
Beyond structural remodeling, IFN-γ disrupts E/I balance by altering neurotransmission. In neuropathic pain, exogenous IFN-γ enhances synaptic transmission from C-fibers to spinal lamina I neurons through microglial activation (Reischer et al., 2020). In MS models, IFN-γ gene delivery promotes microglia-driven glutamate release and paranodal elongation—reversible with NMDA receptor antagonists (Gallego-Delgado et al., 2020). Similarly, in Toxoplasma gondii-infected mice, IFN-γ-primed microglia impair synaptic transmission and neural oscillations, aggravating cognitive deficits. Co-infection with Heligmosomoides polygyrus further amplifies Th1 cytokine production and suppresses synaptic markers such as EAAT2 and GABAAα1, effects mitigated by IFN-γ neutralization, suggesting that IFN-γ drives neuroinflammation-associated synaptic dysfunction (Kann et al., 2022; French et al., 2019).
Despite its deleterious roles, IFN-γ also exerts context-dependent neuroprotective functions. In models of immune dysfunction, IFN-γ enhances GABAergic currents in the prefrontal cortex (PFC) neurons, preventing hyperexcitability and preserving social behavior (Filiano et al., 2016). In AD models, IFN-γ facilitates amyloid-β clearance by restoring autophagy in microglia (He et al., 2020). Environmental enrichment–induced synaptic benefits, including elevated dendritic spine density and LTP, are abrogated by IFN-γ blockade, further supporting its context-specific modulatory role (Qi et al., 2018). Thus, IFN-γ emerges as a double-edged cytokine—detrimental when dysregulated, yet potentially protective under physiological or therapeutic conditions (John and Darcy, 2016).
5 Microglial regulation of the effects of BDNF and TREM2 on synaptic modification
5.1 BDNF
BDNF, a member of the neurotrophin family, plays multifaceted roles in the CNS, contributing to neuronal maturation, circuit refinement, and synaptic modulation. While predominantly produced by neurons and astrocytes, BDNF is also synthesized by microglia, where it exhibits anti-inflammatory properties and contributes to the maintenance of CNS homeostasis (Charlton et al., 2023). Accumulating evidence indicates that BDNF is critically involved in synaptic remodeling during pathological conditions, particularly via its facilitation of hippocampal LTP. Reduced BDNF levels are associated with impaired LTP (Golia et al., 2019), whereas exogenous BDNF administration restores synaptic plasticity in memory-deficient models (White et al., 2016).
The mechanisms underlying BDNF-mediated LTP involve multiple signaling pathways and synaptic regulators. These include activation of cannabinoid type 1 receptors (CB1Rs) (Maglio et al., 2018), upregulation of the RNA-binding protein RBFOX1 (Tomassoni-Ardori et al., 2019), enhanced expression of activity-regulated cytoskeleton-associated protein (Arc/Arg3.1) (Kuipers et al., 2016), and engagement of mature BDNF (mBDNF)/TrkB/CREB signaling cascades (Zhou et al., 2019; Garad et al., 2021; Pan et al., 2019). Moreover, peri-synaptic glial recycling of BDNF has been identified as a critical process modulating its availability at synaptic sites (Vignoli et al., 2016). Microglia-derived BDNF is implicated in both physiological and pathological synaptic plasticity, contributing to neonatal incision-induced spinal LTP (Ding et al., 2018) and cocaine-induced adaptations (Cotto et al., 2018), as well as promoting neuroplasticity and stress resilience (Prowse and Hayley, 2021).
Notably, dysregulation of BDNF signaling can also contribute to neuroinflammation and synaptic impairment. Activation of P2X4 receptors on microglia reduces hippocampal BDNF levels, exacerbates inflammation, and impairs late-phase LTP (L-LTP) (Tanaka et al., 2018; Wei et al., 2024). Conversely, enhanced BDNF/TrkB signaling can shift microglial phenotypes toward anti-inflammatory states and protect against neuronal loss (Hsu et al., 2020). However, BDNF produced by microglia has also been shown to promote pro-inflammatory responses, including the release of cytokines such as IL-1β and TNF-α (Ding et al., 2020). This bidirectional role emphasizes the importance of context-dependent regulation. As noted in Sections 3.1 and 3.3, targeting the interaction between BDNF and microglial-derived cytokines may offer therapeutic potential in neurodegenerative conditions.
At the synaptic level, BDNF enhances NMDAR function by increasing the ratio of GluN2B-containing subunits, a process dependent on proline-rich tyrosine kinase 2 (Pyk2) and heterogeneous nuclear ribonucleoprotein K (hnRNP K) activity (Afonso et al., 2019). It also regulates dendritic spine structure, reversing chronic unpredictable mild stress (CUMS)-induced spine loss and behavioral deficits in rodents (Park et al., 2016; Qiao et al., 2017). In Nhe5-deficient mice, upregulation of BDNF/TrkB signaling correlates with improved cognition (Chen et al., 2017). While numerous studies associate BDNF with changes in dendritic spine morphology, causal relationships, particularly those involving microglial modulation, remain incompletely characterized.
BDNF further modulates microglial activity by preventing excessive synaptic pruning and limiting microglial proliferation in kainic acid-induced injury models (Onodera et al., 2021). Interestingly, in the developing cerebellum, retrograde BDNF–TrkB signaling from Purkinje cells promotes climbing fiber synapse elimination, indicating regional and developmental specificity (Choo et al., 2017). BDNF also regulates the balance between excitatory and inhibitory neurotransmission, with age-dependent alterations in PFC linked to deficits in GABAergic signaling due to disrupted BDNF/NTRK2 pathways (Oh et al., 2016; Meis et al., 2019). In neuropathic pain, microglia-derived BDNF contributes to excitatory/inhibitory imbalance via TrkB and p75 neurotrophin receptor (p75NTR), enhancing dorsal horn excitability (Boakye et al., 2019). Similar interactions involving KCC2 regulation are implicated in ischemia-induced plasticity changes (Cramer et al., 2022). Furthermore, crosstalk between BDNF/TrkB and the endocannabinoid system (ECS) plays a significant role in modulating microglial responses and synaptic strength (Maglio et al., 2018; Pan et al., 2019; Yeh et al., 2017; Wu et al., 2020; Gangarossa et al., 2020).
Collectively, these findings establish BDNF as a critical, yet context-dependent, modulator of microglia–neuron interactions, synaptic architecture, and neurotransmission. Its dual role in promoting neuroplasticity and contributing to inflammation underscores the importance of understanding BDNF signaling in a cell-type- and disease-specific manner.
5.2 TREM2
TREM2 is a microglia-specific type I transmembrane receptor that plays a crucial role in regulating microglial activity, particularly in synaptic pruning and the pathophysiology of neurodegenerative diseases. TREM2 activation promotes the anti-inflammatory microglial phenotype, thereby enhancing phagocytic function, attenuating neuroinflammation, and mitigating synaptic and neuronal loss under pathological conditions (Jiang et al., 2016; Ruganzu et al., 2021; Zhang et al., 2018; Zhang et al., 2017; Jiang et al., 2018; Fracassi et al., 2022). In addition, TREM2 signaling restrains microglial overactivation induced by chronic neuroinflammation (Chen et al., 2020; Krasemann et al., 2017; Ruganzu et al., 2022), and has been implicated in the regulation of neuroplasticity (Raha et al., 2017). Interestingly, TREM2 deficiency enhances LTP without affecting basal synaptic transmission in aged mice (Qu and Li, 2020). Moreover, the AD-associated R47H mutation demonstrates an age-dependent impact on LTP: it initially prevents LTP deficits but later exacerbates them, suggesting a temporally dynamic role of TREM2 in synaptic plasticity (Tran et al., 2023).
In addition to its role in AD, TREM2 modulates microglial activity in other neurodegenerative diseases, including PD and ALS. In PD, TREM2 expression is upregulated in activated microglia surrounding dopaminergic neurons, enhancing phagocytosis and suppressing the excessive release of pro-inflammatory cytokines (Zhang et al., 2018). It has been shown that TREM2 signaling inhibits NLRP3 inflammasome activation and pyroptosis, thereby reducing dopaminergic neuron loss in MPTP induced models. Conversely, TREM2 deficiency results in increased IL-1β and TNF-α production, exacerbating neuroinflammation (Huang et al., 2023). In ALS, TREM2 expression exhibits stage-dependent changes, with upregulation observed in the early and mid-stages of the disease, correlating with a reactive yet neuroprotective microglial phenotype (Jericó et al., 2023). Overall, emerging evidence suggests that TREM2 acts as a key regulatory hub linking innate immune receptor signaling with cytokine-mediated inflammatory pathways, thereby maintaining microglial homeostasis and modulating neuroinflammatory and synaptic outcomes in PD, ALS, and other neurodegenerative diseases (Yeh et al., 2017).
TREM2 also contributes to synaptic pruning. Several studies have shown that TREM2 facilitates microglial engulfment of synaptic components (Fracassi et al., 2022; McQuade et al., 2020; Filipello et al., 2018), with the R47H mutation promoting excessive synaptosome phagocytosis, which may result in abnormal synaptic elimination and accelerated AD progression (Popescu et al., 2022; Das et al., 2023; Penney et al., 2023; Gratuze et al., 2020). In APP/PS1 transgenic mice, early TREM2 deficiency appears to prevent synaptic loss by reducing microglial phagocytic activity, whereas at later stages, its absence impairs amyloid clearance and worsens synaptic dysfunction (Ruganzu et al., 2022; Sheng et al., 2019). Similarly, in a dual-pathology AD model incorporating both β-amyloid and tau, TREM2 deletion disrupts microglial synapse phagocytosis (Dejanovic et al., 2022). External factors, such as chronic alcohol exposure, have also been shown to upregulate TREM2 expression, leading to increased synaptic elimination that correlates with memory impairments and reduced synaptic density (Lan et al., 2022). Mechanistically, TREM2-mediated synaptic remodeling involves p38 MAPK activation, complement pathway engagement, and interactions with phosphatidylserine on neuronal membranes (Tian et al., 2024; Rueda-Carrasco et al., 2023; Zhong et al., 2023; Meng et al., 2024). Notably, astrocytic uptake of synapses may also contribute to these effects, suggesting that TREM2’s role extends beyond direct microglia-synapse interactions (Jay et al., 2019).
Beyond structural remodeling, TREM2 influences neurotransmission. Its deficiency has been associated with elevated excitatory synaptic activity during early development (Filipello et al., 2018). In the context of AD, TREM2 suppresses glutamatergic transmission at early stages but appears to facilitate it at later stages (Sheng et al., 2019). The R47H mutation is further implicated in reducing GABAergic transmission via TNF-α upregulation, aggravating glutamate-induced excitotoxicity (Ren et al., 2020b; Ren et al., 2020a). These observations indicate that TREM2 exerts stage-dependent and context-specific effects on both excitatory and inhibitory transmission.
From a therapeutic standpoint, interventions targeting TREM2 must account for its multifaceted roles in phagocytosis, inflammation resolution, and apoptosis inhibition. While augmenting TREM2 signaling may offer benefits in certain neurodegenerative contexts, inappropriate modulation could inadvertently trigger detrimental pro-inflammatory cascades (Akhter et al., 2021; Kobayashi et al., 2016). Paradoxically, TREM2 loss has been associated with resistance to age-related synaptic and cognitive decline (Qu and Li, 2020), underscoring the nuanced and context-sensitive nature of its functions.
Collectively, TREM2 acts as a dynamic modulator of microglial behavior, synaptic remodeling, and neurotransmission. Its impact is highly dependent on developmental stage, pathological context, and mutation status, emphasizing the importance of precise therapeutic strategies when targeting TREM2-associated pathways.
6 Concluding remarks
Our understanding of microglia, the brain’s resident immune cells, continues to evolve. Once recognized primarily for their role in immune surveillance, microglia are now understood to play essential roles in maintaining neural homeostasis and regulating synaptic architecture. Increasing evidence suggests that hyperactivation of microglia, often accompanied by sustained neuroinflammatory responses, contributes significantly to the pathogenesis of neurological disorders. This review has highlighted recent advances linking dysregulated microglia-mediated synaptic remodeling and aberrant expression of pro-inflammatory cytokines and receptors to both neurodevelopmental and neurodegenerative diseases.
Following CNS injury, activated microglia release pro-inflammatory mediators, initiating an inflammatory cascade that induces structural and functional synaptic alterations, ultimately leading to cognitive and behavioral impairments (Figure 3 and Table 1). Notably, such microglia-driven synaptic disruption may aggravate secondary injury, thereby amplifying neurodegeneration. Consequently, timely and effective suppression of harmful microglial responses is crucial. Nevertheless, microglial responses are highly dynamic and context-dependent, encompassing a spectrum of functional states ranging from pro-inflammatory to restorative and phagocytic profiles. These diverse and adaptable responses can promote either neuroprotection or neurotoxicity depending on the surrounding microenvironment and disease stage. Hence, therapeutic interventions targeting microglia must be carefully designed to minimize adverse effects. Beyond their intrinsic roles, microglia-mediated cytokine signaling orchestrates complex interactions among astrocytes, oligodendrocytes, and peripheral immune cells, collectively shaping synaptic remodeling and influencing the progression of neurodegeneration. A deeper understanding of this intercellular communication network may reveal regulatory mechanisms and therapeutic targets to mitigate microglia-driven neuroinflammatory and neurodegenerative processes.
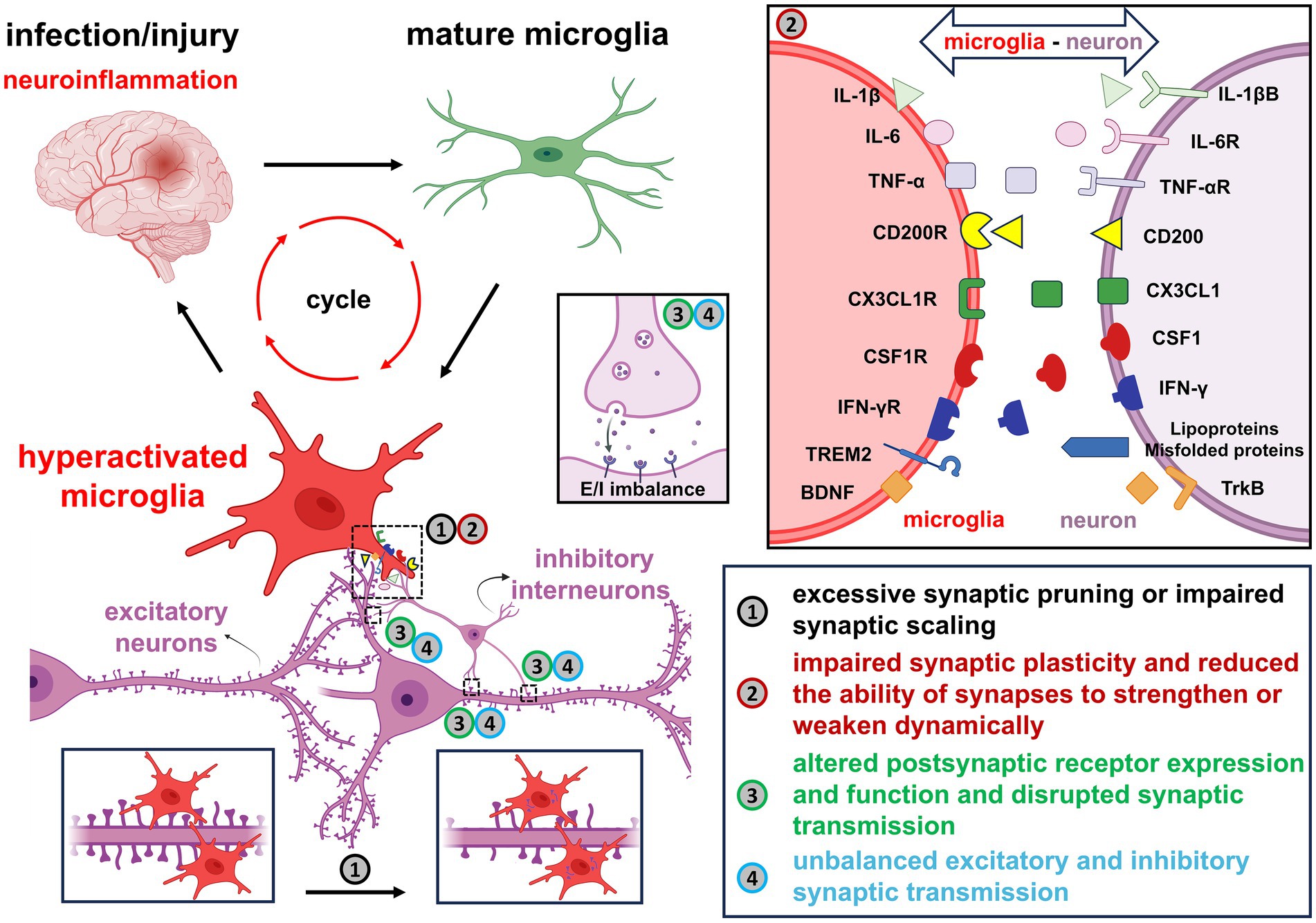
Figure 3. A summary of the impact of overactivated microglia on neuronal synapses under neuroinflammatory conditions. Following infection or injury, microglia transition into hyperactivated pro-inflammatory states and secrete cytokines such as IL-1β, IL-6, TNF-α and IFN-γ, which exacerbate neuroinflammation and contribute to synaptic impairment. Disruption of key neuron–microglia ligand-receptor interactions, such as CD200/CD200R, CX3CL1/CX3CL1R, CSF1/CSF1R, Lipoproteins/Misfolded proteins/TREM2 and BDNF/TrkB, leads to dysregulated synaptic homeostasis. The resulting microglial activation promotes excessive synaptic pruning, impaired synaptic scaling, abnormal receptor expression, and altered synaptic transmission. These changes ultimately disturb the excitatory-inhibitory balance and compromise overall neuronal function.
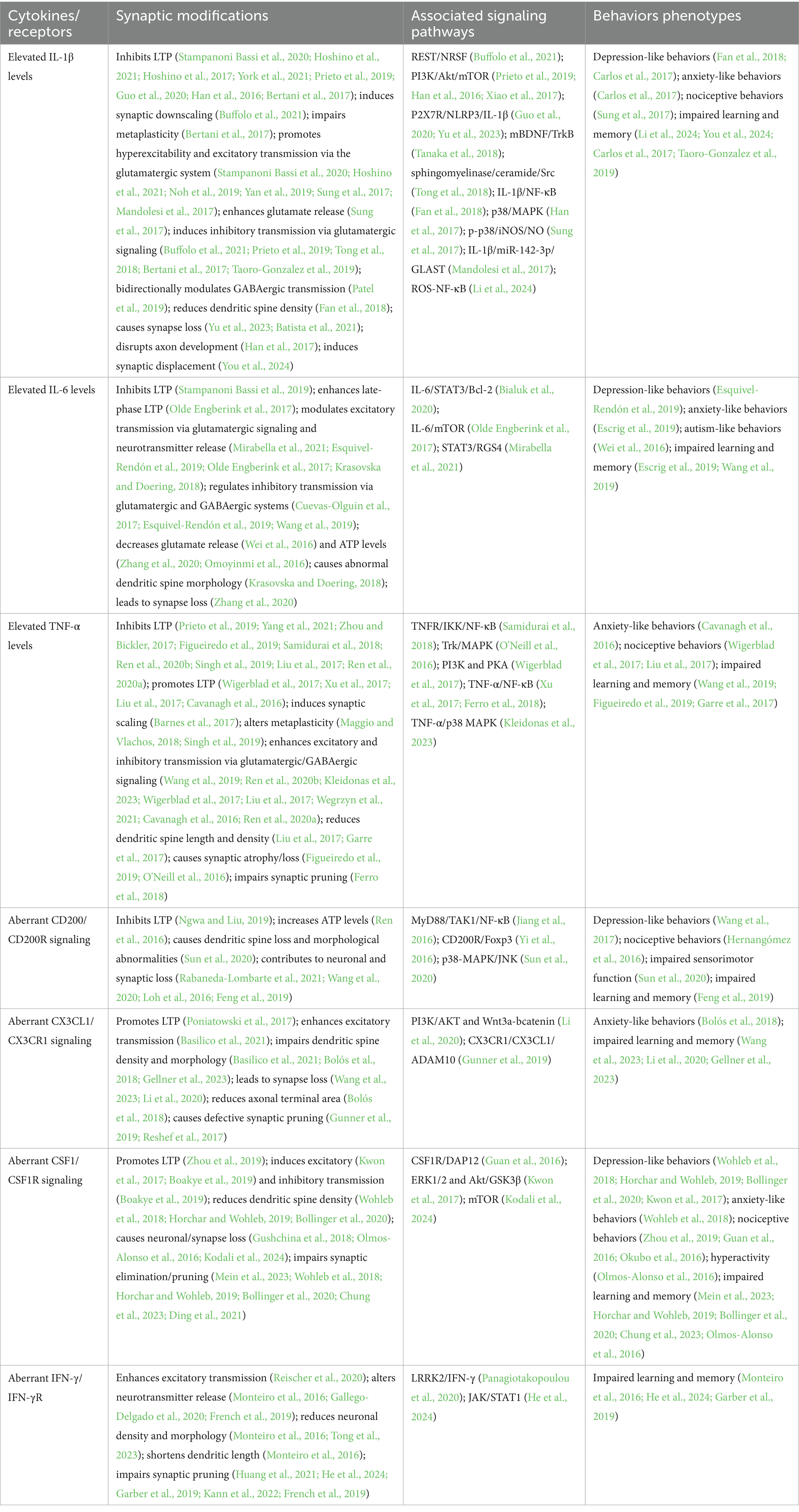
Table 1. Negative regulation of synaptic modifications and behavioral correlates by microglial pro-inflammatory factors/receptors.
In this context, selectively modulating pro-inflammatory cytokines and receptors may offer a more refined strategy to restore synaptic integrity and reduce disease burden. Accomplishing this goal requires a comprehensive understanding of the molecular mechanisms underpinning microglia-driven synaptic remodeling. Future investigations should focus on: (i) elucidating the intracellular and intercellular signaling pathways linked to microglial cytokines and receptors, (ii) identifying optimal cytokine/receptor targets for therapeutic intervention, and (iii) exploring how microglia-astrocyte interactions contribute to synaptic remodeling in both physiological and pathological states.
Author contributions
GY: Data curation, Funding acquisition, Investigation, Writing – original draft. XX: Data curation, Writing – original draft. WG: Data curation, Writing – original draft. XW: Data curation, Writing – review & editing. YZ: Conceptualization, Writing – review & editing. YX: Conceptualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was financially supported by grants from National Natural Science Foundation of China (Grant 82174003 to YX and Grant 82505050 to GY), Shanghai Institute of Traditional Chinese Medicine for mental health (Grant SZB2024201 to GY), and Fellowship of China Postdoctoral Science Foundation (Grant 2024M762109 to GY).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abd-El-Basset, E. M., Rao, M. S., and Alsaqobi, A. (2020). Interferon-gamma and interleukin-1Beta enhance the secretion of brain-derived neurotrophic factor and promotes the survival of cortical neurons in brain injury. Neurosci Insights 15:2633105520947081. doi: 10.1177/2633105520947081
Afonso, P., De Luca, P., Carvalho, R. S., Cortes, L., Pinheiro, P., Oliveiros, B., et al. (2019). BDNF increases synaptic NMDA receptor abundance by enhancing the local translation of Pyk2 in cultured hippocampal neurons. Sci Signal 12:eaav3577. doi: 10.1126/scisignal.aav3577
Akhter, R., Shao, Y., Formica, S., Khrestian, M., and Bekris, L. M. (2021). TREM2 alters the phagocytic, apoptotic and inflammatory response to Abeta42 in HMC3 cells. Mol. Immunol. 131, 171–179. doi: 10.1016/j.molimm.2020.12.035
Barnes, S. J., Franzoni, E., Jacobsen, R. I., Erdelyi, F., Szabo, G., Clopath, C., et al. (2017). Deprivation-induced homeostatic spine scaling in vivo is localized to dendritic branches that have undergone recent spine loss. Neuron 96, 871–882. doi: 10.1016/j.neuron.2017.09.052
Basilico, B., Ferrucci, L., Ratano, P., Golia, M. T., Grimaldi, A., Rosito, M., et al. (2021). Microglia control glutamatergic synapses in the adult mouse hippocampus. Glia 70, 173–195. doi: 10.1002/glia.24101
Batista, A. F., Rody, T., Forny-Germano, L., Cerdeiro, S., Bellio, M., Ferreira, S. T., et al. (2021). Interleukin-1β mediates alterations in mitochondrial fusion/fission proteins and memory impairment induced by amyloid-β oligomers. J. Neuroinflammation 18:54. doi: 10.1186/s12974-021-02099-x
Bertani, I., Iori, V., Trusel, M., Maroso, M., Foray, C., Mantovani, S., et al. (2017). Inhibition of IL-1beta signaling normalizes NMDA-dependent neurotransmission and reduces seizure susceptibility in a mouse model of Creutzfeldt-Jakob disease. J. Neurosci. 37, 10278–10289. doi: 10.1523/JNEUROSCI.1301-17.2017
Bertot, C., Groc, L., and Avignone, E. (2019). Role of CX3CR1 signaling on the maturation of GABAergic transmission and neuronal network activity in the neonate Hippocampus. Neuroscience 406, 186–201. doi: 10.1016/j.neuroscience.2019.03.006
Bialuk, I., Cieślińska, M., Kowalczuk, O., Bonda, T. A., Nikliński, J., and Winnicka, M. M. (2020). IL-6 deficiency attenuates p53 protein accumulation in aged male mouse hippocampus. Biogerontology 21, 29–43. doi: 10.1007/s10522-019-09841-2
Boakye, P. A., Rancic, V., Whitlock, K. H., Simmons, D., Longo, F. M., and Ballanyi, K. (2019). Receptor dependence of BDNF actions in superficial dorsal horn: relation to central sensitization and actions of macrophage colony stimulating factor 1. J Neurophysiol 121, 2308–2322. doi: 10.1152/jn.00839.2018
Bollinger, J. L., Horchar, M. J., and Wohleb, E. S. (2020). Diazepam limits microglia-mediated neuronal remodeling in the prefrontal cortex and associated behavioral consequences following chronic unpredictable stress. Neuropsychopharmacology 45, 1766–1776. doi: 10.1038/s41386-020-0720-1
Bolós, M., Perea, J. R., Terreros-Roncal, J., Pallas-Bazarra, N., Jurado-Arjona, J., Ávila, J., et al. (2018). Absence of microglial CX3CR1 impairs the synaptic integration of adult-born hippocampal granule neurons. Brain Behav. Immun. 68, 76–89. doi: 10.1016/j.bbi.2017.10.002
Buffolo, F., Petrosino, V., Albini, M., Moschetta, M., Carlini, F., Floss, T., et al. (2021). Neuroinflammation induces synaptic scaling through IL-1beta-mediated activation of the transcriptional repressor REST/NRSF. Cell Death Dis. 12:180. doi: 10.1038/s41419-021-03465-6
Carlos, A. J., Tong, L., Prieto, G. A., and Cotman, C. W. (2017). IL-1β impairs retrograde flow of BDNF signaling by attenuating endosome trafficking. J. Neuroinflammation 14:29. doi: 10.1186/s12974-017-0803-z
Cavanagh, C., Tse, Y. C., Nguyen, H. B., Krantic, S., Breitner, J. C. S., Quirion, R., et al. (2016). Inhibiting tumor necrosis factor-alpha before amyloidosis prevents synaptic deficits in an Alzheimer's disease model. Neurobiol. Aging 47, 41–49. doi: 10.1016/j.neurobiolaging.2016.07.009
Charlton, T., Prowse, N., McFee, A., Heiratifar, N., Fortin, T., Paquette, C., et al. (2023). Brain-derived neurotrophic factor (BDNF) has direct anti-inflammatory effects on microglia. Front. Cell. Neurosci. 17:1188672. doi: 10.3389/fncel.2023.1188672
Chen, S., Peng, J., Sherchan, P., Ma, Y., Xiang, S., Yan, F., et al. (2020). TREM2 activation attenuates neuroinflammation and neuronal apoptosis via PI3K/Akt pathway after intracerebral hemorrhage in mice. J. Neuroinflammation 17:168. doi: 10.1186/s12974-020-01853-x
Chen, X., Wang, X., Tang, L., Wang, J., Shen, C., Liu, J., et al. (2017). Nhe5 deficiency enhances learning and memory via upregulating Bdnf/TrkB signaling in mice. Am. J. Med. Genet. B Neuropsychiatr. Genet. 174, 828–838. doi: 10.1002/ajmg.b.32600
Chitu, V., Gokhan, S., Nandi, S., Mehler, M. F., and Stanley, E. R. (2016). Emerging roles for CSF-1 receptor and its ligands in the nervous system. Trends Neurosci. 39, 378–393. doi: 10.1016/j.tins.2016.03.005
Choo, M., Miyazaki, T., Yamazaki, M., Kawamura, M., Nakazawa, T., Zhang, J., et al. (2017). Retrograde BDNF to TrkB signaling promotes synapse elimination in the developing cerebellum. Nat. Commun. 8:195. doi: 10.1038/s41467-017-00260-w
Chung, H.-Y., Wickel, J., Hahn, N., Mein, N., Schwarzbrunn, M., Koch, P., et al. (2023). Microglia mediate neurocognitive deficits by eliminating C1q-tagged synapses in sepsis-associated encephalopathy. Sci. Adv. 9:eabq7806. doi: 10.1126/sciadv.abq7806
Cianciulli, A., Calvello, R., Ruggiero, M., and Panaro, M. A. (2022). Inflammaging and brain: curcumin and its beneficial potential as regulator of microglia activation. Molecules 27. doi: 10.3390/molecules27020341
Cignarella, F., Filipello, F., Bollman, B., Cantoni, C., Locca, A., Mikesell, R., et al. (2020). TREM2 activation on microglia promotes myelin debris clearance and remyelination in a model of multiple sclerosis. Acta Neuropathol. 140, 513–534. doi: 10.1007/s00401-020-02193-z
Cobley, J. N. (2018). Synapse pruning: mitochondrial ROS with their hands on the shears. BioEssays 40:e1800031. doi: 10.1002/bies.201800031
Cotto, B., Li, H., Tuma, R. F., Ward, S. J., and Langford, D. (2018). Cocaine-mediated activation of microglia and microglial MeCP2 and BDNF production. Neurobiol. Dis. 117, 28–41. doi: 10.1016/j.nbd.2018.05.017
Cox, L. M., Calcagno, N., Gauthier, C., Madore, C., Butovsky, O., and Weiner, H. L. (2022). The microbiota restrains neurodegenerative microglia in a model of amyotrophic lateral sclerosis. Microbiome 10:47. doi: 10.1186/s40168-022-01232-z
Cramer, T., Gill, R., Thirouin, Z. S., Vaas, M., Sampath, S., Martineau, F., et al. (2022). Cross-talk between GABAergic postsynapse and microglia regulate synapse loss after brain ischemia. Sci. Adv. 8:eabj0112. doi: 10.1126/sciadv.abj0112
Cuevas-Olguin, R., Esquivel-Rendon, E., Vargas-Mireles, J., Garcia-Oscos, F., Miranda-Morales, M., Salgado, H., et al. (2017). Interleukin 6 trans-signaling regulates basal synaptic transmission and sensitivity to pentylenetetrazole-induced seizures in mice. Synapse 71:e21984. doi: 10.1002/syn.21984
Das, M., Mao, W., Voskobiynyk, Y., Necula, D., Lew, I., Petersen, C., et al. (2023). Alzheimer risk-increasing TREM2 variant causes aberrant cortical synapse density and promotes network hyperexcitability in mouse models. Neurobiol. Dis. 186:106263. doi: 10.1016/j.nbd.2023.106263
Dejanovic, B., Wu, T., Tsai, M. C., Graykowski, D., Gandham, V. D., Rose, C. M., et al. (2022). Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer’s disease mouse models. Nature Aging 2, 837–850. doi: 10.1038/s43587-022-00281-1
Ding, H., Chen, J., Su, M., Lin, Z., Zhan, H., Yang, F., et al. (2020). BDNF promotes activation of astrocytes and microglia contributing to neuroinflammation and mechanical allodynia in cyclophosphamide-induced cystitis. J. Neuroinflammation 17:19. doi: 10.1186/s12974-020-1704-0
Ding, X., Liang, Y. J., Su, L., Liao, F. F., Fang, D., Tai, J., et al. (2018). BDNF contributes to the neonatal incision-induced facilitation of spinal long-term potentiation and the exacerbation of incisional pain in adult rats. Neuropharmacology 137, 114–132. doi: 10.1016/j.neuropharm.2018.04.032
Ding, X., Wang, J., Huang, M., Chen, Z., Liu, J., Zhang, Q., et al. (2021). Loss of microglial SIRPalpha promotes synaptic pruning in preclinical models of neurodegeneration. Nat. Commun. 12:2030. doi: 10.1038/s41467-021-22301-1
Dong, Y., Pu, K. J., Duan, W. J., Chen, H. C., Chen, L. X., and Wang, Y. M. (2018). Involvement of Akt/CREB signaling pathways in the protective effect of EPA against interleukin-1beta-induced cytotoxicity and BDNF down-regulation in cultured rat hippocampal neurons. BMC Neurosci. 19:52. doi: 10.1186/s12868-018-0455-7
El-Gamal, M. I., Al-Ameen, S. K., Al-Koumi, D. M., Hamad, M. G., Jalal, N. A., and Oh, C. H. (2018). Recent advances of Colony-stimulating Factor-1 receptor (CSF-1R) kinase and its inhibitors. J. Med. Chem. 61, 5450–5466. doi: 10.1021/acs.jmedchem.7b00873
Erta, M., Quintana, A., and Hidalgo, J. (2012). Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 8, 1254–1266. doi: 10.7150/ijbs.4679
Escrig, A., Canal, C., Sanchis, P., Fernández-Gayol, O., Montilla, A., Comes, G., et al. (2019). IL-6 trans-signaling in the brain influences the behavioral and physio-pathological phenotype of the Tg2576 and 3xTgAD mouse models of Alzheimer's disease. Brain Behav. Immun. 82, 145–159. doi: 10.1016/j.bbi.2019.08.005
Esquivel-Rendón, E., Vargas-Mireles, J., Cuevas-Olguín, R., Miranda-Morales, M., Acosta-Mares, P., García-Oscos, F., et al. (2019). Interleukin 6 dependent synaptic plasticity in a social defeat-susceptible prefrontal cortex circuit. Neuroscience 414, 280–296. doi: 10.1016/j.neuroscience.2019.07.002
Fan, C., Song, Q., Wang, P., Li, Y., Yang, M., Liu, B., et al. (2018). Curcumin protects against chronic stress-induced dysregulation of neuroplasticity and depression-like behaviors via suppressing IL-1beta pathway in rats. Neuroscience 392, 92–106. doi: 10.1016/j.neuroscience.2018.09.028
Feng, D., Huang, A., Yan, W., and Chen, D. (2019). CD200 dysfunction in neuron contributes to synaptic deficits and cognitive impairment. Biochem. Biophys. Res. Commun. 516, 1053–1059. doi: 10.1016/j.bbrc.2019.06.134
Ferro, A., Qu, W., Lukowicz, A., Svedberg, D., Johnson, A., and Cvetanovic, M. (2018). Inhibition of NF-kappaB signaling in IKKbetaF/F;LysM Cre mice causes motor deficits but does not alter pathogenesis of spinocerebellar ataxia type 1. PLoS One 13:e0200013. doi: 10.1371/journal.pone.0200013
Figueiredo, C. P., Barros-Aragão, F. G. Q., Neris, R. L. S., Frost, P. S., Soares, C., Souza, I. N. O., et al. (2019). Zika virus replicates in adult human brain tissue and impairs synapses and memory in mice. Nat. Commun. 10:3890. doi: 10.1038/s41467-019-11866-7
Filiano, A. J., Xu, Y., Tustison, N. J., Marsh, R. L., Baker, W., Smirnov, I., et al. (2016). Unexpected role of interferon-gamma in regulating neuronal connectivity and social behaviour. Nature 535, 425–429. doi: 10.1038/nature18626
Filipello, F., et al. (2018). The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity 48, 979–991 e978. doi: 10.1016/j.immuni.2018.04.016
Fracassi, A., Marcatti, M., Tumurbaatar, B., Woltjer, R., Moreno, S., and Taglialatela, G. (2022). TREM2-induced activation of microglia contributes to synaptic integrity in cognitively intact aged individuals with Alzheimer's neuropathology. Brain Pathol. 33. doi: 10.1111/bpa.13108
French, T., Düsedau, H. P., Steffen, J., Biswas, A., Ahmed, N., Hartmann, S., et al. (2019). Neuronal impairment following chronic toxoplasma gondii infection is aggravated by intestinal nematode challenge in an IFN-gamma-dependent manner. J. Neuroinflammation 16:159. doi: 10.1186/s12974-019-1539-8
Fu, X. Q., Peng, J., Wang, A. H., and Luo, Z. G. (2020). Tumor necrosis factor alpha mediates neuromuscular synapse elimination. Cell Discov 6:9. doi: 10.1038/s41421-020-0143-5
Gallego-Delgado, P., James, R., Browne, E., Meng, J., Umashankar, S., Tan, L., et al. (2020). Neuroinflammation in the normal-appearing white matter (NAWM) of the multiple sclerosis brain causes abnormalities at the nodes of Ranvier. PLoS Biol. 18:e3001008. doi: 10.1371/journal.pbio.3001008
Gangarossa, G., Perez, S., Dembitskaya, Y., Prokin, I., Berry, H., and Venance, L. (2020). BDNF controls bidirectional endocannabinoid plasticity at Corticostriatal synapses. Cereb. Cortex 30, 197–214. doi: 10.1093/cercor/bhz081
Garad, M., Edelmann, E., and Lessmann, V. (2021). Long-term depression at hippocampal mossy fiber-CA3 synapses involves BDNF but is not mediated by p75NTR signaling. Sci. Rep. 11:8535. doi: 10.1038/s41598-021-87769-9
Garber, C., Soung, A., Vollmer, L. L., Kanmogne, M., Last, A., Brown, J., et al. (2019). T cells promote microglia-mediated synaptic elimination and cognitive dysfunction during recovery from neuropathogenic flaviviruses. Nat. Neurosci. 22, 1276–1288. doi: 10.1038/s41593-019-0427-y
Garre, J. M., Silva, H. M., Lafaille, J. J., and Yang, G. (2017). Cx3cr1(+) monocytes modulate learning and learning-dependent dendritic spine remodeling via TNF-alpha. Nat. Med. 23, 714–722. doi: 10.1038/nm.4340
Gellner, A. K., Reis, J., Fiebich, B. L., and Fritsch, B. (2023). Cx3cr1 deficiency interferes with learning- and direct current stimulation-mediated neuroplasticity of the motor cortex. Eur. J. Neurosci. 59, 177–191. doi: 10.1111/ejn.16206
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Golia, M. T., Poggini, S., Alboni, S., Garofalo, S., Ciano Albanese, N., Viglione, A., et al. (2019). Interplay between inflammation and neural plasticity: both immune activation and suppression impair LTP and BDNF expression. Brain Behav. Immun. 81, 484–494. doi: 10.1016/j.bbi.2019.07.003
Gratuze, M., Leyns, C. E. G., Sauerbeck, A. D., St-Pierre, M. K., Xiong, M., Kim, N., et al. (2020). Impact of TREM2R47H variant on tau pathology-induced gliosis and neurodegeneration. J. Clin. Invest. 130, 4954–4968. doi: 10.1172/JCI138179
Gruol, D. L., Calderon, D., French, K., Melkonian, C., Huitron-Resendiz, S., Cates-Gatto, C., et al. (2023). Neuroimmune interactions with binge alcohol drinking in the cerebellum of IL-6 transgenic mice. Neuropharmacology 228:109455. doi: 10.1016/j.neuropharm.2023.109455
Guan, Z., Kuhn, J. A., Wang, X., Colquitt, B., Solorzano, C., Vaman, S., et al. (2016). Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat. Neurosci. 19, 94–101. doi: 10.1038/nn.4189
Gunner, G., Cheadle, L., Johnson, K. M., Ayata, P., Badimon, A., Mondo, E., et al. (2019). Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat. Neurosci. 22, 1075–1088. doi: 10.1038/s41593-019-0419-y
Guo, D. H., Yamamoto, M., Hernandez, C. M., Khodadadi, H., Baban, B., and Stranahan, A. M. (2020). Visceral adipose NLRP3 impairs cognition in obesity via IL-1R1 on CX3CR1+ cells. J. Clin. Invest. 130, 1961–1976. doi: 10.1172/JCI126078
Gushchina, S., Pryce, G., Yip, P. K., Wu, D., Pallier, P., Giovannoni, G., et al. (2018). Increased expression of colony-stimulating factor-1 in mouse spinal cord with experimental autoimmune encephalomyelitis correlates with microglial activation and neuronal loss. Glia 66, 2108–2125. doi: 10.1002/glia.23464
Han, Q., Lin, Q., Huang, P., Chen, M., Hu, X., Fu, H., et al. (2017). Microglia-derived IL-1beta contributes to axon development disorders and synaptic deficit through p38-MAPK signal pathway in septic neonatal rats. J. Neuroinflammation 14:52. doi: 10.1186/s12974-017-0805-x
Han, T., Qin, Y., Mou, C., Wang, M., Jiang, M., and Liu, B. (2016). Seizure induced synaptic plasticity alteration in hippocampus is mediated by IL-1β receptor through PI3K/Akt pathway. Am. J. Transl. Res. 15, 4499–4509.
Hassan, I., Luo, Q., Majumdar, S., Dominguez, J. M. II, Busik, J. V., and Bhatwadekar, A. D. (2017). Tumor necrosis factor alpha (TNF-alpha) disrupts Kir4.1 channel expression resulting in Muller cell dysfunction in the retina. Invest. Ophthalmol. Vis. Sci. 58, 2473–2482. doi: 10.1167/iovs.16-20712
He, H. Y., Ren, L., Guo, T., and Deng, Y. H. (2019). Neuronal autophagy aggravates microglial inflammatory injury by downregulating CX3CL1/fractalkine after ischemic stroke. Neural Regen. Res. 14, 280–288. doi: 10.4103/1673-5374.244793
He, H., Zhang, X., He, H., Xiao, C., Xu, G., Li, L., et al. (2024). Priming of hippocampal microglia by IFN-γ/STAT1 pathway impairs social memory in mice. Int. Immunopharmacol. 134:112191. doi: 10.1016/j.intimp.2024.112191
He, J., Zhao, C., Dai, J., Weng, C. H., Bian, B. S. J., Gong, Y., et al. (2019). Microglia mediate synaptic material clearance at the early stage of rats with retinitis Pigmentosa. Front. Immunol. 10:912. doi: 10.3389/fimmu.2019.00912
He, Z., Yang, Y., Xing, Z., Zuo, Z., Wang, R., Gu, H., et al. (2020). Intraperitoneal injection of IFN-gamma restores microglial autophagy, promotes amyloid-beta clearance and improves cognition in APP/PS1 mice. Cell Death Dis. 11:440. doi: 10.1038/s41419-020-2644-4
Hernangómez, M., Klusáková, I., Joukal, M., Hradilová-Svíženská, I., Guaza, C., and Dubový, P. (2016). CD200R1 agonist attenuates glial activation, inflammatory reactions, and hypersensitivity immediately after its intrathecal application in a rat neuropathic pain model. J. Neuroinflammation 13:43. doi: 10.1186/s12974-016-0508-8
Hill, S. L., Shao, L., and Beasley, C. L. (2020). Diminished levels of the chemokine fractalkine in post-mortem prefrontal cortex in schizophrenia but not bipolar disorder. World J. Biol. Psychiatry 22, 94–103. doi: 10.1080/15622975.2020.1755451
Hopp, S. C., Lin, Y., Oakley, D., Roe, A. D., DeVos, S. L., Hanlon, D., et al. (2018). The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer's disease. J. Neuroinflammation 15:269. doi: 10.1186/s12974-018-1309-z
Horchar, M. J., and Wohleb, E. S. (2019). Glucocorticoid receptor antagonism prevents microglia-mediated neuronal remodeling and behavioral despair following chronic unpredictable stress. Brain Behav. Immun. 81, 329–340. doi: 10.1016/j.bbi.2019.06.030
Hoshino, K., Hasegawa, K., Kamiya, H., and Morimoto, Y. (2017). Synapse-specific effects of IL-1β on long-term potentiation in the mouse hippocampus. Biomed Res 38, 183–188. doi: 10.2220/biomedres.38.183
Hoshino, K., Uchinami, Y., Uchida, Y., Saito, H., and Morimoto, Y. (2021). Interleukin-1beta modulates synaptic transmission and synaptic plasticity during the acute phase of Sepsis in the senescence-accelerated mouse Hippocampus. Front. Aging Neurosci. 13:637703. doi: 10.3389/fnagi.2021.637703
Hsu, C. C., Kuo, T. W., Liu, W. P., Chang, C. P., and Lin, H. J. (2020). Calycosin preserves BDNF/TrkB signaling and reduces post-stroke neurological injury after cerebral ischemia by reducing accumulation of hypertrophic and TNF-alpha-containing microglia in rats. J. Neuroimmune Pharmacol. 15, 326–339. doi: 10.1007/s11481-019-09903-9
Huang, L., Lafaille, J. J., and Yang, G. (2021). Learning-dependent dendritic spine plasticity is impaired in spontaneous autoimmune encephalomyelitis. Dev. Neurobiol. 81, 736–745. doi: 10.1002/dneu.22827
Huang, P., Zhang, Z., Zhang, P., Feng, J., Xie, J., Zheng, Y., et al. (2023). TREM2 deficiency aggravates NLRP3 inflammasome activation and pyroptosis in MPTP-induced Parkinson’s disease mice and LPS-induced BV2 cells. Mol. Neurobiol. 61, 2590–2605. doi: 10.1007/s12035-023-03713-0
Ising, C., Venegas, C., Zhang, S., Scheiblich, H., Schmidt, S. V., Vieira-Saecker, A., et al. (2019). NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673. doi: 10.1038/s41586-019-1769-z
Jay, T. R., von Saucken, V. E., Muñoz, B., Codocedo, J. F., Atwood, B. K., Lamb, B. T., et al. (2019). TREM2 is required for microglial instruction of astrocytic synaptic engulfment in neurodevelopment. Glia 67, 1873–1892. doi: 10.1002/glia.23664
Jericó, I., Vicuña-Urriza, J., Blanco-Luquin, I., Macias, M., Martinez-Merino, L., Roldán, M., et al. (2023). Profiling TREM2 expression in amyotrophic lateral sclerosis. Brain Behav. Immun. 109, 117–126. doi: 10.1016/j.bbi.2023.01.013
Jiang, Y., Li, Z., Ma, H., Cao, X., Liu, F., Tian, A., et al. (2018). Upregulation of TREM2 ameliorates Neuroinflammatory responses and improves cognitive deficits triggered by surgical trauma in Appswe/PS1dE9 mice. Cell. Physiol. Biochem. 46, 1398–1411. doi: 10.1159/000489155
Jiang, L., Xu, F., He, W., Chen, L., Zhong, H., Wu, Y., et al. (2016). CD200Fc reduces TLR4-mediated inflammatory responses in LPS-induced rat primary microglial cells via inhibition of the NF-kappaB pathway. Inflamm. Res. 65, 521–532. doi: 10.1007/s00011-016-0932-3
Jiang, T., Zhang, Y. D., Chen, Q., Gao, Q., Zhu, X. C., Zhou, J. S., et al. (2016). TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology 105, 196–206. doi: 10.1016/j.neuropharm.2016.01.028
Jiang, C., Zhang, Y., Tang, X., Jing, C., Qiu, S., Li, B., et al. (2021). IL-6 and IL-1beta upregulation and tau protein phosphorylation in response to chronic alcohol exposure in the mouse hippocampus. Neuroreport 32, 851–857. doi: 10.1097/WNR.0000000000001661
John, L. B., and Darcy, P. K. (2016). The double-edged sword of IFN-gamma-dependent immune-based therapies. Immunol. Cell Biol. 94, 527–528. doi: 10.1038/icb.2016.37
Kann, O., Almouhanna, F., and Chausse, B. (2022). Interferon γ: a master cytokine in microglia-mediated neural network dysfunction and neurodegeneration. Trends Neurosci. 45, 913–927. doi: 10.1016/j.tins.2022.10.007
Katsumi, S., Sahin, M. I., Lewis, R. M., Iyer, J. S., Landegger, L. D., and Stankovic, K. M. (2019). Intracochlear perfusion of tumor necrosis factor-alpha induces sensorineural hearing loss and synaptic degeneration in guinea pigs. Front. Neurol. 10:1353. doi: 10.3389/fneur.2019.01353
Kaur, D., Sharma, V., and Deshmukh, R. (2019). Activation of microglia and astrocytes: a roadway to neuroinflammation and Alzheimer's disease. Inflammopharmacology 27, 663–677. doi: 10.1007/s10787-019-00580-x
Kleidonas, D., Kirsch, M., Andrieux, G., Pfeifer, D., Boerries, M., and Vlachos, A. (2023). Microglia modulate TNFα-mediated synaptic plasticity. Glia 71, 2117–2136. doi: 10.1002/glia.24383
Kleidonas, D., and Vlachos, A. (2021). Scavenging tumor necrosis factor α does not affect inhibition of dentate granule cells following in vitro entorhinal cortex lesion. Cells 10:3232. doi: 10.3390/cells10113232
Klein, H. U., Schafer, M., Bennett, D. A., Schwender, H., and De Jager, P. L. (2020). Bayesian integrative analysis of epigenomic and transcriptomic data identifies Alzheimer's disease candidate genes and networks. PLoS Comput. Biol. 16:e1007771. doi: 10.1371/journal.pcbi.1007771
Kobayashi, M., Konishi, H., Sayo, A., Takai, T., and Kiyama, H. (2016). TREM2/DAP12 signal elicits Proinflammatory response in microglia and exacerbates neuropathic pain. J. Neurosci. 36, 11138–11150. doi: 10.1523/JNEUROSCI.1238-16.2016
Kodali, M., Madhu, L. N., Somayaji, Y., Attaluri, S., Huard, C., Panda, P. K., et al. (2024). Residual microglia following short-term PLX5622 treatment in 5xFAD mice exhibit diminished NLRP3 Inflammasome and mTOR signaling, and enhanced autophagy. Aging cell 24:e14398. doi: 10.1101/2024.07.11.603157
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47:e569, 566–581. doi: 10.1016/j.immuni.2017.08.008
Krasovska, V., and Doering, L. C. (2018). Regulation of IL-6 secretion by astrocytes via TLR4 in the fragile X mouse model. Front. Mol. Neurosci. 11:272. doi: 10.3389/fnmol.2018.00272
Kuipers, S. D., Trentani, A., Tiron, A., Mao, X., Kuhl, D., and Bramham, C. R. (2016). BDNF-induced LTP is associated with rapid arc/Arg3.1-dependent enhancement in adult hippocampal neurogenesis. Sci. Rep. 6:21222. doi: 10.1038/srep21222
Kwon, S. H., Han, J. K., Choi, M., Kwon, Y. J., Kim, S. J., Yi, E. H., et al. (2017). Dysfunction of microglial STAT3 alleviates depressive behavior via neuron-microglia interactions. Neuropsychopharmacology 42, 2072–2086. doi: 10.1038/npp.2017.93
Lan, L., Wang, H., Zhang, X., Shen, Q., Li, X., He, L., et al. (2022). Chronic exposure of alcohol triggers microglia-mediated synaptic elimination inducing cognitive impairment. Exp. Neurol. 353:114061. doi: 10.1016/j.expneurol.2022.114061
Lewen, A., Ta, T. T., Cesetti, T., Hollnagel, J. O., Papageorgiou, I. E., Chausse, B., et al. (2020). Neuronal gamma oscillations and activity-dependent potassium transients remain regular after depletion of microglia in postnatal cortex tissue. J. Neurosci. Res. 98, 1953–1967. doi: 10.1002/jnr.24689
Lewitus, G. M., Konefal, S. C., Greenhalgh, A. D., Pribiag, H., Augereau, K., and Stellwagen, D. (2016). Microglial TNF-α suppresses cocaine-induced plasticity and behavioral sensitization. Neuron 90, 483–491. doi: 10.1016/j.neuron.2016.03.030
Li, M., Li, C., Yu, H., Cai, X., Shen, X., Sun, X., et al. (2017). Lentivirus-mediated interleukin-1beta (IL-1beta) knock-down in the hippocampus alleviates lipopolysaccharide (LPS)-induced memory deficits and anxiety- and depression-like behaviors in mice. J. Neuroinflammation 14:190. doi: 10.1186/s12974-017-0964-9
Li, A., Zhao, J., Fan, C., Zhu, L., Huang, C., Li, Q., et al. (2020). Delivery of exogenous proteins by mesenchymal stem cells attenuates early memory deficits in a murine model of Alzheimer's disease. Neurobiol. Aging 86, 81–91. doi: 10.1016/j.neurobiolaging.2019.10.012
Li, C., Zhao, Z., Jin, J., Zhao, C., Zhao, B., and Liu, Y. (2024). NLRP3-GSDMD-dependent IL-1β secretion from microglia mediates learning and memory impairment in a chronic intermittent hypoxia-induced mouse model. Neuroscience 539, 51–65. doi: 10.1016/j.neuroscience.2023.12.006
Li, J., Zhao, R., Li, X., Sun, W., Qu, M., Tang, Q., et al. (2016). Shen-Qi-Jie-Yu-Fang exerts effects on a rat model of postpartum depression by regulating inflammatory cytokines and CD4(+)CD25(+) regulatory T cells. Neuropsychiatr. Dis. Treat. 12, 883–896. doi: 10.2147/NDT.S98131
Lin, L., Chen, X., Zhou, Q., Huang, P., Jiang, S., Wang, H., et al. (2019). Synaptic structure and alterations in the hippocampus in neonatal rats exposed to lipopolysaccharide. Neurosci. Lett. 709:134364. doi: 10.1016/j.neulet.2019.134364
Liu, J., Liu, Y., Chen, J., Hu, C., Teng, M., Jiao, K., et al. (2017). The ROS-mediated activation of IL-6/STAT3 signaling pathway is involved in the 27-hydroxycholesterol-induced cellular senescence in nerve cells. Toxicol. In Vitro 45, 10–18. doi: 10.1016/j.tiv.2017.07.013
Liu, Y., Zhou, L. J., Wang, J., Li, D., Ren, W. J., Peng, J., et al. (2017). TNF-α differentially regulates synaptic plasticity in the Hippocampus and spinal cord by microglia-dependent mechanisms after peripheral nerve injury. J. Neurosci. 37, 871–881. doi: 10.1523/jneurosci.2235-16.2017
Loh, K. H., et al. (2016). Proteomic analysis of unbounded cellular compartments: synaptic clefts. Cell 166:e1221, 1295–1307. doi: 10.1016/j.cell.2016.07.041
Lowery, R. L., Tremblay, M.-E., Hopkins, B. E., and Majewska, A. K. (2017). The microglial fractalkine receptor is not required for activity-dependent plasticity in the mouse visual system. Glia 65, 1744–1761. doi: 10.1002/glia.23192
Luo, P., Chu, S. F., Zhang, Z., Xia, C. Y., and Chen, N. H. (2019). Fractalkine/CX3CR1 is involved in the cross-talk between neuron and glia in neurological diseases. Brain Res. Bull. 146, 12–21. doi: 10.1016/j.brainresbull.2018.11.017
Lyons, A., Minogue, A. M., Jones, R. S., Fitzpatrick, O., Noonan, J., Campbell, V. A., et al. (2017). Analysis of the impact of CD200 on phagocytosis. Mol. Neurobiol. 54, 5730–5739. doi: 10.1007/s12035-016-0223-6
Maggio, N., and Vlachos, A. (2018). Tumor necrosis factor (TNF) modulates synaptic plasticity in a concentration-dependent manner through intracellular calcium stores. J. Mol. Med. (Berl) 96, 1039–1047. doi: 10.1007/s00109-018-1674-1
Maglio, L. E., Noriega-Prieto, J. A., Maraver, M. J., and Fernandez de Sevilla, D. (2018). Endocannabinoid-dependent long-term potentiation of synaptic transmission at rat barrel cortex. Cereb. Cortex 28, 1568–1581. doi: 10.1093/cercor/bhx053
Mandolesi, G., De Vito, F., Musella, A., Gentile, A., Bullitta, S., Fresegna, D., et al. (2017). Mir-142-3p is a key regulator of IL-1β-dependent synaptopathy in neuroinflammation. J. Neurosci. 37, 546–561. doi: 10.1523/jneurosci.0851-16.2017
Manich, G., Recasens, M., Valente, T., Almolda, B., González, B., and Castellano, B. (2019). Role of the CD200-CD200R Axis during homeostasis and Neuroinflammation. Neuroscience 405, 118–136. doi: 10.1016/j.neuroscience.2018.10.030
Mashhadizadeh, S., Farbood, Y., Dianat, M., Khodadadi, A., and Sarkaki, A. (2017). therapeutic effects of ellagic acid on memory, hippocampus electrophysiology deficits, and elevated TNF-α level in brain due to experimental traumatic brain injury. Iran J Basic Med Sci 20, 399–407. doi: 10.22038/IJBMS.2017.8581
Matsumoto, J., Dohgu, S., Takat, F., Machida, T., Bölükbaşi Hatip, F. F., Hatip-Al-Khatib, I., et al. (2018). TNF-alpha-sensitive brain pericytes activate microglia by releasing IL-6 through cooperation between IkappaB-NFkappaB and JAK-STAT3 pathways. Brain Res. 1692, 34–44. doi: 10.1016/j.brainres.2018.04.023
McQuade, A., Kang, Y. J., Hasselmann, J., Jairaman, A., Sotelo, A., Coburn, M., et al. (2020). Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer's disease. Nat. Commun. 11:5370. doi: 10.1038/s41467-020-19227-5
Mein, N., von Stackelberg, N., Wickel, J., Geis, C., and Chung, H.-Y. (2023). Low-dose PLX5622 treatment prevents neuroinflammatory and neurocognitive sequelae after sepsis. J. Neuroinflammation 20:289. doi: 10.1186/s12974-023-02975-8
Meis, S., Endres, T., Munsch, T., and Lessmann, V. (2019). Impact of chronic BDNF depletion on GABAergic synaptic transmission in the lateral amygdala. Int. J. Mol. Sci. 20:174310. doi: 10.3390/ijms20174310
Meng, J., Han, L., Xu, H., Zhang, L., Liu, Z., Zhou, Y., et al. (2024). TREM2 regulates microglial phagocytosis of synapses in innate immune tolerance. Int. Immunopharmacol. 127:111445. doi: 10.1016/j.intimp.2023.111445
Merino, J. J., Muñetón-Gómez, V., Alvárez, M.-I., and Toledano-Díaz, A. (2016). Effects of CX3CR1 and fractalkine chemokines in amyloid beta clearance and p-tau accumulation in Alzheimer’s disease (AD) rodent modelsIs fractalkine a systemic biomarker for AD. Curr Alzheimer Res 13, 403–412. doi: 10.2174/1567205013666151116125714
Milinkeviciute, G., Chokr, S. M., Castro, E. M., and Cramer, K. S. (2021). CX3CR1 mutation alters synaptic and astrocytic protein expression, topographic gradients, and response latencies in the auditory brainstem. J. Comp. Neurol. 529, 3076–3097. doi: 10.1002/cne.25150
Milior, G., Lecours, C., Samson, L., Bisht, K., Poggini, S., Pagani, F., et al. (2016). Fractalkine receptor deficiency impairs microglial and neuronal responsiveness to chronic stress. Brain Behav. Immun. 55, 114–125. doi: 10.1016/j.bbi.2015.07.024
Mirabella, F., Desiato, G., Mancinelli, S., Fossati, G., Rasile, M., Morini, R., et al. (2021). Prenatal interleukin 6 elevation increases glutamatergic synapse density and disrupts hippocampal connectivity in offspring. Immunity 54, 2611–2631.e8. doi: 10.1016/j.immuni.2021.10.006
Monteiro, S., Roque, S., Marques, F., Correia-Neves, M., and Cerqueira, J. J. (2017). Brain interference: revisiting the role of IFNgamma in the central nervous system. Prog. Neurobiol. 156, 149–163. doi: 10.1016/j.pneurobio.2017.05.003
Monteiro, S., Ferreira, F. M., Pinto, V., Roque, S., Morais, M., de Sá-Calçada, D., et al. (2016). Absence of IFNgamma promotes hippocampal plasticity and enhances cognitive performance. Transl. Psychiatry 6:e707. doi: 10.1038/tp.2015.194
Moraes, C. A., Hottz, E. D., Dos Santos Ornellas, D., Adesse, D., de Azevedo, C. T., d’Avila, J. C., et al. (2022). Microglial NLRP3 inflammasome induces excitatory synaptic loss through IL-1β-enriched microvesicle release: implications for sepsis-associated encephalopathy. Mol. Neurobiol. 60, 481–494. doi: 10.1007/s12035-022-03067-z
Ngwa, C., and Liu, F. (2019). CD200-CD200R signaling and diseases: a potential therapeutic target. Int J Physiol Pathophysiol Pharmacol 11, 297–309.
Noh, M. C., Stemkowski, P. L., and Smith, P. A. (2019). Long-term actions of interleukin-1beta on K(+), Na(+) and ca(2+) channel currents in small, IB4-positive dorsal root ganglion neurons; possible relevance to the etiology of neuropathic pain. J. Neuroimmunol. 332, 198–211. doi: 10.1016/j.jneuroim.2019.05.002
Oh, H., Lewis, D. A., and Sibille, E. (2016). The role of BDNF in age-dependent changes of excitatory and inhibitory synaptic markers in the human prefrontal cortex. Neuropsychopharmacology 41, 3080–3091. doi: 10.1038/npp.2016.126
Okubo, M., Yamanaka, H., Kobayashi, K., Dai, Y., Kanda, H., Yagi, H., et al. (2016). Macrophage-Colony stimulating factor derived from injured primary afferent induces proliferation of spinal microglia and neuropathic pain in rats. PLoS One 11:e0153375. doi: 10.1371/journal.pone.0153375
Olde Engberink, A., Hernandez, R., de Graan, P., and Gruol, D. L. (2017). Rapamycin-sensitive late-LTP is enhanced in the Hippocampus of IL-6 transgenic mice. Neuroscience 367, 200–210. doi: 10.1016/j.neuroscience.2017.10.040
Olmos-Alonso, A., Schetters, S. T. T., Sri, S., Askew, K., Mancuso, R., Vargas-Caballero, M., et al. (2016). Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer's-like pathology. Brain 139, 891–907. doi: 10.1093/brain/awv379
Omoyinmi, E., Hamaoui, R., Bryant, A., Jiang, M. C., Athigapanich, T., Eleftheriou, D., et al. (2016). Mitochondrial and oxidative stress genes are differentially expressed in neutrophils of sJIA patients treated with tocilizumab: a pilot microarray study. Pediatr. Rheumatol. Online J. 14:7. doi: 10.1186/s12969-016-0067-7
O'Neill, E., Kwok, B., Day, J. S., Connor, T. J., and Harkin, A. (2016). Amitriptyline protects against TNF-alpha-induced atrophy and reduction in synaptic markers via a Trk-dependent mechanism. Pharmacol. Res. Perspect. 4:e00195. doi: 10.1002/prp2.195
Onodera, J., Nagata, H., Nakashima, A., Ikegaya, Y., and Koyama, R. (2021). Neuronal brain-derived neurotrophic factor manipulates microglial dynamics. Glia 69, 890–904. doi: 10.1002/glia.23934
Onufriev, M. V., Freiman, S. V., Peregud, D. I., Kudryashova, I. V., Tishkina, A. O., Stepanichev, M. Y., et al. (2017). Neonatal Proinflammatory stress induces accumulation of corticosterone and Interleukin-6 in the Hippocampus of juvenile rats: potential mechanism of synaptic plasticity impairments. Biochemistry (Mosc) 82, 275–281. doi: 10.1134/S0006297917030051
Overk, C. R., and Masliah, E. (2014). Pathogenesis of synaptic degeneration in Alzheimer's disease and Lewy body disease. Biochem. Pharmacol. 88, 508–516. doi: 10.1016/j.bcp.2014.01.015
Pan, E., Zhao, Z., and McNamara, J. O. (2019). LTD at mossy fiber synapses onto stratum lucidum interneurons requires TrkB and retrograde endocannabinoid signaling. J Neurophysiol 121, 609–619. doi: 10.1152/jn.00669.2018
Panagiotakopoulou, V., Ivanyuk, D., de Cicco, S., Haq, W., Arsić, A., Yu, C., et al. (2020). Interferon-gamma signaling synergizes with LRRK2 in neurons and microglia derived from human induced pluripotent stem cells. Nat. Commun. 11:5163. doi: 10.1038/s41467-020-18755-4
Paolicelli, R. C., Sierra, A., Stevens, B., Tremblay, M. E., Aguzzi, A., Ajami, B., et al. (2022). Microglia states and nomenclature: a field at its crossroads. Neuron 110, 3458–3483. doi: 10.1016/j.neuron.2022.10.020
Park, S. W., Nhu, L. H., Cho, H. Y., Seo, M. K., Lee, C. H., Ly, N. N., et al. (2016). p11 mediates the BDNF-protective effects in dendritic outgrowth and spine formation in B27-deprived primary hippocampal cells. J. Affect. Disord. 196, 1–10. doi: 10.1016/j.jad.2016.02.010
Patel, R. R., Khom, S., Steinman, M. Q., Varodayan, F. P., Kiosses, W. B., Hedges, D. M., et al. (2019). IL-1beta expression is increased and regulates GABA transmission following chronic ethanol in mouse central amygdala. Brain Behav. Immun. 75, 208–219. doi: 10.1016/j.bbi.2018.10.009
Penney, J., Ralvenius, W. T., Loon, A., Cerit, O., Dileep, V., Milo, B., et al. (2023). iPSC-derived microglia carrying the TREM2 R47H/+ mutation are proinflammatory and promote synapse loss. Glia 72, 452–469. doi: 10.1002/glia.24485
Peters van Ton, A. M., Verbeek, M. M., Alkema, W., Pickkers, P., and Abdo, W. F. (2020). Downregulation of synapse-associated protein expression and loss of homeostatic microglial control in cerebrospinal fluid of infectious patients with delirium and patients with Alzheimer's disease. Brain Behav. Immun. 89, 656–667. doi: 10.1016/j.bbi.2020.06.027
Poniatowski, L. A., Wojdasiewicz, P., Krawczyk, M., Szukiewicz, D., Gasik, R., Kubaszewski, Ł., et al. (2017). Analysis of the role of CX3CL1 (Fractalkine) and its receptor CX3CR1 in traumatic brain and spinal cord injury: insight into recent advances in actions of Neurochemokine agents. Mol. Neurobiol. 54, 2167–2188. doi: 10.1007/s12035-016-9787-4
Popescu, A. S., Butler, C. A., Allendorf, D. H., Piers, T. M., Mallach, A., Roewe, J., et al. (2022). Alzheimer's disease-associated R47H TREM2 increases, but wild-type TREM2 decreases, microglial phagocytosis of synaptosomes and neuronal loss. Glia 71, 974–990. doi: 10.1002/glia.24318
Pozniak, P. D., Darbinyan, A., and Khalili, K. (2016). TNF-alpha/TNFR2 regulatory axis stimulates EphB2-mediated neuroregeneration via activation of NF-kappaB. J. Cell. Physiol. 231, 1237–1248. doi: 10.1002/jcp.25219
Prieto, G. A., Smith, E. D., Tong, L., Nguyen, M., and Cotman, C. W. (2019). Inhibition of LTP-induced translation by IL-1beta reduces the level of newly synthesized proteins in hippocampal dendrites. ACS Chem. Neurosci. 10, 1197–1203. doi: 10.1021/acschemneuro.8b00511
Prieto, G. A., Tong, L., Smith, E. D., and Cotman, C. W. (2019). TNFalpha and IL-1beta but not IL-18 suppresses hippocampal long-term potentiation directly at the synapse. Neurochem. Res. 44, 49–60. doi: 10.1007/s11064-018-2517-8
Prowse, N., and Hayley, S. (2021). Microglia and BDNF at the crossroads of stressor related disorders: towards a unique trophic phenotype. Neurosci. Biobehav. Rev. 131, 135–163. doi: 10.1016/j.neubiorev.2021.09.018
Qi, F., Zuo, Z., Hu, S., Xia, Y., Song, D., Kong, J., et al. (2018). An enriched environment restores hepatitis B vaccination-mediated impairments in synaptic function through IFN-gamma/Arginase1 signaling. Brain Behav. Immun. 71, 116–132. doi: 10.1016/j.bbi.2018.04.003
Qiao, H., An, S. C., Xu, C., and Ma, X. M. (2017). Role of proBDNF and BDNF in dendritic spine plasticity and depressive-like behaviors induced by an animal model of depression. Brain Res. 1663, 29–37. doi: 10.1016/j.brainres.2017.02.020
Qu, W., and Li, L. (2020). Loss of TREM2 confers resilience to synaptic and cognitive impairment in aged mice. J. Neurosci. 40, 9552–9563. doi: 10.1523/JNEUROSCI.2193-20.2020
Rabaneda-Lombarte, N., Serratosa, J., Bové, J., Vila, M., Saura, J., and Solà, C. (2021). The CD200R1 microglial inhibitory receptor as a therapeutic target in the MPTP model of Parkinson's disease. J. Neuroinflammation 18:88. doi: 10.1186/s12974-021-02132-z
Raha, A. A., Henderson, J. W., Stott, S. R., Vuono, R., Foscarin, S., Friedland, R. P., et al. (2017). Neuroprotective effect of TREM-2 in aging and Alzheimer's disease model. J Alzheimer's Dis 55, 199–217. doi: 10.3233/JAD-160663
Reischer, G., Heinke, B., and Sandkuhler, J. (2020). Interferon-gamma facilitates the synaptic transmission between primary afferent C-fibres and lamina I neurons in the rat spinal dorsal horn via microglia activation. Mol. Pain 16:1744806920917249. doi: 10.1177/1744806920917249
Ren, Y., Ye, M., Chen, S., and Ding, J. (2016). CD200 inhibits inflammatory response by promoting KATP Channel opening in microglia cells in Parkinson's disease. Med. Sci. Monit. 22, 1733–1741. doi: 10.12659/msm.898400
Ren, S., Breuillaud, L., Yao, W., Yin, W., Norris, K. A., Zehntner, S. P., et al. (2020b). TNF-alpha-mediated reduction in inhibitory neurotransmission precedes sporadic Alzheimer's disease pathology in young Trem2(R47H) rats. J. Biol. Chem. 296:100089. doi: 10.1074/jbc.RA120.016395
Ren, S., Yao, W., Tambini, M. D., Yin, T., Norris, K. A., and D’Adamio, L. (2020a). Microglia TREM2(R47H) Alzheimer-linked variant enhances excitatory transmission and reduces LTP via increased TNF-alpha levels. eLife 9:e57513. doi: 10.7554/eLife.57513
Reshef, R., Kudryavitskaya, E., Shani-Narkiss, H., Isaacson, B., Rimmerman, N., Mizrahi, A., et al. (2017). The role of microglia and their CX3CR1 signaling in adult neurogenesis in the olfactory bulb. eLife 6:e30809. doi: 10.7554/eLife.30809
Rice, R. A., Pham, J., Lee, R. J., Najafi, A. R., West, B. L., and Green, K. N. (2017). Microglial repopulation resolves inflammation and promotes brain recovery after injury. Glia 65, 931–944. doi: 10.1002/glia.23135
Rizzo, F. R., Musella, A., de Vito, F., Fresegna, D., Bullitta, S., Vanni, V., et al. (2018). Tumor necrosis factor and interleukin-1beta modulate synaptic plasticity during Neuroinflammation. Neural Plast. 2018:8430123. doi: 10.1155/2018/8430123
Roberts, A. J., Khom, S., Bajo, M., Vlkolinsky, R., Polis, I., Cates-Gatto, C., et al. (2019). Increased IL-6 expression in astrocytes is associated with emotionality, alterations in central amygdala GABAergic transmission, and excitability during alcohol withdrawal. Brain Behav. Immun. 82, 188–202. doi: 10.1016/j.bbi.2019.08.185
Rothaug, M., Becker-Pauly, C., and Rose-John, S. (2016). The role of interleukin-6 signaling in nervous tissue. Biochim. Biophys. Acta 1863, 1218–1227. doi: 10.1016/j.bbamcr.2016.03.018
Ru, W., Liu, X., Bae, C., Shi, Y., Walikonis, R., Mo Chung, J., et al. (2019). Microglia mediate HIV-1 gp120-induced synaptic degeneration in spinal pain neural circuits. J. Neurosci. 39, 8408–8421. doi: 10.1523/JNEUROSCI.2851-18.2019
Rueda-Carrasco, J., Sokolova, D., Lee, S. E., Childs, T., Jurčáková, N., Crowley, G., et al. (2023). Microglia-synapse engulfment via PtdSer-TREM2 ameliorates neuronal hyperactivity in Alzheimer's disease models. EMBO J. 42:e113246. doi: 10.15252/embj.2022113246
Ruganzu, J. B., Peng, X., He, Y., Wu, X., Zheng, Q., Ding, B., et al. (2022). Downregulation of TREM2 expression exacerbates neuroinflammatory responses through TLR4-mediated MAPK signaling pathway in a transgenic mouse model of Alzheimer’s disease. Mol. Immunol. 142, 22–36. doi: 10.1016/j.molimm.2021.12.018
Ruganzu, J. B., Zheng, Q., Wu, X., He, Y., Peng, X., Jin, H., et al. (2021). TREM2 overexpression rescues cognitive deficits in APP/PS1 transgenic mice by reducing neuroinflammation via the JAK/STAT/SOCS signaling pathway. Exp. Neurol. 336:113506. doi: 10.1016/j.expneurol.2020.113506
Samidurai, M., Ramasamy, V. S., and Jo, J. (2018). Beta-amyloid inhibits hippocampal LTP through TNFR/IKK/NF-kappaB pathway. Neurol. Res. 40, 268–276. doi: 10.1080/01616412.2018.1436872
Schubert, I., Ahlbrand, R., Winter, A., Vollmer, L., Lewkowich, I., and Sah, R. (2018). Enhanced fear and altered neuronal activation in forebrain limbic regions of CX3CR1-deficient mice. Brain Behav. Immun. 68, 34–43. doi: 10.1016/j.bbi.2017.09.013
Sheng, L., Chen, M., Cai, K., Song, Y., Yu, D., Zhang, H., et al. (2019). Microglial Trem2 induces synaptic impairment at early stage and prevents amyloidosis at late stage in APP/PS1 mice. FASEB J. 33, 10425–10442. doi: 10.1096/fj.201900527R
Sheppard, O., Coleman, M. P., and Durrant, C. S. (2019). Lipopolysaccharide-induced neuroinflammation induces presynaptic disruption through a direct action on brain tissue involving microglia-derived interleukin 1 beta. J. Neuroinflammation 16:106. doi: 10.1186/s12974-019-1490-8
Sidlauskaite, E., Gibson, J. W., Megson, I. L., Whitfield, P. D., Tovmasyan, A., Batinic-Haberle, I., et al. (2018). Mitochondrial ROS cause motor deficits induced by synaptic inactivity: implications for synapse pruning. Redox Biol. 16, 344–351. doi: 10.1016/j.redox.2018.03.012
Singh, A., Jones, O. D., Mockett, B. G., Ohline, S. M., and Abraham, W. C. (2019). Tumor necrosis factor-alpha-mediated metaplastic inhibition of LTP is constitutively engaged in an Alzheimer's disease model. J. Neurosci. 39, 9083–9097. doi: 10.1523/JNEUROSCI.1492-19.2019
Sobue, A., Ito, N., Nagai, T., Shan, W., Hada, K., Nakajima, A., et al. (2018). Astroglial major histocompatibility complex class I following immune activation leads to behavioral and neuropathological changes. Glia 66, 1034–1052. doi: 10.1002/glia.23299
Stampanoni Bassi, M., Iezzi, E., Mori, F., Simonelli, I., Gilio, L., Buttari, F., et al. (2019). Interleukin-6 disrupts synaptic plasticity and impairs tissue damage compensation in multiple sclerosis. Neurorehabil. Neural Repair 33, 825–835. doi: 10.1177/1545968319868713
Stampanoni Bassi, M., Buttari, F., Nicoletti, C. G., et al. (2020). Interleukin-1beta alters Hebbian synaptic plasticity in multiple sclerosis. Int. J. Mol. Sci. 21. doi: 10.3390/ijms21196982
Stefano, G. B., Buttiker, P., Michaelsen, M. M., and Esch, T. (2025). The anatomical and evolutionary impact of pain, pleasure, motivation, and cognition: integrating energy metabolism and the mind–body BERN (behavior, exercise, relaxation, and nutrition) framework. Int. J. Mol. Sci. 26. doi: 10.3390/ijms26125491
Sun, H., He, X., Tao, X., Hou, T., Chen, M., He, M., et al. (2020). The CD200/CD200R signaling pathway contributes to spontaneous functional recovery by enhancing synaptic plasticity after stroke. J. Neuroinflammation 17:171. doi: 10.1186/s12974-020-01845-x
Sung, C. S., Wen, Z. H., Feng, C. W., Chen, C. H., Huang, S. Y., Chen, N. F., et al. (2017). Potentiation of spinal glutamatergic response in the neuron-glia interactions underlies the intrathecal IL-1beta-induced thermal hyperalgesia in rats. CNS Neurosci. Ther. 23, 580–589. doi: 10.1111/cns.12705
Ta, T. T., Dikmen, H. O., Schilling, S., Chausse, B., et al. (2019). Priming of microglia with IFN-gamma slows neuronal gamma oscillations in situ. Proc. Natl. Acad. Sci. USA 116, 4637–4642. doi: 10.1073/pnas.1813562116
Tanaka, N., Cortese, G. P., Barrientos, R. M., Maier, S. F., and Patterson, S. L. (2018). Aging and an immune challenge interact to produce prolonged, but not permanent, reductions in hippocampal L-LTP and mBDNF in a rodent model with features of delirium. ENeuro 5:ENEURO.0009-18.2018. doi: 10.1523/ENEURO.0009-18.2018
Taoro-Gonzalez, L., Cabrera-Pastor, A., and Sancho-Alonso, M.. (2019). Differential role of interleukin-1beta in neuroinflammation-induced impairment of spatial and nonspatial memory in hyperammonemic rats. FASEB J. 33, 9913–9928. doi: 10.1096/fj.201900230RR
Tian, Y., Xiao, X., Liu, W., Cheng, S., Qian, N., Wang, L., et al. (2024). TREM2 improves microglia function and synaptic development in autism spectrum disorders by regulating P38 MAPK signaling pathway. Mol. Brain 17:12. doi: 10.1186/s13041-024-01081-x
Todd, L., Palazzo, I., Suarez, L., Liu, X., Volkov, L., Hoang, T. V., et al. (2019). Reactive microglia and IL1beta/IL-1R1-signaling mediate neuroprotection in excitotoxin-damaged mouse retina. J. Neuroinflammation 16:118. doi: 10.1186/s12974-019-1505-5
Tomasoni, R., Morini, R., Lopez-Atalaya, J. P., Corradini, I., Canzi, A., Rasile, M., et al. (2017). Lack of IL-1R8 in neurons causes hyperactivation of IL-1 receptor pathway and induces MECP2-dependent synaptic defects. eLife 6. doi: 10.7554/eLife.21735
Tomassoni-Ardori, F., Fulgenzi, G., Becker, J., Barrick, C., Palko, M. E., Kuhn, S., et al. (2019). Rbfox1 up-regulation impairs BDNF-dependent hippocampal LTP by dysregulating TrkB isoform expression levels. eLife 8. doi: 10.7554/eLife.49673
Tong, L., Chen, G., Liu, T., Wang, L., Zhang, H., Chen, F., et al. (2023). IFN-γ deficiency in the rostral ventrolateral medulla contributes to stress-induced hypertension by impairing microglial synaptic engulfment. J. Hypertens. 41, 1323–1332. doi: 10.1097/hjh.0000000000003470
Tong, L., Prieto, G. A., and Cotman, C. W. (2018). IL-1beta suppresses cLTP-induced surface expression of GluA1 and actin polymerization via ceramide-mediated Src activation. J. Neuroinflammation 15:127. doi: 10.1186/s12974-018-1158-9
Tran, K. M., Kawauchi, S., Kramár, E. A., Rezaie, N., Liang, H. Y., Sakr, J. S., et al. (2023). A Trem2R47H mouse model without cryptic splicing drives age- and disease-dependent tissue damage and synaptic loss in response to plaques. Mol. Neurodegener. 18:12. doi: 10.1186/s13024-023-00598-4
van der Maten, G., Henck, V., Wieloch, T., and Ruscher, K. (2017). CX3C chemokine receptor 1 deficiency modulates microglia morphology but does not affect lesion size and short-term deficits after experimental stroke. BMC Neurosci. 18:11. doi: 10.1186/s12868-016-0325-0
Vergara, D., Nigro, A., Romano, A., de Domenico, S., Damato, M., Franck, J., et al. (2019). Distinct protein expression networks are activated in microglia cells after stimulation with IFN-gamma and IL-4. Cells 8:580. doi: 10.3390/cells8060580
Vignoli, B., Battistini, G., Melani, R., Blum, R., Santi, S., Berardi, N., et al. (2016). Peri-synaptic glia recycles brain-derived neurotrophic factor for LTP stabilization and memory retention. Neuron 92, 873–887. doi: 10.1016/j.neuron.2016.09.031
Wake, H., Horiuchi, H., Kato, D., Moorhouse, A. J., and Nabekura, J. (2019). Physiological implications of microglia-synapse interactions. Methods Mol. Biol. 2034, 69–80. doi: 10.1007/978-1-4939-9658-2_6
Walker, D. G., Lue, L. F., Tang, T. M., Adler, C. H., Caviness, J. N., Sabbagh, M. N., et al. (2017). Changes in CD200 and intercellular adhesion molecule-1 (ICAM-1) levels in brains of Lewy body disorder cases are associated with amounts of Alzheimer's pathology not alpha-synuclein pathology. Neurobiol. Aging 54, 175–186. doi: 10.1016/j.neurobiolaging.2017.03.007
Wang, H. T., Huang, F. L., Hu, Z. L., Zhang, W. J., Qiao, X. Q., Huang, Y. Q., et al. (2017). Early-life social isolation-induced depressive-like behavior in rats results in microglial activation and neuronal histone methylation that are mitigated by minocycline. Neurotox. Res. 31, 505–520. doi: 10.1007/s12640-016-9696-3
Wang, L., Liu, Y., Yan, S., du, T., Fu, X., Gong, X., et al. (2020). Disease progression-dependent expression of CD200R1 and CX3CR1 in mouse models of Parkinson's disease. Aging Dis. 11, 254–268. doi: 10.14336/AD.2019.0615
Wang, X., Ma, C., Liu, C. Y., Li, G. J., Zhao, D., and Han, D. F. (2019). Neuronal NMDAR currents of the Hippocampus and learning performance in autoimmune anti-NMDAR encephalitis and involvement of TNF-alpha and IL-6. Front. Neurol. 10:684. doi: 10.3389/fneur.2019.00684
Wang, Y., Wernersbach, I., Strehle, J., Li, S., Appel, D., Klein, M., et al. (2022). Early posttraumatic CSF1R inhibition via PLX3397 leads to time- and sex-dependent effects on inflammation and neuronal maintenance after traumatic brain injury in mice. Brain Behav. Immun. 106, 49–66. doi: 10.1016/j.bbi.2022.07.164
Wang, X., Xie, Y., Niu, Y., Wan, B., Lu, Y., Luo, Q., et al. (2023). CX3CL1/CX3CR1 signal mediates M1-type microglia and accelerates high-altitude-induced forgetting. Front Cell Neurosci 17:1189348. doi: 10.3389/fncel.2023.1189348
Wang, C., Xiong, M., Gratuze, M., Bao, X., Shi, Y., Andhey, P. S., et al. (2021). Selective removal of astrocytic strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 109:1657. doi: 10.1016/j.neuron.2021.03.024
Wegrzyn, D., Freund, N., Faissner, A., and Juckel, G. (2021). Poly I:C activated microglia disrupt Perineuronal nets and modulate synaptic balance in primary hippocampal neurons in vitro. Front Synaptic Neurosci 13:637549. doi: 10.3389/fnsyn.2021.637549
Wei, H., Ma, Y., Liu, J., Ding, C., Jin, G., Wang, Y., et al. (2016). Inhibition of IL-6 trans-signaling in the brain increases sociability in the BTBR mouse model of autism. Biochim. Biophys. Acta 1862, 1918–1925. doi: 10.1016/j.bbadis.2016.07.013
Wei, L., Yu, X., Chen, H., and Du, Y. (2024). The role of P2X4R in regulating CA1 hippocampal synaptic impairment in LPS-induced depression. J. Affect. Disord. 362, 595–605. doi: 10.1016/j.jad.2024.07.052
Weyer, M.-P., Strehle, J., Schäfer, M. K. E., and Tegeder, I. (2024). Repurposing of pexidartinib for microglia depletion and renewal. Pharmacol. Ther. 253:108565. doi: 10.1016/j.pharmthera.2023.108565
White, A. O., Kramár, E. A., López, A. J., Kwapis, J. L., Doan, J., Saldana, D., et al. (2016). BDNF rescues BAF53b-dependent synaptic plasticity and cocaine-associated memory in the nucleus accumbens. Nat. Commun. 7:11725. doi: 10.1038/ncomms11725
Wigerblad, G., Huie, J. R., Yin, H. Z., Leinders, M., Pritchard, R. A., Koehrn, F. J., et al. (2017). Inflammation-induced GluA1 trafficking and membrane insertion of ca(2+) permeable AMPA receptors in dorsal horn neurons is dependent on spinal tumor necrosis factor, PI3 kinase and protein kinase a. Exp. Neurol. 293, 144–158. doi: 10.1016/j.expneurol.2017.04.004
Willis, E. F., MacDonald, K. P. A., Nguyen, Q. H., et al. (2020). Repopulating microglia promote brain repair in an IL-6-dependent manner. Cell 180, 833–846. doi: 10.1016/j.cell.2020.02.013
Wohleb, E. S., Terwilliger, R., Duman, C. H., and Duman, R. S. (2018). Stress-induced neuronal Colony stimulating factor 1 provokes microglia-mediated neuronal remodeling and depressive-like behavior. Biol. Psychiatry 83, 38–49. doi: 10.1016/j.biopsych.2017.05.026
Wu, Y., Liu, Q., Guo, B., Ye, F., Ge, J., and Xue, L. (2020). BDNF activates postsynaptic TrkB receptors to induce endocannabinoid release and inhibit presynaptic calcium influx at a Calyx-type synapse. J. Neurosci. 40, 8070–8087. doi: 10.1523/JNEUROSCI.2838-19.2020
Xiao, Z., Peng, J., Wu, L., Arafat, A., and Yin, F. (2017). The effect of IL-1beta on synaptophysin expression and electrophysiology of hippocampal neurons through the PI3K/Akt/mTOR signaling pathway in a rat model of mesial temporal lobe epilepsy. Neurol. Res. 39, 640–648. doi: 10.1080/01616412.2017.1312070
Xu, X. E., Liu, L., Wang, Y. C., Wang, C. T., Zheng, Q., Liu, Q. X., et al. (2019). Caspase-1 inhibitor exerts brain-protective effects against sepsis-associated encephalopathy and cognitive impairments in a mouse model of sepsis. Brain Behav. Immun. 80, 859–870. doi: 10.1016/j.bbi.2019.05.038
Xu, T., Li, D., Zhou, X., Ouyang, H. D., Zhou, L. J., et al. (2017). Oral application of magnesium-L-threonate attenuates vincristine-induced allodynia and hyperalgesia by normalization of tumor necrosis factor-alpha/nuclear factor-kappaB signaling. Anesthesiology 126, 1151–1168. doi: 10.1097/ALN.0000000000001601
Yan, X., Li, F., Maixner, D. W., Yadav, R., Gao, M., Ali, M. W., et al. (2019). Interleukin-1beta released by microglia initiates the enhanced glutamatergic activity in the spinal dorsal horn during paclitaxel-associated acute pain syndrome. Glia 67, 482–497. doi: 10.1002/glia.23557
Yang, G., Chen, L., Gao, Z., and Wang, Y. (2018). Implication of microglia activation and CSF1-CSF1R pathway in lumbar disc degeneration-related back pain. Mol Pain. 14:1744806918811238. doi: 10.1177/1744806918811238
Yang, L., Jin, P., Wang, X., Zhou, Q., Lin, X., and Xi, S. (2018). Fluoride activates microglia, secretes inflammatory factors and influences synaptic neuron plasticity in the hippocampus of rats. Neurotoxicology 69, 108–120. doi: 10.1016/j.neuro.2018.09.006
Yang, X., Li, T., Liu, J., Sun, H., Cheng, L., Song, X., et al. (2023). Effects of minocycline on dendrites, dendritic spines, and microglia in immature mouse brains after kainic acid-induced status epilepticus. CNS Neurosci. Ther. 30. doi: 10.1111/cns.14352
Yang, G., Tong, Y., Wang, X., Zhao, C., Ba, Z., Ahelijiang, R., et al. (2023). Guizhi Fuling capsule relieves memory deficits by inhibition of microglial neuroinflammation through blocking JAK2/STAT3 pathway in presenilin1/2 conditional double knockout mice. Front. Immunol. 14. doi: 10.3389/fimmu.2023.1185570
Yang, Q., Zhang, Y., Zhang, L., Li, X., Dong, R., Song, C., et al. (2021). Combination of tea polyphenols and proanthocyanidins prevents menopause-related memory decline in rats via increased hippocampal synaptic plasticity by inhibiting p38 MAPK and TNF-α pathway. Nutr. Neurosci. 25, 1909–1927. doi: 10.1080/1028415x.2021.1913929
Yang, Y., Zhong, Z., Wang, B., Xia, X., Yao, W., Huang, L., et al. (2019). Early-life high-fat diet-induced obesity programs hippocampal development and cognitive functions via regulation of gut commensal Akkermansia muciniphila. Neuropsychopharmacology 44, 2054–2064. doi: 10.1038/s41386-019-0437-1
Yeh, F. L., Hansen, D. V., and Sheng, M. (2017). TREM2, microglia, and neurodegenerative diseases. Trends Mol. Med. 23, 512–533. doi: 10.1016/j.molmed.2017.03.008
Yeh, M. L., Selvam, R., and Levine, E. S. (2017). BDNF-induced endocannabinoid release modulates neocortical glutamatergic neurotransmission. Synapse 71. doi: 10.1002/syn.21962
Yi, M. H., Zhang, E., Kim, J. J., Baek, H., Shin, N., Kim, S., et al. (2016). CD200R/Foxp3-mediated signalling regulates microglial activation. Sci. Rep. 6:34901. doi: 10.1038/srep34901
Yirmiya, R., and Goshen, I. (2011). Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 25, 181–213. doi: 10.1016/j.bbi.2010.10.015
York, E. M., Zhang, J., Choi, H. B., and MacVicar, B. A. (2021). Neuroinflammatory inhibition of synaptic long-term potentiation requires immunometabolic reprogramming of microglia. Glia 69, 567–578. doi: 10.1002/glia.23913
You, Y., An, D. D., Wan, Y. S., Zheng, B. X., Dai, H. B., Zhang, S. H., et al. (2024). Cell-specific IL-1R1 regulates the regional heterogeneity of microglial displacement of GABAergic synapses and motor learning ability. Cell. Mol. Life Sci. 81:116. doi: 10.1007/s00018-023-05111-0
Yu, Q., Liu, M., Dai, W., Xiong, Y., Mu, X., Xia, M., et al. (2023). The NLRP3 inflammasome is involved in resident intruder paradigm-induced aggressive behaviors in mice. Front. Pharmacol. 14. doi: 10.3389/fphar.2023.974905
Zhang, J., Dong, Y., Lining Huang,, Xu, X., Liang, F., Soriano, S. G., et al. (2020). Interaction of tau, IL-6 and mitochondria on synapse and cognition following sevoflurane anesthesia in young mice. Brain, Behavior, & Immunity - Health 8:100133. doi: 10.1016/j.bbih.2020.100133
Zhang, Y., Feng, S., Nie, K., Li, Y., Gao, Y., Gan, R., et al. (2018). TREM2 modulates microglia phenotypes in the neuroinflammation of Parkinson's disease. Biochem. Biophys. Res. Commun. 499, 797–802. doi: 10.1016/j.bbrc.2018.03.226
Zhang, X., Yan, F., Cui, J., Wu, Y., Luan, H., Yin, M., et al. (2017). Triggering receptor expressed on myeloid cells 2 overexpression inhibits Proinflammatory cytokines in lipopolysaccharide-stimulated microglia. Mediat. Inflamm. 2017, 1–6. doi: 10.1155/2017/9340610
Zhao, X., Cao, F., Liu, Q., Li, X., Xu, G., Liu, G., et al. (2019). Behavioral, inflammatory and neurochemical disturbances in LPS and UCMS-induced mouse models of depression. Behav. Brain Res. 364, 494–502. doi: 10.1016/j.bbr.2017.05.064
Zhong, L., Sheng, X., Wang, W., Li, Y., Zhuo, R., Wang, K., et al. (2023). TREM2 receptor protects against complement-mediated synaptic loss by binding to complement C1q during neurodegeneration. Immunity 56, 1794–1808.e8. doi: 10.1016/j.immuni.2023.06.016
Zhou, R., and Bickler, P. (2017). interaction of isoflurane, tumor necrosis factor-alpha and beta-amyloid on long-term potentiation in rat hippocampal slices. Anesth Analg 124, 582–587. doi: 10.1213/ANE.0000000000001698
Zhou, L. J., et al. (2019). Microglia are indispensable for synaptic plasticity in the spinal dorsal horn and chronic pain. Cell Rep. 27, 3844–3859 e3846. doi: 10.1016/j.celrep.2019.05.087
Keywords: microglia, neuroinflammation, cytokines/receptors, synaptic modification, synaptic plasticity, synaptic pruning, neurodegeneration
Citation: Yang G, Xu X, Gao W, Wang X, Zhao Y and Xu Y (2025) Microglia-orchestrated neuroinflammation and synaptic remodeling: roles of pro-inflammatory cytokines and receptors in neurodegeneration. Front. Cell. Neurosci. 19:1700692. doi: 10.3389/fncel.2025.1700692
Edited by:
Francisco M. Nadal-Nicolás, National Eye Institute (NIH), United StatesReviewed by:
Luana Chagas, Fluminense Federal University, BrazilMarie Karam, Spanish National Research Council (CSIC), Spain
Copyright © 2025 Yang, Xu, Gao, Wang, Zhao and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Xu, eWluZ3h1NjEyQHNodXRjbS5lZHUuY24=; Yan Zhao, eWFuemhhbzMxOUBzaHV0Y20uZWR1LmNu
†These authors have contributed equally to this work