 Yaroslav I. Molkov1,2*
Yaroslav I. Molkov1,2* Florent Krust3†
Florent Krust3† Russell Jeter1,2†
Russell Jeter1,2† Tommy Stell1Mohammed A. Y. Mohammed1Frédéric Brocard3‡
Tommy Stell1Mohammed A. Y. Mohammed1Frédéric Brocard3‡ Ilya A. Rybak4‡
Ilya A. Rybak4‡- 1Department of Mathematics and Statistics, Georgia State University, Atlanta, GA, United States
- 2Neuroscience Institute, Georgia State University, Atlanta, GA, United States
- 3Institut de Neurosciences de la Timone, Aix Marseille University, Centre National de la Recherche Scientifique (CNRS), Marseille, France
- 4Department of Neurobiology and Anatomy, Drexel University College of Medicine, Philadelphia, PA, United States
Spinal motoneurons are the final output of spinal circuits that engage skeletal muscles to generate motor behaviors. Many motoneurons exhibit bistable behavior, alternating between a quiescent resting state and a self-sustained firing mode, classically attributed to plateau potentials driven by persistent inward currents. This intrinsic property is important for normal movement control, but can become dysregulated, causing motor function deficits, like spasticity. Here we use a conductance-based single-compartment model, together with mouse spinal slice recordings, to investigate the ionic interactions underlying motoneuron bistability. We show that synergistic interactions among high-voltage-activated L-type Ca2+ current (ICaL), calcium-induced calcium release (CICR) and the Ca2+-activated non-specific cation current (ICAN) constitute a minimal mechanistic core that produces plateau potentials and bistable firing. Within this framework, the persistent sodium current (INaP) promotes plateau generation, in contrast to the Ca2+-dependent K+ current (IKCa) which opposes it. These results delineate ionic dependencies at the level of interactions rather than spatial localization and provide a tractable basis for interpreting altered motoneuron excitability in disease.
Introduction
Motoneurons show a variety of non-linear intrinsic behaviors that determine their input-output properties (Binder et al., 2020). Among them, bistability allows motoneurons to toggle between two stable states: a quiescent resting state, and a self-sustained firing state characterized by a regular pattern of action potentials generated in absence of synaptic drive. A short excitatory input can trigger a transition from the quiescent state to the active state, whereas a brief inhibitory input can induce the opposite transition (Hultborn et al., 1975; Hounsgaard et al., 1984). The underlying mechanism of the bistability involves formation of plateau potentials, long-lasting membrane depolarizations, during which the motoneuron firing is maintained (Hounsgaard et al., 1988a; Hounsgaard and Mintz, 1988). Persistent firing described in motor units in intact animals (Eken et al., 1989; Eken and Kiehn, 1989; Kiehn and Eken, 1997) as well as in humans (Gorassini et al., 1998, 1999; Collins et al., 2001) provides indirect evidence that motoneuron bistability is an important feature of motor control contributing to many behaviors. It has been suggested that bistability is involved in postural control lessening the requirement for continuous synaptic drive (Hounsgaard et al., 1988a; Lee and Heckman, 1998a; Heckman et al., 2009). Consistent with this, limiting motoneuron bistability was shown to impair postural control (Bos et al., 2021).
Early studies proposed that bistability primarily resulted from a persistent component of the calcium current (ICaL) (Hounsgaard and Mintz, 1988; Hounsgaard and Kiehn, 1989) conducted through Cav1.3 channels (Carlin et al., 2000; Simon et al., 2003; Zhang et al., 2006). However, subsequent research has shown that bistability results from a more complex interaction of several ionic currents (Bouhadfane et al., 2013), including the calcium-activated non-specific cation current (ICAN) through the Transient Receptor Potential Melastatin 5 (TRPM5) channels (Bos et al., 2021) and the persistent sodium current (INaP) via Nav1.6 channels (Drouillas et al., 2023). The emerging picture is a causal sequence of cellular processes: the initial membrane depolarization activates INaP, triggering firing and calcium influx; ICaL increases intracellular calcium, which then activates calcium-induced calcium release (CICR) and recruits ICAN, leading to further depolarizing of the membrane. This cascade of depolarization and triggered currents leads to self-sustained firing, reinforcing the positive feedback loop underlying bistability. The contribution of these intrinsic processes to motoneuron bistability is closely tied to cell size with larger motoneurons exhibiting both higher current expression and a greater propensity for bistable behavior (Harris-Warrick et al., 2024).
Bistability is modulated by neuromodulatory and biophysical contexts. Monoamines released from supraspinal centers can promote and unmask bistability in motoneurons with its latent or partial expression (Conway et al., 1988; Hounsgaard et al., 1988a; Hounsgaard and Kiehn, 1989; Perrier and Delgado-Lezama, 2005). Temperature also modulates bistability: values above 30 °C unmask plateau potentials by recruiting thermosensitive currents mediated by TRPM5 channels (Bos et al., 2021).
Beyond its physiological role, bistability has critical implications in pathology. Following spinal cord injury, excessive bistability in motoneurones has been linked to spasticity, a disabling condition characterized by involuntary muscle contractions (Bennett et al., 2001; Heckman et al., 2008) arising from dysregulated intrinsic ionic currents that amplify self-sustained firing and disrupt motor control (Brocard et al., 2016; Murray et al., 2010).
Despite significant progress, the integration of multiple ionic mechanisms underlying bistability remains poorly understood. Most prior studies often examined individual currents in isolation, such as INaP, ICaLor ICANwithout considering how their non-linear interactions jointly generate and stabilize bistable firing. In this study, we combine computational modeling, using a conductance-based single-compartment motoneuron model, with mouse spinal slice recordings to dissect how these currents, considered separately and in combination, control the emergence, maintenance and modulation of bistability in motoneurons. Our goal is to identify a minimal mechanistic core of motoneuron bistability at the level of ionic interactions.
Methods
Modeling methods
Motoneuron model
We used a conductance-based single-compartment mathematical model of a motoneuron that includes the main spike-generating channels, fast sodium (INaF) and potassium rectifier (IKdr), as well as the persistent sodium (INaP), slowly inactivating potassium (IKv1.2), high-voltage activated calcium (ICaL), Ca2+-activated, non-specific cation (ICAN, associated with TRPM5 channels) and Ca2+-dependent potassium (IKCa, associated with SK channels) current.
The voltage dynamics are described by the current balance equation:
where V is membrane potential in mV, t is time in ms, C is membrane capacitance (C = 1 μF·cm−2), and the right hand side of the equation contains all transmembrane currents described as follows.
Fast sodium current (Booth et al., 1997):
The persistent sodium current was assumed to be non-inactivating. Activation was assumed instantaneous and non-inactivating. Activation dependence on voltage was taken from (Brocard et al., 2013):
Persistent sodium current is assumed non-inactivating, i.e. hNaP = 1.
Potassium rectifier current (Booth et al., 1997):
Slowly inactivating potassium current (Bos et al., 2018):
High-voltage calcium L (ICaL) current (Jasinski et al., 2013):
Calcium-activated non-specific cation (ICAN) current (Toporikova and Butera, 2011):
Calcium-dependent potassium current (IKCa) (Booth et al., 1997). This current was modified to be instantaneous:
Leak current:
The two currents described above (ICAN and IKCa) depend on the intracellular Ca2+ concentration [Ca2+]i in mM (denoted by Ca in the equations). The Ca2+ concentration increases directly from the influx of calcium ions through calcium channels (captured by ICaLin the mathematical model) and indirectly from the release of calcium ions from intracellular stores via a calcium-induced calcium release (CICR) mechanism. Additionally, they are pumped out by the Ca-ATP pumps.
The dynamics of intracellular calcium concentration (Ca) in our model are described by the differential equation:
On the right hand side of this equation, the first term represents calcium influx through high-threshold, voltage-gated calcium channels (ICaL) which open during action potentials. Here f = 0.01 defines the ratio of entered Ca2+ ions remaining unbound; the coefficient α = (2·F·δ)−1 converts inward ICaL current to Ca2+ concentration rate of change; here F is Faraday's constant (F = 9.648 · 104 C/mol) and δ is the thickness (0.1 μm) of the shell adjacent to the membrane. Based on these parameters, α = 5·10−4 mM·cm2·ms−1·μA−1.
The second term describes an increase of cytoplasmic Ca2+ concentration through the CICR mechanism, where the rate of calcium release from internal stores is proportional to the intracellular Ca2+ concentration (defined by kCICR). The third term describes the action of calcium pumps (both plasma membrane and SERCA) which rapidly remove calcium from the cytoplasm, with a time constant τCa = 10 ms.
Qualitative analysis
To investigate the bistable behavior of spinal motoneurons, we implemented a current ramp simulation protocol designed to probe the transitions between quiescent and self-sustained firing states. This approach leverages a linearly varying injected current (Iinj) to systematically explore the system's response across a range of input intensities, making it an effective tool for detecting hysteresis and state-dependent dynamics in neuronal models.
The protocol was designed as follows: the injected current was initially set to zero and then increased linearly to a predetermined maximum value (ascending phase) over a specified duration. Subsequently, the current was decreased linearly back to zero (descending phase) at the same rate. This bidirectional ramp allowed us to identify two key transition points: the current threshold at which the system shifts from silence to spiking during the ascending phase (Iup) and the lower threshold at which spiking ceases during the descending phase (Idown). Bistability is indicated when Idown is less than Iup, revealing a range of current values where the system can stably maintain either state, depending on its prior condition. This hysteresis reflects the non-linear properties of the model and its history-dependent behavior. To ensure the reliability of these thresholds, the ramp time was progressively increased until the current thresholds for transition to spiking (Iup) and for return to silence (Idown) stabilized. , confirming that the observed transitions were not influenced by transient dynamics. Ramp durations of several seconds per phase were typically sufficient; longer ramps produced nearly identical Iup and Idown values, confirming that ramp velocity no longer influenced the hysteresis width.
The results of this protocol are presented in injected current–voltage (I–V) bifurcation diagrams. These diagrams depict the steady-state voltage response as a function of the injected current, delineating the regions corresponding to quiescence, repetitive spiking, and bistability. The upward and downward branches correspond to the ascending and descending phases of the current ramp, respectively, thereby illustrating the distinct transition thresholds (Iup and Idown) and the hysteresis loop between them. For interpretation, the I–V bifurcation diagram provides a compact representation of how the system's qualitative behavior changes with input strength, serving as a graphical summary of the underlying non-linear dynamics and the coexistence of multiple stable states.
Equilibrium points of our single-compartment model, which represent the steady-state solutions of the system, were incorporated into the bifurcation diagrams. These points were determined by setting the net membrane current (the right-hand side of Equation 1) to zero. For each fixed value of injected current (Iinj), the gating variables and intracellular calcium concentration were assigned their steady-state values. The corresponding equilibrium membrane potential was then numerically obtained by solving the aforementioned equation for V. By collecting these steady-state points across a range of Iinj, one or more branches of the equilibrium voltage were formed.
To further validate the protocol's ability to detect bistability, we employed a complementary current step simulation inspired by experimental approaches. Starting from a baseline of Iinj = 0, the current was stepped to an intermediate value within the suspected bistable range (Idown<Iinj<Iup), then increased to a level above Iup, and subsequently returned to the intermediate value before returning to zero. This sequence demonstrated that, at the intermediate current level, the system's state, silent or spiking, depended on its prior activation history, reinforcing the findings of the ramp protocol.
The current ramp simulations also supported parametric analyses by varying key model parameters and plotting the resulting bifurcation diagrams. These diagrams mapped the system's equilibrium points and oscillatory regimes as functions of Iinj, providing a visual representation of the bistable region. The protocol's design, with its carefully adjusted ramp duration, ensured that the gradual variation of input current effectively captured the boundaries of this region, offering a robust technical framework for studying the conditions under which bistability emerges and persists in the model.
Simulations
Simulations were performed using custom-written C++ and Julia software. Integration was performed by the Dormand-Prince 5(4) method using (Boost Development Team, n.d.). Source code written in C++ and Julia for the model and examples of the ramping protocols can be found in the Github repository associated with this manuscript (Jeter and Molkov, 2025).
Experimental methods
Experimental model
Mice (C57/Bl6 background) were housed under a 12 h light/dark cycle with ad libitum access to water and food. Room temperature was kept between 21 and 24 °C and between 40 and 60% relative humidity. All animal care and use were conformed to the French regulations (Décret 2010-118) and approved by the local ethics committee (Comité d'Ethique en Neurosciences INT-Marseille, CE71 Nb A1301404, authorization Nb #50133-2024060612594852).
In vitro preparations
For the slice preparation, mice were cryoanaesthetized (P5-P7) or anesthetized (P8-P11) with intraperitoneal injection of a mixture of ketamine/xylazine (100 and 10 mg/kg, respectively). They were then decapitated, eviscerated and the spinal cord removed by laminectomy, and placed in a Sylgard-lined petri dish with ice-cold (+4 °C) artificial CSF (aCSF) solution composed of the following (in mM): 252 sucrose, 3 KCl, 1.25 KH2PO4, 4 MgSO4, 0.2 CaCl2, 26 NaHCO3, 25 D-glucose, pH 7.4. The meninges were removed and the lumbar spinal cord was then introduced into a 1% agar solution, quickly cooled, mounted in a vibrating microtome (Leica, VT1000S) and sliced (325 μm) through the L4–5 lumbar segments. Slices were immediately transferred into the holding chamber filled with bubbled (95% O2 and 5% CO2) aCSF solution composed of (in mM): 120 NaCl, 3 KCl, 1.25 NaH2PO4, 1.3 MgSO4, 1.2 CaCl2, 25 NaHCO3, 20 D-glucose, pH 7.4, 30–32 °C. After a 30–60 min resting period, individual slices were transferred to a recording chamber continuously perfused with aCSF heated to 32–34 °C.
In vitro recordings
Whole-cell patch-clamp recordings were performed using a Multiclamp 700B amplifier (Molecular Devices) from L4-L5 motoneurons with the largest soma (>400 μm2) located in the lateral ventral horn. These cells are the most likely to correspond to large, fast-type motoneurons, which are those most prone to expressing bistable behavior and plateau potentials (Harris-Warrick et al., 2024). A total of 17 motoneurons that met the inclusion criteria described below were successfully recorded from 12 neonatal mice. Motoneurons were isolated from most rapid synaptic inputs with a combination of kynurenic acid (1.5 mM), picrotoxin (100 μM) and strychnine (1 μM) to block glutamatergic, fast GABAergic and glycinergic synapses, respectively. Patch electrodes (2–4 MΩ) were pulled from borosilicate glass capillaries (1.5 mm OD, 1.12 mm ID; World Precision Instruments) on a Sutter P-97 puller (Sutter Instruments Company) and filled with an intracellular solution (in mM): 140 K+-gluconate, 5 NaCl, 2 MgCl2, 10 HEPES, 0.5 EGTA, 2 ATP, 0.4 GTP, pH 7.3. Pipette and neuronal capacitive currents were canceled and, after breakthrough, the series resistance was compensated and monitored. Recordings were digitized on-line and filtered at 20 kHz through a Digidata 1550B interface using Clampex 10.7 software (Molecular Devices). All experiments were designed to gather data within a stable period (i.e., at least 2 min after establishing whole-cell access).
Drug list
All solutions were oxygenated with 95% O2/5% CO2. All salt compounds, as well as veratridine (40 nM), apamin (200 nM), kynurenate (1.5 mM), picrotoxin (100 μM), strychnine (1 μM) were obtained from Sigma-Aldrich.
Data quantification
Electrophysiological data analyses were analyzed off-line with Clampfit 10.7 software (Molecular Devices). For intracellular recordings, several basic criteria were set to ensure optimum quality of intracellular recordings. Only cells exhibiting a stable resting membrane potential, access resistance ( ≤ 20 MΩ with no > 20% variation) and an action potential amplitude (measured from threshold to peak) larger than 40 mV under normal aCSF were considered. Passive membrane properties of cells were measured by determining from the holding potential the largest voltage deflections induced by small current pulses that avoided activation of voltage-sensitive currents. We determined input resistance by the slope of linear fits to voltage responses evoked by small positive and negative current injections. The peak amplitude of the slow afterdepolarization (slow ADP or sADP) was defined as the difference between the holding potential and the peak voltage deflection after the burst of spikes. The sADP area was measured between the end of the stimulus pulse and the onset of the hyperpolarizing pulse (delta= 7.5 s). If necessary, using bias currents, the pre-pulse membrane potential was maintained at the holding potential fixed in the control condition. Bistable properties were investigated using a 2 s depolarizing current pulses of varying amplitudes (0.8–2 nA). To assess the ability of a motoneuron to express bistability, the holding current was gradually increased in 25 pA increments thereby shifting the membrane potential (Vh) toward more depolarized values before delivering the depolarizing current pulse. This protocol, previously described and illustrated in detail in our earlier work [e.g., Supplementary Figure 1C in (Bos et al. 2021)], was repeated until the neuron reached its spiking threshold. A cell was considered as bistable when (1) the pre-stimulus membrane potential remained hyperpolarized below the spiking threshold (downstate), (2) the post-stimulus membrane potential remained depolarized above the spike threshold (upstate), and (3) the membrane potential could return to downstate after a brief hyperpolarizing pulse. To quantify the extent of bistability, we measured both the voltage (V) range between the most hyperpolarized holding potential (Vh min, from which a plateau could still be induced) and the most depolarized holding potential (Vh max, at which the plateau could be maintained), and the corresponding range of injected currents (ΔI) over which bistable behavior was observed.
Statistics
When two conditions (control vs. drugs) were compared, we used the Wilcoxon matched pairs test. For all statistical analyses, the data met the assumptions of the test and the variance between the statistically compared groups was similar. The level of significance was set at p < 0.05. Statistical analyses were performed using Graphpad Prism 7 software.
Results
Calcium dynamics and calcium-dependent currents
The role of calcium-induced calcium release (CICR) mechanism
A brief excitatory current pulse into a motoneuron triggers a train of action potentials, causing a substantial increase in intracellular calcium levels ([Ca2+]i). This increase is driven by influx via voltage-gated calcium channels, further amplified by calcium-induced calcium release (CICR) from internal stores. The resulting [Ca2+]i accumulation activates two Ca2+-dependent currents with opposing effects: the depolarizing calcium-activated non-specific cation current (ICAN) and the hyperpolarizing calcium-dependent potassium current (IKCa).
We first tested the hypothesis that CICR is essential for amplifying intracellular calcium to levels sufficient for activating calcium-dependent currents underlying bistability, a key step in the ionic cascade proposed in the Introduction. We modeled [Ca2+]i dynamics and compared scenarios with and without CICR.As described in Methods, the [Ca2+]i dynamics in our model are governed by the differential (Equation 2). The calcium clearance pump operates with a time constant of τCa = 10 ms, which, in the absence of CICR, would clear all calcium introduced by a spike before the next spike. The term kCICR·Ca in Equation 2 captures the CICR mechanism, where the rate of calcium release from internal stores is directly proportional to the current calcium concentration Ca with coefficient kCICR.
For simplicity, the intracellular calcium dynamics described by Equation 2 can be expressed as:
where τeff represents the effective time constant
The gain kCICR must not exceed 1/τCa to prevent negative τeff, which would lead to an infinite increase in calcium. In our model, kCICR is set at 0.096 ms−1, yielding an effective time constant τeff = 250 ms. This value was chosen phenomenologically to match typical slow afterdepolarization decay rates observed in our in vitro data and prior studies.
This prolonged τeff (compared to τCa) reflects how the CICR mechanism substantially slows calcium clearance, resulting in [Ca2+]i build-up during repetitive firing. As shown in Figure 1, calcium levels sharply increase in response to a rectangular current pulse, strongly activating ICAN and IKCa. Without CICR (kCICR = 0), pumps clear calcium rapidly between spikes, preventing significant accumulation during repetitive firing. Consequently, in the absence of sustained elevations in intracellular Ca2+, neither ICAN nor IKCa currents are activated to a significant degree.
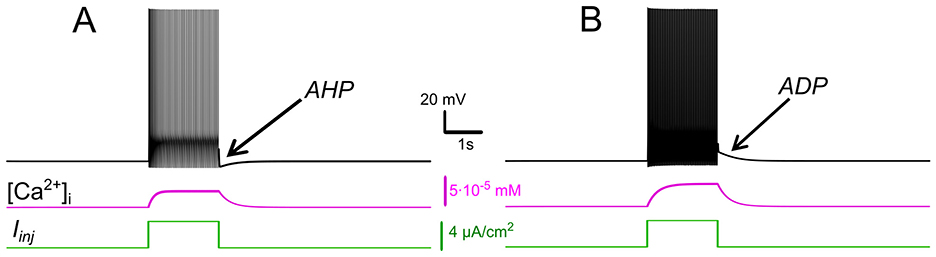
Figure 1. Effects of IKCa vs. effects of ICAN. Membrane potential (black) and intracellular Ca2+ (magenta) in response to step current (green). Intracellular Ca2+ concentration increases during spiking activity and then slowly adapts. (A) When IKCa is stronger than ICAN(gKCa= 0.5 mS, gCAN = 0 mS), this leads to after-hyperpolarization (AHP). (B) When ICAN is stronger than IKCa (gKCa=0.5 mS, gCAN=0.7 mS), this leads to after-depolarization (ADP) immediately following spiking activity.
These analyses support our hypothesis, demonstrating that CICR slows calcium clearance and is necessary for the sustained [Ca2+]i elevation.
Interplay between ICAN and IKCa: afterdepolarization vs. afterhyperpolarization
Intracellular recordings have shown that motoneurons demonstrate two post-stimulus behaviors: a slow afterdepolarization (sADP) after brief excitatory inputs, typical for a bistable motoneuron type, and an afterhyperpolarization (AHP) observed in a non-bistable motoneuron type (Harris-Warrick et al., 2024). We hypothesized that the balance between ICAN and IKCa determines post-stimulus behavior and bistability, with ICAN dominance favoring sADP and bistability, while IKCa dominance promotes AHP, aligning with observed differences in motoneuron subtypes. To test this, we simulated responses to depolarizing current pulses, varying the relative strengths of these currents.
Our simulations show that when IKCa predominates, Ca2+ -dependent potassium efflux produces hyperpolarization of the membrane and a decrease in excitability. This effect promotes a prominent post-stimulus AHP supported by sustained [Ca2+]i elevation (Figure 1A). Conversely, when ICANpredominates, the elevation of [Ca2+]i activates this current, leading to a depolarizing influx of sodium, that in turn increases the firing frequency during the pulse and yields an sADP afterward (Figure 1B). As [Ca2+]i declines, the membrane potential slowly relaxes back toward its resting values.
The sADP may be directly linked to neuronal bistability because it provides a sustained depolarizing drive that maintains the neuron in a high-activity state through positive feedback. Therefore, these model behaviors support our hypothesis and provide a mechanistic interpretation: ICAN biases toward sADP and plateau maintenance, whereas IKCa biases toward AHP and termination of spiking.
Mechanisms of ICAN-based bistability and their modulation
ICAN-based bistability
We then hypothesized that ICAN provides a robust mechanism for bistability via a positive feedback loop sustaining depolarization post-stimulus. We tested this using ramp and step current protocols in the model, analyzing bifurcation diagrams as gCAN varied. Ramp and step protocols were chosen because they are the standard experimental approaches used to investigate motoneuron bistability: ramps reveal firing hysteresis, whereas steps assess plateau potentials and self-sustained firing (Hounsgaard and Kiehn, 1985; Hounsgaard and Mintz, 1988; Hounsgaard et al., 1988a; Lee and Heckman, 1998b,a; Li and Bennett, 2003).
Our model simulations support that the expression of ICANprovides a robust route to bistability. The process begins with the opening of voltage-gated calcium channels during each action potential. While this influx alone is insufficient to fully activate ICAN, it triggers CICR, which amplifies [Ca2+]i (see below).
The elevated [Ca2+]i activates ICANestablishing a positive feedback loop: ICANsustains depolarization, promoting continuous firing; spikes further elevate [Ca2+]i via Ca2+ entry and CICR, and thus reinforce ICANactivation. This self-perpetuating mechanism allows the motoneuron to remain in a high-activity state (persistent spiking) even after the initial stimulus is removed, creating bistability: the neuron can operate in either a quiescent state or an active spiking state.
The 2-parameter bifurcation diagram (Figure 2A) shows how ICAN controls the appearance and the extent of the bistability region. For each gCAN, we replicated the experimental ramp current protocol in the model as described in Methods. To identify bistability, we used a triangular current ramp, consisting of two phases, ascending (“up”) and descending (“down”). These correspond to the increasing and decreasing portions of the injected current, respectively. No bistability is seen at gCAN = 0 (Figures 2A, B). It is appearing when gCAN starts increasing (gCAN >0, Figures 2A, C). Particularly, at gCAN = 0.5 mS/cm2, the voltage–current relation during a linear Iinjramp (Figure 2C) reveals distinct up (Iup) and down (Idown) thresholds. At gCAN = 0, Idown and Iupcoincide (no hysteresis; Figure 2B). Once gCAN becomes large enough, the interval (Idown<Iinj<Iup) opens and bistability emerges. This is evident as the current required to trigger spiking during the ramp-up exceeds the current at which spiking stops during ramp-down (Figure 2C). This hysteresis widens with further increases in gCAN (Figure 2A), reflecting the strengthening of the ICAN-mediated positive feedback loop, which supports the self-sustained spiking state.
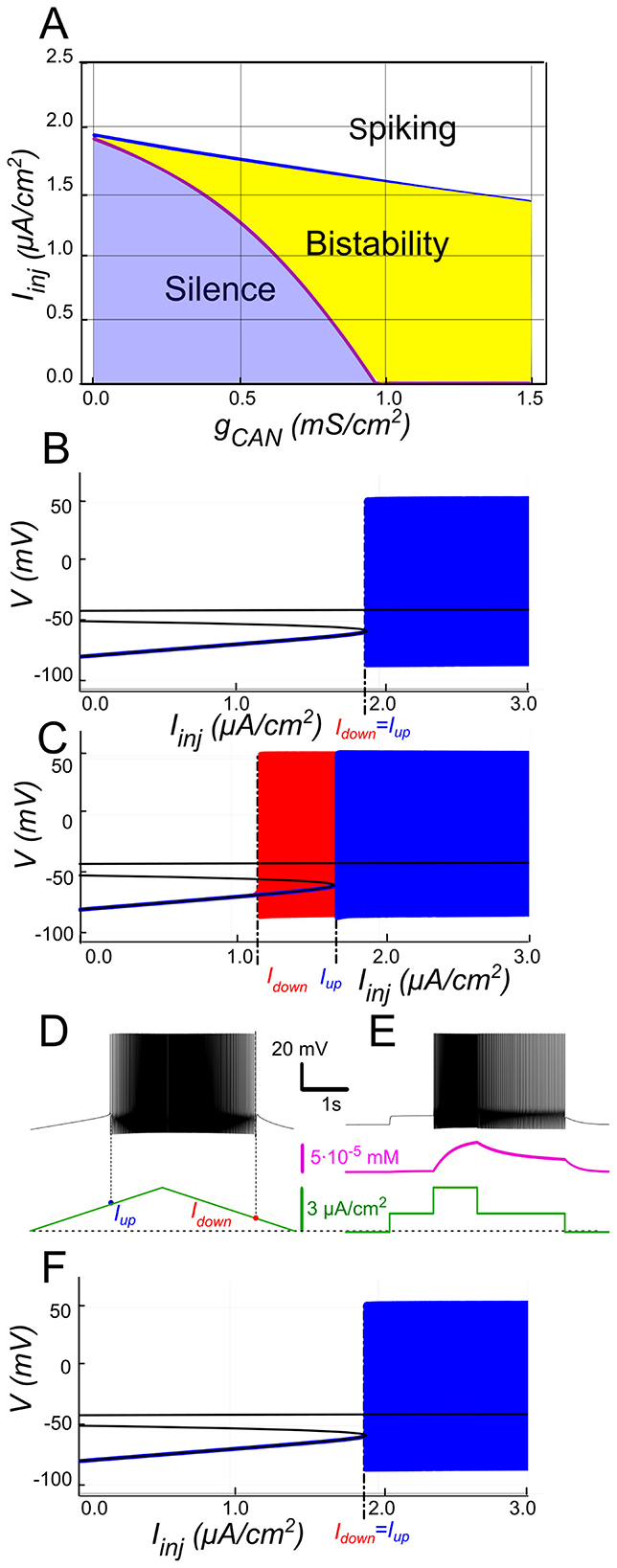
Figure 2. ICAN-induced bistability and its disappearance after CICR blockade. (A) Parameter plane (gCAN, Iinj) partitioned into regions of different behaviors (gKCa= gNaP= 0). The upper and lower boundaries of the bistability region represent the dependence of Iup (blue line) and Idown (red line) on gCAN, respectively. With no ICAN (gCAN = 0) the transitions from silence to spiking and back occur at the same values signifying no bistability. As gCAN increases, Idown becomes smaller than Iup, with the bistability range progressively expanding. (B, C) Bifurcation diagrams showing different behaviors of the system at gCAN = 0 and gCAN = 0.5 mS/cm2 across a range of the injected current values, Iinj. Black lines depict the system's unstable equilibrium states. Blue traces represent silence or spiking during the ascending current ramp; red traces indicate firing during the descending current ramp (when different from the ascending ramp). When Iinj<Iup there is a stable hyperpolarized state (stable node). If Iinj is increased over Iup, the system transitions to spiking, covering the voltage range shown in red and blue. This stable spiking regime exists in the range Iinj > Idown. When Iinj is reduced below Idown, the limit cycle representing spiking disappears, and the system transitions to the low voltage stable fixed point. Between these bifurcation points the hyperpolarized state (silence) coexists with the stable limit cycle (spiking) shown in red. (D, E) Bistability revealed with ramp and step protocols. (D) Iinj was linearly increased from 0 to 3 μA/cm2 and back (green). Note that spiking started at higher current than the transition back to silence (Iup > Idown). (E) Iinj was held piecewise constant at 0 first, then increased to 1.5 μA/cm2, then to 3, then reduced back to 1.5, and, finally, to 0 (see the black trace at the bottom). At Iinj = 1.5 μA/cm2 the system exhibits spiking or silence depending on whether it was active or not during the previous stage. Note the difference in the intracellular calcium concentration levels (green trace). (F) Same representation as in C but with intracellular calcium release blocked (kCICR = 0). Note lack of bistability.
In Figures 2B, C blue and red traces correspond to ascending and descending phases of the current ramp, respectively. For Iinj<Iup= 1.7 μA/cm2, the system displays two attracting regimes separated by an unstable equilibrium (saddle): a stable hyperpolarized state corresponding to the resting state (silent) and a stable spiking state (limit cycle) (Figure 2C). As Iinj exceeds Iup= 1.7 μA/cm2, the low potential stable branch of the V-nullcline merges with the saddle and vanishes via a fold bifurcation, and the system transitions to a stable limit cycle representing a repetitive spiking regime. During the descending phase of the ramp, spiking persists until Iinj decreases to Idown = 1.1 μA/cm2, which is less than Iup (Figures 2C, D), yielding a hysteresis interval (ΔI > 0 with Idown<Iup), indicative of bistability.
To further probe bistability, we implemented a step protocol inspired by experimental methods (Figure 2E). Starting at Iinj = 0 in a silent state, we applied an intermediate pulse within the bistable range (Idown<Iinj<Iup), then increased it above Iupto induce firing, before returning to the intermediate current (Figure 2E). This protocol showed that, at the same intermediate current value, the motoneuron could either continue firing or remain silent, depending on whether it was previously activated.
The above computational findings support our hypothesis, identifying ICAN as a key determinant of the bistable behavior through self-sustained spiking.
The role of CICR
We further hypothesized that CICR is required to raise [Ca2+]i enough for ICAN activation and bistability, as transient influx through voltage-gated calcium channels alone is insufficient. To test this, we set kCICR = 0 and repeated ramp protocols. As shown in Figure 2F, when CICR is blocked the ICAN-based bistability collapses (ramp hysteresis vanishes) because the transient rise in [Ca2+]i during spiking activity is almost completely abolished (not shown), as Ca2+ influx through voltage-gated channels alone is rapidly cleared by pumps. Consequently, ICAN is not recruited, and the system behaves equivalently to simulations where gCAN is set to zero (Figure 2B). In other words, CICR effectively slows Ca2+ clearance (τeff in the hundreds of ms) and is necessary in this framework to maintain the Ca2+-dependent depolarizing drive provided by ICAN. This supports our hypothesis, emphasizing the role of CICR in maintaining the ICAN-driven feedback loop.
The role of IKCa in modulating ICAN-based bistability
Hypothesizing that IKCa opposes ICAN-based bistability by hyperpolarizing the membrane, we analyzed the model's bifurcation maps varying gKCa and gCAN, and validated model predictions with apamin experiments in slices.
Our model supports that IKCahas a negative effect on ICAN-based bistability. As [Ca2+]i increases, SK-type K+ channels (mediating IKCa) open, allowing intracellular K+ ions to exit the cell. This outward current hyperpolarizes the membrane, decreasing excitability, and preventing the self-sustaining depolarization provided by ICAN. Bifurcation analyses (Figure 3) summarize this interplay. At low gCAN, a regime where IKCadominates the Ca2+-dependent response, the neuron transitions between spiking and silence at the same current threshold during ascending and descending current ramps, with this common threshold only weakly influenced by gCAN (Figure 3A). Increasing gCAN lowers the spiking onset threshold and, once gCAN reaches approximately 1 mS/cm2 (Figure 3A), a hysteresis interval opens (ΔI > 0 when Idown<Iup), indicating the emerging bistability. This is evident as the current required to trigger spiking during the ramp-up exceeds the current at which spiking stops during ramp-down, with the current range ΔI supporting bistability, progressively expanding as gCAN increases further. Additionally, the specific gCAN value at which this bifurcation occurs depends linearly on gKCa (Figure 3B), suggesting that the bistability arises when ICAN begins to dominate over IKCa. Equivalently, reducing gKCa at fixed gCAN unmasks bistability (Figures 3C–F).
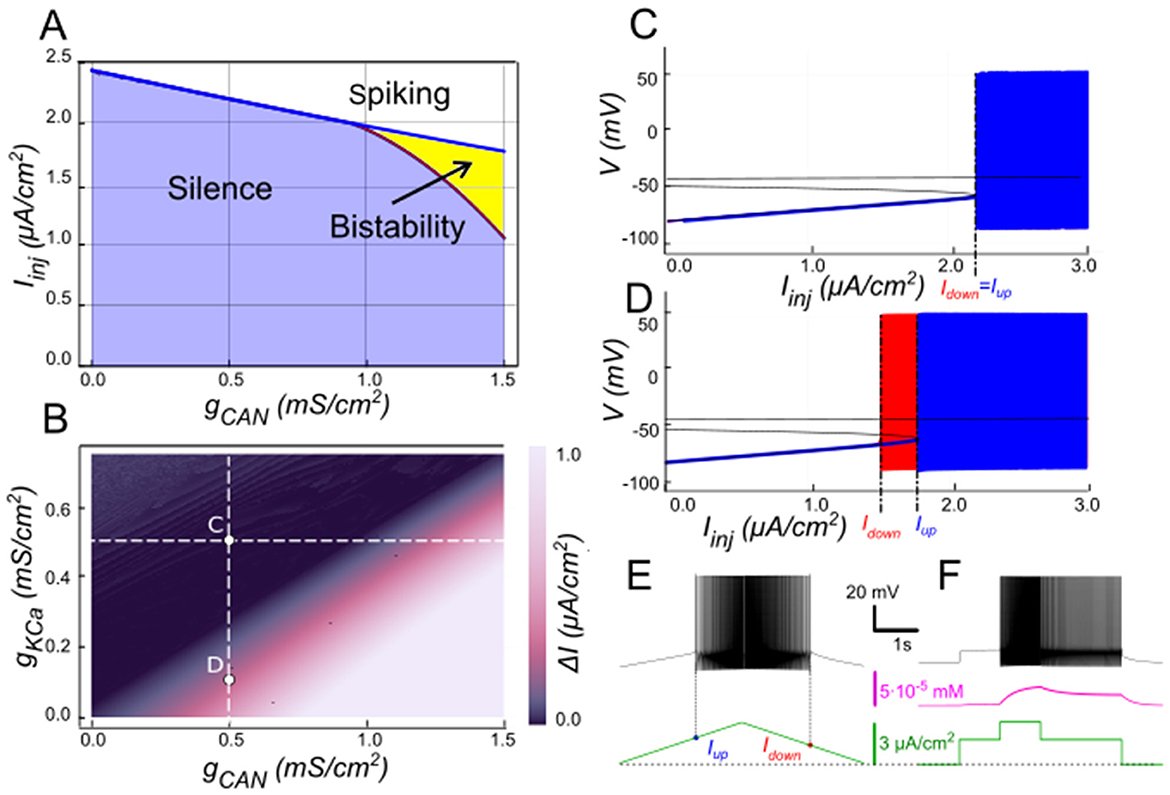
Figure 3. Modulation of ICAN-dependent bistability by IKCa. (A) Bifurcation diagram similar to Figure 2A, constructed for gKCa = 0.5 mS/cm2. Note, that unlike in Figure 2, bistability emerges once gCAN exceeds 1 mS/cm2. (B) Bistability range ΔIinj depending on gCAN and gKCa conductances. Bistability range (defined as Iup – Idown) is color coded. Bistability exists in the lower right part of the diagram. Note near linear dependence of gCAN bistability threshold on gKCa. White dashed line shows the value of gKCa used to construct the bifurcation diagram in (A). (C, D) Bifurcation diagrams showing possible behaviors of the system at the parameter values labeled correspondingly in (B). (C) At higher gKCa value (gKCa = 0.5) transitions from quiescence to spiking and back occur at the same injected current value (Iup = Idown), indicating no bistability. (D) When gKCa is lowered to 0.1, the transition from spiking to quiescence occurs at a lower injected current than the transition from quiescence to spiking (Idown<Iup), so spiking shown in red coexists with the silent regime. (C, D) Blue indicates activity during the ascending current ramp and red indicates activity during the descending current ramp. (E, F) Ramp (left) and step (right) current injection protocols, illustrating bistability revealed in (D). The intermediate current step is between Idown and Iup. The system's state depends on whether it was active or not at the previous step, exhibiting bistable behavior.
We tested these predictions by performing patch-clamp recordings of lumbar motoneurons, focusing on the effects of apamin (200 nM), a selective blocker of IKCa. We measured the parameters of the sADP induced by a brief depolarization of the motoneurons. Application of apamin significantly increased the amplitude and area of the sADP, indicating a larger depolarizing response when IKCa is reduced (Figures 4A–C). In addition, apamin also enhanced the capacity of motoneurons for bistable behavior. Specifically, motoneurons were able to express plateau potentials from more hyperpolarized holding potentials (Figure 4D), reflected by a significant shift in Vhmin (defined as the most negative holding potential from which a plateau could be induced) from −59.7 mV (+/– 5.4) mV to −61.9 (+/– 5.2) mV (p < 0.05), accompanied by increases in the voltage range (ΔV = Vhmax – Vhmin, where Vhmax is the most depolarized holding potential at which a plateau could be maintained before reaching action potential threshold) from 4.9 (+/– 3.3) mV to 8.2 (+/– 1.5) mV (p < 0.05; Figure 4E) and the current range (ΔI) over which bistability was observed from 76.3 (+/−25.9) pA to 109.6 (+/−46.4) pA (p < 0.05; Figure 4F). Together, these results support the prediction of the model that reducing IKCa expands the bistable regime by reducing the after-hyperpolarizing influence opposing ICAN-driven depolarization (Figures 1, 3). These results support our hypothesis, underscoring the antagonistic interplay between ICAN and IKCa in shaping motoneuron excitability and providing a mechanistic basis for bistable behaviors.
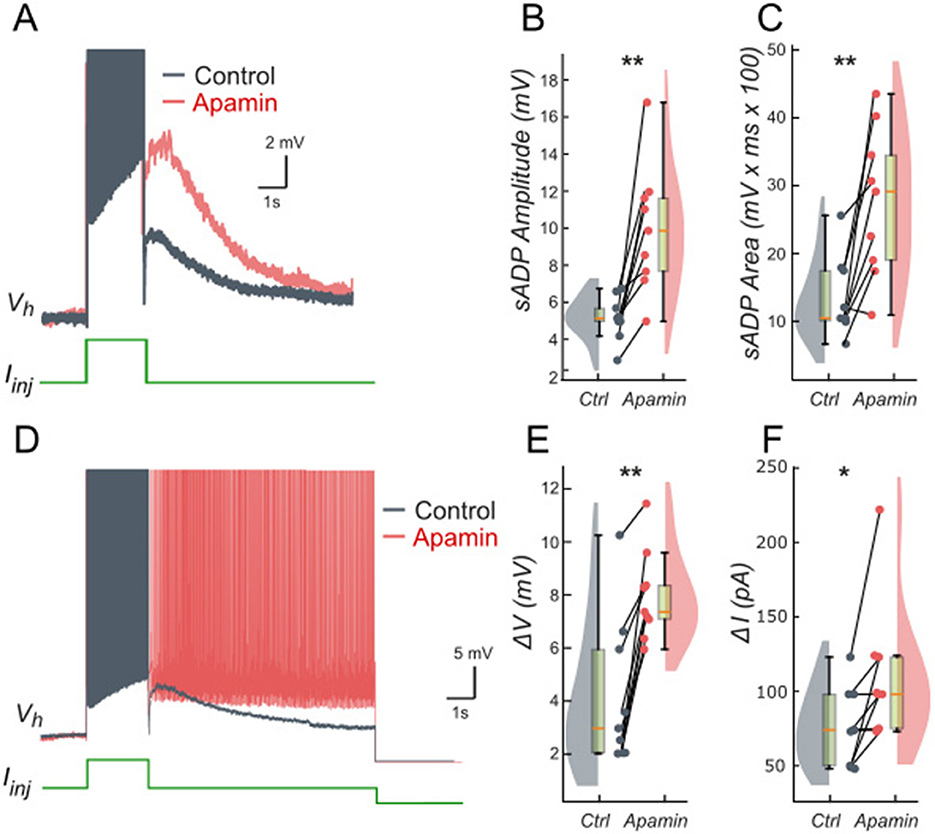
Figure 4. Ca2+-activated K+ current (IKCa) limits the slow afterdepolarization and membrane bistability in lumbar motoneurons. (A) Superimposed voltage traces recorded in the same motoneuron during a brief (2 s) depolarizing current step (bottom) under control conditions (black) and after bath application of apamin (200 nM; red). (B, C) Quantification of the sADP amplitude (B) and area (C). (D) Superimposed voltage traces recorded from the same holding potential (Vh) in response to a 2-s depolarizing current pulse before (black) and after apamin (red). (E, F) Quantification of bistability through ΔV and ΔI. ΔV and ΔI represent the range of holding potentials and holding currents, respectively, over which self-sustained firing can be observed. Each is defined as the difference between the most depolarized and the most hyperpolarized value (potential for ΔV, current for ΔI) at which self-sustained firing is triggered or maintained (see Methods). Paired data from individual motoneurons (n = 9) are linked and overlaid on violin and box-and-whisker plots. *P < 0.05, **P < 0.01, two-tailed Wilcoxon signed-rank test.
Modulation of ICAN-based bistability by extracellular potassium concentration
Because IKCa depends on the K+driving force, we hypothesized that elevated [K+]o enhances bistability by depolarizing the potassium reversal potential EK and reducing IKCa's opposition to ICAN. EK is set by the Nernst equation which provides the potassium ion equilibrium potential based on the ratio of intracellular [K+]i to extracellular [K+]o potassium concentration (see Methods). Increasing extracellular potassium [K+]o depolarizes EK thereby reducing the outward driving force through IKCa. This weakens the hyperpolarizing influence of IKCa that counteracts the ICAN-mediated depolarizing feedback.
We explored this interaction by varying [K+]o and gCAN in the model while holding gKCa at 0.5 mS/cm2. The resulting two-parameter map (Figure 5B) shows the bistable range (color-coded width ΔI = Iup – Idown, black indicating no bistability) as a function of gCAN and [K+]o. At low [K+]o (e.g., physiologically normal levels of 4 mM), EK is strongly negative and IKCa efficiently counteracts ICAN, so relatively large gCAN (e.g., ~1 mS/cm2, Figure 3A) is required for bistability. As [K+]o increases, EK depolarizes, weakening IKCa and the bistable interval opens at progressively lower gCAN (Figure 5A). For instance, at gCAN = 0.9 mS/cm2 the model is not bistable at [K+]o = 4 mM (Figure 5C), but becomes bistable at [K+]o = 8 mM (Figures 5D, E), with the hysteresis width further expanding as [K+]o rises (Figure 5B). As elsewhere, we corroborated bistable firing behavior using a step protocol showing different coexisting stable regimes at identical inputs (Figure 5F).
![Simulated diagrams reveal that raising [K.]o promotes ICAN-based bistability. Panel A shows bifurcation results at [K.]o = 6 mM: hysteresis begins at lower gCAN than in normal (4 mM) conditions. Panel B maps bistability range (Iup – Idown) versus gCAN and [K.]o, where hotter colors mark larger hysteresis and black indicates none. Panels C–D compare voltage behaviors at normal and elevated potassium, showing bistability only when [K.]o = 8 mM. Ramp (E) and step (F) current injections confirm that higher extracellular potassium lowers the threshold for bistability emergence, suggesting that depolarizing ionic environments stabilize ICAN-driven self-sustained activity in motoneurons.](https://www.frontiersin.org/files/Articles/1710893/fncel-19-1710893-HTML/image_m/fncel-19-1710893-g005.jpg)
Figure 5. Modulation of ICAN-dependent bistability by extracellular potassium concentration ([K+]o). (A) Bifurcation diagram similar to Figure 3A, constructed for gKCa = 0.5 mS/cm2, but at elevated [K+]o = 6 mM instead of physiologically normal 4 mM. Note, that compared to Figure 3A, bistability emerges at lower gCAN. (B) Color-coded bistability range (Iup – Idown) depending on gCAN and [K+]o. Black area corresponds to no bistability. White dashed line shows the [K+]o value used in (A). gCAN bifurcation value reduces as [K+]o increases, therefore an increase in [K+]o can lead to bistability emergence, as shown in (C, D). If gCAN = 0.9 mS/cm2, at [K+]o = 4 mM (physiologically normal value) no bistability exists (B), but if [K+]o is raised to 8 mM, bistability emerges (C), as illustrated by ramp (E) and step (F) current protocols. (C, D) Blue indicates activity during the ascending current ramp and red indicates activity during the descending current ramp.
Our findings support the hypothesis, suggesting relevance to physiological [K+]o fluctuations: during sustained activity or in certain pathological conditions, elevation of [K+]o can occur, which, by decreasing the effectiveness of IKCa, would favor expression of the ICAN-driven positive feedback and broaden the bistable operating range. More generally, these findings illustrate how intrinsic mechanisms of excitability can be tuned by extracellular milieu, here via the dependence of EK on [K+]o.
The role of INaP in modulating ICAN-based bistability
The persistent sodium current (INaP) is well-known for amplifying neuronal excitability by providing a sustained depolarizing drive at subthreshold voltages (Crill, 1996). Hypothesizing INaP facilitates ICAN-based bistability, we examined its interactions with ICAN using our computational model (Figure 6). The two-parameter bifurcation map in Figure 6B depicts the bistability range (color-coded as the hysteresis width ΔI = Iup – Idown) as a function of gNaP and gCAN. Overall, the bistable interval expands with increasing gNaP, indicating that INaP enhances the robustness of bistability. For instance, at moderately low gCAN values (e.g., 0.9 mS/cm2; Figure 6B), where ICAN alone fails to produce bistability (Figure 6C), elevating gNaP from zero to 0.45 mS/cm2 uncovers a clear hysteresis interval (Figures 6B, D–F), enabling the coexistence of silent and spiking states over a range of injected currents.
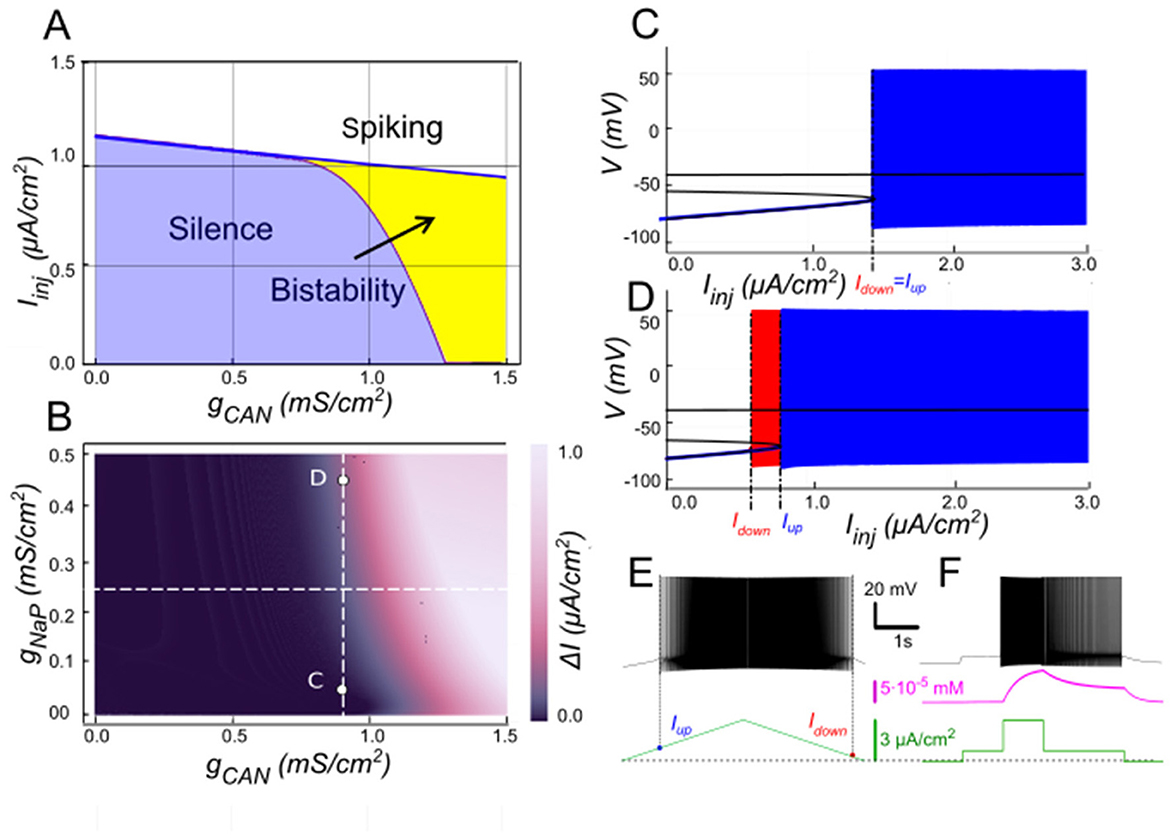
Figure 6. Modulation of ICAN-dependent bistability by INaP. (A) Bifurcation diagram similar to Figure 3A, constructed for gKCa = 0.5 mS/cm2, but at gNaP = 0.25 mS/cm2 instead of zero. Note, that compared to Figure 3A, bistability emerges at lower gCAN. (B) Color-coded bistability range (Iup – Idown) depending on gCAN and gNaP with gKCa fixed at 0.5 mS/cm2. Black area corresponds to no bistability. gCAN bifurcation value reduces as gNaP increases, therefore an increase in gNaP can lead to bistability emergence, as shown in (C, D) where blue indicates activity during the ascending current ramp and red indicates activity during the descending current ramp. If gCAN = 0.9 mS/cm2, at gNaP = 0 no bistability exists (C), but if gNaP is raised to 0.45 mS/cm2, bistability emerges (D), as illustrated by ramp (E) and step current protocols (F).
This effect aligns with the core ICAN-driven positive feedback loop underpinning bistability: (i) spiking activity opens voltage-gated calcium channels, leading to Ca2+ influx; (ii) this influx triggers CICR, amplifying the cytosolic Ca2+ signal; (iii) elevated [Ca2+]i then activates ICAN, and (iv) the resulting depolarisation accelerates spiking promoting additional Ca2+ entry and closing the self-reinforcing loop. Amplifying any element of this loop can elevate its overall gain, tipping the system toward bistability.
Here, INaP contributes by delivering a tonic depolarizing current that lowers the voltage threshold for spike initiation and Ca2+ entry. This facilitates the recruitment of ICAN during the onset of activity and bolsters its ability to sustain the high-activity state once engaged, even at lower gCAN levels. For example, at gCAN = 0.9 mS/cm2 and gNaP = 0.45 mS/cm2, the model displays robust bistability (Figures 6D–F), whereas reducing gNaP to 0 eliminates it (Figure 6C). However, at lower ICAN expression (e.g., at gCAN = 0.5 mS/cm2), INaP-evoked depolarization is insufficient to achieve the high-gain regime needed for bistability at any gNaP.
We tested these predictions pharmacologically in patch-clamp recorded lumbar motoneurons using veratridine (40 nM), which at low molar nanomolar concentrations enhances INaP (Alkadhi and Tian, 1996; Tazerart et al., 2008). Following a short depolarizing current pulse, veratridine increased the amplitude and area of the sADP (Figures 7A–C), indicating a larger depolarizing tail when INaP is enhanced. Veratridine also facilitated bistability: self-sustained spiking activity were triggered from more hyperpolarized holding potentials (Figure 7D), with Vh shifting from −57.5 mV (+/– 9.5) to −62.1 mV (+/−9.0) (p < 0.01). Both the voltage range (ΔV) and the current range (ΔI) supporting bistability increased: ΔV shifts from 3.3 mV (+/– 1.9) to 7.3 mV (+/– 2.4) (p < 0.05; Figure 7E) and ΔI shifts from 58.5 pA (+/– 22.3) to 126.0 pA (+/−59.1) (p < 0.05; Figure 7F).
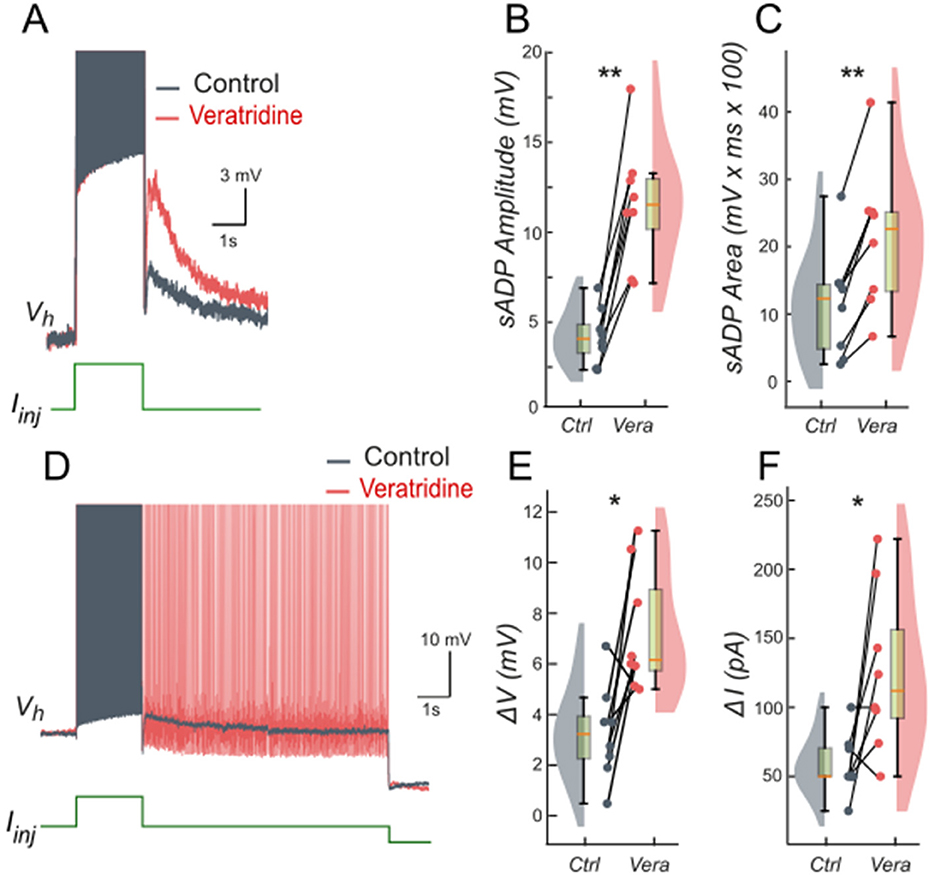
Figure 7. Persistent Na+ current (INaP) facilitates the slow afterdepolarization and membrane bistability in lumbar motoneurons. (A) Superimposed voltage traces recorded in the same motoneuron during a brief (2 s) depolarizing current step (bottom) under control conditions (black) and after bath application of veratridine (40 nM; red). (B, C) Quantification of the sADP amplitude (B) and area (C). (D) Superimposed voltage traces recorded from the same holding potential (Vh = −65 mV) in response to a 2-s depolarizing current pulse before (black) and after (red) veratridine. (E, F) Quantification of bistability through ΔV and ΔI. ΔV and ΔI represent the range of holding potentials and holding currents, respectively, over which self-sustained firing can be observed. Each is defined as the difference between the most depolarized and the most hyperpolarized value (potential for ΔV, current for ΔI) at which self-sustained firing is triggered or maintained (see Methods). Paired data from individual motoneurons (n = 8) are linked and overlaid on violin and box-and-whisker plots. *P < 0.05, **P < 0.01, two-tailed Wilcoxon signed-rank test.
Taken together, these findings support the hypothesis that increasing INaP facilitates and broadens the operating window of ICAN-driven bistability by enhancing depolarization while ICAN remains the principal maintenance mechanism once engaged. This synergy between ICAN and INaP helps explain how modest changes in persistent Na+ conductance can markedly reshape motoneuron firing regimes.
Bistability based on INaP and the role of [K+]o
INaP could theoretically sustain bistability independently of ICAN under conditions that maintain its activation between action potentials. For instance, if the inter-spike membrane potential remains above INaP's deactivation threshold, INaP would provide a continuous depolarizing drive, creating a self-reinforcing loop where subthreshold depolarization promotes spiking, and the resulting activity further engages INaP without full reset. Hypothetically, this mechanism might enable the neuron to toggle between quiescent and self-sustained firing states purely through sodium-based persistence, highlighting a potential alternative pathway for bistability in scenarios where calcium-dependent processes are minimized or absent.
In our model, when gCAN = 0, INaP alone could not support bistability in the parameter range examined (Figure 8A). The bifurcation diagram (Figure 8D) elucidates this. At low injected current (Iinj < 0.85 μA/cm2), the system's sole stable state is a low-voltage resting state (stable node). As Iinj surpasses this threshold, the node merges with a saddle point and subsequently ceases, leading to a stable limit cycle, which represents a spiking regime. A strong hyperpolarization follows each spike, dropping below the resting potential. This large post-spike hyperpolarization fully deactivates INaP, explaining its inability to maintain bistability independently. Consequently, when the injected current is decreased, the silent regime reappears via the same bifurcation, demonstrating an absence of hysteresis.
![Color-coded parameter maps and voltage traces show how combining INaP and elevated [K.]o produces bistability. Panel A: at baseline [K.]o = 4 mM, neurons transition from silent to spiking without hysteresis. Panel B: at 12 mM [K.]o, bistability appears once gNaP > 0.15 mS/cm². Panel C summarizes the bistable range versus both parameters: higher [K.]o lowers the gNaP threshold. Panels D–E compare bifurcation diagrams at 4 mM and 12 mM potassium; only the latter shows hysteresis. Ramp (F) and step (G) simulations confirm sustained firing dependent on prior state, showing that depolarizing extracellular conditions promote INaP-driven bistable activity.](https://www.frontiersin.org/files/Articles/1710893/fncel-19-1710893-HTML/image_m/fncel-19-1710893-g008.jpg)
Figure 8. Bistability based on INaP and the role of [K+]o. (A) Activity regimes of the model neuron depending on the injected current (Iinj) and the conductance of persistent sodium current (gNaP) at baseline K+ extracellular concentration ([K+]o = 4 mM). At all values of gNaP as the injected current changes, the model transitions from silence to spiking and back with no hysteresis which indicates no bistability. (B) Activity patterns of the model neuron depending on the injected current and gNaP conductance at [K+]o = 12 mM. Once gNaP exceeds approximately 0.15 mS/cm2, bistability emerges. (C) Adjusting the sodium persistent inward current (INaP) conductance (gNaP) and extracellular potassium concentration ([K+]o) in a model neuron reveals bistable regimes. The range of injected current where bistability occurs is shown in color, with black indicating no bistability. Higher [K+]o levels require smaller gNaP for bistability, suggesting that increased [K+]o can induce bistability in neurons with otherwise insufficient gNaP expression. (D) At gNaP= 0.4 mS/cm2 and [K+]o = 4 mM no bistability is observed. (E) However, as [K+]o is increased to 12 mM, bistability emerges, as illustrated by ramp (F) and step current protocols (G). (D, E) Blue indicates activity during the ascending current ramp and red indicates activity during the descending current ramp.
The post-spike hyperpolarization is mediated by K+ outward currents and thus can be modulated by [K+]o, which sets the reversal potential EK. Raising [K+]o depolarizes EK , thereby reducing the driving force of outward currents and diminishing the post-spike hyperpolarization. Under these conditions, INaP is less completely deactivated, allowing bistability to emerge (Figures 8B, E).
As shown in Figure 8E, when [K+]o = 12 mM and Iinj crosses a value of 0.8 μA/cm2, the resting state is lost through the same saddle-node bifurcation, but unlike at normal [K+]o, the trajectory joins a pre-existing spiking limit cycle with attenuated post-spike hyperpolarization (Figures 8E, F). When Iinj is reduced, spiking persists down to 0.45 μA/cm2, where the limit cycle intersects with the saddle point and disappears through a saddle-loop (homoclinic) bifurcation, and the system returns to resting state (Figures 8E, F). Thus when Iinj falls between 0.45 and 0.85 μA/cm2 both rest and spiking coexist, indicating a bistable regime (Figure 8G). A distinctive feature of this type of bistability is that the resting potential lies below the voltage range of the limit cycle (spiking).
The presence and extent of bistable behavior depends jointly on gNaP and [K+]o. At [K+]o = 12 mM, bistability emerges once gNaP exceeds ~0.2 mS/cm2 (Figure 8B), with the current range supporting bistability expanding as gNaP increases. Mapping across parameters (Figure 8C) shows that for gNaP < 0.5 mS/cm2 bistability requires [K+]o > 10 mM; conversely, the bistability gNaP threshold decreases as [K+]o increases. For instance, with gNaP = 0.25 mS/cm2, bistability emerges once [K+]o exceeds ~12 mM.
Together, these results partially support the hypothesis, indicating that while under normal conditions INaP alone is insufficient to produce bistability, elevated [K+]o reduces outward current-mediated hyperpolarization, potentially enabling bistable firing.
The role of slowly inactivating potassium current (IKv1.2)
Recent work indicates that the potassium current mediated by Kv1.2 channels (IKv1.2) is prevalent in bistable motoneurons (Harris-Warrick et al., 2024), although its direct contribution to bistable behavior has not been fully clarified. Kv1.2 channels inactivate very slowly during repetitive firing. In principle, such slow inactivation generates a positive feedback loop: as IKv1.2 gradually decreases during ongoing spiking, the cell becomes more excitable and can persist in an active state. On the other hand, when the neuron is silent, Kv1.2 channels are fully open and IKv1.2 can strongly oppose the initial depolarization. Therefore, we tested the hypotheses that IKv1.2 alone can support bistability.
To clarify IKv1.2 contribution, we simulated our computational model while varying both Iinj and IKv1.2's maximal conductance, gKv1.2. In these simulations, IKv1.2 alone did not produce bistability between resting and spiking states thus invalidating the hypothesis. Instead, the model generated a regime of periodic bursting, alternating between spiking and quiescent phases, over an extremely narrow range of Iinj.
A signature of IKv1.2 is its effect on firing dynamics during constant depolarization: rather than stabilizing tonic firing, it induced a delayed excitation with ramping spike frequency (Bos et al., 2018). To isolate this effect, we removed both ICAN and INaP (gNaP = gCAN = 0). Under these conditions (Figure 9B), a rectangular current pulse elicited an initial fast depolarization followed by a slow secondary depolarization. If the stimulus was strong enough, this slow drift brought the neuron to a spiking threshold and then spiking began, with the firing rate progressively accelerating. This behavior reflects very slow inactivation kinetics of IKv1.2. Initially, IKv1.2 activates quickly, temporarily opposing depolarization, but then inactivates with a time constant of ~2.5 s (see Methods), progressively reducing its own inhibitory effect and permitting further depolarization and firing.
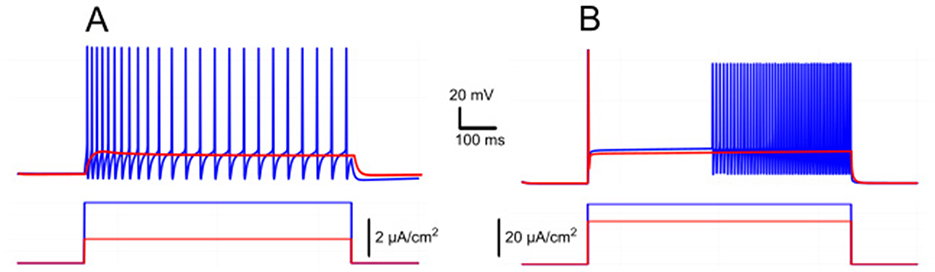
Figure 9. Ramp/delayed excitation vs. spike frequency adaptation in response to rectangular current injection. (A) Model response with IKCa alone (gKv1.2 = 0, gKCa = 1 mS/cm2, gNaP = gCAN = 0), displaying spike frequency adaptation as IKCa activation hyperpolarizes the neuron, reducing firing rate over time. (B) Model response with IKv1.2 alone (gKv1.2 = 2 mS/cm2, gKCa = 0, gNaP = gCAN = 0), showing delayed excitation and a ramping firing rate due to slow inactivation of IKv1.2. The membrane potential exhibits an initial jump followed by gradual depolarization, leading to progressively increasing spike frequency.
By contrast, in absence of IKv1.2 but with IKCa present, the model showed spike frequency adaptation (Figure 9A). Here, firing decreased over time because IKCa hyperpolarized the membrane. When both currents (IKv1.2 and IKCa) were present, the firing dynamics (whether it shows ramping or adaptation) depended on their relative balance. This interaction can lead to complex dynamics where the slow inactivation of IKv1.2 competes with the calcium-dependent potassium currents tendency to stabilize or reduce firing frequency.
Discussion
In this study, we used a single-compartment computational model of spinal motoneurons to dissect the ionic mechanisms underlying bistability. Our results identify a minimal core mechanism based on the synergistic interactions among ICaL, CICR, and ICAN strongly modulated by IKCa, INaP and [K+]o. These findings offer new insights into how bistable firing is generated and regulated in motoneurons.
Motoneuron bistability critically depends on the ICaL-CICR-ICAN loop
Early studies attributed plateau potentials and bistability to persistent L-type calcium currents (Schwindt and Crill, 1980; Hounsgaard et al., 1988a,b; Hounsgaard and Mintz, 1988; Hounsgaard and Kiehn, 1989; Hounsgaard and Kjaerulff, 1992; Svirskis and Hounsgaard, 1997), a view reinforced by computational models requiring dendritic ICaL to replicate these bistable behaviors (Booth and Rinzel, 1995; Booth et al., 1997; Carlin et al., 2000; ElBasiouny et al., 2005; Bui et al., 2006; Carlin et al., 2009; Kim and Jones, 2011). Immunohistochemical data further supported this interpretation by revealing the dendritic distribution of L-type Cav1.3 channels (Simon et al., 2003; Zhang et al., 2008). More recent findings have revealed a complementary mechanism, in which ICaL primarily acts as a trigger, while other currents, notably the ICAN, play a central role in mediating plateau potentials and bistability (Bos et al., 2021), as observed in motoneurons from neonatal and young adult murine models (Harris-Warrick et al., 2024).
Our computational findings reinforce and extend this view. The model shows that initial Ca2+ entry through ICaL is insufficient to sustain bistability due to fast Ca2+ removal from the cytosol. The necessary amplification arises from CICR, linking ICaL-mediated Ca2+ influx to intracellular stores. Accordingly, CICR surpasses the capacity of Ca2+ pumps to rapidly clear cytoplasmic Ca2+, and therefore enables sustained intracellular Ca2+, long-lasting ICAN activation and bistability. Consistent with this, motoneuron bistability is abolished when CICR is inhibited, even though ICaL currents are still present (Bouhadfane et al., 2013; Bos et al., 2021; Harris-Warrick et al., 2024). Together, ICaL, CICR, and ICAN emerge from the findings as a functional triad instrumental in motoneuron bistability. As such, the triad may provide a general feed-forward mechanism for plateau generation across different structures of the CNS, owing to the fact that ICAN also supports motor, sensory and memory-related plateaus (Fraser and MacVicar, 1996; Morisset and Nagy, 1999; Di Prisco et al., 2000; Yan et al., 2009; Toporikova and Butera, 2011; Jasinski et al., 2013).
Suppression of ICAN -based bistability by IKCa
The model adds a key regulatory layer to motoneuron bistability by clarifying the interplay between ICAN and IKCa. IKCa dominance, thought to be driven by Ca2+ influx through dendritic L-type calcium channels (Mousa and Elbasiouny, 2020) inhibits ICAN-driven plateaus and prevents their formation. In line with the model, reducing IKCa with apamin facilitates the expression of bistability and can unmask latent plateau potentials (Hounsgaard and Mintz, 1988; Hounsgaard and Kiehn, 1993). The relative contributions of ICAN and IKCa may tune the propensity to bistability across motoneuron subtypes. Bistability appears more frequently observed in large motoneurons (Harris-Warrick et al., 2024), a finding that, according to the assumption, would reflect a higher ICAN/IKCa ratio. This interpretation aligns with established physiological distinctions where large motoneurons, in line with size-dependent differential expression of SK channels (Deardorff et al., 2013), display brief AHPs, whereas small motoneurons exhibit prolonged AHPs (Kernell, 1965; Gustafsson and Pinter, 1984). In addition, large motoneurons generate stronger ICAN than small motoneurons (Harris-Warrick et al., 2024).
Our model also identifies [K+]o as a critical factor influencing IKCa efficacy. Elevating [K+]o depolarizes EK, weakening IKCa and shifting the balance toward ICAN, thereby enhancing bistability (Figure 4). Disruption of K+ buffering through astrocytic Kir4.1 dysfunction in spinal cord injury (Olsen et al., 2010; Benson et al., 2023; Barbay et al., 2025), is likely to elevate [K+]o, paralleling findings from epilepsy studies (Djukic et al., 2007; Tong et al., 2014). A subsequent shift toward ICAN is predicted to strengthen bistability and worsen spasticity (Bennett et al., 2001; Brocard et al., 2016).
Facilitation of ICAN -mediated bistability by INaP
While ICAN is the main driver of bistability, INaP acts as an essential modulator, extending the conditions under which bistability occurs. In our simulations, increasing INaP lowers the threshold for ICAN-mediated bistability, enabling sustained firing even when ICAN alone is insufficient (Figure 6). Due to subthreshold depolarization (Crill, 1996) that enables repetitive spiking (Kuo et al., 2006), INaP biases the system toward ICaL-CICR-ICAN engagement and plateau generation. In line with this role, riluzole, an established inhibitor of INaP, reliably suppresses self-sustained firing in bistable motoneurons (Bouhadfane et al., 2013; Drouillas et al., 2023). At first glance, this might imply that plateau potentials depend directly on INaP. However, the persistence of TTX-resistant sADP after rizulole application (Bouhadfane et al., 2013; Drouillas et al., 2023) suggests that ICAN provides the essential substrate for bistable behavior, while INaP serves as a facilitator. The INaP and ICAN interaction extends beyond motoneurons; in the preBotzinger complex, for example, the two currents cooperate to produce rhythmic bursting (Jasinski et al., 2013; Phillips et al., 2019, 2022).
Under physiological conditions, INaP alone unlikely supports bistability because hyperpolarizing potassium currents deactivate INaP between spikes (Figure 8). Elevating [K+]o mitigates this hyperpolarization by depolarizing EK, and allows INaP to create bistability at higher gNaP (Figure 8C). This has been clearly demonstrated in our simulations, where a pure INaP-based bistability emerges as [K+]o approaches or exceeds 12 mM. This scenario may be relevant in spinal cord injury (SCI), where INaP is enhanced (Bennett et al., 2001; Li et al., 2004; Harvey et al., 2006; Brocard et al., 2016). In conjunction with high [K+]o, this enhancement can strengthen bistability and promote spasticity. Consequently, riluzole (Rilutek), originally developed for amyotrophic lateral sclerosis (ALS), is being explored to target spasticity in SCI patients (Cotinat et al., 2023).
The role of slowly inactivating potassium current (IKv1.2)
In spinal motoneurons, IKv1.2imposes an initial brake on excitability and then relaxes over seconds, yielding delayed spiking and a characteristic ramping of discharge during sustained depolarization (Bos et al., 2018). Because this property scales with motoneuron size, larger α-motoneurons show stronger delayed excitation and ramping (Harris-Warrick et al., 2024). Yet, despite the greater prevalence of bistability in larger motoneurons, evidence for a generative role of Kv1.2 remains elusive. Our simulations clarify this point. Varying IKv1.2in isolation never produced robust switching between silent and self-sustained firing states. Instead, it generated narrow-band bursting around a tight input window, while the hallmark hysteresis of bistability was absent. Mechanistically, IKv1.2lacks the positive feedback needed to maintain a depolarized up-state. It is an outward conductance that weakens with use, modulating access to the plateau regime but not providing the sustaining inward drive. In contrast, stable bistability in both our model and experiments requires the ICaL-CICR- ICAN triad, a view reinforced by the identification of Trpm5 as the principal Na+-permeable carrier of ICAN underlying motoneuron plateaus (Bos et al., 2021). When TRPM5/ICAN is suppressed, slow afterdepolarization and plateaus collapse even though IKv1.2is intact, directly demonstrating that Kv1.2 is permissive rather than generative for bistability.
Serotonin and bistability
Brainstem-derived monoamines are central to motoneuron excitability and bistability (Heckman et al., 2008). In decerebrate cats, bistability depends on descending monoaminergic drive (Conway et al., 1988; Hounsgaard et al., 1988a; Lee and Heckman, 1998b, 1999). Acute spinalization removes these inputs and thereby reduces bistable properties of motoneurons, whereas monoamine reintroduction restores plateau potentials (Hounsgaard et al., 1988a). Among these modulators, serotonin is especially effective. In vertebrates, exogenous serotonin enhances excitability and bistability (Hounsgaard and Kiehn, 1985, 1989).
Mechanistically, serotonin acts primarily through 5-HT2 receptors to amplify ionic currents that promote bistability. By increasing dendritic L-type calcium currents (Hounsgaard and Kiehn, 1989; Perrier and Hounsgaard, 2003; Perrier and Delgado-Lezama, 2005; Perrier and Cotel, 2008) it can promote calcium build-up and ICAN activation as described in our model. Serotonin also shifts INaP activation toward hyperpolarized potentials, thereby amplifying neuronal excitability (Li and Bennett, 2003; Harvey et al., 2006). The shift enhances INaP and thus facilitates ICAN-mediated bistability. Finally, serotonin also reduces outward currents, notably IKCa, thereby facilitating high-frequency firing (Grunnet et al., 2004). This serotonin-evoked reduction of IKCa may lead to bistability in spinal motoneurons (Hounsgaard et al., 1988b; Hounsgaard and Kiehn, 1993), as demonstrated in our present modeling and experimental results.
The role of serotonin becomes especially evident during SCI, not because of its direct action, but rather because of its sudden loss following disruption of descending inputs. Initial serotonin depletion produces motoneurons hypofunction, otherwise known as “spinal shock” (Schadt and Barnes, 1980), mirroring the loss of bistability in our model upon blockade of ICAN or CICR. However, with time, excitability and plateau potentials re-emerge, representing an electrophysiological correlate of chronic spasticity and hyperreflexia (Bennett et al., 2001). The rebound is attributed to plasticity in serotonin receptor signaling. Most notably, 5-HT2B and 5-HT2C receptors become constitutively active which restores persistent inward currents (e.g., INaP) and plateau firing even without serotonergic input (Li and Bennett, 2003; Murray et al., 2010, 2011; D'Amico et al., 2013; Tysseling et al., 2017). Consistent with this, our model shows that increased INaP strengthens ICAN-driven bistability.
Limitations and future research directions
Dendritic calcium currents have been repeatedly linked to motoneuron plateaus and bistability (Booth et al., 1997; Carlin et al., 2000; Simon et al., 2003; ElBasiouny et al., 2005; Bui et al., 2006; Zhang et al., 2008). Our single-compartment computational model reproduces key features of motoneuron bistability, yet its simplified architecture cannot capture the full spatial complexity of motoneurons. By collapsing dendrites into one compartment, our results indicate that a simplified representation can nevertheless sustain bistability (Booth et al., 1997; Carlin et al., 2000; ElBasiouny et al., 2005; Bui et al., 2006) Indeed, dendritic ICaL may amplify calcium signaling and could shape the expression and robustness of plateaus. On the other hand, this simplification does not preclude physiological relevance, as somatic L-type calcium channels can themselves generate prolonged tail currents in motoneurons (Moritz et al., 2007). Such findings support the view that critical aspects of motoneuron bistability can also emerge from intrinsic soma-based mechanisms.
Our future work will thus include the construction and use of multi-compartment models to explore the role of spatial distribution of ICaL, CICR, and ICAN for testing how dendrite- vs. soma-localized mechanisms affect bistability. In this context, establishing the precise subcellular localization of ICAN channels remains a priority but progress is limited by the lack of highly specific antibodies to TRPM5, the presumed molecular correlate of ICAN (Bos et al., 2021). Our analysis centers at L-type calcium channels only, but motoneurons also express T-type and N-type calcium channels (Umemiya and Berger, 1995; Hivert et al., 1995; Viana et al., 1997). Since these currents can complement or replace L-type currents in generating plateaus (Bouhadfane et al., 2013), future models may be instrumental in evaluating their contributions.
In addition to SK-mediated AHPs, motoneurons also display an ultra-slow afterhyperpolarization (usAHP) generated by the electrogenic Na+/K+-ATPase pump. This current, activated by intracellular Na+ accumulation during repetitive firing, produces a long-lasting hyperpolarization that transiently decreases motoneuron excitability. Such pump-mediated usAHPs have been described in lumbar motoneurons (Picton et al., 2017a,b; Sharples et al., 2025). Although not included in the present model, it would be interesting to test in future work how the inclusion of a Na+/K+-pump component could influence plateau potentials and bistability in motoneurons.
In summary, our minimal model captures the core features of bistable firing, with the ICaL-CICR-ICAN triad emerging as a central mechanism, while leaving open the contribution of dendritic processes. Future anatomically detailed models and experiments will be needed to resolve the effects of channels' spatial distributions across motoneuron compartments.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://doi.org/10.5281/zenodo.15527835.
Ethics statement
The animal study was approved by Comité d'Ethique en Neurosciences INT-Marseille, CE71 Nb A1301404, authorization Nb 2018110819197361. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
YM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. FK: Data curation, Formal analysis, Investigation, Validation, Visualization, Writing – review & editing. RJ: Formal analysis, Investigation, Software, Supervision, Validation, Visualization, Writing – review & editing. TS: Software, Visualization, Writing – review & editing. MM: Investigation, Software, Visualization, Writing – review & editing. FB: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. IR: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the ANR MotoBis grant (ANR-24-CE16-1548-01) to FB and the NIH/NINDS grant (R01 NS130799) to IR.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Gen AI was used in the creation of this manuscript. The authors used an AI tool exclusively for grammar and phrasing purposes.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alkadhi, K. A., and Tian, L. M. (1996). Veratridine-enhanced persistent sodium current induces bursting in CA1 pyramidal neurons. Neuroscience 71, 625–632. doi: 10.1016/0306-4522(95)00488-2
Barbay, T., Pecchi, E., Ramirez-Franco, J., Ivanov, A., Brocard, F., Rouach, N., et al. (2025). Functional contribution of astrocytic Kir4.1 channels to spasticity after spinal cord injury. Brain 48:awaf147. doi: 10.1101/2024.10.11.617793
Bennett, D. J., Li, Y., and Siu, M. (2001). Plateau potentials in sacrocaudal motoneurons of chronic spinal rats, recorded in vitro. J. Neurophysiol. 86, 1955–1971. doi: 10.1152/jn.2001.86.4.1955
Benson, C. A., King, J. F., Kauer, S. D., Waxman, S. G., and Tan, A. M. (2023). Increased astrocytic GLT-1 expression in tripartite synapses is associated with SCI-induced hyperreflexia. J. Neurophysiol. 130, 1358–1366. doi: 10.1152/jn.00234.2023
Binder, M. D., Powers, R. K., and Heckman, C. J. (2020). Non-linear input-output functions of motoneurons. Physiology 35, 31–39. doi: 10.1152/physiol.00026.2019
Boost Development Team (n.d.). Boost C++ libraries (Version 1.86). (ver. 1.86). [C++]. Available online at: https://www.boost.org/ (Accessed October 27, 2025).
Booth, V., and Rinzel, J. (1995). A minimal, compartmental model for a dendritic origin of bistability of motoneuron firing patterns. J. Comput. Neurosci. 2, 299–312. doi: 10.1007/BF00961442
Booth, V., Rinzel, J., and Kiehn, O. (1997). Compartmental model of vertebrate motoneurons for Ca2+-dependent spiking and plateau potentials under pharmacological treatment. J. Neurophysiol. 78, 3371–3385. doi: 10.1152/jn.1997.78.6.3371
Bos, R., Drouillas, B., Bouhadfane, M., Pecchi, E., Trouplin, V., Korogod, S. M., et al. (2021). Trpm5 channels encode bistability of spinal motoneurons and ensure motor control of hindlimbs in mice. Nat. Commun. 12:6815. doi: 10.1038/s41467-021-27113-x
Bos, R., Harris-Warrick, R. M., Brocard, C., Demianenko, L. E., Manuel, M., Zytnicki, D., et al. (2018). Kv1.2 Channels promote non-linear spiking motoneurons for powering up locomotion. Cell Rep. 22, 3315–3327. doi: 10.1016/j.celrep.2018.02.093
Bouhadfane, M., Tazerart, S., Moqrich, A., Vinay, L., and Brocard, F. (2013). Sodium-mediated plateau potentials in lumbar motoneurons of neonatal rats. J. Neurosci. 33, 15626–15641. doi: 10.1523/JNEUROSCI.1483-13.2013
Brocard, C., Plantier, V., Boulenguez, P., Liabeuf, S., Bouhadfane, M., Viallat-Lieutaud, A., et al. (2016). Cleavage of Na+ channels by calpain increases persistent Na+ current and promotes spasticity after spinal cord injury. Nat. Med. 22, 404–411. doi: 10.1038/nm.4061
Brocard, F., Shevtsova, N. A., Bouhadfane, M., Tazerart, S., Heinemann, U., Rybak, I. A., et al. (2013). Activity-dependent changes in extracellular Ca2+ and K+ reveal pacemakers in the spinal locomotor-related network. Neuron 77, 1047–1054. doi: 10.1016/j.neuron.2013.01.026
Bui, T. V., Ter-Mikaelian, M., Bedrossian, D., and Rose, P. K. (2006). Computational estimation of the distribution of L-type Ca(2+) channels in motoneurons based on variable threshold of activation of persistent inward currents. J. Neurophysiol. 95, 225–241. doi: 10.1152/jn.00646.2005
Carlin, K. P., Bui, T. V., Dai, Y., and Brownstone, R. M. (2009). Staircase currents in motoneurons: insight into the spatial arrangement of calcium channels in the dendritic tree. J. Neurosci. 29, 5343–5353. doi: 10.1523/JNEUROSCI.5458-08.2009
Carlin, K. P., Jones, K. E., Jiang, Z., Jordan, L. M., and Brownstone, R. M. (2000). Dendritic L-type calcium currents in mouse spinal motoneurons: implications for bistability. Eur. J. Neurosci. 12, 1635–1646. doi: 10.1046/j.1460-9568.2000.00055.x
Collins, D. F., Burke, D., and Gandevia, S. C. (2001). Large involuntary forces consistent with plateau-like behavior of human motoneurons. J. Neurosci. 21, 4059–4065. doi: 10.1523/JNEUROSCI.21-11-04059.2001
Conway, B. A., Hultborn, H., Kiehn, O., and Mintz, I. (1988). Plateau potentials in alpha-motoneurones induced by intravenous injection of L-dopa and clonidine in the spinal cat. J. Physiol. 405, 369–384. doi: 10.1113/jphysiol.1988.sp017337
Cotinat, M., Boquet, I., Ursino, M., Brocard, C., Jouve, E., Alberti, C., et al. (2023). Riluzole for treating spasticity in patients with chronic traumatic spinal cord injury: study protocol in the phase ib/iib adaptive multicenter randomized controlled RILUSCI trial. PLoS ONE 18:e0276892. doi: 10.1371/journal.pone.0276892
Crill, W. E. (1996). Persistent sodium current in mammalian central neurons. Annu. Rev. Physiol. 58, 349–362. doi: 10.1146/annurev.ph.58.030196.002025
D'Amico, J. M., Murray, K. C., Li, Y., Chan, K. M., Finlay, M. G., Bennett, D. J., et al. (2013). Constitutively active 5-HT2/α1 receptors facilitate muscle spasms after human spinal cord injury. J. Neurophysiol. 109, 1473–1484. doi: 10.1152/jn.00821.2012
Deardorff, A. S., Romer, S. H., Deng, Z., Bullinger, K. L., Nardelli, P., Cope, T. C., et al. (2013). Expression of postsynaptic Ca2+-activated K+ (SK) channels at C-bouton synapses in mammalian lumbar-motoneurons. J. Physiol. 591, 875–897. doi: 10.1113/jphysiol.2012.240879
Di Prisco, G. V., Pearlstein, E., Le Ray, D., Robitaille, R., and Dubuc, R. (2000). A cellular mechanism for the transformation of a sensory input into a motor command. J. Neurosci. 20, 8169–8176. doi: 10.1523/JNEUROSCI.20-21-08169.2000
Djukic, B., Casper, K. B., Philpot, B. D., Chin, L-. S., and McCarthy, K. D. (2007). Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. 27, 11354–11365. doi: 10.1523/JNEUROSCI.0723-07.2007
Drouillas, B., Brocard, C., Zanella, S., Bos, R., and Brocard, F. (2023). Persistent Nav1.1 and Nav1.6 currents drive spinal locomotor functions through non-linear dynamics. Cell Rep. 42:113085. doi: 10.1016/j.celrep.2023.113085
Eken, T., Hultborn, H., and Kiehn, O. (1989). Possible functions of transmitter-controlled plateau potentials in alpha motoneurones. Progr. Brain Res. 80, 257–267; discussion 239-242. doi: 10.1016/S0079-6123(08)62219-0
Eken, T., and Kiehn, O. (1989). Bistable firing properties of soleus motor units in unrestrained rats. Acta Physiol. Scand. 136, 383–394. doi: 10.1111/j.1748-1716.1989.tb08679.x
ElBasiouny, S. M., Bennett, D. J., and Mushahwar, V. K. (2005). Simulation of dendritic CaV 1.3 channels in cat lumbar motoneurons: spatial distribution. J. Neurophysiol. 94, 3961–3974. doi: 10.1152/jn.00391.2005
Fraser, D. D., and MacVicar, B. A. (1996). Cholinergic-dependent plateau potential in hippocampal CA1 pyramidal neurons. J. Neurosci. 16, 4113–4128. doi: 10.1523/JNEUROSCI.16-13-04113.1996
Gorassini, M., Bennett, D. J., Kiehn, O., Eken, T., and Hultborn, H. (1999). Activation patterns of hindlimb motor units in the awake rat and their relation to motoneuron intrinsic properties. J. Neurophysiol. 82, 709–717. doi: 10.1152/jn.1999.82.2.709
Gorassini, M. A., Bennett, D. J., and Yang, J. F. (1998). Self-sustained firing of human motor units. Neurosci. Lett. 247, 13–16. doi: 10.1016/S0304-3940(98)00277-8
Grunnet, M., Jespersen, T., and Perrier, J-. F. (2004). 5-HT1A receptors modulate small-conductance Ca2+-activated K+ channels. J. Neurosci. Res. 78, 845–854. doi: 10.1002/jnr.20318
Gustafsson, B., and Pinter, M. J. (1984). Relations among passive electrical properties of lumbar alpha-motoneurones of the cat. J. Physiol. 356, 401–431. doi: 10.1113/jphysiol.1984.sp015473
Harris-Warrick, R. M., Pecchi, E., Drouillas, B., Brocard, F., and Bos, R. (2024). Effect of size on expression of bistability in mouse spinal motoneurons. J. Neurophysiol. 131, 577–588. doi: 10.1152/jn.00320.2023
Harvey, P. J., Li, Y., Li, X., and Bennett, D. J. (2006). Persistent sodium currents and repetitive firing in motoneurons of the sacrocaudal spinal cord of adult rats. J. Neurophysiol. 96, 1141–1157. doi: 10.1152/jn.00335.2005
Heckman, C. J., Johnson, M., Mottram, C., and Schuster, J. (2008). Persistent inward currents in spinal motoneurons and their influence on human motoneuron firing patterns. Neurosci. Rev. J. Bring. Neurobiol. Neurol. Psychiatry 14, 264–275. doi: 10.1177/1073858408314986
Heckman, C. J., Mottram, C., Quinlan, K., Theiss, R., and Schuster, J. (2009). Motoneuron excitability: the importance of neuromodulatory inputs. Clin. Neurophysiol. 120, 2040–2054. doi: 10.1016/j.clinph.2009.08.009
Hivert, B., Bouhanna, S., Diochot, S., Camu, W., Dayanithi, G., Henderson, C. E., et al. (1995). Embryonic rat motoneurons express a functional P-type voltage-dependent calcium channel. Int. J. Dev. Neurosci. 13, 429–436. doi: 10.1016/0736-5748(95)00026-D
Hounsgaard, J., Hultborn, H., Jespersen, B., and Kiehn, O. (1984). Intrinsic membrane properties causing a bistable behaviour of alpha-motoneurones. Exp. Brain Res. 55, 391–394. doi: 10.1007/BF00237290
Hounsgaard, J., Hultborn, H., Jespersen, B., and Kiehn, O. (1988a). Bistability of alpha-motoneurones in the decerebrate cat and in the acute spinal cat after intravenous 5-hydroxytryptophan. J. Physiol. 405, 345–367. doi: 10.1113/jphysiol.1988.sp017336
Hounsgaard, J., and Kiehn, O. (1985). Ca++ dependent bistability induced by serotonin in spinal motoneurons. Exp. Brain Res. 57, 422–425. doi: 10.1007/BF00236551
Hounsgaard, J., and Kiehn, O. (1989). Serotonin-induced bistability of turtle motoneurones caused by a nifedipine-sensitive calcium plateau potential. J. Physiol. 414, 265–282. doi: 10.1113/jphysiol.1989.sp017687
Hounsgaard, J., and Kiehn, O. (1993). Calcium spikes and calcium plateaux evoked by differential polarization in dendrites of turtle motoneurones in vitro. J. Physiol. 468, 245–259. doi: 10.1113/jphysiol.1993.sp019769
Hounsgaard, J., Kiehn, O., and Mintz, I. (1988b). Response properties of motoneurones in a slice preparation of the turtle spinal cord. J. Physiol. 398, 575–589. doi: 10.1113/jphysiol.1988.sp017058
Hounsgaard, J., and Kjaerulff, O. (1992). Ca2+-Mediated plateau potentials in a subpopulation of interneurons in the ventral horn of the turtle spinal cord. Eur. J. Neurosci. 4, 183–188. doi: 10.1111/j.1460-9568.1992.tb00865.x
Hounsgaard, J., and Mintz, I. (1988). Calcium conductance and firing properties of spinal motoneurones in the turtle. J. Physiol. 398, 591–603. doi: 10.1113/jphysiol.1988.sp017059
Hultborn, H., Wigström, H., and Wängberg, B. (1975). Prolonged activation of soleus motoneurones following a conditioning train in soleus Ia afferents—A case for a reverberating loop? Neurosci. Lett. 1, 147–152. doi: 10.1016/0304-3940(75)90030-0
Jasinski, P. E., Molkov, Y. I., Shevtsova, N. A., Smith, J. C., and Rybak, I. A. (2013). Sodium and calcium mechanisms of rhythmic bursting in excitatory neural networks of the pre- B ötzinger complex: a computational modelling study. Eur. J. Neurosci. 37, 212–230. doi: 10.1111/ejn.12042
Jeter, R., and Molkov, Y. (2025). Jeterlab/Ionic-Mechanisms-Underlying-Bistability-in-Spinal-Motoneurons (Version Pre-Release) [Computer software]. Zenodo.
Kernell, D. (1965). The Limits of Firing Frequency in Cat Lumbosacral Motoneurones Possessing Different Time Course of Afterhyperpolarization. Acta Physiol. Scand. 65, 87–100. doi: 10.1111/j.1748-1716.1965.tb04252.x
Kiehn, O., and Eken, T. (1997). Prolonged firing in motor units: evidence of plateau potentials in human motoneurons? J. Neurophysiol. 78, 3061–3068. doi: 10.1152/jn.1997.78.6.3061
Kim, H., and Jones, K. E. (2011). Asymmetric electrotonic coupling between the soma and dendrites alters the bistable firing behaviour of reduced models. J. Comput. Neurosci. 30, 659–674. doi: 10.1007/s10827-010-0284-x
Kuo, J. J., Lee, R. H., Zhang, L., and Heckman, C. J. (2006). Essential role of the persistent sodium current in spike initiation during slowly rising inputs in mouse spinal neurones. J. Physiol. 574, 819–834. doi: 10.1113/jphysiol.2006.107094
Lee, R. H., and Heckman, C. J. (1998a). Bistability in spinal motoneurons in vivo: systematic variations in persistent inward currents. J. Neurophysiol. 80, 583–593. doi: 10.1152/jn.1998.80.2.583
Lee, R. H., and Heckman, C. J. (1998b). Bistability in spinal motoneurons in vivo: systematic variations in rhythmic firing patterns. J. Neurophysiol. 80, 572–582. doi: 10.1152/jn.1998.80.2.572
Lee, R. H., and Heckman, C. J. (1999). Enhancement of bistability in spinal motoneurons in vivo by the noradrenergic α1 agonist methoxamine. J. Neurophysiol. 81, 2164–2174. doi: 10.1152/jn.1999.81.5.2164
Li, Y., and Bennett, D. J. (2003). Persistent sodium and calcium currents cause plateau potentials in motoneurons of chronic spinal rats. J. Neurophysiol. 90, 857–869. doi: 10.1152/jn.00236.2003
Li, Y., Gorassini, M. A., and Bennett, D. J. (2004). Role of persistent sodium and calcium currents in motoneuron firing and spasticity in chronic spinal rats. J. Neurophysiol. 91, 767–783. doi: 10.1152/jn.00788.2003
Morisset, V., and Nagy, F. (1999). Ionic basis for plateau potentials in deep dorsal horn neurons of the rat spinal cord. J. Neurosci. 19, 7309–7316. doi: 10.1523/JNEUROSCI.19-17-07309.1999
Moritz, A. T., Newkirk, G., Powers, R. K., and Binder, M. D. (2007). Facilitation of somatic calcium channels can evoke prolonged tail currents in rat hypoglossal motoneurons. J. Neurophysiol. 98, 1042–1047. doi: 10.1152/jn.01294.2006
Mousa, M. H., and Elbasiouny, S. M. (2020). Dendritic distributions of L-type Ca2+ and SKL channels in spinal motoneurons: a simulation study. J. Neurophysiol. 124, 1285–1307. doi: 10.1152/jn.00169.2020
Murray, K. C., Nakae, A., Stephens, M. J., Rank, M., D'Amico, J., Harvey, P. J., et al. (2010). Recovery of motoneuron and locomotor function after spinal cord injury depends on constitutive activity in 5-HT2C receptors. Nat. Med. 16, 694–700. doi: 10.1038/nm.2160
Murray, K. C., Stephens, M. J., Ballou, E. W., Heckman, C. J., and Bennett, D. J. (2011). Motoneuron excitability and muscle spasms are regulated by 5-HT2B and 5-HT2C receptor activity. J. Neurophysiol. 105, 731–748. doi: 10.1152/jn.00774.2010
Olsen, M. L., Campbell, S. C., McFerrin, M. B., Floyd, C. L., and Sontheimer, H. (2010). Spinal cord injury causes a wide-spread, persistent loss of Kir4.1 and glutamate transporter 1: benefit of 17 beta-oestradiol treatment. Brain J. Neurol. 133, 1013–1025. doi: 10.1093/brain/awq049
Perrier, J-. F., and Cotel, F. (2008). Serotonin differentially modulates the intrinsic properties of spinal motoneurons from the adult turtle. J. Physiol. 586, 1233–1238. doi: 10.1113/jphysiol.2007.145706
Perrier, J-. F., and Delgado-Lezama, R. (2005). Synaptic release of serotonin induced by stimulation of the raphe nucleus promotes plateau potentials in spinal motoneurons of the adult turtle. J. Neurosci. 25, 7993–7999. doi: 10.1523/JNEUROSCI.1957-05.2005
Perrier, J-. F., and Hounsgaard, J. (2003). 5-HT2 receptors promote plateau potentials in turtle spinal motoneurons by facilitating an L-type calcium current. J. Neurophysiol. 89, 954–959. doi: 10.1152/jn.00753.2002
Phillips, R. S., John, T. T., Koizumi, H., Molkov, Y. I., and Smith, J. C. (2019). Biophysical mechanisms in the mammalian respiratory oscillator re-examined with a new data-driven computational model. Elife 8:e41555. doi: 10.7554/eLife.41555.018
Phillips, R. S., Koizumi, H., Molkov, Y. I., Rubin, J. E., and Smith, J. C. (2022). Predictions and experimental tests of a new biophysical model of the mammalian respiratory oscillator. Elife 11:e74762. doi: 10.7554/eLife.74762.sa2
Picton, L. D., Nascimento, F., Broadhead, M. J., Sillar, K. T., and Miles, G. B. (2017a). Sodium pumps mediate activity-dependent changes in mammalian motor networks. J. Neurosci. 37, 906–921. doi: 10.1523/JNEUROSCI.2005-16.2016
Picton, L. D., Zhang, H., and Sillar, K. T. (2017b). Sodium pump regulation of locomotor control circuits. J. Neurophysiol. 118, 1070–1081. doi: 10.1152/jn.00066.2017
Schadt, J. C., and Barnes, C. D. (1980). Motoneuron membrane changes associated with spinal shock and the Schiff-Sherrington phenomenon. Brain Res. 201, 373–383. doi: 10.1016/0006-8993(80)91041-0
Schwindt, P. C., and Crill, W. E. (1980). Properties of a persistent inward current in normal and TEA-injected motoneurons. J. Neurophysiol. 43, 1700–1724. doi: 10.1152/jn.1980.43.6.1700
Sharples, S. A., Nisbet, S. J., Broadhead, M. J., Bo Jensen, D., Sorrell, F. L., Meehan, C. F., et al. (2025). Intrinsic mechanisms contributing to the biophysical signature of mouse gamma motoneurons. J. Physiol. 0, 1–26. doi: 10.1113/JP289631
Simon, M., Perrier, J-. F., and Hounsgaard, J. (2003). Subcellular distribution of L-type Ca2+ channels responsible for plateau potentials in motoneurons from the lumbar spinal cord of the turtle. Eur. J. Neurosci. 18, 258–266. doi: 10.1046/j.1460-9568.2003.02783.x
Svirskis, G., and Hounsgaard, J. (1997). Depolarization-induced facilitation of a plateau-generating current in ventral horn neurons in the turtle spinal cord. J. Neurophysiol. 78, 1740–1742. doi: 10.1152/jn.1997.78.3.1740
Tazerart, S., Vinay, L., and Brocard, F. (2008). The persistent sodium current generates pacemaker activities in the central pattern generator for locomotion and regulates the locomotor rhythm. J. Neurosci. 28, 8577–8589. doi: 10.1523/JNEUROSCI.1437-08.2008
Tong, X., Ao, Y., Faas, G. C., Nwaobi, S. E., Xu, J., Haustein, M. D., et al. (2014). Astrocyte Kir4.1 ion channel deficits contribute to neuronal dysfunction in Huntington's disease model mice. Nat. Neurosci. 17, 694–703. doi: 10.1038/nn.3691
Toporikova, N., and Butera, R. J. (2011). Two types of independent bursting mechanisms in inspiratory neurons: an integrative model. J. Comput. Neurosci. 30, 515–528. doi: 10.1007/s10827-010-0274-z
Tysseling, V. M., Klein, D. A., Imhoff-Manuel, R., Manuel, M., Heckman, C. J., Tresch, M. C., et al. (2017). Constitutive activity of 5-HT2C receptors is present after incomplete spinal cord injury but is not modified after chronic SSRI or baclofen treatment. J. Neurophysiol. 118, 2944–2952. doi: 10.1152/jn.00190.2017
Umemiya, M., and Berger, A. J. (1995). Single-channel properties of four calcium channel types in rat motoneurons. J. Neurosci. 15, 2218–2224. doi: 10.1523/JNEUROSCI.15-03-02218.1995
Viana, F., Van den Bosch, L., Missiaen, L., Vandenberghe, W., Droogmans, G., Nilius, B., et al. (1997). Mibefradil (Ro 40-5967) blocks multiple types of voltage-gated calcium channels in cultured rat spinal motoneurones. Cell Calcium 22, 299–311. doi: 10.1016/S0143-4160(97)90068-3
Yan, H-. D., Villalobos, C., and Andrade, R. (2009). TRPC channels mediate a muscarinic receptor-induced afterdepolarization in cerebral cortex. J. Neurosci. 29, 10038–10046. doi: 10.1523/JNEUROSCI.1042-09.2009
Zhang, M., Møller, M., Broman, J., Sukiasyan, N., Wienecke, J., Hultborn, H., et al. (2008). Expression of calcium channel CaV 1.3 in cat spinal cord: light and electron microscopic immunohistochemical study. J. Compar. Neurol. 507, 1109–1127. doi: 10.1002/cne.21595
Keywords: motoneuron, bistability, modeling, plateau potential, spinal cord
Citation: Molkov YI, Krust F, Jeter R, Stell T, Mohammed MAY, Brocard F and Rybak IA (2025) Ionic mechanisms underlying bistability in spinal motoneurons: insights from a computational model. Front. Cell. Neurosci. 19:1710893. doi: 10.3389/fncel.2025.1710893
Received: 22 September 2025; Accepted: 24 October 2025;
Published: 11 November 2025.
Edited by:
Haruyuki Kamiya, Hokkaido University, JapanReviewed by:
Simon Arthur Sharples, Cincinnati Children's Hospital Medical Center, United StatesHiroshi Kuba, Nagoya University, Japan
Copyright © 2025 Molkov, Krust, Jeter, Stell, Mohammed, Brocard and Rybak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaroslav I. Molkov, eW1vbGtvdkBnc3UuZWR1
†These authors have contributed equally to this work
‡These authors share senior authorship