 Małgorzata Trofimiuk-Müldner1*†
Małgorzata Trofimiuk-Müldner1*† Bartosz Domagała2
Bartosz Domagała2 Grzegorz Sokołowski1
Grzegorz Sokołowski1 Anna Skalniak1
Anna Skalniak1 Alicja Hubalewska-Dydejczyk1
Alicja Hubalewska-Dydejczyk1- 1Chair and Department of Endocrinology, Jagiellonian University Medical College, Kraków, Poland
- 2Department of Endocrinology, Endocrine Oncology and Nuclear Medicine, University Hospital in Kraków, Kraków, Poland
Introduction: Up to 5% of all pituitary tumors are hereditary e.g. due to MEN1 or aryl hydrocarbon receptor-interacting protein (AIP) genes mutations.
Objectives: The study was aimed at the assessment of the frequency and characteristics of AIP-mutation related tumors in patients with apparently sporadic pituitary macroadenomas in the Polish population.
Materials and methods: The study included 131 patients (57 males, 74 females; median age 42 years) diagnosed with pituitary macroadenomas, and with a negative family history of familial isolated pituitary adenoma (FIPA) or multiple endocrine neoplasia type 1 (MEN1) syndromes. Sanger sequencing was used for the assessment of AIP gene variants. The study was approved by the Ethics Board of JUMC.
Results: AIP variants were identified in five of the 131 included subjects (3.8%): one diagnosed with Cushing’s disease, two with acromegaly, and two with non-secreting adenomas. Patients harboring hereditary AIP gene alterations did not differ from the rest of the study group in median age at diagnosis (41.0 vs. 42.5 years, P=0.8), median largest tumor diameter (25 vs. 24 mm, P=0.6), gender distribution (60.0% vs. 56.3% females, P=0.8), secreting tumor frequency (60.0% vs. 67.5%, P=0.7), or acromegaly diagnosis frequency (40.0% vs.37.3%, P=0.9).
Conclusions: In our series of apparently sporadic pituitary macroadenomas, AIP gene variant carriers did not differ substantially from patients with negative genetic testing. A risk factor-centred approach to AIP genetic screening may result in missing germline variants. Considering the clinical impact of such genetic variants and their relatively low penetrance, it is, however, doubtful if general genetic screening benefits the whole cohort of pituitary macroadenoma patients and their families.
Introduction
Pituitary tumors, if autopsy and radiological imaging are taken into consideration, occur in about 16.7% of the general population (1). The prevalence of clinically significant lesions is estimated at one case per approximately 1000 individuals (2, 3). Most of pituitary adenomas are sporadic (4). Only up to 5% of them are hereditary and can be caused, e.g. by MEN1 or aryl hydrocarbon receptor-interacting protein (AIP) gene mutations, and present as a part of multiple endocrine neoplasia type 1 (MEN1) or familial isolated pituitary adenoma (FIPA) syndromes (4, 5).
The AIP gene is a suppressor gene encoding a 330 amino acid protein involved in the cAMP-phosphodiesterases pathway (6–11). The most common AIP variants (AIPvar) are nonsense and missense mutations, deletions, insertions, splice-site and promoter mutations, and large deletions (6, 7). Most of them may result in a truncated protein or, less frequently, affect the tetratricopeptide repeat (TPR) domains or the C-terminal α-helix (6–8, 12, 13). Furthermore, in patients with germline AIPvar, loss of heterozygosity (LOH) has been found in the tumor tissue at the site of the AIP gene in the 11q13 region (6, 8). Some of the AIPvar are rare alterations without pathogenic effects, and no impact on protein function. Differentiating between these issues is important because rare genetic changes are also found in healthy controls (7). Whenever AIPvar is used in this manuscript, it refers to pathogenic or possibly pathogenic variants of AIP.
About 90% of AIPvar-related pituitary tumors are macroadenomas (14), mostly (80%) somatotropinomas and prolactinomas (6, 14, 15). Adenomas in patients carrying pathogenic AIPvar are characterized by larger size, younger age at diagnosis (<30 years), aggressive growth, and resistance to treatment (4, 5, 14, 16). Low AIP protein expression is a better predictor of GH-secreting tumor aggressiveness than high Ki-67 index or p53 expression (4, 17, 18).
Our investigation was aimed at the assessment of the frequency and characteristics of pathogenic AIPvar-related tumors in a population of Polish patients with apparently sporadic pituitary macroadenomas.
Materials and methods
This was a single-center study. Inclusion criteria were: (a) diagnosis of pituitary macroadenoma, (b) age >18 years, (c) negative family history of MEN1 syndrome, familial isolated pituitary adenomas (FIPA), and other hereditary syndromes with pituitary involvement, (d) informed consent to genetic testing. Between 2013 and 2019, 1134 patients with pituitary adenomas were hospitalized at the Department of Endocrinology specializing in adult care. Finally, after assessment for eligibility, 131 patients were included (Figure 1). Data on patients’ sex, age at diagnosis, tumor size, tumor type (clinical manifestation), pituitary surgery, and other treatments were recorded.

Figure 1 Flow chart of the patients’ inclusion.
Written informed consent was obtained from all participants. The study design was approved by the Ethics Board of the Jagiellonian University (KBET/119/B/2013).
Genetic testing was performed between 2013 and 2019. DNA was isolated from peripheral blood samples. DNA sequencing was done using the Sanger method on an ABI3500 Genetic Analyzer. Analysis of the results was performed in SeqScape v2.7. All AIP coding exons and adjacent splice sites were analyzed. For variants, the clinical significance and prevalence in the population were determined based on publicly accessible databases (NCBI ClinVar, HGMD, VarSome), with NM_003977 used as a reference sequence (19–21).
Statistical analysis was performed with IBM SPSS Statistics 28.0. The Mann-Whitney U test was used for comparing the groups due to non-Gaussian distribution of data. P values <0.05 were considered statistically significant.
Results
The study included 131 pituitary macroadenoma patients (57 males – 43.5%) diagnosed at 18-75 years (median age 42 years). Forty-two patients were diagnosed with GH-secreting tumors (32.1%), 21 (16.0%) with prolactinomas, 11 (8.4%) with ACTH-secreting tumors, 6 (4.6%) with gonadotropinomas, 1 (0.8%) with TSH-oma, and 7 (5.3%) with plurihormonal pituitary adenomas. The remaining 43 (32.8%) tumors were non-secreting.
Patients without AIP gene germline variants
This group comprised 126 patients (96.2% of the study group), 55 males (43.7%). The median age at diagnosis was 42.5 years (range 18-75 years). The median largest tumor diameter in this group was 24 mm. 101 (80.2%) of AIPvar-negative (AIPvar(–)) patients underwent neurosurgical procedures (mostly transsphenoidal adenomectomy); the surgery was curative in 42 patients (41.6%). Among 47 patients with GH-secreting and plurihormonal tumors, 22 required prolonged treatment with somatostatin analogues (SSA).
Patients with AIP gene germline variants
A germline AIPvar was identified in five patients (3.8%): two with c.47G>A (p.Arg16His), two with c.911G>A (p.Arg304Gln) and one with c.684G>A (p.Gln228=). One patient was diagnosed with Cushing’s disease (9.1% of 11 ACTH-producing adenomas), two with acromegaly (4.8% of 42 GH-producing adenomas), and two with non-secreting adenomas (4.7% of 43 non-secreting tumors). There was no difference in secreting tumor (P=0.7), or acromegaly frequency (P=0.9) as compared to the group without pathogenic AIPvar. This group consisted of two males and three females (no difference in gender distribution from the AIPvar(–) group, P=0.8). The patients’ median age at diagnosis was 41 years (range 23-74 years) and did not differ from the AIPvar(–) group (p=0.8). The median largest tumor diameter (25 mm) also did not differ significantly (P=0.6). Four out of 5 patients were treated surgically (80%), and the surgery was curative in one patient. One out of two patients diagnosed with acromegaly was treated with somatostatin analogues.
A brief description of the AIPvar positive (AIPvar(+)) patients and the predicted impact of the detected AIPvar are presented in Table 1 and Table 2, respectively.
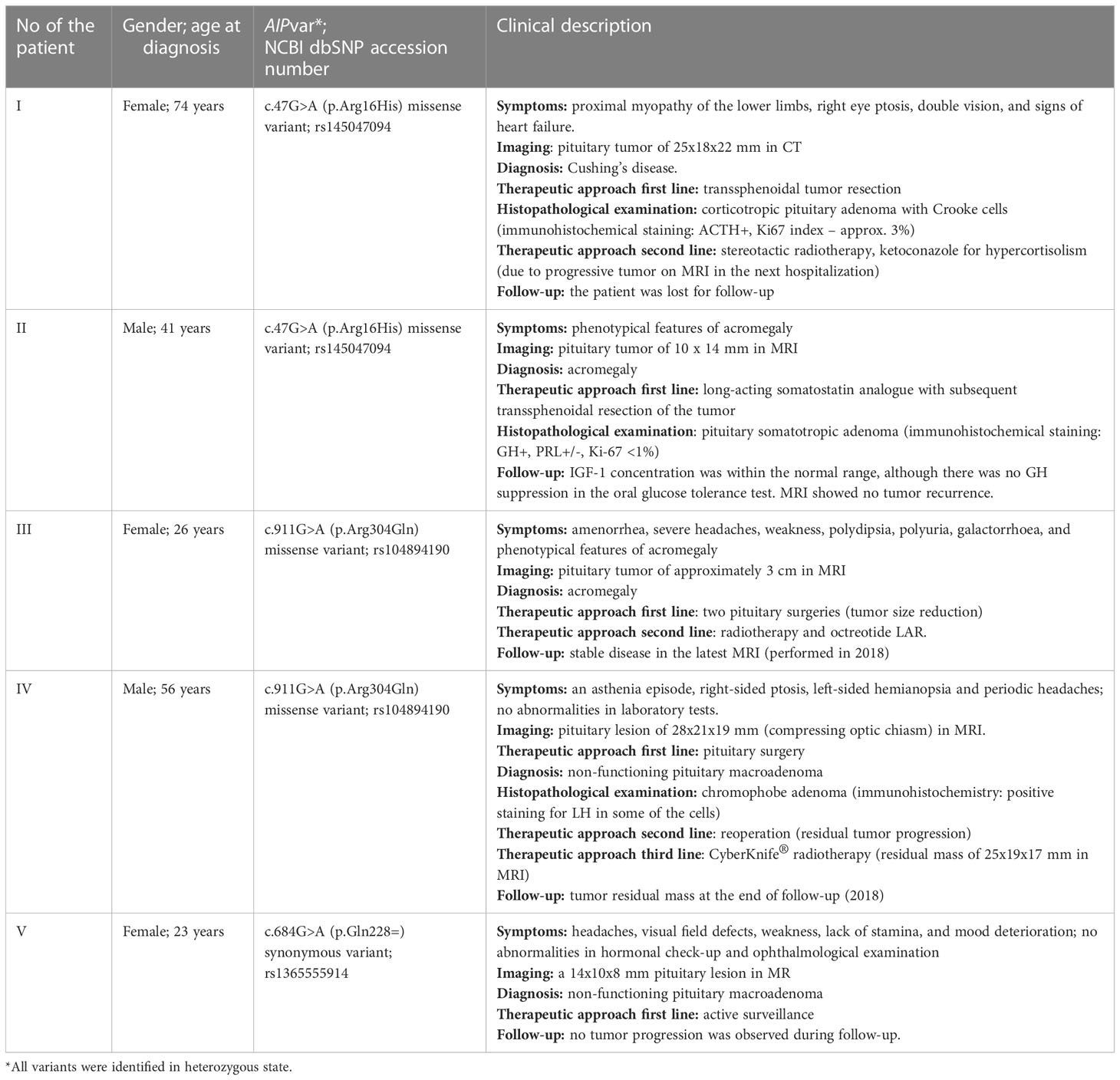
Table 1 Clinical description of AIPvar(+) patients.
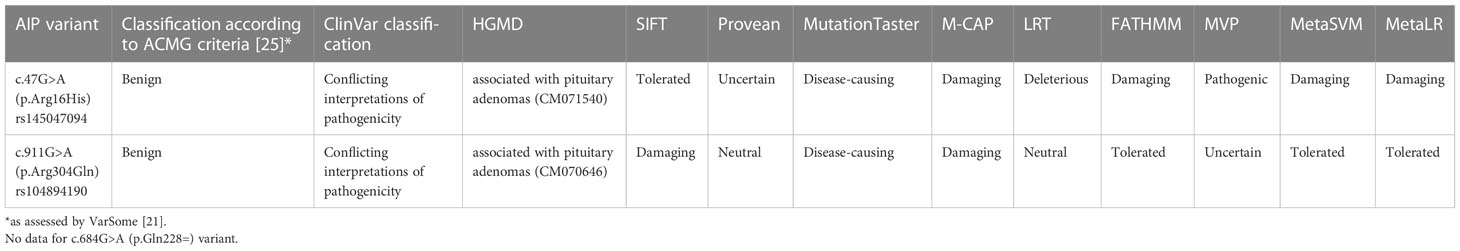
Table 2 Predicted impact of the detected AIP variants: disease-related classifications and in silico assessment of the impact on the AIP protein (according do prediction tools).
Family screening
Two of the five AIPvar(+) patients agreed for their families to be offered AIP genetic testing. The AIP alteration c.911G>A was found in the asymptomatic mother of patient III, who proved negative on hormonal check-ups and imaging. The second tested person was the underaged son of patient II. He carried an AIPvar of unknown clinical significance, c.47G>A, and has not been clinically screened yet.
Discussion
In this study, we wanted to investigate the frequency and characteristics of germline AIPvar in Polish patients with apparently sporadic pituitary macroadenomas, followed up in the tertiary academic clinical center. We have found AIP variants that are very rare and/or have been described in the databases NCBI ClinVar or HGMD as pathogenic or of unknown significance in 3.8% of the studied group. Similar frequencies of germline AIPvar were found in a study by Cazabat et al. They have found germline AIPvar in 16 (3.6%) out of 443 (aged 4-87 years, both with micro- and macroadenomas) (22). The prevalence of AIPvar(+) patients might be higher in younger (≤30 years) cohorts. In a study by Hernandez et al., 8.4% of young-onset sporadic pituitary adenoma cases harbored pathogenic or likely pathogenic AIPvar (15). In a sporadic cohort of patients diagnosed with macroadenomas ≤30 years or with pituitary adenomas ≤18 years of age, Marques et al. found AIP alterations in 6.8% of cases (23). In contrast, in a Spanish cohort of 235 apparently sporadic adenomas in patients ≤30 years of age, pathogenic AIPvar were detected in 3.8 of cases (24). The higher incidence of pathogenic mutations in some of the above-mentioned studies is mainly because the pediatric population was not included in our analysis. It should be noted that pathogenicity assessment may be based on different criteria, parameters, and tools. The missense variants identified in our study, designated as pathogenic or of unknown significance in the clinical databases NCBI ClinVar and HGMD, have been classified as (likely) benign according to ACMG 2015 criteria (25), as assessed by VarSome. The different assessment of pathogenicity may also be the cause of different variant frequency identification in the studies mentioned above.
Based on the collected data, we have found that our patients with AIPvar did not differ substantially from the rest of the study group. In our study, the median age at diagnosis in AIPvar(+) patients was 41.0 vs. 42.5 years in the rest of the group. This is inconsistent with most of the available data. In the series by Marques et al., patients with pathogenic AIPvar were 8 years younger at the onset of symptoms and 6 years younger at diagnosis (23). 65% of them were younger than 19 years, and 87% were younger than 30 years. Daly et al. compared patients with or without AIPvar and recorded a larger difference in age at diagnosis: 25.7 vs. 38.8 years, respectively (13). A similar age difference was found by Cazabat et al. (22): 23.5 vs. 40.9 years, respectively; none of the AIPvar carriers was older than 40 years. This contrasts with our group, in which 3 out of 5 patients were older than 40 years. It may be argued that patients were younger at the onset of the disease, nevertheless, the oldest was over 70 years old when diagnosed with Cushing’s disease. Data may also be distorted by a large number of patients excluded due to lack of consent to participate in the study, exclusion of children and adolescents, and a delay in referring patients to the Endocrinology Department by other medical specialists due to the lack of specific symptoms.
We noted no difference in median tumor diameter, whereas, in published studies, the size difference is significant in favor of AIPvar-related tumors, 24.6 ± 10.7 mm vs. 14.5 ± 10.1 mm in genetically unaltered cases (13). This may be because we only enrolled macroadenoma patients, and large tumors are predictive of germline AIPvar (26). In the sporadic pituitary tumors cohort reported by Hernandez-Ramirez et al., there was no difference in the proportion of giant adenomas between AIPvar positive and negative patients (15). All AIPvar(+) sporadic cases had macroadenomas (in contrast to 86.3% in AIPvar(–) group), and presented more frequently extrasellar extension (95% vs. 58.9%, respectively).
The available data indicates a small predominance of males (12, 16, 26) or an equal number of males and females (15, 27) among AIPvar carries. In a large international collaborative study, AIPvar carriers were predominantly males (63.5%) (16). Similar results were obtained in a smaller study of sporadic pituitary tumors (around 61% AIPvar carriers being males) (12). Interestingly, male and female patients showed no phenotypic differences (12, 16). The gender proportions in our study were inverted: 3 AIPvar(+) females to 2 males (similarly as in the whole screened group).
We have also noticed a deviation from other cohorts in the tumor types distribution. In our study, GH-secreting tumors were the most common (40% of AIPvar(+) patients and 37% of the rest of the group). The data indicate that GH-secreting tumors appear more often in AIPvar(+) cases, up to 78.1% of some studied cohorts (14, 16, 26). The proportion of AIPvar(+) acromegalic patients was similar (4.8% vs. 4.1%) as reported by Cazabat et al., and higher in the case of ACTH-producing (9.1% vs. 6.8% in Cazabat’s study) or non-secreting/gonadotropin-secreting adenomas (4.7% vs. 0.9%, respectively) (22). We have not found any AIPvar(+) patient with prolactinoma (in contrast to 4.6% of all prolactin secreting tumors in Cazabat study (22)). The group of Hernandez-Ramirez did not find any AIPvar(+) patients with Cushing’s disease, functioning gonadotropinomas, or TSH-omas (15). In their cohort of sporadic AIPvar(+) cases, all patients were diagnosed with acromegaly. In a sporadic cohort of young patients, AIPvar-related tumors accounted for 10.5% of somatotropinomas, 1.3% of prolactinomas, and none of the non-functioning adenomas (23).
Our group of AIPvar(+) patients differs from those reported in the literature in the clinical presentation (Table 1) and age at diagnosis. This may be explained by the small group size and selection bias, particularly screening-out patients diagnosed during childhood, who comprise a large proportion of the other cohorts (22). Most of the patients who agreed to participate were referred for hospital workup, which may explain the underrepresentation of prolactinomas.
Surgery and SSA treatment are less effective in AIPvar(+) cases (5, 16). Leontiou et al. evaluated the response to SSA in AIPvar(+) patients and found that 53% had a poor response to therapy (27). In contrast, resistance to SSA therapy is noted in about 25% of AIPvar (–) patients (27–29). In another study, more than one-third of patients with AIPvar-related somatotropinomas underwent two or more surgical interventions. Furthermore, only 11% achieved disease control during post-operative SSA treatment; tumor shrinkage was observed in 16% of cases (5). In Marqes et al. study AIPvar-related adenomas more frequently required multimodal and multiple treatments (23). Similar tumor behavior was noticed in our group. Only one patient was followed without any interventions so far. Most of the presented cases, like in other reports (4, 5, 14, 16), required a multimodal therapeutic approach, which frequently did not result in proper disease control. The considered reasons for treatment resistance in AIPvar(+) tumors are defective Gai2 or ZAC1 pathways, mediating SST2 receptors function (18, 30). It is likely that decreased AIP expression is the cause of the poor response of GH-releasing tumors to SSA treatment (18, 30, 31).
In our study, we also wanted to assess the impact of the three AIPvar types detected in our group, on the course of the disease and compare the results with the available literature.
Patients I and II harbored the c.47G>A (p.Arg16His) missense AIPvar (rs145047094). In the non-Finnish European population, the variant occurs with a frequency of 0.34%, according to the gnomAD v2.1.1 database (32). The pathogenicity interpretation according to ACMG guidelines (25) (assessed by VarSome (21)), ClinVar and HGMD databases, as well as in silico classifications of the variant are summarized in Table 2.
The summary of published data on the clinical significance of the c.47G>A variant is presented in Table 3. Most authors have interpreted this alteration as a very rare variant without a clear causative effect (Table 3). Interestingly, this variant was noted in patients with Cushing’s disease from Poland, as well as in a young patient with Cushing’s disease in Cazabat et al. cohort (22, 33). Similarly, patient I in our group was diagnosed with Cushing’s disease due to an aggressive type of adenoma (Crooke cells in IHC staining: ACTH+, Ki67 about 3%), refractory to treatment. On the other hand, in patient II with acromegaly, the adenoma showed no signs of increased aggressiveness on histopathological examination; however, he did not fulfil all acromegaly remission criteria. This variant was also found in the patient’s son, but he is yet to be evaluated clinically. Regarding offspring age, only longitudinal observation may prove if the variant will be pathogenic. Therefore, the impact of the c.47G>A variant on pituitary tumorigenesis still needs to be elucidated.
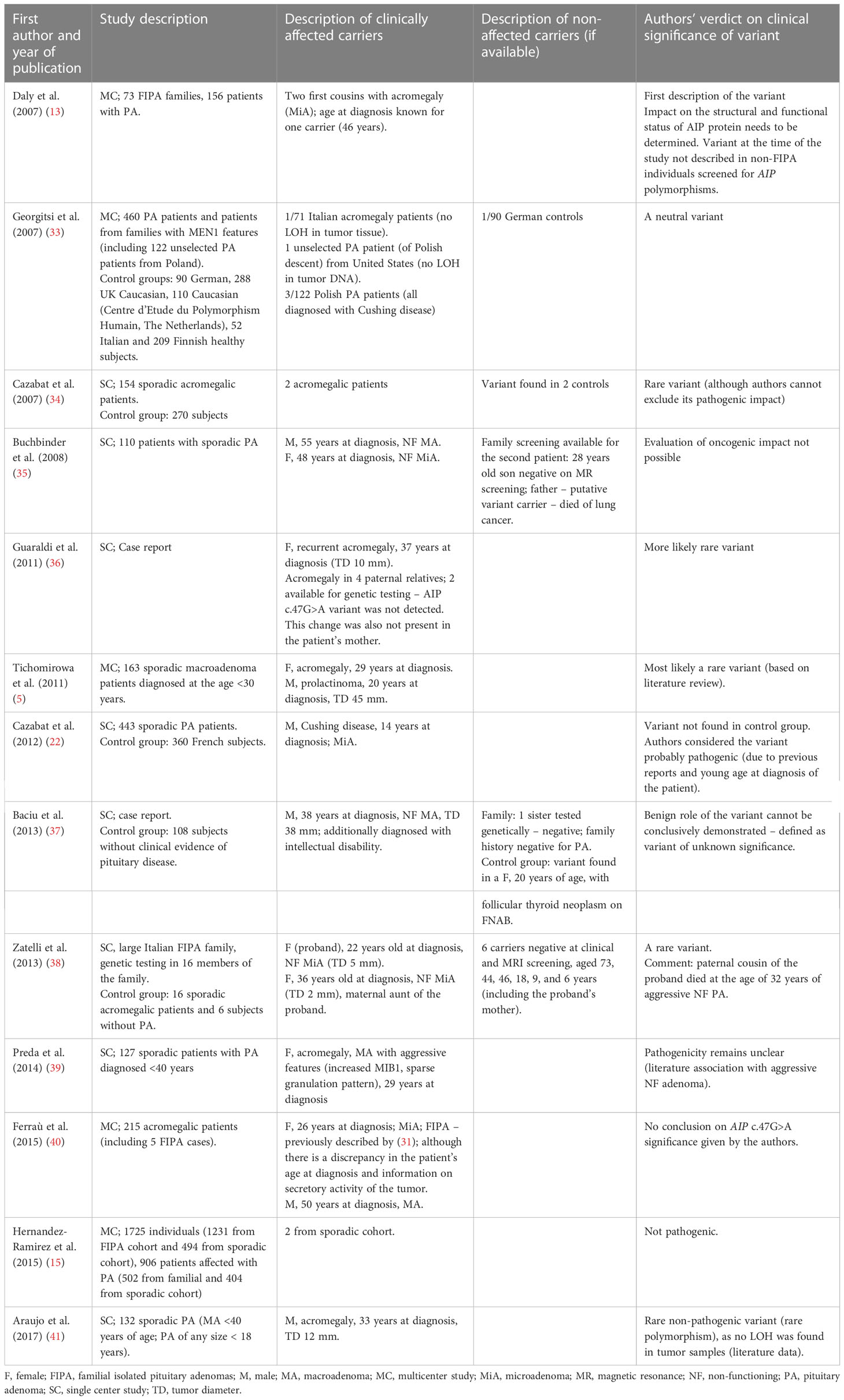
Table 3 Published data on clinical significance of AIP c.47G>A variant.
While examining the mechanisms of the pathogenicity of the rs145047094 variant, Baciu et al. concluded that pathogenic variants which lead to premature stop codons, cause truncating AIP proteins and affect important functional domains; however, missense changes can have both pathological and mild effects (37). Pituitary tumorigenesis may require other pathology, for example, the LOH in adenoma cells, which was not observed in the case of the above-mentioned variant (13, 33–35). It has also been suggested that this variant lacks functional impact due to the weak binding effect of PDE4A5 (6). In a latest work of Garcia-Rendueles et al., who performed a series of functional in vitro analyses, N-term and C-term AIP point variants were proven to impact the molecular interactions of AIP and block the RET-apoptotic pathway. Based on those criteria, the variant p.Arg16His was classified as pathogenic (42).
Another missense variant, c.911G>A (p.Arg304Gln), was detected in our study in a 26-year-old acromegalic woman and a 56-year-old man with non-secreting adenoma (patients III and IV). It is registered in NCBI dbSNP under the accession number rs104894190. For pathogenicity classification and in silico analyses, see Table 2. This variant occurs at a CpG island hotspot (5–7). The clinical significance of the c.911G>A variant is equivocal (Table 4).
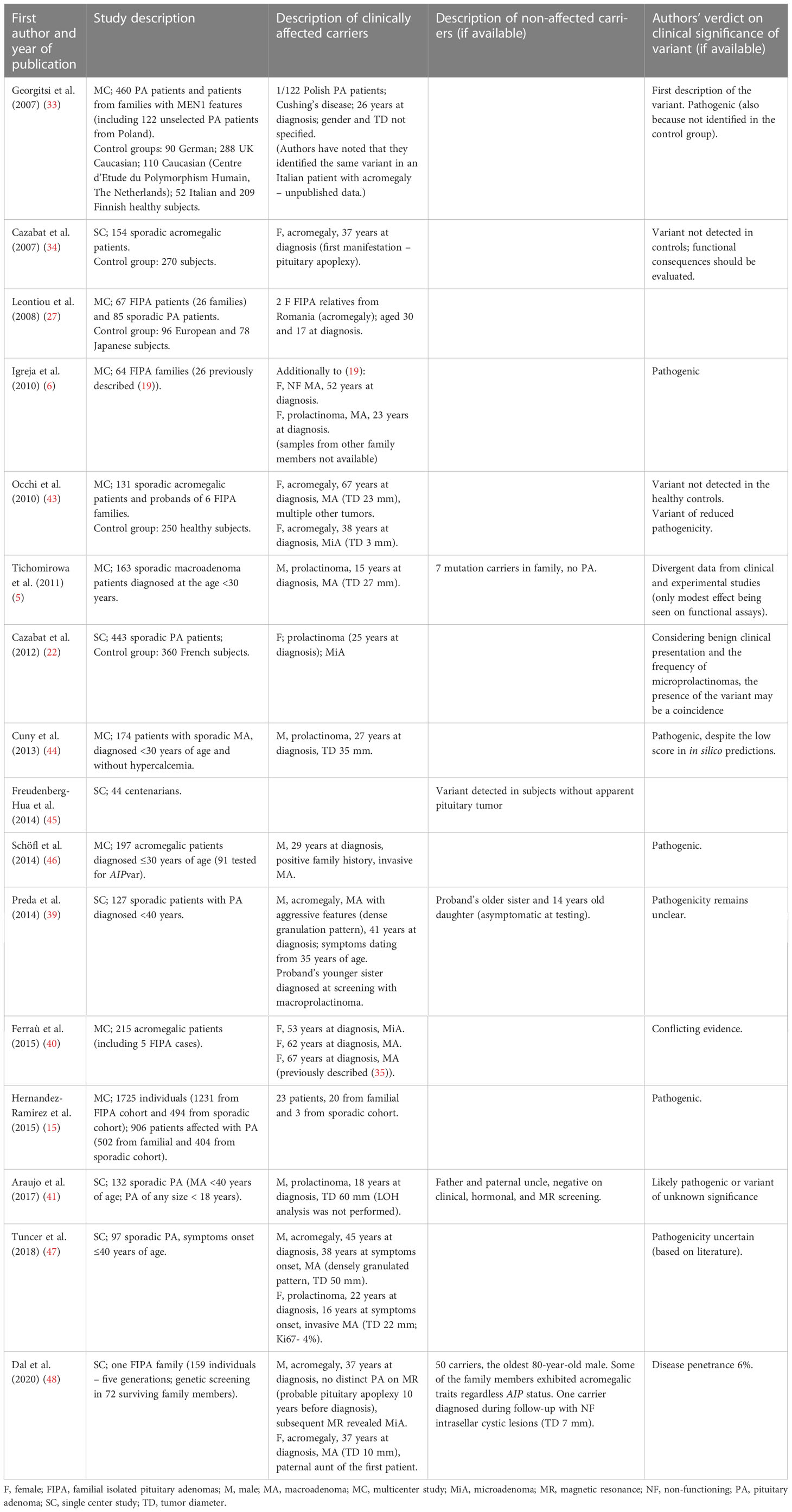
Table 4 Published data on clinical significance of AIP c.911G>A variant.
Dal et al. have postulated the role of other genes in the development of pituitary tumors in AIPvar carriers, namely PDE11A (associated with adrenal tumorigenesis) and ALG (coding a protein essential for glycoprotein folding and stability) (48). Patient III from our study had a treatment-resistant tumor and required two surgeries, radiotherapy, and SSA treatment. The mother of the patient had the c.911G>A (p.Arg304Gln) variant but she was negative on biochemical check-up and imaging, which may question the variant’s clinical pathogenicity. Patient IV required double surgery and stereotactic radiotherapy, which may suggest that the variant impacts the clinical course of pituitary adenomas. While examining the mechanism of the mutation some authors found that this variant did not significantly reduce PDE4A5 binding (6), other concluded that it may impact AIP and aryl hydrocarbon receptor (AHR) interactions (33) or may interfere with protein stability or folding (49) without directly affecting the protein-protein interaction (6). Other authors observed that this AIP alteration did not disrupt chaperone binding and did not show a significant reduction in β-galactosidase activity, which may reduce its pathogenic effect (6, 50). Hernández-Ramírez et al. also concluded that the pathogenicity of the c.911G>A variant is uncertain (51). Aflorei et al. in their in vivo tests using Drosophila melanogaster models, decided that both the p.Arg16His and p.Arg304Gln variants should be assessed as non-pathogenic (52). Dal et al. estimated the penetrance of c.911G>A variant at 6% (48). In the study by Garcia-Rendueles et al., the functional outcome in transfected cells was similar as in the case of the variant p.Arg16His, therefore, the disease-causing potential of the variant p.Arg304Gln was also proven positive in this study (42).
The last variant was c.684G>A (rs1365555914), which was found in patient V. The pituitary tumor in this case was smaller than in the previous patients (I-IV), and subsequent pituitary MRIs did not reveal its progression over the years. The c.684G>A variant itself does not introduce an amino acid change (CAG>CAA, Lys>Lys), however, due to the additional common variant rs641081 (c.682C>A) in homozygous state in the patient, the final codon at position 228 in the protein in this patient changed from CAG to AAA, leading to the amino acid alteration p.Gln228Lys. The p.Gln228Lys alteration caused by the common variant rs641081 alone has been classified as benign, and the more prevalent allele A (observed in a homozygous state in the patient) occurs in the non-Finnish European population with a frequency of 99.8%. The variant rs1365555914 detected in the patient is extremely rare, with no information on its frequency available and zero allele counts reported in the gnomAD database for any population. This rare variant does not change the coded amino acid with any of the alleles of the rs641081 variant and, although there is a noticeable difference in the codon usage frequency for the reference glutamine (35.5 vs 14.1 per thousand for CAG and CAA, respectively), the usage frequency for lysine codons with the rs1365555914 variant, as observed in the patient, are very similar: 31.8 vs 27.5 per thousand for AAG and AAA, respectively (53). It is, therefore, not clear whether or not this variant is of clinical significance for the patient. However, we report it due to its rarity and because it was the only suspected variant identified in the genetic screening of the patient diagnosed at a young age.
The above-discussed AIP alterations illustrate the difficulties in genetic testing results interpretation. The inconsistency in assessing the pathogenic impact of AIP variants is particularly seen in simplex cases (54). Negative family history may be caused by reduced penetration of the variant or lack of information about the family (55). The penetration of pituitary adenoma in AIPvar carriers is described in the range 12-30% (14, 55). Some studies even indicate that the pathogenic variant type has little effect on penetration (15, 55).
The question remains, what significance for clinical management each type of AIPvar has. Daly et al. asked whether dividing AIP variants into non-pathogenic or pathogenic is useful (56). It is unclear whether AIP alterations are always the main factor responsible for tumor development. It may be that AIP variants only facilitate tumor formation. Variants considered clinically pathogenic may be reclassified as innocent on further analysis (56). Therefore, rare alterations deserve special attention. In our study, regardless of the specific type of the variant and its pathogenicity described in literature, most of the AIPvar-related tumors were aggressive and usually resistant to standard treatment. Other data suggest that the type of changes in the AIP-encoded protein caused by mutation may be relevant for disease course and the decision on family member screening. According to Hernandez-Ramirez et al., truncating mutations are related to a younger age at diagnosis and the onset of symptoms, and a more common occurrence of pediatric cases (15). In this study, there was no difference between truncating and nontruncating mutations in the proportion of acromegaly, the number of patients per family, maximum tumor diameter, or extrasellar expansion.
During the genetic screening, new variants/mutations of the AIP gene may be found, and their pathogenicity usually needs to be proven. For example, in the study by Cazabat et al. (22), previously not described variants were found in half of sporadic AIPvar(+) patients. The authors performed parental screening in 7 out of 16 AIPvar(+) patients. In all of them, one of the parents was an asymptomatic carrier. Of note, no pituitary adenoma was found in parents-carriers, who agreed to clinical evaluation (22). Ten new, likely pathogenic mutations were also reported by Hernandez-Ramirez’ group (15).
The important question is the probability of pituitary adenoma development in asymptomatic AIPvar carriers. In a group of 160 apparently unaffected AIPvar carriers in the Hernandez-Ramirez study, pituitary adenoma was established in 11.3% of patients (15). Half of them exhibited GH oversecretion, the rest were diagnosed with non-functioning adenomas. Only five out of 18 prospective cases were diagnosed with macroadenomas. The authors concluded that depending on the applied clinical screening, up to 25% of apparently unaffected carriers may develop pituitary adenoma. Marques et al. prospectively followed 187 apparently unaffected AIPvar carriers identified by testing of the first-degree relatives of AIPvar(+) FIPA and sporadic adenoma patients (23). 88.2% of them did not develop a pituitary adenoma during the mean 5.9 ± 3.3 years follow-up. 19 of 22 adenomas were diagnosed at first screening (in 8 cases, retrospective signs which may be attributed to the pituitary tumor were noted), and 3 cases were recognized during subsequent follow-up. The prospectively recognized pituitary adenomas were smaller, 68% being microadenomas, and were associated with lower rates of hypopituitarism at diagnosis, extrasellar extension, or cavernous sinus invasion. Such patients were less frequently operated and none of them required radiotherapy.
Finally, it is worth asking which patients should be screened, considering the varying course of the disease and low disease penetration in patients with AIPvar. Genetic screening was most commonly indicated in the case of (12, 14, 26): meeting the criteria of FIPA, or pituitary adenoma diagnosed in <18 year-olds, or pituitary macroadenoma diagnosed in <30 year-olds. The probability of detecting new AIP alterations in the fifth decade of life is low (2, 12, 15). Published data suggest that only 13.2% of AIPvar(+) patients had an onset of illness after 30 years of age (15). Therefore, unselected screening is probably not a cost-effective method (22, 27, 57–59). In large unselected cohorts, AIP pathogenic or likely-pathogenic variants occurred in 3.6%-8.3% of included patients (22, 27, 57, 59, 60). If young populations were considered, the incidence increased from 11.7% in patients <30 years to 20.5% in pediatrics (5). Although in our group, AIP variants were detected in older patients with an aggressive course of the disease, the benefit for the individual patient over 40-50 years of age from genetic screening is negligible if the family history is negative. It seems that the detection of an AIPvar in this setting does not impact the patients’ management, as the AIP-encoded protein is not a therapeutic target currently. Even if a more aggressive course of adenomas in AIP germline variant-carriers may be predicted, the treatment modalities did not differ from those applied in large to giant, invading or drug-resistant pituitary tumors unrelated to AIP alterations.
In a systematic review by van den Broek et al. (26), the following recommendations were proposed based on available studies. The authors strongly recommend against routine genetic testing in sporadic pituitary adenoma. A weak recommendation for AIP mutation analysis in patients with sporadic pituitary adenomas 30 years old or younger, especially those diagnosed with acromegaly and gigantism, was made based on low-quality evidence.
Conclusion
In conclusion, in our series of apparently sporadic pituitary macroadenomas, AIPvar carriers were identified in 3.8% of the study group and did not differ substantially from patients with negative genetic testing. Therefore, routine genetic screening for AIP variants in non-selected adult pituitary adenoma patients seems currently ineffective. It seems that in clinical practice, a targeted screening approach limited to patients at risk of AIP-related pituitary adenomas should be applied. If an AIP variant is detected, genetic testing should be discussed with the proband’s family members (particularly symptomatic and younger ones), as additional data may improve our understanding of the clinical significance of detected genetic alteration.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
Study design: MT-M. Data collection: MT-M, GS. Data analysis: MT-M, BD, AS. Manuscript drafting: MT-M, BD, AS. Manuscript revision: MT-M, BD, GS, AS, AH-D. All authors contributed to the article and approved the submitted version.
Funding
The study was funded by the Jagiellonian University Medical College grant No K/ZDS/003795.
Acknowledgments
The authors acknowledge Jakub Piątkowski, for his help in DNA sequencing and analysis.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ezzat S, Asa SL, Couldwell WT, Barr CE, Dodge WE, Vance ML, et al. The prevalence of pituitary adenomas: A systematic review. Cancer (2004) 101(3):613–9. doi: 10.1002/cncr.20412
2. Williams F, Hunter S, Bradley L, Chahal HS, Storr HL, Akker SA, et al. Clinical experience in the screening and management of a large kindred with familial isolated pituitary adenoma due to an aryl hydrocarbon receptor interacting protein (AIP) mutation. J Clin Endocrinol Metab (2014) 99(4):1122–31. doi: 10.1210/jc.2013-2868
3. Daly AF, Rixhon M, Adam C, Dempegioti A, Tichomirowa MA, Beckers A. High prevalence of pituitary adenomas: A cross-sectional study in the province of liege, Belgium. J Clin Endocrinol Metab (2006) 91(12):4769–75. doi: 10.1210/jc.2006-1668
4. Gadelha MR, Trivellin G, Hernández Ramírez LC, Korbonits M. Genetics of pituitary adenomas. Front Horm Res (2013) 41:111–40. doi: 10.1159/000345673
5. Tichomirowa MA, Barlier A, Daly AF, Jaffrain-Rea ML, Ronchi C, Yaneva M, et al. High prevalence of AIP gene mutations following focused screening in young patients with sporadic pituitary macroadenomas. Eur J Endocrinol (2011) 165(4):509–15. doi: 10.1530/EJE-11-0304
6. Igreja S, Chahal HS, King P, Bolger GB, Srirangalingam U, Guasti L, et al. Characterization of aryl hydrocarbon receptor interacting protein (AIP) mutations in familial isolated pituitary adenoma families. Hum Mutat (2010) 31(8):950–60. doi: 10.1002/humu.21292
7. Chahal HS, Chapple JP, Frohman LA, Grossman AB, Korbonits M. Clinical, genetic and molecular characterization of patients with familial isolated pituitary adenomas (FIPA). Trends Endocrinol Metab TEM (2010) 21(7):419–27. doi: 10.1016/j.tem.2010.02.007
8. Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, Raitila A, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science (2006) 312(5777):1228–30. doi: 10.1126/science.1126100
9. Schernthaner-Reiter MH, Trivellin G, Stratakis CA. Interaction of AIP with protein kinase a (cAMP-dependent protein kinase). Hum Mol Genet (2018) 27(15):2604–13. doi: 10.1093/hmg/ddy166
10. Schernthaner-Reiter MH, Trivellin G, Stratakis CA. Chaperones, somatotroph tumors and the cyclic AMP (cAMP)-dependent protein kinase (PKA) pathway. Mol Cell Endocrinol (2020) 499:110607. doi: 10.1016/j.mce.2019.110607
11. Bizzi MF, Bolger GB, Korbonits M, Ribeiro-Oliveira A Jr. Phosphodiesterases and cAMP pathway in pituitary diseases. Front Endocrinol (Lausanne) (2019) 10:141. doi: 10.3389/fendo.2019.00141
12. Korbonits M, Storr H, Kumar AV. Familial pituitary adenomas - who should be tested for AIP mutations? Clin Endocrinol (Oxf) (2012) 77(3):351–6. doi: 10.1111/j.1365-2265.2012.04445.x
13. Daly AF, Vanbellinghen JF, Khoo SK, Jaffrain-Rea ML, Naves LA, Guitelman MA, et al. Aryl hydrocarbon receptor-interacting protein gene mutations in familial isolated pituitary adenomas: Analysis in 73 families. J Clin Endocrinol Metab (2007) 92(5):1891–6. doi: 10.1210/jc.2006-2513
14. Marques P, Korbonits M. Genetic aspects of pituitary adenomas. Endocrinol Metab Clin North Am (2017) 46(2):335–74. doi: 10.1016/j.ecl.2017.01.004
15. Hernández-Ramírez LC, Gabrovska P, Dénes J, Trivellin G, Tilley D, Ferrau F, et al. Landscape of familial isolated and young-onset pituitary adenomas: Prospective diagnosis in AIP mutation carriers. J Clin Endocrinol Metab (2015) 100(9):E1242–1254. doi: 10.1210/jc.2015-1869
16. Daly AF, Tichomirowa MA, Petrossians P, Heliövaara E, Jaffrain-Rea ML, Barlier A, et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: An international collaborative study. J Clin Endocrinol Metab (2010) 95(11):E373–383. doi: 10.1210/jc.2009-2556
17. Chahal HS, Stals K, Unterländer M, Balding DJ, Thomas MG, Kumar AV, et al. AIP mutation in pituitary adenomas in the 18th century and today. N Engl J Med (2011) 364(1):43–50. doi: 10.1056/NEJMoa1008020
18. Kasuki Jomori de Pinho L, Vieira Neto L, Armondi Wildemberg LE, Gasparetto EL, Marcondes J, de Almeida Nunes B, et al. Low aryl hydrocarbon receptor-interacting protein expression is a better marker of invasiveness in somatotropinomas than ki-67 and p53. Neuroendocrinology (2011) 94(1):39–48. doi: 10.1159/000322787
19. Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res (2018) 4;46(D1):D1062–7. doi: 10.1093/nar/gkx1153
20. Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, et al. Human gene mutation database (HGMD): 2003 update. Hum Mutat (2003) 21(6):577–81. doi: 10.1002/humu.10212
21. Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: The human genomic variant search engine. Bioinforma Oxf Engl (2019) 35(11):1978–80. doi: 10.1093/bioinformatics/bty897
22. Cazabat L, Bouligand J, Salenave S, Bernier M, Gaillard S, Parker F, et al. Germline AIP mutations in apparently sporadic pituitary adenomas: Prevalence in a prospective single-center cohort of 443 patients. J Clin Endocrinol Metab (2012) 97(4):E663–670. doi: 10.1210/jc.2011-2291
23. Marques P, Caimari F, Hernández-Ramírez LC, Collier D, Iacovazzo D, Ronaldson A, et al. Significant benefits of AIP testing and clinical screening in familial isolated and young-onset pituitary tumors. J Clin Endocrinol Metab (2020) 105(6):e2247–60. doi: 10.1210/clinem/dgaa040
24. Martínez de LaPiscina I, Portillo Najera N, Rica I, Gaztambide S, Webb SM, Santos A, et al. Clinical and genetic characteristics in patients under 30 years with sporadic pituitary adenomas. Eur J Endocrinol (2021) 185(4):485–96. doi: 10.1530/EJE-21-0075
25. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med Off J Am Coll Med Genet (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
26. van den Broek MFM, van Nesselrooij BPM, Verrijn Stuart AA, van Leeuwaarde RS, Valk GD. Clinical relevance of genetic analysis in patients with pituitary adenomas: A systematic review. Front Endocrinol (2019) 10:837. doi: 10.3389/fendo.2019.00837
27. Leontiou CA, Gueorguiev M, van der Spuy J, Quinton R, Lolli F, Hassan S, et al. The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. J Clin Endocrinol Metab (2008) 93(6):2390–401. doi: 10.1210/jc.2007-2611
28. Melmed S. Medical progress: Acromegaly. N Engl J Med (2006) 355(24):2558–73. doi: 10.1056/NEJMra062453
29. Cozzi R, Montini M, Attanasio R, Albizzi M, Lasio G, Lodrini S, et al. Primary treatment of acromegaly with octreotide LAR: A long-term (up to nine years) prospective study of its efficacy in the control of disease activity and tumor shrinkage. J Clin Endocrinol Metab (2006) 91(4):1397–403. doi: 10.1210/jc.2005-2347
30. Kasuki L, Vieira Neto L, Wildemberg LE, Colli LM, de Castro M, Takiya CM, et al. AIP expression in sporadic somatotropinomas is a predictor of the response to octreotide LAR therapy independent of SSTR2 expression. Endocr Relat Cancer (2012) 19(3):L25–9. doi: 10.1530/ERC-12-0020
31. Chahal HS, Trivellin G, Leontiou CA, Alband N, Fowkes RC, Tahir A, et al. Somatostatin analogs modulate AIP in somatotroph adenomas: The role of the ZAC1 pathway. J Clin Endocrinol Metab (2012) 97(8):E1411–20. doi: 10.1210/jc.2012-1111
32. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature (2020) 581(7809):434–43. doi: 10.1038/s41586-020-2308-7
33. Georgitsi M, Raitila A, Karhu A, Tuppurainen K, Mäkinen MJ, Vierimaa O, et al. Molecular diagnosis of pituitary adenoma predisposition caused by aryl hydrocarbon receptor-interacting protein gene mutations. Proc Natl Acad Sci U.S.A. (2007) 104(10):4101–5. doi: 10.1073/pnas.0700004104
34. Cazabat L, Libè R, Perlemoine K, René-Corail F, Burnichon N, Gimenez-Roqueplo AP, et al. Germline inactivating mutations of the aryl hydrocarbon receptor-interacting protein gene in a large cohort of sporadic acromegaly: Mutations are found in a subset of young patients with macroadenomas. Eur J Endocrinol (2007) 157(1):1–8. doi: 10.1530/EJE-07-0181
35. Buchbinder S, Bierhaus A, Zorn M, Nawroth PP, Humpert P, Schilling T. Aryl hydrocarbon receptor interacting protein gene (AIP) mutations are rare in patients with hormone secreting or non-secreting pituitary adenomas. Exp Clin Endocrinol Diabetes Off J Ger Soc Endocrinol Ger Diabetes Assoc (2008) 116(10):625–8. doi: 10.1055/s-2008-1065366
36. Guaraldi F, Salvatori R. Familial isolated pituitary adenomas: From genetics to therapy. Clin Transl Sci (2011) 4(1):55–62. doi: 10.1111/j.1752-8062.2010.00254.x
37. Baciu I, Radian S, Capatina C, Botusan I, Aflorei D, Stancu C, et al. The p.R16H (C.47G>A) AIP gene variant in a case with invasive non-functioning pituitary macroadenoma and screening of a control cohort. Acta Endocrinol (Copenh) (2013) IX:97–108. doi: 10.4183/aeb.2013.97
38. Zatelli MC, Torre ML, Rossi R, Ragonese M, Trimarchi F, degli Uberti E, et al. Should aip gene screening be recommended in family members of FIPA patients with R16H variant? Pituitary (2013) 16(2):238–44. doi: 10.1007/s11102-012-0409-5
39. Preda V, Korbonits M, Cudlip S, Karavitaki N, Grossman AB. Low rate of germline AIP mutations in patients with apparently sporadic pituitary adenomas before the age of 40: a single-centre adult cohort. Eur J Endocrinol (2014) 171(5):659–66. doi: 10.1530/EJE-14-0426
40. Ferraù F, Romeo PD, Puglisi S, Ragonese M, Torre ML, Scaroni C, et al. Analysis of GPR101 and AIP genes mutations in acromegaly: A multicentric study. Endocrine (2016) 54(3):762–7. doi: 10.1007/s12020-016-0862-4
41. Araujo PB, Kasuki L, de Azeredo Lima CH, Ogino L, Camacho AHS, Chimelli L, et al. AIP mutations in Brazilian patients with sporadic pituitary adenomas: A single-center evaluation. Endocr Connect (2017) 6(8):914–25. doi: 10.1530/EC-17-0237
42. Garcia-Rendueles AR, Chenlo M, Oroz-Gonjar F, Solomou A, Mistry A, Barry S, et al. RET signalling provides tumorigenic mechanism and tissue specificity for AIP-related somatotrophinomas. Oncogene (2021) 40(45):6354–68. doi: 10.1038/s41388-021-02009-8
43. Occhi G, Trivellin G, Ceccato F, De Lazzari P, Giorgi G, Demattè S, et al. Prevalence of AIP mutations in a large series of sporadic Italian acromegalic patients and evaluation of CDKN1B status in acromegalic patients with multiple endocrine neoplasia. Eur J Endocrinol (2010) 163(3):369–76. doi: 10.1530/EJE-10-0327
44. Cuny T, Pertuit M, Sahnoun-Fathallah M, Daly A, Occhi G, Odou MF, et al. Genetic analysis in young patients with sporadic pituitary macroadenomas: Besides AIP don't forget MEN1 genetic analysis. Eur J Endocrinol (2013) 168(4):533–41. doi: 10.1530/EJE-12-0763
45. Freudenberg-Hua Y, Freudenberg J, Vacic V, Abhyankar A, Emde AK, Ben-Avraham D, et al. Disease variants in genomes of 44 centenarians. Mol Genet Genomic Med (2014) 2(5):438–50. doi: 10.1002/mgg3.86
46. Schöfl C, Honegger J, Droste M, Grussendorf M, Finke R, Plöckinger U, et al. Frequency of AIP gene mutations in young patients with acromegaly: A registry-based study. J Clin Endocrinol Metab (2014) 99(12):E2789–93. doi: 10.1210/jc.2014-2094
47. Tuncer FN, Çiftçi Doğanşen S, Serbest E, Tanrıkulu S, Ekici Y, Bilgiç B, et al. Screening of AIP gene variations in a cohort of Turkish patients with young-onset sporadic hormone-secreting pituitary adenomas. Genet Test Mol biomark (2018) 22(12):702–8. doi: 10.1089/gtmb.2018.0133
48. Dal J, Nielsen EH, Klose M, Feldt-Rasmussen U, Andersen M, Vang S, et al. Phenotypic and genotypic features of a large kindred with a germline AIP variant. Clin Endocrinol (Oxf) (2020) 93(2):146–53. doi: 10.1111/cen.14207
49. Hernández-Ramírez LC, Martucci F, Morgan RM, Trivellin G, Tilley D, Ramos-Guajardo N, et al. Rapid proteasomal degradation of mutant proteins is the primary mechanism leading to tumorigenesis in patients with missense AIP mutations. J Clin Endocrinol Metab (2016) 101(8):3144–54. doi: 10.1210/jc.2016-1307
50. Morgan RM, Hernández-Ramírez LC, Trivellin G, Zhou L, Roe SM, Korbonits M, et al. Structure of the TPR domain of AIP: Lack of client protein interaction with the c-terminal α-7 helix of the TPR domain of AIP is sufficient for pituitary adenoma predisposition. PloS One (2012) 7(12):e53339. doi: 10.1371/journal.pone.0053339
51. Hernández-Ramírez LC, Stratakis CA. Genetics of cushing’s syndrome. Endocrinol Metab Clin North Am (2018) 47(2):275–97. doi: 10.1016/j.ecl.2018.02.007
52. Aflorei ED, Klapholz B, Chen C, Radian S, Dragu AN, Moderau N, et al. In vivo bioassay to test the pathogenicity of missense human AIP variants. J Med Genet (2018) 55(8):522–9. doi: 10.1136/jmedgenet-2017-105191
53. Alexaki A, Kames J, Holcomb DD, Athey J, Santana-Quintero LV, Lam PVN, et al. Codon and codon-pair usage tables (CoCoPUTs): Facilitating genetic variation analyses and recombinant gene design. J Mol Biol (2019) 431(13):2434–41. doi: 10.1016/j.jmb.2019.04.021
54. Stiles CE, Korbonits M, et al. Familial isolated pituitary adenoma, in: Endotext. MDText.com, inc (2000). Available at: http://www.ncbi.nlm.nih.gov/books/NBK278949/ (Accessed January 24, 2021).
55. Marques P, Barry S, Ronaldson A, Ogilvie A, Storr HL, Goadsby PJ, et al. Emergence of pituitary adenoma in a child during surveillance: Clinical challenges and the family members’ view in an AIP mutation-positive family. Int J Endocrinol (2018), 8581626. doi: 10.1155/2018/8581626
56. Daly AF, Beckers A. The role of AIP mutations in pituitary adenomas: 10 years on. Endocrine (2017) 55(2):333–5. doi: 10.1007/s12020-016-1194-0
57. Beckers A, Aaltonen LA, Daly AF, Karhu A. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr Rev (2013) 34(2):239–77. doi: 10.1210/er.2012-1013
58. Toledo RA, Lourenço DM, Liberman B, Cunha-Neto MB, Cavalcanti MG, Moyses CB, et al. Germline mutation in the aryl hydrocarbon receptor interacting protein gene in familial somatotropinoma. J Clin Endocrinol Metab (2007) 92(5):1934–7. doi: 10.1210/jc.2006-2394
59. Barlier A, Vanbellinghen JF, Daly AF, Silvy M, Jaffrain-Rea ML, Trouillas J, et al. Mutations in the aryl hydrocarbon receptor interacting protein gene are not highly prevalent among subjects with sporadic pituitary adenomas. J Clin Endocrinol Metab (2007) 92(5):1952–5. doi: 10.1210/jc.2006-2702
Keywords: aryl hydrocarbon receptor-interacting protein, AIP, pituitary, adenoma, mutation
Citation: Trofimiuk-Müldner M, Domagała B, Sokołowski G, Skalniak A and Hubalewska-Dydejczyk A (2023) AIP gene germline variants in adult Polish patients with apparently sporadic pituitary macroadenomas. Front. Endocrinol. 14:1098367. doi: 10.3389/fendo.2023.1098367
Received: 14 November 2022; Accepted: 23 January 2023;
Published: 10 February 2023.
Edited by:
Eva Christine Coopmans, Leiden University Medical Center (LUMC), NetherlandsReviewed by:
Sergei I. Bannykh, Cedars Sinai Medical Center, United StatesMarta Araujo-Castro, Ramón y Cajal University Hospital, Spain
Adrian Daly, University Hospital of Liège, Belgium
Robert Formosa, Queen Mary University of London, United Kingdom
Copyright © 2023 Trofimiuk-Müldner, Domagała, Sokołowski, Skalniak and Hubalewska-Dydejczyk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Małgorzata Trofimiuk-Müldner, malgorzata.trofimiuk@uj.edu.pl
†ORCID: Małgorzata Trofimiuk-Müldner, https://orcid.org/0000-0001-5247-9760