 Gengyang Li
Gengyang Li Mason Peng1
Mason Peng1 Qingguo Huang
Qingguo Huang Ching-Hua Huang
Ching-Hua Huang Yongsheng Chen
Yongsheng Chen Gary Hawkins
Gary Hawkins Ke Li
Ke Li- 1College of Engineering, University of Georgia, Athens, GA, United States
- 2Department of Crop and Soil Science, College of Agricultural and Environmental Sciences, Griffin, GA, United States
- 3School of Civil and Environmental Engineering, Georgia Institute of Technology, Atlanta, GA, United States
Per- and polyfluoroalkyl substances (PFAS) have drawn public concern recently due to their toxic properties and persistence in the environment, making it urgent to eliminate PFAS from contaminated water. Electrochemical oxidation (EO) has shown great promise for the destructive treatment of PFAS with direct electron transfer and hydroxyl radical (⋅OH)-mediated indirect reactions. One of the most popular electrodes is Magnéli phase titanium suboxides. However, the degradation mechanisms of PFAS are still unsure and are under investigation now. The main methodology is the first-principal density functional theory (DFT) computation, which is recently used to explore the degradation mechanisms and interpret by-product formation during PFAS mineralization. From the literature review, the main applications of DFT computation for studying PFAS degradation mechanisms by EO include bond dissociation energy, absorption energy, activation energy, and overpotential η for oxygen evolution reactions. The main degradation mechanisms and pathways of PFAS in the EO process include mass transfer, direct electron transfer, decarboxylation, peroxyl radical generation, hydroxylation, intramolecular rearrangement, and hydrolysis. In the recent 4 years, 11 papers performed DFT computation to explore the possible PFAS degradation mechanisms and pathways in the EO process. This paper’s objectives are to: 1) summarize the main degradation mechanisms of PFAS degradation in EO; 2) review the application of DFT computation for studying PFAS degradation mechanisms during EO; process; 3) review the possible degradation pathways of perfluorooctane sulfonoic acid (PFOS) and per-fluorooctanoic acid (PFOA) during EO process.
1 Introduction
Per- and polyfluoroalkyl substances (PFAS) have drawn public concern recently due to their toxic properties and environmental persistence (Rozen and Filler, 1985). PFAS substances are a broad class of emerging environmental contaminants used heavily by the industrial, commercial, and manufacturing sectors in previous decades, like surface coating, lubricants, and firefighting foams (Crone et al., 2019; Rahman et al., 2014). PFAS are highly persistent and bioaccumulated in the environment within biota and humans. Because of this, PFAS species have been consistently detected within the environment, in food products, and in humans (Hori et al., 2004). Various potential health effects have been attributed to PFAS exposure, particularly PFOS and PFOA, which are highly toxic and suspected carcinogens (Wanninayake, 2021).
It is urgent to eliminate PFAS from the contaminated water. The occurrence of PFAS is in many water bodies, including wastewater treatment plants, surface water, groundwater, and drinking water treatment plants (Wang et al., 2022a). PFAS is disposed of in water and wastewater sources at high levels, up to 120 μg/L (Vo et al., 2020). However, the remediation and treatment of PFAS-contaminated water are challenging because of its highly stable spiral structure and carbon−fluorine (C-F) bonds (106–124 kcal/mol) (Lin et al., 2018).
Many studies have recently shown the promise of EO for the destructive treatment of PFAS (Schaefer et al., 2017; Wang Z. et al., 2025; Zhao et al., 2025). It is increasingly recognized favorably as a next-generation water treatment technology for a few advantages, such as easy operation under ambient conditions, high degradation efficiency of the contaminants, and ease of manipulation and automation. Electrochemical treatments utilize electrolytic cell composed of at least one anode, one cathode, and a source of electrons (Figure 1) (Ryan et al., 2021). Oxidation reactions occur on the anode and reduction reactions occur on the cathode, with the type of electrode materials determining the specific reactions occurring. Sufficient thermodynamic energy (e.g., working electrode potential) must be available for these reactions to occur (Ryan et al., 2021). Some important parameters in EO influence the degradation efficiency of PFAS, such as the anode material, current density, solution pH, initial PFAS concentration, electrolyte, plate distance and electrical connectors in the EO system (Mirabediny et al., 2023; Wang Y. et al., 2025).
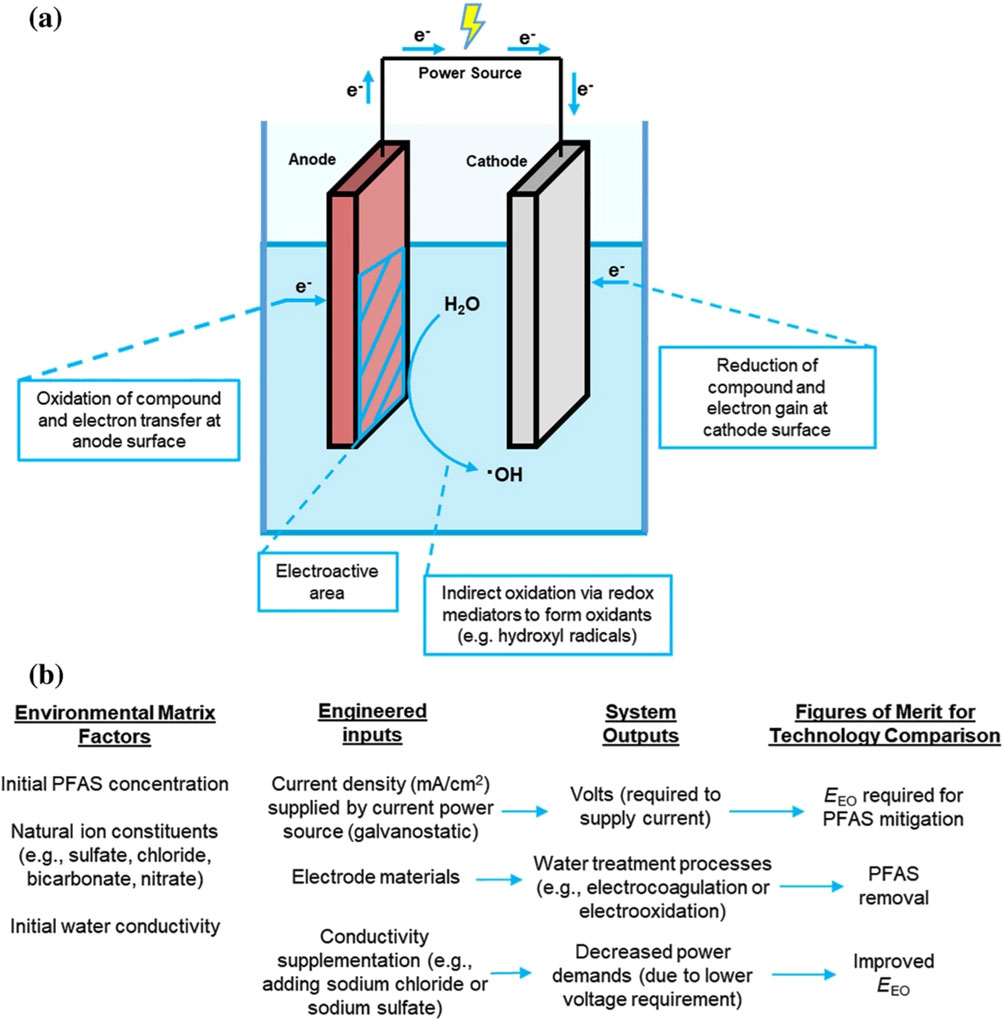
Figure 1. (a) Electrolytic cell schematic for electrochemical water treatment reactors. In these cells, oxidation reactions occur at the anode and electrons flow to the cathode, where reduction reactions occur. In these systems, the current density (mA/cm2) is determined by the amount of current (mA) that passes through the electroactive surface area. (b) Additional information is provided for environmental inputs to the electrolytic cell, in addition to engineered inputs and corresponding system outputs and figures of merit for comparing technology. EEO = electrical energy per order (Ryan et al., 2021).
To date, there are various electrode materials available, including boron-doped diamond (BDD), Magnéli phase titanium suboxides (TSO), lead dioxide (PbO2), and stannous dioxide (SnO2) (Wang et al., 2022b). Of note, electrode materials such as SnO2 and PbO2 may not have feasible applications for drinking water treatment due to the potential risk of secondary metal contamination (such as lead), which is recommended to be excluded from water infrastructure. Several studies also investigated how to improve the degradation efficiency by doping, such as Ce3+ -doped Ti4O7 electrode (Lin et al., 2021), Pd -doped Ti4O7 electrode (Huang et al., 2020), and Nb-doped TiO2 electrode (Ko et al., 2021). Furthermore, reactive electrochemical membrane (REM) systems of TSO are also developed to break the limitation of mass transfer (Wang et al., 2022b). Also, Ma et al. demonstrated a hybrid electrocatalyst of Magnéli phase titanium oxide (Ti4O7)/Ti3C2Tx MXene for degrading PFOA and Perfluorobutanoic acid (PFBA) with EO (Ma et al., 2024).
The first principal DFT calculations are useful computational tools for exploring the degradation mechanism and interpreting certain by-product formation during PFAS mineralization (Yamijala et al., 2022). From 2013 to 2025, a decade, there are around twenty papers investigating the mechanism and pathway of PFAS degradation by the EO process. Most focus on PFOA, PFOS, and Perfluorobutanesulfonic acid (PFBS). Among these studies, 11 papers performed density functional theory (DFT) calculations to explore the possible chemical reactions and pathways of PFASs in the aqueous and on anodes (Chen et al., 2021; Cheng et al., 2021; Wang C. et al., 2022). DFT is recently used to calculate parameters such as, the bond dissociation energy (BDE) of PFAS chains, the adsorption energy of PFAS ion on anodes, the activation energy of PFAS radical, hydroxyl radical, and C-S breaking, as well oxygen evolution reactions (OER) for water splitting (Eslamibidgoli and Eikerling, 2015). Therefore, these DFT calculation parameters can be used as indicators to explore the degradation mechanism and interpret the formation of certain by-products during PFAS mineralization. Several review papers on removing PFAS with electrochemical techniques have recently been published (Ryan et al., 2021; Zhou et al., 2024). Specifically, some reviews discussed the recent advancements in EO, the PFAS degradation mechanisms, and the key factors that influence PFAS removal efficiency, such as anode material, current density, electrolyte, pH, initial PFAS concentration, and other coexisting pollutants (Alalm and Boffito, 2022; Zhou et al., 2024). However, to the best of our knowledge, no existing review has comprehensively examined how density functional theory (DFT) calculations contribute to understanding PFAS degradation mechanisms. This review aims to bridge that gap by synthesizing insights from both experimental studies and DFT calculations to provide a more mechanistic understanding of PFAS degradation. By integrating theoretical modeling with empirical findings, this work offers a deeper perspective on degradation pathways and reaction mechanisms, particularly for PFOA and PFOS in the EO process. This approach not only complements existing studies but also enhances predictive capabilities for optimizing PFAS removal strategies.
2 PFAS degradation mechanism by electrochemical oxidation
The underlying PFAS destruction mechanism for EO is proposed as a combination of the direct electron transfer (DET) and hydroxyl radical (⋅OH)-mediated indirect reactions (Wang et al., 2022b). In the direct path, the direct electron withdrawal from the organics to the electrode is also the same as the oxidation of organics by holes (h+) on the electrode surface. Some research has supported this mechanism by comparing experimentally measured kinetics to activation barriers calculated using density functional theory (DFT) (Chen et al., 2022b; Shi et al., 2019). In the ⋅OH -mediated indirect oxidation process, the ⋅OH is also initiated by the DET of H2O oxidation on the electrode surface. During EO, the ⋅OH is produced at the anode surface that can only diffuse in a thin layer (<1 μm) near the anode surface due to its short lifespan. In contrast, the DET reaction occurs directly on the electrode surface. Also, many reactions happen during the degradation in the EO process, such as mass transfer of PFAS to anodes, decarboxylation of PFCAs, hydroxylation and hydrolysis of by-products, intramolecular rearrangement, and peroxyl radical participated oxidation.
2.1 Mass transfer
Degradation via the electrochemical advanced oxidation process is constrained by the mass transfer of PFAS from the bulk solution to the anode surface, which occurs only at or near the anode surface (Trellu et al., 2018). The mass transfer coefficients can be affected by electricity current, the reactive surface area, flow rate at consistent temperature and pressure. To overcome the mass transfer limitation, a reactive electrochemical membrane operation was suggested, which has improved inter-phase mass transfer through the filtration of polluted samples using porous material that functions simultaneously as a membrane and anode (Liu and Vecitis, 2012). Consequently, even a single run of water through the membrane can enhance the overall electrochemical oxidation proficiency for treating PFAS.
It has been shown that the most promising way to avoid diffusion, overcome mass transfer limitations, and to increase the electroactive surface area is by using porous electrodes in flow-through configurations (Wang Y. Y. et al., 2022). experimented and compared the PFOS adsorption in REM operation with the batch reactor. The results indicated PFOS adsorption in REM operation was stronger than in the batch reactor because the inner pore surfaces were made available for PFOS adsorption during REM operation. Furthermore, the adsorption of PFOS in the micro-Ti4O7 (10 MPa) material was much stronger than that in the nano-Ti4O7 (10 MPa) material. It was suggested that the pores of 2–4 nm are most favorable for PFOS diffusion and adsorption because the molecular length of PFOS is 1.32 nm. (Deng et al., 2015). The surface area of the pores in the micro-Ti4O7 (10 MPa) anode within the range of 2–4 nm is 0.355 m2 g−1, while pores of this range are absent in the nano-Ti4O7 (10 MPa) anode.
2.2 Direct electron transfer
In the direct path, the direct electron withdrawal from the organics to the electrode is also the same as the oxidation of organics by electron holes (h+) on the electrode surface. During the EO process, electron holes destabilize PFCAs (perfluoroalkyl carboxylic acids) and PFSAs (perfluoroalkyl sulfonic acids) into radicals by direct electron transfer Equations 1, 2 Some research has supported this mechanism by comparing experimentally measured kinetics to activation barriers calculated using DFT (Chen et al., 2022b; Shi et al., 2019). The DET that causes the transformation of PFCAs or PFSAs ion to a free radical via losing an electron to the anode surface was proposed to be the rate-limiting step in their degradation.
At the same time, water in the solution can also be also oxidized by electron holes (h+) with DEF to form hydroxyl radical (∙OH) and hydrogen ion (Equation 3).
2.3 Decarboxylation
For PFCAs, direct defluorination through nucleophilic substitution is very difficult due to the strong C−F bond. Decarboxylation is a main PFCA degradation process, in which removes a carboxyl group and releases carbon dioxide (CO2). Usually, decarboxylation refers to a reaction of carboxylic acids, removing a carbon atom from a carbon chain. Previous studies indicated that perfluoroalkyl acids (PFAAs), such as the oxidative degradation of PFOA and PFOS, began in the elimination of the end group. The electron transfer process of carboxylic acid occurred on the anode first, which belongs to the electrochemical process and generates carboxylic radical (
2.4 Peroxyl radical generation
Apart from ·OH, the
2.5 Hydroxylation
Hydroxyl radical (⋅OH) is a strong oxidative radical that plays an important role in reacting with PFAS degradation by-products and contribute to PFAS mineralization. Though ⋅OH is not strong enough to break the C-F bond, the attack of ⋅OH is essential to PFAS degradation (radical chain propagation), otherwise PFAS⋅ may react with water to revert to PFAS (H. Shi et al., 2019).
Furthermore, after losing a CF2 unit, the new perfluoro radical (
The rate-limiting step in the indirect path is assumed to be the electrochemical production of hydroxyl free radicals (⋅OH) at the electrode surface as an intermediate of water oxidation, which then reacts with organic contaminants in water. Essentially, the ⋅OH -mediated indirect oxidation process is also initiated by the DET of H2O oxidation on the electrode surface.
2.6 Intramolecular rearrangement
Defluorination can also be caused by intramolecular rearrangement (Equation 9).
2.7 Hydrolysis
Hydrolysis plays an important role in the degradation of some small molecules in the EO processes (Li et at., 2004). Since H2O was the dominant species in the solution, hydrolysis might also contribute to the destruction of organic pollutants. For PFCAs, after
3 DFT computation applications for PFAS degradation mechanisms investigation
DFT computation has gained significant attention in investigating PFAS degradation mechanisms due to its ability to provide detailed insights into molecular structures, energetics, intermediates, and reaction pathways. The electronic structure is assessed using a potential acting on the system electrons in modern DFT techniques. This DFT potential is created by adding the exterior potentials, which are completely governed by the system’s structure and elemental makeup representing interelectronic interactions. DFT can accurately predict the molecular structures of PFAS compounds, including their bond lengths, angles, and electronic configurations. This information is crucial for understanding the reactivity and stability of PFAS molecules during degradation processes. DFT calculations can determine the energetics of various degradation reactions involved in PFAS transformation, such as hydrolysis, photolysis, and oxidative degradation. By calculating reaction energies, activation barriers, and reaction pathways, DFT helps identify the most favorable degradation pathways and mechanisms.
DFT-based simulations have frequently been performed for applications relating to bond dissociation energy (BDE) of PFAS chains, the activation energy of chemical degradation and the adsorption energy for PFAS mass transfer on anodes (Chen et al., 2022a; Jinyu Gao et al., 2023; Li et al., 2021; Wang Y. Y. et al., 2022) (Figure 2). The bonds with lower BDE are more likely to be cleaved by ROS than those with higher BDE (Bentel et al., 2019; Chen et al., 2022a). Higher adsorption energy of PFAS ions on anodes is favorable for mass transfer (Li et al., 2021). Lower activation energy of PFAS direct oxidation indicates the PFAS− would be easily oxidized on the surface during the direct electron transfer (Wang et al., 2022b). Higher overpotential η for OER means it will be harder to oxide ⋅OH, hence suppressing OER and holding more ⋅OH during the indirectly oxidation for PFAS.
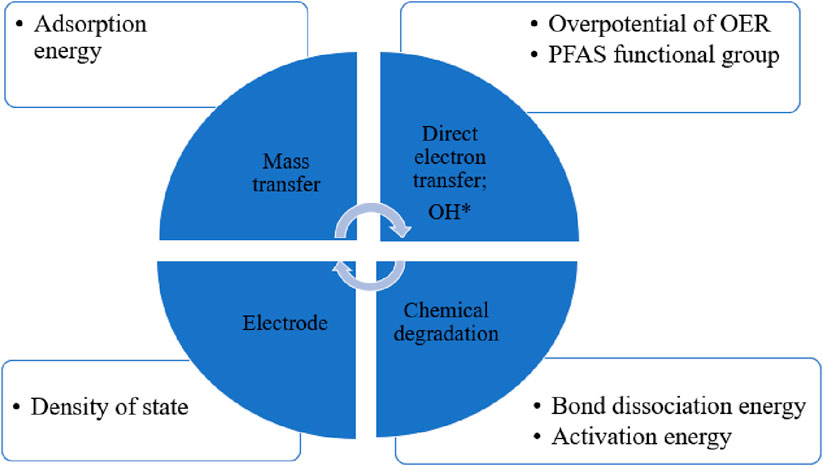
Figure 2. DFT quantification descriptors for electrodes and PFAS degradation mechanism.
3.1 Adsorption energy
The adsorption energy is one of the most basic parameters for understanding the behavior of a molecule on a surface. It can be defined as the energy changing when a molecule is adsorbed on a solid surface. The formula used to calculate the adsorption energy of PFAS–on an anode supercell during the EO process is described as follows:
where
The adsorption energy of a hydroxyl radical (⋅OH) on anode supercell during the EO process (
where,
Table 3 shows some reported DFT adsorption energy calculations for PFOA, PFOS, and PFBS degradation in EO process. Higher adsorption energy indicates stronger adsorption on the anode (Li et al., 2021). calculated the adsorption energy on the Ti4O7 cluster surface with DolM3. The results decrease in the following order: ⋅OH > C4F9SO3−(PFBS−) ∼ C8F17SO3−(PFOS−) > H2O. This suggests that ⋅OH formed from water oxidation strongly adsorbs on the Ti4O7 cluster surface. The adsorption energies of ⋅OH on a Ti4O7 surface are much larger than that of H2O on a Ti4O7 surface. This illustrates that hydroxyl radicals, once formed by water oxidation, are more likely bound on a Ti4O7 surface rather than diffusing into the bulk solution or reacting with PFOS−.
The adsorption energy of different anodes can also be compared. Higher chemical adsorption energies on the anode indicate that the anode drives a much stronger affinity towards the chemical, thus favoring its electrochemical degradation on the anode surface (Wang et al., 2022b). performed and compared the adsorption energies with DolM3 of PFOS on the Ti4O7 cluster (∼-2.5 eV) and on the Ti9O17 cluster (−1.5 eV∼0.2 eV). The greater adsorption energies of PFOS on the Ti4O7 cluster indicate that the Ti4O7 cluster drives a much stronger affinity towards PFOS, thus favoring its electrochemical degradation that occurs on the anode surface.
(Wang et al., 2020) also performed molecular simulation based on density functional theory (DFT) using VASP to specifically probe the interactions of Cl atom and OH moieties on Ti4O7 (110) and BDD (001), thus providing insights into the formation of Cl⋅ and HO⋅ radicals on these anodes (Wang et al., 2020). calculated the adsorption energy of Cl atom and OH moieties adsorbed on Ti4O7 (110) and BDD (001) surfaces during PFOS degradation with VASP.
The value of the OH adsorbate on the Ti4O7 electrode is −5.13 eV, considerably lower than those on BDD (−7.89 eV) in Table 3. Generally, a lower adsorption energy means a higher tendency of desorption to form radicals. Thus, the OH on the Ti4O7 electrode has a higher reactivity than those on the BDD. This contributes to the higher PFOS removal rate on Ti4O7 than on BDD, since HO− plays an important role on its degradation. The data in Table 3 also show that the adsorption energy of Cl and OH adsorbates are close to Ti4O7 (110). However, on BDD (001), Cl adsorbate shows significantly lower adsorption energy (−4.16/-3.85 eV) than OH adsorbate (−7.891/-8.80 eV). This suggests that Cl− is more easily released than HO− on BDD (001).
3.2 Density of state
The density of states (DOS) is one of the most important concepts for understanding the physical properties of materials because it provides a simple way to characterize complex electronic structures. Key aspects that underlie electrical and optical properties of materials are visually apparent from the DOS, including the band gap and effective masses of carriers. Density functional theory is a powerful and effective tool that can be used for predicting the density of state of a material. Density functional theory is widely used to predict and calculate the electronic properties of crystal structures with good accuracy (Bharti et al., 2021). The assumption of DFT that the energy of a system can be calculated using the function of the electronic density rather than solving the complex many-body Schrodinger equations reduces the computational difficulty and time significantly.
The density of states (DOS) plays a crucial role in electron transfer processes. A higher DOS implies more available states for electrons to occupy, facilitating electron transfer by providing a larger pool of states near the Fermi level for electron donation and acceptance. In summary, the density of states directly affects electron transfer by influencing the availability of electronic states, energy level alignment, interface properties, activation energies, and charge carrier transport in materials. Understanding and controlling DOS profiles are essential for optimizing electron transfer rates, designing functional materials, and developing advanced electronic and optoelectronic devices. In semiconductors and metals, the DOS near the conduction band (CB) and valence band (VB) significantly influence electron transfer. A higher DOS near the CB promotes electron conduction and transport, while a higher DOS near the VB facilitates hole conduction and valence band transitions.
Ma et al. first calculated the DOS for Ti4O7 and the composite structure of Ti4O7/MXene surface in order to compare the electron transport properties of these two electrodes (Ma et al., 2024). Figure 3a shows that the Ti4O7 conduction band parts are constructed by mainly Ti d states and a small part of O p states (Ma et al., 2024). The Ti4O7 valence bonding is constructed by mostly O p states and a small part of Ti d states, in agreement with previous DFT simulations of the electronic construction for a Magnéli phase TiO2 (Ma et al., 2024). Figure 3b shows that both the conduction band position and valence band position of Ti4O7/MXene moved down significantly, which narrowed the band gap and thus enhanced electrical conductivity and interfacial electron transfer (Ma et al., 2024). This demonstrates that the charge transfer to the PFAS are more favorable on Ti4O7/MXene (Ma et al., 2024).
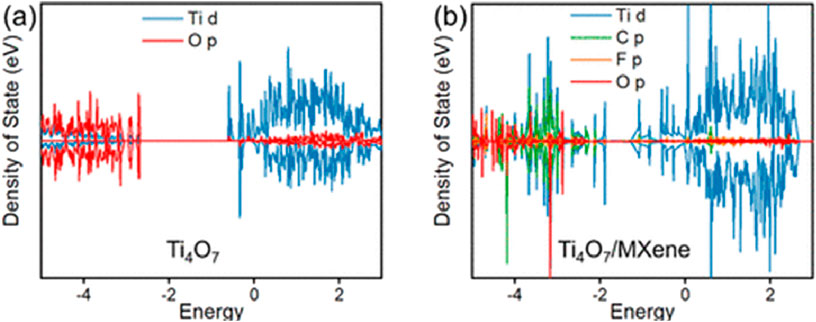
Figure 3. Density of states (DOS) of (a) Ti4O7 and (b) Ti4O7/MXene surface (Ma et al., 2024).
3.3 Overpotential for oxygen evolution reaction (OER)
In the EO process, the rate-limiting step in the indirect path is assumed to be the electrochemical production of hydroxyl free radicals (⋅OH) at the electrode surface as an intermediate of water oxidation, which then reacts with organic contaminants in water. Essentially, the ⋅OH mediated indirect oxidation process is also initiated by the DET of H2O oxidation on the electrode surface.
Overpotential η for OER (⋅OH ↔ O⋅ + H+ + e−) can be an indicator. Higher η means it will be harder to oxidize ⋅OH, hence suppressing OER and holding more ⋅OH during the indirect PFAS oxidation (Man et al., 2011). The descriptor ΔGO − ΔGOH has been verified to well describe the trend of OER activities on various metal oxide surfaces (Huang et al., 2019).
DFT calculations are used to estimate the overpotential η for OER for different materials as simulated above. Using the previously validated relationship for a wide range of oxide surfaces, η in V relative to SHE for each material as a function of the descriptor GO∗ − G⋅OH:
3.2 eV is the average value of ΔGHOO∗ - ΔGHO∗ of OER on oxide surfaces in university (Man et al., 2011). For the ideal case, ΔGHO∗ is 1.23 eV at pH = 0 and T = 298 K. The descriptor ΔG represents the difference in binding free energies of the critical intermediate species O* and ⋅OH. This is calculated based on total energies of systems consisting of the O* or ⋅OH species bound to each material surface as determined by DFT. The theoretical overpotential is independent of the pH or the potential values. Therefore, the calculations/analysis performed for the free energies is at standard conditions (pH = 0, T = 298.15 K) and U = 0.
The results of DFT calculations were reported to compare Ti4O7, TiO2, and Nb-doped TiO2 materials for effective ⋅OH production with Quantum Espresso version 6.6 (Ko et al., 2021). They simulated the Ti4O7 (120), TiO2 (110), and 12.5 at% Nb-doped TiO2 (110) anode supercell. Ti4O7 anode had the highest overpotential η for OER, 2.0 V. Overpotential η for OER of 12.5 at% Nb-doped TiO2 anode (1.4 ± 0.49 V) is higher than TiO2 (1.3 V) (Table 1).

Table 1. Overpotential η for OER calculated for anodes in EO process by DFT computation.
3.4 Bond-dissociation energy
The bond-dissociation energy is one measure of the strength of a chemical bond. For PFAS degradation, several studies performed C-F BDE and C-C BDE calculations. Theoretical calculations on the C-F BDE of PFAS structures can reveal relationships among the rate of decay defluorination, the position and number of C−F bonds with low BDE, and fluoroalkyl chain length (Bentel et al., 2019). The bonds with lower BDE are more likely to be cleaved by ROS than those with higher BDE. Therefore, it also can be used to suggest and interpret the formation of certain by-products during PFAS mineralization.
Bentel et al. calculated BDEs for all of the C−F BDEs (kcal mol−1) for PFCAs (C2-C12) using the GAUSSIAN 09 software package with DFT in conjunction with an SDM polarizable continuum model. As expected, BDEs of all primary C−F bonds (i.e., bonds on the terminal −CF3; 117.8–123.4 kcal mol−1) are higher than all secondary C−F bonds (i.e., bonds on −CF2−; 106.4–113.6 kcal mol−1) (Table 2). In general, lower BDEs for both primary and secondary C−F bonds are observed in PFAS with longer fluoroalkyl chains. This trend may explain why the rate of parent compound degradation was faster for the longer chain PFCAs and PFSAs, where more −CF2− functional groups in the middle of the fluoroalkyl chains have low BDEs (typically ≤107.5 kcal mol−1) (Bentel et al., 2019).

Table 2. C-F BDEs for C7F15COO− (Note: The first C-F is on the terminal −CF3.).
BDE calculations can also help explore the primary pathway. Past studies revealed that C7F15COO⋅ easily underwent a decarboxylation reaction to form C7F15 ⋅. To test whether it was the primary pathway (Chen et al., 2022b), calculated and compared the C−C BDEs and C−F BDEs for both C7F15COO− and C7F15COO⋅ to get the change for BDEs after C7F15COO− lost one electron using Gaussian 16 with M06-2X-D3 (0)/6–311+G (d,p). Their results showed all C−F bonds (108.7–125.6 kcal mol−1) were much higher than those for C−C bonds (86.3–92.6 kcal mol−1) (Table 2). This indicated that C−F bonds were much harder to break than C−C bonds, and the cleavage of C−F bonds should not be the primary pathway. So, decarboxylation of C7F15COO⋅ may be the primary pathway. After C7F15COO− lost one electron to form C7F15COO⋅, the BDEs for all of the C−C bonds were lower, and bond 1 (with functional group COO−) had the lowest BDE (−25.7 kcal mol−1). This suggested that all of the C−C bonds significantly weakened after C7F15COO− lost one electron, and the cleavage of bond 1 was most likely to occur. Bond 1 had a BDE of −25.7 kcal mol−1. A negative BDE indicated that bond 1 was remarkably unstable and would easily dissociate to release CO2 and form C7F15 ⋅.
3.5 Activation energy
In chemistry, activation energy is the minimum amount of energy that must be provided for compounds to result in a chemical reaction. Table 3 also shows some reported DFT activation energy calculations for PFOA, PFOS, or PFBS degradation in the EO process. It is found that the activation energies of PFBS degradation by possible hydroxyl radical attacks were prohibitively high (Carter and Farrell, 2008), while those of direct electron transfer (DET) were lower and comparable to experimentally measured values. The density functional simulations were performed to calculate the reaction energies and activation barriers for PFBS oxidation by hydroxyl radicals and by direct electron transfer on BDD anode (Liao and Farrell, 2009). The activation energy determined from the transition state was 1.275 eV by the reaction of hydroxyl radicals at different sites on the PFBS molecule. The activation energy determined was 0.0964 ± 0.0311 eV with the experimental temperature dependence of the PFBS reaction rates method. The low activation energy of PFAS direct oxidation indicates the PFAS− would be easily oxidized on the surface during direct electron transfer.
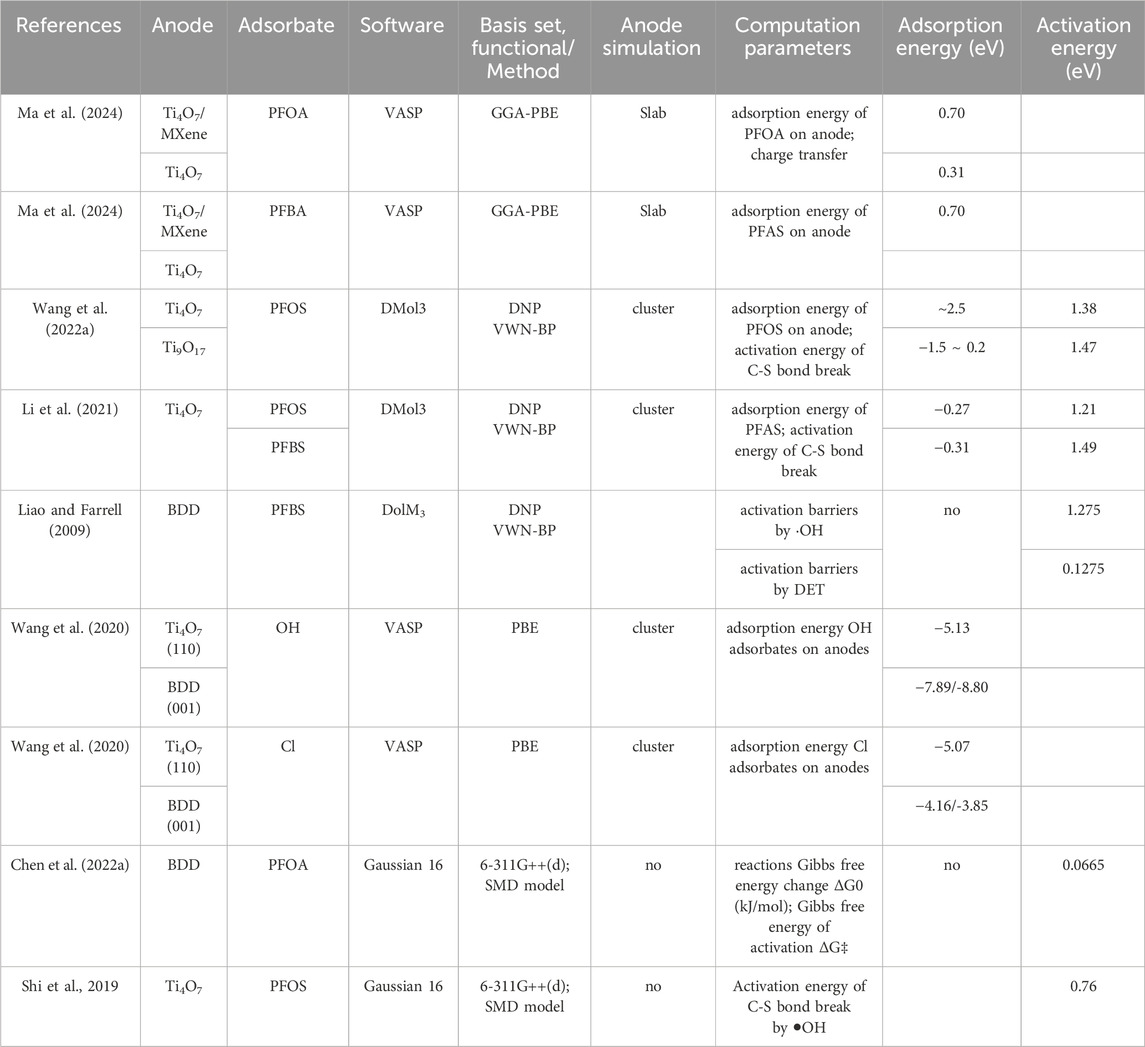
Table 3. Adsorption energy and activation energy reported for PFOA, PFOS, and PFBS degradation during EO process with DFT computation.
Some studies also calculated the activation energy of PFOS by hydroxyl radicals. Shi et al. (2019) contacted DFT simulation of PFOS on an inert anode by calculating its activation energy as a function of anodic potential. It indicates that the DET of PFOS occurs at 3.67 V/SHE without an activation barrier, higher than that for water oxidation (2.74 V/SHE) reported in an earlier study. Therefore, the DET reaction of PFOS would be slower and weaker than water oxidation on an inert anode such as titanium suboxide or BDD. Their calculation revealed that C8F17SO3• may react with water to form C8F17SO3− via an energetically favorable reaction. Therefore, once C8F17SO3• is formed by PFOS oxidation during EO via DET, it can spontaneously transform back to C8F17SO3− in water solution. It is proposed that C8F17SO3• tends to undergo desulfurization, accompanied by a reaction with ⋅OH to form C8F17OH.
3.6 Limitations of the DFT calculation
While DFT calculations provide invaluable insights into PFAS degradation mechanisms in electrochemical oxidation, they are subject to several limitations that must be considered when interpreting results.
One key limitation of DFT is the accuracy of its exchange-correlation functionals. Standard generalized gradient approximation (GGA) functionals, such as PBE or B3LYP, may introduce errors in calculating bond dissociation energies (BDEs) and reaction barriers, particularly for complex reaction systems involving radical intermediates. Hybrid functionals and dispersion-corrected methods can improve accuracy, but their computational cost is significantly higher.
Another major limitation is the treatment of solvation effects. PFAS degradation occurs in aqueous environments, yet many DFT studies employ implicit solvation models (e.g., SMD), which approximate solvent effects rather than explicitly modeling solute-solvent interactions. This can lead to inaccuracies in adsorption energy predictions and reaction barrier calculations. Explicit solvation models, while more accurate, demand significantly higher computational resources.
DFT calculations also struggle with modeling dynamic reaction pathways. Most studies rely on static energy calculations to infer degradation mechanisms, but the actual reaction process involves multiple intermediate steps, solvent interactions, and surface effects that are best captured by molecular dynamics (MD) simulations or hybrid quantum mechanics/molecular mechanics (QM/MM) approaches. For example, the interaction between PFAS radicals and hydroxyl radicals (⋅OH) on an anode surface is highly dynamic and influenced by local electrochemical conditions, which are difficult to model with standard DFT methods.
Moreover, the choice of computational parameters, such as basis sets and pseudopotentials, can introduce systematic errors. Small basis sets may underestimate binding energies, while large basis sets increase computational demand without always improving accuracy. Similarly, pseudopotential selection can affect the accuracy of adsorption energy calculations on metal-oxide anode surfaces, impacting the predicted reactivity of PFAS species.
Finally, scaling DFT findings to real-world electrochemical conditions presents challenges. Experimental electrochemical oxidation occurs under varying pH, electrode potential, and ionic strength conditions, which influence reaction kinetics and pathways. However, most DFT studies are conducted at fixed oxidation states and vacuum or simplified solvated conditions, making direct comparison with experimental data difficult.
To address these limitations, future research should incorporate higher-level post-DFT methods, such as coupled-cluster theory for improved energy accuracy, and hybrid approaches integrating molecular dynamics simulations to capture solvent effects and reaction kinetics more realistically. Additionally, experimental validation through spectroscopic techniques, such as in situ Raman spectroscopy or X-ray photoelectron spectroscopy (XPS), can provide critical benchmarks for refining computational predictions (Chen et al., 2024).
By acknowledging these limitations and refining computational approaches, DFT calculations can continue to play a crucial role in elucidating PFAS degradation mechanisms while improving their predictive reliability for practical electrochemical treatment application.
4 Degradation pathways of some PFAS species
4.1 PFOA degradation pathways
Based on the literature review, three possible PFOA degradation pathways in the EO process have been proposed: pathways a), b) and c) in Figure 4. These three possible pathways have two common initial steps. The first common step involvesthe direct oxidation of C7F15COO− to generate C7F15COO⋅ (Equation 12) as C7F15COO− remains the dominant PFOA species in the solution throughout the electrolysis. The second step is the decarboxylation of C7F15COO⋅ to form C7F15⋅ (Equation 13), which is supported by the C-C BDEs DFT calculations by Chen et al. (in 2.1 BDE section).
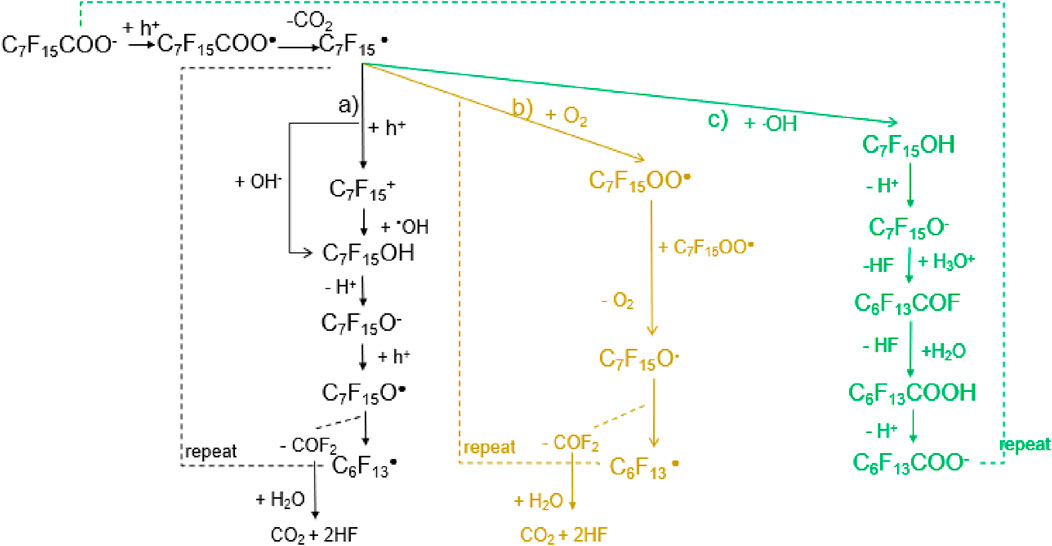
Figure 4. The possible PFOA degradation pathways on the anode in the EO proces. (a) Direct hydroxylation pathway (black): This involves hydroxyl radical attack, leading to sequential transformations, including the formation of alcohol (C7F15OH), ketone (C7F15O•), and further fragmentation to shorter-chain perfluorinated species. (b) Oxygenation pathway (yellow): In the presence of molecular oxygen (O2), perfluoroalkyl peroxy radicals (C7F15OO•) form, which subsequently degrade into smaller perfluorinated fragments through oxidative cleavage. (c) Hydroxyl radical-mediated pathway (green): The introduction of hydroxyl radicals promotes successive transformations, leading to the formation of carboxylic acids (C6F13COOH) and ultimately mineralization into carbon dioxide (CO2) and fluoride ions (HF).
Pathway a) is widely considered the primary route for PFOA mineralization in the EO process (Chen et al., 2022b). DFT calculations of the reactions’ standard Gibbs free energy change (ΔG0) and Gibbs free energy of activation (ΔG‡) indicate that the reaction of C7F15⋅ with ⋅OH to form C7F15OH (Equation 14) is highly favorable, with a barrierless reaction and a ΔG0 of −419.1 kJ/mol. Alternatively, the direct oxidation of C7F15⋅ to C7F15+ (Equation 15) followed by its reaction with OH− to form C7F15OH (Equation 16) is also energetically feasible. Given the significantly exergonic nature of both pathways (ΔG0 of −459.9 kJ/mol for Equation 16), hydroxylation appears to be the dominant mechanism.
OnceC7F15OH formed, it dissociates into C7F15O− can due to the low pKa (∼2) in the solution (Zhang et al., 2019). Accordingly, the majority of C7F15OH species would become C7F15O− in the solution. In the subsequent degradation process, C7F15O− can react with h+, H2O, and HO•. Chen et al. found that the formation of C7F15O• via hole oxidation (Equation 17) is favorable due to its low activation energy (ΔG‡ = 5.11 kJ/mol). C7F15O• then undergoes cleavage to form C6F13• and COF2 (Equation 18), a reaction with a ΔG0 of −90.6 kJ/mol and a low activation energy (ΔG‡ = 13.3 kJ/mol), making it a highly likely step in the overall degradation process. C6F13• subsequently undergoes similar reactions, leading to sequential cleavage until complete mineralization to CO2 and HF through hydrolysis.
Pathway b), proposed by Niu et al. (2013), suggests that C7F15• reacts with O2 to form C7F15OO• (Equation 19), which then decomposes into C7F15O• and O2 (Equation 20) (Niu et al., 2013). Since electrolysis generates a significant amount of O2 at the anode, this reaction could occur in competition with the hydroxylation pathway in pathway a). However, the competition between these two routes is influenced by reaction kinetics and the relative concentrations of reactive oxygen species (ROS) and molecular oxygen. Although the formation of peroxides provides an alternative route for C7F15O• generation, it is unclear whether this mechanism is dominant under typical electrochemical conditions.
Pathway c), as shown in Figure 4, proposes that C7F15O− can react with H3O+ to form C6F13COF and release HF (Equation 21). The relatively low ΔG0 (−63.6 kJ/mol) suggests that this reaction is thermodynamically feasible and could compete with the hole oxidation pathway (Equation 17). However, the subsequent hydrolysis of C6F13COF to C6F13COOH (Equation 22) has a high activation energy (ΔG‡ = 159.8 kJ/mol), making it less favorable compared to the sequential cleavage mechanism in pathway a). This suggests that while pathway c) may occur in parallel, it is unlikely to be the primary mineralization pathway due to its slower reaction kinetics.
While all three pathways contribute to PFOA degradation, pathway a) is likely the dominant route due to its lower energy barriers and strong exergonic reactions. The direct interaction of C7F15• with •OH is particularly favorable, given the abundance of hydroxyl radicals in electrochemical oxidation. Pathway b), involving O2, may become more significant under conditions where hydroxyl radical production is limited or oxygen concentration is high. Pathway c) is less favorable for complete mineralization due to the high activation energy of its hydrolysis step, though it may still contribute to intermediate transformation.
In practical applications, the dominant pathway will depend on operational factors such as anode material, applied potential, pH, and the presence of competing oxidants. Higher anode potentials and catalytic surfaces that enhance hydroxyl radical production will likely favor pathway a), whereas systems with high oxygen evolution may shift the balance toward pathway b). Further experimental validation is needed to quantify the relative contributions of each pathway under real-world conditions.
4.2 PFOS degradation pathways
There are two proposed possible PFOS degradation pathways on the anode in the EO process by Shi et al. (2019), Pierpaoli et al. (2021) in Figure 5. However, we found the second pathway proposed by Pierpaoli et al. unreasonable.
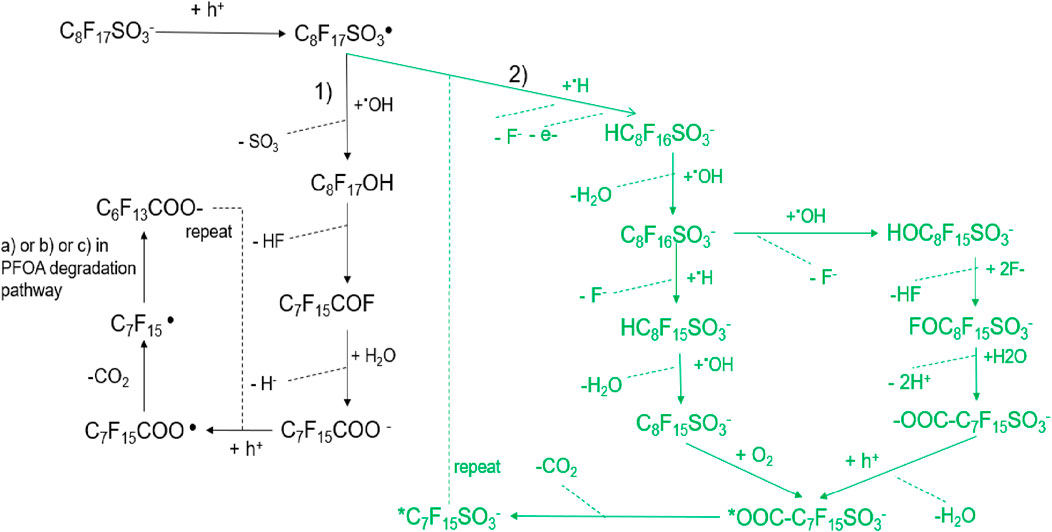
Figure 5. The possible PFOS degradation pathways on the anode in the EO process. 1) Oxidative Decay Pathway (Black, Left Side): PFOS ion (C8F17SO3−) undergoes direct electron transfer, forming the perfluorinated sulfonyl radical (C8F17SO3•); The radical can lose a sulfonate SO3) group, generating C8F17OH; Further transformations through dehydrofluorination and hydrolysis produce ketone (C7F15COF) and shorter perfluorinated carboxylates (C7F15COO−), which follow similar degradation steps as observed in PFOA breakdown; The cycle repeats until the perfluorinated chains are broken down into smaller fragments, eventually forming CO2 and HF. 2) Reductive and Hydroxylation Pathway (Green, Right Side): PFOS undergoes direct electron transfer and hydrogenation, leading to the formation of partially fluorinated sulfonates (HC8F16SO3−); Hydrolysis and hydroxyl radical (•OH) attack further drive defluorination, forming species like C8F16SO3− and HOC8F15SO3−; Successive F- and HF eliminations lead to oxidative fragmentation, producing perfluorinated carboxylates (-OOC-C7F15SO3−) and eventually mineralized products CO2, HF and H2O.
Shi et al. (2019) ’s study explored PFOS degradation with DFT calculations in the EO process and they found the role of hydroxyl free radicals in PFOS. Combining the experiment analysis and DFT theoretical studies they proposed the 1) PFOS degradation pathways in Figure 2. By DFT calculations, they also found the role of hydroxyl free radicals in PFOS degradation (Equation 23) during EO that C8F17SO3⋅ may react with water to form C8F17SO3− via an energetically favorable reaction (Equation 24). Therefore, once C8F17SO3⋅ is formed by PFOS oxidation during EO via DET, it can transform back to C8F17SO3− spontaneously in water solution. It is proposed that C8F17SO3⋅ tends to undergo desulfurization, accompanied by a reaction with ⋅OH to form C8F17OH (Equation 25). It is proposed that C8F17OH formed in Equation 25 decomposes to C7F15CFO (Equation 26), and then rapidly hydrolyze to C7F15COO− (Equation 27). C7F15COO− may further go through the decarboxylation cycle stepwise like PFOA degradation pathways (a, b or c pathways in Figure 4), leading to rip off of a CF2 unit each step until complete mineralization.
However Pierpaoli et al. (2021), proposed a free hydrogen radical eliminates a fluoride atom from PFSAs or sulfonated radicals by H-F substitution followed by the transformation to new sulfonated products by hydrogen abstraction, based on the identification of by-product with the UHPLC-ESI-MS technique. They observed the perfluooroheptyl sulphate ion and radical (C7F15SO3−, ⋅ C7F15SO3−), as well C6F13COO-, C5F11COO−, and C4F9COO−.The 2) pathway in Figure 5 includes radical reaction, decarboxylation, hydrolysis, and hydroxylation for PFOS anodic degradation as follows. The elimination of two fluoride ions giving the observed
At the same time, the
However, only after hydrolysis of the formed product (
However, the proposed PFOS degradation pathway by Pierpaoli has been critiqued for issues related to charge balance, as noted in their study. Some of the reactions are not balanced such as there are three negative charges on the right side and only one on the left in reaction Equation 28. Furthermore, the formation of hydrogen radical is not mentioned in this paper. There may be some wandering hydrogen radicals in the system, but it’s doubtful that the hydrogen radicals are reactive enough to eliminate a fluoride atom from PFSA. So, it is not a reasonable possible pathway for PFOS degradation. While the authors did not analyze in depth the rationale behind their pathway proposal or potential directions for its improvement. Further exploration is necessary to refine the mechanistic understanding and address the rationality of the proposed transformations.
5 Comparative analysis of electrode materials
A comparative analysis of electrode materials is essential for understanding the effectiveness of electrochemical oxidation (EO) in PFAS degradation. Various anode materials, including boron-doped diamond (BDD), Magnéli phase titanium suboxides (TSO), lead dioxide (PbO2), and stannous dioxide (SnO2), have been extensively studied. Each material offers distinct advantages and limitations in terms of degradation efficiency, selectivity, energy consumption, and potential secondary contamination.
BDD electrodes are widely regarded as one of the most effective materials for PFAS degradation due to their high oxygen evolution potential, which minimizes competition from side reactions. This allows for the generation of hydroxyl radicals (⋅OH) that can efficiently break down PFAS molecules. However, BDD electrodes are expensive to manufacture and have limited scalability for large-scale water treatment applications.
TSO electrodes, particularly Ti4O7, have gained attention as a cost-effective alternative to BDD. Studies have shown that Magnéli phase Ti4O7 anodes provide a high degree of PFAS mineralization while maintaining good stability. Additionally, the porous structure of TSO electrodes enhances mass transfer, which can improve degradation kinetics. However, their lower oxidation potential compared to BDD can result in partial degradation and the formation of persistent byproducts.
PbO2 and SnO2 electrodes have also been investigated for PFAS degradation due to their strong oxidizing properties. PbO2 anodes demonstrate high degradation efficiency but pose a significant environmental risk due to potential lead leaching, making them unsuitable for drinking water applications. SnO2 electrodes, particularly when doped with elements like antimony (Sb-SnO2), have shown promise in PFAS treatment. However, their long-term stability remains a concern, as SnO2-based electrodes tend to degrade over extended operation times.
Recent advancements in electrode materials include doping strategies and hybrid electrode systems. For example, Nb-doped TiO2 has been explored as a means to enhance the electrochemical activity of Ti-based electrodes while maintaining stability. Similarly, composite materials, such as Ti4O7/MXene hybrids, have been shown to improve electrical conductivity and PFAS adsorption, leading to more efficient degradation pathways.
From an application standpoint, the choice of electrode material depends on the specific treatment objectives. For high-efficiency mineralization with minimal byproduct formation, BDD remains the gold standard, albeit at a higher cost. For scalable and cost-effective alternatives, TSO electrodes provide a balance between efficiency and affordability. PbO2 and SnO2 electrodes, while effective, require careful consideration of stability and potential secondary contamination. Hybrid and doped materials represent promising directions for future research, offering opportunities to optimize both performance and economic feasibility.
Future studies should focus on optimizing electrode surface properties, enhancing long-term stability, and integrating EO with complementary treatment technologies. Additionally, life cycle assessments comparing the environmental and economic impacts of different electrode materials would provide valuable insights for real-world implementation.
6 Practical implications
The insights presented in this study have significant implications for the development of more effective electrochemical oxidation (EO) systems for PFAS remediation. Understanding the mechanistic pathways of PFAS degradation allows for the strategic optimization of electrode materials, operational conditions, and system design to enhance treatment efficiency.
• Optimization of Electrode Materials–The findings highlight the critical role of electrode composition in PFAS degradation. Electrodes with high overpotential for oxygen evolution, should be prioritized to promote direct electron transfer and hydroxyl radical generation. Future research should focus on doping strategies and surface modifications to improve electrocatalytic performance and stability.
• Operational Condition Enhancement–The study suggests that electrolyte composition, current density, and applied potential significantly influence PFAS degradation efficiency (Wang Y. et al., 2025). Maintaining optimal current densities while avoiding excessive energy consumption is crucial. Further research on pH adjustments, electrolyte additives, and pulsed voltage application could improve selectivity and degradation rates.
• System Design Considerations–The integration of EO with complementary treatment processes, such as adsorption or photochemical oxidation, could enhance PFAS removal efficiency (Gomri et al., 2025). Reactor design modifications, including flow cell optimization and electrode spacing adjustments, may further improve mass transfer and degradation kinetics. Pilot-scale testing should be prioritized to assess scalability and long-term performance.
By translating these mechanistic insights into practical design and operational strategies, this study provides a foundation for advancing EO systems as a viable technology for PFAS remediation in groundwater and wastewater treatment applications.
7 Conclusion and insights for future study
This review elucidates the primary PFAS degradation mechanisms during the EO process, highlighting the critical role of density functional theory (DFT) calculations in understanding reaction pathways. The applications of DFT in mechanism investigation are demonstrated, including bond dissociation energy, adsorption energy of PFAS, activation energy of PFAS and hydroxyl radicals, and the overpotential of oxygen evolution reactions. Furthermore, possible PFOA and PFOS degradation pathways are discussed in detail.
From the literature review, the main PFAS degradation mechanisms in the EO process include mass transfer, direct electron transfer, decarboxylation, peroxyl radical generation, hydroxylation, intramolecular rearrangement, and hydrolysis. While previous studies demonstrate that EO is highly effective for PFAS degradation, significant challenges remain that hinder its large-scale application. These challenges include high energy consumption, the generation of persistent intermediate byproducts, competition between degradation pathways, and the influence of anode material properties on reaction kinetics. To address these limitations, the following targeted recommendations are proposed:
7.1 Optimizing anode materials to enhance degradation efficiency and reduce energy consumption
The choice of anode material significantly affects EO efficiency by influencing hydroxyl radical generation, electron transfer rates, and energy demands. Future research should focus on developing high-activity, low-cost anode materials with improved selectivity for PFAS oxidation. Boron-doped diamond (BDD) electrodes have demonstrated high oxidation potential but are costly, while mixed metal oxide (MMO) anodes are more affordable but less efficient. Novel composite anodes, such as doped Ti/IrO2 or graphene-modified electrodes, could be explored to achieve a balance between efficiency and cost-effectiveness. Additionally, optimizing the anode surface structure to enhance reactive species generation and reduce overpotentials could further improve performance.
7.2 Enhancing electrolyte conditions and reactor design for energy efficiency
The energy consumption of EO is strongly influenced by electrolyte composition and reactor design. Future studies should investigate the impact of electrolyte pH, ionic strength, and the presence of co-contaminants on PFAS degradation kinetics. Electrolytes with lower resistivity can reduce energy losses, while buffer systems could help maintain optimal reaction conditions. Furthermore, novel reactor designs, such as flow-through electrochemical cells or gas-diffusion electrodes, could enhance mass transfer and improve energy efficiency. The integration of EO with complementary processes, such as adsorption or membrane filtration, could also help reduce overall treatment energy costs by concentrating PFAS before EO treatment.
7.3 Expanding investigations beyond PFOA and PFOS to other PFAS species
Most research has focused on PFOA and PFOS degradation pathways, but short-chain PFAS and emerging alternatives may exhibit different electrochemical behavior. Future studies should systematically examine the degradation kinetics of a broader range of PFAS compounds and explore whether short-chain PFAS require different operational conditions or anode materials.
7.4 Combining DFT calculations with experimental studies for more accurate mechanistic insights
DFT calculations are valuable for predicting reaction pathways, but experimental validation is essential for practical implementation. Future research should focus on integrating computational models with real-time electrochemical experiments to refine degradation pathway predictions. This could involve in situ spectroscopic techniques, such as Raman spectroscopy, to track PFAS degradation intermediates and confirm theoretical predictions.
In conclusion, while EO remains a promising approach for PFAS degradation, addressing energy consumption, optimizing electrode materials, and refining mechanistic understanding are critical for its large-scale application. Future research should focus on material innovations, reactor optimization, and hybrid treatment approaches to enhance both efficiency and feasibility.
Author contributions
GL: Data curation, Investigation, Writing – original draft, Formal Analysis. MP: Writing – review and editing, Conceptualization, Data curation, Formal Analysis. QH: Supervision, Writing – review and editing, Conceptualization. C-HH: Supervision, Writing – review and editing. YC: Supervision, Writing – review and editing. GH: Supervision, Writing – review and editing. KL: Conceptualization, Formal Analysis, Methodology, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported in part by U.S. Environmental Protection Agency Grant R840080.
Acknowledgments
It has not been formally reviewed by EPA. EPA does not endorse any products or commercial services mentioned in this publication.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The views expressed in this document are solely those of the authors and do not necessarily reflect those of the Agency.
References
Bentel, M., Yu, Y. C., Xu, L. H., Li, Z., Wong, B., Men, Y. J., et al. (2019). Defluorination of per- and polyfluoroalkyl substances (PFASs) with hydrated electrons: structural dependence and implications to PFAS remediation and management. Abstr. Pap. Am. Chem. Soc. 258. doi:10.1021/acs.est.8b06648
Bharti, , Ahmed, G., Kumar, Y., and Sharma, S. (2021). DFT computation of quantum capacitance of pure and doped niobium nitrides for supercapacitor applications. Ceram. Int. 47 (13), 18948–18955. doi:10.1016/j.ceramint.2021.03.237
Carter, K. E., and Farrell, J. (2008). Oxidative destruction of perfluorooctane sulfonate using boron-doped diamond film electrodes. Environ. Sci. and Technol. 42 (16), 6111–6115. doi:10.1021/es703273s
Chen, C., Ma, Q. Q., Liu, F. Z., Gao, J. A., Li, X. Y., Sun, S. B., et al. (2021). Photocatalytically reductive defluorination of perfluorooctanoic acid (PFOA) using Pt/La2Ti2O7 nanoplates: experimental and DFT assessment. J. Hazard. Mater. 419, 126452. doi:10.1016/j.jhazmat.2021.126452
Chen, Y., Yang, Y., Cui, J., Zhang, H., and Zhao, Y. (2024). Decoding PFAS contamination via Raman spectroscopy: a combined DFT and machine learning investigation. J. Hazard. Mater. 465, 133260. doi:10.1016/j.jhazmat.2023.133260
Chen, Z. F., Wang, X. J., Feng, H. L., Chen, S. H., Niu, J. F., Di, G. L., et al. (2022a). Electrochemical advanced oxidation of perfluorooctanoic acid: mechanisms and process optimization with kinetic modeling. Environ. Sci. and Technol. 56, 14409–14417. doi:10.1021/acs.est.2c02906
Chen, Z. F., Wang, X. J., Feng, H. L., Chen, S. H., Niu, J. F., Di, G. L., et al. (2022b). Electrochemical advanced oxidation of perfluorooctanoic acid: mechanisms and process optimization with kinetic modeling. Environ. Sci. and Technol. 56 (20), 14409–14417. doi:10.1021/acs.est.2c02906
Cheng, Z. W., Chen, Q. C., Liu, Z. K., Liu, J. Y., Liu, Y. W., Liu, S. Q., et al. (2021). Interpretation of reductive PFAS defluorination with quantum chemical parameters. Environ. Sci. and Technol. Lett. 8 (8), 645–650. doi:10.1021/acs.estlett.1c00403
Crone, B. C., Speth, T. F., Wahman, D. G., Smith, S. J., Abulikemu, G., Kleiner, E. J., et al. (2019). Occurrence of per- and polyfluoroalkyl substances (PFAS) in source water and their treatment in drinking water. Crit. Rev. Environ. Sci. Technol. 49 (24), 2359–2396. doi:10.1080/10643389.2019.1614848
Deng, S. B., Nie, Y., Du, Z. W., Huang, Q., Meng, P. P., Wang, B., et al. (2015). Enhanced adsorption of perfluorooctane sulfonate and perfluorooctanoate by bamboo-derived granular activated carbon. J. Hazard. Mater. 282, 150–157. doi:10.1016/j.jhazmat.2014.03.045
Eslamibidgoli, M. J., and Eikerling, M. H. (2015). Electrochemical Formation of reactive oxygen species at Pt (111)-A density functional theory study. Acs Catal. 5 (10), 6090–6098. doi:10.1021/acscatal.5b01154
Gao, J., Liu, Z., Chen, Z., Rao, D., Che, S., Gu, C., et al. (2023). Photochemical degradation pathways and near-complete defluorination of chlorinated polyfluoroalkyl substances. Nat. water 1, 381–390. doi:10.1038/s44221-023-00046-z
Gar Alalm, M., and Boffito, D. C. (2022). Mechanisms and pathways of PFAS degradation by advanced oxidation and reduction processes: a critical review. Chem. Eng. J. 450, 138352. doi:10.1016/j.cej.2022.138352
Gomri, C., Makhoul, E., Koundia, F. N., Petit, E., Raffy, S., Bechelany, M., et al. (2025). Electrochemical advanced oxidation combined to electro-Fenton for effective treatment of perfluoroalkyl substances “PFAS” in water using a Magnéli phase-based anode. Nanoscale Adv. 7 (1), 261–268. doi:10.1039/d4na00626g
Hori, H., Hayakawa, E., Einaga, H., Kutsuna, S., Koike, K., Ibusuki, T., et al. (2004). Decomposition of environmentally persistent perfluorooctanoic acid in water by photochemical approaches. Environ. Sci. and Technol. 38 (22), 6118–6124. doi:10.1021/es049719n
Huang, D., Wang, K., Niu, J., Chu, C., Weon, S., Zhu, Q., et al. (2020). Amorphous Pd-loaded Ti4O7 electrode for direct anodic destruction of perfluorooctanoic acid. Environ. Sci. Technol. 54 (17), 10954–10963. doi:10.1021/acs.est.0c03800
Huang, X., Wang, J., Tao, H. B., Tian, H., and Xu, H. (2019). An essential descriptor for the oxygen evolution reaction on reducible metal oxide surfaces. Chem. Sci. 10 (11), 3340–3345. doi:10.1039/c8sc04521f
Ko, J. S., Le, N. Q., Schlesinger, D. R., Zhang, D. J., Johnson, J. K., and Xia, Z. Y. (2021). Author Correction: novel niobium-doped titanium oxide towards electrochemical destruction of forever chemicals. Sci. Rep. 11 (1), 19014. doi:10.1038/s41598-021-98715-0
Li, L., Wang, Y., and Huang, Q. (2021). First-principles study of the degradation of perfluorooctanesulfonate and perfluorobutanesulfonate on a Magnéli phase Ti4O7 anode. ACS ES&T Water 1 (8), 1737–1744. doi:10.1021/acsestwater.1c00086
Liao, Z. H., and Farrell, J. (2009). Electrochemical oxidation of perfluorobutane sulfonate using boron-doped diamond film electrodes. J. Appl. Electrochem. 39 (10), 1993–1999. doi:10.1007/s10800-009-9909-z
Lin, H., Niu, J., Liang, S., Wang, C., Wang, Y., Jin, F., et al. (2018). Development of macroporous Magnéli phase Ti4O7 ceramic materials: as an efficient anode for mineralization of poly- and perfluoroalkyl substances. Chem. Eng. J. 354, 1058–1067. doi:10.1016/j.cej.2018.07.210
Lin, H., Xiao, R., Xie, R., Yang, L., Tang, C., Wang, R., et al. (2021). Defect engineering on a Ti4O7 electrode by Ce(3+) doping for the efficient electrooxidation of perfluorooctanesulfonate. Environ. Sci. Technol. 55 (4), 2597–2607. doi:10.1021/acs.est.0c06881
Liu, H., and Vecitis, C. D. (2012). Reactive transport mechanism for organic oxidation during electrochemical filtration: mass-transfer, physical adsorption, and electron-transfer. J. Phys. Chem. C 116 (1), 374–383. doi:10.1021/jp209390b
Ma, Q. Q., Gao, J. A., Cheng, K. Y., Young, J., Zhao, M. Q., Ronen, A., et al. (2024). Electrooxidation of perfluorocarboxylic acids by an interfacially engineered Magnéli phase titanium oxide (Ti4O7) electrode with MXene. ACS ES&T Eng. 4, 1102–1112. doi:10.1021/acsestengg.3c00571
Man, I. C., Su, H. Y., Calle-Vallejo, F., Hansen, H. A., Martinez, J. I., Inoglu, N. G., et al. (2011). Universality in oxygen evolution electrocatalysis on oxide surfaces. Chemcatchem 3 (7), 1159–1165. doi:10.1002/cctc.201000397
Mirabediny, M., Sun, J., Yu, T. T., Akermark, B., Das, B., and Kumar, N. (2023). Effective PFAS degradation by electrochemical oxidation methods-recent progress and requirement. Chemosphere 321, 138109. doi:10.1016/j.chemosphere.2023.138109
Niu, J. F., Lin, H., Gong, C., and Sun, X. M. (2013). Theoretical and experimental insights into the electrochemical mineralization mechanism of perfluorooctanoic acid. Environ. Sci. and Technol. 47 (24), 14341–14349. doi:10.1021/es402987t
Phong Vo, H. N., Ngo, H. H., Guo, W. S., Hong Nguyen, T. M., Li, J. X., Liang, H., et al. (2020). Poly?and perfluoroalkyl substances in water and wastewater: a comprehensive review from sources to remediation. J. Water Process Eng. 36, 101393. doi:10.1016/j.jwpe.2020.101393
Pierpaoli, M., Szopińska, M., Wilk, B. K., Sobaszek, M., Łuczkiewicz, A., Bogdanowicz, R., et al. (2021). Electrochemical oxidation of PFOA and PFOS in landfill leachates at low and highly boron-doped diamond electrodes. J. Hazard. Mater. 403, 123606. doi:10.1016/j.jhazmat.2020.123606
Rahman, M. F., Peldszus, S., and Anderson, W. B. (2014). Behaviour and fate of perfluoroalkyl and polyfluoroalkyl substances (PFASs) in drinking water treatment: a review. Water Res. 50, 318–340. doi:10.1016/j.watres.2013.10.045
Rozen, S., and Filler, R. (1985). α-Fluorocarbonyl compounds and related chemistry. Tetrahedron, 41(7), 1111–1153. doi:10.1016/S0040-4020(01)96514-7
Ryan, D. R., Mayer, B. K., Baldus, C. K., Mcbeath, S. T., Wang, Y., and Mcnamara, P. J. (2021). Electrochemical technologies for per- and polyfluoroalkyl substances mitigation in drinking water and water treatment residuals. AWWA Water Sci. 3 (5). doi:10.1002/aws2.1249
Schaefer, C. E., Andaya, C., Burant, A., Condee, C. W., Urtiaga, A., Strathmann, T. J., et al. (2017). Electrochemical treatment of perfluorooctanoic acid and perfluorooctane sulfonate: insights into mechanisms and application to groundwater treatment. Chem. Eng. J. 317, 424–432. doi:10.1016/j.cej.2017.02.107
Shi, H., Wang, Y., Li, C., Pierce, R., Gao, S., and Huang, Q. (2019). Degradation of perfluorooctanesulfonate by reactive electrochemical membrane composed of magneli phase titanium suboxide. Environ. Sci. Technol. 53 (24), 14528–14537. doi:10.1021/acs.est.9b04148
Trellu, C., Chaplin, B. P., Coetsier, C., Esmilaire, R., Cerneaux, S., Causserand, C., et al. (2018). Electro-oxidation of organic pollutants by reactive electrochemical membranes. Chemosphere 208, 159–175. doi:10.1016/j.chemosphere.2018.05.026
Wang, C., Zhang, T. N., Yin, L. F., Ni, C. S., Ni, J. P., and Hou, L. A. (2022d). Enhanced perfluorooctane acid mineralization by electrochemical oxidation using Ti3+ self-doping TiO2 nanotube arrays anode. Chemosphere 286, 131804. doi:10.1016/j.chemosphere.2021.131804
Wang, L., Lu, J., Li, L., Wang, Y., and Huang, Q. (2020). Effects of chloride on electrochemical degradation of perfluorooctanesulfonate by Magneli phase Ti4O7 and boron doped diamond anodes. Water Res. 170, 115254. doi:10.1016/j.watres.2019.115254
Wang, Y., Kim, J., Huang, C.-H., Hawkins, G. L., Li, K., Chen, Y., et al. (2022a). Occurrence of per- and polyfluoroalkyl substances in water: a review. Environ. Sci. Water Res. and Technol. 8 (6), 1136–1151. doi:10.1039/d1ew00851j
Wang, Y., Li, L., Wang, Y., Shi, H., Wang, L., and Huang, Q. (2022b). Electrooxidation of perfluorooctanesulfonic acid on porous Magnéli phase titanium suboxide Anodes: impact of porous structure and composition. Chem. Eng. J. 431, 133929. doi:10.1016/j.cej.2021.133929
Wang, Y., Wang, Y., Dong, S., and Huang, Q. (2025a). The impact of anions on electrooxidation of perfluoroalkyl acids by porous Magnéli phase titanium suboxide anodes. Plos One 20 (1), e0317696. doi:10.1371/journal.pone.0317696
Wang, Y. Y., Pierce, R., Shi, H. N., Li, C. G., and Huang, Q. G. (2022c). Electrochemical degradation of perfluoroalkyl acids by titanium suboxide anodes (vol 6, pg 144, 2020). Environ. Science-Water Res. and Technol. 8 (2), 443. doi:10.1039/d1ew90044g
Wang, Z., You, X., Lan, L., Huang, G., Zhu, T., Tian, S., et al. (2025b). Electrocatalytic oxidation of hexafluoropropylene oxide homologues in water using a boron-doped diamond electrode. Environ. Technol. 46 (8), 1280–1291. doi:10.1080/09593330.2024.2382937
Wanninayake, D. M. (2021). Comparison of currently available PFAS remediation technologies in water: a review. J. Environ. Manag. 283, 111977. doi:10.1016/j.jenvman.2021.111977
Yamijala, S. S. R. K. C., Shinde, R., Hanasaki, K., Ali, Z. A., and Wong, B. M. (2022). Photo-induced degradation of PFASs: excited-state mechanisms from real-time time-dependent density functional theory. J. Hazard. Mater. 423, 127026. doi:10.1016/j.jhazmat.2021.127026
Zhang, Y. Y., Moores, A., Liu, J. X., and Ghoshal, S. (2019). New insights into the degradation mechanism of perfluorooctanoic acid by persulfate from density functional theory and experimental data. Environ. Sci. and Technol. 53 (15), 8672–8681. doi:10.1021/acs.est.9b00797
Zhao, L., Pu, R., Deng, S., Lin, L., Mantzavinos, D., Naidu, R., et al. (2025). Enhanced electrochemical degradation of per-and polyfluoroalkyl substances (PFAS) by activating persulfate on boron-doped diamond (BDD) anode. Sep. Purif. Technol. 359, 130459. doi:10.1016/j.seppur.2024.130459
Keywords: electrochemical oxidation, per-and poly-fluoroalkyl substances (PFAS), density functional theory (DFT) computation, degradation mechanisms, degradation pathways
Citation: Li G, Peng M, Huang Q, Huang C-H, Chen Y, Hawkins G and Li K (2025) A review on the recent mechanisms investigation of PFAS electrochemical oxidation degradation: mechanisms, DFT calculation, and pathways. Front. Environ. Eng. 4:1568542. doi: 10.3389/fenve.2025.1568542
Received: 30 January 2025; Accepted: 24 March 2025;
Published: 09 April 2025.
Edited by:
Shujuan Zhang, Nanjing University, ChinaReviewed by:
Wang Zunyao, Nanjing University, ChinaBingdang Wu, Suzhou University of Science and Technology, China
Copyright © 2025 Li, Peng, Huang, Huang, Chen, Hawkins and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ke Li, bHVrZWxpQHVnYS5lZHU=