 Angela M. Arensdorf
Angela M. Arensdorf Danilo Diedrichs
Danilo Diedrichs D. Thomas Rutkowski
D. Thomas Rutkowski- 1Department of Anatomy and Cell Biology, University of Iowa Carver College of Medicine, Iowa City, IA, USA
- 2Department of Mathematics and Computer Science, Wheaton College, Wheaton, IL, USA
- 3Department of Internal Medicine, University of Iowa Carver College of Medicine, Iowa City, IA, USA
The mammalian unfolded protein response (UPR) is propagated by three ER-resident transmembrane proteins, each of which initiates a signaling cascade that ultimately culminates in production of a transcriptional activator. The UPR was originally characterized as a pathway for upregulating ER chaperones, and a comprehensive body of subsequent work has shown that protein synthesis, folding, oxidation, trafficking, and degradation are all transcriptionally enhanced by the UPR. However, the global reach of the UPR extends to genes involved in diverse physiological processes having seemingly little to do with ER protein folding, and this includes a substantial number of mRNAs that are suppressed by stress rather than stimulated. Through multiple non-canonical mechanisms emanating from each of the UPR pathways, the cell dynamically regulates transcription and mRNA degradation. Here we highlight these mechanisms and their increasingly appreciated impact on physiological processes.
Introduction
The ER is best known as the gateway to the secretory pathway. As the site of synthesis for nascent secretory proteins and resident lumenal and transmembrane proteins of the endomembrane system, the ER shepherds the folding, oxidation, modification, and assembly of approximately one-third of the cellular proteome—or more in cell types specialized for protein secretion such as antibody-secreting plasma B lymphocytes or endocrine or exocrine cells (Huh et al., 2003; Tagliavacca et al., 2003). As such, the ER is replete with chaperones, cochaperones, oxidases, and thiol isomerases to facilitate protein folding, and utilizes an elaborate quality control system to recognize terminally misfolded proteins and purge them from the ER for degradation (Araki and Nagata, 2012). With this system in place, the low level of protein misfolding that arises because of the inherent error rate in the process can presumably be managed. However, the quality control machinery can be overwhelmed either by an overload of nascent client proteins or by any exogenous disruption to the protein folding and trafficking system—so-called “ER stress” (Ron and Walter, 2007). The consequences of a pervasive defect in ER protein folding can be grave for both the cell and the organism. A cell with overwhelmed ER quality control machinery will, at best, fail to maintain secretory pathway integrity and to effectively sense and respond to the extracellular milieu. At worst, accumulated misfolded proteins might seed the formation of toxic protein aggregates (Matus et al., 2011). Thus, the cell has in the UPR a signal transduction system that augments the protein folding capacity of the ER. While the UPR improves ER function by several short-term mechanisms, it ultimately culminates in gene regulation for longer-lasting enhancement of the ER folding environment. Classically, this regulation constitutes a self-contained system in which ER stress leads to transcriptional induction of genes encoding ER chaperones and other proteins that grease the wheels of secretory pathway function, thereby alleviating ER stress and shutting the response off (Travers et al., 2000). However, it is now becoming clear that the UPR is much more deeply entwined in cellular physiology than this simple view would suggest. In mammals, it regulates genes involved in a number of cellular processes that have little on their face to do with ER function, including metabolism and inflammation (Fu et al., 2012; Garg et al., 2012). The regulation of many of these genes cannot be explained by the canonical mechanisms of UPR signaling. The aim of this review is to highlight emerging concepts in the non-canonical regulation of mRNA expression by the UPR. Rather than providing an exhaustive account of all possible means by which mRNA abundance might be controlled by the UPR, here we describe the general principles by which such regulation can occur and provide illustrative examples that emphasize the diverse physiological consequences of such pathways.
The Canonical UPR
The idea that there must be a signal transduction pathway emanating from the ER first emerged from the observation that expression of misfolded influenza hemagglutinin (HA) in mammalian cultured cells led to upregulation of the ER chaperones Bip (aka GRP78, the product of the Hspa5 gene) and GRP94 (the product of the Hsp90b1 gene) (Kozutsumi et al., 1988). This finding allowed previous reports of Bip and Grp94 induction in response to chemical perturbants (Drummond et al., 1987; Kim and Lee, 1987) to be tied specifically to disrupted ER protein folding. The general applicability of the phenomenon was extended to other misfolded ER client proteins (Dorner et al., 1989), and to the upregulation of ER oxidases and thiol isomerases as well (Dorner et al., 1990). As with many fundamental cellular processes, an analogous response was soon discovered in yeast (Normington et al., 1989), and a cis-acting unfolded protein response element (UPRE) within the yeast Bip (aka KAR2) promoter was discovered (Mori et al., 1992; Kohno et al., 1993). The ER stress-responsiveness of KAR2 served as the springboard for a classic series of genetic and biochemical studies describing the mechanistic basis of what had become known as the unfolded protein response (Cox et al., 1993; Mori et al., 1993; Cox and Walter, 1996; Sidrauski et al., 1996; Sidrauski and Walter, 1997). Together, these studies identified Ire1p (for Inositol-requiring enzyme) as an ER-resident transmembrane kinase that became autophosphorylated during ER stress, activating a cytosolic endoribonuclease activity that catalyzed the removal of an inhibitory intron from HAC1 mRNA. This splicing event allowed the mRNA to be translated into the Hac1p transcription factor, which subsequently bound to the KAR2 UPRE and stimulated KAR2 transcription. A survey of the breadth of UPR targets (Travers et al., 2000) took advantage of then-new microrarray technology, and revealed two fundamental features of the yeast UPR that have heavily influenced the subsequent portrayal of the response in both yeast and mammals: (1) It is predominantly an inductive response, with a bias for upregulated genes; and (2) its scope is not limited solely to ER chaperones and phospholipid synthesis enzymes, as had been originally thought, but encompasses genes involved in other aspects of secretory pathway function, including ER-associated protein degradation (ERAD), vesicular trafficking, and protein translocation into the ER, among other processes. Thus, even though the response was more expansive than anticipated, it could still be considered as a discrete transcriptional program designed to upregulate the expression of genes with a common cis-element, all of which influenced ER protein folding either directly or indirectly.
Once Ire1p was described in yeast, its mammalian homologs (IRE1α and an intestine-specific paralog IRE1β) were discovered (Tirasophon et al., 1998; Wang et al., 1998), as was a gene encoding a transmembrane protein with a lumenal domain homologous to IRE1α but a cytosolic aspect homologous to other kinases such as PKR that phosphorylate the translation initiation factor eIF2α. The protein encoded by this gene was named PERK (PKR-like ER kinase) (Shi et al., 1998; Harding et al., 1999). And, much as the UPRE had been instrumental in identification of Ire1p in yeast, so also a mammalian ER stress response element (ERSE) in the promoters of Bip and Grp94 was characterized and used to identify ATF6 (of the activating transcription factor family) as another ER-resident stress sensor (Yoshida et al., 1998; Haze et al., 1999). All three pathways are conserved throughout metazoa, but the PERK and ATF6 pathways appear to assume more modest roles in invertebrates such as flies and worms (Shen et al., 2001; Ryoo and Steller, 2007).
As for yeast Ire1p, activation of each of the three mammalian UPR pathways culminates in production of a transcriptional activator and attendant rearrangement of chromatin structure (Baumeister et al., 2005; Donati et al., 2006; Gal-Yam et al., 2006) and recruitment of RNA Polymerase II (Sela et al., 2012) to stimulate gene transcription. These pathways have been reviewed exhaustively elsewhere (Schröder and Kaufman, 2005) and so will only be described briefly here. Mammalian IRE1α is activated by autophosphorylation and catalyzes the splicing of Xbp1 mRNA, resulting in excision of a 26-base intron and thus allowing in-frame translation of the downstream transcriptional activation domain of XBP1 (Yoshida et al., 2001a; Lee et al., 2002). Translocation and activity of the bZIP protein produced from spliced Xbp1 mRNA are regulated by the protein produced by the unspliced mRNA (Lee et al., 2003a; Tirosh et al., 2006), by the regulatory subunits of PI3 Kinase (Park et al., 2010; Winnay et al., 2010), and by acetylation (Wang et al., 2011). In the nucleus, XBP1 binds to UPRE sequences (distinct from yeast UPRE elements) in, among others, the promoters of genes encoding ERAD factors (Yoshida et al., 2001b; Lee et al., 2003b; Yoshida et al., 2003; Yamamoto et al., 2007). XBP1 can also bind to additional non-UPRE sequences and regulate genes involved in phospholipid biosynthesis (Sriburi et al., 2007), lipogenesis (Lee et al., 2008) and myogenic differentiation (Acosta-Alvear et al., 2007). Mice lacking XBP1 die prenatally due to liver defects (Reimold et al., 2000), and mice lacking IRE1α die even earlier with both liver and lymphocyte differentiation defects (Zhang et al., 2005) that might be secondary to placental failure (Iwawaki et al., 2009). Liver-specific rescue of XBP1 deficiency only postpones death into the neonatal period, when dysmorphogenesis of the exocrine pancreas leads to digestive failure (Lee et al., 2005).
PERK activation and autophosphorylation lead to eIF2α phosphorylation which, while having the immediate (and transient) effect of inhibiting the translation of most mRNAs, stimulates translation of the Atf4 mRNA due to the presence of upstream open reading frames (uORFs) in the Atf4 5' UTR (Lu et al., 2004; Vattem and Wek, 2004). This effect is a consequence of the inefficient ribosome assembly brought on by eIF2α phosphorylation, which allows certain mRNAs—2 to 8 percent by one estimate—to be translationally stimulated rather than inhibited, based on the presence of uORFs (Ventoso et al., 2012; Barbosa et al., 2013). Like XBP1, ATF4 is a bZIP transcription factor; it binds to amino acid response elements (AAREs) in target gene promoters. eIF2α can be phosphorylated by other kinases in response to various cellular stresses, and this pathway of signal transduction is known as the integrated stress response (ISR) (Harding et al., 2003). Perk−/− mice develop progressive postnatal diabetes and exocrine pancreas disruption (Harding et al., 2001).
Finally, ATF6 is an ER-localized transmembrane transcription factor. ER stress releases it from the ER to the Golgi, where it is cleaved by regulated intramembrane proteolysis (RIP) to liberate the transcriptionally active cytosolic domain—itself a bZIP family member—that dimerizes with the constitutive factors NFY and YY1 and binds to ERSE and ERSE-II sequences in target genes (Li et al., 2000; Ye et al., 2000; Yoshida et al., 2000; Kokame et al., 2001; Baumeister et al., 2005). ATF6 can also heterodimerize with XBP1 on UPRE sites (Yamamoto et al., 2007); indeed, the mammalian UPRE was first identified by virtue of its binding by ATF6 (Wang et al., 2000). Mice lacking ATF6 are overtly normal (Wu et al., 2007; Yamamoto et al., 2007), and no basal phenotype has yet been reported, although this surprising absence might be due to functional redundancy between ATF6 and its paralog ATF6β (discussed in more detail later).
The phenotypes of mice with constitutive deletions of UPR components mesh with the narrative of the UPR as a self-contained program for maintaining ER protein folding homeostasis. Where basal phenotypes are evident, disrupted cell types in the various animals show evidence of grossly altered ER structure and impaired secretory pathway function. In addition, constitutive deletion of Xbp1, Atf4, and Atf6 allowed the transcriptional programs downstream of each to be examined by microarray in the most convenient cell type, mouse embryonic fibroblasts (MEFs)—the cell type in which most of the basic pathways of the UPR were elucidated. Similarly to yeast, MEFs respond to ER stress with an upregulation of genes encoding ER chaperones and cochaperones, ERAD factors, lipid synthesis enzymes, and other proteins of importance to secretory pathway function and general protein biosynthesis. Subsets of these genes were found to depend on each of the three UPR-regulated bZIP transcription factors. Although there is considerable overlap in the sets of genes regulated by these factors, to a first approximation ATF4 coordinates the upregulation of genes involved in protein anabolism and redox defense (Harding et al., 2003), XBP1 appears to regulate ERAD (Lee et al., 2003b), and ATF6 contributes to upregulation of chaperones and ERAD factors (Wu et al., 2007; Adachi et al., 2008). For the purposes of this review, we shall refer to this mechanistic framework, culminating in production of XBP1, ATF4, and ATF6—along with the direct actions of these factors on target genes—as the canonical UPR, and it is indicated in green in the accompanying figure to highlight the non-canonical mechanisms that are the focus of this review (Figure 1).
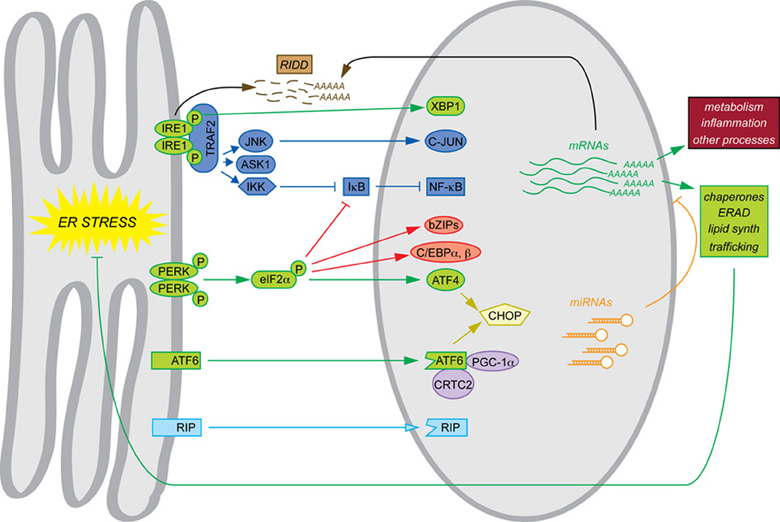
Figure 1. Canonical and non-canonical pathways of mRNA regulation. Examples of each of the pathways of mRNA regulation discussed in this review are shown. The canonical UPR pathways are shown in green. Also depicted are transltional regulation (red), scaffolding (dark blue), transcriptional cascades (yellow), cofactor titration (purple), alternate RIP substrates (light blue), RIDD (brown), and miRNAs (orange). Together, these processes result in the regulation not only of ER protein folding function (green) but also other cellular processes such as metabolism and inflammation (maroon).
A deeper look at these microarray studies reveals that the transcriptional output of the UPR is not so simple and self-contained. The emphasizing of the UPR as a program for transcriptional induction meant that the downregulated genes were not characterized in each of these analyses; yet between one quarter and one half of mRNAs regulated by ER stress are actually suppressed, with few mechanisms to account for them. In contrast to upregulated genes, the suppressed genes cluster among a number of cellular processes having apparently little to do with ER protein folding (Arensdorf and Rutkowski, 2013). Further, even among genes upregulated by ER stress, only a relatively small number can be tied definitively to ATF4, XBP1, and/or ATF6. The mechanisms responsible for regulation of the majority of genes during ER stress even in a presumptively “generic” cell type such as the MEF are not understood, and quite possibly fall outside the scope of the canonical UPR.
Temporal Dynamics of mRNA Regulation
There are four ways in which mRNA abundance might be regulated by the UPR: stimulation or inhibition of transcription, and enhancement or compromise of mRNA stability. The historical view of the UPR as a program for upregulation of ER chaperones has shone the most attention on the first mechanism, but the other three contribute as well. Indeed, they probably collectively contribute to a substantial fraction of the observed changes in mRNA expression upon ER stress, or perhaps even the majority of it. The mechanism by which an mRNA is regulated has implications for the timing and persistence of that event and, by extension, the window of time during which the protein product translated from that mRNA is able to influence cellular function.
To illustrate how the mode of regulation impacts the kinetics of mRNA expression, we provide here a simple computational model of a gene regulatory event, where the expression of a target gene is controlled by the expression of an upstream factor that can either stimulate transcription of the target gene, inhibit transcription, stimulate degradation of the target gene mRNA, or inhibit degradation (Figure 2A). The regulatory step is modeled as a non-cooperative interaction obeying simple Michaelis-Menten kinetics. Transcriptional effects depend upon the synthesis rate constant of the target transcript, the concentration of the upstream factor, and the affinity constant of that factor for its target gene. mRNA stability effects depend upon the same values, and also, as with any first-order decay process, on the concentration of the target mRNA itself. (The equations and parameters are given in the Supplemental Material).
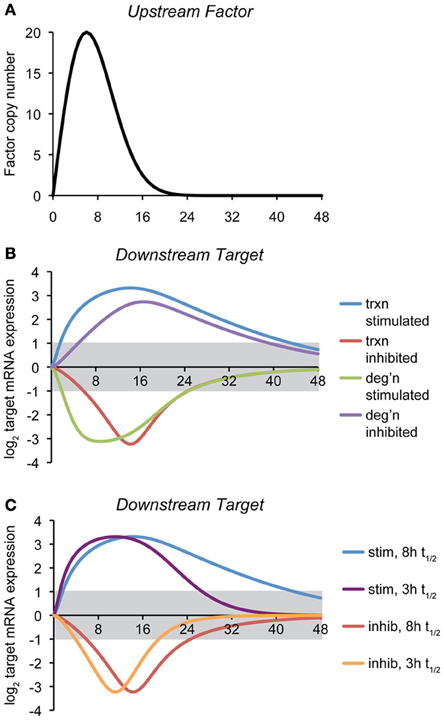
Figure 2. Temporal dynamics of mRNA regulation by different mechanisms. (A) A computational model was created in which the expression of a downstream target mRNA is directly controlled by the induction of an upstream factor, the expression of which is shown. The behavior of this factor is modeled loosely off of the dynamics of ATF4, ATF6, and XBP1 upon a level of ER stress to which cells can successfully adapt (Rutkowski et al., 2006). (B) Expression of the downstream target mRNA was modeled based on the upstream factor either stimulating or inhibiting transcription, or stimulating or inhibiting degradation, under conditions where maximal expression of the factor approaches the saturation level. For this simulation, the intrinsic (i.e., unregulated) half-life of target mRNA was chosen to be 8 h, and the parameters were then chosen to elicit 10-fold maximal regulation in expression, and varying the rate- and affinity-constants did not change the essential behavior of the model. The gray region indicates changes in mRNA levels that are less than two-fold (the threshold most frequently used to identify regulated genes in microarray experiments). The various curves illustrate two principles: (1) the window of time in which the expression of an mRNA will appear to be regulated (based on the two-fold criterion) depends on the mechanism of regulation, and (2) downregulation of mRNA either by inhibition of transcription or stimulation of degradation is necessarily shorter-lived than is upregulation, implying that changes in expression of downregulated genes might be easily overlooked. (C) The effect on transcriptional regulation of decreasing the half-life of the target mRNA form ~8 to ~3 h is shown. While the median half-life of cellular mRNA is 8–9 h, many of those encoding transcription factors have shorter half-lives (Schwanhausser et al., 2011). This includes that of Chop, which is itself transcriptionally regulated by the UPR.
The point of this exercise is to illustrate how—all other variables being held constant—the mechanism of mRNA regulation influences the rapidity with which the regulation is executed and its persistence. Thus, the hypothetical scenario shown here proposes that the action of the upstream factor has an effect on the target mRNA that results in its regulation (either up- or down-) by approximately ten-fold at its peak. With that stipulation, it then tests how the expression of an mRNA with an otherwise fixed rate of synthesis and rate of degradation behave in response to the expression of the upstream factor.
For this particular example, we modeled the response so that its peak effect was close to its saturation level, but the results were similar when the association/dissociation rate constants were varied over a wide range. This analysis reveals two salient features that are largely independent of the actual parameters (rate constants and affinity constants) chosen: First, mRNA levels can be more rapidly altered by stimulatory processes (of either transcription or degradation) than by inhibitory ones. Second, downregulated genes as a group return to basal expression levels more rapidly than do upregulated genes (Figure 2B). Further, the stability of the target mRNA influences the window of time when its expression is regulated, with shorter half-lives causing mRNAs to more directly mirror the expression of the upstream controlling factor (Figure 2C). In other words, while factors intrinsic to an mRNA (its synthesis and degradation rate constants) influence the window of time in which the mRNA is expressed, “stretching” its expression curve to the right or left, the extrinsic mode of regulation determines whether the expression of an mRNA experiences a lag either as stress is first experienced (i.e., if transcription or degradation is inhibited) or when the response begins to resolve (i.e., when transcription or degradation is stimulated). These observations imply that varied mechanisms for mRNA regulation ensure that the UPR is a dynamically evolving amalgam of outputs rather than a single output that simply varies in intensity over time. Practically, they also imply that the timing of mRNA regulatory events will reveal important clues about the mechanisms responsible.
Non-Canonical Pathways of Transcriptional Regulation
Non-canonical transcriptional outputs may arise as offshoots of the framework of the canonical UPR by three general mechanisms: (1) UPR pathway branching that results in the production of additional transcriptional regulators; (2) transcriptional cascades that expand the repertoire of targeted genes; and (3) alteration of the activity of constitutively expressed transcription factors through competition or cooperativity in the nucleus. In addition, UPR transcriptional output can be enhanced by the existence of parallel non-canonical stress-sensing pathways. Below, we provide examples of each of these modes of transcriptional regulation.
Non-Canonical Regulation Emanating From Canonical Pathways
The PERK and IRE1α cascades of the UPR in particular present multiple points at which additional signaling cascades could be initiated. First, both molecules are kinases, raising the possibility that other substrates exist. Both also self-associate during activation (Bertolotti et al., 2000), potentially forming stress-dependent scaffolds that can seed the assembly of signaling modules and that culminate in transcriptional regulation. In addition, the effects of eIF2α phosphorylation on protein synthesis potentially allow for the production of transcription factors in addition to ATF4.
Translational regulation
Phosphroylation of eIF2α initially suppresses the translation of 90% of cellular mRNA, which decreases to 50% within the first 3 h of stress (Ventoso et al., 2012). The effect of this suppression on the expression level of a given protein depends on the half-life of that protein and the duration of eIF2α phosphorylation, which is regulated by both constitutive (CreP) and inducible (GADD34) phosphatases (Jousse et al., 2003; Marciniak et al., 2004). Accordingly, the expression of proteins with short half-lives diminishes more rapidly than does that of long-lived proteins, as demonstrated initially for the cell cycle regulator Cyclin D1, the loss of which upon eIF2α phosphorylation leads to cell cycle arrest (Brewer et al., 1999). Translational inhibition offers the opportunity to transiently alter the composition of the transcription factor network based on the expression of both the factors themselves and upstream proteins that regulate transcriptional cascades. While transcription factors as a class tend to have short half-lives, the range of their half-lives nonetheless spans an order of magnitude or more, meaning that inhibition of protein synthesis will have a more pronounced effect on the protein levels of some transcription factors than others (Schwanhausser et al., 2011, 2013).
UPR activation converges on inflammatory signaling in part through translation-dependent regulation of NF-κB. Members of the NF-κB/Rel family of transcription factors (i.e., NFKB1, NFKB2, c-REL, RELA, RELB) dimerize to form the NF-κ B transcriptional complex. The transcriptional activity of this complex is determined by its composition (Elsharkawy et al., 2010); and the complex regulates the transcription of genes involved in immunoregulation, growth regulation, inflammation, carcinogenesis and apoptosis (Hoesel and Schmid, 2013). NF-κB is sequestered in the cytoplasm by IκB; in order for NF-κB to be activated, the inhibitory subunit must be removed through phosphorylation and degradation (Ahn and Aggarwal, 2005).
PERK—through the phosphorylation of eIF2α—affects NF-κ B signaling by suppressing the translation of Iκ B family members (i.e., NFKBIE, NFKBIB), which have a shorter half-life than NF-κ B/Rel family members (Jiang et al., 2003; Deng et al., 2004). Thus, eIF2α phosphorylation decreases the amount of IκB relative to NF-κB. In addition, the translation of certain NF-κ B/Rel family members is suppressed during ER stress (i.e., REL, RELA, RELB) while others are not (i.e., NFKB1, NFKB2) (Ventoso et al., 2012), suggesting that eIF2α phosphorylation might not simply stimulate NF-κB activity, but might instead regulate the formation of specific NF-κ B complexes. The physiological significance of NF-κ B activation during ER stress is unknown; however, a number of NF-κ B target genes (Pahl, 1999) are found among ER stress-regulated genes, pointing to a contribution of NF-κB to UPR transcriptional output.
While NF-κB activity appears to be regulated passively by eIF2α phosphorylation through simple loss of an unstable inhibitor, translational control is also used to stimulate the translation of specific proteins, including several transcription factors beyond ATF4. Indeed, while the majority of mRNA translation is suppressed by eIF2α phosphorylation, the translation of ~2–8% of cellular transcripts is increased (Ventoso et al., 2012). Among the transcripts whose translation is stimulated are a number of transcription factors, including several among the bZIP family in addition to ATF4. These include ATF5 (Zhou et al., 2008), ATF3, CHOP, JUN, JUNB, FOS, FOSB, and CREB1 (Ventoso et al., 2012). How the translation of these transcripts is regulated during conditions of eIF2α phosphorylation is an area of active research; however, the presence of alternative uORFs within these transcripts is thought to dictate their translation during ER stress (Morris and Geballe, 2000).
In the case of the constitutively expressed transcription factors C/EBPα and C/EBPβ, translational control allows for the production of a truncated inhibitory form at the expense of a full-length activating form. C/EBP family members are bZIP transcription factors that participate in diverse physiological processes including differentiation, proliferation, metabolism, and inflammation (Ramji and Foka, 2002). Both α and β forms possess a cluster of potential translation initiation sites that are highly conserved among mammals. One of the start codons within this cluster is out of frame with respect to the others, and introduces a short open reading frame just upstream of the start codons that initiate translation of the full-length forms of α and β. The presence of this uORF is essential for translation of the truncated forms of both proteins, which is stimulated by eIF2α phosphorylation (Calkhoven et al., 2000; Wethmar et al., 2010). The truncated versions of both proteins have intact DNA binding domains but lack transactivation domains, making them potentially dominant-negative inhibitors of transcription (Descombes and Schibler, 1991).
Like Atf4, Cebpa, and Cebpb mRNAs both have uORFs that regulate the translation of alternate isoforms, but their expression appears to be controlled by a somewhat different mechanism. While the mechanism of C/EBPα translational regulation by eIF2α phosphorylation is still somewhat unclear, the expression of the long (aka Liver Activating Protein or LAP) and short (Liver Inhibiting Protein or LIP) forms of C/EBPβ are regulated by both eIF2α phosphorylation and dephosphorylation. Synthesis of the LIP form of C/EBPβ is translationally inhibited by eIF2α phosphorylation, with levels diminishing starkly because of its short half-life (Li et al., 2008). However, translational recovery promotes both increased synthesis and increased stability of the LIP form, causing its expression to predominate over that of LAP at later times after the induction of ER stress (Li et al., 2008; Arensdorf and Rutkowski, 2013). Thus, C/EBPβ, and probably C/EBPα as well, are more indirectly tied to eIF2α phosphorylation than is ATF4.
Translational regulation of both C/EBPα and C/EBPβ has been linked to diminished lipogenesis and improved glucose tolerance in mice fed a high-fat diet (Oyadomari et al., 2008). Mice with liver-specific overexpression of a constitutively active fragment of GADD34 that were raised on a high fat diet were resistant to weight gain and showed enhanced insulin sensitivity and reduced hepatic triglyceride accumulation. The reduced steady-state phosphorylation of eIF2α seen in these animals corresponded with lower expression of both C/EBPα and C/EBPβ (only the long forms of each were studied) and their downstream target genes, including the lipogenic mediator PPARγ. While a direct role for C/EBPα and β was not tested in GADD34-overexpressing mice, these findings illustrate a potentially significant physiological consequence of non-canonical ER stress-mediated transcription—namely, that eIF2α phosphorylation promotes hepatic lipid accumulation through enhanced or altered synthesis of C/EBPα and β.
C/EBPβ regulates inflammatory cascades in diverse cell types; it is alternatively named NF-IL6 based on its ability to regulate expression of the pro-inflammatory cytokine IL-6 (Akira et al., 1990). Preferential production of the LIP form of C/EBPβ during ER stress in cultured cells was found to result in transcriptional repression of a number of inflammatory genes, including IL4RA, which is an essential component of the IL-4 and IL-13 receptors (Arensdorf and Rutkowski, 2013). Most cell types express either the IL-4 or IL-13 receptor, and signaling through IL4RA stimulates pro-inflammatory processes as diverse as B cell proliferation, IgE class-switching, TH2 cell differentiation, smooth muscle contraction, mucus hypersecretion, eosinophil requirement, fibrotic deposition, and chemokine expression (Hershey, 2003; Wynn, 2003; Wills-Karp and Finkelman, 2008; Holgate, 2011). Remarkably, while ER stress suppressed IL-4/IL-13-dependent downstream signaling, this suppression was lost in Cebpb−/− cells, indicating that the translational regulation of C/EBPβ can influence responsiveness to inflammatory signals. This finding suggests that UPR or ISR activation could influence the natural history of parasitic infections and allergic responses, both of which are accompanied by extensive IL-4 and IL-13 signaling (McKenzie, 2000).
Scaffolding
Both PERK and IRE1α self-associate during ER stress (Bertolotti et al., 2000). For yeast Ire1p, this self-association leads to the formation of oligomeric Ire1p clusters (Korennykh et al., 2009). While the dynamics of self-association for mammalian IRE1α and PERK are less understood, it seems likely that, here too, higher ordered multimeric complexes of both proteins form, resulting in the creation of potential stress-specific scaffold supports on the cytosolic face of the ER membrane (Li et al., 2010). IRE1α can seed the formation of cytosolic signaling modules, at least some of which culminate in transcriptional regulation (Hetz and Glimcher, 2009).
Among the proteins recruited to phosphorylated and oligomerized IRE1α is TNF receptor-associated factor 2 (TRAF2) (Urano et al., 2000). This association is thought to be required for activation of several kinases including IKK, JNK, ASK1, p38 MAPK, and ERK that contribute to cell fate decisions (i.e., survival vs. apoptosis) during ER stress (Urano et al., 2000; Nishitoh et al., 2002; Nguyen et al., 2004; Hu et al., 2006; Li et al., 2010; Tam et al., 2012). For IKK regulation, the subsequent activation of NF-κB (IKK phosphorylates IκB, leading to its degradation) was found to lead to induction of TNF-α expression (Hu et al., 2006). Likewise, JNK activation leads to phosphorylation of the bZIP transcription factor C-JUN (Urano et al., 2000), which has been implicated in ER stress-mediated regulation in cultured neurons of the gene encoding methylenetetrahydrofolate reductase, which participates in folate and homocysteine metabolism (Leclerc and Rozen, 2008), and of Gpt1 and Got1 in the liver, which encode the liver enzymes AST and ALT that are released from the liver upon damage (Josekutty et al., 2013). These examples notwithstanding, however, the contributions of IRE1α/TRAF2-dependent signaling to the sum of ER stress-mediated transcriptome control are poorly understood.
PERK also has the potential to recruit other molecules upon autophosphorylation and oligomerization, but there are fewer known parallel pathways arising from PERK activation than from IRE1α. PERK activation is necessary and sufficient for phosphorylation of the transcription factor NRF2 [Nuclear Factor (Erythroid-Derived 2)-Like 2] and can phosphorylate NRF2 in vitro (Cullinan et al., 2003). Cells lacking PERK or NRF2 do not effectively upregulate the NRF2-target genes Gclc or Nqo1 upon ER stress (Cullinan and Diehl, 2004). PERK was also found to be required for activation of MAP kinase- and phospholipase C-dependent gene expression in response to ER calcium depletion (Liang et al., 2006a). However, as with IRE1α-dependent signaling modules, the global contribution of these pathways to mRNA regulation during ER stress is not clear.
Transcriptional Cascades
The output of the UPR is dramatically expanded by hierarchically arranged gene regulatory networks, in which the expression of subordinate transcription factors is targeted for regulation by the canonical UPR factors. Beyond conferring stress-responsiveness to genes not directly bound by ATF4, XBP1, or ATF6, this expansion also allows for transcriptional suppression, to the extent that ATF4, XBP1, or ATF6 enhance the expression of repressive transcription factors.
The best-characterized example of a secondary UPR-regulated transcription factor is C/EBP Homologous Protein (CHOP). CHOP is a direct target of ATF4 and ATF6 (Ma et al., 2002), and its translation is stimulated by eIF2α phosphorylation (Palam et al., 2011). Phosphorylation of CHOP by p38 MAP kinase appears to be required for its full activity (Wang and Ron, 1996). A member of the C/EBP family of transcriptional regulators, CHOP can form heterodimers with other C/EBP proteins, and was proposed to act as a dominant-negative inhibitor of C/EBPα and β in particular (Ron and Habener, 1992). It is now clear that CHOP possesses both activating and repressing potential, and so its effect on the transcriptome is complex.
CHOP is strongly functionally associated with cell death; both cells and animals lacking CHOP are protected from a diverse array of stressful stimuli (Zinszner et al., 1998; Oyadomari and Mori, 2004). Due at least in part to the very short half-life of both the protein and its mRNA, CHOP expression is strongly correlated with the ER stress burden in real-time (Rutkowski et al., 2006). Hence, the window of time in which CHOP can exert a direct effect on the transcriptome is tightly controlled, as one might expect a priori for a factor that potentiates cell death. As a transcriptional regulator rather than a conventional pro-apoptotic effector, CHOP promotes cell death through the regulation of several classes of downstream genes. CHOP has been proposed to regulate the expression of both anti-apoptotic and pro-apoptotic genes of the Bcl2 family (McCullough et al., 2001; Puthalakath et al., 2007). In addition, CHOP regulates expression of the bZIP factor ATF5, which is itself translationally regulated by eIF2α phosphorylation (Watatani et al., 2008; Zhou et al., 2008), and the targets of ATF5 include the pro-apoptotic protein NOXA (Teske et al., 2013). However, ChIP-seq analysis revealed that its direct targets are most prominently enriched for genes involved in protein synthesis, which are co-regulated by ATF4 (Han et al., 2013). Among these genes is GADD34, which indicates that CHOP controls a negative feedback loop allowing for the dephosphorylation of eIF2α and resumption of protein synthesis, even if the ER is ill-equipped to handle nascent protein influx (Marciniak et al., 2004). A consequence of restored protein synthesis is increased production of reactive oxygen species (ROS), likely resulting from oxidative protein folding in the ER; indeed, CHOP also regulates expression of the ER oxidase ERO1α (Li et al., 2009), indicating that CHOP promotes oxidative folding in tandem with increased ER influx. Blunting either ROS accumulation or protein synthesis neuters CHOP's pro-apoptotic potential (Marciniak et al., 2004; Malhotra et al., 2008; Li et al., 2009).
The strong association of CHOP with cell death in both cell and animal models raises the question of whether CHOP is intrinsically apoptotic, or instead whether this role is only manifested in the context of severe stress, masking other roles for CHOP in maintaining normal physiologic homeostasis. Indeed, the restoration of protein synthesis following stress is essential to maintain vital cellular functions, and the existence of a constitutive phosphatase ensures that even Chop−/− or Gadd34−/− cells are able to resume protein synthesis (Harding et al., 2009; Tsaytler et al., 2011); CHOP, therefore, merely accelerates the process, and it is possible that the kinds of stresses encountered in normal (i.e., non-pathologic) physiology are sufficiently mild that the benefits to cellular function of restoring protein synthesis outweigh the cost of increased ROS production. In addition, while CHOP induction is largely suppressed in some instances of physiological UPR induction such as B lymphocyte differentiation (Gass et al., 2002) and toll-like receptor ligation (Woo et al., 2009), it occurs in others such as feeding after a fast (Pfaffenbach et al., 2010).
CHOP likely contributes directly to the suppression of several metabolic transcriptional regulators during ER stress in the liver (Chikka et al., 2013). This finding suggests that CHOP might serve a role in regulating lipid metabolism in vivo, which is consistent with the steatosis observed in Chop−/− mice (Maris et al., 2012). To the extent that CHOP (or other secondary stress-regulated transcription factors) regulates metabolic transcription factors such as Cebpa, Ppara, and Srebf1c, there exists a multistep gene regulatory network during ER stress in hepatocytes that culminates in changes in the expression of genes encoding rate-limiting enzymes of intermediary metabolism (Rutkowski et al., 2008). CHOP also promotes inflammation (Maris et al., 2012; DeZwaan-McCabe et al., 2013; Malhi et al., 2013), although whether this is a consequence of direct CHOP action on inflammatory genes, or instead a secondary consequence of the other functions of CHOP is not yet clear.
The actions of CHOP on the transcriptome and on the accompanying physiological processes highlight the ability of the UPR to expand its reach through the regulation of secondary transcription factor expression, but CHOP is certainly not the only transcription factor whose transcription is regulated by ER stress. In fact, a search through several published microarrays from ER stress-treated MEFs (Marciniak et al., 2004; Wu et al., 2007; Rutkowski et al., 2008) reveals expression changes for several dozen transcription factors and cofactors, including both activators and repressors (Table 1). It follows that such transcriptional cascading will create a temporal hierarchy of gene regulation, with the earliest regulated genes being most proximally connected to UPR pathways. Such cascading likely contributes to the regulation of metabolic genes in the liver [(Arensdorf et al., 2013) this issue].
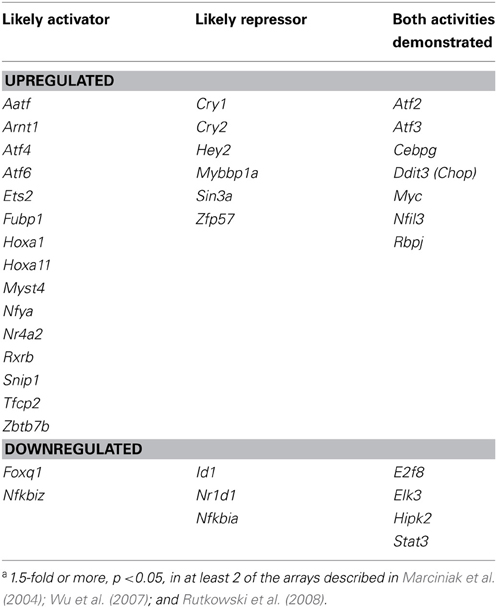
Table 1. Transcription factors and cofactors whose mRNA expression is regulated by the UPRa.
Heteromeric Interactions and Cofactor Titration
The major transcription factors of the UPR, both canonical and secondary, are bZIPs (ATF4, ATF6, XBP1, CHOP, JUN, ATF3, ATF5). The dozens of members of this family form homotypic and heterotypic dimers, typically within functionally related subclasses (Vinson et al., 2006). Thus, the complement of genes regulated during ER stress can be influenced by the formation of novel regulatory complexes not possible when the UPR is inactive, containing one UPR-regulated member and one constitutively expressed member. For instance, as a C/EBP family member, CHOP can interact with C/EBPα and C/EBPβ, altering the transactivation potential of each of these (Fawcett et al., 1996; Ubeda et al., 1996; Chiribau et al., 2010). Likewise, ATF6 was recently shown to interact with C/EBPβ upon stimulation with the inflammatory cytokine IFN-γ to transcriptionally upregulate the autophagy-promoting gene Dapk1 (Gade et al., 2012).
Stress-regulated transcription factors can also influence global gene expression beyond the genes they directly regulate through the titration of coregulatory molecules shared with constitutive transcription factors. Such a mechanism was demonstrated recently for the coactivating factor CRTC2, which was shown to lose its costimulatory interaction with the gluconeogenic bZIP transcription factor CREB in favor of an interaction with ATF6 (Wang et al., 2009). The consequence of this titration was inhibition of hepatic gluconeogenesis during acute ER stress, and gluconeogenesis and hyperglycemia could be suppressed in diabetic animals by ATF6 overexpression.
ATF6 has also been shown to interact with the coactivator PGC-1α. In skeletal muscle, this interaction promoted the full upregulation of canonical UPR target genes upon exercise (Wu et al., 2011). Likewise, a PGC-1α/ATF6 interaction stimulated expression of the ERRγ orphan nuclear receptor/transcription factor in a hepatocyte cell line (Misra et al., 2013). Roles for ERRγ in glucose and alcohol metabolism in the liver have recently emerged (Kim et al., 2012, 2013). PGC-1α has been implicated in the transcriptional regulation of many key metabolic processes including gluconeogenesis, fatty acid oxidation, and mitochondrial biogenesis, based on its ability to coactivate a number of transcription factors (Lin et al., 2005). Therefore, it is possible that ER stress will disrupt or otherwise influence the regulation of such gene networks based on competition for PGC-1α binding, although this has not yet been demonstrated.
Expansion of UPR Signaling Pathways
The status of PERK, IRE1, and ATF6 as canonical UPR regulators arises from the primacy of their early discoveries and their ubiquity. However, a number of other stress-signaling pathways have since been discovered that extend the scope of the UPR in both general and context-specific ways.
While IRE1α is ubiquitously expressed, its paralog IRE1β, expressed in mucin-producing cells of the gut (Bertolotti et al., 2001) and airway (Martino et al., 2013), was identified around the same time (Wang et al., 1998). Like its paralog, IRE1β can catalyze the splicing of Xbp1 (Calfon et al., 2002). Mice lacking IRE1β are sensitive to colitis induced by dextran sodium sulfate challenge (Bertolotti et al., 2001), as are animals lacking XBP1 (Kaser et al., 2008), ATF6, or the ER cochaperone p58IPK/ERDJ6 (Cao et al., 2013). The phenotypic similarities among these animals suggests that, like its paralog, IRE1β contributes to ER homeostasis largely through Xbp1 splicing and upregulation of the ER folding and quality control machinery. However, the endonuclease domain of IRE1β displays less activity toward Xbp1 than does that of IRE1α and has an enhanced specificity for 28s rRNA, which can contribute to suppression of protein synthesis (Imagawa et al., 2008). In addition, in contrast to Ire1α−/− animals, Ire1β−/− mice showed elevated expression of mucin 2 mRNA, impaired MUC2 secretion, and exacerbated ER stress, including, paradoxically, increased Xbp1 splicing (Tsuru et al., 2013). These findings suggest that IRE1α and β have at least partially separable functions, and raise the question of how strong a contribution IRE1β makes to mRNA regulation in the cells where it is expressed.
IRE1β aside, the reach of the UPR has been most expanded by the discovery of ER localized proteins that, like ATF6, are activated by regulated intramembrane proteolysis. First among these was a paralog of ATF6 known as ATF6β (ATF6 is also known as ATF6α) that is 36 percent identical to ATF6 over 93 percent of its length (Haze et al., 2001). Cells lacking ATF6β show no apparent defect in upregulation of canonical UPR target genes (Yamamoto et al., 2007). However, like ATF6, ATF6β binds to ERSE sequences in conjunction with NFY (Yoshida et al., 2001a). Mice lacking both ATF6 and ATF6β die during embryogenesis (Yamamoto et al., 2007), as do similarly manipulated medaka fish (Ishikawa et al., 2012). Overexpression of Bip could partially rescue impaired notochord development in these fish, suggesting that ATF6 and ATF6β converge on chaperone mRNA regulation, albeit with somewhat different kinetics (Haze et al., 2001). Whether ATF6β regulates the expression of any unique genes is not yet known.
In addition to ATF6 and ATF6β, there are at least 5 additional ER-resident transmembrane bZIP transcription factors that are cleaved by RIP, including CREBH, Luman, OASIS, BBF2H7, and CREB4 [reviewed in (Asada et al., 2011)]. These 5 proteins are not highly homologous to each other or to ATF6, and each is expressed in a unique subset of tissues, but they are all known or thought to be cleaved by S1P (Raggo et al., 2002; Murakami et al., 2006; Stirling and O'Hare, 2006; Zhang et al., 2006). These proteins can be activated by conventional ER stress and/or can regulate the expression of UPR target genes through traditional ER stress-responsive cis-acting sequences (Kondo et al., 2005; Liang et al., 2006b; Stirling and O'Hare, 2006; Zhang et al., 2006; Kondo et al., 2007). However, they may be activated more strongly by physiological signals that influence ER homeostasis in ways other than the simple perturbation of or excess demand upon the protein folding machinery, and might also be retained in the ER by distinct mechanisms; at least CREBH appears to be retained by virtue of its cytosolic membrane-proximal segment rather than by lumenal Bip binding as for ATF6 (Llarena et al., 2010). In addition, they also appear to regulate expression of distinct sets of genes, suggesting that they diversify the responsiveness of the UPR and the scope of genes that it regulates in various tissues rather than simply augmenting these processes.
The predominantly liver-specific RIP substrate CREBH illustrates the complex relationship between these substrates and the canonical UPR. Crebh mRNA is upregulated by conventional ER stress in the liver, and cleavage of the protein is modestly stimulated as well; the cleaved form is capable of associating with ATF6 and upregulating UPRE- or ERSE-dependent reporters (Zhang et al., 2006). However, both its expression and cleavage are also strongly induced by inflammatory stimuli such as LPS exposure or IL-6 treatment, and the genes encoding the inflammatory modulators CRP and SAP were also identified as likely CREBH targets. While inflammatory stimuli such as LPS appear capable of inducing ER stress, they spawn a modified eIF2α-independent response (Woo et al., 2009), suggesting that, even if CREBH is activated simply by the accumulation of unfolded proteins in the ER, it would be so in the context of a modified UPR. CREBH also regulates the expression of hepicidin, leading to dysregulation of iron homeostasis upon ER stress in wild-type mice but not Crebh−/− animals (Vecchi et al., 2009). Therefore, CREBH can also contribute to the expansion of mRNA expression even during exposure to a conventional ER stressor. More recently, CREBH was shown to directly regulate genes involved in gluconeogenesis (Lee et al., 2010) and lipid metabolism (Zhang et al., 2012). The latter of these processes showed a stronger CREBH dependence in the context of a high-fat atherogenic diet, raising the question of the extent to which physiological stimuli such as obesity impact gene expression through the canonical UPR vs. through pathways that enlist the action of molecules like CREBH to produce a unique response.
Knockout of each of the RIP substrates [or, in the case of Luman, a regulator of the pathway, LRF; (Martyn et al., 2012)] yields a discrete phenotype attributable to dysfunction of the major tissue in which the substrate is expressed (Asada et al., 2011). In contrast, mice lacking ATF6, which have compromised ER chaperone induction upon ER stress, show no apparent basal phenotype (Wu et al., 2007; Yamamoto et al., 2007). Therefore, simple failure to upregulate ER chaperones to the maximum extent is not sufficient to elicit a phenotype, making it unlikely that the RIP substrates merely augment chaperone induction; more likely, the phenotypes induced by their deletion are attributable to non-redundant actions on specific genes, be they chaperones or something else entirely. However, their activation signals and the global effects on transcriptome regulation remain unclear.
mRNA Stability
Although each UPR pathway culminates in production of a transcription factor, mRNA abundance can just as readily be regulated by enhanced or diminished stability. Indeed, one estimate of the relative contribution of transcriptional and post-transcriptional mechanisms to mRNA abundance during ER stress—based on the comparison of mRNA levels in nuclear run-off assays against total mRNA pools—suggested that ~75 percent of mRNAs were regulated at least in part at the level of stability (Kawai et al., 2004). Pathways linking ER stress to mRNA stability are much less understood than transcriptional mechanisms, but are emerging as important influences on UPR output and physiological responses.
Gene Regulation by RIDD
A subset of ER-localized mRNAs are degraded directly by the endonuclease activity of IRE1 in a process called regulated IRE1-dependent decay (RIDD). The process was first observed in Drosophila cells, in which a subset of mRNAs was rapidly suppressed by ER stress in an IRE1α-dependent but XBP1-independent manner (Hollien and Weissman, 2006). This group of mRNAs was highly enriched for those encoding proteins with in-frame ER targeting sequences, but the cleavage of these mRNAs was otherwise non-sequence-specific. It is possible that RIDD represents an ancient activity of IRE1, since Ire1p in the fission yeast S. pombe catalyzes a RIDD activity but not a HAC1 splicing activity (Kimmig et al., 2012). More recently, a sequence necessary for mRNA cleavage by IRE1α in vitro was identified (Oikawa et al., 2010), although it is not yet clear whether these findings extend to RIDD targets in vivo as well. These findings led to the idea that activated IRE1α directly cleaves some mRNAs that are brought to proximity with the ER membrane by virtue of their association with translating ribosomes synthesizing signal peptide- or signal anchor-encoding proteins. This finding was subsequently extended to mammalian cells (Han et al., 2009; Hollien et al., 2009). The RIDD and Xbp1 splicing activities of IRE1α are functionally separable (Han et al., 2009; Hollien et al., 2009), implying that each process plays a distinct role in UPR-mediated control over the transcriptome. Where these functions overlap—i.e., in the expression of ER chaperones and other ER-localized proteins that facilitate recovery from stress but whose mRNAs are RIDD targets by virtue of their localization—the transcriptional induction must be sufficient to overcome degradation by RIDD.
Although the RIDD pathway has not yet been as well characterized in mammalian cells, its targets in that context are involved in processes ranging from signaling cascades (e.g., Pdgfrb, Efnb2, Ncam1, Raptor) to transcription (e.g., Pbxip1, Hoxb4, Srsf3) to lysosomal degradation (e.g., Bloc1s1, Tpp1, Hgsnat) to xenobiotic metabolism (Cytochrome p450s-encoding genes) and energy production (e.g., Oxct1) (Hollien et al., 2009; Hur et al., 2012). RIDD appears now not to be solely limited to genes encoding ER-translocated proteins but includes mRNAs encoding cytosolic factors as well (Hollien et al., 2009; Oikawa et al., 2010; Ventoso et al., 2012). This ability of the ER-tethered IRE1α to degrade mRNAs encoding cytosolic proteins might arise from the localization of specific cytosolic mRNAs in the process of translation to the ER membrane (Stephens et al., 2005); indeed, localization of an mRNA to the ER membrane, irrespective of whether it encodes a protein of the endomembrane system, appears sufficient to target that mRNA for RIDD for the large majority of mRNAs, at least in insect cells (Gaddam et al., 2012). In addition, even mRNAs not stably associated with the ER membrane can still be targeted for RIDD if they contain an Xbp1-like stem-loop structure that allows them to associate directly with IRE1 (Moore et al., 2013).
Irrespective of whether RIDD acts on specific mRNAs or more generally on most of those associated with the ER membrane, its activation has distinct physiological consequences. IRE1β (but not α) might control efflux of absorbed lipids through its RIDD activity. Lipids absorbed from the diet are packaged by enterocytes into lipoprotein particles known as chylomicrons, and a key step in this packaging is lipidation of apolipoproteins in the ER by microsomal triglyceride transfer protein (MTTP) (Hussain, 2000). IRE1β was found to cleave Mttp mRNA, and Ire1β−/− mice on a high fat diet had elevated MTTP expression, elevated chylomicron production, and hyperlipidemia (Iqbal et al., 2008). The RIDD pathway also can regulate lipid metabolism in the liver; IRE1α-dependent degradation of genes involved in lipogenesis and lipoprotein synthesis was elicited as a feedback mechanism in mice lacking XBP1 (So et al., 2012). An analogous pathway of feedback elicited IRE1α-dependent suppression of proinsulin processing in pancreatic β cells of Xbp1−/− mice (Lee et al., 2011). These findings are consistent with the idea that RIDD is most active on ER-localized mRNAs, since lipoprotein formation, lipogenesis, and proinsulin processing all take place in the ER or at the ER membrane. A conservative estimate puts the frequency of RIDD targets at ~5 percent of all ER stress-regulated mRNAs in cultured mammalian cells (Hollien et al., 2009), and it might in fact be substantially higher (Gaddam et al., 2012). Whether RIDD is more or less active during physiological stimuli in vivo and whether its specificity for certain groups of substrates can be meaningfully regulated are not yet understood.
mRNA Regulation by miRNAS
Another common source of mRNA regulation occurs through microRNAs (miRNAs). miRNAs are short (~22 nt) single-stranded RNAs which bind to complementary mRNAs and promote their degradation or, less frequently, inhibit their translation (Valencia-Sanchez et al., 2006). The UPR-mediated regulation of miRNA is a rapidly emerging area of investigation and a potential mechanism for fine-tuning of mRNA abundance. The pathways leading from UPR activation to miRNA regulation and the consequences of this regulation for mRNA abundance and downstream cellular processes have been reviewed extensively in (Maurel and Chevet, 2013), to which we direct the reader for details. Each of the three canonical UPR pathways transcriptionally regulates the expression of discrete miRNAs (Bartoszewski et al., 2011; Belmont et al., 2012; Byrd et al., 2012; Chitnis et al., 2012; Gupta et al., 2012). To date, the best-described functions of ER stress-regulated miRNAs are in tuning UPR sensitivity (Byrd et al., 2012; Maurel et al., 2013; Zhang et al., 2013) or in regulating cell proliferation and apoptosis during stress (Chitnis et al., 2012; Duan et al., 2012; Gupta et al., 2012; Muratsu-Ikeda et al., 2012).
As an emerging area of study, the physiological roles of ER stress-mediated miRNA regulation are largely unknown; however, it was recently demonstrated that IRE1α activation causes degradation of miR-17, which in turn leads to upregulation of the miR-17 target mRNA encoding thioredoxin-interacting protein (TXNIP) (Lerner et al., 2012). TXNIP promoted inflammasome assembly, caspase-1 activation, and cell death, and Txnip−/− mice were protected from pancreatic β cell death induced by production of misfolded insulin. Given the ability of miRNAs to coordinately regulate the stability of many target mRNAs, it seems likely that this mechanism will emerge as a major contributor to noncanonical UPR output, and that further physiological roles will be discovered.
ER stress has been associated with changes in the stability of individual mRNAs through undetermined mechanisms (Pereira et al., 2010; Park et al., 2012), and so it is possible that other pathways for regulating mRNA turnover exist as well. One suggested mechanism is the sequestration of translationally inhibited mRNAs in stress granules (Kimball et al., 2003). mRNA stabilization through sequestration would thus decouple mRNA abundance from protein abundance (Kawai et al., 2004). This possibility illustrates the caution that must be exercised when interpreting changes in mRNA abundance in general: mRNA and protein expression are only loosely correlated (Gygi et al., 1999), and understanding the mechanisms of mRNA regulation by the UPR only illuminates one component in the regulation of gene expression.
A Way Forward: Dissecting UPR-Responsive Gene Regulatory Networks
The abundance of non-canonical mechanisms of mRNA regulation by the UPR suggest that the number of mRNAs whose regulation is attributable to direct binding by XBP1, ATF4, or ATF6 is likely to represent only a small portion of all the regulated genes. How, then, can the complex gene regulatory network of the UPR be best studied moving forward? The explosion of high throughput methodology and increasingly sophisticated bioinformatic tools holds promise for both “top-down” and “bottom-up” approaches to this problem. The application of microarray technology to the yeast UPR first revealed the complexity of the transcriptional response (Travers et al., 2000); similar microarray-based approaches (Harding et al., 2003; Lee et al., 2003b; Wu et al., 2007) in mammals revealed the dependencies of subsets of genes on each UPR pathway, but could not separate direct from indirect influences. Next-generation sequencing methodologies, including mRNA-seq and ChIP-seq, will now be used to piece together regulatory hierarchies; these techniques were recently combined to elucidate the gene networks regulated by XBP1 (Acosta-Alvear et al., 2007) and CHOP and ATF4 (Han et al., 2013). A complementary approach will be to find groups of genes that are coordinately regulated and use bioinformatic analysis to predict previously hidden upstream regulators. As proof-of-principle, we have shown that the temporal organization of metabolic gene regulation upon ER stress in the liver identifies the transcription factor HNF4α as a key link between UPR activation and the expression of genes involved in lipid metabolism [(Arensdorf et al., 2013); this issue]. Such functional genomics approaches have until recently been restricted to studies in simple organisms like yeast. However, the ability to probe and experimentally manipulate the entire mammalian genome has now made these techniques feasible in higher eukaryotes as well (Kampmann et al., 2013), and these approaches have been used to understand secretory pathway function (Bassik et al., 2013) and the ERAD network (Christianson et al., 2011).
Although best known as the gateway to the secretory pathway, the ER participates in many cellular processes that have little or nothing to do with protein folding per se. While augmentation of the ER protein folding capacity is certainly a significant consequence of UPR activation, it remains to be seen whether most of the genes regulated by the UPR ultimately redound to this capacity, or whether the UPR has been co-opted in the homeostatic regulation of other cellular processes—particularly those, such as lipid metabolism, that involve the ER. Deciphering the pathways leading from UPR activation to mRNA regulation will allow the functional significance of non-canonical UPR signaling mechanisms to be understood in the contexts of normal and pathological physiology.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fgene.2013.00256/abstract
References
Acosta-Alvear, D., Zhou, Y., Blais, A., Tsikitis, M., Lents, N. H., Arias, C., et al. (2007). XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 27, 53–66. doi: 10.1016/j.molcel.2007.06.011
Adachi, Y., Yamamoto, K., Okada, T., Yoshida, H., Harada, A., and Mori, K. (2008). ATF6 Is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct. Funct. 33, 75–89. doi: 10.1247/csf.07044
Ahn, K. S., and Aggarwal, B. B. (2005). Transcription Factor NF-κB: a sensor for Smoke and Stress Signals. Ann. N.Y. Acad. Sci. 1056, 218–233. doi: 10.1196/annals.1352.026
Akira, S., Isshiki, H., Sugita, T., Tanabe, O., Kinoshita, S., Nishio, Y., et al. (1990). A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO J. 9, 1897–1906.
Araki, K., and Nagata, K. (2012). Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 3, a007526. doi: 10.1101/cshperspect.a015438
Arensdorf, A. M., and Rutkowski, D. T. (2013). Endoplasmic reticulum stress impairs IL-4/IL-13 signaling through C/EBPbeta-mediated transcriptional suppression. J. Cell. Sci. 126, 4026–4036. doi: 10.1242/jcs.130757
Arensdorf, A. M., McCabe, D. D., Kaufman, R. J., and Rutkowski, D. T. (2013). Temporal clustering of gene expression links the metabolic transcription factor HNF4α to the ER stress-dependent gene regulatory network. Front. Genet. 4:188. doi: 10.3389/fgene.2013.00188
Asada, R., Kanemoto, S., Kondo, S., Saito, A., and Imaizumi, K. (2011). The signalling from endoplasmic reticulum-resident bZIP transcription factors involved in diverse cellular physiology. J. Biochem. 149, 507–518. doi: 10.1093/jb/mvr041
Barbosa, C., Peixeiro, I., and Romao, L. (2013). Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 9:e1003529. doi: 10.1371/journal.pgen.1003529
Bartoszewski, R., Brewer, J. W., Rab, A., Crossman, D. K., Bartoszewska, S., Kapoor, N., et al. (2011). The unfolded protein response (UPR)-activated transcription factor X-box-binding protein 1 (XBP1) induces microRNA-346 expression that targets the human antigen peptide transporter 1 (TAP1) mRNA and governs immune regulatory genes. J. Biol. Chem. 286, 41862–41870. doi: 10.1074/jbc.M111.304956
Bassik, M. C., Kampmann, M., Lebbink, R. J., Wang, S., Hein, M. Y., Poser, I., et al. (2013). A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell 152, 909–922. doi: 10.1016/j.cell.2013.01.030
Baumeister, P., Luo, S., Skarnes, W. C., Sui, G., Seto, E., Shi, Y., et al. (2005). Endoplasmic reticulum stress induction of the Grp78/BiP promoter: activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol. Cell. Biol. 25, 4529–4540. doi: 10.1128/MCB.25.11.4529-4540.2005
Belmont, P. J., Chen, W. J., Thuerauf, D. J., and Glembotski, C. C. (2012). Regulation of microRNA expression in the heart by the ATF6 branch of the ER stress response. J. Mol. Cell. Cardiol. 52, 1176–1182. doi: 10.1016/j.yjmcc.2012.01.017
Bertolotti, A., Wang, X., Novoa, I., Jungreis, R., Schlessinger, K., Cho, J. H., et al. (2001). Increased sensitivity to dextran sodium sulfate colitis in IRE1beta-deficient mice. J. Clin. Invest. 107, 585–593. doi: 10.1172/JCI11476
Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P., and Ron, D. (2000). Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332. doi: 10.1038/35014014
Brewer, J. W., Hendershot, L. M., Sherr, C. J., and Diehl, J. A. (1999). Mammalian unfolded protein response inhibits cyclin D1 translation and cell-cycle progression. Proc. Natl. Acad. Sci. U.S.A. 96, 8505–8510. doi: 10.1073/pnas.96.15.8505
Byrd, A. E., Aragon, I. V., and Brewer, J. W. (2012). MicroRNA-30c-2* limits expression of proadaptive factor XBP1 in the unfolded protein response. J. Cell Biol. 196, 689–698. doi: 10.1083/jcb.201201077
Calfon, M., Zeng, H., Urano, F., Till, J. H., Hubbard, S. R., Harding, H. P., et al. (2002). IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96. doi: 10.1038/415092a
Calkhoven, C. F., Muller, C., and Leutz, A. (2000). Translational control of C/EBPalpha and C/EBPbeta isoform expression. Genes Dev. 14, 1920–1932.
Cao, S. S., Zimmermann, E. M., Chuang, B. M., Song, B., Nwokoye, A., Wilkinson, J. E., et al. (2013). The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology 144, 989–1000. doi: 10.1053/j.gastro.2013.01.023
Chikka, M. R., McCabe, D. D., Tyra, H. M., and Rutkowski, D. T. (2013). C/EBP homologous protein (CHOP) contributes to suppression of metabolic genes during endoplasmic reticulum stress in the liver. J. Biol. Chem. 288, 4405–4415. doi: 10.1074/jbc.M112.432344
Chiribau, C. B., Gaccioli, F., Huang, C. C., Yuan, C. L., and Hatzoglou, M. (2010). Molecular symbiosis of CHOP and C/EBP beta isoform LIP contributes to endoplasmic reticulum stress-induced apoptosis. Mol. Cell. Biol. 30, 3722–3731. doi: 10.1128/MCB.01507-09
Chitnis, N. S., Pytel, D., Bobrovnikova-Marjon, E., Pant, D., Zheng, H., Maas, N. L., et al. (2012). miR-211 is a prosurvival microRNA that regulates chop expression in a PERK-dependent manner. Mol. Cell 48, 353–364. doi: 10.1016/j.molcel.2012.08.025
Christianson, J. C., Olzmann, J. A., Shaler, T. A., Sowa, M. E., Bennett, E. J., Richter, C. M., et al. (2011). Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol. 14, 93–105. doi: 10.1038/ncb2383
Cox, J. S., and Walter, P. (1996). A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 87, 391–404. doi: 10.1016/S0092-8674(00)81360-4
Cox, J. S., Shamu, C. E., and Walter, P. (1993). Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206. doi: 10.1016/0092-8674(93)90648-A
Cullinan, S. B., and Diehl, J. A. (2004). PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 279, 20108–20117. doi: 10.1074/jbc.M314219200
Cullinan, S. B., Zhang, D., Hannink, M., Arvisais, E., Kaufman, R. J., and Diehl, J. A. (2003). Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 23, 7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003
Deng, J., Lu, P. D., Zhang, Y., Scheuner, D., Kaufman, R. J., Sonenberg, N., et al. (2004). Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell. Biol. 24, 10161–10168. doi: 10.1128/MCB.24.23.10161-10168.2004
Descombes, P., and Schibler, U. (1991). A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell 67, 569–579. doi: 10.1016/0092-8674(91)90531-3
DeZwaan-McCabe, D., Riordan, J. D., Arensdorf, A. M., Icardi, M. S., Dupuy, A. J., and Rutkowski, D. T. (2013). The stress-regulated transcription factor CHOP promotes hepaatic inflammatory gene expression, fibrosis, and oncogenesis. PLoS Genet. 9, e1003937. doi: 10.1371/journal.pgen.1003937
Donati, G., Imbriano, C., and Mantovani, R. (2006). Dynamic recruitment of transcription factors and epigenetic changes on the ER stress response gene promoters. Nucleic Acids Res. 34, 3116–3127. doi: 10.1093/nar/gkl304
Dorner, A. J., Wasley, L. C., and Kaufman, R. J. (1989). Increased synthesis of secreted proteins induces expression of glucose-regulated proteins in butyrate-treated Chinese hamster ovary cells. J. Biol. Chem. 264, 20602–20607.
Dorner, A. J., Wasley, L. C., Raney, P., Haugejorden, S., Green, M., and Kaufman, R. J. (1990). The stress response in Chinese hamster ovary cells. Regulation of ERp72 and protein disulfide isomerase expression and secretion. J. Biol. Chem. 265, 22029–22034.
Drummond, I. A., Lee, A. S., Resendez, E. Jr., and Steinhardt, R. A. (1987). Depletion of intracellular calcium stores by calcium ionophore A23187 induces the genes for glucose-regulated proteins in hamster fibroblasts. J. Biol. Chem. 262, 12801–12805.
Duan, Q., Wang, X., Gong, W., Ni, L., Chen, C., He, X., et al. (2012). ER stress negatively modulates the expression of the miR-199a/214 cluster to regulates tumor survival and progression in human hepatocellular cancer. PLoS ONE 7:e31518. doi: 10.1371/journal.pone.0031518
Elsharkawy, A. M., Oakley, F., Lin, F., Packham, G., Mann, D. A., and Mann, J. (2010). NF-κ B p50:p50:HDAC-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes. J. Hepatol. 53, 519–527. doi: 10.1016/j.jhep.2010.03.025
Fawcett, T. W., Eastman, H. B., Martindale, J. L., and Holbrook, N. J. (1996). Physical and functional association between GADD153 and CCAAT/enhancer-binding protein beta during cellular stress. J. Biol. Chem. 271, 14285–14289. doi: 10.1074/jbc.271.24.14285
Fu, S., Watkins, S. M., and Hotamisligil, G. S. (2012). The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 15, 623–634. doi: 10.1016/j.cmet.2012.03.007
Gaddam, D., Stevens, N., and Hollien, J. (2012). Comparison of mRNA localization and regulation during endoplasmic reticulum stress in Drosophila cells. Mol. Biol. Cell 24, 14–20. doi: 10.1091/mbc.E12-06-0491
Gade, P., Ramachandran, G., Maachani, U. B., Rizzo, M. A., Okada, T., Prywes, R., et al. (2012). An IFN-gamma-stimulated ATF6-C/EBP-beta-signaling pathway critical for the expression of death associated protein kinase 1 and induction of autophagy. Proc. Natl. Acad. Sci. U.S.A. 109, 10316–10321. doi: 10.1073/pnas.1119273109
Gal-Yam, E. N., Jeong, S., Tanay, A., Egger, G., Lee, A. S., and Jones, P. A. (2006). Constitutive nucleosome depletion and ordered factor assembly at the GRP78 promoter revealed by single molecule footprinting. PLoS Genet. 2:e160. doi: 10.1371/journal.pgen.0020160
Garg, A. D., Kaczmarek, A., Krysko, O., Vandenabeele, P., Krysko, D. V., and Agostinis, P. (2012). ER stress-induced inflammation: does it aid or impede disease progression? Trends Mol. Med. 18, 589–598. doi: 10.1016/j.molmed.2012.06.010
Gass, J. N., Gifford, N. M., and Brewer, J. W. (2002). Activation of an unfolded protein response during differentiation of antibody-secreting B cells. J. Biol. Chem. 277, 49047–49054. doi: 10.1074/jbc.M205011200
Gupta, S., Read, D. E., Deepti, A., Cawley, K., Gupta, A., Oommen, D., et al. (2012). Perk-dependent repression of miR-106b-25 cluster is required for ER stress-induced apoptosis. Cell Death Dis 3:e333. doi: 10.1038/cddis.2012.74
Gygi, S. P., Rochon, Y., Franza, B. R., and Aebersold, R. (1999). Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19, 1720–1730.
Han, D., Lerner, A. G., Vande Walle, L., Upton, J. P., Xu, W., Hagen, A., et al. (2009). IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138, 562–575. doi: 10.1016/j.cell.2009.07.017
Han, J., Back, S. H., Hur, J., Lin, Y. H., Gildersleeve, R., Shan, J., et al. (2013). ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 15, 481–490. doi: 10.1038/ncb2738
Harding, H. P., Zeng, H., Zhang, Y., Jungries, R., Chung, P., Plesken, H., et al. (2001). Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol. Cell 7, 1153–1163. doi: 10.1016/S1097-2765(01)00264-7
Harding, H. P., Zhang, Y., and Ron, D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274. doi: 10.1038/16729
Harding, H. P., Zhang, Y., Scheuner, D., Chen, J. J., Kaufman, R. J., and Ron, D. (2009). Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc. Natl. Acad. Sci. U.S.A. 106, 1832–1837. doi: 10.1073/pnas.0809632106
Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633. doi: 10.1016/S1097-2765(03)00105-9
Haze, K., Okada, T., Yoshida, H., Yanagi, H., Yura, T., Negishi, M., et al. (2001). Identificaiton of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem. J. 355(Pt. 1), 19–28. doi: 10.1042/0264-6021:3550019
Haze, K., Yoshida, H., Yanagi, H., Yura, T., and Mori, K. (1999). Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 10, 3787–3799. doi: 10.1091/mbc.10.11.3787
Hershey, G. K. K. (2003). IL-13 receptor and signaling pathways: an evolving web. J. Allergy Clin. Immunol. 111, 677–690. doi: 10.1067/mai.2003.1333
Hetz, C., and Glimcher, L. H. (2009). Fine-tuning of the unfolded protein response: assembling the IRE1alpha interactome. Mol. Cell 35, 551–561. doi: 10.1016/j.molcel.2009.08.021
Hoesel, B., and Schmid, J. A. (2013). The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 12, 86. doi: 10.1186/1476-4598-12-86
Holgate, S. T. (2011). Pathophysiology of asthma: what has our current understanding taught us about new therapeutic approaches. J. Allergy Clin. Immunol. 128, 495–505. doi: 10.1016/j.jaci.2011.06.052
Hollien, J., and Weissman, J. S. (2006). Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104–107. doi: 10.1126/science.1129631
Hollien, J., Lin, J. H., Li, H., Stevens, N., Walter, P., and Weissman, J. S. (2009). Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 186, 323–331. doi: 10.1083/jcb.200903014
Hu, P., Han, Z., Couvillon, A. D., Kaufman, R. J., and Exton, J. H. (2006). Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol. Cell. Biol. 26, 3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006
Huh, W. K., Falvo, J. V., Gerke, L. C., Carroll, A. S., Howson, R. W., Weissman, J. S., et al. (2003). Global analysis of protein localization in budding yeast. Nature 425, 686–691. doi: 10.1038/nature02026
Hur, K. Y., So, J. S., Ruda, V., Frank-Kamenetsky, M., Fitzgerald, K., Koteliansky, V., et al. (2012). IRE1alpha activation protects mice against acetaminophen-induced hepatotoxicity. J. Exp. Med. 209, 307–318. doi: 10.1084/jem.20111298
Hussain, M. M. (2000). A proposed model for the assembly of chylomicrons. Atherosclerosis 148, 1–15. doi: 10.1016/S0021-9150(99)00397-4
Imagawa, Y., Hosoda, A., Sasaka, S., Tsuru, A., and Kohno, K. (2008). RNase domains determine the functional difference between IRE1alpha and IRE1beta. FEBS Lett. 582, 656–660. doi: 10.1016/j.febslet.2008.01.038
Iqbal, J., Dai, K., Seimon, T., Jungreis, R., Oyadomari, M., Kuriakose, G., et al. (2008). IRE1beta inhibits chylomicron production by selectively degrading MTP mRNA. Cell Metab. 7, 445–455. doi: 10.1016/j.cmet.2008.03.005
Ishikawa, T., Okada, T., Ishikawa-Fujiwara, T., Todo, T., Kamei, Y., Shigenobu, S., et al. (2012). ATF6alpha/beta-mediated adjustment of ER chaperone levels is essential for development of the notochord in medaka fish. Mol. Biol. Cell 24, 1387–1395. doi: 10.1091/mbc.E12-11-0830
Iwawaki, T., Akai, R., Yamanaka, S., and Kohno, K. (2009). Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc. Natl. Acad. Sci. U.S.A. 106, 16657–16662. doi: 10.1073/pnas.0903775106
Jiang, H. Y., Wek, S. A., McGrath, B. C., Scheuner, D., Kaufman, R. J., Cavener, D. R., et al. (2003). Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol. Cell. Biol. 23, 5651–5663. doi: 10.1128/MCB.23.16.5651-5663.2003
Josekutty, J., Iqbal, J., Iwawaki, T., Kohno, K., and Hussain, M. M. (2013). Microsomal triglyceride transfer protein inhibition induces endoplasmic reticulum stress and increases gene transcription via Ire1alpha/cJun to enhance plasma ALT/AST. J. Biol. Chem. 288, 14372–14383. doi: 10.1074/jbc.M113.459602
Jousse, C., Oyadomari, S., Novoa, I., Lu, P., Zhang, Y., Harding, H. P., et al. (2003). Inhibition of a constitutive translation initiation factor 2alpha phosphatase, CReP, promotes survival of stressed cells. J. Cell Biol. 163, 767–775. doi: 10.1083/jcb.200308075
Kampmann, M., Bassik, M. C., and Weissman, J. S. (2013). Integrated platform for genome-wide screening and construction of high-density genetic interaction maps in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 110, E2317–E2326. doi: 10.1073/pnas.1307002110
Kaser, A., Lee, A. H., Franke, A., Glickman, J. N., Zeissig, S., Tilg, H., et al. (2008). XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 134, 743–756. doi: 10.1016/j.cell.2008.07.021
Kawai, T., Fan, J., Mazan-Mamczarz, K., and Gorospe, M. (2004). Global mRNA stabilization preferentially linked to translational repression during the endoplasmic reticulum stress response. Mol. Cell. Biol. 24, 6773–6787. doi: 10.1128/MCB.24.15.6773-6787.2004
Kim, D. K., Kim, Y. H., Jang, H. H., Park, J., Kim, J. R., Koh, M., et al. (2013). Estrogen-related receptor gamma controls hepatic CB1 receptor-mediated CYP2E1 expression and oxidative liver injury by alcohol. Gut 62, 1044–1054. doi: 10.1136/gutjnl-2012-303347
Kim, D. K., Ryu, D., Koh, M., Lee, M. W., Lim, D., Kim, M. J., et al. (2012). Orphan nuclear receptor estrogen-related receptor gamma (ERRgamma) is key regulator of hepatic gluconeogenesis. J. Biol. Chem. 287, 21628–21639. doi: 10.1074/jbc.M111.315168
Kim, I., Shu, C. W., Xu, W., Shiau, C. W., Grant, D., Vasile, S., et al. (2009). Chemical biology investigation of cell death pathways activated by endoplasmic reticulum stress reveals cytoprotective modulators of ASK1. J. Biol. Chem. 284, 1593–1603. doi: 10.1074/jbc.M807308200
Kim, Y. K., and Lee, A. S. (1987). Transcriptional activation of the glucose-regulated protein genes and their heterologous fusion genes by beta-mercaptoethanol. Mol. Cell. Biol. 7, 2974–2976.
Kimball, S. R., Horetsky, R. L., Ron, D., Jefferson, L. S., and Harding, H. P. (2003). Mammalian stress granules represent sites of accumulation of stalled translation initiation complexes. Am. J. Physiol. Cell Physiol. 284, C273–C284. doi: 10.1152/ajpcell.00314.2002
Kimmig, P., Diaz, M., Zheng, J., Williams, C. C., Lang, A., Aragon, T., et al. (2012). The unfolded protein response in fission yeast modulates stability of select mRNAs to maintain protein homeostasis. Elife 1:e00048. doi: 10.7554/eLife.00048
Kohno, K., Normington, K., Sambrook, J., Gething, M. J., and Mori, K. (1993). The promoter region of the yeast KAR2 (BiP) gene contains a regulatory domain that responds to the presence of unfolded proteins in the endoplasmic reticulum. Mol. Cell. Biol. 13, 877–890.
Kokame, K., Kato, H., and Miyata, T. (2001). Identification of ERSE-II, a new cis-acting element responsible for the ATF6-dependent mammalian unfolded protein response. J. Biol. Chem. 276, 9199–9205. doi: 10.1074/jbc.M010486200
Kondo, S., Murakami, T., Tatsumi, K., Ogata, M., Kanemoto, S., Otori, K., et al. (2005). OASIS, a CREB/ATF-family member, modulates UPR signalling in astrocytes. Nat. Cell Biol. 7, 186–194. doi: 10.1038/ncb1213
Kondo, S., Saito, A., Hino, S., Murakami, T., Ogata, M., Kanemoto, S., et al. (2007). BBF2H7, a novel transmembrane bZIP transcription factor, is a new type of endoplasmic reticulum stress transducer. Mol. Cell. Biol. 27, 1716–1729. doi: 10.1128/MCB.01552-06
Korennykh, A. V., Egea, P. F., Korostelev, A. A., Finer-Moore, J., Zhang, C., Shokat, K. M., et al. (2009). The unfolded protein response signals through high-order assembly of Ire1. Nature 457, 687–693. doi: 10.1038/nature07661
Kozutsumi, Y., Segal, M., Normington, K., Gething, M. J., and Sambrook, J. (1988). The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332, 462–464. doi: 10.1038/332462a0
Leclerc, D., and Rozen, R. (2008). Endoplasmic reticulum stress increases the expression of methylenetetrahydrofolate reductase through the IRE1 transducer. J. Biol. Chem. 283, 3151–3160. doi: 10.1074/jbc.M708598200
Lee, A. H., Chu, G. C., Iwakoshi, N. N., and Glimcher, L. H. (2005). XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. EMBO J. 24, 4368–4380. doi: 10.1038/sj.emboj.7600903
Lee, A. H., Heidtman, K., Hotamisligil, G. S., and Glimcher, L. H. (2011). Dual and opposing roles of the unfolded protein response regulated by IRE1alpha and XBP1 in proinsulin processing and insulin secretion. Proc. Natl. Acad. Sci. U.S.A. 108, 8885–8890. doi: 10.1073/pnas.1105564108
Lee, A. H., Iwakoshi, N. N., Anderson, K. C., and Glimcher, L. H. (2003a). Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl. Acad. Sci. U.S.A. 100, 9946–9951. doi: 10.1073/pnas.1334037100
Lee, A. H., Iwakoshi, N. N., and Glimcher, L. H. (2003b). XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 23, 7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003
Lee, A. H., Scapa, E. F., Cohen, D. E., and Glimcher, L. H. (2008). Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320, 1492–1496. doi: 10.1126/science.1158042
Lee, K., Tirasophon, W., Shen, X., Michalak, M., Prywes, R., Okada, T., et al. (2002). IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 16, 452–466. doi: 10.1101/gad.964702
Lee, M. W., Chanda, D., Yang, J., Oh, H., Kim, S. S., Yoon, Y. S., et al. (2010). Regulation of hepatic gluconeogenesis by an ER-bound transcription factor, CREBH. Cell Metab. 11, 331–339. doi: 10.1016/j.cmet.2010.02.016
Lerner, A. G., Upton, J. P., Praveen, P. V., Ghosh, R., Nakagawa, Y., Igbaria, A., et al. (2012). IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 16, 250–264. doi: 10.1016/j.cmet.2012.07.007
Li, G., Mongillo, M., Chin, K. T., Harding, H., Ron, D., Marks, A. R., et al. (2009). Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 186, 783–792. doi: 10.1083/jcb.200904060
Li, H., Korennykh, A. V., Behrman, S. L., and Walter, P. (2010). Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. U.S.A. 107, 16113–16118. doi: 10.1073/pnas.1010580107
Li, M., Baumeister, P., Roy, B., Phan, T., Foti, D., Luo, S., et al. (2000). ATF6 as a transcription activator of the endoplasmic reticulum stress element: thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol. Cell. Biol. 20, 5096–5106. doi: 10.1128/MCB.20.14.5096-5106.2000
Li, Y., Bevilacqua, E., Chiribau, C. B., Majumder, M., Wang, C., Croniger, C. M., et al. (2008). Differential control of the CCAAT/enhancer-binding protein beta (C/EBPbeta) products liver-enriched transcriptional activating protein (LAP) and liver-enriched transcriptional inhibitory protein (LIP) and the regulation of gene expression during the response to endoplasmic reticulum stress. J. Biol. Chem. 283, 22443–22456. doi: 10.1074/jbc.M801046200
Liang, S. H., Zhang, W., McGrath, B. C., Zhang, P., and Cavener, D. R. (2006a). PERK (eIF2alpha kinase) is required to activate the stress-activated MAPKs and induce the expression of immediate-early genes upon disruption of ER calcium homoeostasis. Biochem. J. 393(Pt 1), 201–209.
Liang, G., Audas, T. E., Li, Y., Cockram, G. P., Dean, J. D., Martyn, A. C., et al. (2006b). Luman/CREB3 induces transcription of the endoplasmic reticulum (ER) stress response protein Herp through an ER stress response element. Mol. Cell. Biol. 26, 7999–8010. doi: 10.1128/MCB.01046-06
Lin, J., Handschin, C., and Spiegelman, B. M. (2005). Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 1, 361–370. doi: 10.1016/j.cmet.2005.05.004
Llarena, M., Bailey, D., Curtis, H., and O'Hare, P. (2010). Different mechanisms of recognition and ER retention by transmembrane transcription factors CREB-H and ATF6. Traffic 11, 48–69. doi: 10.1111/j.1600-0854.2009.00997.x
Lu, P. D., Harding, H. P., and Ron, D. (2004). Translation reinitiation at alternative open reading frames regulates gene expression in an integrated stress response. J. Cell Biol. 167, 27–33. doi: 10.1083/jcb.200408003
Ma, Y., Brewer, J. W., Diehl, J. A., and Hendershot, L. M. (2002). Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J. Mol. Biol. 318, 1351–1365. doi: 10.1016/S0022-2836(02)00234-6
Malhi, H., Kropp, E. M., Clavo, V. F., Kobrossi, C. R., Han, J., Mauer, A. S., et al. (2013). C/EBP homologous protein-induced macrophage apoptosis protects mice from steatohepatitis. J. Biol. Chem. 288, 18624–18642. doi: 10.1074/jbc.M112.442954
Malhotra, J. D., Miao, H., Zhang, K., Wolfson, A., Pennathur, S., Pipe, S. W., et al. (2008). Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. U.S.A. 105, 18525–18530. doi: 10.1073/pnas.0809677105
Marciniak, S. J., Yun, C. Y., Oyadomari, S., Novoa, I., Zhang, Y., Jungreis, R., et al. (2004). CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 18, 3066–3077. doi: 10.1101/gad.1250704
Maris, M., Overbergh, L., Gysemans, C., Waget, A., Cardozo, A. K., Verdrengh, E., et al. (2012). Deletion of C/EBP homologous protein (Chop) in C57Bl/6 mice dissociates obesity from insulin resistance. Diabetologia 55, 1167–1178. doi: 10.1007/s00125-011-2427-7
Martino, M. B., Jones, L., Brighton, B., Ehre, C., Abdulah, L., Davis, C. W., et al. (2013). The ER stress transducer IRE1beta is required for airway epithelial mucin production. Mucosal Immunol 6, 639–654. doi: 10.1038/mi.2012.105
Martyn, A. C., Choleris, E., Gillis, D. J., Armstrong, J. N., Amor, T. R., McCluggage, A. R., et al. (2012). Luman/CREB3 recruitment factor regulates glucocorticoid receptor activity and is essential for prolactin-mediated maternal instinct. Mol. Cell. Biol. 32, 5140–5150. doi: 10.1128/MCB.01142-12
Matus, S., Glimcher, L. H., and Hetz, C. (2011). Protein folding stress in neurodegenerative diseases: a glimpse into the ER. Curr. Opin. Cell Biol. 23, 239–252. doi: 10.1016/j.ceb.2011.01.003
Maurel, M., and Chevet, E. (2013). Endoplasmic reticulum stress signaling: the microRNA connection. Am. J. Physiol. Cell Physiol. 304, C1117–C1126. doi: 10.1152/ajpcell.00061.2013
Maurel, M., Dejeans, N., Taouji, S., Chevet, E., and Grosset, C. F. (2013). MicroRNA-1291-mediated silencing of IRE1alpha enhances Glypican-3 expression. RNA 19, 778–788. doi: 10.1261/rna.036483.112
McCullough, K. D., Martindale, J. L., Klotz, L. O., Aw, T. Y., and Holbrook, N. J. (2001). Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 21, 1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001
McKenzie, A. N. (2000). Regulation of T helper type 2 cell immunity by interleukin-4 and interleukin-13. Pharmacol. Ther. 88, 143–151. doi: 10.1016/S0163-7258(00)00088-7
Misra, J., Kim, D. K., Choi, W., Koo, S. H., Lee, C. H., Back, S. H., et al. (2013). Transcriptional cross talk between orphan nuclear receptor ERRgamma and transmembrane transcription factor ATF6alpha coordinates endoplasmic reticulum stress response. Nucleic Acids Res. 41, 6960–6974. doi: 10.1093/nar/gkt429
Moore, K. A., Plant, J. J., Gaddam, D., Craft, J., and Hollien, J. (2013). Regulation of sumo mRNA during endoplasmic reticulum stress. PLoS One 8:e75723. doi: 10.1371/journal.pone.0075723
Mori, K., Ma, W., Gething, M. J., and Sambrook, J. (1993). A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 74, 743–756. doi: 10.1016/0092-8674(93)90521-Q
Mori, K., Sant, A., Kohno, K., Normington, K., Gething, M. J., and Sambrook, J. F. (1992). A 22 bp cis-acting element is necessary and sufficient for the induction of the yeast KAR2 (BiP) gene by unfolded proteins. EMBO J. 11, 2583–2593.
Morris, D. R., and Geballe, A. P. (2000). Upstream Open Reading Frames as Regulators of mRNA Translation. Mol. Cell. Biol. 20, 8635–8642. doi: 10.1128/MCB.20.23.8635-8642.2000
Murakami, T., Kondo, S., Ogata, M., Kanemoto, S., Saito, A., Wanaka, A., et al. (2006). Cleavage of the membrane-bound transcription factor OASIS in response to endoplasmic reticulum stress. J. Neurochem. 96, 1090–1100. doi: 10.1111/j.1471-4159.2005.03596.x
Muratsu-Ikeda, S., Nangaku, M., Ikeda, Y., Tanaka, T., Wada, T., and Inagi, R. (2012). Downregulation of miR-205 modulates cell susceptibility to oxidative and endoplasmic reticulum stresses in renal tubular cells. PLoS ONE 7:e41462. doi: 10.1371/journal.pone.0041462
Nguyen, D. T., Kebache, S., Fazel, A., Wong, H. N., Jenna, S., Emadali, A., et al. (2004). Nck-dependent activation of extracellular signal-regulated kinase-1 and regulation of cell survival during endoplasmic reticulum stress. Mol. Biol. Cell 15, 4248–4260. doi: 10.1091/mbc.E03-11-0851
Nishitoh, H., Matsuzawa, A., Tobiume, K., Saegusa, K., Takeda, K., Inoue, K., et al. (2002). ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 16, 1345–1355. doi: 10.1101/gad.992302
Normington, K., Kohno, K., Kozutsumi, Y., Gething, M. J., and Sambrook, J. (1989). S. cerevisiae encodes an essential protein homologous in sequence and function to mammalian BiP. Cell 57, 1223–1236. doi: 10.1016/0092-8674(89)90059-7
Oikawa, D., Tokuda, M., Hosoda, A., and Iwawaki, T. (2010). Identification of a consensus element recognized and cleaved by IRE1 alpha. Nucleic Acids Res. 38, 6265–6273. doi: 10.1093/nar/gkq452
Oyadomari, S., Harding, H. P., Zhang, Y., Oyadomari, M., and Ron, D. (2008). Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 7, 520–532. doi: 10.1016/j.cmet.2008.04.011
Oyadomari, S., and Mori, M. (2004). Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11, 381–389. doi: 10.1038/sj.cdd.4401373
Pahl, H. L. (1999). Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18, 6853–6866. doi: 10.1038/sj.onc.1203239
Palam, L. R., Baird, T. D., and Wek, R. C. (2011). Phosphorylation of eIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J. Biol. Chem. 286, 10939–10949. doi: 10.1074/jbc.M110.216093
Park, S. H., Choi, H. J., Yang, H., Do, K. H., Kim, J., Kim, H. H., et al. (2012). Two in-and-out modulation strategies for endoplasmic reticulum stress-linked gene expression of pro-apoptotic macrophage-inhibitory cytokine 1. J. Biol. Chem. 287, 19841–19855. doi: 10.1074/jbc.M111.330639
Park, S. W., Zhou, Y., Lee, J., Lu, A., Sun, C., Chung, J., et al. (2010). The regulatory subunits of PI3K, p85alpha and p85beta, interact with XBP-1 and increase its nuclear translocation. Nat. Med. 16, 429–437. doi: 10.1038/nm.2099
Pereira, E. R., Liao, N., Neale, G. A., and Hendershot, L. M. (2010). Transcriptional and post-transcriptional regulation of proangiogenic factors by the unfolded protein response. PLoS ONE 5:e12521. doi: 10.1371/journal.pone.0012521
Pfaffenbach, K. T., Nivala, A. M., Reese, L., Ellis, F., Wang, D., Wei, Y., et al. (2010). Rapamycin inhibits postprandial-mediated X-Box-binding protein-1 splicing in rat liver. J. Nutr. 140, 879–884. doi: 10.3945/jn.109.119883
Puthalakath, H., O'Reilly, L. A., Gunn, P., Lee, L., Kelly, P. N., Huntington, N. D., et al. (2007). ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129, 1337–1349. doi: 10.1016/j.cell.2007.04.027
Raggo, C., Rapin, N., Stirling, J., Gobeil, P., Smith-Windsor, E., O'Hare, P., et al. (2002). Luman, the cellular counterpart of herpes simplex virus VP16, is processed by regulated intramembrane proteolysis. Mol. Cell. Biol. 22, 5639–5649. doi: 10.1128/MCB.22.16.5639-5649.2002
Ramji, D. P., and Foka, P. (2002). CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem. J. 365(Pt. 3), 561–575. doi: 10.1042/BJ20020508
Reimold, A. M., Etkin, A., Clauss, I., Perkins, A., Friend, D. S., Zhang, J., et al. (2000). An essential role in liver development for transcription factor XBP-1. Genes Dev. 14, 152–157.
Ron, D., and Habener, J. F. (1992). CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 6, 439–453. doi: 10.1101/gad.6.3.439
Ron, D., and Walter, P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. 8, 519–529. doi: 10.1038/nrm2199
Rutkowski, D. T., Arnold, S. M., Miller, C. N., Wu, J., Li, J., Gunnison, K. M., et al. (2006). Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 4:e374. doi: 10.1371/journal.pbio.0040374
Rutkowski, D. T., Wu, J., Back, S. H., Callaghan, M. U., Ferris, S. P., Iqbal, J., et al. (2008). UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev. Cell 15, 829–840. doi: 10.1016/j.devcel.2008.10.015
Ryoo, H. D., and Steller, H. (2007). Unfolded protein response in Drosophila: why another model can make it fly. Cell Cycle 6, 830–835. doi: 10.4161/cc.6.7.4064
Schröder, M., and Kaufman, R. J. (2005). The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739–789. doi: 10.1146/annurev.biochem.73.011303.074134
Schwanhausser, B., Busse, D., Li, N., Dittmar, G., Schuchhardt, J., Wolf, J., et al. (2011). Global quantification of mammalian gene expression control. Nature 473, 337–342. doi: 10.1038/nature10098
Schwanhausser, B., Busse, D., Li, N., Dittmar, G., Schuchhardt, J., Wolf, J., et al. (2013). Corrigendum: global quantification of mammalian gene expression control. Nature 495, 126–127. doi: 10.1038/nature11848
Sela, D., Chen, L., Martin-Brown, S., Washburn, M. P., Florens, L., Conaway, J. W., et al. (2012). Endoplasmic reticulum stress-responsive transcription factor ATF6alpha directs recruitment of the Mediator of RNA polymerase II transcription and multiple histone acetyltransferase complexes. J. Biol. Chem. 287, 23035–23045. doi: 10.1074/jbc.M112.369504
Shen, X., Ellis, R. E., Lee, K., Liu, C. Y., Yang, K., Solomon, A., et al. (2001). Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell 107, 893–903. doi: 10.1016/S0092-8674(01)00612-2
Shi, Y., Vattem, K. M., Sood, R., An, J., Liang, J., Stramm, L., et al. (1998). Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 18, 7499–7509.
Sidrauski, C., Cox, J. S., and Walter, P. (1996). tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell 87, 405–413. doi: 10.1016/S0092-8674(00)81361-6
Sidrauski, C., and Walter, P. (1997). The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039. doi: 10.1016/S0092-8674(00)80369-4
So, J. S., Hur, K. Y., Tarrio, M., Ruda, V., Frank-Kamenetsky, M., Fitzgerald, K., et al. (2012). Silencing of lipid metabolism genes through IRE1alpha-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 16, 487–499. doi: 10.1016/j.cmet.2012.09.004
Sriburi, R., Bommiasamy, H., Buldak, G. L., Robbins, G. R., Frank, M., Jackowski, S., et al. (2007). Coordinate regulation of phospholipid biosynthesis and secretory pathway gene expression in XBP-1(S)-induced endoplasmic reticulum biogenesis. J. Biol. Chem. 282, 7024–7034. doi: 10.1074/jbc.M609490200
Stephens, S. B., Dodd, R. D., Brewer, J. W., Lager, P. J., Keene, J. D., and Nicchitta, C. V. (2005). Stable ribosome binding to the endoplasmic reticulum enables compartment-specific regulation of mRNA translation. Mol. Biol. Cell 16, 5819–5831. doi: 10.1091/mbc.E05-07-0685
Stirling, J., and O'Hare, P. (2006). CREB4, a transmembrane bZip transcription factor and potential new substrate for regulation and cleavage by S1P. Mol. Biol. Cell 17, 413–426. doi: 10.1091/mbc.E05-06-0500
Tagliavacca, L., Anelli, T., Fagioli, C., Mezghrani, A., Ruffato, E., and Sitia, R. (2003). The making of a professional secretory cell: architectural and functional changes in the ER during B lymphocyte plasma cell differentiation. Biol. Chem. 384, 1273–1277. doi: 10.1515/BC.2003.141
Tam, A. B., Mercado, E. L., Hoffmann, A., and Niwa, M. (2012). ER stress activates NF-kappaB by integrating functions of basal IKK activity, IRE1 and PERK. PLoS ONE 7:e45078. doi: 10.1371/journal.pone.0045078
Teske, B. F., Fusakio, M. E., Zhou, D., Shan, J., McClintick, J. N., Kilberg, M. S., et al. (2013). CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell 24, 2477–2490. doi: 10.1091/mbc.E13-01-0067
Tirasophon, W., Welihinda, A. A., and Kaufman, R. J. (1998). A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824. doi: 10.1101/gad.12.12.1812
Tirosh, B., Iwakoshi, N. N., Glimcher, L. H., and Ploegh, H. L. (2006). Rapid turnover of unspliced Xbp-1 as a factor that modulates the unfolded protein response. J. Biol. Chem. 281, 5852–5860. doi: 10.1074/jbc.M509061200
Travers, K. J., Patil, C. K., Wodicka, L., Lockhart, D. J., Weissman, J. S., and Walter, P. (2000). Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101, 249–258. doi: 10.1016/S0092-8674(00)80835-1
Tsaytler, P., Harding, H. P., Ron, D., and Bertolotti, A. (2011). Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 332, 91–94. doi: 10.1126/science.1201396
Tsuru, A., Fujimoto, N., Takahashi, S., Saito, M., Nakamura, D., Iwano, M., et al. (2013). Negative feedback by IRE1beta optimizes mucin production in goblet cells. Proc. Natl. Acad. Sci. U.S.A. 110, 2864–2869. doi: 10.1073/pnas.1212484110
Ubeda, M., Wang, X. Z., Zinszner, H., Wu, I., Habener, J. F., and Ron, D. (1996). Stress-induced binding of the transcriptional factor CHOP to a novel DNA control element. Mol. Cell. Biol. 16, 1479–1489.
Urano, F., Wang, X., Bertolotti, A., Zhang, Y., Chung, P., Harding, H. P., et al. (2000). Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666. doi: 10.1126/science.287.5453.664
Valencia-Sanchez, M. A., Liu, J., Hannon, G. J., and Parker, R. (2006). Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 20, 515–524. doi: 10.1101/gad.1399806
Vattem, K. M., and Wek, R. C. (2004). Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 101, 11269–11274. doi: 10.1073/pnas.0400541101
Vecchi, C., Montosi, G., Zhang, K., Lamberti, I., Duncan, S. A., Kaufman, R. J., et al. (2009). ER stress controls iron metabolism through induction of hepcidin. Science 325, 877–880. doi: 10.1126/science.1176639
Ventoso, I., Kochetov, A., Montaner, D., Dopazo, J., and Santoyo, J. (2012). Extensive translatome remodeling during ER stress response in mammalian cells. PLoS ONE 7:e35915. doi: 10.1371/journal.pone.0035915
Vinson, C., Acharya, A., and Taparowsky, E. J. (2006). Deciphering B-ZIP transcription factor interactions in vitro and in vivo. Biochim. Biophys. Acta 1759, 4–12. doi: 10.1016/j.bbaexp.2005.12.005
Wang, F. M., Chen, Y. J., and Ouyang, H. J. (2011). Regulation of unfolded protein response modulator XBP1s by acetylation and deacetylation. Biochem. J. 433, 245–252. doi: 10.1042/BJ20101293
Wang, X. Z., Harding, H. P., Zhang, Y., Jolicoeur, E. M., Kuroda, M., and Ron, D. (1998). Cloning of mammalian Ire1 reveals diversity in the ER stress response. EMBO J. 17, 5708–5717. doi: 10.1093/emboj/17.19.5708
Wang, X. Z., and Ron, D. (1996). Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science 272, 1347–1349. doi: 10.1126/science.272.5266.1347
Wang, Y., Shen, J., Arenzana, N., Tirasophon, W., Kaufman, R. J., and Prywes, R. (2000). Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J. Biol. Chem. 275, 27013–27020.
Wang, Y., Vera, L., Fischer, W. H., and Montminy, M. (2009). The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature 460, 534–537. doi: 10.1038/nature08111
Watatani, Y., Ichikawa, K., Nakanishi, N., Fujimoto, M., Takeda, H., Kimura, N., et al. (2008). Stress-induced translation of ATF5 mRNA is regulated by the 5'-untranslated region. J. Biol. Chem. 283, 2543–2553. doi: 10.1074/jbc.M707781200
Wethmar, K., Begay, V., Smink, J. J., Zaragoza, K., Wiesenthal, V., Dorken, B., et al. (2010). C/EBPbetaDeltauORF mice–a genetic model for uORF-mediated translational control in mammals. Genes Dev. 24, 15–20. doi: 10.1101/gad.557910
Wills-Karp, M., and Finkelman, F. D. (2008). Untangling the complex web of IL-4 and IL-13-mediated signaling pathways. Sci. Signal 1, 1–4. doi: 10.1126/scisignal.1.51.pe55
Winnay, J. N., Boucher, J., Mori, M. A., Ueki, K., and Kahn, C. R. (2010). A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat. Med. 16, 438–445. doi: 10.1038/nm.2121
Woo, C. W., Cui, D., Arellano, J., Dorweiler, B., Harding, H., Fitzgerald, K. A., et al. (2009). Adaptive suppression of the ATF4-CHOP branch of the unfolded protein response by toll-like receptor signalling. Nat. Cell Biol. 11, 1473–1480. doi: 10.1038/ncb1996
Wu, J., Ruas, J. L., Estall, J. L., Rasbach, K. A., Choi, J. H., Ye, L., et al. (2011). The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1alpha/ATF6alpha complex. Cell Metab. 13, 160–169. doi: 10.1016/j.cmet.2011.01.003
Wu, J., Rutkowski, D. T., Dubois, M., Swathirajan, J., Saunders, T., Wang, J., et al. (2007). ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 13, 351–364. doi: 10.1016/j.devcel.2007.07.005
Wynn, T. A. (2003). IL-13 Effector Functions. Annu. Rev. Immunol. 21, 425–456. doi: 10.1146/annurev.immunol.21.120601.141142
Yamamoto, K., Sato, T., Matsui, T., Sato, M., Okada, T., Yoshida, H., et al. (2007). Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 13, 365–376. doi: 10.1016/j.devcel.2007.07.018
Ye, J., Rawson, R. B., Komuro, R., Chen, X., Dave, U. P., Prywes, R., et al. (2000). ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 6, 1355–1364. doi: 10.1016/S1097-2765(00)00133-7
Yoshida, H., Haze, K., Yanagi, H., Yura, T., and Mori, K. (1998). Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 273, 33741–33749. doi: 10.1074/jbc.273.50.33741
Yoshida, H., Matsui, T., Hosokawa, N., Kaufman, R. J., Nagata, K., and Mori, K. (2003). A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 4, 265–271. doi: 10.1016/S1534-5807(03)00022-4
Yoshida, H., Okada, T., Haze, K., Yanagi, H., Yura, T., Negishi, M., et al. (2001a). Endoplasmic reticulum stress-induced formation of transcription factor complex ERSF including NF-Y (CBF) and activating transcription factors 6alpha and 6beta that activates the mammalian unfolded protein response. Mol. Cell. Biol. 21, 1239–1248. doi: 10.1128/MCB.21.4.1239-1248.2001
Yoshida, H., Matsui, T., Yamamoto, A., Okada, T., and Mori, K. (2001b). XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891. doi: 10.1016/S0092-8674(01)00611-0
Yoshida, H., Okada, T., Haze, K., Yanagi, H., Yura, T., Negishi, M., et al. (2000). ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 20, 6755–6767. doi: 10.1128/MCB.20.18.6755-6767.2000
Zhang, C., Wang, G., Zheng, Z., Maddipati, K. R., Zhang, X., Dyson, G., et al. (2012). Endoplasmic reticulum-tethered transcription factor cAMP responsive element-binding protein, hepatocyte specific, regulates hepatic lipogenesis, fatty acid oxidation, and lipolysis upon metabolic stress in mice. Hepatology 55, 1070–1082. doi: 10.1002/hep.24783
Zhang, K., Shen, X., Wu, J., Sakaki, K., Saunders, T., Rutkowski, D. T., et al. (2006). Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 124, 587–599. doi: 10.1016/j.cell.2005.11.040
Zhang, K., Wong, H. N., Song, B., Miller, C. N., Scheuner, D., and Kaufman, R. J. (2005). The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J. Clin. Invest. 115, 268–281.
Zhang, W. G., Chen, L., Dong, Q., He, J., Zhao, H. D., Li, F. L., et al. (2013). mmu-miR-702 functions as an anti-apoptotic mirtron by mediating ATF6 inhibition in mice. Gene 531, 235–242. doi: 10.1016/j.gene.2013.09.005
Zhou, D., Palam, L. R., Jiang, L., Narasimhan, J., Staschke, K. A., and Wek, R. C. (2008). Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J. Biol. Chem. 283, 7064–7073. doi: 10.1074/jbc.M708530200
Keywords: ER stress, unfolded protein response (UPR), mRNA stability, Transcription Factors, gene regulatory networks (GRN)
Citation: Arensdorf AM, Diedrichs D and Rutkowski DT (2013) Regulation of the transcriptome by ER stress: non-canonical mechanisms and physiological consequences. Front. Genet. 4:256. doi: 10.3389/fgene.2013.00256
Received: 24 September 2013; Accepted: 08 November 2013;
Published online: 02 December 2013.
Edited by:
Kezhong Zhang, Wayne State University, USACopyright © 2013 Arensdorf, Diedrichs and Rutkowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: D. Thomas Rutkowski, Department of Anatomy and Cell Biology, University of Iowa Carver College of Medicine, 51 Newton Rd., Iowa City, 52242 IA, USA e-mail:dGhvbWFzLXJ1dGtvd3NraUB1aW93YS5lZHU=