 Martin Braud1
Martin Braud1 David A. Magee2
David A. Magee2 Stephen D. E. Park3
Stephen D. E. Park3 Tad S. Sonstegard4
Tad S. Sonstegard4 Sinead M. Waters5
Sinead M. Waters5 David E. MacHugh2,6
David E. MacHugh2,6 Charles Spillane1*
Charles Spillane1*- 1Genetics and Biotechnology Lab, Plant and AgriBiosciences Research Centre, School of Natural Sciences, National University of Ireland Galway, University Road, Galway, Ireland
- 2Animal Genomics Laboratory, UCD School of Agriculture and Food Science, University College Dublin, Dublin, Ireland
- 3IdentiGEN Ltd, Unit 2, Trinity Enterprise Centre, Dublin, Ireland
- 4Recombinetics, Inc., St. Paul, MN, USA
- 5Animal and Bioscience Research Department, Animal and Grassland Research and Innovation Centre, Teagasc, Dunsany, Ireland
- 6UCD Conway Institute of Biomolecular and Biomedical Research, University College Dublin, Dublin, Ireland
The domestication of cattle from the now-extinct wild aurochs (Bos primigenius) involved selection for physiological and behavioral traits, with underlying genetic factors that remain largely unknown. Non-coding microRNAs have emerged as key regulators of the spatio-temporal expression of target genes controlling mammalian growth and development, including in livestock species. During the domestication process, selection of mutational changes in miRNAs and/or miRNA binding sites could have provided a mechanism to generate some of the traits that differentiate domesticated cattle from wild aurochs. To investigate this, we analyzed the open reading frame DNA sequence of 19,994 orthologous protein-coding gene pairs from extant Bos taurus genomes and a single extinct B. primigenius genome. We identified miRNA binding site polymorphisms in the 3′ UTRs of 1,620 of these orthologous genes. These 1,620 genes with altered miRNA binding sites between the B. taurus and B. primigenius lineages represent candidate domestication genes. Using a novel Score Site ratio metric we have ranked these miRNA-regulated genes according to the extent of divergence between miRNA binding site presence, frequency and copy number between the orthologous genes from B. taurus and B. primigenius. This provides an unbiased approach to identify cattle genes that have undergone the most changes in miRNA binding (i.e., regulation) between the wild aurochs and modern-day cattle breeds. In addition, we demonstrate that these 1,620 candidate domestication genes are enriched for roles in pigmentation, fertility, neurobiology, metabolism, immunity and production traits (including milk quality and feed efficiency). Our findings suggest that directional selection of miRNA regulatory variants was important in the domestication and subsequent artificial selection that gave rise to modern taurine cattle.
Introduction
Since their domestication from wild aurochs (Bos primigenius) some 10,000 years ago, cattle (Bos taurus and Bos indicus) have been continuously exposed to both natural and artificial selection (Bradley and Magee, 2006; Magee et al., 2014). These selection processes, coupled with mutation, genetic drift and admixture have produced the multitude of extant humpless taurine and humped zebu cattle breeds (Food and Agriculture Organization, 2015). However, the underlying genetic factors (including genes, regulatory elements and DNA sequence polymorphisms) contributing to phenotypic traits selected during and after cattle domestication remain largely unknown (Elsik et al., 2009; Canavez et al., 2012; Kemper and Goddard, 2012; Womack, 2012; Gutierrez-Gil et al., 2015; Utsunomiya et al., 2015).
Many studies have highlighted the importance of microRNAs (miRNAs)—short non-coding RNAs that post-transcriptionally modulate gene expression—in regulating a wide range of biological processes in vertebrate species, including domestic livestock (McDaneld, 2009; Liu et al., 2010; Fatima and Morris, 2013; Wang et al., 2013). In mammals, miRNA targeting has predominantly been associated with the 3′ UTR region of transcripts derived from open reading frames (ORFs), typically leading to down-regulation through triggering RNA degradation, RNA instability and/or reduced translation (Winter et al., 2009; Krol et al., 2010; Huntzinger and Izaurralde, 2011).
DNA sequence polymorphisms occurring within the targeted miRNA binding site of genes can influence phenotypic traits of economic importance in mammalian livestock (Clop et al., 2006; Sasaki et al., 2013; Hou et al., 2015; Rong et al., 2015; An et al., 2016). Hence, we hypothesized that DNA sequence polymorphisms affecting miRNA-mediated gene regulation are likely associated with pre- and post-domestication differences in cattle, particularly for traits under natural and artificial selection within the ∼10,000 year time frame of cattle domestication (Larson and Fuller, 2014; Larson et al., 2014; MacHugh et al., 2016).
Using sequence data from the recently published B. primigenius genome (Park et al., 2015) and sequenced genomes of modern-day B. taurus (Zimin et al., 2009), we developed a ranking metric [Score Site ratio (SSr)] to identify the miRNA-targeted bovine genes that display the greatest sequence difference in their miRNA binding sites between domesticated taurine cattle breeds and wild aurochs. Using this approach we have identified genes displaying high levels of miRNA binding site DNA sequence differences (polymorphisms) between orthologous gene pairs where one gene is from the wild aurochs and the other is from domesticated taurine cattle. Genes displaying post-domestication variation in miRNA binding sites are shown to be involved in mammalian growth and development (e.g., ephrin signaling and androgen signaling) and cellular function and metabolism (e.g., cholesterol biosynthesis). We consider that some of the miRNA binding site polymorphisms identified here, which are either absent or specific to the aurochs genome, modulate expression of candidate domestication genes associated with traits that have been selected during or subsequent to domestication.
Materials and Methods
Aurochs Genome Sequence Data
All B. primigenius genome sequence data was retrieved from the Sequence Read Archive database (accession number SRX1266623). The laboratory and bioinformatics methods used for DNA extraction, purification and sequence analysis of the CPC98 aurochs specimen have been described by us previously (Park et al., 2015). The genome sequence data is from an archeologically verified B. primigenius specimen retrieved in 1998 from Carsington Pasture Cave in Derbyshire, England, and radiocarbon dated to 6,738 ± 68 calibrated (cal.) years before present (yBP). The authenticity of the CPC98 genome sequence was verified using a number of different approaches that are detailed by Park et al. (2015). The high-quality single nucleotide polymorphisms (SNPs) that were used for the analyses described here were identified using a stringent computational workflow fully described in Park et al. (2015).
Identification of B. primigenius 3′ UTR Sequences
To identify miRNA binding sites in target genes, 3′ UTR sequences and associated bovine genome coordinates were obtained from Ensembl using the Biomart retrieval tool (Kinsella et al., 2011) and the B. taurus UMD3.1 reference genome assembly (Zimin et al., 2009). 3′ UTR sequence information from B. taurus UMD3.1 annotation has been used to predict and retrieve the 3′ UTR sequences from the B. primigenius genome consensus sequence, using the coordinates associated with each B. taurus 3′ UTR. All B. taurus and B. primigenius 3′ UTR pairs were then used to identify sequence polymorphisms within putative miRNA binding sites for B. taurus and B. primigenius. In addition, the SNP data from the sequencing of 49 B. taurus cattle samples from the Park et al. (2015) aurochs genome study have been used to confirm the fixation of the SNPs identified between B. taurus and B. primigenius across different breeds (Angus, Holstein, Jersey, Limousin, Romagnola, Fleckvieh, N’Dama).
Identification of miRNA Complementary Binding Sites in 3′ UTR of Target Genes Using TargetScan
To identify miRNA target binding sites in B. taurus and B. primigenius 3′ UTR sequences, the TargetScan 6.1 software package was used (Lewis et al., 2005). Targetscan has been previously shown to be an accurate predictor of miRNA binding sites (Baek et al., 2008; Ulitsky et al., 2010). The prediction score for each miRNA binding site incorporates the evolutionary conservation of a predicted miRNA binding site among different species (Friedman et al., 2009). It also takes into account the presence of many structural motifs in mRNA sequences related to miRNA binding to infer a particular score for each site.
The TargetScan 6.1 package consists of two main Perl scripts: targetscan_60.pl and targetscan_60_context_scores.pl. The first script, targetscan_60.pl, identifies only the seed region recognition motif composed of six nucleotides (positions 2–7 in the mature miRNA) and one additional flanking nucleotide (position 1 or 8 of the mature miRNA) to identify four types of seed region [six nucleotides, seven nucleotides (6 + 1 adenosine), seven nucleotides, and eight nucleotides (7 + 1 adenosine)]. The script targetscan_60.pl takes as its input the sequence alignments of the 3′ UTRs and the list of miRNA seed regions for B. taurus and B. primigenius miRNAs. For each alignment the script returns an output containing the miRNA seed regions that have been identified in the 3′ UTR and their corresponding positions. The output of this script was parsed using a custom Python script that reads through the output files and extracts the information contained in each line of the file. This includes the binding site nucleotide location in the 3′ UTR and the group of species that have a site at the same position in the 3′ UTR multiple alignments. B. taurus and B. primigenius seed regions that were identified, yet contained ambiguous nucleotides in the B. primigenius 3′ UTR were removed from the analysis. Any seed sites overlapping with a previously identified seed site for the same miRNA were also removed.
The second Targetscan Perl script, targetscan_60_context_scores.pl, generates a score for each site taking into account the position of the seed region in the 3′ UTR, its nucleotide composition, the pairing of the miRNA in its 13–16 position with the UTR and the composition of the flanking region of the binding site. The Targetscan context score corresponds to a prediction, based on experimental miRNA repression expressed as log-ratios (Grimson et al., 2007; Garcia et al., 2011). For this script, the results files from the first Targetscan script (cataloged as either a gain, a loss, an increase or a decrease of miRNA target sites), the alignments and the list of mature miRNA sequences specific to either B. taurus or B. primigenius were used as inputs. To parse the results, a custom Python script was used to generate two results files: a file containing genes with miRNA binding sites polymorphic between aurochs and domestic taurine cattle and a second file with shared miRNA binding sites across the two taxa. All results in which ambiguous nucleotides were present in the UTR target region (complementary to the 3′ end region of the mature miRNA) were removed. For miRNAs with multiple binding sites, the score for each gene/miRNA pair was generated by summing the individual context scores (calculated by TargetScan for each site).
To rank the targeted genes, we calculated an additional score, termed the SSr), which facilitated analysis of the extent to which miRNA binding sites were polymorphic between B. taurus and B. primigenius, relative to those that were common to both B. taurus and B. primigenius. The SSr was calculated by summing all context scores for each predicted miRNA binding site across each 3′ UTR and multiplying this sum by the ratio of the number of polymorphic sites over the number of shared sites between B. taurus and B. primigenius. The SSr statistic is calculated using the following equation:
where n is the number of miRNA binding sites identified for a particular gene UTR; scorei is the score for each predicted site; psn represents the number of miRNA binding sites polymorphic between B. taurus and B. primigenius for a particular gene; and csn represents the number of miRNA binding sites common to B. taurus and B. primigenius for a particular gene.
The SSr score was used to rank genes according to the extent of their miRNA site polymorphisms differentiating B. primigenius and B. taurus. This provided a basis for ranking of the genes according to the extent of dysregulation of their miRNA binding sites between aurochs and modern cattle. In addition, the SSr statistic provides a combined weighting for each gene, which combines both the number of different miRNA binding sites (ratio of number of sites) and their possible impact (sum of TargetScan scores) between B. taurus and B. primigenius and facilitates normalization of the number of discovered sites, which can depend of the size of the 3′ UTR.
Analyses of Biological Pathways Involved in Differential miRNA Targeting of B. taurus Genes Relative to B. primigenius Genes
To identify the biological pathways that are most affected by miRNA binding site variation between the 3′ UTRs of B. taurus and B. primigenius genes, Ingenuity Systems Pathway Analysis (IPA – www.ingenuity.com) was used. B. taurus genes with variant miRNA binding sites (when compared with B. primigenius) were used as the input to IPA. The IPA core analysis evaluates enrichment of the selected genes in different pathways, returning a P-value for each pathway identified, which was then used to rank enriched pathways and identify the most statistically significant (P-value < 0.05). A pathway score was then assigned to each significant pathway corresponding to the sum of the SSr score for each of the genes associated with a particular pathway. Significant pathways were then assigned a summed SSr score to produce a ranked list of the pathways containing genes with significant miRNA binding site differences between B. taurus and B. primigenius.
Analysis of Transition/Transversion Ratios
To determine the transition/transversion ratio (ti/tv) in the 3′ UTR miRNA binding site SNPs, the fixed SNPs identified in the miRNA binding sites were sorted into two mutational classes based on the variants observed: a transition class (A↔G, C↔T) and a transversion class (A↔C, A↔T, C↔G, G↔T). Using a custom python script, the number of variants were summed for each group and the ti/tv ratio was calculated.
Results
DNA Sequence Differences between the 3′ UTRs of B. taurus and B. primigenius Genes
Of the 19,994 annotated protein-coding genes in the taurine cattle genome (assembly UMD3.1), 11,761 have an annotated 3′ UTR (59%). To identify all SNPs differentiating the 3′ UTRs of B. taurus and B. primigenius genes, we compared these 11,761 3′ UTR sequences from B. taurus with the corresponding mapped regions in B. primigenius. We determined that 3,066 (26%) of these genes contained one or more SNPs, and identified a total of 6,355 SNPs that were different between B. taurus and B. primigenius in the 3,066 3′ UTRs. Analysis of the numbers of SNPs in the 3′ UTR of each gene yielded a mean of 1.92 SNPs per 3′ UTR, with a median value of 1.00 SNPs per 3′ UTR and a maximum of 27 B. taurus/B. primigenius SNPs for the methionine adenosyltransferase I, alpha gene (MATA1).
DNA Sequence Polymorphisms Differentiating B. taurus and B. primigenius 3′ UTRs Generate Altered miRNA Binding Sites
Alteration of miRNA binding sites in 3′ UTRs of target genes can disrupt the regulatory effects of existing miRNAs, and/or can generate new miRNA binding sites. To investigate this further, we screened for SNPs that differentiate the 3′ UTRs of B. taurus and B. primigenius genes. From the 3,066 3′ UTRs exhibiting SNPs differentiating the two taxa, we identified a total of 2,634 SNPs located in miRNA binding sites within 1,620 genes. Notably, DNA sequence polymorphisms in the 3′ UTR of the protein kinase, AMP-activated, gamma 3 non-catalytic subunit gene (PRKAG3) produced the highest number of miRNA binding site changes across all of these 1,620 genes. For the PRKAG3 3′ UTR, our analysis demonstrated that 23 different miRNAs have a modification of their binding potential (gain or loss). These 23 miRNAs were associated with 15 distinct miRNA-binding sites that differ between the B. taurus and B. primigenius PRKAG3 genes.
For our analysis of polymorphisms in 3′ UTRs, individual miRNA binding sites were classified as a “gain” when SNPs between B. taurus and B. primigenius generate a new miRNA binding sites in B. taurus 3′ UTRs, and as a “loss” when they lead to loss of an existing miRNA binding site in the B. taurus genome. In some cases, a polymorphic miRNA binding site can be considered simultaneously as a “gain” and a “loss” when one or more SNPs generate new binding sites for one particular miRNA and eliminate a binding site for another miRNA. An additional scenario we considered is when a miRNA gains a new binding site in B. taurus but also has another common binding site between B. taurus and B. primigenius. We categorize such miRNA binding site copy number “amplification” as an “increase,” while a reduction of binding site copy number it is categorized as a “decrease.”
Among the 1,620 genes with SNPs in 3′ UTR miRNA binding sites, we determined that 892 genes have lost their miRNA binding sites including up to 13 losses for the membrane-spanning four-domains, subfamily A, member seven gene (MS4A7) in the B. taurus lineage. We also identified 885 genes that have gained a miRNA binding site in B. taurus, which is not present in B. primigenius including a gain of up to 11 new miRNA binding sites for the 3-hydroxy-3-methylglutaryl-CoA synthase 2 gene (HMGCS2). There is an overlap of 322 genes that have both a gain and loss of miRNA binding sites in B. taurus. We also determined that 306 genes in B. taurus have a number of miRNA-binding sites that have increased in copy number for at least one miRNA (up to seven miRNA binding sites for the ubiquitin specific peptidase 33 gene, USP33), while 250 genes have a decrease in miRNA binding site copy number up to 11 miRNA binding sites for the MAT1A gene in comparison to B. primigenius (Figure 1; Supplementary Table S1).
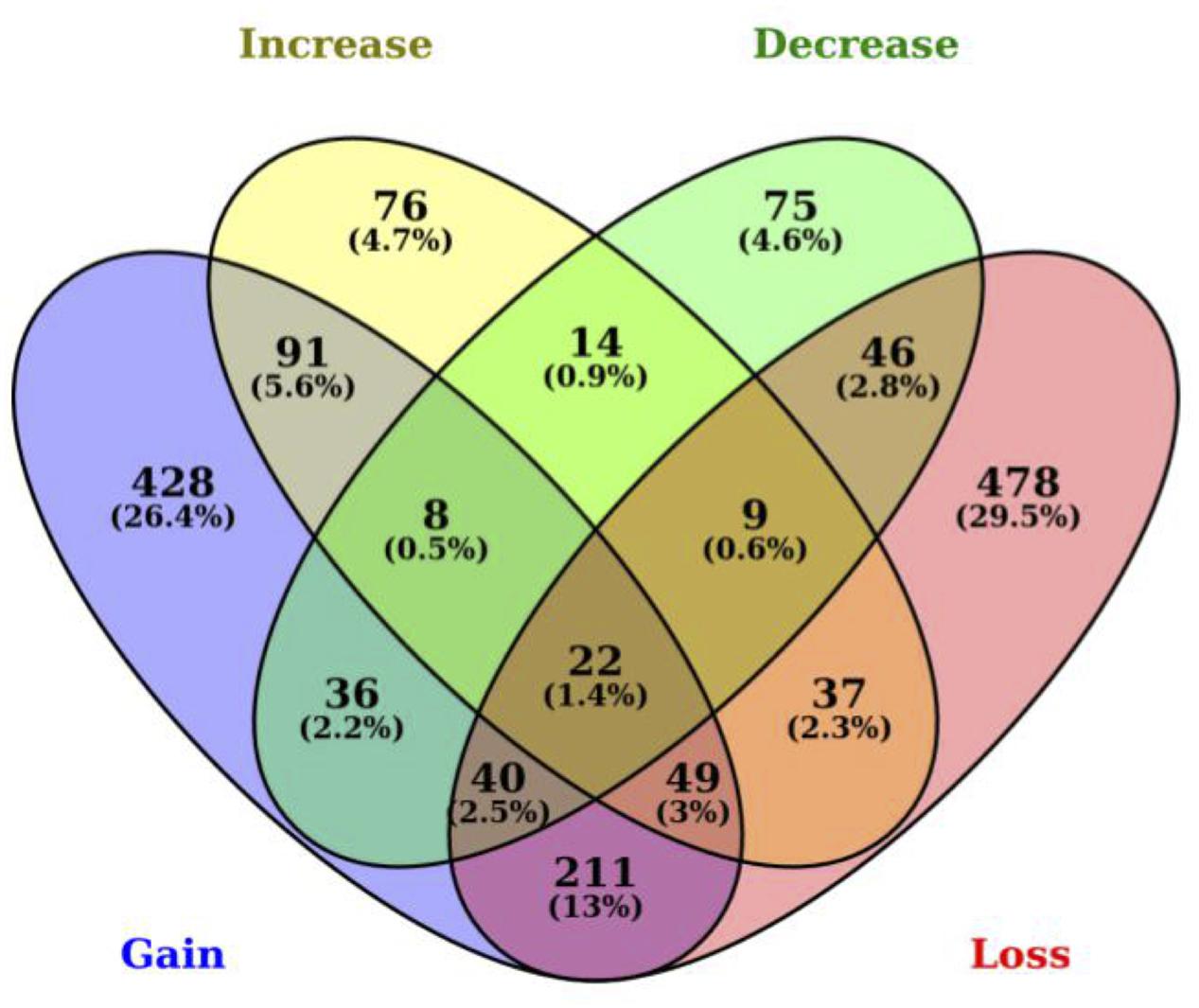
FIGURE 1. Polymorphisms between Bos taurus and Bos primigenius in 3′ UTRs generate or disrupt miRNA binding sites. Venn diagram representing the number of genes with 3′ UTR polymorphisms between B. taurus and B. primigenius. MiRNA binding sites are represented as gained (blue), lost (yellow), increased (green), or decreased (red). The intersection represents SNPs in 3′ UTRs that generate a binding site for one miRNA while disrupting another miRNA binding site in the same gene (within the same miRNA-binding site region).
Using SSr Scores to Rank the 3′ UTRs that have undergone the Most Extensive miRNA Binding Site Modifications between B. primigenius and B. taurus
We hypothesized that selection processes (both natural and artificial) during and after the domestication of cattle may have led to new allelic variants at miRNA binding sites of genes that are associated with anatomical, physiological and behavioral trait variation in cattle. To identify miRNA-targeted genes (and associated cellular pathways) that displayed the most miRNA targeting divergence between B. taurus and B. primigenius, we used the SSr score metric to rank all of the genes. Several of the top 10 SSr-ranked genes were found to be involved in key metabolic and physiological processes. These included: (1) the guanylate cyclase activator 2B gene (GUCA2B), in which the binding sites for five miRNAs (bta-miR-541, bta-miR-2433, bta-miR-1777a, bta-miR-2454, and bta-miR-2327) were absent in B. taurus but present in B. primigenius, with only one common binding site between both; (2) the HMGCS2 gene, in which the binding sites for 11 miRNAs were present in B. taurus but absent in B. primigenius and also the number of binding sites increased for two miRNAs in B. taurus; and (3) the MS4A7 gene, in which the binding sites for 13 miRNAs were absent in B. taurus and present in B. primigenius, which represented the most significant loss of potential miRNA binding sites in our entire data set.
We also identified three genes important for metabolism: the glutathione peroxidase 5 gene (GPX5), the casein kappa gene (CSN3) and the MAT1A gene, which displayed DNA sequence differences between the B. taurus and B. primigenius lineages in their respective 3′ UTR miRNA binding sites (Table 1).

TABLE 1. Genes with polymorphic miRNA binding sites in candidate miRNA-regulated cattle domestication genes.
Identification of Networks, Pathways, and Biological Functions Enriched in Genes with miRNA Target Sites Polymorphic between B. taurus and B. primigenius
We used Ingenuity Systems Pathway Analysis (IPA) to gain further insight into the biological functions, canonical pathways and biological networks enriched for genes with miRNA binding sites polymorphic between B. taurus and B. primigenius. For this, we uploaded the 1,620 genes identified by SSr analysis and ranked the IPA-identified functions, canonical pathways and networks based on enrichment P-values generated by IPA. Using this approach, we identified 25 networks, 74 pathways, and 500 functions that are enriched for genes with miRNA binding sites polymorphic between B. taurus and B. primigenius.
The two top-ranked networks we identified were metabolism and neurological diseases. Among the significantly enriched canonical pathways differentially regulated by the miRNAs were pathways involved in immunobiology (for example, the IL-1, CXCL4, and NFAT pathways), physiology and development (for example, the ephrin signaling pathway and the PTEN and PI3K/AKT signaling pathways), and reproductive physiology (for example, the androgen signaling pathway) (Figure 2).
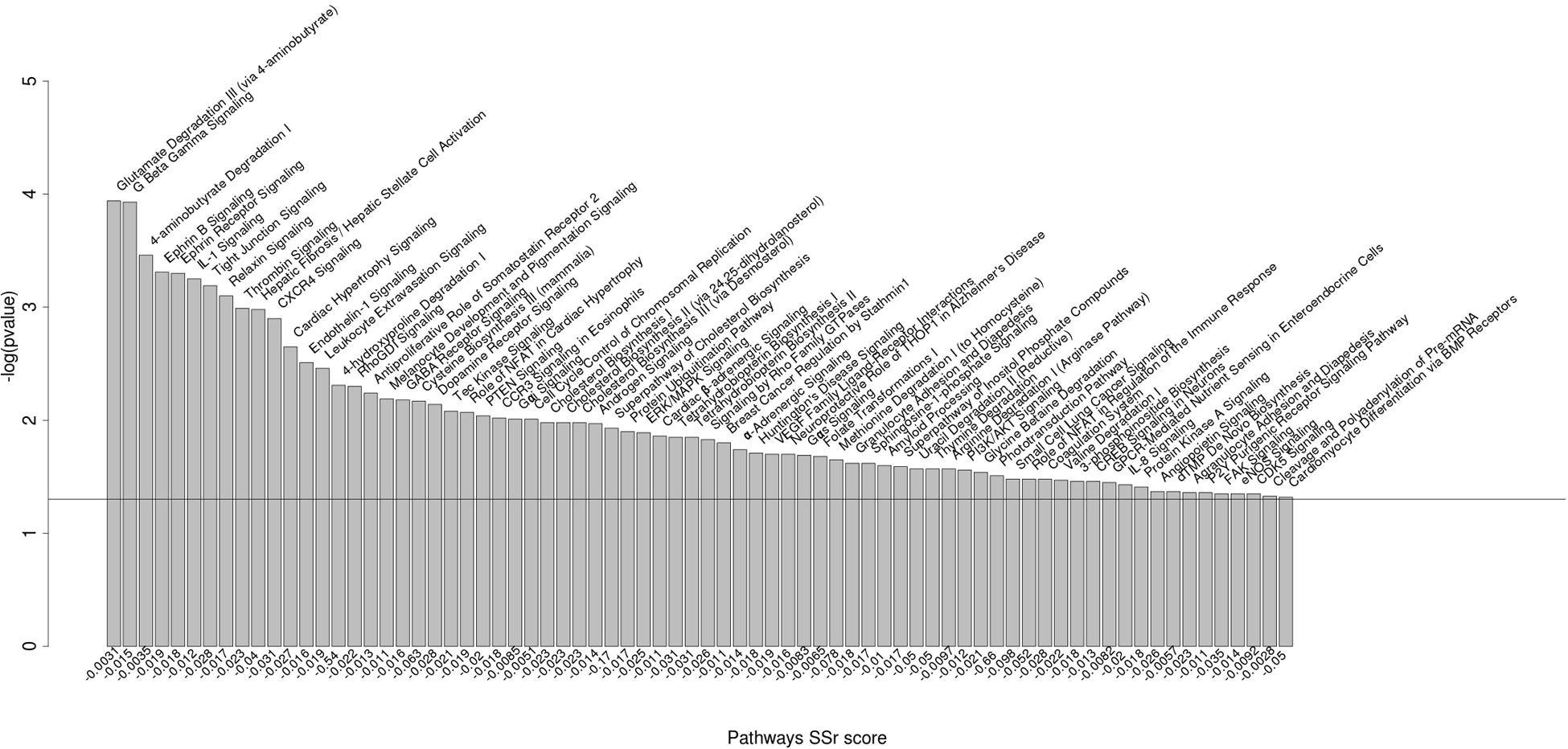
FIGURE 2. Biological pathways significantly enriched for miRNA polymorphic target genes. Biological pathways significantly enriched for genes having SNPs in miRNA binding sites between B. primigenius and B. taurus [P-value < 0.05; -log10(P-value) > 1.3]. The pathways significantly enriched are represented by –log10(P-value) in the ordinate. The ratio of genes corresponds to the number of candidate genes over the number of background genes in one pathway (ranked by P-value and SSr score).
The Polymorphic miRNA Binding Sites in Aurochs 3′ UTRs are not Enriched for Transitions due to Post-mortem Deamination
The analysis of ancient DNA sequence data can be complicated where post-mortem deamination leading to C→T or G→A transition substitutions occurs (Hofreiter et al., 2001; Briggs et al., 2007). To investigate whether the 2,634 SNPs (that were polymorphic in the miRNA binding sites in aurochs 3′ UTRs) could be enriched for transitions due to post-mortem deamination, the ti/tv ratio was calculated for the set of 2,634 SNPs located in miRNA binding sites and was determined to be 1.93:1.00. Previously, for the entire CPC98 aurochs genome, we examined 2,009,261 biallelic SNPs (73.3% of which were homozygous) and determined a transition to transversion (ti/tv) ratio of 2.19:1.00 (Park et al., 2015), which is very similar to ti/tv ratios obtained for female and male Holstein genome sequences: 2.18:1.00 and 2.16:1.00, respectively (Koks et al., 2013, 2014). The lower ti/tv ratio detected for the 3′ UTR miRNA SNPs is consistent with published ti/tv ratios calculated for mammalian 3′ UTRs (Chen et al., 2015), and suggests that post-mortem deamination is not a confounding factor for SNPs within miRNA binding sites located in aurochs 3′ UTRs.
Discussion
Analyses of whole genome sequence data from mammalian livestock (and domesticated companion animals) and their wild ancestors have begun to reveal microevolutionary processes, key genetic variants and biological pathways underlying animal domestication (Orlando et al., 2013; Schubert et al., 2014; Der Sarkissian et al., 2015; Librado et al., 2015; Park et al., 2015; Skoglund et al., 2015; Frantz et al., 2016; MacHugh et al., 2016). MicroRNAs play key roles in regulating a wide range of biological processes at the post-transcriptional level. Indeed, DNA sequence variation in miRNA-binding sites of mammalian genes can have a profound influence on phenotypic variation.
In this study, we hypothesized that genome-wide comparison of miRNA-binding sites in the 3′ UTR of genes of domestic B. taurus with miRNA binding sites in the 3′ UTR of orthologs in the wild ancestor, B. primigenius, would identify which genes have undergone the most change in relation to miRNA-regulation between wild and domesticated cattle. We consider that genes which have undergone the most divergence in miRNA binding sites between wild and domestic cattle represent candidate miRNA-regulated domestication loci, which may be associated with phenotypic traits subject to selection during and after the domestication process.
Our analysis of B. taurus and B. primigenius genome sequences has identified 2,634 SNPs in the 3′ UTR miRNA binding sites of 1,620 bovine genes that differentiate domestic cattle and the wild aurochs. These SNPs were demonstrated to have a low ti/tv ratio, indicating that post-mortem cytosine deamination is not a significant confounding factor in establishing the veracity of these sequence polymorphisms. To identify the 3′ UTRs that have undergone the greatest extent of miRNA binding site divergence between B. taurus and B. primigenius we developed a novel SSr metric. Using the SSr ranking approach we have identified candidate miRNA-regulated domestication genes and pathways with roles in immunology, metabolism, physiology, development and fertility.
Neurological Development Functions
Modifications to neurological development likely led to tameness and facilitated human handling of cattle, for instance through increased docility (Jensen, 2014; Jensen and Wright, 2014). In our previous study of the B. primigenius genome, we demonstrated that the bovine phytanoyl-CoA 2-hydroxylase interacting protein gene (PHYHIP), which is involved in neurodevelopment, is targeted differentially by a miRNA gene variant (i.e., not a miRNA binding site variant in the target gene) that is polymorphic between aurochs and domestic cattle (Park et al., 2015). In this study, we further demonstrate that Eph/ephrin intracellular signaling pathways, involved in brain development (Lai and Ip, 2009; Hruska and Dalva, 2012; Cramer and Miko, 2016), are enriched in genes with miRNA target sites that are highly polymorphic between aurochs and cattle (Figure 2). Given the selection during domestication for behavioral traits (such as docility), we consider that variation in the mRNA-based regulation (i.e., both miRNA genes and miRNA binding sites in 3′ UTRs) of these neurological genes could have been subject to artificial selection.
Metabolism Functions
Metabolism is an important biological process that likely was modified during and after domestication, as cattle adapted to new diets and feeding regimens associated with increasingly sophisticated animal husbandry practices (Noe-Nygaard et al., 2005). In this context, we have identified three genes and two pathways, which are related to metabolism and have undergone extensive miRNA-based regulation modification in B. taurus relative to B. primigenius. For instance, the 3-hydroxy-3-methylglutaryl-CoA synthase 2 gene (HMGCS2) is targeted by miRNAs in B. taurus but not in B. primigenius, and is ranked in the top 10 genes by SSr score (Table 1). The HMGCS2 protein product is a mitochondrial enzyme involved in ketone metabolism, which varies depending on the availability of dietary carbohydrates (Kostiuk et al., 2010). We also identified miRNA binding site differences between B. taurus and B. primigenius in the MAT1A gene, which is expressed exclusively in mature mammalian liver cells where it is involved in regulation of hepatic function (Mato et al., 2002). The MAT1A gene product also plays a role in S-adenosylmethionine formation, which has been linked in knockdown mice to hepatic regeneration (Chen et al., 2004). Polymorphisms in MAT1A have also been associated with levels of homocysteine in dietary fatty acids (Huang et al., 2012).
Our SSr-ranking approach also demonstrated that the PRKAG3 gene displays extensive miRNA binding site polymorphisms between B. taurus and B. primigenius. Previous work has shown that PRKAG3 sequence variants have been associated with differences in porcine skeletal muscle glycogen levels (Milan et al., 2000) and also meat production and quality traits in both domestic pigs and cattle (Reardon et al., 2010; Ryan et al., 2012; Uimari and Sironen, 2014; Zhang et al., 2015; Bongiorni et al., 2016). Finally, the insulin receptor signaling and valine degradation pathways, both of which are associated with metabolism, are also highly enriched for genes with miRNA binding site polymorphisms between B. taurus and B. primigenius. We consider that differential miRNA-regulatory evolution of metabolic genes and pathways between B. taurus and B. primigenius may underlie adaptations to changing diets under captivity and animal husbandry.
Immunological Functions
Under most models of animal domestication, increased confinement and interaction with humans results in increased exposure of early domesticates to novel pathogens, potentially leading to increased selection pressure on genes regulating immune functions (Cleaveland et al., 2001; Harper and Armelagos, 2013; Reperant et al., 2013). In this context, we discovered that the MS4A7 gene is the gene that has lost the most miRNA binding sites in domestic taurine cattle compared to aurochs. MS4A7 encodes a member of the membrane-spanning 4A (MS4A) protein family (Eon Kuek et al., 2016), which is expressed in B lymphocytes (Pant et al., 2006; Zuccolo et al., 2013). We have also identified an enrichment of genes with miRNA binding site variants belonging to the nuclear factor of activated T-cells (NFATs) pathway, which is involved in the establishment of innate immune responses to infection (Fric et al., 2012; Zanoni and Granucci, 2012). It is possible that the changes in miRNA regulation of these immune genes between wild and domesticated cattle could be a consequence of domestication and subsequent natural and artificial selection.
Reproduction, Fertility, and Lactation Functions
Reproduction, fertility, and related biological processes (such as lactation) have been profoundly affected by domestication and human-mediated artificial selection of livestock (Larson and Fuller, 2014; Larson et al., 2014; Zeder, 2015). In this context, we identified two genes and four biological pathways that are ranked highly by SSr score (Table 1). For instance, GPX5 is ranked 28th and is targeted by eight additional miRNAs in domestic taurine cattle compared to aurochs. The GPX5 protein plays an important role in the maintenance of spermatozoa DNA integrity (Chabory et al., 2010). GUCA2B which encodes guanylate cyclase activator 2B (uroguanylin), is our highest ranked gene with five less miRNA binding sites in domestic cattle compared to aurochs. GUCA2B is an activator of guanylate cyclase (encoded by GUCY2C), a membrane protein that modulates cyclic guanosine monophosphate (cGMP) levels, thereby regulating water homeostasis in the intestine and kidneys (Kita et al., 1994; Sindic and Schlatter, 2006). Guanylate cyclase also has a well-characterized role in vasodilatation and blood pressure regulation (Buys and Sips, 2014). Furthermore, a microarray analysis identified variation of GUCA2B expression levels in mice and shows that this variation has an effect on pregnancy and fertility (McConaha et al., 2011).
The androgen signaling pathways, which were enriched in genes containing miRNA-binding site polymorphisms between domestic taurine cattle and aurochs, also have a significant impact on both male and female fertility (Smith and Walker, 2014; Walters, 2015). In addition to these two genes, we determined that five pathways related to fertility and reproduction are enriched in genes displaying miRNA binding site variations between B. taurus and B. primigenius. The PTEN and PI3K/AKT signaling pathways display significant enrichments that could be linked to fertility. Conjugated linoleic acid has been shown to increase the expression of PTEN and PI3K/AKT regulating granulosa cell proliferation and steroidogenesis in buffaloes, which could affect ovulation and therefore fertility (Sharma and Singh, 2012). Another significantly enriched pathway, the IL-8 pathway is activated during theca cell differentiation and in pre-ovulatory follicle differentiation (Walsh et al., 2012). Finally, one of the highest ranked genes showing loss of miRNA targets in domestic taurine cattle (rank 52/1,606), CSN3, encodes casein kappa, a protein associated with the composition and nutritional properties of milk (Caroli et al., 2009; Holt et al., 2013; Pereira, 2014).
Conclusion
It is important to note that the British B. primigenius genome used for this study, although having some admixture with modern-day cattle, is not the main contributor to European cattle breeds (as the major progenitor is known to be B. primigenius from the Fertile Crescent domestication center). As we only had one genome of aurochs available we compared this genome to multiple cattle genomes. As more aurochs genome sequence data becomes available some of the miRNA variants identified here as specific to the aurochs lineages may be found to be present in other aurochs genomes (e.g., in aurochs from the Fertile Crescent). Nonetheless, our study provides the first reference point for ongoing and future investigations of the contribution of miRNA variants to cattle domestication.
Overall, we identify a range of candidate miRNA-regulated domestication genes that may underlie some of the phenotypic traits that distinguish domesticated taurine cattle (B. taurus) from wild aurochs (B. primigenius). These genes have significant miRNA binding site polymorphism divergence between both taxa, which we propose could influence neurobiological, metabolic, immunobiological and reproductive phenotypes associated with the transition from wild aurochs to domesticated taurine cattle.
Author Contributions
Conceived and designed the project: CS, MB. Managed and oversaw research project: CS. Provided the Aurochs genome sequence and SNP data: SP, DAM, DEM, TS. Analyzed SNPs in miRNA binding site: MB. Wrote the paper: MB, CS. Discussed and edited the paper: CS, MB, DAM, SP, TS, SW, DEM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer DN declared a shared affiliation, though no other collaboration, with one of the authors TS to the handling Editor, who ensured that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
This research was financed by a Teagasc Walsh Fellowship to MB (2010062); Investigator Grants from Science Foundation Ireland (SFI/08/IN.1/B2038 and SFI/13/IA/1820); a Research Stimulus Grant from the Department of Agriculture, Food and the Marine (No: RSF 06 406); and a European Union Framework 7 Project Grant (No: KBBE-211602-MACROSYS).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fgene.2017.00003/full#supplementary-material
References
An, X., Song, Y., Bu, S., Ma, H., Gao, K., Hou, J., et al. (2016). Association of polymorphisms at the microRNA binding site of the caprine KITLG 3′-UTR with litter size. Sci. Rep. 6:25691. doi: 10.1038/srep25691
Baek, D., Villen, J., Shin, C., Camargo, F. D., Gygi, S. P., and Bartel, D. P. (2008). The impact of microRNAs on protein output. Nature 455, 64–71. doi: 10.1038/nature07242
Bongiorni, S., Gruber, C. E., Bueno, S., Chillemi, G., Ferre, F., Failla, S., et al. (2016). Transcriptomic investigation of meat tenderness in two Italian cattle breeds. Anim. Genet. 47, 273–287. doi: 10.1111/age.12418
Bradley, D. G., and Magee, D. A. (2006). Genetics and the Origins of Domestic Cattle. Berkeley, CA: University of California Press.
Briggs, A. W., Stenzel, U., Johnson, P. L., Green, R. E., Kelso, J., Prufer, K., et al. (2007). Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. U.S.A. 104, 14616–14621. doi: 10.1073/pnas.0704665104
Buys, E., and Sips, P. (2014). New insights into the role of soluble guanylate cyclase in blood pressure regulation. Curr. Opin. Nephrol. Hypertens 23, 135–142. doi: 10.1097/01.mnh.0000441048.91041.3a
Canavez, F. C., Luche, D. D., Stothard, P., Leite, K. R., Sousa-Canavez, J. M., Plastow, G., et al. (2012). Genome sequence and assembly of Bos indicus. J. Hered. 103, 342–348. doi: 10.1093/jhered/esr153
Caroli, A. M., Chessa, S., and Erhardt, G. J. (2009). Invited review: milk protein polymorphisms in cattle: effect on animal breeding and human nutrition. J. Dairy Sci. 92, 5335–5352. doi: 10.3168/jds.2009-2461
Chabory, E., Damon, C., Lenoir, A., Henry-Berger, J., Vernet, P., Cadet, R., et al. (2010). Mammalian glutathione peroxidases control acquisition and maintenance of spermatozoa integrity. J. Anim. Sci. 88, 1321–1331. doi: 10.2527/jas.2009-2583
Chen, L., Zeng, Y., Yang, H., Lee, T. D., French, S. W., Corrales, F. J., et al. (2004). Impaired liver regeneration in mice lacking methionine adenosyltransferase 1A. FASEB J. 18, 914–916. doi: 10.1096/fj.03-1204fje
Chen, X., Paranjape, T., Stahlhut, C., McVeigh, T., Keane, F., Nallur, S., et al. (2015). Targeted resequencing of the microRNAome and 3′UTRome reveals functional germline DNA variants with altered prevalence in epithelial ovarian cancer. Oncogene 34, 2125–2137. doi: 10.1038/onc.2014.117
Cleaveland, S., Laurenson, M. K., and Taylor, L. H. (2001). Diseases of humans and their domestic mammals: pathogen characteristics, host range and the risk of emergence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356, 991–999. doi: 10.1098/rstb.2001.0889
Clop, A., Marcq, F., Takeda, H., Pirottin, D., Tordoir, X., Bibe, B., et al. (2006). A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat. Genet. 38, 813–818. doi: 10.1038/ng1810
Cramer, K. S., and Miko, I. J. (2016). Eph-ephrin signaling in nervous system development. F1000Res. 5:F1000. doi: 10.12688/f1000research.7417.1
Der Sarkissian, C., Ermini, L., Schubert, M., Yang, M. A., Librado, P., Fumagalli, M., et al. (2015). Evolutionary genomics and conservation of the endangered Przewalski’s horse. Curr. Biol. 25, 2577–2583. doi: 10.1016/j.cub.2015.08.032
Elsik, C. G., Tellam, R. L., Worley, K. C., Gibbs, R. A., Muzny, D. M., Weinstock, G. M., et al. (2009). The genome sequence of taurine cattle: a window to ruminant biology and evolution. Science 324, 522–528. doi: 10.1126/science.1169588
Eon Kuek, L., Leffler, M., Mackay, G. A., and Hulett, M. D. (2016). The MS4A family: counting past 1, 2 and 3. Immunol. Cell Biol. 94, 11–23. doi: 10.1038/icb.2015.48
Fatima, A., and Morris, D. G. (2013). MicroRNAs in domestic livestock. Physiol. Genomics 45, 685–696. doi: 10.1152/physiolgenomics.00009.2013
Food and Agriculture Organization (2015). “The second report on the state of the world’s animal genetic resources for food and agriculture,” in Commission on Genetic Resources for Food and Agriculture Assessments, eds B.D. Scherf and D. Pilling (Rome: Food and Agriculture Organization).
Frantz, L. A., Mullin, V. E., Pionnier-Capitan, M., Lebrasseur, O., Ollivier, M., Perri, A., et al. (2016). Genomic and archaeological evidence suggest a dual origin of domestic dogs. Science 352, 1228–1231. doi: 10.1126/science.aaf3161
Fric, J., Zelante, T., Wong, A. Y., Mertes, A., Yu, H. B., and Ricciardi-Castagnoli, P. (2012). NFAT control of innate immunity. Blood 120, 1380–1389. doi: 10.1182/blood-2012-02-404475
Friedman, R. C., Farh, K. K., Burge, C. B., and Bartel, D. P. (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92–105. doi: 10.1101/gr.082701.108
Garcia, D. M., Baek, D., Shin, C., Bell, G. W., Grimson, A., and Bartel, D. P. (2011). Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat. Struct. Mol. Biol. 18, 1139–1146. doi: 10.1038/nsmb.2115
Grimson, A., Farh, K. K., Johnston, W. K., Garrett-Engele, P., Lim, L. P., and Bartel, D. P. (2007). MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell. 27, 91–105. doi: 10.1016/j.molcel.2007.06.017
Gutierrez-Gil, B., Arranz, J. J., and Wiener, P. (2015). An interpretive review of selective sweep studies in Bos taurus cattle populations: identification of unique and shared selection signals across breeds. Front. Genet. 6:167. doi: 10.3389/fgene.2015.00167
Harper, K. N., and Armelagos, G. J. (2013). Genomics, the origins of agriculture, and our changing microbe-scape: time to revisit some old tales and tell some new ones. Am. J. Phys. Anthropol. 152(Suppl. 57), 135–152. doi: 10.1002/ajpa.22396
Hofreiter, M., Jaenicke, V., Serre, D., von Haeseler, A., and Paabo, S. (2001). DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 29, 4793–4799. doi: 10.1093/nar/29.23.4793
Holt, C., Carver, J. A., Ecroyd, H., and Thorn, D. C. (2013). Invited review: caseins and the casein micelle: their biological functions, structures, and behavior in foods. J. Dairy Sci. 96, 6127–6146. doi: 10.3168/jds.2013-6831
Hou, J., An, X., Song, Y., Gao, T., Lei, Y., and Cao, B. (2015). Two mutations in the caprine MTHFR 3′UTR regulated by microRNAs are associated with milk production traits. PLoS ONE 10:e0133015. doi: 10.1371/journal.pone.0133015
Hruska, M., and Dalva, M. B. (2012). Ephrin regulation of synapse formation, function and plasticity. Mol. Cell. Neurosci. 50, 35–44. doi: 10.1016/j.mcn.2012.03.004
Huang, T., Tucker, K., Lee, Y., Crott, J., Parnell, L., Shen, J., et al. (2012). MAT1A variants modulate the effect of dietary fatty acids on plasma homocysteine concentrations. Nutr. Metab. Cardiovasc. Dis. 22, 362–368. doi: 10.1016/j.numecd.2010.07.015
Huntzinger, E., and Izaurralde, E. (2011). Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat. Rev. Genet. 12, 99–110. doi: 10.1038/nrg2936
Jensen, P. (2014). Behavior genetics and the domestication of animals. Annu. Rev. Anim. Biosci. 2, 85–104. doi: 10.1146/annurev-animal-022513-114135
Jensen, P., and Wright, D. (2014). “Behavioral genetics and animal domestication,” in Genetics and the Behavior of Domestic Animals, 2nd Edn, eds T. Grandin and M. J. Deesing (San Diego, CA: Academic Press), 41–79.
Kemper, K. E., and Goddard, M. E. (2012). Understanding and predicting complex traits: knowledge from cattle. Hum. Mol. Genet. 21, R45–R51. doi: 10.1093/hmg/dds332
Kinsella, R. J., Kahari, A., Haider, S., Zamora, J., Proctor, G., Spudich, G., et al. (2011). Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (Oxford) 2011, bar030. doi: 10.1093/database/bar030
Kita, T., Smith, C. E., Fok, K. F., Duffin, K. L., Moore, W. M., Karabatsos, P. J., et al. (1994). Characterization of human uroguanylin: a member of the guanylin peptide family. Am. J. Physiol. 266, F342–F348.
Koks, S., Lilleoja, R., Reimann, E., Salumets, A., Reemann, P., and Jaakma, U. (2013). Sequencing and annotated analysis of the Holstein cow genome. Mamm. Genome 24, 309–321. doi: 10.1007/s00335-013-9464-0
Koks, S., Reimann, E., Lilleoja, R., Lattekivi, F., Salumets, A., Reemann, P., et al. (2014). Sequencing and annotated analysis of full genome of Holstein breed bull. Mamm. Genome 25, 363–373. doi: 10.1007/s00335-014-9511-5
Kostiuk, M. A., Keller, B. O., and Berthiaume, L. G. (2010). Palmitoylation of ketogenic enzyme HMGCS2 enhances its interaction with PPARalpha and transcription at the Hmgcs2 PPRE. FASEB J. 24, 1914–1924. doi: 10.1096/fj.09-149765
Krol, J., Loedige, I., and Filipowicz, W. (2010). The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 11, 597–610. doi: 10.1038/nrg2843
Lai, K. O., and Ip, N. Y. (2009). Synapse development and plasticity: roles of ephrin/Eph receptor signaling. Curr. Opin. Neurobiol. 19, 275–283. doi: 10.1016/j.conb.2009.04.009
Larson, G., and Fuller, D. Q. (2014). The evolution of animal domestication. Annu. Rev. Ecol. Evol. Syst. 45, 115–136. doi: 10.1146/annurev-ecolsys-110512-135813
Larson, G., Piperno, D. R., Allaby, R. G., Purugganan, M. D., Andersson, L., Arroyo-Kalin, M., et al. (2014). Current perspectives and the future of domestication studies. Proc. Natl. Acad. Sci. U.S.A. 111, 6139–6146. doi: 10.1073/pnas.1323964111
Lewis, B. P., Burge, C. B., and Bartel, D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. doi: 10.1016/j.cell.2004.12.035
Librado, P., Der Sarkissian, C., Ermini, L., Schubert, M., Jonsson, H., Albrechtsen, A., et al. (2015). Tracking the origins of Yakutian horses and the genetic basis for their fast adaptation to subarctic environments. Proc. Natl. Acad. Sci. U.S.A. 112, E6889–E6897. doi: 10.1073/pnas.1513696112
Liu, H. C., Hicks, J. A., Trakooljul, N., and Zhao, S. H. (2010). Current knowledge of microRNA characterization in agricultural animals. Anim. Genet. 41, 225–231. doi: 10.1111/j.1365-2052.2009.01995.x
MacHugh, D. E., Larson, G., and Orlando, L. (2016). Taming the past: ancient DNA and the study of animal domestication. Annu. Rev. Anim. Biosci. doi: 10.1146/annurev-animal-022516-022747 [Epub ahead of print].
Magee, D. A., MacHugh, D. E., and Edwards, C. J. (2014). Interrogation of modern and ancient genomes reveals the complex domestic history of cattle. Anim. Front. 4, 7–22. doi: 10.2527/af.2014-0017
Mato, J. M., Corrales, F. J., Lu, S. C., and Avila, M. A. (2002). S-Adenosylmethionine: a control switch that regulates liver function. FASEB J. 16, 15–26. doi: 10.1096/fj.01-0401rev
McConaha, M. E., Eckstrum, K., An, J., Steinle, J. J., and Bany, B. M. (2011). Microarray assessment of the influence of the conceptus on gene expression in the mouse uterus during decidualization. Reproduction 141, 511–527. doi: 10.1530/rep-10-0358
McDaneld, T. G. (2009). MicroRNA: mechanism of gene regulation and application to livestock. J. Anim. Sci. 87, E21–E28. doi: 10.2527/jas.2008-1303
Milan, D., Jeon, J. T., Looft, C., Amarger, V., Robic, A., Thelander, M., et al. (2000). A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science 288, 1248–1251. doi: 10.1126/science.288.5469.1248
Noe-Nygaard, N., Price, T. D., and Hede, S. U. (2005). Diet of aurochs and early cattle in southern Scandinavia: evidence from N-15 and C-13 stable isotopes. J. Archaeol. Sci. 32, 855–871. doi: 10.1016/j.jas.2005.01.004
Orlando, L., Ginolhac, A., Zhang, G., Froese, D., Albrechtsen, A., Stiller, M., et al. (2013). Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 499, 74–78. doi: 10.1038/nature12323
Pant, P. V., Tao, H., Beilharz, E. J., Ballinger, D. G., Cox, D. R., and Frazer, K. A. (2006). Analysis of allelic differential expression in human white blood cells. Genome Res. 16, 331–339. doi: 10.1101/gr.4559106
Park, S. D., Magee, D. A., McGettigan, P. A., Teasdale, M. D., Edwards, C. J., Lohan, A. J., et al. (2015). Genome sequencing of the extinct Eurasian wild aurochs, Bos primigenius, illuminates the phylogeography and evolution of cattle. Genome Biol. 16, 234. doi: 10.1186/s13059-015-0790-2
Pereira, P. C. (2014). Milk nutritional composition and its role in human health. Nutrition 30, 619–627. doi: 10.1016/j.nut.2013.10.011
Reardon, W., Mullen, A. M., Sweeney, T., and Hamill, R. M. (2010). Association of polymorphisms in candidate genes with colour, water-holding capacity, and composition traits in bovine M. longissimus and M. semimembranosus. Meat Sci. 86, 270–275. doi: 10.1016/j.meatsci.2010.04.013
Reperant, L. A., Cornaglia, G., and Osterhaus, A. (2013). “The importance of understanding the human-animal interface from early hominins to global citizens,” in One Health: The Human-Animal-Environment Interfaces in Emerging Infectious Diseases: The Concept and Examples of a One Health Approach, eds J. S. Mackenzie, M. Jeggo, P. Daszak, and J. A. Richt (Berlin: Springer), 49–81.
Rong, E., Zhang, Z., Qiao, S., Yang, H., Yan, X., Li, H., et al. (2015). Functional characterization of a single nucleotide polymorphism in the 3′ untranslated region of sheep DLX3 gene. PLoS ONE 10:e0137135. doi: 10.1371/journal.pone.0137135
Ryan, M. T., Hamill, R. M., O’Halloran, A. M., Davey, G. C., McBryan, J., Mullen, A. M., et al. (2012). SNP variation in the promoter of the PRKAG3 gene and association with meat quality traits in pig. BMC Genet. 13:66. doi: 10.1186/1471-2156-13-66
Sasaki, S., Ibi, T., Watanabe, T., Matsuhashi, T., Ikeda, S., and Sugimoto, Y. (2013). Variants in the 3′ UTR of general Transcription factor IIF, polypeptide 2 affect female calving efficiency in Japanese Black cattle. BMC Genet. 14:41. doi: 10.1186/1471-2156-14-41
Schubert, M., Jonsson, H., Chang, D., Der Sarkissian, C., Ermini, L., Ginolhac, A., et al. (2014). Prehistoric genomes reveal the genetic foundation and cost of horse domestication. Proc. Natl. Acad. Sci. U.S.A. 111, E5661–E5669. doi: 10.1073/pnas.1416991111
Sharma, I., and Singh, D. (2012). Conjugated linoleic acids attenuate FSH- and IGF1-stimulated cell proliferation; IGF1,GATA4, and aromatase expression; and estradiol-17beta production in buffalo granulosa cells involving PPARgamma, PTEN, and PI3K/Akt. Reproduction 144, 373–383. doi: 10.1530/rep-12-0079
Sindic, A., and Schlatter, E. (2006). Cellular effects of guanylin and uroguanylin. J. Am. Soc. Nephrol. 17, 607–616. doi: 10.1681/asn.2005080818
Skoglund, P., Ersmark, E., Palkopoulou, E., and Dalen, L. (2015). Ancient wolf genome reveals an early divergence of domestic dog ancestors and admixture into high-latitude breeds. Curr. Biol. 25, 1515–1519. doi: 10.1016/j.cub.2015.04.019
Smith, L. B., and Walker, W. H. (2014). The regulation of spermatogenesis by androgens. Semin. Cell Dev. Biol . 30, 2–13. doi: 10.1016/j.semcdb.2014.02.012
Uimari, P., and Sironen, A. (2014). A combination of two variants in PRKAG3 is needed for a positive effect on meat quality in pigs. BMC Genet. 15:29. doi: 10.1186/1471-2156-15-29
Ulitsky, I., Laurent, L. C., and Shamir, R. (2010). Towards computational prediction of microRNA function and activity. Nucleic Acids Res. 38:e160. doi: 10.1093/nar/gkq570
Utsunomiya, Y. T., Perez, O., Brien, A. M., Sonstegard, T. S., Solkner, J., and Garcia, J. F. (2015). Genomic data as the “hitchhiker’s guide” to cattle adaptation: tracking the milestones of past selection in the bovine genome. Front. Genet. 6:36. doi: 10.3389/fgene.2015.00036
Walsh, S. W., Fair, T., Browne, J. A., Evans, A. C., and McGettigan, P. A. (2012). Physiological status alters immunological regulation of bovine follicle differentiation in dairy cattle. J. Reprod. Immunol. 96, 34–44. doi: 10.1016/j.jri.2012.07.002
Walters, K. A. (2015). Role of androgens in normal and pathological ovarian function. Reproduction 149, R193–R218. doi: 10.1530/rep-14-0517
Wang, X., Gu, Z., and Jiang, H. (2013). MicroRNAs in farm animals. Animal 7, 1567–1575. doi: 10.1017/s1751731113001183
Winter, J., Jung, S., Keller, S., Gregory, R. I., and Diederichs, S. (2009). Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat. Cell Biol. 11, 228–234. doi: 10.1038/ncb0309-228
Womack, J. E. (2012). First steps: bovine genomics in historical perspective. Anim. Genet. 43(Suppl. 1), 2–8. doi: 10.1111/j.1365-2052.2012.02382.x
Zanoni, I., and Granucci, F. (2012). Regulation and dysregulation of innate immunity by NFAT signaling downstream of pattern recognition receptors (PRRs). Eur. J. Immunol. 42, 1924–1931. doi: 10.1002/eji.201242580
Zeder, M. A. (2015). Core questions in domestication research. Proc. Natl. Acad. Sci. U.S.A. 112, 3191–3198. doi: 10.1073/pnas.1501711112
Zhang, C., Wang, Z., Bruce, H., Kemp, R. A., Charagu, P., Miar, Y., et al. (2015). Genome-wide association studies (GWAS) identify a QTL close to PRKAG3 affecting meat pH and colour in crossbred commercial pigs. BMC Genet. 16:33. doi: 10.1186/s12863-015-0192-1
Zimin, A. V., Delcher, A. L., Florea, L., Kelley, D. R., Schatz, M. C., Puiu, D., et al. (2009). A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 10, R42. doi: 10.1186/gb-2009-10-4-r42
Keywords: microRNA, polymorphism, evolution, domestication, agriculture, Bos primigenius, Bos taurus, ancient DNA
Citation: Braud M, Magee DA, Park SDE, Sonstegard TS, Waters SM, MacHugh DE and Spillane C (2017) Genome-Wide microRNA Binding Site Variation between Extinct Wild Aurochs and Modern Cattle Identifies Candidate microRNA-Regulated Domestication Genes. Front. Genet. 8:3. doi: 10.3389/fgene.2017.00003
Received: 10 July 2016; Accepted: 09 January 2017;
Published: 31 January 2017.
Edited by:
Martien Groenen, Wageningen University and Research Centre, NetherlandsReviewed by:
Dan Nonneman, Agricultural Research Service (USDA), USALaurent Frantz, University of Oxford, UK
Copyright © 2017 Braud, Magee, Park, Sonstegard, Waters, MacHugh and Spillane. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Charles Spillane, Y2hhcmxlcy5zcGlsbGFuZUBudWlnYWx3YXkuaWU=