 Yunpeng Cao1
Yunpeng Cao1 Yahui Han2
Yahui Han2 Dandan Meng1Guohui Li1
Dandan Meng1Guohui Li1 Dahui Li1
Dahui Li1 Muhammad Abdullah2Qing Jin1Yi Lin1Yongping Cai1*
Muhammad Abdullah2Qing Jin1Yi Lin1Yongping Cai1*- 1School of Life Sciences, Anhui Agricultural University, Hefei, China
- 2State Key Laboratory of Tea Plant Biology and Utilization, Anhui Agricultural University, Hefei, China
WUSCHEL-related homeobox (WOX) family is one of the largest group of transcription factors (TFs) specifically found in plant kingdom. WOX TFs play an important role in plant development processes and evolutionary novelties. Although the roles of WOXs in Arabidopsis and rice have been well-studied, however, little are known about the relationships among the main clades in the molecular evolution of these genes in Rosaceae. Here, we carried out a genome-wide analysis and identified 14, 10, 10, and 9 of WOX genes from four Rosaceae species (Fragaria vesca, Prunus persica, Prunus mume, and Pyrus bretschneideri, respectively). According to evolutionary analysis, as well as amino acid sequences of their homodomains, these genes were divided into three clades with nine subgroups. Furthermore, due to the conserved structural patterns among these WOX genes, it was proposed that there should exist some highly conserved regions of microsynteny in the four Rosaceae species. Moreover, most of WOX gene pairs were presented with the conserved orientation among syntenic genome regions. In addition, according to substitution models analysis using PMAL software, no significant positive selection was detected, but type I functional divergence was identified among certain amino acids in WOX protein. These results revealed that the relaxed purifying selection might be the main driving force during the evolution of WOX genes in the tested Rosaceae species. Our result will be useful for further precise research on evolution of the WOX genes in family Rosaceae.
Introduction
The WUSCHEL-related homeobox (WOX) gene family encodes a group of plant-specific transcription factors (TFs), which belongs to the homeodomain (HD) TF superfamily (Deveaux et al., 2008; Zhang et al., 2015). There are 15 and 13 members of the WOX family in Arabidopsis thaliana and rice (Oryza sativa) genomes, respectively (Haecker et al., 1991; Graaff et al., 2009; Xin et al., 2010). WOX TFs have been reported to play important role in plant development, such as regulating dynamic balance of stem cell division and differentiation, embryo development, and post-embryonic development (Palovaara et al., 2010; Yadav et al., 2010; Ueda et al., 2011). Bioinformatics analysis showed that WOX homology sequences were found in the genomes of Selaginella, Bryophyta and Chlorophyta, but not in the Rhodophyta genome, indicating that WOX family might originate from green algae (Mukherjee et al., 2009; Lian et al., 2014). According to the phylogenetic analysis among A. thaliana and Petunia hybrida, tomato (Solanum lycopersicum) and rice (O. sativa), the WOX family was divided into three separate clades: WUS/modern clade, the intermediate clade, and the ancient clade (Haecker et al., 1991). Research on the structural characteristics of WOX members showed that the evolutionary branch members contained a specific WUS box (T-L-X-L-F-P-X-X, where X represents an amino acid) (Haecker et al., 1991). WUS box is an essential component for WUS regulation of stem tip meristem stem cell homeostasis and floral meristem morphogenesis (Ikeda and Ohme-Takagi, 2009). Moreover, AtWUS and AtWOX5 within modern/WUS clade, can redundantly maintain the apical stem cells under undifferentiated status (Sarkar et al., 2007); AtWOX4 can influent the process of secondary growth by modulating the activity of vascular cambium (Hirakawa et al., 2010); AtWOX1/3 can coordinate the development of paraxial and distal ends during the leaf development; the primordial initiation and development within meristem were terminated by overexpression of AtWOX6. Within the intermediate clade, AtWOX9 can maintain the growth and division of meristematic cells (Wu et al., 2005); AtWOX11 was specifically expressed in the cambium, and can promote the formation of adventitious roots (Zhao et al., 2009). Within the ancient clade, AtWOX13 can promote the formation of embryonic placenta during fruit development (Romera-Branchat et al., 2013); AtWOX14 and AtWOX4 can redundantly regulate the differentiation of vascular meristem (Etchells et al., 2013). These studies suggest that the WOX gene family is widely involved in the regulation of plant meristem. The members of WOX gene family appear to be functionally diverse. Although this gene family in some model plants, such as Arabidopsis and rice, has been studied on a phylogenetic scale, a comprehensive molecular evolutionary study remains elusive in Rosaceae species. Recently, a number of researches on application of comparative genome in analysis of evolution and function of the gene family have been reported (Cao et al., 2016a). Similar to other highly conserved genes, the WOX gene and its flanking sequences are likely to be conserved with microsynteny, which can promote the transfer of genetic knowledge among the related species of Rosaceae. The genomes of pear (Pyrus bretschneideri), peach (Prunus persica), mei (Prunus mume), and strawberry (Fragaria vesca) were published in Shulaev et al. (2011), Zhang et al. (2012), Verde et al. (2013), and Wu et al. (2013), respectively. Therefore, the availability of whole-genome sequences for four members of three Rosaceae subfamilies (Fragaria, Prunus, and Pyrus) have enabled us to explore the selection regimes under which WOX genes have diversified during the radiation of the Rosaceae. The present research could lead to a better understanding of WOX gene family on evolutionary history and diversification in Rosaceae.
Materials and Methods
Database Search
WOX genes were identified from the genome data representing the four Rosaceae species (P. bretschneideri, P. persica, F. vesca, and P. mume, respectively). Two different methods were used to identify WOX genes in the P. bretschneideri, P. persica, F. vesca, and P. mume genome: (1) BLASTP search using Arabidopsis and rice WOX protein sequences according to previous research methods (Cao et al., 2016a,d), and (2) the screening of Hidden Markov Model profile (PF00046) in four Rosaceae genome using DNAtools software with an e-value cut off of 0.001 (Gehring, 1992). All candidate WOX proteins were confirmed to have a complete WOX domain using both Pfam (Punta et al., 2011), SMART dadabases (Letunic et al., 2012) and InterProScan database (Zdobnov and Apweiler, 2001).
Phylogenetic Trees Construction
The multiple alignment of WOX proteins in five plant species (P. bretschneideri, P. persica, F. vesca, P. mume, and A. thaliana) was performed using CLUSTAL_X software (Thompson et al., 1997). Subsequently, we constructed NJ (neighbor-joining) tree using MEGA version 5.1 software (Tamura et al., 2011) with the following parameters: bootstrap (1000 replicate), pairwise deletion and Poisson correction. At the same time, we used ML (maximum-likelihood) and ME (Minimum-evolution) methods to generate the phylogenetic trees to validate the topologies.
Exon–Intron Structural Analysis and Identification of Conserved Motifs
The online program Gene Structure Display Server (Hu et al., 2014) was used to detect the exon–intron structure of cDNAs and genomic DNA sequences. Subsequently, the MEME (Multiple Em for Motif Elicitation, Version 4.11.1) program (Bailey et al., 2015) was used to obtain the motifs in all candidate WOX proteins, with the parameters: the maximum number of motifs at 20, and the optimum motif width between six and 200 residues. Furthermore, the Pfam database (Punta et al., 2011), SMART software (Letunic et al., 2012), and InterProScan database (Zdobnov and Apweiler, 2001) were used to annotate these structure motifs.
Microsynteny Analysis
According to the comparisons of the specific regions containing WOX genes, we carried out microsynteny analysis across the four Rosaceae species. Similarly, the WOX genes of P. bretschneideri, P. persica, P. mume, and F. vesca were categorized based on their classification in the evolutionary tree. Subsequently, all WOX genes in P. bretschneideri, P. persica, F. vesca, and P. mume were set as anchor sites, according to their physical location. Then the flanking protein-coding genes of the WOX gene in one species were compared with those in other species. The criterion for dividing an interspecific synteny block is to locate three or more conserved homologous genes within 100 KB between genomes (BLASTP E-value < 10-10) (Wang et al., 2012).
Selective Pressure and Functional Divergence Analysis
To future understand whether the WOX genes have undergone positive selection during evolution, maximum likelihood codon models (site models and branch-site models) in PAML software (Yang, 2007) were performed. Three pairs of models (M0 vs. M3, M1a vs. M2a, and M7 vs. M8) were utilized to detect positive selection sites. In the free site models, M0 (one ratio), M1a (neutral), M2a (selection), M3 (discrete), M7 (beta), and M8 (beta and ω) were evaluated by the likelihood ratio test (LRT). The LRT was used to judge which model was more suitable in the two models, and the amino acids sites with positive selection were obtained by the Bayesian method of PAML software (Yang, 2007).
Functional divergence analysis of amino acid sequence data was performed using Diverge 2.0 combined with constructed phylogenetic tree (Gu, 1999, 2006; Gu et al., 2013). The type I functional divergence led to a change of functional limitation, which was highly correlated with the evolution rate after gene duplication (Gu, 1999, 2006; Gu et al., 2013). Type II functional divergence did not result in a change in the functional limitation of the members after gene duplication, but the change of physical and chemical properties of amino acid residues (Gu, 1999, 2006; Gu et al., 2013).
cis-Acting Elements Analysis
To identify putative cis-elements in promoter regions of WOX genes, the PlantCARE database (Lescot et al., 2002) was used. 2000 bp genomic sequence upstream of the start codon (ATG) was used for cis-acting elements analysis.
Expression Profiles of FvWOX Genes
The normalized data (Fragments Per Kilobase Exon model per Million mapped fragments, FPKM) during F. vesca development was reported by Darwish et al. (2013), and available from SGR GBrowse. A gene was thought to be expressed if the FPKM value was greater than or equal to 0 FPKM in at least one of the 14 tissues. Subsequently, the transcriptome data of FvWOXs was visualized using the R software1.
Results
Identification and Chromosomal Distribution of WOX Genes in Rosaceae
For identification of WOXs gene families, the genome data of tested Rosaceae species was subjected to HMM and BLASTP searches. As a result it is revealed, presence of 9, 10, 10, and 14 WOX genes in P. mume, P. bretschneideri, P. persica, and F. vesca, respectively (Supplementary Table S1). These WOX genes were named according to method of Haecker et al. (1991). For this purpose, the phylogenetic tree was carried out based on multiple sequence alignments for the full-length WOX protein sequences of tested Rosaceae species and A. thaliana. The identified WOX genes of these tested four Rosaceae species were renamed according to the evolutionary relationship as shown in Supplementary Table S1. Subsequently, the distribution of these WOX genes on chromosomes was identified, based on genomic annotation information. As shown in Figure 1, it is discovered that the WOX genes were unevenly distributed among the chromosomes in each of tested four species. In the P. bretschneideri genome, four of WOX genes were distributed on chromosome 15, while remaining distributed on chromosomes 3, 5, 6, 12, and 17. In F. vesca, five and four of 14 WOX genes were distributed among chromosomes 3 and 5, respectively. In both P. persica and P. mume, three WOX genes distributed were found on one chromosome (no. 7 and no. 8, respectively), with others scattered across different chromosomes (Figure 1).
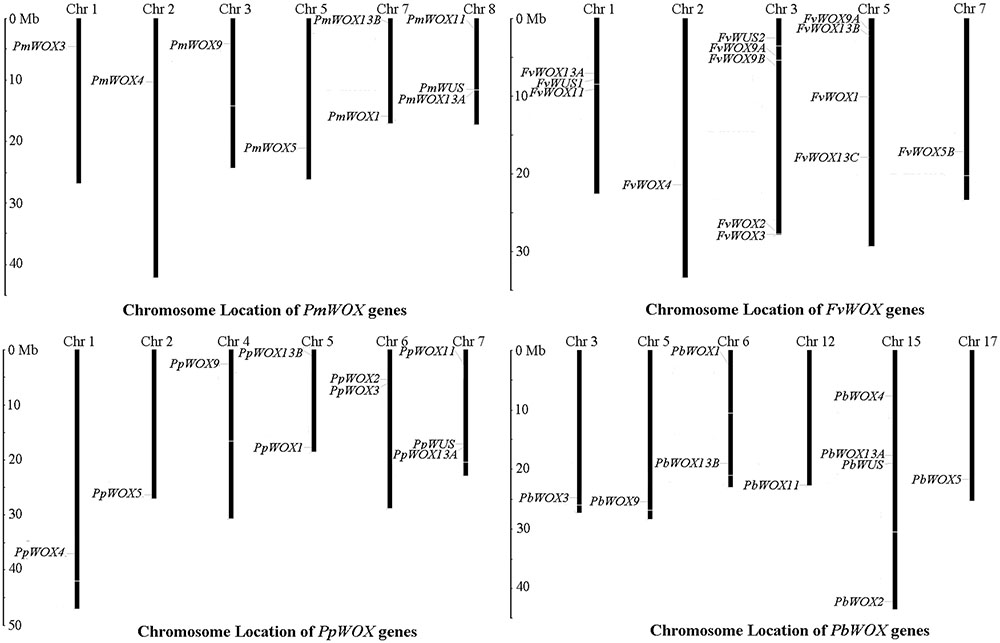
FIGURE 1. Chromosomal location of WOX genes from Pyrus bretschneideri, Prunus persica, Fragaria vesca and Prunus mume. The chromosome number was represented by the top of each chromosome.
Evolution of WOX Genes in Rosaceae
To investigate the possible evolutionary history of the WOX genes in the tested Rosaceae species, we carried out a joint phylogenetic analysis using three methods; ME, ML, and NJ. Based on previous report that WOX13 subfamily was an ancient member in the WOX gene family (Deveaux et al., 2008), the WOX13 subfamily was selected as an outgroup to root phylogenetic tree. All the tree topologies generated by the three methods (ME, ML, and NJ) were largely consistent with each other, with only minor changes in internal branches (Figure 2 and Supplementary Figures S1, S2). Therefore, only NJ phylogenetic tree was used in the following analysis. Previous studies on WOX genes have confirmed that motifs FYWFQNH, FYWFQNR, and YNWFQNR were representative markers for the WUS/modern clade, intermediate clade and ancient clade, respectively (Graaff et al., 2009; Ge et al., 2016). Confining the previous results (Deveaux et al., 2008; Xin et al., 2010), our evolutionary analysis exposed a total of 58 members of the WOX genes in P. bretschneideri, P. persica, F. vesca, P. mume along with A. thaliana. These 58 members of WOX genes were divided into three clades and nine subfamilies. The Modern clade contained a total of six subfamilies (WUS, WOX1, WOX2, WOX3, WOX4, and WOX5), and Intermediate clade included two subfamilies (WOX9 and WOX11), while Ancient clade just had a WOX13 subfamily, which were consistent with the evolutionary relationships of WOXs in other species (Xin et al., 2010; Hedman et al., 2013; Nardmann and Werr, 2013; Lian et al., 2014). Remarkably, we found that all subfamilies contained at least one WOX member from each of the four Rosaceae species (Figure 2). These results imply that rapid duplication of WOX genes occurred before these dicotyledonous species were diverged.
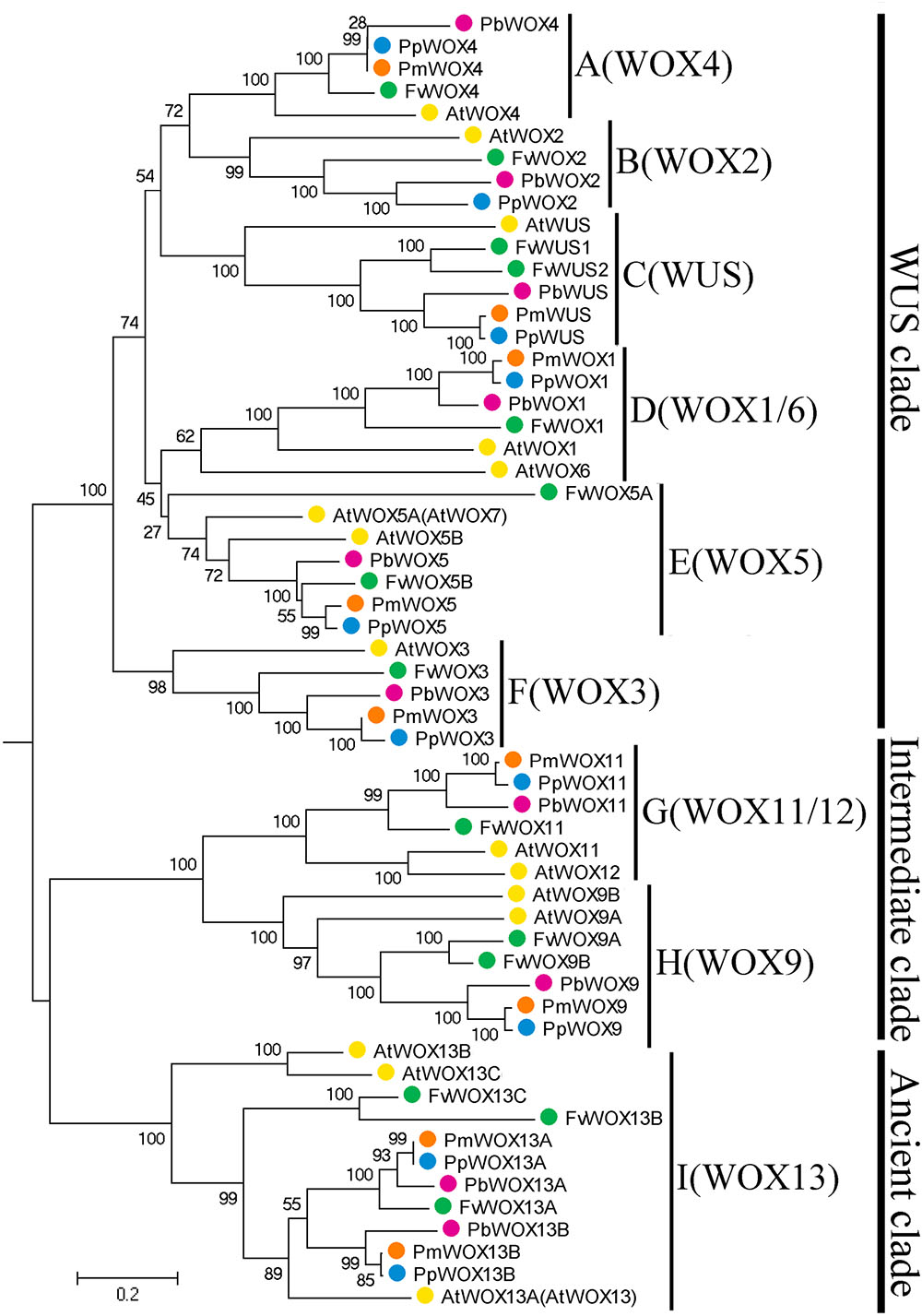
FIGURE 2. Neighbor-Joining tree of WOX family members in four Rosaceae species, F. vesca (Fv, green), P. mume (Pm, orange), P. persica (Pp, blue), and P. bretschneideri (Pb, red). Numbers indicate bootstrap support for branches. The clade I WOX genes (and only this group) are found both in some green algae and in all land plants, and so provide a root for this tree.
Phylogenetic analysis revealed that three pairs of paralogous genes were found among the WOX genes, which were consistent with the previous notion that most members of the WOX gene family are represented by pairs of orthologous genes. As shown in Figure 3, up to eight pairs of orthologous WOX genes were shared by P. persica and P. mume, whereas only two pairs of orthologous WOX genes found between P. bretschneideri and P. persica. However, no orthologous WOX genes were found between F. vesca and other species. These results were consistent with the evolutionary relationships among these four Rosaceae species (Dickinson et al., 2007; Cao et al., 2016b).
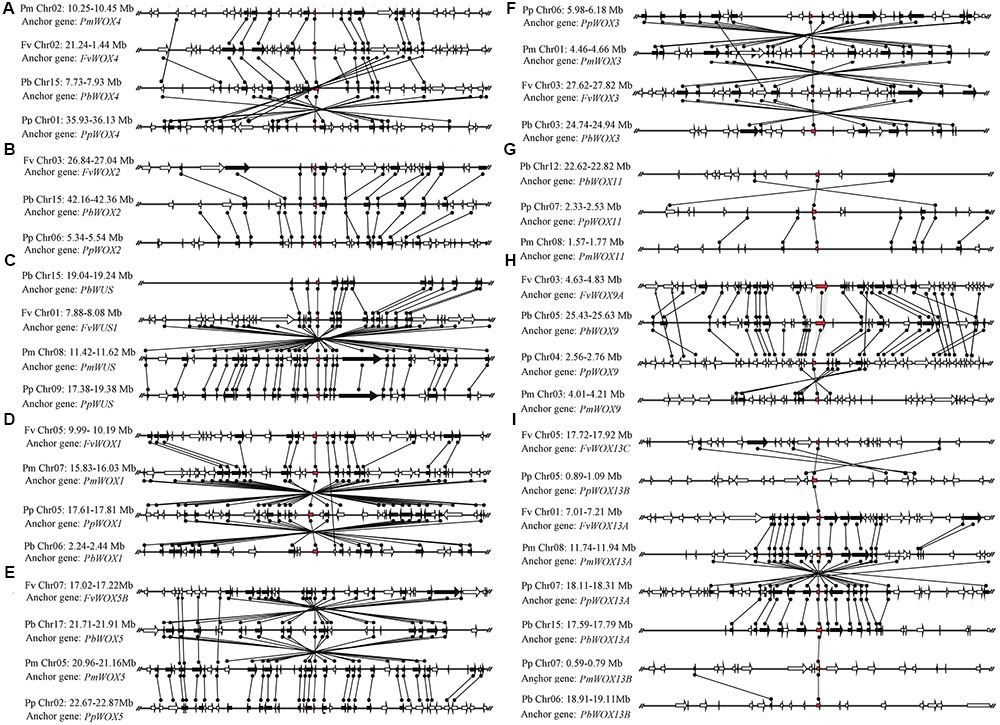
FIGURE 3. Extensive microsynteny of WOX regions across P. bretschneideri (Pb), P. persica (Pp), F. vesca (Fv), and P. mume (Fm) chromosomes. The gene’s orientation on strands was indicated by the triangle. Remarkably, the relative positions of all flanking protein-coding genes were defined by anchored WOX genes, highlighted in red. Subsequently, we used black lines to connect the homologous genes on two fragments. All genes are numbered from left to right, in order, for each segment. The (A–I) subfamilies in figure were consistent with those in Figure 2.
Analysis of Exon–Intron Structure and the Conserved Motifs
Previous studies have shown that gene structural diversity is an important resource for the evolution of multigene families (Liu et al., 2009; Cao et al., 2016c). To understand the structural diversity of the WOX genes in Rosaceae, gene structures of PbWOXs, PpWOXs, PmWOXs, and FvWOXs were deduced. It is revealed that these WOX genes contained different numbers of exons as shown in Figure 4A. For example, FvWOX11 only contained one exon, while FvWOX9A contained the largest number of exons (5). Moreover, 16, 15, and 3 of WOX genes contained two, three and four exons, respectively. These results suggested that the functional diversity of WOX genes may be in consequence due to exon loss or gain during the evolution of the WOX gene family. Subsequently, gene structures of the WOX paralogous and orthologous gene pairs were further analyzed. Among these genes, we found that the exon number of seven gene pairs had changed, including FvWOX9A/FvWOX9B, FvWOX13B/FvWOX13C, PmWOX3/PpWOX3, PbWOX4/PpWOX4, PmWUS/PpWUS, PmWOX11/PpWOX11, and PmWOX13B/PpWOX13B. By comparing among these seven gene pairs, it was found that one exon was lost in FvWOX9B, PmWOX3, PpWOX4, PpWUS, PmWOX11 and PmWOX13B, while one exon was obtained in FvWOX9A, PpWOX3, PbWOX4, PmWOXWUS, PpWOX11, and PpWOX13B. It may happen during the long evolutionary period. Previous studies have proposed that introns could be specifically inserted and remained in the plant genome during evolution (Rogozin et al., 2003; Carmel et al., 2007; Cao et al., 2016d). In our study, these phenomenon were observed, which might explain the functional differences and diversity of closely related WOX genes, such as PbWOX3 and FvWOX3, PbWOX13B, and FvWOX13A (Figure 4A).
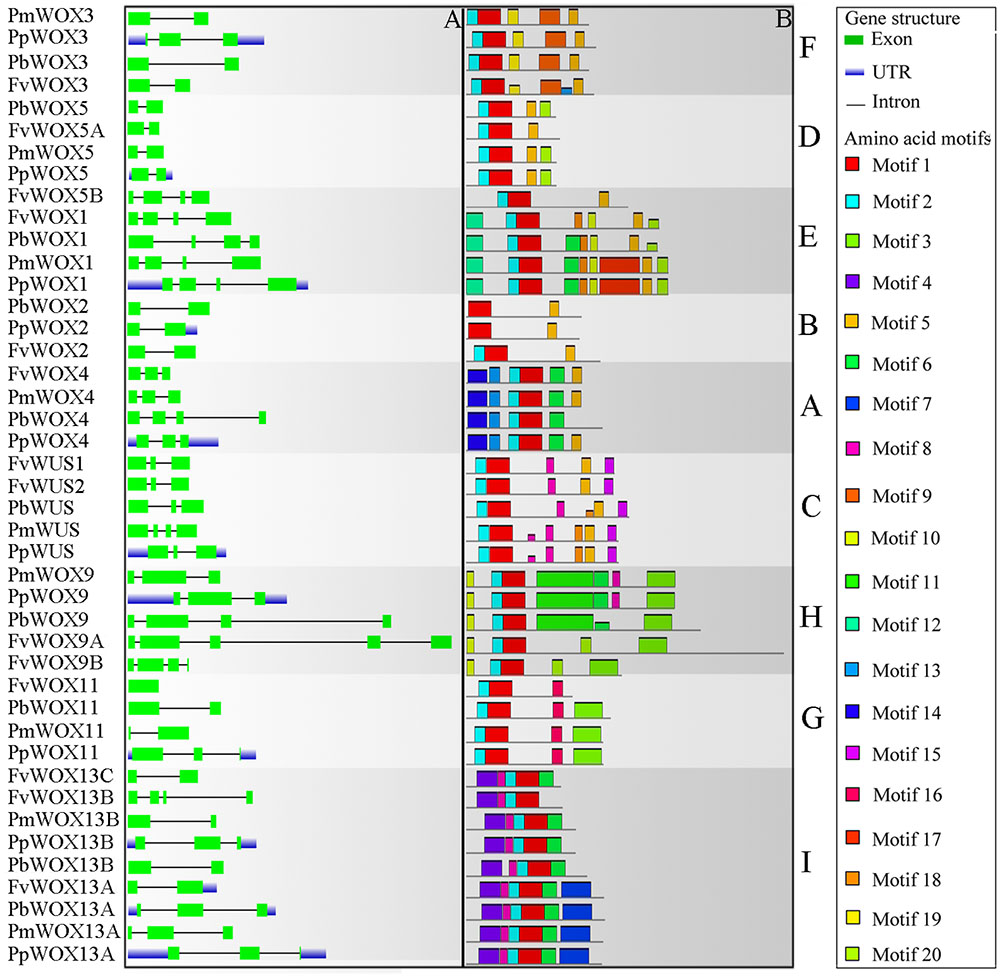
FIGURE 4. Exon/intron structure (A) and motif compositions (B) of the Rosaceae WOX genes. Relative protein or gene lengths can be estimated by gray bars. Untranslated regions (UTRs), exons and introns are represented by blue lines, green boxes and gray lines, respectively. Motif sequences were shown in Supplementary Table S2. The A-I subfamilies in figure were consistent with those in Figure 2.
Furthermore, it was observed that 20 of the conserved motifs were found in the 43 WOX proteins using MEME website (Supplementary Table S2). These motifs were annotated by using Pfam and SMART. Motif 1, present in all subfamilies, was identified to encode for a conserved homodomain. In addition to the homodomain, most of the WOX members within the same clade shared the similar motif compositions as shown in Figure 4B. These results reinforced the classification of WOX subfamilies. However, several motifs were unique to the proteins in some clades. For example, Motif 4 was unique to Ancient clade (clade I: WOX13 subfamily), Motif 5 to WUS clade (clades A–F) and Motif 15 to clade C (Supplementary Table S2 and Figure 4B). To some extent, these specific motifs may play an important role in the clade or subfamily, as well as contribution to the functional divergence of WOX genes.
Sequence Analysis of WOX Domains
Based on their amino acid sequences, the newly identified WOX gene family members were found to contain the conserved homeodomain by multiple sequence alignment of Cluxa2.0 with default parameters. The conserved homeodomain was selected for the visualized results by ESPript 3 (Gouet et al., 2005). The homeodomain structures of these four species were highly similar with each other. They contained a helix-loop-helix-turn-helix structure with either 65 or 66 amino acid residues. A total of 11 conserved sites (Q, L and Y in helix1; I, V, W, F, N, K, and R in helix3) of homeodomain reported previously (Gehring, 1992; Xin et al., 2010), were also conservative in the WOX proteins of the Rosaceae species (Figure 5). These findings suggested that these amino acid residues could play an important role in their functions. In addition to the previously reported conserved amino acid sites, other conserved amino acid sites have been identified in this study, such as P, L, and I in helix 2, Q and F in helix 3, as well as G in the turn region. Interestingly, an extra Y residue was found in the homeodomain of PbWUS, PmWUS, PpWUS, FvWUS and AtWUS, compared with other members of WOX gene family in P. bretschneideri, P. persica, P. mume and F. vesca and A. thaliana. Similar finding have been reported by Mayer et al. (1998) and Xin et al. (2010), that the homeodomains of A. thaliana WUS, O. sativa WUS, Z. mays WUS1, Z. mays WUS2, S. bicolor WUS and P. trichocarpa WUS were composed of 66 amino acid residues containing an extra Y residue by multiple sequence alignment, which indicates that this residue might play an important role on the function of WUS TF. Remarkably, in Arabidopsis, AtWOX5 (without Y residue between Helix1 and Loop) could replace AtWUS (containing Y residue) to maintain the dynamic balance of stem cells in the shoot apical meristem (Sarkar et al., 2007)
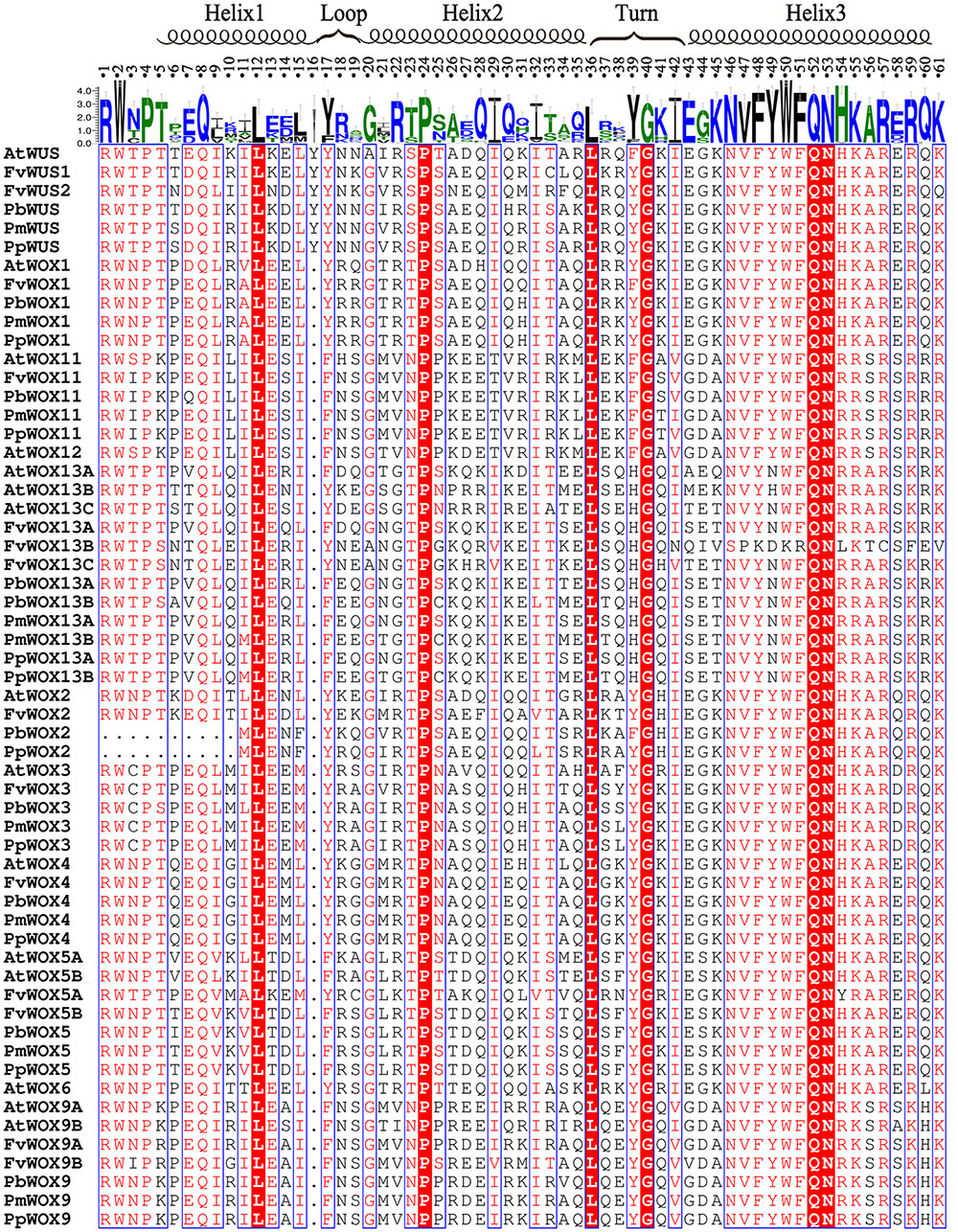
FIGURE 5. Alignment of the WOX homodomain sequences in five plant species. Highly conserved residues of homodomains were represented by the red shaded blocks in the five tested species. The secondary structure was indicated according to Mayer et al. (1998).
It was reported previously that the WUS protein contains three functional domains, including WUS-box, acidic region and EAR-like motif (Xin et al., 2010; Xiaoxu et al., 2016). These functional domains significantly contribute to its function as a TF (Xin et al., 2010; Xiaoxu et al., 2016). In present study, Motif 5 (WUS box: amino acids, TLLFP) was observed to be in the presence of all WOX proteins in WUX clade (clades A–F) (Supplementary Table S2 and Figure 4B). In clade C, the Motif 15 (EAR-like motif: amino acids, SLELSL) was found in all WOX proteins. However, no acidic region was identified in all WOX proteins (Supplementary Table S2 and Figure 4B). These results were consistent with previous findings that acidic region may be an important function domain only in Arabidopsis WUS gene (Xin et al., 2010; Xiaoxu et al., 2016).
Microsynteny Analysis of WOX Genes
Microsynteny has been surveyed in different species to understand the position of the homologous genes (orthology or paralogy) (Cannon et al., 2003; Yan et al., 2004; Cao et al., 2016a). In this study, microsynteny analysis was carried out for identification of homologous relationships within the WOX genes in P. bretschneideri, P. persica, F. vesca, and P. mume (Figure 3). Additionally, to measure the linkages and molecular history among WOX genes, a stepwise gene-by-gene reciprocal comparison was performed. In general, if the flanking genes in the chromosome region of the target gene contained three or more pairs of genes that are collinear, they could be considered as the conserved microsynteny (Lin et al., 2014; Cao et al., 2016a).
Primarily, the intraspecies microsynteny was investigated among four Rosaceae species. However, it was revealed that no collinear WOX genes were observed (Figure 3). These results suggested that independent duplication events were the main expansion pattern of WOX gene family members. Consequently, we analyzed the relationship of the WOX genes within each interspecies. The results exposed that the nine clades containing 38 WOX genes were found, among which 10 were from P. persica and F. vesca, 9 from P. bretschneideri and P. mume, respectively. Then several higher levels of microsynteny found in subfamilies A–G. Among these microsynteny some were remarkably inverted, duplicated such as PpWOX3/PmWOX3, PbWOX1/PpWOX1, and FvWOX13C/PpWOX13B (Figure 3). Usually, genome segments in the same group may evolve from a single sequence, which led to species differentiation (Tripoli et al., 2005; Jing et al., 2016). However, sequence fragments from the same group are considered to be homologous genes, and their genetic evolution resulted in species segregation (Tripoli et al., 2005; Jing et al., 2016). Remarkably, with the construction of the phylogenetic tree, the conservation of microsynteny in different families gradually emerged. Furthermore, some flanking genes were not conserved in each microsyntenic group. Therefore, it was speculated that these new genes were later than this duplication event. Interestingly, several lower levels of microsynteny were also found, such as PpWOX13B/FvWOX13A and PbWOX13A/PmWOX13B in clade I, PbWOX11/PpWOX11 and PpWOX11/PmWOX11 in clade H (Figure 3). These results strongly suggested that the ancient large-scale duplications could follow by gene rearrangement and loss.
Analysis of Selection Pressures and Functional Divergence
To investigate whether WOX genes have undergone strong selection pressures in the evolution of WOX gene family, we used site and branch-site models in the CODEML program of PAML software to detect positive selection sites (Yang, 2007). However, no positive selection was detected among these genes (Supplementary Table S3). These finding imply that relaxed purifying selection might play a major role in the evolution of WOX genes. Nardmann and Werr (2013) have shown that the WOX gene expansion was resulted from the increased complexity of plant morphology (Nardmann and Werr, 2013). These results proposed that the novel members after gene expansion were retained with partly overlapping expression domains and functions. These results are similar with findings of Nardmann and Werr (2013) who reported that with the relaxed purifying, dosage effects will lead to a selective advantage (Nardmann and Werr, 2013).
For further investigation to comprehend significant differences in selection pressures among WUS clade, intermediate clade and ancient clade, the branch-site models were performed using PAML software. It was exposed that no significant positive selection observed in the different branches of WOX gene family (Supplementary Table S4). This result was in contrast to the previous report that some significant positive selection sites were fixed in WOX genes of peanut (Wang et al., 2015). Because significant positive selection usually exerts its effects only in few sites and in a short period of evolutionary processes, it is difficult to detect positive selection. Thus, the selected signal could be diluted by the purifying selection (Zhang, 2005). However, as the WOX coding regions are highly conserved among members of orthologous families, the absence of strong positive selection was expected in Rosaceae species.
Due to the fact that significant positive selection could only detect a limited number of adaptive selection events, we performed a functional divergence analysis according to method used by Cao et al. (2016a). The DIVERGE software was used to calculate functional divergence of type I or II between gene clades in WOX genes with posterior analysis. In general, type I functional divergence usually resulted in a specific amino acid selectivity change, i.e., evolutionary rate change. The type II functional divergence only led to the change of physical and chemical properties of amino acids, which were occurred after gene duplication. In present study, to avoid the emergence of false positives, the sites with a posterior probability Q K > 0.9 were set as the key amino acid sites arising the functional differences according to previous experimentation reported (Yin et al., 2013; Cao et al., 2016a). Our results showed that five key amino acid sites (144, 154, 161, 165, and 166) were identified as type I functional divergence between Ancient and Modern (Table 1), while just one key site (152) between Intermediate and Modern (Table 1). The chi-square test (x 2) found that the P-values of Ancient/Modern and Intermediate/Modern were less than 0.05, reaching a significant level. Interestingly, among these three clades, no specific type II functional divergence site (Q K > 0.9) was detected (Supplementary Table S5), suggesting that the physicochemical properties of amino acid sequences between these Rosaceae WOX genes were highly identical.
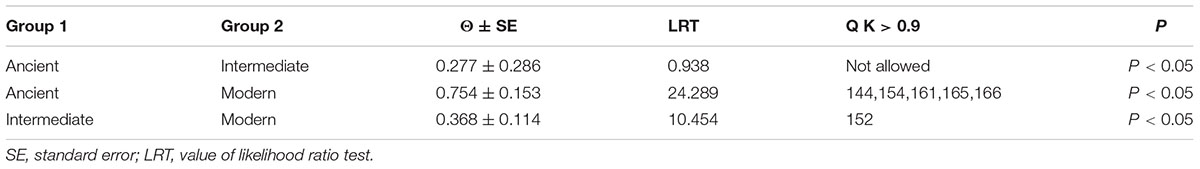
TABLE 1. Analysis of type I functional divergence.
cis-Acting Element Analysis of WOX Genes
Two thousand bp sequences of upstream from start codon (ATG) among the putative WOX genes, were used for analysis of WOX promoters by searching, against the PlantCARE website. Consequently, we detected various types of cis-acting elements in the promoter region of 43 WOX genes (Supplementary Table S6). These results indicated that the same type of WOX might carry out different functions. MBS and ABRE elements were found to be distributed in promoter region of most WOX genes, implying that WOX genes were transcriptionally regulated upon salt stress and dehydration. Remarkably, we found that the cis-elements exhibit significant differences in the promoter regions of duplicated WOX genes. These results indicated that the duplicated WOX genes may exhibit different regulation features.
Expression Profiles of F. vesca WOX Genes
To explore the role of the WOX gene family in F. vesca development process, the expression of the FvWOX genes was explored. The results showed that their expression levels were divergent from each other, indicating that they may be functionally active among all tissues except FvWOX5B (Supplementary Figure S3), which was located in Pollen with no expression. At the same time, most of FvWOX genes exhibited developmental stage-specificity, such as higher expression of FvWOX13A, FvWOX3, FvWOX13B, and FvWOX1 in flowering, and FvWOX4, FvWOX5, and FvWOX5A in embryo (Supplementary Figure S3). Surprisingly, we found that FvWOX13A, FvWOX9A, and FvWOX1 were highly expressed among all tissues, indicating that these genes were persistent and very important during development process of F. vesca.
Discussion
In present study, 43 WOX genes from four Rosaceae species were identified. It is observed that no direct relevance between genome sizes and the number of WOX gene family members. For example, there was no significant variety in the genome size of P. bretschneideri (271.9 Mb) (Wu et al., 2013) and F. vesca (240 Mb) (Shulaev et al., 2011), the number of WOX genes have been obviously changed. On the contrary, the number of WOX genes of the P. persica (224.6 Mb) (Verde et al., 2013) and P. mume (201 Mb) (Zhang et al., 2012) had a corresponding relationship with their genome sizes. In addition, we also noted that P. bretschneideri undergoes two genome-wide duplication events compared with those from P. persica, P. mume, and F. vesca (Wu et al., 2013). Nevertheless, the members of the WOX gene family among these four species did not change significantly. These findings indicate that the recent genome-wide duplication event did not contribute to the expansion of P. bretschneideri WOX gene family numbers. These results were supported by microsynteny analysis (Figure 3).
Previous studies suggested that the WOX gene family was divided into three major clades; the ancient clade were mainly present in land plants and green algae, while the intermediate and modern clades were only present in ferns and seed plants (Deveaux et al., 2008; Graaff et al., 2009; Nardmann and Werr, 2012, 2013). In present study, we found that all WOX genes from four Rosaceae species were distributed in the three clades, and was supported by the result of exon–intron and conserved domains analysis. At the same time, we also found that each clade contained its specific conserved motifs, implying these specific conserved motifs were likely required for subfamily-specific functions, such as Motif 5 to WUS clade (clades A–F). In the WOX gene family, the modern/WUS clade and intermediate clade were evolved from the ancient clade. It is well-known that gene sequence divergence, recombination, and duplications were considered to be the main driving forces for the evolution of gene families (Lin et al., 2014). In our study, the selection pressure was analyzed by using PAML program (Yang, 2007). In general, values of dn/ds (ω) >1, =1, and <1 represents positive selection, neutral evolution and purifying selection on the target gene, respectively. In this study, we found that the ω value of WOX genes was 0.07304 in M0 model (Supplementary Table S3). These results implied that WOX genes from four Rosaceae mainly underwent purifying selection during evolution, which was consistent with the hypothesis that highly conserved genes remain in the genome due to purifying selection. For example, the conserved WOX clade genes were all retained from green alga to seed plants (Nei, 2007).
Hedman et al. (2013) found that most conifer Picea abies WOX genes expressed at high levels in all developmental stages, while a few PaWOXs expression were low in specific tissues (Hedman et al., 2013). Zhang et al. (2015) reported that 10 Citrullus lanatus WOX genes were expressed in almost all tissues (Na et al., 2015). In our study, we found that the most of the FvWOX genes were expressed in different tissues. Among them, FvWOX4, FvWOX5, FvWOX5A, and FvWOX9B were mainly expressed in embryo stage with a very low expression for these genes in other tissues, which implied that these genes might have the same function as the key regulation factor AtWOX9A and AtWOX9B which was involved in the maintenance of the SAM (Wu et al., 2005; Skylar et al., 2010). The high expression of FvWOX13A, FvWOX13B, and FvWOX13C (Ancient clade) in flower tissue implied it had an important role similar to AtWOX13A and AtWOX13B in floral transition (Deveaux et al., 2008).
In this work, we identified 43 WOX genes in four Rosaceae species. These genes were divided into three well-supported clades (ancient, modern/WUS, intermediate) with nine subgroups. We also found that WOX genes phylogenetic relationship was supported by the presence of gene structure and conserved motif distribution. Our study demonstrated the existence of extensive microsynteny between WOX genes by comparing the WOX genes across four Rosaceae genomic sequences. The results showed that the maintenance of gene copy number after a whole genome duplication event was the main force to shape the WOX family evolution, with the purifying selection and a period of possibly relaxed constraint. Functional divergence was detected among the ancient, intermediate, and modern clades, which leaded to functional constraints, especially different evolutionary rates, after gene duplication. Furthermore, the expression profile of FvWOX gene identified that these genes play crucial roles in the floral transition during strawberry growth and development. The comprehensive analysis of the WOX family genes and the preliminary results presented here will be useful in the selection of appropriate candidate genes for further research on biological functions of WOX genes in strawberry.
Author Contributions
YuC and YH conceived and designed the experiments; YuC, QJ, and YH performed the experiments; YuC, YH, and DM analyzed the data; YuC, YH, DL, GL, MA, YL, and YoC contributed reagents/materials/analysis tools; YuC and YH wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by The National Natural Science Foundation of China (grant 31640068) and 2017 Graduate innovation fund of Anhui Agriculture University (2017yjs-31).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fgene.2017.00078/full#supplementary-material
FIGURE S1 | Maximum-Likelihood tree of WOX family members in four Rosaceae species.
FIGURE S2 | Minimum-Evolution tree of WOX family members in four Rosaceae species.
FIGURE S3 | Expression patterns of FvWOX genes during strawberry growth and development.
Footnotes
References
Bailey, T. L., Johnson, J., Grant, C. E., and Noble, W. S. (2015). The MEME suite. Nucleic Acids Res. 43, W39–W46. doi: 10.1093/nar/gkv416
Cannon, S. B., Mccombie, W. R., Sato, S., Tabata, S., Denny, R., Palmer, L., et al. (2003). Evolution and microsynteny of the apyrase gene family in three legume genomes. Mol. Genet. Genomics 270, 347–361. doi: 10.1007/s00438-003-0928-x
Cao, Y., Han, Y., Jin, Q., Lin, Y., and Cai, Y. (2016a). Comparative genomic analysis of the GRF genes in Chinese pear (Pyrus bretschneideri Rehd), poplar (Populous), grape (Vitis vinifera), Arabidopsis and rice (Oryza sativa). Front. Plant Sci. 7:1750.
Cao, Y., Han, Y., Li, D., Lin, Y., and Cai, Y. (2016b). MYB transcription factors in chinese pear (Pyrus bretschneideri Rehd.): genome-wide identification, classification, and expression profiling during fruit development. Front. Plant Sci. 7:577. doi: 10.3389/fpls.2016.00577
Cao, Y., Han, Y., Li, D., Lin, Y., and Cai, Y. (2016c). Systematic analysis of the 4-coumarate:coenzyme a ligase (4CL) related genes and expression profiling during fruit development in the chinese pear. Genes 7:89.
Cao, Y., Han, Y., Meng, D., Li, D., Jin, Q., Lin, Y., et al. (2016d). Structural, evolutionary, and functional analysis of the class III peroxidase gene family in chinese pear (Pyrus bretschneideri). Front. Plant Sci. 7:1874.
Carmel, L., Wolf, Y. I., Rogozin, I. B., and Koonin, E. V. (2007). Three distinct modes of intron dynamics in the evolution of eukaryotes. Genome Res. 17, 1034–1044. doi: 10.1101/gr.6438607
Darwish, O., Slovin, J. P., Kang, C., Hollender, C. A., Geretz, A., Houston, S., et al. (2013). SGR: an online genomic resource for the woodland strawberry. BMC Plant Biol. 13:223. doi: 10.1186/1471-2229-13-223
Deveaux, Y., Toffanonioche, C., Claisse, G., Thareau, V., Morin, H., Laufs, P., et al. (2008). Genes of the most conserved WOX clade in plants affect root and flower development in Arabidopsis. BMC Evol. Biol. 8:291. doi: 10.1186/1471-2148-8-291
Dickinson, T. A., Lo, E., and Talent, N. (2007). Polyploidy, reproductive biology, and Rosaceae: understanding evolution and making classifications. Plant Syst. Evol. 266, 59–78. doi: 10.1007/s00606-007-0541-2
Etchells, J. P., Provost, C. M., Mishra, L., and Turner, S. R. (2013). WOX4 and WOX14 act downstream of the PXY receptor kinase to regulate plant vascular proliferation independently of any role in vascular organisation. Development 140, 2224–2234. doi: 10.1242/dev.091314
Ge, Y., Liu, J., Zeng, M., He, J., Qin, P., Huang, H., et al. (2016). Identification of WOX family genes in Selaginella kraussiana for studies on stem cells and regeneration in lycophytes. Front. Plant Sci. 7:93. doi: 10.3389/fpls.2016.00093
Gehring, W. J. (1992). The homeobox in perspective. Trends Biochem. Sci. 17, 277–280. doi: 10.1016/0968-0004(92)90434-B
Gouet, P., Robert, X., and Courcelle, E. (2005). ESPript/ENDscript: sequence and 3D information from protein structures. Acta Crystallogr. 61, 42–43. doi: 10.1107/S0108767305098211
Graaff, E. V. D., Laux, T., and Rensing, S. A. (2009). The WUS homeobox-containing (WOX) protein family. Genome Biol. 10:248. doi: 10.1186/gb-2009-10-12-248
Gu, X. (1999). Statistical methods for testing functional divergence after gene duplication. Mol. Biol. Evol. 16, 1664–1674. doi: 10.1093/oxfordjournals.molbev.a026080
Gu, X. (2006). A simple statistical method for estimating type-II (cluster-specific) functional divergence of protein sequences. Mol. Biol. Evol. 23, 1937–1945. doi: 10.1093/molbev/msl056
Gu, X., Zou, Y., Su, Z., Huang, W., Zhou, Z., Arendsee, Z., et al. (2013). An update of DIVERGE software for functional divergence analysis of protein family. Mol. Biol. Evol. 30, 1713–1719. doi: 10.1093/molbev/mst069
Haecker, A., Grosshardt, R., Geiges, B., Sarkar, A., Breuninger, H., Herrmann, M., et al. (1991). Expression dynamics of WOX genes mark cell fate decisions during early embryonic patterning in Arabidopsis thaliana. Development 131, 657–668. doi: 10.1242/dev.00963
Hedman, H., Zhu, T., Von, A. S., and Sohlberg, J. J. (2013). Analysis of the WUSCHEL-RELATED HOMEOBOX gene family in the conifer picea abies reveals extensive conservation as well as dynamic patterns. BMC Plant Biol. 13:89. doi: 10.1186/1471-2229-13-89
Hirakawa, Y., Kondo, Y., and Fukuda, H. (2010). TDIF peptide signaling regulates vascular stem cell proliferation via the WOX4 homeobox gene in Arabidopsis. Plant Cell 22, 2618–2629. doi: 10.1105/tpc.110.076083
Hu, B., Jin, J., Guo, Y. A., Zhang, H., Luo, J., and Gao, G. (2014). GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31:1296. doi: 10.1093/bioinformatics/btu817
Ikeda, M., and Ohme-Takagi, M. (2009). Arabidopsis WUSCHEL is a bifunctional transcription factor that acts as a repressor in stem cell regulation and as an activator in floral patterning. Plant Cell 21, 3493–3505. doi: 10.1105/tpc.109.069997
Jing, J., Kong, J., Qiu, J., Zhu, H., Peng, Y., and Jiang, H. (2016). High level of microsynteny and purifying selection affect the evolution of WRKY family in Gramineae. Dev. Genes Evol. 226, 15–25. doi: 10.1007/s00427-015-0523-2
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van De Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Letunic, I., Doerks, T., and Bork, P. (2012). SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res. 40, D302–D305. doi: 10.1093/nar/gkr931
Lian, G., Ding, Z., Wang, Q., Zhang, D., and Xu, J. (2014). Origins and evolution of WUSCHEL-related homeobox protein family in plant kingdom. Sci. World J. 2014, 534140–534140. doi: 10.1155/2014/534140
Lin, Y., Cheng, Y., Jin, J., Jin, X., Jiang, H., Yan, H., et al. (2014). Genome duplication and gene loss affect the evolution of heat shock transcription factor genes in legumes. PLoS ONE 9:e102825. doi: 10.1371/journal.pone.0102825
Liu, S. L., Zhuang, Y., Zhang, P., and Adams, K. L. (2009). Comparative analysis of structural diversity and sequence evolution in plant mitochondrial genes transferred to the nucleus. Mol. Biol. Evol. 26, 875–891. doi: 10.1093/molbev/msp011
Mayer, K. F., Schoof, H., Haecker, A., Lenhard, M., Jürgens, G., and Laux, T. (1998). Role of WUSCHEL in regulating stem cell fate in the Arabidopsis shoot meristem. Cell 95, 805–815. doi: 10.1016/S0092-8674(00)81703-1
Mukherjee, K., Brocchieri, L., and Bürglin, T. R. (2009). A comprehensive classification and evolutionary analysis of plant homeobox genes. Mol. Biol. Evol. 26, 2775–2794. doi: 10.1093/molbev/msp201
Na, Z., Xing, H., Bao, Y., Bo, W., Liu, L., Dai, L., et al. (2015). Genome-wide identification and expression profiling of WUSCHEL-related homeobox (WOX) genes during adventitious shoot regeneration of watermelon (Citrullus lanatus). Acta Physiol. Plant. 37, 1–12.
Nardmann, J., and Werr, W. (2012). The invention of WUS-like stem cell-promoting functions in plants predates leptosporangiate ferns. Plant Mol. Biol. 78, 123–134. doi: 10.1007/s11103-011-9851-4
Nardmann, J., and Werr, W. (2013). Symplesiomorphies in the WUSCHEL clade suggest that the last common ancestor of seed plants contained at least four independent stem cell niches. New Phytol. 199, 1081–1092. doi: 10.1111/nph.12343
Nei, M. (2007). The new mutation theory of phenotypic evolution. Proc. Natl. Acad. Sci. U.S.A. 104, 12235–12242. doi: 10.1073/pnas.0703349104
Palovaara, J., Hallberg, H., Stasolla, C., and Hakman, I. (2010). Comparative expression pattern analysis of WUSCHEL-related homeobox 2 (WOX2) and WOX8/9 in developing seeds and somatic embryos of the gymnosperm Picea abies. New Phytol. 188, 122–135. doi: 10.1111/j.1469-8137.2010.03336.x
Punta, M., Coggill, P. C., Eberhardt, R. Y., Mistry, J., Tate, J., Boursnell, C., et al. (2011). The Pfam protein families database. Nucleic Acids Res. 40, D290–D301. doi: 10.1093/nar/gkr1065
Rogozin, I. B., Wolf, Y. I., Sorokin, A. V., Mirkin, B. G., and Koonin, E. V. (2003). Remarkable interkingdom conservation of intron positions and massive, lineage-specific intron loss and gain in eukaryotic evolution. Curr. Biol. 13, 1512–1517. doi: 10.1016/S0960-9822(03)00558-X
Romera-Branchat, M., Ripoll, J. J., Yanofsky, M. F., and Pelaz, S. (2013). The WOX13 homeobox gene promotes replum formation in the Arabidopsis thaliana fruit. Plant J. 73, 37–49. doi: 10.1111/tpj.12010
Sarkar, A. K., Luijten, M., Miyashima, S., Lenhard, M., Hashimoto, T., Nakajima, K., et al. (2007). Conserved factors regulate signalling in Arabidopsis thaliana shoot and root stem cell organizers. Nature 446, 811–814. doi: 10.1038/nature05703
Shulaev, V., Sargent, D. J., Crowhurst, R. N., Mockler, T. C., Folkerts, O., Delcher, A. L., et al. (2011). The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 43, 109–116. doi: 10.1038/ng.740
Skylar, A., Hong, F., Chory, J., Weigel, D., and Wu, X. (2010). STIMPY mediates cytokinin signaling during shoot meristem establishment in Arabidopsis seedlings. Development 137, 541–549. doi: 10.1242/dev.041426
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. doi: 10.1093/nar/25.24.4876
Tripoli, G., D’elia, D., Barsanti, P., and Caggese, C. (2005). Comparison of the oxidative phosphorylation (OXPHOS) nuclear genes in the genomes of Drosophila melanogaster, Drosophila pseudoobscura and Anopheles gambiae. Genome Biol. 6:R11. doi: 10.1186/gb-2005-6-2-r11
Ueda, M., Zhang, Z., and Laux, T. (2011). Transcriptional activation of Arabidopsis axis patterning genes WOX8/9 links zygote polarity to embryo development. Dev. Cell 20, 264–270. doi: 10.1016/j.devcel.2011.01.009
Verde, I., Abbott, A. G., Scalabrin, S., Jung, S., Shu, S., Marroni, F., et al. (2013). The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 45, 487–494. doi: 10.1038/ng.2586
Wang, P., Li, C., Li, C., Zhao, C., Xia, H., Zhao, S., et al. (2015). Identification and expression dynamics of three WUSCHEL related homeobox 13 (WOX13) genes in peanut. Dev. Genes Evol. 225, 221–233. doi: 10.1007/s00427-015-0506-3
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Wu, J., Wang, Z., Shi, Z., Zhang, S., Ming, R., Zhu, S., et al. (2013). The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 23, 396–408. doi: 10.1101/gr.144311.112
Wu, X., Dabi, T., and Weigel, D. (2005). Requirement of homeobox gene STIMPY/WOX9 for Arabidopsis meristem growth and maintenance. Curr. Biol. 15, 436–440. doi: 10.1016/j.cub.2004.12.079
Xiaoxu, L., Cheng, L., Wei, L., Zenglin, Z., Xiaoming, G., Hui, Z., et al. (2016). Genome-wide identification, phylogenetic analysis and expression profiling of the WOX family genes in Solanum lycopersicum. Hereditas 38, 444–460. doi: 10.16288/j.yczz.15-499
Xin, Z., Jie, Z., Liu, J., Yin, J., and Zhang, D. (2010). Genome-wide analysis of WOX Gene Family in Rice, Sorghum, Maize, Arabidopsis and Poplar. J. Integr. Plant Biol. 52, 1016–1026. doi: 10.1111/j.1744-7909.2010.00982.x
Yadav, R. K., Tavakkoli, M., and Reddy, G. V. (2010). WUSCHEL mediates stem cell homeostasis by regulating stem cell number and patterns of cell division and differentiation of stem cell progenitors. Development 137, 3581–3589. doi: 10.1242/dev.054973
Yan, H. H., Mudge, J., Kim, D. J., Shoemaker, R. C., Cook, D. R., and Young, N. D. (2004). Comparative physical mapping reveals features of microsynteny between Glycine max, Medicago truncatula, and Arabidopsis thaliana. Genome 47, 141–155. doi: 10.1139/g03-106
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yin, G., Xu, H., Xiao, S., Qin, Y., Li, Y., Yan, Y., et al. (2013). The large soybean (Glycine max) WRKY TF family expanded by segmental duplication events and subsequent divergent selection among subgroups. BMC Plant Biol. 13:148. doi: 10.1186/1471-2229-13-148
Zdobnov, E. M., and Apweiler, R. (2001). InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. doi: 10.1093/bioinformatics/17.9.847
Zhang, J. (2005). Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 22, 2472–2479. doi: 10.1093/molbev/msi237
Zhang, Q., Chen, W., Sun, L., Zhao, F., Huang, B., Yang, W., et al. (2012). The genome of Prunus mume. Nat. Commun. 3:1318. doi: 10.1038/ncomms2290
Zhang, Y., Yue, J., Liu, Z., and Zhu, Y. X. (2015). ROW1 maintains quiescent centre identity by confining WOX5 expression to specific cells. Nat. Commun. 6:6003. doi: 10.1038/ncomms7003
Keywords: WOX genes, phylogenetic analysis, microsynteny, selection, functional divergence
Citation: Cao Y, Han Y, Meng D, Li G, Li D, Abdullah M, Jin Q, Lin Y and Cai Y (2017) Genome-Wide Analysis Suggests the Relaxed Purifying Selection Affect the Evolution of WOX Genes in Pyrus bretschneideri, Prunus persica, Prunus mume, and Fragaria vesca. Front. Genet. 8:78. doi: 10.3389/fgene.2017.00078
Received: 10 March 2017; Accepted: 29 May 2017;
Published: 15 June 2017.
Edited by:
Rosalba Giugno, University of Verona, ItalyReviewed by:
Manoj Prasad, National Institute of Plant Genome Research, IndiaRosario Muleo, Università degli Studi della Tuscia, Italy
Copyright © 2017 Cao, Han, Meng, Li, Li, Abdullah, Jin, Lin and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongping Cai, c3dreDEyQGFoYXUuZWR1LmNu