 Raúl A. Ortiz-Merino1†
Raúl A. Ortiz-Merino1† Javier A. Varela2†
Javier A. Varela2† Aisling Y. Coughlan1Hisashi Hoshida3Wendel B. da Silveira4Caroline Wilde5Niels G. A. Kuijpers6
Aisling Y. Coughlan1Hisashi Hoshida3Wendel B. da Silveira4Caroline Wilde5Niels G. A. Kuijpers6 Jan-Maarten Geertman6
Jan-Maarten Geertman6 Kenneth H. Wolfe1
Kenneth H. Wolfe1 John P. Morrissey2*
John P. Morrissey2*- 1School of Medicine, UCD Conway Institute, University College Dublin, Dublin, Ireland
- 2School of Microbiology, Centre for Synthetic Biology and Biotechnology, Environmental Research Institute, APC Microbiome Institute, University College Cork, Cork, Ireland
- 3Department of Applied Chemistry, Graduate School of Sciences and Technology for Innovation, Yamaguchi University, Yamaguchi, Japan
- 4Department of Microbiology, Universidade Federal de Viçosa, Viçosa, Brazil
- 5Lallemand Inc., Montreal, QC, Canada
- 6Heineken Supply Chain, Zoeterwoude, Netherlands
Kluyveromyces marxianus is traditionally associated with fermented dairy products, but can also be isolated from diverse non-dairy environments. Because of thermotolerance, rapid growth and other traits, many different strains are being developed for food and industrial applications but there is, as yet, little understanding of the genetic diversity or population genetics of this species. K. marxianus shows a high level of phenotypic variation but the only phenotype that has been clearly linked to a genetic polymorphism is lactose utilisation, which is controlled by variation in the LAC12 gene. The genomes of several strains have been sequenced in recent years and, in this study, we sequenced a further nine strains from different origins. Analysis of the Single Nucleotide Polymorphisms (SNPs) in 14 strains was carried out to examine genome structure and genetic diversity. SNP diversity in K. marxianus is relatively high, with up to 3% DNA sequence divergence between alleles. It was found that the isolates include haploid, diploid, and triploid strains, as shown by both SNP analysis and flow cytometry. Diploids and triploids contain long genomic tracts showing loss of heterozygosity (LOH). All six isolates from dairy environments were diploid or triploid, whereas 6 out 7 isolates from non-dairy environment were haploid. This also correlated with the presence of functional LAC12 alleles only in dairy haplotypes. The diploids were hybrids between a non-dairy and a dairy haplotype, whereas triploids included three copies of a dairy haplotype.
Introduction
The yeast Kluyveromyces marxianus is best-known because of its frequent association with traditional dairy products such as kefir and cheese (Lachance, 2011; Gethins et al., 2016; Coloretti et al., 2017). This association with fermented dairy beverages, a consequence of its capacity to use the milk sugar lactose as a carbon source, has led to inclusion of K. marxianus on GRAS (FDA) and QPS (EU) lists of safe micro-organism for use in foods (Lane and Morrissey, 2010; Ricci et al., 2017). The yeast is also regularly isolated from non-dairy environments (e.g., decaying fruit) and is part of the natural flora involved in production of Agave-based alcoholic beverages such as tequila and mezcal (Lappe-Oliveras et al., 2008; Verdugo Valdez et al., 2011). In the latter case, the production of enzymes that degrade plant fructans to simpler sugars (inulinases) undoubtedly contributes to its growth in this environment (Arrizon et al., 2012). The capacity of K. marxianus to utilise a broad array of sugars also creates potential for biotechnological applications (Fonseca et al., 2008; Lane and Morrissey, 2010), which is illustrated by the many studies exploring potential for bioethanol production from diverse substrates such as whey permeate, crop plants, and lignocellulosic biomass (Nonklang et al., 2009; Guimarães et al., 2010; Wu et al., 2016; Kobayashi et al., 2017). This yeast is used commercially for production of the flavour molecule 2-phenylethanol, and there is considerable interest in development of K. marxianus as a cell factory for production of other bioflavours (Morrissey et al., 2015). K. marxianus is also distinguished by thermotolerance (Lane et al., 2011), and the fastest reported growth rate of any eukaryote (Groeneveld et al., 2009). Recent years have seen increasing interest in new applications such as production of biomolecules (Hughes et al., 2017; Lin et al., 2017), biocatalysis (Oliveira et al., 2017; Wang et al., 2017) and heterologous protein production (Gombert et al., 2016; Lee et al., 2017).
One of the interesting aspects of the nascent development of K. marxianus as an important yeast for biotechnology is the wide variety of strains that are being used, both for research and for application. This contrasts with the traditional yeast, Saccharomyces cerevisiae, where, until recently, there was a very strong focus on a relatively narrow set of model strains. While giving access to the broad diversity that exists within any species, the non-reliance on model strains also creates challenges since findings with one isolate are not automatically transferrable to other isolates. This is illustrated well by studies that demonstrate wide variance in tolerance to different external stresses (Lane et al., 2011; Rocha et al., 2011). Indeed, it has emerged that even a trait such as lactose utilisation, long considered one of the defining characteristics of K. marxianus, is not universal, and many strains exhibit very poor growth on lactose, a phenotype that was shown to be due to polymorphisms in the LAC12 gene, which encodes a permease responsible for transport of lactose into the cell (Varela et al., 2017). Although recent studies on sugar transport and physiology are starting to address the deficit (Fonseca et al., 2013; Signori et al., 2014; Beniwal et al., 2017; Dias et al., 2017; Diniz et al., 2017), it is true to say that a lot of the underlying knowledge about the biology of K. marxianus is based on inference of similarity with its sister species, Kluyveromyces lactis, which was developed as a model for studying lactose-positive yeasts since the 1960's (Fukuhara, 2006). The genome of K. lactis was sequenced more than a decade ago (Souciet et al., 2000), with a more recent functional reannotation and genome scale model that provides a deeper understanding of the core metabolism of this species (Dias et al., 2012, 2014). Notwithstanding the utility of a related species for comparison, the many metabolic and physiological differences between K. lactis and K. marxianus necessitate independent studies of K. marxianus to provide the comprehensive understanding of its genetics and physiology that will underpin future developments in fundamental biology and biotechnology.
Genomic and transcriptomic studies have started to shed light on K. marxianus and a growing number of genome sequences of K. marxianus strains are now available (Jeong et al., 2012; Silveira et al., 2014; Inokuma et al., 2015; Lertwattanasakul et al., 2015; Quarella et al., 2016). As yet, however, there has not been a systematic comparison of the sequenced K. marxianus genomes, nor a comparison to the single K. lactis genome that is in the public domain. In contrast to K. lactis, whose genome comprises 6 chromosomes, several studies have reported that K. marxianus has a full complement of 8 chromosomes, with many areas of local synteny between the species. There is strong conservation of the mating type locus (Lane et al., 2011) and thus K. marxianus could be expected to be capable of mating type switching and mating in a manner similar to K. lactis (Barsoum et al., 2010; Rajaei et al., 2014). Based on information to date, however, there does appear to be a fundamental difference in life-cycles. Studies of natural isolates of K. lactis suggest that this yeast is primarily a haploid (haplontic) species. Mating is induced by depletion of nitrogen or phosphate in the environment, and zygotes formed by mating usually sporulate immediately (although diploids can be maintained in the lab, e.g., by selection for auxotrophic markers) (Schaffrath and Breunig, 2000; Zonneveld and Steensma, 2003; Booth et al., 2010; Rodicio and Heinisch, 2013). In contrast, analysis of the mating-type locus of natural and culture collection K. marxianus isolates identified both haploid and diploid strains (Lane et al., 2011; Fasoli et al., 2016).
To put the phenotypic diversity of K. marxianus into context, it is important to characterise its genomic diversity and to assess the population structure of the species. There have been some pre-whole genome sequence studies that addressed this question using different methods. Pulsed-field gel electrophoresis studies suggested that there were variable numbers of chromosomes in K. marxianus strains (Belloch et al., 1998; Fasoli et al., 2015), a finding not in accordance with genome sequence data, which has consistently indicated 8 chromosomes. Mitochondrial DNA haplotypes and variation at some genomic loci was used to try to determine population structure in a collection from Italian cheeses (Fasoli et al., 2016). That particular study identified variations in population structure and proposed the occurrence of homozygous and heterozygous strains. A Multi Locus Sequence Typing (MLST) method was developed to further explore the diversity in that collection and in this case, the analysis was extended to other strains that are sequenced or available in culture collections (Tittarelli et al., 2018). MLST analysis did not identify distinct sub-populations but while the method was very diagnostic for strain identification, the surprisingly high level of heterozygosity in diploid strains reduced resolution to a level too low for population-type analysis. Analysis of population structure in diploid yeasts is challenging and, in many cases, has relied on SNPs identified in genome sequences derived from haploids or from completely homozygous diploids (made by self-mating of single spore derivatives) (Liti et al., 2009; Schacherer et al., 2009; Strope et al., 2015). In highly heterozygous species, this method may not generate an accurate view of the relationships among haplotypes or among strains.
In this study, we set out to explore the genomic diversity of K. marxianus by analysing whole-genome data from 14 strains isolated from different sources. Some of these strains had been previously sequenced and published, whereas others were sequenced for this study. To take heterozygosity into account, raw sequence reads were used to allow analysis of single nucleotide polymorphisms (SNPs) between strains. The results indicate a high degree of variation among isolates, in both ploidy and heterozygosity, and show a correlation between ploidy and environmental niche. Our work raises important questions about the life cycle of K. marxianus, and emphasizes the need to take ploidy and heterozygosity into account when considering using K. marxianus for biotechnological purposes.
Materials and Methods
Yeast Strains, Growth, and Phenotypic Analysis
The 14 K. marxianus strains analysed in this study are listed in Table 1. Two strains (DMKU3-1042 and UFS-Y2791) were not available for phenotypic assessment but the remaining 12 strains were obtained from the sources indicated in Table 1 and were routinely cultured at 30°C in YPD medium (10 g/L yeast extract, 20 g/L bactopeptone, 20 g/L glucose). For lactose utilisation tests, yeast strains were first grown overnight in 5 mL minimal media (MM) supplemented with 2% glucose (Fonseca et al., 2007). Cells from the overnight cultures were harvested by centrifugation, washed twice with 5 mL of water and used to inoculate MM supplemented with 2% lactose to an OD600 of 0.1. These cultures were incubated for 15 h, when the final OD600 was determined. Lactose concentration was determined by HPLC at 0 and 15 h and used to calculate lactose consumption as previously described (Varela et al., 2017). Experiments were performed in triplicate with error bars showing standard deviation.
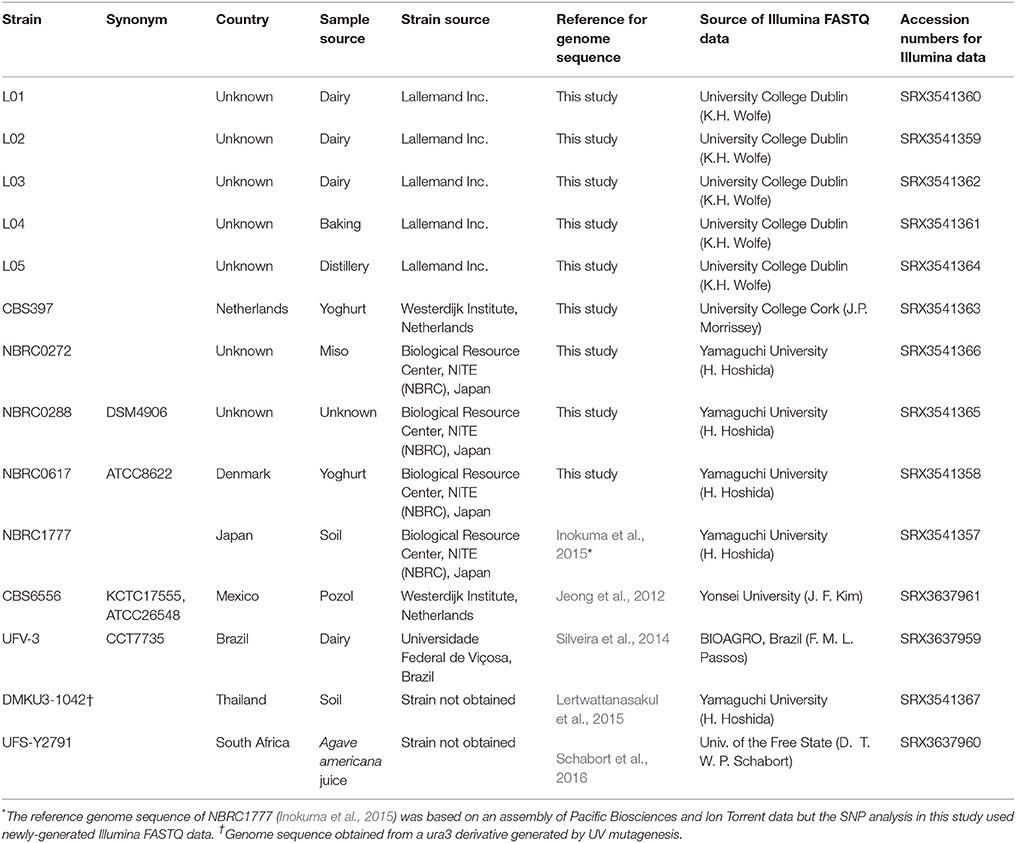
Table 1. Sources of K. marxianus strains and genomes analysed in this study.
Flow Cytometry
DNA content was determined by flow cytometry using SYTOX green (Thermo-Fisher) as previously described (Haase and Reed, 2002). Yeast strains were grown in YPD at 30°C with 200 rpm agitation in a New Brunswick Innova 40/40 R orbital shaker (Eppendorf, Hamburg, Germany). Cultures were harvested by centrifugation and resuspended in 1 mL sterile water. Cells were then washed, resuspended in 400 μL sterile water and fixed by adding 950 μL 100% ethanol. The suspensions were incubated overnight at 4°C, then centrifuged and washed in 50 mM sodium citrate (pH 7.2). The cells were resuspended in 500 μL RNAse A solution (0.25 mg/mL RNAse A, 50 mM sodium citrate pH 7.2) and incubated for 1 h at 37°C. Then, 100 μL of 20 mg/mL Proteinase K was added to each sample and the tubes were incubated at 50°C for 2 h. Finally, 500 μL of SYTOX Green solution (4 μM SYTOX Green, 50 mM sodium citrate pH 7.2) was added to each tube. Samples were analysed using a BD FACSCelesta system (BD Biosciences, CA, USA) and the data was processed using FlowJo software v10 (BD Biosciences, CA, USA).
Genome Data, Sequencing, and Read Mapping
The genomes of five strains (NBRC1777, CBS6556, UFV-3, DMKU3-1042, and UFS-Y2791) had previously been published and some of the authors kindly made the source Illumina FASTQ data available for this analysis. The 11 strains sequenced in this study are indicated in Table 1, accession numbers are provided for all strains and references are given when applicable. All strains were sequenced on Illumina HiSeq 2000 or 2500 instruments after Truseq genomic library preparation. Details of the sequencing data type and coverage for all 14 strains, including those sequenced elsewhere, are summarised in Table 2. We performed quality control checks for all libraries with FastQC v. 0.10.1 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.) Reads for strain CBS6556 were trimmed using skewer v. 0.2.2 (Jiang et al., 2014) with parameters -q 20 -m pe -l 70. For BLAST analyses, de novo assemblies of each newly sequenced genome were made using SPAdes 3.5.0 (Bankevich et al., 2012).
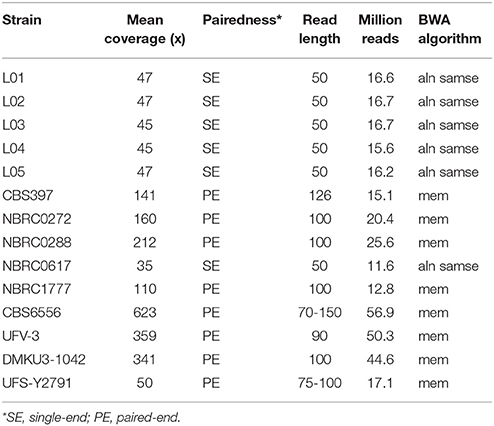
Table 2. Summary of Illumina sequencing strategies and coverage for 14 strains used in SNP analysis.
The NBRC1777 genome sequence was selected as a reference because of the high quality of its assembly into 8 chromosomes, and because our analysis confirmed the haploid nature of this strain (Inokuma et al., 2015). Sequencing libraries from all strains were aligned to the NBRC1777 reference using the Burrows-Wheeler Aligner (BWA) v. 0.7.9a-r786 (Li and Durbin, 2009) with default parameters. The BWA “mem” alignment algorithm was used for libraries with read length ≥100 bp, the “aln” alignment algorithm for libraries with read length <100 bp, and the alignment modes were set to “samse” and “sampe” for single-end and paired-end data respectively. Samtools v. 0.1.19-44428cd (Li et al., 2009) was used to remove unmapped reads from the BWA output files and to generate indexes for downstream steps. Picard tools v. 2.0.1 (http://broadinstitute.github.io/picard) function AddOrReplaceReadGroups was used to add identifiers to the BAM files, followed by MarkDuplicates to mark and discard PCR duplicates. Indel realignment and coverage calculation were performed using the RealignerTargetCreator, IndelRealigner, and DepthOfCoverage tools from the Genome Analysis Tool Kit (GATK) v. 3.5-0-g36282e4 (Van der Auwera et al., 2013). Mean coverage was calculated omitting a 19 kb region on chromosome 5 that contains the array encoding the rRNA genes. Coverage plots were obtained by calculating the average in 10-kb windows. Segment means were calculated using the R Bioconductor package DNAcopy v 1.50.1 (DOI: 10.18129/B9.bioc.DNAcopy).
Variant Calling
“Variable sites” were defined as the set of sites in the genome that contain a non-reference base, hereafter called “variants,” in at least one of the 14 strains. Variant calling in the 14 strains was done using the GATK tool HaplotypeCaller in DISCOVERY and GVCF modes, requiring a minimum quality score of 20. The output files were then used for multi-sample analysis using the GenotypeGVCF tool and a custom Perl script was used to remove all variants that had low genotype quality (GQ < 20), or had low approximate read depth (below 10% of the mean coverage for the sample excluding the rDNA locus and telomeric regions). For every remaining variable site, the output from GATK enabled us to calculate the empirical allele frequencies of the reference base (designated fA) and the variant (designated fB and referred as alternative allele frequencies), which sum to 1. Empirical allele frequencies were calculated at each variable site on each strain by dividing the allelic depth of the variant by the approximate read depth observed at that site (AD and DP fields in GATK's HaplotypeCaller). Each variant remaining after the filtering steps and having an fB > 0.15 is considered to be SNPs. Accordingly, variable sites were only used for further analysis when having a SNP. If fA ≥ 0.85 the strain was called homozygous for the reference base (AA) at this variable site, and if fB ≥ 0.85 it was called homozygous for the alternative base (BB). Sites with intermediate allele frequencies (0.15 < fA < 0.85) were called heterozygous (AB). Using these thresholds, a small number of SNPs were called in some strains later shown to be haploid. These apparent SNPs clustered in sub-telomeric regions known to contain repetitive DNA and are likely to be technical artefacts due to misaligning of reads to different repeats or to copy number variants. Note that these calls were only made for variable sites; the genomes also contain a much larger number of invariant sites that are considered identical among all strains and are therefore AA.
Nucleotide diversity (π) and average SNP density were calculated with VariScan v 2.0.3 (Hutter et al., 2006) using a non-overlapping window size of 1 kb along all chromosomes. In the case of heterozygous variants, only the variant with highest fB was used. Sites considered for the analysis of the 14 strains were required to show variation in a minimum number of 4 strains. SnpEff v 4.3s (Cingolani et al., 2012) was used to produce summary statistics and annotate the SNPs using the public NBRC1777 genome annotation as a reference (Inokuma et al., 2015).
Phylogenetic Analysis
Because the data consisted of a mixture of strains with different ploidies, we developed a custom method for phylogenetic analysis of haplotypes. This method is based on a window approach similar to our previous development for an interspecies hybrid (Schröder et al., 2016). Homozygosity and heterozygosity were first assessed in 1 kb windows of the genome of each diploid strain. For each 1 kb window in each strain, the total numbers of variable sites that were called with each genotype (#AA, #BB, and #AB) in the window were calculated. The whole window was then classified as either heterozygous for the two haplotypes if #AB ≥ 3, or homozygous otherwise. Homozygous windows were then classified as either homozygous for the alternative haplotype if #BB ≥ 9, or homozygous for the reference haplotype otherwise. The cut-off values of 3 and 9 were chosen based on analysis of the distributions of window frequencies, using strain L01 as a test case (Figure S1). Each 1 kb window of the genome was only used if it was heterozygous in all five diploid strains, and the regions with aberrant allele frequencies in NBRC0272 were excluded (chromosome 6). Concatenated nucleotide sequences of the shared heterozygous windows were extracted from the NBRC1777 genome and used to infer the sequences of alleles in each strain, depending on its ploidy.
For haploid strains, the variant was used to replace the reference base at the corresponding position of the variable site in the concatenated sequence. For diploid strains, two different putative A and B alleles were first generated and used as a template for base replacement depending on the type of variable site. For homozygous alternative (BB) sites, the variant was used for replacement in both A and B alleles. For heterozygous (AB) variable sites, the variant was only used for replacement in the B allele. For triploid strains, putative alleles 1, 2, and 3 were first generated and used as a template for base replacement depending on the types of variable sites and their allele frequencies. For homozygous alternative (BB) sites, the variant was used for replacement in all three putative alleles. For heterozygous (AB) sites, the variant was used for replacement in both alleles 2 and 3 if fB > 0.6, or for replacement only in allele 3 if fB < 0.4. The phylogenetic tree of the inferred and concatenated haplotype sequences was generated using PhyML v. 3.1 (Guindon et al., 2010) selecting for the best of NNI and SPR methods, using five random starts, and with empirical estimation of base frequencies and proportions of invariable sites (parameters: –search BEST –rand_start –n_rand_starts 5 -f e –v e). The tree was visualized using FigTree v. 1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/.)
Data Availability
The Illumina sequences generated and analyzed for this study can be found in the NCBI Sequence Read Archive under the accession number SRP128575, strain-specific accessions are provided in Table 1.
Results
K. marxianus Displays a High Level of Genomic Variation
The genome sequences of 14 strains of K. marxianus were analysed for SNPs to determine the extent of variability in this species. The strains selected for analysis included the five strains with published whole genome sequences (at the time of the study) and 9 other strains from different collections (Table 1). The five sequenced strains (NBRC1777, CBS6556, UFV3, DMKU3-1042, and UFS-Y2791) have been phenotypically analysed to different extents by a number of research teams and are of interest for biotechnological applications (Nonklang et al., 2008; Fonseca et al., 2013; Costa et al., 2014; Schabort et al., 2016; Nambu-Nishida et al., 2017). This is also the case for the previously unsequenced strains CBS397, NBRC0272, NBRC0288, and NBRC0617, which were obtained from national culture collections (Lane et al., 2011; Foukis et al., 2012; Yarimizu et al., 2013). The additional five strains (L01–L05) are from the in-house culture collection of the Lallemand company and there are no published data available for these strains. The original source of isolation is known for 13/14 strains and is almost evenly divided between dairy and non-dairy environments. The genome sequences of the 9 previously un-sequenced strains were obtained as described in methods and thus all 14 genomes could be analysed and compared. It should be noted that although all 14 strains were sequenced using Illumina technology, this was performed by different laboratories using a diversity of sequencing strategies and thus the depth of coverage is quite variable (Table 2).
We used the genome sequence of strain NBRC1777, which was assembled into eight complete chromosomes using Pacific Biosciences technology, as the reference for SNP analysis (Inokuma et al., 2015). GATK software was used to identify sequence variants present in the Illumina reads from all 14 strains relative to this reference. Variable sites were defined as the set of sites in the genome that contain a non-reference base in at least one of the 14 strains. For each variable site in each strain, the empirical allele frequency in the Illumina reads from that strain was calculated (see Methods). Depending on frequency, the variant was classified as homozygous SNP (fB ≥ 0.85), or heterozygous SNP (0.15 < fB < 0.85). Variants appearing at frequencies below 0.15 were assumed to be due to sequencing errors and were ignored. Only 249 SNPs were identified in the Illumina data from the reference NBRC1777, confirming that the reference sequence is accurate and this strain is haploid. All other strains contained more than 30,000 SNPs relative to the reference (Table 3). The highest number of SNPs is in strain UFS-Y2791 (Schabort et al., 2016) and corresponds to 3.0% nucleotide sequence divergence in the 10.9 Mb genome. From the numbers of heterozygous and homozygous (non-reference) SNPs, the other strains fall into three groups: one group with low numbers of heterozygous SNPs (<2000), one group with more heterozygous than homozygous SNPs, and one group with fewer heterozygous than homozygous SNPs. Below, we show that these three groups are haploid, diploid, and triploid strains, respectively.
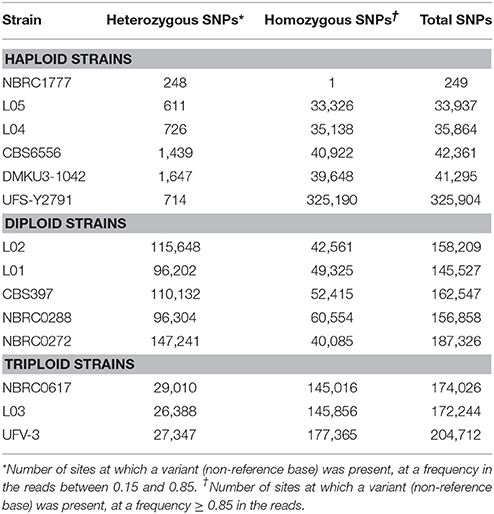
Table 3. SNPs identified in 14 K. marxianus strains.
In total, 667,472 variable sites, comprising 597,466 SNPs, and 70,006 indels were found. Indels were not analysed further, and, after filtering (see methods) a subset of 571,339 SNPs was retained for analysis. SNP diversity in K. marxianus is relatively high, with average pairwise difference between strains (π) of 12 × 10−3. The average density of SNPs in K. marxianus is 7.6 SNPs/kb in coding regions and 11.1 SNPs/kb in intergenic regions (Table S1). Of the 359,354 variants located within the coding regions of genes, 71.6% were predicted to be silent, 28.1% missense, and 0.2% (745) nonsense mutations, when compared against the NBRC1777 annotation (Inokuma et al., 2015).
K. marxianus Isolates Show Different Ploidy States
The distribution of allele frequencies for each strain was assessed using a graphical method similar to that used in a recent study in S. cerevisiae (Zhu et al., 2016). Histograms (Figure 1A), of the distribution of allele frequencies at all the variable sites in the genome created three sets of strains that corresponded to those identified on the basis of the numbers of heterozygous and homozygous SNPs (Table 3). The five strains with the highest numbers of heterozygous SNPs in (L02, L01, CBS397, NBRC0288, and NBRC0272) all show a symmetrical peak of allele frequencies centred on 0.5, suggesting that they are diploid. The three strains with high numbers of both homozygous and heterozygous SNPs (NBRC0617, L03, and UFV-3) all show bimodal distributions, with peaks at 0.33 and 0.66 for the frequency of the variant, suggesting that they are triploid. In contrast, in the six strains UFS-Y2791, DMKU3-1042, L05, L04, CBS6556, and NBRC1777, only a low number of sites were designated as heterozygous, and these sites show little pattern in their frequency distributions. These data are most consistent with a haploid genome.
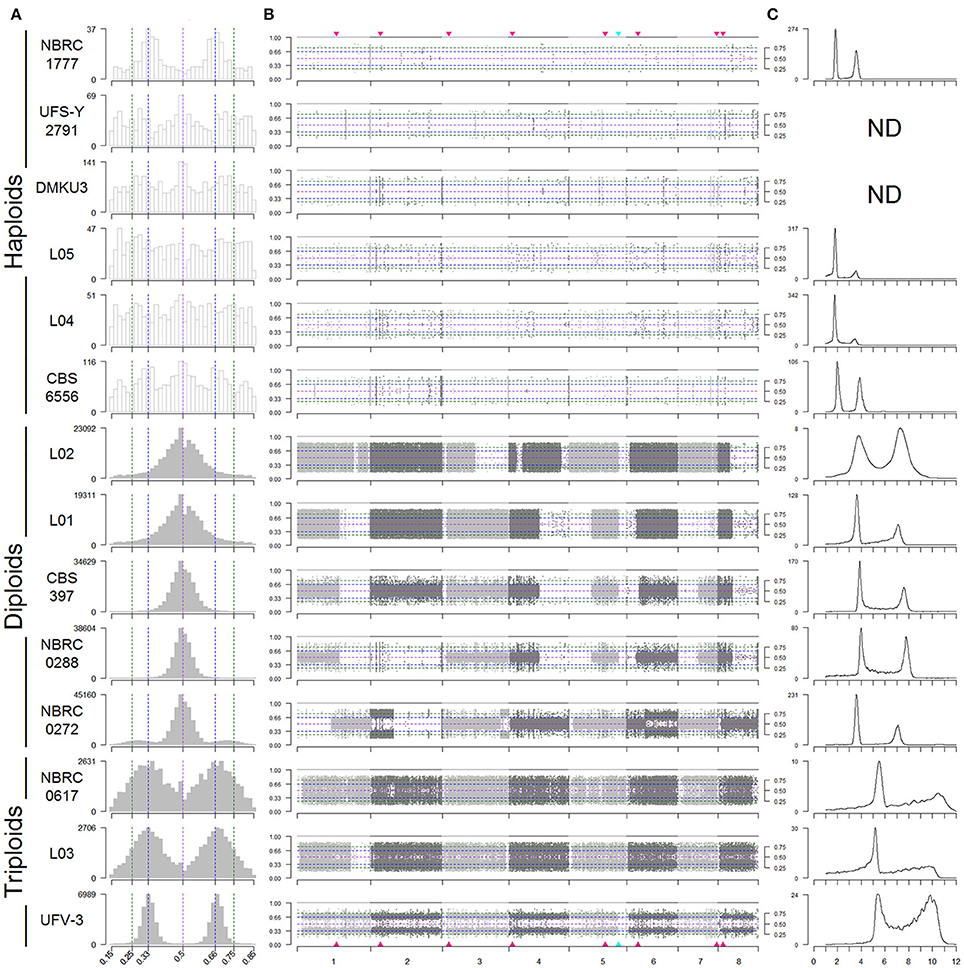
Figure 1. Variable ploidy in Kluyveromyces marxianus strains. Strain names are shown on the left. (A) Histograms of the alternative allele frequencies of variant (non-reference) bases, for SNPs designated as heterozygous (sites with alternative allele frequencies fB between 0.15 and 0.85). Histograms are coloured grey if at least 10% of the SNPs in a strain are heterozygous. Dashed vertical lines mark frequencies of 0.5 (purple), 0.33/0.66 (blue), and 0.25/0.75 (green). Bin sizes are 2% intervals. (B) Plots of alternative allele frequencies along the 8 chromosomes, for each strain. Horizontal dashed lines mark frequencies as in (A). Light and dark gray points indicate SNPs on different chromosomes. Red triangles mark the locations of centromeres, and the blue triangle marks the ribosomal DNA locus. Allele frequencies ≥ 0.85 are shown as 1. Alternative allele frequencies ≤ 0.15 are not shown. (C) Flow cytometry of DNA content. The Y-axis shows numbers of cells, and the X-axis shows SYTOX Green fluorescence signal intensity (arbitrary units) which is proportional to DNA content. Flow cytometry was not carried out for UFS-Y2791 and DMKU3-1042.
To provide an independent measurement of ploidy, DNA content in each strain was measured by flow cytometry (Figure 1C). Each analysed strain shows a bimodal distribution of DNA content, corresponding to the G1 and G2 phases of the cell cycle. For the haploid strains, the DNA content is 1n (in G1 phase) and 2n (in G2 phase), where n is the DNA content of the haploid genome. For the diploids, it is 2n and 4n, and for the triploids it is 3n and 6n. The patterns observed were consistent with the designation based on allele frequencies and confirmed that this set of strains was comprised of 6 haploid, 5 diploid and 3 triploid strains.
Common Patterns of Loss of Heterozygosity in Diploid Strains
For each SNP, its allele frequency vs. its chromosomal location in the reference assembly was plotted to determine whether heterozygosity was uniformly distributed (Figure 1B). For haploid strains, the low level of variation does not show any particular pattern, whereas in all five diploid strains, large regions of the genome with loss of heterozygosity (LOH) are apparent. In heterozygous regions of the genome, allele frequency should be distributed about 0.5 but there are regions where no variability is seen. These predominantly white areas in the plots indicate stretches of chromosome that are homozygous in that strain (Figure 1B). For example, strain L02 is heterozygous through most of its genome but shows homozygosity on the right half of chromosome 3 and over most of chromosome 8. In the diploids, most chromosomes are heterozygous over at least some of their length, but chromosome 2 in NBRC0288 and chromosome 7 in L01 are essentially completely homozygous. Three diploid strains L01, CBS397 and NBRC0288 exhibit almost identical extents of partial LOH on chromosomes 1, 3 (left end), 4, 5, 6, and 8 (Figure 1B), but different patterns on chromosomes 2 and 7. On chromosome 6, the region of LOH is slightly larger in CBS397 than in L01 and NBRC0288 (it crosses the centromere only in CBS397). Strain L02 shares two LOH boundaries with this group of three strains, on chromosome 5 (the boundary occurs at the rDNA locus) and chromosome 3 (left end). Only one region of LOH is visible in our triploids, on chromosome 1 in strain L03. This LOH region occupies ~4% of the L03 genome, whereas in the diploid strains the LOH regions total 25–51% of the genome by length.
Copy Number Variation and Partial Aneuploidy in Multiple Strains
Read coverage in 10 kb windows was determined across the genome of each strain to investigate whether any of the strains displayed aneuploidy (Figure 2). In this analysis, a value of zero indicates no variation between expected and actual numbers of reads, whereas higher or lower numbers could indicate DNA duplications or deletions. Although the SNP and flow cytometry results (Figure 1) indicated that there are three groups of strains with genomes that are primarily haploid, diploid, and triploid, there is also evidence in multiple strains of partial aneuploidy, segmental duplications, or deletions that alter the copy number of some parts of the genome. These possible aneuploidies do not correspond to the regions of LOH described in Figure 1, indicating that these are distinct phenomena.
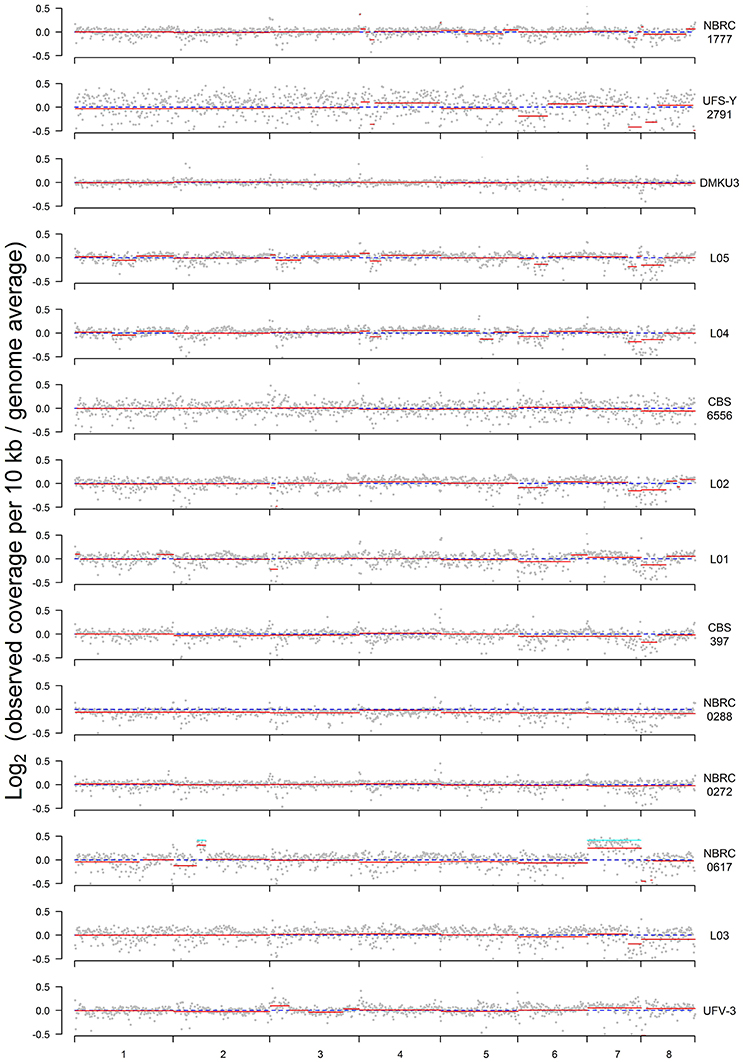
Figure 2. Plots of sequence coverage in each strain. The Y-axis is log2 of the ratio between the observed and expected coverage, for 10-kb windows through the genome; a value of zero (dashed blue line) indicates no difference. Expected coverage is based on the average in the whole genome. Red lines show the segmental means for consecutive 10-kb windows calculated using the Bioconductor package DNAcopy. Cyan lines for NBRC0617 indicate the value expected for the 1.33-fold increase in coverage that would result from a fourth copy of a region in a triploid.
The clearest case of aneuploidy is in strain NBRC0617, which has an extra copy of chromosome 7 (Figure 2). The analysis of ratios shows that reads from chromosome 7 are present at 1.18x the expected frequency (log2 value 0.249) in this strain. Since NBRC0617 is primarily triploid, a fourth copy of a chromosome should increase the read coverage on that chromosome by ~4/3 = 1.33-fold (cyan line in Figure 2) relative to the genome average, and many of the 10-kb windows in chromosome 7 are not significantly different from this value (Figure S2A). Furthermore, the distribution of allele frequency values for SNPs on chromosome 7 of NBRC0617 shows a peak at 0.25 (Figure S2B), which is consistent with the presence of four copies of the chromosome, and contrasts with the peaks at 0.33 and 0.66 that are seen when the whole genome of NBRC0617 is considered (Figure 1A). NBRC0617 also shows increased copy number of a circa 150 kb segment of chromosome 2 (coordinates 420–570 kb) (Figure 2). Within this segment, allele frequencies of 0.25 and 0.75 are visible (Figure S2B), indicating that it is present in 4 copies. With the current data, we are unable to determine the precise structure of the chromosomal rearrangement that increased the segment's copy number. The region immediately to the left of it (0–420 kb) may be present at a reduced copy number.
Some intriguing possible examples of copy number variation are seen in strain NBRC0272, which was designated as a diploid based on its overall allele frequency and flow cytometry patterns. Examination of allele frequencies indicates that there are three genomic regions (in chromosomes 2, 3, and 6) present at higher copy numbers (Figure 1B). This can be seen in more detail in the allele frequency plots of those chromosomes for this strain (Figure S3). Chromosomes 2 (left end) and 3 (right end) contain SNPs with 0.25/0.75 allele frequencies, indicating the presence of four copies. Chromosome 6 (right end) contains SNPs with allele frequency peaks close to 0.33/0.66, indicating three copies, and contrasting with the peaks at 0.5 on the left part of this chromosome. Since the flow cytometry (Figure 1C) shows that the total DNA content of NBRC0272 is close to diploid, it is concluded that the extra copies of these three chromosomal regions must be the result of segmental duplications and not extra copies of the whole chromosome. Puzzlingly, however, these putative segmental duplications were not apparent in the coverage plot of NBRC0272 (Figure 2).
Phylogenetic Analysis Separates Dairy From Non-dairy Haplotypes
The presence of strains with different ploidy in the dataset presents a problem for phylogenetic analysis. In studies on diploid eukaryotes such as mammals, the standard approaches for constructing phylogenies of individuals from SNP data either exclude all heterozygous sites, or randomly choose one of the alleles at these sites (Lischer et al., 2014). In our preliminary analyses of the K. marxianus data, it was noticed that in the diploid strains, one allele was often very similar to the NBRC1777 reference sequence, but the other allele was considerably different. We were therefore motivated to construct a phylogenetic tree of the K. marxianus strains that kept the alleles separate—namely, a tree of haplotypes rather than a tree of strains.
To make a tree of haplotypes, we used a method previously developed to investigate the pathogenic yeast Candida orthopsilosis, which is an interspecies hybrid (Schröder et al., 2016). In each of the 5 diploid K. marxianus strains, each region of the genome was classified as either heterozygous, homozygous for the “A” haplotype (the haplotype more similar to the NBRC1777 reference), or homozygous for the “B” haplotype (the haplotype less similar to the reference) (Figure 3; see Methods for details). Approximately 18% of the genome was heterozygous in all 5 diploid strains, and we then extracted the sequences of the “A” and “B” haplotypes from these regions in each diploid. The aberrant (trisomic or tetrasomic) regions of the NBRC0272 genome were excluded from this dataset. A similar process was used to estimate the sequences of the three haplotypes present in each of the three triploid strains in these regions (see Materials and Methods). In the phylogenetic tree of haplotypes then generated, each haploid strain appears once, each diploid strain appears twice, and each triploid strain appears three times (Figure 4). Three clades are evident, but Clade 3 contains only the haploid strain UFS-Y2791. Despite the high divergence of UFS-Y2791 from the other strains, a phylogenetic tree using K. lactis and Lachancea thermotolerans as outgroups confirms that it is indeed a strain of K. marxianus (Figure 4, inset). All other K. marxianus haplotypes lie in Clades 1 and 2. Clade 1 contains all the haploid strains except the outlier UFS-Y2791, and the “A” haplotypes of each of the five diploid strains. Clade 2 contains the “B” haplotypes of the diploid strains, and all three haplotypes of the triploid strains.
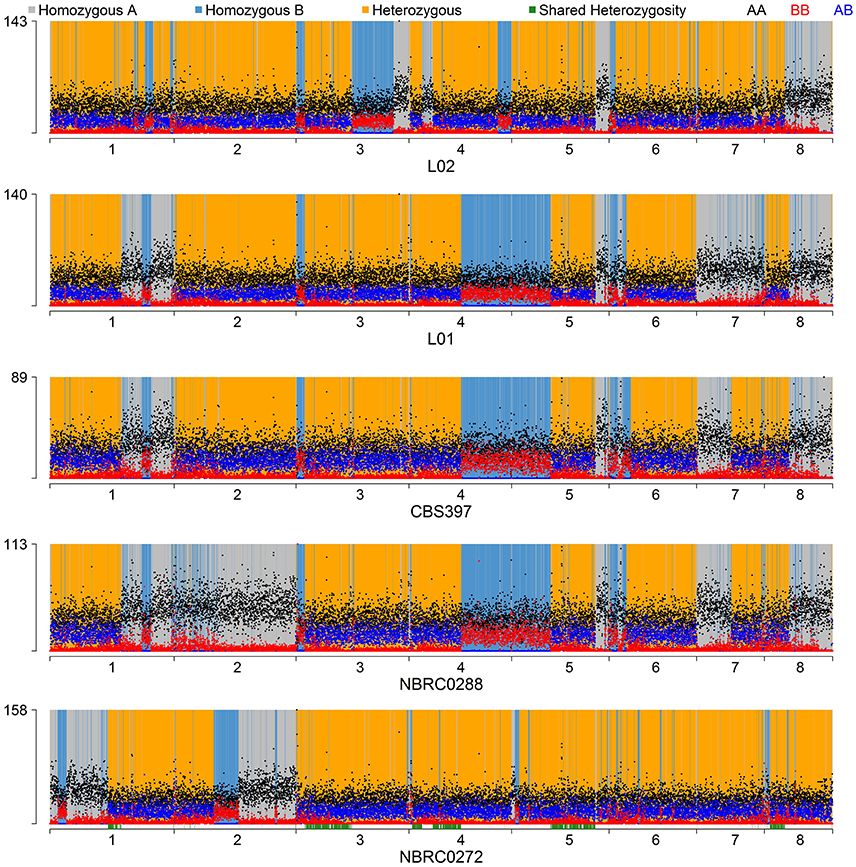
Figure 3. Haplotype assignment in diploid strains. For each 1-kb window through the genome, dots show the number of variable sites in the window with genotypes AA (black dots), BB (red dots), or AB (blue dots). Each window was then classified as either heterozygous (yellow background), homozygous for the B haplotype (blue background), or homozygous for the reference A haplotype (grey background) following rules as described in Methods and Figure S1. The green bars along the bottom axis show the regions (18%) that were heterozygous in all diploids and used for the haplotype phylogeny analysis.
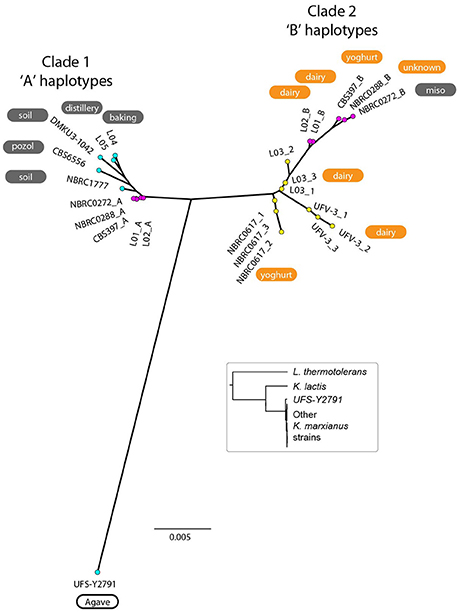
Figure 4. Phylogenetic tree of K. marxianus haplotypes. Coloured dots indicate the ploidy of strains as haploid (cyan; 1 haplotype per strain), diploid (magenta; 2 haplotypes per strain), or triploid (yellow; 3 haplotypes per strain). The environmental source of each strain is shown (for diploid strains, the source is only labeled on the “B” haplotype). Orange backgrounds indicate Lac+ strains, grey backgrounds indicate Lac− strains, and white backgrounds indicate Lac phenotypes not tested. The tree was constructed from SNP data from regions that are heterozygous in diploids, totalling 18% of the genome. Inset, phylogenetic tree constructed from DNA sequences of GCN1 (an arbitrarily chosen large gene), confirming that UFS-Y2791 is a strain of K. marxianus.
Lactose Consumption Phenotypes and LAC12 Genotypes
When the environments from which the strains were isolated are considered, an unexpected relationship is apparent between environment, ploidy and clade (Figure 4). The six strains from “dairy” environments (and strain NBRC0288, from an unknown source) are all either diploid or triploid whereas, with the exception of strain NBRC0272 (isolated from miso), all the strains from “non-dairy” environments are haploid (Table 1). Since the use of lactose as a sugar source is considered important for growth of dairy yeasts, the capacity of the strains to grow on lactose was assessed to determine whether a similar pattern would emerge (Figure 5). Indeed, none of the tested haploid strains used lactose, whereas all the diploid and triploid strains were Lac+, again with the exception of NBRC0272, which is diploid but Lac−. Although DMKU3-1042 was not available for this study, our previous work has demonstrated that it is Lac− (Varela et al., 2017). We also previously established that the variable ability of K. marxianus strains to consume lactose is explained by polymorphism of a single gene, the lactose transporter LAC12, and that functional and non-functional (in terms of lactose transport) alleles of this gene differ by 13 key amino acid substitutions (Varela et al., 2017). BLAST searches against de novo assemblies of the genomes showed that the key polymorphisms in all the Lac+ strains in Figure 5 match were an exact match to the functional LAC12+ allele, except for NBRC0617 which matched in 11 positions. Similarly, all the Lac− strains exactly matched the non-functional LAC12 allele, except for NBRC0272, which diverged at a single amino acid (Figure S4). None of the diploid strains is a LAC12+/− heterozygote, due to LOH at the left end of chromosome 3 where LAC12 is located (the gene is only 15 kb from the telomere). Thus, although all five diploids are AB heterozygous for most of chromosome 3, the four Lac+ diploids are “BB” homozygous and the Lac− diploid strain NBRC0272 is “AA” homozygous in this region (Figure 3). In addition to the polymorphisms associated with a non-functional allele, the LAC12 gene in NBRC0272 contains an internal stop codon (Figure S4).
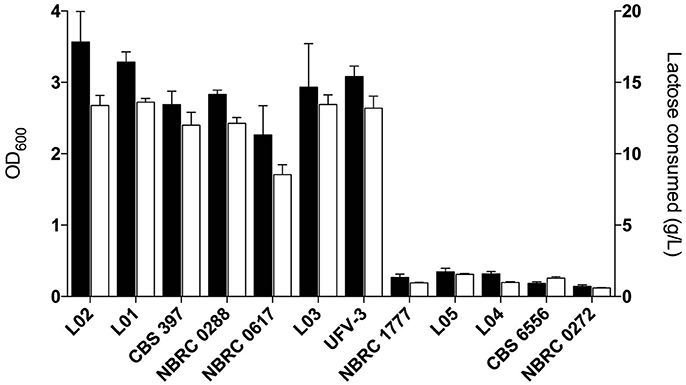
Figure 5. Growth and lactose consumption by K. marxianus strains. Growth in MM + 2 % lactose and lactose consumption are shown in black and white bars, respectively. Lactose consumption was calculated by subtracting the final and initial lactose concentrations in the medium.
Discussion
Ploidy in K. marxianus Distinguishes Dairy and Non-dairy Strains
Although this study relied on a relatively small set of strains (14), it delivered some remarkable insights into the life-cycle of K. marxianus. Our previous report that natural isolates of this yeast can be either haploid or diploid (Lane et al., 2011) was confirmed and then extended by the discovery of three triploid strains. The sample size is too small to draw statistical conclusions but it can be said that haploids, diploids, and triploids were present at roughly equal frequencies (43, 36, and 21% respectively) in this set. This appears to contrast with K. lactis, which is considered to be haploid, though it must be borne in mind that there are as yet no published production-level studies with that yeast. Variable ploidy is not uncommon in yeasts; for example, among 144 mainly clinical isolates of S. cerevisiae, the basal ploidy levels (i.e., ignoring aneuploid chromosomes) were haploid (11%), diploid (57%), triploid (16%), and tetraploid (16%) (Zhu et al., 2016).
The most striking finding was that all the isolates from a dairy environment were either diploid or triploid, whereas non-dairy isolates were haploid. Furthermore, it was possible to distinguish two genomic haplotypes, described here as “A” and “B” that mapped 13/14 strains into distinct clades (Clades 1 and 2). The 14th strain, UFS-Y2791 may represent a third clade. All the dairy isolates contained at least one of the B haplotype genomes, suggesting that this is a dairy-niche associated genome. In the case of the three triploid strains, the B genome was represented three times, whereas the diploid strains contained one A haplotype genome and one B haplotype genome. The sequence divergence (~2%) between the “A” and “B” haplotypes indicates that the diploid dairy strains were probably formed by mating between haploid representatives of Clades 1 and 2, as opposed to any other mechanism of ploidy change. Nevertheless, we were unable to examine MAT locus genotypes because the sequence assemblies are too fragmented in the MAT/HML/HMR regions. The phylogeny of the haplotypes indicates that the diploid dairy strains were formed by at least two independent matings between parents from the A and B clades, and that the A parents in these matings were very closely related to each other (much more so than the B parents). Triploids may have arisen by self-mating of B-haplotype (clade 2) strains, with one scenario being mating of a BB diploid with a B haploid to form a triploid; other routes to a triploid are also possible. It is implicit in these scenarios that B-haplotype haploid strains should also exist, though none were found in the current study. It is also notable that in our recent study developing an MLST method for K. marxianus, all 57 strains that were listed as coming from (6 different) dairy environments were heterozygous and therefore presumably diploid (Tittarelli et al., 2018). In fact, in that study, only 13/83 strains were homozygous in the regions included in the MLST. It is noted that one well-studied strain in the literature is K. marxianus CBS397 and this study and those of Fasoli et al. (2016) and Tittarelli et al. (2018) show that this strain is diploid (Fasoli et al., 2016; Tittarelli et al., 2018), which contrasts with what appears to be a previous erroneous suggestion based on long range PCR of the MAT locus that it was haploid (Lane et al., 2011).
The data suggest that the B-haplotype is a dairy-associated genome. It was gratifying, therefore, to identify one locus in this haplotype that confers a growth advantage in milk, the LAC12 gene. Our previous work identified positions in the Lac12p where the functional protein had one particular amino acid and the non-functional protein a different one (Varela et al., 2017). In six of the strains with a B-haplotype genome, there was an exact match to this functional sequence and in the seventh (NBRC0617), there was a match in 11 positions (Figure S4). Since all these strains grew on lactose as a sole sugar source, this now allows us to propose that the number of amino acid positions that distinguish a functional and non-functional lactose-transporting Lac12p protein can be refined to these 11 amino acids though confirmation would require functional tests. The Lac− strain with the B-haplotype genome was NBRC0272, which is homozygous for the non-functional LAC12 allele. This strain was isolated from a non-dairy environment (miso) and the most likely explanation is that it arose like the other diploids as a hybrid between an A and a B strain but since lactose transport was not required in its niche, it was possible for it to lose the B-haplotype LAC12 allele through a LOH event at the left end of chromosome 3 whereas the other diploids lost the non-functional A-haplotype LAC12 allele via a similar LOH event (Figure 4).
The situation in K. marxianus seems to resemble a pattern seen in Saccharomyces and Zygosaccharomyces species, where strains used in industrial processes or isolated from industrial environments are often polyploids or interspecies hybrids, whereas “natural” isolates (e.g., from non-anthropogenic environments) tend to be haploid or homozygous diploid (Hittinger, 2013; Suh et al., 2013; Wendland, 2014; Ortiz-Merino et al., 2017). This pattern is thought to reflect selection toward stress tolerance in the industrial environment, but toward maintenance of the ability to mate and sporulate in natural environments. If this is also the case with K. marxianus, it could be expected that the AB diploids display enhanced stress tolerance, at least over B-haplotype haploid strains in a dairy environment. Experiments to date have not succeeded in identifying any correlations between ploidy and stress tolerance (data not shown), but more studies that also include B-haplotype haploids are required to further address this question. As mentioned, B-haplotype strains have not yet been positively identified but there are Lac+ candidates worth investigating, for example, K. marxianus NCYC1424, shown to be homozygous by Tittarelli et al. (2018), and K. marxianus NCYC1429, which appears to be haploid based on genetic crossing (Varela et al., 2017).
Sequence Diversity, Aneuploidy, and LOH in K. marxianus
One of the aims of this study was to assess if the wide phenotypic diversity that has been observed in K. marxianus was reflected in its genome diversity. The large number of SNPs (>500 k) observed in the set of K. marxianus strains used in our study shows relatively high SNP diversity. We found an average pairwise difference (π) of 12 × 10−3, which is comparable with reported values from other yeasts; for example, 4 × 10−3 in S. cerevisiae, 12 × 10−3 in S. uvarum, and 17 × 10−3 in L. kluyveri (Peter and Schacherer, 2016).
Previous studies with S. cerevisiae isolates showed variable ploidy (from 1 to 4 copies of the genome), aneuploidy (unequal copy numbers of different chromosomes), or variation in the copy number of segments of chromosomes (Hose et al., 2015; Strope et al., 2015; Zhu et al., 2016). Similar to S. cerevisiae but unlike L. kluyveri (Friedrich et al., 2015), all these phenomena were observed in this study of K. marxianus. The most unambiguous example of aneuploidy in K. marxianus is the presence of an extra copy of chromosome 7 in NBRC0617, but, as described in the results, there are multiple other likely cases of aneuploidies or copy number variation that would need to be investigated in more detail.
There are also quite extensive regions of LOH in the diploid strains (25–51% by genome length) but not in the triploids. LOH arises when the genome homogenises in a region and it is expected to be rarer in triploid strains, because it will only be apparent if all three copies of a genomic region were homogenised. The shared patterns of LOH in different diploid K. marxianus isolates was unexpected. This observation could indicate that these strains are closely related and are mitotic descendants of a recent diploid common ancestor that had already lost heterozygosity in the shared regions. Alternatively, it could indicate that this species mostly reproduces by mitosis and rarely goes through meiosis and sporulation, at least in the dairy environment. Nonetheless, the divergence between the K. marxianus clades is low enough that it would not be expected to cause problems in meiosis. In S. paradoxus, crosses between strains with sequence divergence of up to 4.6% can still produce viable gametes, as long as the genomes are collinear (Liti et al., 2006). It should also be considered that dairy is probably not the original niche for K. marxianus and the strains that we studied were most likely selected during a fermentation process. This could have specifically selected hybrids between A and B clade strains, and may also promote LOH. Other than the preservation of the functional lactose transporting allele of LAC12 (B-haplotype), there was not an obvious preference for either the A-haplotype or the B-haplotype during LOH events (Figure 3). Because lactose utilisation confers a benefit during growth in milk, one can speculate that the LOH of the region containing the functional LAC12 gene is an adaptive response. It is not possible to say, however, whether or not other selective pressures played a role in determining the overall patterns of LOH.
Implications for Biotechnology
This study focused on a small set of strains of biotechnological interest and therefore may not be fully representative of the species diversity. Indeed, one strain (UFS-Y2791) was far more diverse than the others, suggesting that there is further diversity to be accessed. Given that UFS-Y2791 was isolated from agave juice (in South Africa), it will be interesting to see whether strains associated with tequila/mezcal fermentation (also from agave) in Mexico show any relationship to this strain. The divergence between strains used, either deliberately or traditionally, in the food biotechnology sector is very significant in comparison to those isolated from “natural” environments. Perhaps the natural state of K. marxianus is haploid like its sister K. lactis, and diploids only arise after biotechnological selection. It is possible that diploids will have advantages, though, other than for lactose utilisation, these are not yet apparent. Haploid strains are much easier to engineer and manipulate so, for most biotechnological applications, it may be preferable to choose Clade 1 (A haplotype) strains. Nonetheless, divergent alleles in Clade 2 (B haplotype) may also be functionally important (for example LAC12) so this will still need to be considered in future studies.
Author Contributions
RO-M and JV: contributed equally to the paper, they carried out the bulk of the experimental and bioinformatic analysis and wrote the manuscript; AC: contributed with strain handling and sample preparation for DNA sequencing; CW, NK, J-MG, WdS, and HH: sequenced K. marxianus strains for the study; KW and JM: conceived the study, supervised the research, analysed and interpreted data and contributed to writing the manuscript.
Conflict of Interest Statement
CW was employed by company Lallemand Inc and J-MG and NK by Heineken.
The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
RO-M and JV were supported by the YEASTCELL Marie Curie ITN project which received funding from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7/2007-2013/ under REA grant agreement n° 606795. RO-M was also partially supported by CONACyT, Mexico (fellowship number 440667). AC was supported by Science Foundation Ireland (13/IA/1910). This study was supported in part by the Adaptable and the Advanced Low Carbon Technology R&D Program (JST, Japan). We thank Kevin Byrne for computational support and colleagues listed in Table 1 for providing raw Illumina source files of genome sequences.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2018.00094/full#supplementary-material
Figure S1. Method of classifying 1-kb genomic windows into haplotypes. The cutoff values chosen are marked by triangles in all panels (windows with ≥3 AB SNPs were classified as AB heterozygous windows; windows with ≥9 BB SNPs were classified as BB heterozygous windows; other windows were classified as AA homozygous windows). The data plotted are for the diploid strain L01. (A) Histogram of numbers of SNPs of each type, in all 1-kb windows in the genome. (B) Heatmap showing the distribution of numbers of AB and BB SNPs per window. Cells in the matrix show numbers of windows. The cutoff values were chosen to coincide with minima on the two axes. (C) Distribution of numbers of BB SNPs in windows that have zero AB SNPs.
Figure S2. Allele frequencies and sequence coverage in three strains on (A) chromosome 7 and (B) chromosome 2. The left and middle panels show distributions of allele frequency on each chromosome, as in Figure 1. The centromere is marked by a vertical red line. The right panels show log2 ratios between observed and expected sequence coverage, as in Figure 2. Cyan lines for NBRC0617 indicate the value expected for the 1.33-fold increase in coverage that would result from a fourth copy of a region in a triploid. The vertical orange lines in (B) mark the 150 kb region from with increased coverage in NBRC0617 (coordinates 420–570 kb).
Figure S3. Allele frequencies on each chromosome of NBRC0272. Details are as in Figures 1A,B. In the plots on the right, red vertical lines mark centromeres and blue lines mark the rDNA array.
Figure S4. Multiple alignment of Lac12p sequences from the strains used in this study. Amino acid sequences shown were derived from the haplotypes of each strain. Haplotypes from diploid and triploid strains are denoted by letters and numbers, respectively. The residues marked in color are those associated with either functional or non-functional alleles. The positions marked in blue were previously part of the set that distinguished alleles but these are not conserved in all haplotype B Lac12p. Those in red are those that still distinguish based on functionality and an additional differentiating AA at position 475 (pink shading) that was overlooked in the previous study is also marked. Thus, based on sequences currently available, there are 11 differentiating amino acids. The stop codon in the NBRC0272_B sequence at position 139 is indicated with a green asterisk.
Table S1. SNP density.
References
Arrizon, J., Morel, S., Gschaedler, A., and Monsan, P. (2012). Fructanase and fructosyltransferase activity of non-Saccharomyces yeasts isolated from fermenting musts of Mezcal. Bioresour. Technol. 110, 560–565. doi: 10.1016/j.biortech.2012.01.112
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barsoum, E., Martinez, P., and Aström, S. U. (2010). Alpha3, a transposable element that promotes host sexual reproduction. Genes Dev. 24, 33–44. doi: 10.1101/gad.557310
Belloch, C., Barrio, E., García, M. D., and Querol, A. (1998). Inter- and intraspecific chromosome pattern variation in the yeast genus Kluyveromyces. Yeast 14, 1341–1354.
Beniwal, A., Saini, P., Kokkiligadda, A., and Vij, S. (2017). Physiological growth and galactose utilization by dairy yeast Kluyveromyces marxianus in mixed sugars and whey during fermentation. 3 Biotech 7:349. doi: 10.1007/s13205-017-0985-1
Booth, L. N., Tuch, B. B., and Johnson, A. D. (2010). Intercalation of a new tier of transcription regulation into an ancient circuit. Nature 468, 959–963. doi: 10.1038/nature09560
Cingolani, P., Platts, A., Wang le, L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Coloretti, F., Chiavari, C., Luise, D., Tofalo, R., Fasoli, G., Suzzi, G., et al. (2017). Detection and identification of yeasts in natural whey starter for Parmigiano Reggiano cheese-making. Int. Dairy J. 66(Suppl. C), 13–17. doi: 10.1016/j.idairyj.2016.10.013
Costa, D. A., de Souza, C. J., Costa, P. S., Rodrigues, M. Q., dos Santos, A. F., Lopes, M. R., et al. (2014). Physiological characterization of thermotolerant yeast for cellulosic ethanol production. Appl. Microbiol. Biotechnol. 98, 3829–3840. doi: 10.1007/s00253-014-5580-3
Dias, O., Basso, T. O., Rocha, I., Ferreira, E. C., and Gombert, A. K. (2017). Quantitative physiology and elemental composition of Kluyveromyces lactis CBS 2359 during growth on glucose at different specific growth rates. Antonie Van Leeuwenhoek 111, 183–195. doi: 10.1007/s10482-017-0940-5
Dias, O., Gombert, A. K., Ferreira, E. C., and Rocha, I. (2012). Genome-wide metabolic (re-) annotation of Kluyveromyces lactis. BMC Genomics 13:517. doi: 10.1186/1471-2164-13-517
Dias, O., Pereira, R., Gombert, A. K., Ferreira, E. C., and Rocha, I. (2014). iOD907, the first genome-scale metabolic model for the milk yeast Kluyveromyces lactis. Biotechnol. J. 9, 776–790. doi: 10.1002/biot.201300242
Diniz, R. H. S., Villada, J. C., Alvim, M. C. T., Vidigal, P. M. P., Vieira, N. M., Lamas-Maceiras, M., et al. (2017). Transcriptome analysis of the thermotolerant yeast Kluyveromyces marxianus CCT 7735 under ethanol stress. Appl. Microbiol. Biotechnol. 101, 6969–6980. doi: 10.1007/s00253-017-8432-0
Fasoli, G., Barrio, E., Tofalo, R., Suzzi, G., and Belloch, C. (2016). Multilocus analysis reveals large genetic diversity in Kluyveromyces marxianus strains isolated from Parmigiano Reggiano and Pecorino di Farindola cheeses. Int. J. Food Microbiol. 233, 1–10. doi: 10.1016/j.ijfoodmicro.2016.05.028
Fasoli, G., Tofalo, R., Lanciotti, R., Schirone, M., Patrignani, F., Perpetuini, G., et al. (2015). Chromosome arrangement, differentiation of growth kinetics and volatile molecule profiles in Kluyveromyces marxianus strains from Italian cheeses. Int. J. Food Microbiol. 214, 151–158. doi: 10.1016/j.ijfoodmicro.2015.08.001
Fonseca, G. G., de Carvalho, N. M., and Gombert, A. K. (2013). Growth of the yeast Kluyveromyces marxianus CBS 6556 on different sugar combinations as sole carbon and energy source. Appl. Microbiol. Biotechnol. 97, 5055–5067. doi: 10.1007/s00253-013-4748-6
Fonseca, G. G., Gombert, A. K., Heinzle, E., and Wittmann, C. (2007). Physiology of the yeast Kluyveromyces marxianus during batch and chemostat cultures with glucose as the sole carbon source. FEMS Yeast Res. 7, 422–435. doi: 10.1111/j.1567-1364.2006.00192.x
Fonseca, G. G., Heinzle, E., Wittmann, C., and Gombert, A. K. (2008). The yeast Kluyveromyces marxianus and its biotechnological potential. Appl. Microbiol. Biotechnol. 79, 339–354. doi: 10.1007/s00253-008-1458-6
Foukis, A., Stergiou, P. Y., Theodorou, L. G., Papagianni, M., and Papamichael, E. M. (2012). Purification, kinetic characterization and properties of a novel thermo-tolerant extracellular protease from Kluyveromyces marxianus IFO 0288 with potential biotechnological interest. Bioresour. Technol. 123, 214–220. doi: 10.1016/j.biortech.2012.06.090
Friedrich, A., Jung, P., Reisser, C., Fischer, G., and Schacherer, J. (2015). Population genomics reveals chromosome-scale heterogeneous evolution in a protoploid yeast. Mol. Biol. Evol. 32, 184–192. doi: 10.1093/molbev/msu295
Fukuhara, H. (2006). Kluyveromyces lactis- a retrospective. FEMS Yeast Res. 6, 323–324. doi: 10.1111/j.1567-1364.2005.00012.x
Gethins, L., Rea, M. C., Stanton, C., Ross, R. P., Kilcawley, K., O'Sullivan, M., et al. (2016). Acquisition of the yeast Kluyveromyces marxianus from unpasteurised milk by a kefir grain enhances kefir quality. FEMS Microbiol. Lett. 363:fnw165. doi: 10.1093/femsle/fnw165
Gombert, A. K., Madeira, J. V. Jr., Cerdán, M. E., and González-Siso, M. I. (2016). Kluyveromyces marxianus as a host for heterologous protein synthesis. Appl. Microbiol. Biotechnol. 100, 6193–6208. doi: 10.1007/s00253-016-7645-y
Groeneveld, P., Stouthamer, A. H., and Westerhoff, H. V. (2009). Super life–how and why ‘cell selection’ leads to the fastest-growing eukaryote. FEBS J. 276, 254–270. doi: 10.1111/j.1742-4658.2008.06778.x
Guimarães, P. M., Teixeira, J. A., and Domingues, L. (2010). Fermentation of lactose to bio-ethanol by yeasts as part of integrated solutions for the valorisation of cheese whey. Biotechnol. Adv. 28, 375–384. doi: 10.1016/j.biotechadv.2010.02.002
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Haase, S. B., and Reed, S. I. (2002). Improved flow cytometric analysis of the budding yeast cell cycle. Cell Cycle 1, 132–136. doi: 10.4161/cc.1.2.114
Hittinger, C. T. (2013). Saccharomyces diversity and evolution: a budding model genus. Trends Genet. 29, 309–317. doi: 10.1016/j.tig.2013.01.002
Hose, J., Yong, C. M., Sardi, M., Wang, Z., Newton, M. A., and Gasch, A. P. (2015). Dosage compensation can buffer copy-number variation in wild yeast. Elife 4:e05462. doi: 10.7554/eLife.05462
Hughes, S. R., Qureshi, N., López-Núñez, J. C., Jones, M. A., Jarodsky, J. M., Galindo-Leva, L. Á., et al. (2017). Utilization of inulin-containing waste in industrial fermentations to produce biofuels and bio-based chemicals. World J. Microbiol. Biotechnol. 33:78. doi: 10.1007/s11274-017-2241-6
Hutter, S., Vilella, A. J., and Rozas, J. (2006). Genome-wide DNA polymorphism analyses using VariScan. BMC Bioinformatics 7:409. doi: 10.1186/1471-2105-7-409
Inokuma, K., Ishii, J., Hara, K. Y., Mochizuki, M., Hasunuma, T., and Kondo, A. (2015). Complete genome sequence of Kluyveromyces marxianus NBRC1777, a nonconventional thermotolerant yeast. Genome Announc 3:e00389-15. doi: 10.1128/genomeA.00389-15
Jeong, H., Lee, D. H., Kim, S. H., Kim, H. J., Lee, K., Song, J. Y., et al. (2012). Genome sequence of the thermotolerant yeast Kluyveromyces marxianus var. marxianus KCTC 17555. Eukaryot. Cell 11, 1584–1585. doi: 10.1128/EC.00260-12
Jiang, H., Lei, R., Ding, S. W., and Zhu, S. (2014). Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15:182. doi: 10.1186/1471-2105-15-182
Kobayashi, Y., Sahara, T., Suzuki, T., Kamachi, S., Matsushika, A., Hoshino, T., et al. (2017). Genetic improvement of xylose metabolism by enhancing the expression of pentose phosphate pathway genes in Saccharomyces cerevisiae IR-2 for high-temperature ethanol production. J. Ind. Microbiol. Biotechnol. 44, 879–891. doi: 10.1007/s10295-017-1912-5
Lachance, M.-A. (2011). “Chapter 35-Kluyveromyces van der Walt (1971) A2-Kurtzman, Cletus P,” in The Yeasts 5th Edn, eds J. W. Fell and T. Boekhout (London: Elsevier), 471–481.
Lane, M. M., Burke, N., Karreman, R., Wolfe, K. H., O'Byrne, C. P., and Morrissey, J. P. (2011). Physiological and metabolic diversity in the yeast Kluyveromyces marxianus. Antonie Van Leeuwenhoek 100, 507–519. doi: 10.1007/s10482-011-9606-x
Lane, M. M., and Morrissey, J. P. (2010). Kluyveromyces marxianus: a yeast emerging from its sister's shadow. Fungal Biol. Rev. 24, 17–26. doi: 10.1016/j.fbr.2010.01.001
Lappe-Oliveras, P., Moreno-Terrazas, R., Arrizón-Gaviño, J., Herrera-Suárez, T., García-Mendoza, A., and Gschaedler-Mathis, A. (2008). Yeasts associated with the production of Mexican alcoholic nondistilled and distilled Agave beverages. FEMS Yeast Res. 8, 1037–1052. doi: 10.1111/j.1567-1364.2008.00430.x
Lee, J. W., In, J. H., Park, J. B., Shin, J., Park, J. H., Sung, B. H., et al. (2017). Co-expression of two heterologous lactate dehydrogenases genes in Kluyveromyces marxianus for l-lactic acid production. J. Biotechnol. 241, 81–86. doi: 10.1016/j.jbiotec.2016.11.015
Lertwattanasakul, N., Kosaka, T., Hosoyama, A., Suzuki, Y., Rodrussamee, N., Matsutani, M., et al. (2015). Genetic basis of the highly efficient yeast Kluyveromyces marxianus: complete genome sequence and transcriptome analyses. Biotechnol. Biofuels 8:47. doi: 10.1186/s13068-015-0227-x
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lin, Y. J., Chang, J. J., Lin, H. Y., Thia, C., Kao, Y. Y., Huang, C. C., et al. (2017). Metabolic engineering a yeast to produce astaxanthin. Bioresour. Technol. 245(Pt A), 899–905. doi: 10.1016/j.biortech.2017.07.116
Lischer, H. E., Excoffier, L., and Heckel, G. (2014). Ignoring heterozygous sites biases phylogenomic estimates of divergence times: implications for the evolutionary history of microtus voles. Mol. Biol. Evol. 31, 817–831. doi: 10.1093/molbev/mst271
Liti, G., Barton, D. B., and Louis, E. J. (2006). Sequence diversity, reproductive isolation and species concepts in Saccharomyces. Genetics 174, 839–850. doi: 10.1534/genetics.106.062166
Liti, G., Carter, D. M., Moses, A. M., Warringer, J., Parts, L., James, S. A., et al. (2009). Population genomics of domestic and wild yeasts. Nature 458, 337–341. doi: 10.1038/nature07743
Morrissey, J. P., Etschmann, M. M., Schrader, J., and de Billerbeck, G. M. (2015). Cell factory applications of the yeast Kluyveromyces marxianus for the biotechnological production of natural flavour and fragrance molecules. Yeast 32, 3–16. doi: 10.1002/yea.3054
Nambu-Nishida, Y., Nishida, K., Hasunuma, T., and Kondo, A. (2017). Development of a comprehensive set of tools for genome engineering in a cold- and thermo-tolerant Kluyveromyces marxianus yeast strain. Sci. Rep. 7:8993. doi: 10.1038/s41598-017-08356-5
Nonklang, S., Abdel-Banat, B. M., Cha-aim, K., Moonjai, N., Hoshida, H., Limtong, S., et al. (2008). High-temperature ethanol fermentation and transformation with linear DNA in the thermotolerant yeast Kluyveromyces marxianus DMKU3-1042. Appl. Environ. Microbiol. 74, 7514–7521. doi: 10.1128/AEM.01854-08
Nonklang, S., Ano, A., Abdel-Banat, B. M., Saito, Y., Hoshida, H., and Akada, R. (2009). Construction of flocculent Kluyveromyces marxianus strains suitable for high-temperature ethanol fermentation. Biosci. Biotechnol. Biochem. 73, 1090–1095. doi: 10.1271/bbb.80853
Oliveira, S. S. S., Bello, M. L., Rodrigues, C. R., Azevedo, P. L., Ramos, M. C. K. V., Aquino-Neto, F. R., et al. (2017). Asymmetric bioreduction of beta-ketoesters derivatives by Kluyveromyces marxianus: influence of molecular structure on the conversion and enantiomeric excess. An. Acad. Bras. Cienc. 89, 1403–1415. doi: 10.1590/0001-3765201720170118
Ortiz-Merino, R. A., Kuanyshev, N., Braun-Galleani, S., Byrne, K. P., Porro, D., Branduardi, P., et al. (2017). Evolutionary restoration of fertility in an interspecies hybrid yeast, by whole-genome duplication after a failed mating-type switch. PLoS Biol. 15:e2002128. doi: 10.1371/journal.pbio.2002128
Peter, J., and Schacherer, J. (2016). Population genomics of yeasts: towards a comprehensive view across a broad evolutionary scale. Yeast 33, 73–81. doi: 10.1002/yea.3142
Quarella, S., Lovrovich, P., Scalabrin, S., Campedelli, I., Backovic, A., Gatto, V., et al. (2016). Draft genome sequence of the probiotic yeast Kluyveromyces marxianus fragilis B0399. Genome Announc. 4:e00923-16. doi: 10.1128/genomeA.00923-16
Rajaei, N., Chiruvella, K. K., Lin, F., and Aström, S. U. (2014). Domesticated transposase Kat1 and its fossil imprints induce sexual differentiation in yeast. Proc. Natl. Acad. Sci. U.S.A. 111, 15491–15496. doi: 10.1073/pnas.1406027111
Ricci, A., Allende, A., Bolton, D., Chemaly, M., Davies, R., Girones, R., et al. (2017). Update of the list of QPS-recommended biological agents intentionally added to food or feed as notified to EFSA 5: suitability of taxonomic units notified to EFSA until September 2016. EFSA J. 15:4663. doi: 10.2903/j.efsa.2017.4663
Rocha, S. N., Abrahão-Neto, J., and Gombert, A. K. (2011). Physiological diversity within the Kluyveromyces marxianus species [corrected]. Antonie Van Leeuwenhoek 100, 619–630. doi: 10.1007/s10482-011-9617-7
Rodicio, R., and Heinisch, J. J. (2013). Yeast on the milky way: genetics, physiology and biotechnology of Kluyveromyces lactis. Yeast 30, 165–177. doi: 10.1002/yea.2954
Schabort, D. T. W. P., Letebele, P. K., Steyn, L., Kilian, S. G., and du Preez, J. C. (2016). Differential RNA-seq, multi-network analysis and metabolic regulation analysis of Kluyveromyces marxianus reveals a compartmentalised response to xylose. PLoS ONE 11:e0156242. doi: 10.1371/journal.pone.0156242
Schacherer, J., Shapiro, J. A., Ruderfer, D. M., and Kruglyak, L. (2009). Comprehensive polymorphism survey elucidates population structure of Saccharomyces cerevisiae. Nature 458, 342–345. doi: 10.1038/nature07670
Schaffrath, R., and Breunig, K. D. (2000). Genetics and molecular physiology of the yeast Kluyveromyces lactis. Fungal Genet. Biol. 30, 173–190. doi: 10.1006/fgbi.2000.1221
Schröder, M. S., Martinez de San Vicente, K., Prandini, T. H., Hammel, S., Higgins, D. G., Bagagli, E., et al. (2016). Multiple origins of the pathogenic yeast Candida orthopsilosis by separate hybridizations between two parental species. PLoS Genet. 12:e1006404. doi: 10.1371/journal.pgen.1006404
Signori, L., Passolunghi, S., Ruohonen, L., Porro, D., and Branduardi, P. (2014). Effect of oxygenation and temperature on glucose-xylose fermentation in Kluyveromyces marxianus CBS712 strain. Microb. Cell Fact. 13:51. doi: 10.1186/1475-2859-13-51
Silveira, W. B., Diniz, R. H., Cerdán, M. E., González-Siso, M. I., Souza Rde, A., Vidigal, P. M., et al. (2014). Genomic sequence of the yeast Kluyveromyces marxianus CCT 7735 (UFV-3), a highly lactose-fermenting yeast isolated from the Brazilian Dairy Industry. Genome Announc 2:e01136-14. doi: 10.1128/genomeA.01136-14
Souciet, J., Aigle, M., Artiguenave, F., Blandin, G., Bolotin-Fukuhara, M., Bon, E., et al. (2000). Genomic exploration of the hemiascomycetous yeasts: 1. A set of yeast species for molecular evolution studies. FEBS Lett. 487, 3–12. doi: 10.1016/S0014-5793(00)02272-9
Strope, P. K., Skelly, D. A., Kozmin, S. G., Mahadevan, G., Stone, E. A., Magwene, P. M., et al. (2015). The 100-genomes strains, an S. cerevisiae resource that illuminates its natural phenotypic and genotypic variation and emergence as an opportunistic pathogen. Genome Res. 25, 762–774. doi: 10.1101/gr.185538.114
Suh, S. O., Gujjari, P., Beres, C., Beck, B., and Zhou, J. (2013). Proposal of Zygosaccharomyces parabailii sp. nov. and Zygosaccharomyces pseudobailii sp. nov., novel species closely related to Zygosaccharomyces bailii. Int. J. Syst. Evol. Microbiol. 63(Pt 5), 1922–1929. doi: 10.1099/ijs.0.048058-0
Tittarelli, F., Varela, J. A., Gethins, J., Stanton, C., Ross, C., Suzzi, P., et al. (2018). Development and implementation of multilocus sequence typing to study diversity of the yeast Kluyveromyces marxianus in Italian cheeses. Microb. Genomics doi: 10.1099/mgen.0.000153. [Epub ahead of print].
Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43
Varela, J. A., Montini, N., Scully, D., Van der Ploeg, R., Oreb, M., Boles, E., et al. (2017). Polymorphisms in the LAC12 gene explain lactose utilisation variability in Kluyveromyces marxianus strains. FEMS Yeast Res. 17:fox021. doi: 10.1093/femsyr/fox021
Verdugo Valdez, A., Segura Garcia, L., Kirchmayr, M., Ramírez Rodríguez, P., González Esquinca, A., Coria, R., et al. (2011). Yeast communities associated with artisanal mezcal fermentations from Agave salmiana. Antonie Van Leeuwenhoek 100, 497–506. doi: 10.1007/s10482-011-9605-y
Wang, Y. J., Ying, B. B., Shen, W., Zheng, R. C., and Zheng, Y. G. (2017). Rational design of Kluyveromyces marxianus ZJB14056 aldo-keto reductase KmAKR to enhance diastereoselectivity and activity. Enzyme Microb. Technol. 107, 32–40. doi: 10.1016/j.enzmictec.2017.07.012
Wendland, J. (2014). Lager yeast comes of age. Eukaryot. Cell 13, 1256–1265. doi: 10.1128/EC.00134-14
Wu, W. H., Hung, W. C., Lo, K. Y., Chen, Y. H., Wan, H. P., and Cheng, K. C. (2016). Bioethanol production from taro waste using thermo-tolerant yeast Kluyveromyces marxianus K21. Bioresour. Technol. 201, 27–32. doi: 10.1016/j.biortech.2015.11.015
Yarimizu, T., Nonklang, S., Nakamura, J., Tokuda, S., Nakagawa, T., Lorreungsil, S., et al. (2013). Identification of auxotrophic mutants of the yeast Kluyveromyces marxianus by non-homologous end joining-mediated integrative transformation with genes from Saccharomyces cerevisiae. Yeast 30, 485–500. doi: 10.1002/yea.2985
Zhu, Y. O., Sherlock, G., and Petrov, D. A. (2016). Whole genome analysis of 132 clinical Saccharomyces cerevisiae strains reveals extensive ploidy variation. G3 (Bethesda). 6, 2421–2434. doi: 10.1534/g3.116.029397
Keywords: lactose transport, non-conventional yeast, yeast evolution, industrial yeast, dairy, Kluyveromyces, LAC12
Citation: Ortiz-Merino RA, Varela JA, Coughlan AY, Hoshida H, Silveira WB, Wilde C, Kuijpers NGA, Geertman J-M, Wolfe KH and Morrissey JP (2018) Ploidy Variation in Kluyveromyces marxianus Separates Dairy and Non-dairy Isolates. Front. Genet. 9:94. doi: 10.3389/fgene.2018.00094
Received: 20 January 2018; Accepted: 05 March 2018;
Published: 21 March 2018.
Edited by:
Isabel Sá-Correia, Instituto Superior Técnico, Universidade de Lisboa, PortugalReviewed by:
Amparo Querol, Consejo Superior de Investigaciones Científicas (CSIC), SpainJosé Manuel Guillamón, Consejo Superior de Investigaciones Científicas (CSIC), Spain
Copyright © 2018 Ortiz-Merino, Varela, Coughlan, Hoshida, Silveira, Wilde, Kuijpers, Geertman, Wolfe and Morrissey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John P. Morrissey, ai5tb3JyaXNzZXlAdWNjLmll
†These authors have contributed equally to this work.