 Anna Morgan1*
Anna Morgan1* Stefania Lenarduzzi2Stefania Cappellani2Vanna Pecile2Marcello Morgutti2
Stefania Lenarduzzi2Stefania Cappellani2Vanna Pecile2Marcello Morgutti2 Eva Orzan2
Eva Orzan2 Sara Ghiselli2Umberto Ambrosetti3Marco Brumat1Poornima Gajendrarao2Martina La Bianca2
Sara Ghiselli2Umberto Ambrosetti3Marco Brumat1Poornima Gajendrarao2Martina La Bianca2 Flavio Faletra2Enrico Grosso4Fabio Sirchia2Alberto Sensi5
Flavio Faletra2Enrico Grosso4Fabio Sirchia2Alberto Sensi5 Claudio Graziano6Marco Seri6Paolo Gasparini1,2
Claudio Graziano6Marco Seri6Paolo Gasparini1,2 Giorgia Girotto1,2
Giorgia Girotto1,2- 1Department of Medicine, Surgery and Health Sciences, University of Trieste, Trieste, Italy
- 2IRCCS Materno Infantile Burlo Garofolo, Trieste, Italy
- 3Audiologia, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, University of Milan, Milan, Italy
- 4Medical Genetics Unit, Azienda Ospedaliero-Universitaria Città della Salute e della Scienza di Torino, Turin, Italy
- 5Medical Genetics Unit, Department of Clinical Pathology, Azienda Unità Sanitaria Locale (AUSL) della Romagna, Cesena, Italy
- 6Unit of Medical Genetics, S. Orsola-Malpighi Hospital, Bologna, Italy
Hereditary hearing loss (HHL) is a common disorder characterized by a huge genetic heterogeneity. The definition of a correct molecular diagnosis is essential for proper genetic counseling, recurrence risk estimation, and therapeutic options. From 20 to 40% of patients carry mutations in GJB2 gene, thus, in more than half of cases it is necessary to look for causative variants in the other genes so far identified (~100). In this light, the use of next-generation sequencing technologies has proved to be the best solution for mutational screening, even though it is not always conclusive. Here we describe a combined approach, based on targeted re-sequencing (TRS) of 96 HHL genes followed by high-density SNP arrays, aimed at the identification of the molecular causes of non-syndromic HHL (NSHL). This strategy has been applied to study 103 Italian unrelated cases, negative for mutations in GJB2, and led to the characterization of 31% of them (i.e., 37% of familial and 26.3% of sporadic cases). In particular, TRS revealed TECTA and ACTG1 genes as major players in the Italian population. Furthermore, two de novo missense variants in ACTG1 have been identified and investigated through protein modeling and molecular dynamics simulations, confirming their likely pathogenic effect. Among the selected patients analyzed by SNP arrays (negative to TRS, or with a single variant in a recessive gene) a molecular diagnosis was reached in ~36% of cases, highlighting the importance to look for large insertions/deletions. Moreover, copy number variants analysis led to the identification of the first case of uniparental disomy involving LOXHD1 gene. Overall, taking into account the contribution of GJB2, plus the results from TRS and SNP arrays, it was possible to reach a molecular diagnosis in ~51% of NSHL cases. These data proved the usefulness of a combined approach for the analysis of NSHL and for the definition of the epidemiological picture of HHL in the Italian population.
Introduction
Hearing loss (HL) is a frequent disease affecting 1–3 in every 1,000 live births (Ječmenica et al., 2015). It is typically described based on its clinical presentation and can be classified by a number of different factors, such as the age of onset, severity, etiology, and pathobiology (Rehm and Morton, 1999; Dror and Avraham, 2009; Alford et al., 2014; Smith et al., 2014; Parker and Bitner-Glindzicz, 2015).
Overall, 60–70% of cases have a genetic etiology (Nishio et al., 2015) and can be further categorized as to whether the gene causes only hearing loss (non-syndromic or NSHL) or multiple clinical features (syndromic). NSHL accounts for the vast majority of hereditary HL cases and includes, according to the pattern of inheritance, autosomal recessive cases (~80%, labeled as “DFNB”), autosomal dominant (~20%, labeled as “DFNA”) X-linked or mitochondrial cases (<1%) (Stelma and Bhutta, 2014). In Italy, from 20 to 40% of cases are caused by mutations in GJB2 gene (Cama et al., 2009; Primignani et al., 2009; Salvago et al., 2014), making it the major player. However, as expected, considering the unique and complex structure of the inner ear, many other genes have been found to be involved in the hearing phenotype. To date, 158 NSHL loci (60 DFNA loci, 88 DFNB loci, 6 X-linked loci, 2 modifier loci, 1 Y-linked locus, and 1 locus for auditory neuropathy), and 95 genes (27 DFNA genes, 56 DFNB genes, 8 DFNA/DFNB genes, and 4 X-linked genes) have been reported as causative (Hereditary Hearing Loss Homepage; http://hereditaryhearingloss.org/). In this light, a combined approach based on an accurate clinical characterization and on different analytical technologies represents the most effective strategy for the identification of the molecular cause of NSHL, which is essential for proper genetic counseling, recurrence risk estimation, prognosis, and therapeutic options.
Here, we report the results obtained on a large cohort of unrelated patients (N = 103), negative for GJB2 mutations, applying a targeted re-sequencing panel of 96 HHL genes followed by SNP arrays in negative cases. This approach allowed: (1) the characterization of 31% of all NSHL cases, leading to an overall detection rate of ~51% (together with GJB2), (2) the identification of an extremely rare case of uniparental disomy (UPD) in HHL (i.e., the first one in LOXHD1 gene), (3) the demonstration of the importance of copy number variants (CNVs), and (4) the identification of 17 new alleles.
Materials and Methods
Ethical Statement
All patients provided written informed consent forms for both genetic counseling and molecular genetic testing prior to enrolment. Written informed consent was obtained from the next of kin on behalf of the minors/children involved in this study. The study was approved by the Institutional Review Board of IRCCS Burlo Garofolo, Trieste, Italy. All research was conducted according to the ethical standard as defined by the Helsinki Declaration.
Patients: Clinical Evaluation and Sample Collection
A total of 103 Italian unrelated NSHL patients have been recruited in the following centers (ENTs or Medical Genetics): Trieste (IRCCS Burlo Garofolo), Milano (IRCCS Cà Granda—Ospedale Maggiore Policlinico), Torino (A.O.U. Citta della Salute e della Scienza), Cesena (AUSL Romagna–Cesena), and Bologna (Policlinico S.Orsola-Malpighi). Inclusion criteria were: (1) absence of vestibular signs, (2) no obvious syndromic features, (3) absence of diabetes, cardiovascular diseases, visual problems, and neurological disorders, (4) absence of mutations in GJB2, GJB6, and MTRNR1 genes.
All participants underwent pure tone audiometric testing (PTA) or auditory brainstem response (ABR) in order to characterize the severity of HL according to the International guidelines described by Clark (1981).
Based on the pedigree structure, cases were divided into sporadic (N = 57) and familial (N = 46), the latter being classified as autosomal recessive (AR) (N = 4), autosomal dominant (AD) (N = 14), X-linked dominant (N = 1), and Y-linked (N = 1). The remaining 27 cases had an unclear pattern of inheritance.
At least three to four individuals per family have been analyzed by sequencing (both affected and healthy), and for the sporadic cases both the proband and the parents have been sequenced.
Targeted Resequencing (TRS) Hereditary Hearing Loss Panel
The 96 hearing loss genes panel described by Vozzi et al. has been used in this study (Vozzi et al., 2014). DNA libraries were constructed using Ion AmpliSeqTM Library Kit 2.0 (Thermo Fisher Scientific), and run on the Ion Torrent Personal Genome Machine (PGM) System (Thermo Fisher Scientific), according to manufacturer's instructions.
Data Analysis
TRS data were analyzed with Ion Torrent SuiteTM v4.0 software, set up with standardized parameters. Single Nucleotides Variations (SNVs) and Small Insertions and Deletions (INDELs) were collected into a standardized Variant Call Format (VCF) version 4.1 (Danecek et al., 2011). SNVs and INDELS were then annotated with ANNOVAR (Wang et al., 2010) using human genome build 19 (hg19) as the reference.
SNVs leading to synonymous amino acids substitutions not predicted as damaging and not affecting highly conserved residues were excluded, as well as SNVs/INDELs with quality score (QUAL) <20 and called in off-target regions.
A comparison between the identified genetic variants and data reported in NCBI dbSNP build150 (http://www.ncbi.nlm.nih.gov/SNP/) as well as in gnomAD (http://gnomad.broadinstitute.org/), and NHLBI Exome Sequencing Project (ESP) Exome Variant Server 1 led to the exclusion of those variants previously reported as polymorphism. In particular, a Minor Allele Frequency (MAF) cut off of 0.01 and one of 0.001 were used for recessive and dominant cases, respectively.
The pathogenicity of known genetic variants was evaluated using ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/), Deafness Variation Database (http://deafnessvariationdatabase.org/) as well as The Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php).
On the other hand, for novel variants, several in silico tools, such as PolyPhen-2 (Adzhubei et al., 2013), SIFT (Ng and Henikoff, 2003), MutationTaster (Schwarz et al., 2010), LRT (Chun and Fay, 2009), CADD score (Kircher et al., 2014) were used to evaluate the pathogenicity of the variant identified. Moreover, the evolutionary conservation of residues across species was evaluated by PhyloP (Pollard et al., 2010) and GERP (Cooper et al., 2005) scores.
Human Splicing Finder (HSF) version 2.4.1 (http://www.umd.be/HSF/) (Desmet et al., 2009) and Splice Site Prediction by Neural Network (NNSPLICE) version 9 (www.fruitfly.org) were used to predict the effect of the splice-site mutations.
We manually investigated the raw sequence reads for all the candidate pathogenic variants using the Integrative Genomics Viewer (IGV) (Thorvaldsdottir et al., 2013) with the purpose of excluding likely false positive calls due to read misalignment.
Finally, on a patient-by-patient basis, identified variants were discussed in the context of phenotypic data at interdisciplinary meetings and the most likely disease-causing SNVs/INDELs were analyzed by direct Sanger sequencing on a 3500 Dx Genetic Analyzer (Applied Biosystems), using ABI PRISM 3.1 Big Dye terminator chemistry (Applied Biosystems).
Sanger sequencing was employed also to perform the segregation analysis within the family.
SNP Arrays Analysis
SNP arrays analysis was performed using the Human OmniExpress Exome-8 Bead Chip (Illumina Inc., San Diego, CA, United States) containing 960,919 loci derived from phases I, II and III of the International HapMap project. The array includes over 274,000 functional exonic markers, delivering unparalleled coverage of putative functional exonic variant selected from 12,000 individual exome and whole-genome sequences. A total of 200 ng of gDNA (50 ng/μl) for each sample was processed according to Illumina's Infinium HD Assay Super protocol. Normalization of raw image intensity data, genotype clustering, and individual sample genotype calls were performed using Illumina's Genome Studio software v2011.1 (cnvpartition 3.2.0). The CNVs were mapped to the human reference genome hg19 and annotated with UCSC RefGene. Allele detection and genotype calling were performed with Genome Studio software, B allele frequencies (BAFs) and log R ratios were exported as text files for PennCNV analysis (Wang et al., 2007).
Protein Modeling and Molecular Dynamics Simulation
The protein structure for the modeling of two de novo variants of ACTG1 was collected from the protein data bank (PDB, http://www.rcsb.org/pdb/) (PDB ID: 5JLH) (von der Ecken et al., 2016). The wild-type (WT) structure was used to generate the mutational models p.(Thr66Ile) and p.(Arg183Gln) with PyMOL (www.pymol.org). The WT and mutant structures were used in GROMACS (Groningen Machine for Chemical Simulation V 4.5.4 package) (Van Der Spoel et al., 2005; Hess et al., 2008) for molecular dynamics simulations. The GROMOS96 (van Gunsteren., 1996) force field was applied to the structures. Energy minimization was carried out using the steepest descent algorithm with a tolerance of 2,000 kJ/mol/nm. The energy-minimized structures were used for the molecular dynamics simulations. The SPC3 (Berendsen et al., 1981) water model was used for solvation in a cubic box (0.8 nm) with periodic boundary conditions applied in all directions. The simulation systems were neutralized by adding counter ions. A twin range cut-off was used for long-range interactions: 0.8 nm for van der Waals interactions and 1.4 nm for electrostatic interactions. All bond lengths were constrained with the LINCS algorithm (Hess et al., 1997). The SETTLE algorithm (Miyamoto and Kollman, 1992) was applied to constrain the geometry of the water molecules. The energy minimized system was subjected to 100 ps equilibration followed by 10 ns production molecular dynamics simulations with a time-step of 2 fs at constant temperature (300 K), pressure (1 atm) and number of particles, without any position restraints (Berendsen et al., 1984). The collected trajectories were analyzed using the tools within GROMACS and the structures were analyzed using PyMOL. In addition, the representative structures were selected from molecular dynamics simulations for the structural analyses using the cluster analysis (Krishnamoorthy et al., 2018) and surface mapping calculations.
Results
Targeted Re-sequencing (TRS)
For the 96 genes under investigation an average 95% of the targeted region was covered at least 20X, with a 337 mean-depth. A total of 170 Mbp of raw sequence data was produced per individual. We identified an average of 468 genetic variants (SNVs/INDELs) per subject. After applying the filtering pipeline described in the “Methods” section, an average of 17 residual SNVs/INDELs for each subject were obtained.
A likely pathogenic variant was found in 27 cases (15 familial out of 46 and 12 sporadic out of 57) (Table 1) leading to an overall detection rate of ~33% of the familial cases and ~21% of the sporadic ones. A summary of the phenotypic and genetic data of the TRS positive cases is reported in Tables 1,2 respectively.
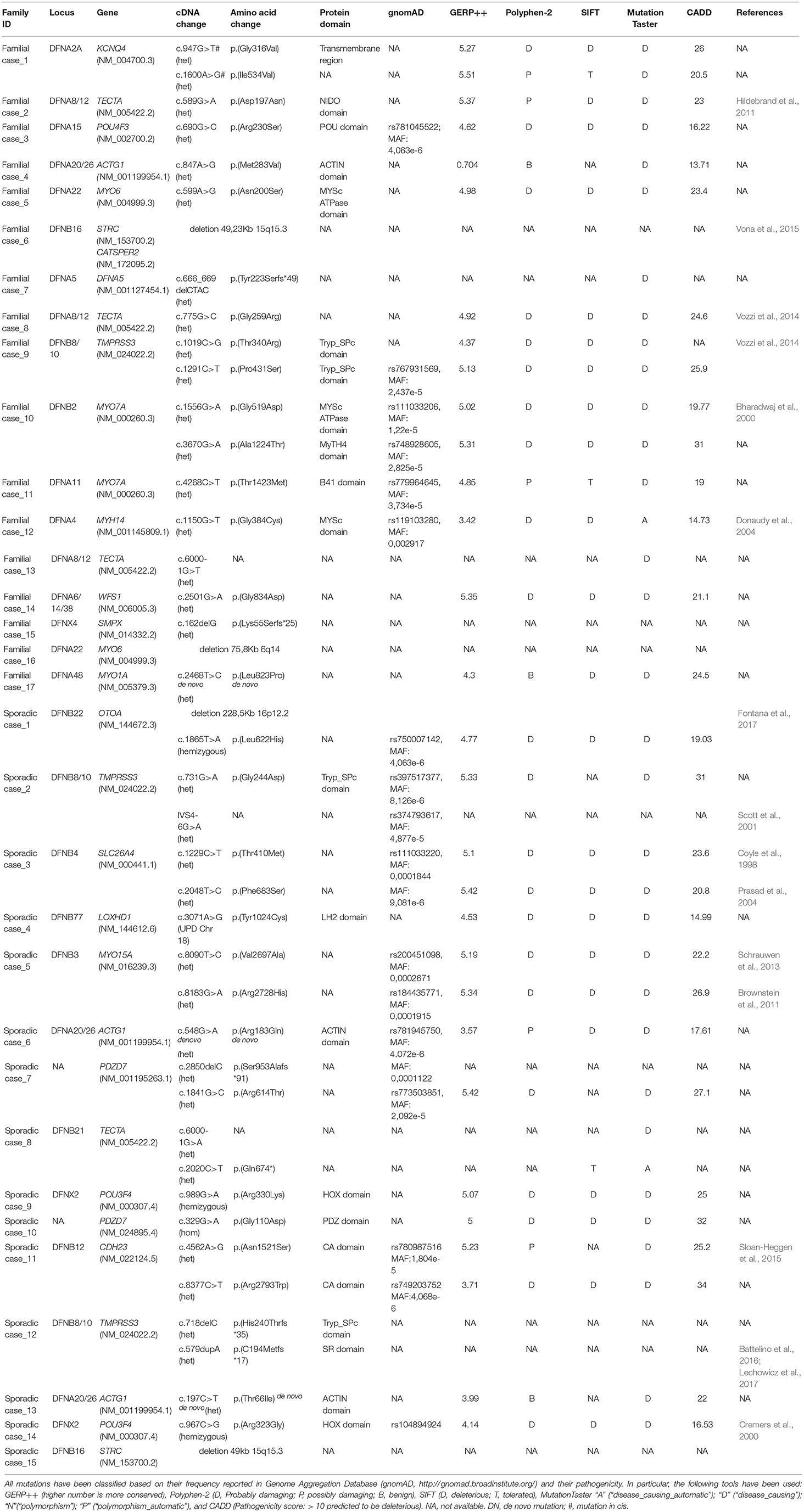
Table 1. List of likely causative variants identified by TRS.
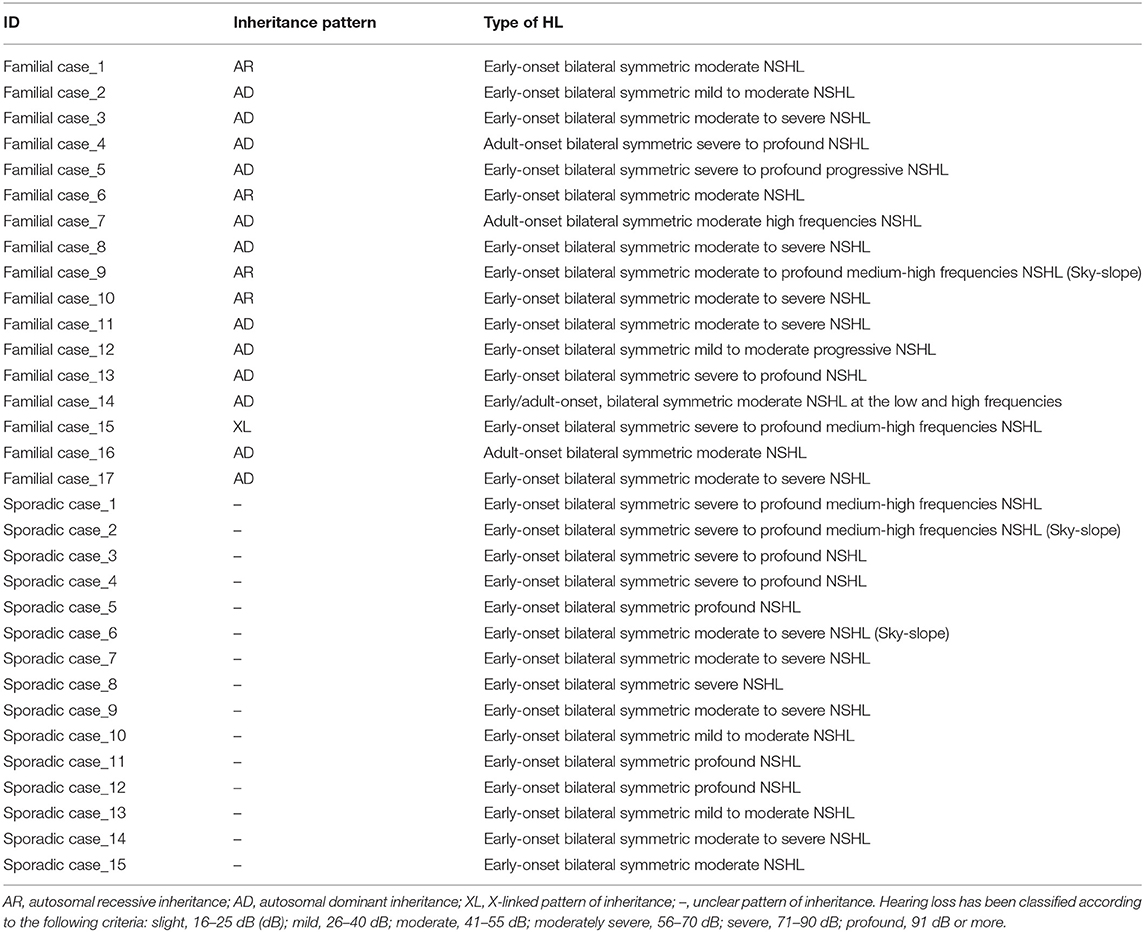
Table 2. Inheritance pattern and phenotypic data of all families and sporadic cases positive to TRS and/or SNP array.
Causative mutations in the following genes were identified: ACTG1, CDH23, DFNA5, KCNQ4, LOXHD1, MYH14, MYO1A, MYO6, MYO7A, MYO15A, OTOA, PDZD7, POU3F4, POU4F3, SLC26A4, SMPX, TECTA, TMPRSS3, and WFS1 (Figure 1).
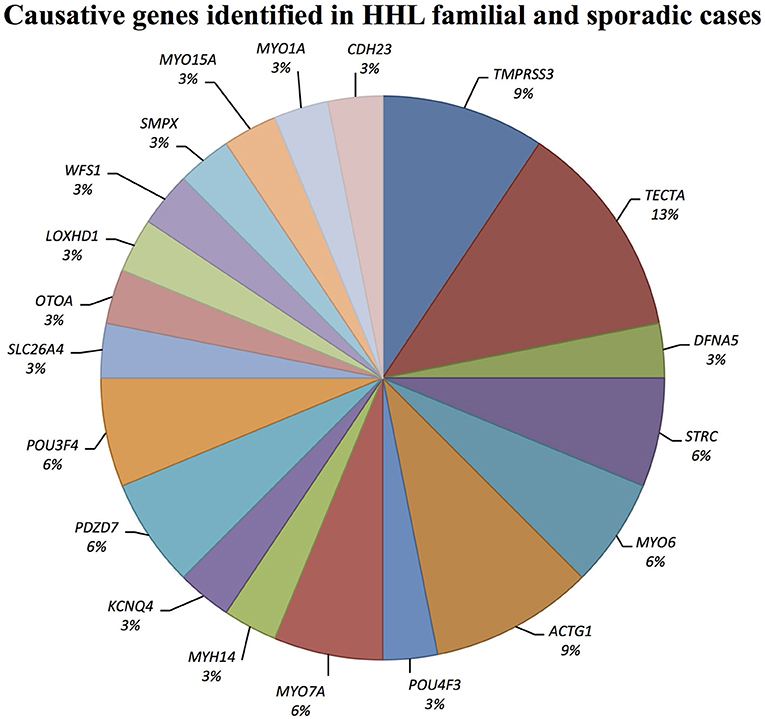
Figure 1. Causative genes identified in our cohort of Italian patients. The graph shows the distribution of the causative genes detected by TRS and SNP arrays.
A detailed summary of pathogenic variants identified in our screening is reported in Table 1. Overall, 23 already known mutations and 16 novel variants have been detected as here detailed: 30 missense (two de novo in ACTG1, and one in MYO1A), four frameshift deletions, one frameshift insertion, one non-sense, three variants affecting splice-sites.
Moreover, since 14 familiar and sporadic cases were carrying only one causative allele (i.e., an already known pathogenic mutation or a variant predicted as highly damaging) in an autosomal recessive gene (Table 3), we hypothesized that a second mutation, or a large CNV, has been missed in trans. In this light, these individuals have been further investigated with SNP arrays and the most interesting results are reported below.
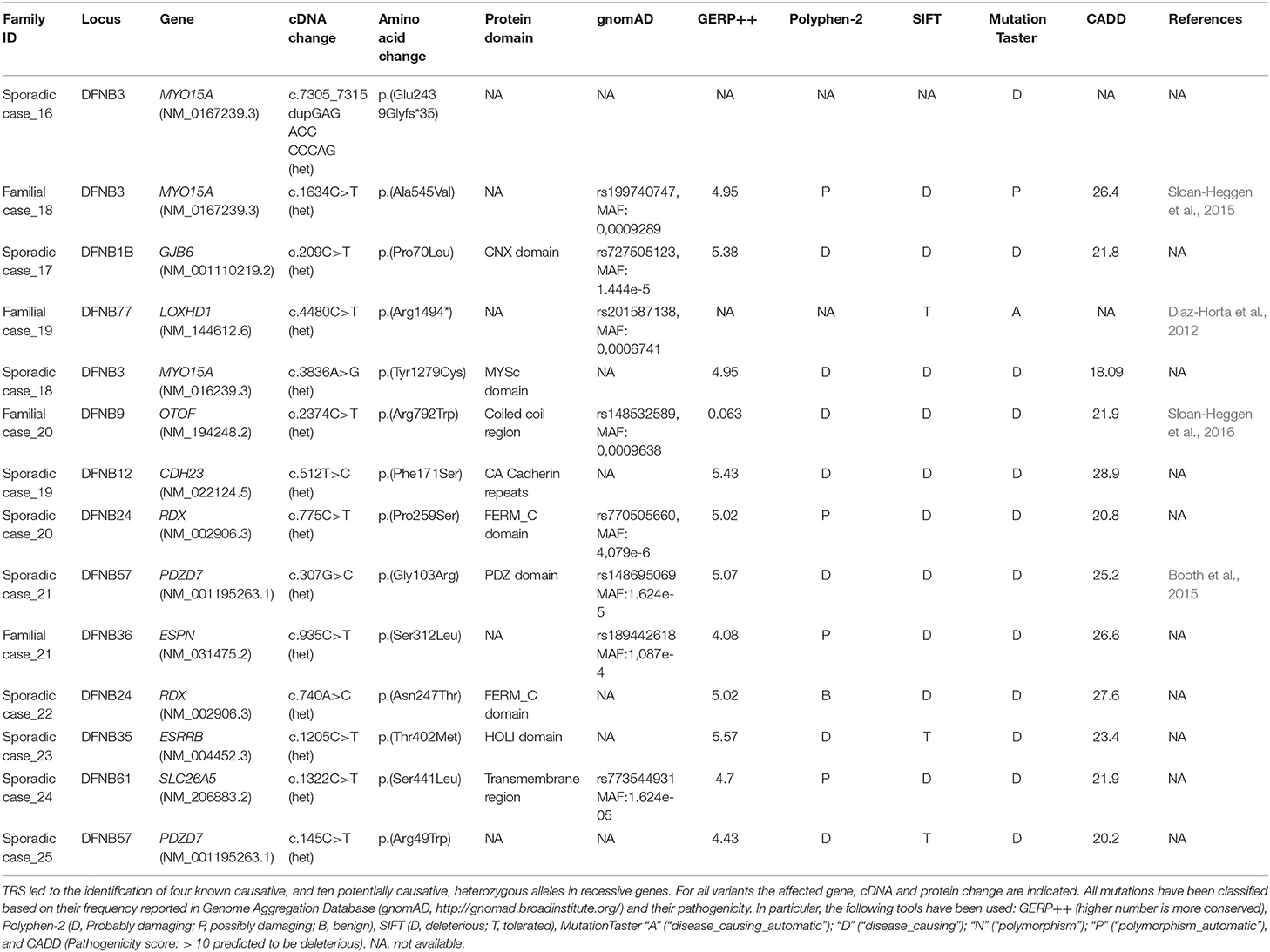
Table 3. List of likely damaging alleles identified by TRS in recessive genes.
TECTA
Combining previous data (Vozzi et al., 2014) with the results of the present study, TECTA (NM_005422.2) has been identified as the major NSHL gene in the Italian population (after GJB2), characterizing 13% of positive cases (with both autosomal dominant and recessive pattern of inheritance; Figure 2). Four different alleles in Familial case_8 [c.775G>C; p.(Gly259Arg)], Familial case_13 (c.6000-1G>T) and Sporadic case_8 [c.6000-1G>A; c.2020C>T; p.(Gln674*)] (Table 1), plus one already described [c.589G>A; p.(Asp197Asn)] in Vozzi et al. (Familial case_2) have been identified.
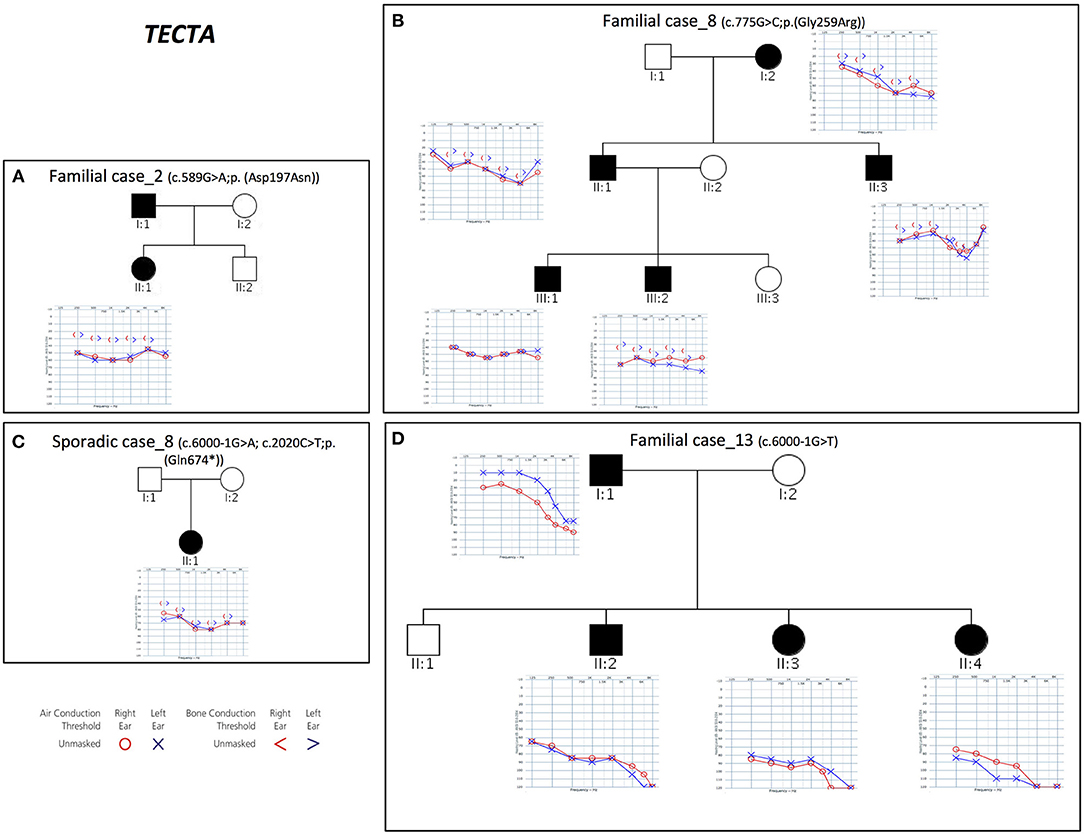
Figure 2. Pedigrees and audiograms of patients carrying variants in TECTA gene. (A–D) Phenotypic details of all patients carrying pathogenic variants in TECTA gene. Filled symbols represent affected individuals.
All patients show an early onset symmetrical NSHL, with different degrees of severity (Table 2 and Figure 2). The identified variants, predicted as damaging by several in silico predictor tools, affect functional domains of the α-tectorin (the entactin (ENT)-like, zonadhesin (ZA), and zona pellucida (ZP) domains) and involve highly conserved residues. Two of these changes [c.589G>A, p.(Asp197Asn); c.775G>C, p.(Gly259Arg)] were already known for causing NSHL, while the remaining three [c.6000-1G>T; c.6000-1G>A; c.2020C>T, p.(Gln674*)] have been identified in the present study.
Two different splicing variants affecting the same nucleotide have been identified in Familial case_13 and Sporadic case_8. Since TECTA gene is expressed at low levels in peripheral blood, it was not possible to test their effect on mRNA processing directly on the patients samples, however, according to the prediction of Human Splicing Finder software (http://www.umd.be/HSF3/) the c.6000-1G>T allele causes the total loss of the acceptor splice site, while the c.6000-1G>A leads to the loss of the acceptor splice site, together with the creation of a new site one nucleotide after.
ACTG1
Three alleles in ACTG1 (NM_001199954.1) have been identified: two are novel, [c.847A>G; p.(Met283Val) in Familial case_4, c.197C>T, p.(Thr66Ile) in Sporadic case_13] while one was already described [c.548G>A; p.(Arg183Gln) in Sporadic case_6]. All variants affect the actin-binding domain of ACTG1 protein. Patients display various degrees of severity of hearing loss (from mild-moderate, to severe-profound) and different age of onset (both adult-onset and early-onset), as expected from autosomal dominant NSHL (Table 2, Figure 3). Overall mutations in ACTG1 characterize 11% of all cases in our cohort (Table 1).
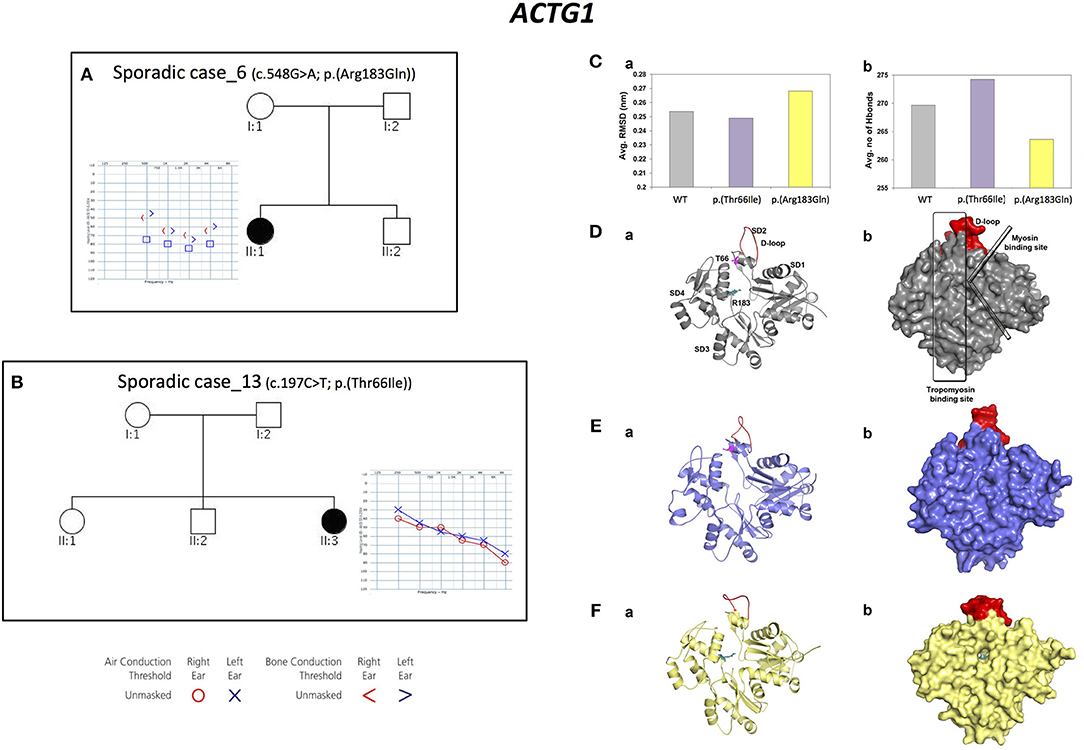
Figure 3. Phenotypic features of patients carrying variants in ACTG1 gene and molecular modeling of ACTG1 de novo variants. (A,B) Pedigrees and audiograms of patients carrying de novo variants in ACTG1. Filled symbols represent affected individuals. (C) Trajectory based analyses of the WT and mutants (a) structural stability and (b) intra-molecular interactions, (D–F) (a) Cartoon and (b) surface models for the representative structures from molecular dynamics simulations. Here, WT is labeled with structural features and the p.(Thr66Ile) and p.(Arg183Gln) mutations are labeled as sticks colored in pink and teal, respectively. The structurally and functionally important D-loop is shown in red.
Since ACTG1 de novo mutations are a rare cause of NSHL (Wang et al., 2018), protein modeling and molecular dynamics simulations of these two variants have been performed.
The structural stability of the WT and of the ACTG1 mutants was dynamically calculated using root mean square deviation (RMSD). Results showed that the deviation pattern was different for the mutants compared to the WT (Figure 3C-a); in particular, the p.(Thr66Ile) mutant deviated less than the WT, while the p.(Arg183Gln) mutant deviated more than the WT. This behavior correlates with the average number of hydrogen bonds, which are needed for protein's structural stability (Figure 3C-b). Here, p.(Thr66Ile) displayed an increased number of hydrogen bonds compared to both the WT and the p.(Arg183Gln) mutant and this explains the stable/rigid structural nature of p.(Thr66Ile). On the other hand, the p.(Arg183Gln) mutant showed a decrease in the number of hydrogen bonds that might result in an increased protein flexibility (Figure 3C-a).
The representative structures from molecular dynamics simulations showed that the mutational spots for both Thr66 and Arg183 are located in the binding region of the tropomyosin (Figure 3D). Results showed that the mutant p.(Thr66Ile), which is very close to the D-loop, causes a conformational change of the loop itself that alters the binding surface, while in the case of p.(Arg183Gln), both the tropomyosin-binding region and the D-loop are involved leading to a potential cavity on the binding surface (Figures 3E,F).
These results proved that both variants affect a key-binding region, introducing few critical changes in the structure.
POU4F3 and SMPX
Three families with an X-linked pattern of inheritance have been detected (Figure 4).
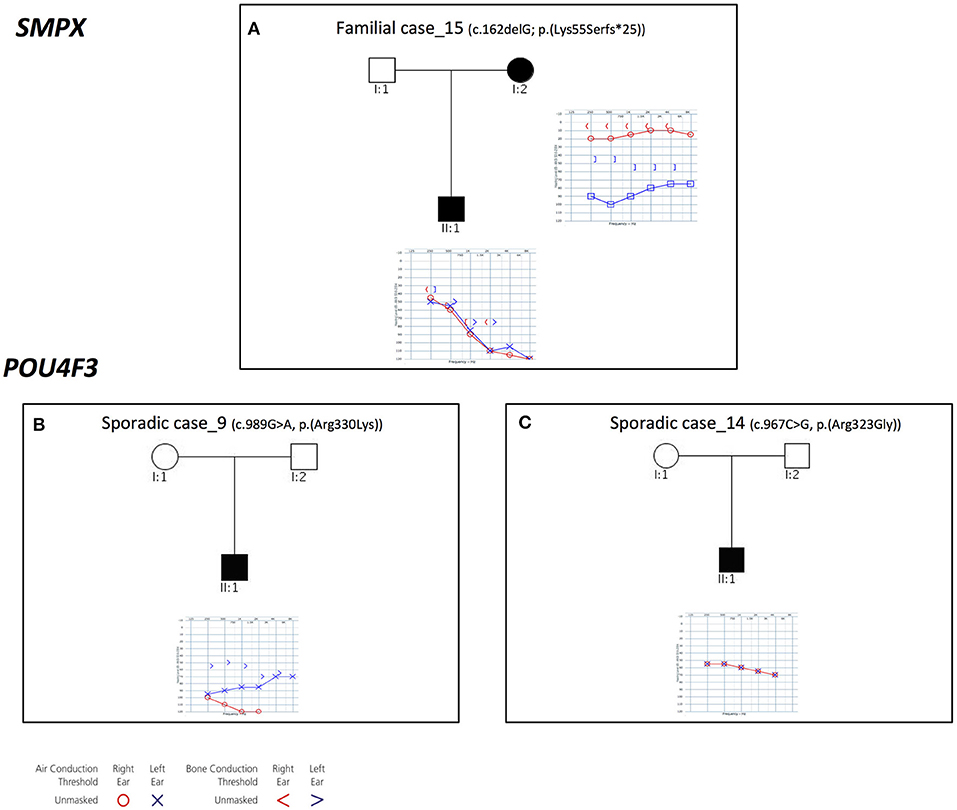
Figure 4. Pedigrees and audiograms of patients with X-linked HHL. (A) Patients of Familial case_15 carry a frameshift deletion in SMPX gene. (B-C) Sporadic case_9 and 14 carry two missense variant in POU4F3 gene. Filled symbols represent affected individuals.
Familial case_15 carries a novel frameshift deletion [c.162delG; p.(Lys55Serfs*25)] in SMPX gene (NM_014332.2), known for causing dominant X-linked NSHL (Table 1), The proband presents post-lingual bilateral symmetric severe to profound medium-high frequencies HL, while the mother shows a monolateral profound to severe HL (Figure 4A). As expected, no preferential X-inactivation has been detected in subject I:2.
POU4F3 (NM_000307.4), known for causing recessive X-linked HL, was found to be mutated in Sporadic Case_9 (c.989G>A, p.(Arg330Lys) and Sporadic Case_14 (c.967C>G, p.(Arg323Gly) (Table 1). Both variants affect the HOX domain of the protein and are predicted to be damaging. In particular, the c.967C>G, p.(Arg323Gly) allele, was already known to cause hearing loss (Cremers et al., 2000). Both patients display early-onset bilateral symmetric moderate to severe hearing loss, with perilymphatic gusher occurred during cochleostomy (Figures 4B,C).
SNP Arrays
Cases negative to TRS, or those showing a single variant in recessive genes, were analyzed with SNP arrays.
LOXHD1
In the present study we identified the second case of UPD ever described associated with HL, in a patient (Sporadic case_4) showing an early-onset bilateral symmetric severe to profound NSHL (Figure 5A). Briefly, TRS revealed a novel homozygous mutation in LOXHD1 (NM_144612.6) [c.3071A>G; p.(Tyr1024Cys)], apparently segregating only from the father. The variant was predicted as damaging by all in silico predictor tools and affected the LH2 domain of the protein. SNP array analysis identified one run of homozygosity bigger than 8 Mb in length, spanning LOXHD1 gene on chromosome 18. Analysis of informative SNPs in parental samples and their comparison with the patient's genotype confirmed the presence of a paternal UPD. Moreover, a deep analysis of SNPs on the whole chromosome 18 confirmed the presence of both the small isodisomy segment spanning the LOXHD1 gene plus the presence of heterodisomy on the remaining parts of Chr18 (Figure 5B).
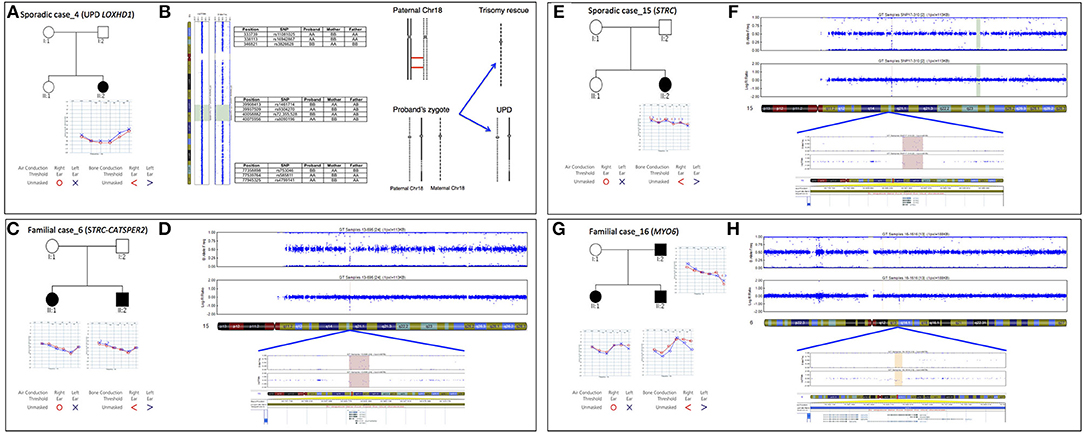
Figure 5. Pedigree, audiograms, and SNP array data of cases carrying pathogenic CNVs. (A) Pedigree and audiometric features of Sporadic case_4. Filled symbols represent affected individuals. (B) SNP array analysis of LOXHD1 gene: results of SNP array on the proband show a UPD with ROH region (reported in green) suggesting a recombination event originated in meiosis I. The log R ratio of the analysis is consistent with normal copy number. Some of the markers in both the telomeric regions 18p and 18q plus in the ROH region are also indicated. (C) Pedigree and audiometric features of Familial case_6. Filled symbols represent affected individuals. (D) SNP array analysis: the image shows a double deletion (CNV = 0) of 49.23 Kb in the q15.3 region of chromosome 15 (highlighted in red). The deletion includes STRC and CATSPER2 genes. (E) Pedigree and audiometric features of Sporadic case_15. Filled symbols represent affected individuals. (F) SNP array analysis: the graph shows a double deletion (CNV = 0) of 49 Kb in the q15.3 region of chromosome 15 (highlighted in red). Deletion includes STRC gene. (G) Pedigree and audiometric features of Familial case_16. Filled symbols represent affected individuals. (H) SNP arrays analysis: the image shows a deletion (CNV = 1) of 75.8 Kb in the q14.1 region of chromosome 6 (highlighted in orange). In the deletion region is possible to see a loss of heterozygosity in the B allele frequency and a lowering of the log ratio under the zero (about −0.66). This deletion includes MYO6 gene.
OTOA
In Sporadic case_1, showing early-onset bilateral symmetric severe to profound medium-high frequencies hearing loss, a homozygous missense variant [c.1865T>A; p.(Leu622His), rs750007142] in OTOA gene (NM_144672.3) was detected. The variant, predicted as damaging by all in silico predictor tools, apparently segregated only from the father, thus the clinical case was further investigated by SNP array. Data analysis led to the identification of a large (~228.5 Kb) heterozygous submicroscopic 16p12.2 deletion inherited from the mother (Fontana et al., 2017).
STRC and CATSPER2
In Familial Case_6 and Sporadic Case_15 two different deletion involving STRC gene (NM_153700.2) have been identified (Figures 5C–F). The first one belongs to an AR family composed of 4 members, 2 affected siblings (a 5-year old girl and a 3-year old boy) and their normal hearing parents. Both children showed a bilateral moderate symmetric SNHL characterized by a pre-lingual onset (Figure 5C). A 49.23 Kb deletion on chromosome 15 spanning through STRC and CATSPER2 (NM_172095.2) genes was identified (Figure 5D). In the second case, a 10 y.o. girl affected by early-onset bilateral symmetric moderate NSHL, with no familiarity for HL (Figure 5E), an homozygous deletion of 49 Kb involving only STRC gene has been detected (Figure 5F).
MYO6
In Familial Case_16 SNP arrays led to the identification of a novel heterozygous deletion affecting MYO6 gene, known for causing autosomal dominant NSHL. The family is composed of two affected siblings (37 and 35 y.o., respectively), the healthy mother (64 y.o.) and the affected father (80 y.o.). All affected individuals display adult onset bilateral moderate symmetric SNHL (Figure 5G) and carry a deletion of 75.8 Kb in MYO6 gene (Figure 5H).
Overall, the combination of an accurate clinical characterization, TRS and SNP arrays, led to the identification of the molecular cause of hearing loss in 31% of cases (37% of familial cases and 26.3% of sporadic cases). Moreover, among the 14 patients analyzed with SNP array (negative to TRS, or with a single variant in a recessive gene) we reached a molecular diagnosis in ~36% of cases (5 out of 14).
Discussion
Nowadays, the use of targeted re-sequencing in routine clinical diagnosis seems to be one of the most accurate approaches for the molecular diagnosis of highly heterogeneous genetic diseases, such as inherited deafness. Nevertheless, this approach has some limitations, not being able to accurately detect rearrangements such as deletions and duplications (i.e., CNVs) which might be involved in causing a significant proportion of genetic disorders (Yang et al., 2013). Moreover, recent studies have also suggested that runs of homozygosity (ROH) are much more frequent than previously recognized and, in some cases, can unravel UPD (Wang et al., 2015).
To overcome this limitation and to further increase the detection rate of HHL cases, we refined our strategy performing high-density SNP arrays in TRS-negative cases. Results confirmed that a multi-step integrated approach based on TRS followed by SNP arrays, is extremely powerful in advancing the molecular characterization of HHL. In particular, the largest study of Italian HHL patients so far carried out, demonstrates the importance of SNP arrays analysis in detecting the first case of UPD in LOXHD1 gene and confirming the importance of genomic rearrangements in the etiopathogenesis of hearing loss.
Combining TRS and SNP arrays, our strategy allowed to characterize 31% of the ~65% of cases negative to GJB2 mutations (leading to an overall detection rate -including GJB2– of ~51%). These results seem to be in agreement with previous data reported by Shearer and Smith (2015) and Sloan-Heggen et al. (2015) in which a detection rate of 39% (including GJB2 gene) in a much larger cohort is described. Moreover, results of the present study demonstrated a higher detection rate in familial cases (37%), mainly characterized by autosomal dominant inheritance, compared to sporadic cases (26.3%). Also in this case, these data were in agreement with those reported in the literature (71% for familial cases and 37% for sporadic cases) (Shearer et al., 2011; Sloan-Heggen et al., 2016).
Our study highlights the importance of TECTA as the second most frequently mutated gene in the Italian population, following GJB2. Patients affected by TECTA mutations display an early onset symmetrical NSHL, with different degrees of severity (from mild-moderate, to severe-profound). According to literature data, we identified different genotype-phenotype correlations depending on the inheritance pattern and on which of the functional domain of TECTA was affected (Hildebrand et al., 2011). Thus, as expected, Sporadic Case_8, affected by autosomal recessive NSHL, carried a splicing and a non-sense variant, and displayed a moderately severe to severe hearing phenotype (Asgharzade et al., 2017). As regards dominant families, the phenotype of the patients was much more variable and not always reflecting data of previous studies leading to new genotype-phenotype links to be further investigated (Choi et al., 2014).
Another gene frequently mutated in our cohort of patients turned out to be ACTG1. Two out of the three mutations identified in this gene were detected as “de novo.” So far, several mutations in ACTG1 gene have been described and analyzed by protein modeling (van Wijk et al., 2003; Rendtorff et al., 2006) although de novo ACTG1 variants seem to be a rare cause of NSHL (Wang et al., 2018). In this light, we further investigated their potential role by protein modeling and molecular dynamics simulations demonstrating a significant modification of two functional regions of the protein, the tropomyosin, and myosin binding sites and the D-loop. Considering that the binding of tropomyosin and myosin to actin is a key mechanism for regulating the normal function of the complex (Rayment et al., 1993; Behrmann et al., 2012) and that an alteration in actin filament regulation is an important factor in deafness caused by ACTG1 mutations, these de novo variants likely alter the protein activity, leading to deleterious effects (Lee et al., 2018).
Furthermore, another relevant finding of this study is the high prevalence of X-linked forms, which are expected to account for an inconsiderable part of the genetic forms (Smith et al., 2005) and that have been detected in three cases of both dominant and recessive X-linked NSHL, involving SMPX and POU3F4 genes. Among them, a careful attention should be directed to SMPX gene whose mutations have been recently reported to lead to a mild bilateral HL phenotype in females and a severe to profound early-onset HL in the affected males (Niu et al., 2017). Alternatively, as in the case of our family, the mother shows a monolateral severe to profound (sky-slope) HL while the proband displays a post-lingual bilateral symmetric severe to profound medium-high frequencies NSHL, as described in only one case worldwide (Weegerink et al., 2011). For this reason, during genetic counseling, it would be very important to carefully verify the monolateral clinical manifestations in affected females as an indicator of an X-linked form of HL.
Finally, our data highlight the huge importance of CNVs discovery in TRS negative cases, or in patients carrying only one mutated allele in recessive genes. In fact, among the 14 families analyzed by SNP arrays, a causative CNV was identified in 5 of them, explaining ~36% of cases.
In particular, this approach allowed the detection of a paternal UPD in chromosome 18, involving LOXHD1 gene. This represents the second example of UPD associated to NSHL, in addition to that one described in GJB2, the most common mutated HL gene. In fact, the only cases of UPD so far described as causative of hearing loss, affect GJB2 gene (Yan et al., 2007).
Furthermore, CNVs analysis confirmed the significant contribution of STRC deletions in hearing loss as recently described in other populations (Plevova et al., 2017). STRC deletions are reported in approximately 1% of mixed deafness populations (Francey et al., 2012; Hoppman et al., 2013), making it as a major contributor to congenital hearing impairment, and can cause autosomal recessive NSHL (Verpy et al., 2001) or Deafness-infertility syndrome (DIS) in males if the adjacent CATSPER2 gene is also involved in the deletion (Zhang et al., 2009). Considering that in Familial Case_6 the deletion also involved CATSPER2 gene, it will be important to evaluate future fertility problems in the male sibling (since now he is only 3 years old).
Overall, our results proved that the combination of an accurate clinical evaluation, TRS of known NSHL genes and SNP arrays can effectively enhance, in a cost-effective way, the genetic characterization of NSHL, leading to the identification of new mutations/CNVs and thus helping in the clinical management of patients.
Author Contributions
AM: samples quality control, data production and analysis, study design assessment; SL: data production and analysis; SC: Copy Number Variation (CNV) analysis; VP: Copy Number Variation (CNV) analysis; MM: data production and analysis; EO: samples recruitment, patients' clinical evaluation; SG: samples recruitment, patients' clinical evaluation; UA: samples recruitment, patients' clinical evaluation; MB: data analysis; PoG: protein modeling and molecular dynamics simulation; MB: data production; FF: patients' clinical evaluation; EG: samples recruitment, patients' clinical evaluation; FS: patients' clinical evaluation; AS: samples recruitment, patients' clinical evaluation; CG: samples recruitment, patients' clinical evaluation; MS: samples recruitment, patients' clinical evaluation; PaG: patients' clinical evaluation, data analysis, study design assessment; GG: data analysis, patients' clinical evaluation, study design assessment. All authors contributed in writing or revising the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This Research was supported by the Ministry of Health, IRCCS Burlo Garofolo RC-5x1000 to PaG and RBSI14AG8P (SIR2014) to GG. We gratefully acknowledge the all the families that participate to the study and Prof. Alfredo Brusco and Dr. Elisa Giorgio for the X-inactivation analysis in Familial case_15.
Footnotes
1. ^[Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA]. Available online at: http://evs.gs.washington.edu/EVS/
References
Adzhubei, I., Jordan, D. M., and Sunyaev, S. R. (2013). Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 76:7.20. doi: 10.1002/0471142905.hg0720s76
Alford, R. L., Arnos, K. S., Fox, M., Lin, J. W., Palmer, C. G., Pandya, A., et al. (2014). American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genet. Med. 16, 347–355. doi: 10.1038/gim.2014.2
Asgharzade, S., Tabatabaiefar, M. A., Modarressi, M. H., Ghahremani, M. H., Reiisi, S., Tahmasebi, P., et al. (2017). A novel TECTA mutation causes ARNSHL. Int. J. Pediatr. Otorhinolaryngol. 92, 88–93. doi: 10.1016/j.ijporl.2016.11.010
Battelino, S., Klancar, G., Kovac, J., Battelino, T., and Trebusak Podkrajsek, K. (2016). TMPRSS3 mutations in autosomal recessive nonsyndromic hearing loss. Eur. Arch. Otorhinolaryngol. 273, 1151–1154. doi: 10.1007/s00405-015-3671-0
Behrmann, E., Müller, M., Penczek, P. A., Mannherz, H. G., Manstein, D. J., and Raunser, S. (2012). Structure of the Rigor actin-tropomyosin-myosin complex. Cell 150, 327–338. doi: 10.1016/j.cell.2012.05.037
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A., and Haak, J. R. (1984). Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. doi: 10.1063/1.448118
Berendsen, H. J. C., Postma, J. P. M., Van Gunsteren, W. F., and Hermans, J. (1981). “Interaction models for water in relation to protein hydration,” in Intermolecular Forces. The Jerusalem Symposia on Quantum Chemistry and Biochemistry, ed B. Pullman (Dordrecht: Springer), 331–342.
Bharadwaj, A. K., Kasztejna, J. P., Huq, S., Berson, E. L., and Dryja, T. P. (2000). Evaluation of the myosin VIIA gene and visual function in patients with usher syndrome type I. Exp. Eye Res. 71, 173–181. doi: 10.1006/exer.2000.0863
Booth, K. T., Azaiez, H., Kahrizi, K., Simpson, A. C., Tollefson, W. T. A., Sloan, C. M., et al. (2015). PDZD7 and hearing loss: more than just a modifier. Am. J. Med. Genet. Part A 167, 2957–2965. doi: 10.1002/ajmg.a.37274
Brownstein, Z., Friedman, L. M., Shahin, H., Oron-Karni, V., Kol, N., Rayyan, A., et al. (2011). Targeted genomic capture and massively parallel sequencing to identify genes for hereditary hearing loss in middle eastern families. Genome Biol. 12:R89. doi: 10.1186/gb-2011-12-9-r89
Cama, E., Melchionda, S., Palladino, T., Carella, M., Santarelli, R., Genovese, E., et al. (2009). Hearing loss features in GJB2 biallelic mutations and GJB2/GJB6 digenic inheritance in a large Italian cohort. Int. J. Audiol. 48, 12–17. doi: 10.1080/14992020802400654
Choi, B. Y., Kim, J., Chung, J., Kim, A. R., Mun, S. J., Kang, S., et al. (2014). Whole-exome sequencing identifies a novel genotype-phenotype correlation in the entactin domain of the known deafness gene TECTA. PLoS ONE 9:e97040. doi: 10.1371/journal.pone.0097040
Chun, S., and Fay, J. C. (2009). Identification of deleterious mutations within three human genomes. Genome Res. 19, 1553–1561. doi: 10.1101/gr.092619.109
Clark, J. G. (1981). Uses and Abuses of Hearing Loss Classification. ASHA 23, 493–500. Available online at: http://www.ncbi.nlm.nih.gov/pubmed/7052898 (Accessed December 9, 2016).
Cooper, G. M., Stone, E. A., Asimenos, G. N. I. S. C., Comparative Sequencing Program, Green, E. D., Batzoglou, S., et al. (2005). Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 15, 901–913. doi: 10.1101/gr.3577405
Coyle, B., Reardon, W., Herbrick, J. A., Tsui, L. C., Gausden, E., Lee, J., et al. (1998). Molecular analysis of the PDS gene in Pendred syndrome. Hum. Mol. Genet. 7, 1105–12. doi: 10.1093/hmg/7.7.1105
Cremers, F. P., Cremers, C. W., and Ropers, H. H. (2000). The ins and outs of X-linked deafness type 3. Adv. Otorhinolaryngol. 56, 184–95. doi: 10.1159/000059101
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Desmet, F.-O., Hamroun, D., Lalande, M., Collod-Beroud, G., Claustres, M., and Beroud, C. (2009). Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37:e67. doi: 10.1093/nar/gkp215
Diaz-Horta, O., Duman, D., Foster, J., Sirmac,i, A., Gonzalez, M., Mahdieh, N., et al. (2012). Whole-exome sequencing efficiently detects rare mutations in autosomal recessive non-syndromic hearing loss. PLoS ONE 7:e50628. doi: 10.1371/journal.pone.0050628
Donaudy, F., Snoeckx, R., Pfister, M., Zenner, H.-P., Blin, N., Di Stazio, M., et al. (2004). Nonmuscle myosin heavy-chain gene MYH14 Is expressed in cochlea and mutated in patients affected by autosomal dominant hearing impairment (DFNA4). Am. J. Hum. Genet. 74, 770–776. doi: 10.1086/383285
Dror, A. A., and Avraham, K. B. (2009). Hearing loss: mechanisms revealed by genetics and cell biology. Annu. Rev. Genet. 43, 411–437. doi: 10.1146/annurev-genet-102108-134135
Fontana, P., Morgutti, M., Pecile, V., Lenarduzzi, S., Cappellani, S., Falco, M., et al. (2017). A novel OTOA mutation in an Italian family with hearing loss. Gene Rep. 9, 111–114. doi: 10.1016/j.genrep.2017.10.002
Francey, L. J., Conlin, L. K., Kadesch, H. E., Clark, D., Berrodin, D., Sun, Y., et al. (2012). Genome-wide SNP genotyping identifies the Stereocilin (STRC) gene as a major contributor to pediatric bilateral sensorineural hearing impairment. Am. J. Med. Genet. A 158A, 298–308. doi: 10.1002/ajmg.a.34391
Hess, B., Bekker, H., Berendsen, H. J. C., and Fraaije, J. G. E. M. (1997). 3 LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H
Hess, B., Kutzner, C., Van Der Spoel, D., and Lindahl, E. (2008). GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447. doi: 10.1021/ct700301q
Hildebrand, M. S., Morín, M., Meyer, N. C., Mayo, F., Modamio-Hoybjor, S., Mencía, A., et al. (2011). DFNA8/12 caused by TECTA mutations is the most identified subtype of non-syndromic autosomal dominant hearing loss. Hum. Mutat. 32, 825–834. doi: 10.1002/humu.21512
Hoppman, N., Aypar, U., Brodersen, P., Brown, N., Wilson, J., Babovic-Vuksanovic, D., et al. (2013). Genetic testing for hearing loss in the United States should include deletion/duplication analysis for the deafness/infertility locus at 15q15.3. Mol. Cytogenet. 6:19. doi: 10.1186/1755-8166-6-19
Ječmenica, J., Bajec-Opančina, A., and Ječmenica, D. (2015). Genetic hearing impairment. Childs. Nerv. Syst. 31, 515–519. doi: 10.1007/s00381-015-2628-3
Kircher, M., Witten, D. M., Jain, P., O'Roak, B. J., Cooper, G. M., and Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315. doi: 10.1038/ng.2892
Krishnamoorthy, N., Tseng, Y., Gajendrarao, P., Sarathchandra, P., McCormack, A., Carubelli, I., et al. (2018). A strategy to enhance secretion of extracellular matrix components by stem cells: relevance to tissue engineering. Tissue Eng. Part A 24, 145–156. doi: 10.1089/ten.tea.2017.0060
Lechowicz, U., Gambin, T., Pollak, A., Podgorska, A., Stawinski, P., Franke, A., et al. (2017). Iterative Sequencing And Variant Screening (ISVS) as a novel pathogenic mutations search strategy - application for TMPRSS3 mutations screen. Sci. Rep. 7:2543. doi: 10.1038/s41598-017-02315-w
Lee, C. G., Jang, J., and Jin, H.-S. (2018). A novel missense mutation in the ACTG1 gene in a family with congenital autosomal dominant deafness: a case report. Mol. Med. Rep. 17, 7611–7617. doi: 10.3892/mmr.2018.8837
Miyamoto, S., and Kollman, P. A. (1992). Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962. doi: 10.1002/jcc.540130805
Ng, P. C., and Henikoff, S. (2003). SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–4. doi: 10.1093/nar/gkg509
Nishio, S. -Y, Hattori, M., Moteki, H., Tsukada, K., Miyagawa, M., Naito, T., et al. (2015). Gene expression profiles of the cochlea and vestibular endorgans: localization and function of genes causing deafness. Ann. Otol. Rhinol. Laryngol. 124, 6S−48S. doi: 10.1177/0003489415575549
Niu, Z., Feng, Y., Mei, L., Sun, J., Wang, X., Wang, J., et al. (2017). A novel frameshift mutation of SMPX causes a rare form of X-linked nonsyndromic hearing loss in a Chinese family. PLoS One 12:e0178384. doi: 10.1371/journal.pone.0178384
Parker, M., and Bitner-Glindzicz, M. (2015). Genetic investigations in childhood deafness. Arch. Dis. Child. 100, 271–278. doi: 10.1136/archdischild-2014-306099
Plevova, P., Paprskarova, M., Tvrda, P., Turska, P., Slavkovsky, R., and Mrazkova, E. (2017). STRC deletion is a frequent cause of slight to moderate congenital hearing impairment in the Czech Republic. Otol. Neurotol. 38, e393–e400. doi: 10.1097/MAO.0000000000001571
Pollard, K. S., Hubisz, M. J., Rosenbloom, K. R., and Siepel, A. (2010). Detection of non-neutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121. doi: 10.1101/gr.097857.109
Prasad, S., Kölln, K. A., Cucci, R. A., Trembath, R. C., Camp, G., and Van Smith, R. J. H. (2004). Pendred syndrome and DFNB4-mutation screening of SLC26A4 by denaturing high-performance liquid chromatography and the identification of eleven novel mutations. Am. J. Med. Genet. Part A 124A, 1–9. doi: 10.1002/ajmg.a.20272
Primignani, P., Trotta, L., Castorina, P., Lalatta, F., Sironi, F., Radaelli, C., et al. (2009). Analysis of the GJB2 and GJB6 genes in Italian patients with non-syndromic hearing loss: frequencies, novel mutations, genotypes, and degree of hearing loss. Genet. Test. Mol. Biomarkers 13, 209–217. doi: 10.1089/gtmb.2008.0086
Rayment, I., Holden, H. M., Whittaker, M., Yohn, C. B., Lorenz, M., Holmes, K. C., et al. (1993). Structure of the actin-myosin complex and its implications for muscle contraction. Science 261, 58–65. doi: 10.1126/science.8316858
Rehm, H. L., and Morton, C. C. (1999). A new age in the genetics of deafness. Genet. Med. 1, 295–302; quiz 303. doi: 10.1097/00125817-199909000-00009
Rendtorff, N. D., Zhu, M., Fagerheim, T., Antal, T. L., Jones, M., Teslovich, T. M., et al. (2006). A novel missense mutation in ACTG1 causes dominant deafness in a Norwegian DFNA20/26 family, but ACTG1 mutations are not frequent among families with hereditary hearing impairment. Eur. J. Hum. Genet. 14, 1097–1105. doi: 10.1038/sj.ejhg.5201670
Salvago, P., Martines, E., La Mattina, E., Mucia, M., Sammarco, P., Sireci, F., et al. (2014). Distribution and phenotype of GJB2 mutations in 102 Sicilian patients with congenital non-syndromic sensorineural hearing loss. Int. J. Audiol. 53, 558–563. doi: 10.3109/14992027.2014.905717
Schrauwen, I., Sommen, M., Corneveaux, J. J., Reiman, R. A., Hackett, N. J., Claes, C., et al. (2013). A sensitive and specific diagnostic test for hearing loss using a microdroplet PCR-based approach and next generation sequencing. Am. J. Med. Genet. Part A 161, 145–152. doi: 10.1002/ajmg.a.35737
Schwarz, J. M., Rödelsperger, C., Schuelke, M., and Seelow, D. (2010). MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576. doi: 10.1038/nmeth0810-575
Scott, H. S., Kudoh, J., Wattenhofer, M., Shibuya, K., Berry, A., Chrast, R., et al. (2001). Insertion of β-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat. Genet. 27, 59–63. doi: 10.1038/83768
Shearer, A. E., Hildebrand, M. S., Sloan, C. M., and Smith, R. J. H. (2011). Deafness in the genomics era. Hear. Res. 282, 1–9. doi: 10.1016/j.heares.2011.10.001
Shearer, A. E., and Smith, R. J. H. (2015). Massively parallel sequencing for genetic diagnosis of hearing loss: the new standard of care. Otolaryngol. Head. Neck Surg. 153, 175–182. doi: 10.1177/0194599815591156
Sloan-Heggen, C. M., Babanejad, M., Beheshtian, M., Simpson, A. C., Booth, K. T., Ardalani, F., et al. (2015). Characterising the spectrum of autosomal recessive hereditary hearing loss in Iran. J. Med. Genet. 52, 823–829. doi: 10.1136/jmedgenet-2015-103389
Sloan-Heggen, C. M., Bierer, A. O., Shearer, A. E., Kolbe, D. L., Nishimura, C. J., Frees, K. L., et al. (2016). Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 135, 441–450. doi: 10.1007/s00439-016-1648-8
Smith, R. J., Bale, J. F., and White, K. R. (2005). Sensorineural hearing loss in children. Lancet 365, 879–890. doi: 10.1016/S0140-6736(05)71047-3
Smith, R. J., Shearer, A. E., Hildebrand, M. S., and Van Camp, G. (2014). Deafness and Hereditary Hearing Loss Overview. Available online at: http://www.ncbi.nlm.nih.gov/books/NBK1434/ (Accessed December 21, 2014).
Stelma, F., and Bhutta, M. F. (2014). Non-syndromic hereditary sensorineural hearing loss: review of the genes involved. J. Laryngol. Otol. 128, 13–21. doi: 10.1017/S0022215113003265
Thorvaldsdottir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192. doi: 10.1093/bib/bbs017
Van Der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A. E., and Berendsen, H. J. C. (2005). GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718. doi: 10.1002/jcc.20291
van Gunsteren, W. F. (1996). Biomolecular Simulation : the GROMOS96 Manual und User Guide. Vdf, Hochschulverlag an der ETH Available online at: https://books.google.it/books/about/Biomolecular_Simulation.html?id=0TbJAAAACAAJ&redir_esc=y (Accessed April 24, 2018).
van Wijk, E., Krieger, E., Kemperman, M. H., De Leenheer, E. M. R., Huygen, P. L. M., Cremers, C. W. R. J., et al. (2003). A mutation in the gamma actin 1 (ACTG1) gene causes autosomal dominant hearing loss (DFNA20/26). J. Med. Genet. 40, 879–84. doi: 10.1136/jmg.40.12.879
Verpy, E., Masmoudi, S., Zwaenepoel, I., Leibovici, M., Hutchin, T. P., Del Castillo, I., et al. (2001). Mutations in a new gene encoding a protein of the hair bundle cause non-syndromic deafness at the DFNB16 locus. Nat. Genet. 29, 345–349. doi: 10.1038/ng726
von der Ecken, J., Heissler, S. M., Pathan-Chhatbar, S., Manstein, D. J., and Raunser, S. (2016). Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature 534, 724–728. doi: 10.1038/nature18295
Vona, B., Hofrichter, M. A. H., Neuner, C., Schröder, J., Gehrig, A., Hennermann, J. B., et al. (2015). DFNB16 is a frequent cause of congenital hearing impairment: implementation of STRC mutation analysis in routine diagnostics. Clin. Genet. 87, 49–55. doi: 10.1111/cge.12332
Vozzi, D., Morgan, A., Vuckovic, D., D'Eustacchio, A., Abdulhadi, K., Rubinato, E., et al. (2014). Hereditary hearing loss: a 96 gene targeted sequencing protocol reveals novel alleles in a series of Italian and Qatari patients. Gene 542, 209–216. doi: 10.1016/j.gene.2014.03.033
Wang, H., Guan, J., Lan, L., Yu, L., Xie, L., Liu, X., et al. (2018). A novel de novo mutation of ACTG1 in two sporadic non-syndromic hearing loss cases. Sci. China. Life Sci. 61, 729–732. doi: 10.1007/s11427-017-9165-2
Wang, J.-C., Ross, L., Mahon, L. W., Owen, R., Hemmat, M., Wang, B. T., et al. (2015). Regions of homozygosity identified by oligonucleotide SNP arrays: evaluating the incidence and clinical utility. Eur. J. Hum. Genet. 23, 663–671. doi: 10.1038/ejhg.2014.153
Wang, K., Li, M., Hadley, D., Liu, R., Glessner, J., Grant, S. F. A., et al. (2007). PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 17, 1665–1674. doi: 10.1101/gr.6861907
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164–e164. doi: 10.1093/nar/gkq603
Weegerink, N. J. D., Huygen, P. L. M., Schraders, M., Kremer, H., Pennings, R. J. E., and Kunst, H. P. M. (2011). Variable degrees of hearing impairment in a Dutch DFNX4 (DFN6) family. Hear. Res. 282, 167–177. doi: 10.1016/j.heares.2011.08.010
Yan, D., Ouyang, X. M., Angeli, S. I., Du, L. L., and Liu, X. Z. (2007). Paternal uniparental disomy of chromosome 13 causing homozygous 35delG mutation of theGJB2 gene and hearing loss. Am. J. Med. Genet. Part A 143A, 385–386. doi: 10.1002/ajmg.a.31553
Yang, T.-L., Guo, Y., Papasian, C. J., and Deng, H.-W. (2013). “Copy Number Variation,” in Genetics of Bone Biology and Skeletal Disease, eds R. Thakker, M. Whyte, and J. Eisman (Elsevier), 123–132.
Keywords: targeted re-sequencing, SNP arrays, hereditary hearing loss, italian families, molecular diagnosis
Citation: Morgan A, Lenarduzzi S, Cappellani S, Pecile V, Morgutti M, Orzan E, Ghiselli S, Ambrosetti U, Brumat M, Gajendrarao P, La Bianca M, Faletra F, Grosso E, Sirchia F, Sensi A, Graziano C, Seri M, Gasparini P and Girotto G (2018) Genomic Studies in a Large Cohort of Hearing Impaired Italian Patients Revealed Several New Alleles, a Rare Case of Uniparental Disomy (UPD) and the Importance to Search for Copy Number Variations. Front. Genet. 9:681. doi: 10.3389/fgene.2018.00681
Received: 03 May 2018; Accepted: 07 December 2018;
Published: 21 December 2018.
Edited by:
Haranatha R. Potteti, University of Illinois at Chicago, United StatesReviewed by:
Bruna De Felice, Università degli Studi della Campania Luigi Vanvitelli Caserta, ItalyNoor Ahmad Shaik, King Abdulaziz University, Saudi Arabia
Copyright © 2018 Morgan, Lenarduzzi, Cappellani, Pecile, Morgutti, Orzan, Ghiselli, Ambrosetti, Brumat, Gajendrarao, La Bianca, Faletra, Grosso, Sirchia, Sensi, Graziano, Seri, Gasparini and Girotto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Morgan, YW5uYS5tb3JnYW5AYnVybG8udHJpZXN0ZS5pdA==